

BACKGROUND
Inborn errors of immunity (IEIs) can hide in plain sight among patients with a lifelong history of allergic disorders, such as asthma. Especially in cases of mild antibody deficiency and immune dysregulation, in the absence of definitive IEI diagnosis, the smoldering disease process may go unnoticed and lead to organ damage (eg, bronchiectasis).1,2 As routine evaluation of several hundred genes linked to IEIs becomes increasingly attainable and affordable, specific diagnoses can be promptly elucidated, allowing early initiation of therapies best suited for the underlying disease. The success of this process depends on close collaboration between the community and academic specialists, as demonstrated by this case. Our patient was initially followed for moderate persistent asthma, specific antibody deficiency, lymphopenia, and clinical signs of mild immune dysregulation. This report will review the pathway to diagnosis of partial recombination activating gene 1 (RAG1) deficiency (pRD), which not only warranted immunoglobulin replacement therapy but also opened the door for consideration of hematopoietic cell transplantation (HCT).
CASE REPORT
A 34-year-old woman with a history of moderate persistent asthma and frequent upper respiratory tract infections was evaluated by a pulmonologist after developing, at the age of 32 years, multilobar pneumonia causing respiratory failure, requiring mechanical ventilation, and demonstrating bilateral bronchiectasis, confirmed by computed tomography (Figure 1). During her hospitalization, extensive infectious evaluation including bronchoalveolar lavage fluid staining (eg, gram, gomori methenamine silver, and acid-fast bacteria) and bronchoalveolar lavage cultures did not reveal a pathogen. On further inquiry of her medical history, she reported episodes of purpura and bruising as a child that had not been fully evaluated, long-standing history of alopecia areata and vitiligo since puberty, and a recent diagnosis of infertility. The patient had short stature, which was considered to be familial; however, she denied any history of stunting in childhood, recurrent episodes of diarrhea, or poor weight gain. No occupation or hobbies associated with an increased risk of fungal exposure were reported. The patient was a lifetime nonsmoker.
FIGURE 1.
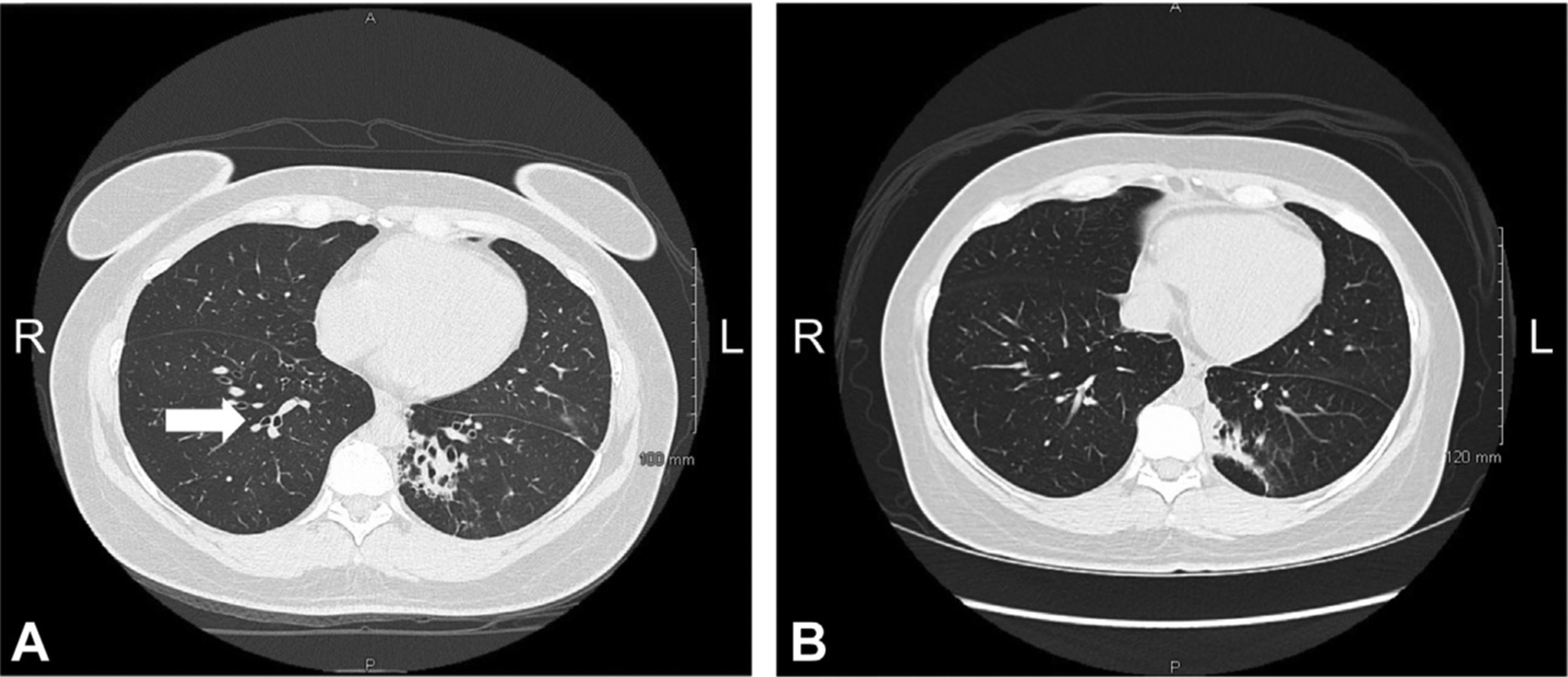
High-resolution computed tomography of the chest. (A) Axial view at initial presentation showing bilateral lower lobe central bronchiectasis and left lower lobe consolidation. Arrow: Signet-ring sign. (B) Axial view at 2-year outpatient follow-up showing medial left basilar wedge-shaped consolidation with central bronchiectasis and bronchiolectasis and associated calcified granuloma.
Physical examination revealed universal hair thinning, with areas of patchy alopecia on the occipital and parietal scalp (Figure 2, A). Several depigmented skin patches on the face, neck, trunk, and upper and lower extremities (especially noticeable on the dorsum of her hands and medial aspect of both thighs) were present (Figure 2, B). She had bilateral scant wheezing. No lymphadenopathies or hepatosplenomegaly were appreciated. Her pulmonary function testing revealed an obstructive pattern (FEV1/forced vital capacity: 45%) with concomitant decreased FEV1 and forced vital capacity (32% and 70% of predicted, respectively). Laboratory evaluation revealed normal immunoglobulin levels (IgG, IgA, IgM) with decreased IgG subclasses (IgG2 and IgG4), normal alpha-1 antitrypsin level, and undetectable IgE, arguing against allergic broncho-pulmonary aspergillosis (Table I). Subsequent interventions focused on pulmonary hygiene and clearance. Because of concern for a possible allergic or immunologic process, she was also referred to a local allergist.
FIGURE 2.
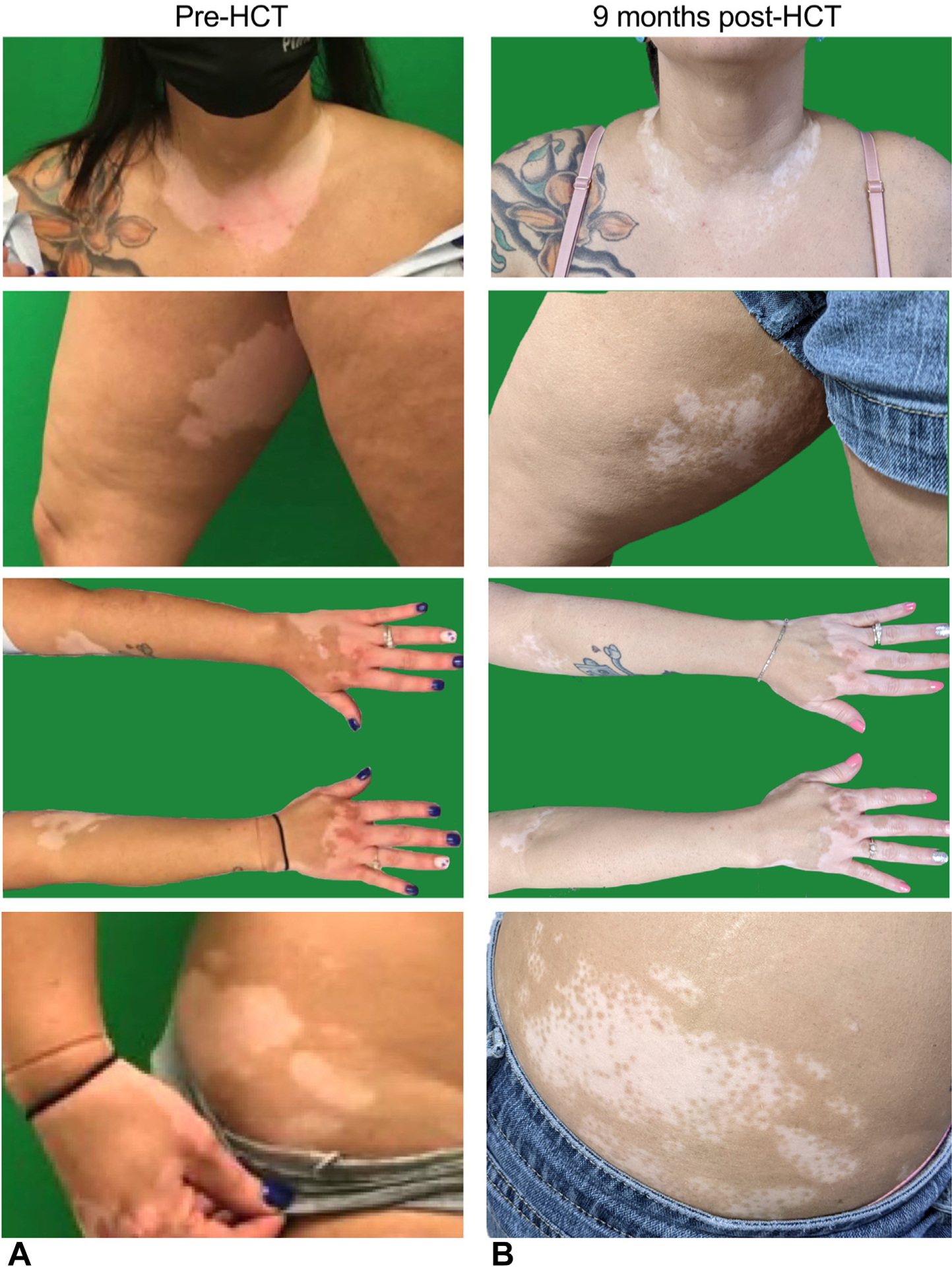
Changes in presentation of vitiligo. Vitiligo presentation (A) before HCT and (B) 9 months after HCT on the neck, thigh, and forearms, and at the level of the iliac crest.
TABLE I.
Immunologic phenotyping laboratory findings before and 1 y and 18 mo after HCT
| Parameter evaluated | Pre–allogenic HCT | Post–allogenic HCT (1 y) | Post–allogenic HCT (18 mo) | Normal range |
|---|---|---|---|---|
| Lymphocyte subsets (103 cells/μL) | ||||
| WBC | 2,650 (L) | 5,700 | 4,370 | 4,400–8,100 |
| Lymphocyte | 613 (L) | 1,180 | 1,540 | 1,400–3,300 |
| CD3+ absolute | 500 (L) | 608 (L) | 839 | 823–2,353 |
| CD4+ absolute | 280 (L) | 325 (L) | 456 | 467–2,353 |
| CD8+ absolute | 204 | 268 | 360 | 201–746 |
| CD19+ absolute | 21 (L) | 411 | 464 | 52–527 |
| CD56+ absolute | 86 (L) | 160 | 239 | 126–841 |
| CD4+CD45RA+ naïve absolute (% total CD4) | 67 (23%) (L) | 72 (22%) (L) | 95 (21%) (L) | 210–750 |
| CD8+CD45RA+ naïve absolute (% total CD8) | 90 (44%) (L) | 150 (56%) (L) | 154 (43%) (L) | 170–560 |
| Immunoglobulins (mg/dL; IU/mL*) | ||||
| IgG (†on/‡Off IgG replacement therapy) | 1,047‡/1,530† | 1191‡ | 1074‡ | 552–1,631 |
| IgM | 76–118 | 33 (L) | 42 | 33–293 |
| IgA | 98–77 | 20 (L) | 45 (L) | 65–421 |
| IgG1 (pre–IgG replacement therapy) | 793 | NA | NA | 382–929 |
| IgG2 (pre–IgG replacement therapy) | 86 (L) | NA | NA | 241–700 |
| IgG3 (pre–IgG replacement therapy) | 62 | NA | NA | 22–178 |
| IgG4 (pre–IgG replacement therapy) | 0.7 (L) | NA | NA | 4–086 |
| IgE | 2–2.31 | 251 | 57 | 1.03–161.3* |
| Vaccine antibody titers (IU/mL) | ||||
| Streptococcal pneumoniae; 14 serotype testing (before and after vaccine booster) | Prebooster: 3 of 14 serotypes | (Off IgRT postimmunizations) | NA | ≥1.3 |
| Postbooster: 4 of 14 serotypes | 12 of 12 serotypes | |||
| Diphtheria toxoid IgG | 0.66 IU/mL | >2 | NA | ≥0.1 IU/mL |
| Tetanus toxoid IgG | 0.23 IU/mL | 2.5 IU/mL | NA | ≥0.1 IU/mL |
| SARS-CoV-2 IgG (spike protein) | Positive | Positive, 4027 IU/mL | NA | |
| Lymphocyte proliferation | ||||
| Proliferation to antigens | ||||
| Candida antigen, induced, CPM | 30,172 | NA | NA | |
| Candida antigen, induced, SI | 17 | NA | NA | ≥3 SI |
| Tetanus toxoid, induced, CPM | 599 | NA | NA | |
| Tetanus toxoid, induced, SI | <3 (L) | NA | NA | ≥3 SI |
| Tuberculin, induced, CPM | 650 | NA | NA | |
| Tuberculin, induced, SI | <3 (L) | NA | NA | ≥3 SI |
| Lymphocyte viability | NA | 83.1 | NA | ≥75.0% |
| Candida, % of CD45 | NA | 6 | NA | ≥5.7% |
| Candida, % of CD3 | NA | 11.3 | NA | ≥3.0% |
| Tetanus toxoid, % of CD45 | NA | 31.8 | NA | ≥5.2% |
| Tetanus toxoid, % of CD3 | NA | 43.5 | NA | ≥3.3% |
| Proliferation to mitogens | ||||
| Lymphocyte viability | 70.2 (L) | 81.3 | NA | ≥75.0% |
| PWM, induced, % of CD45 | 27.9 | 19.7 | NA | ≥4.5% |
| PWM, induced, % of CD3 | 28.3 | 28.8 | NA | ≥3.5% |
| PWM, induced, % of CD19 | 35.7 | 21.9 | NA | ≥3.9% |
| PHA, induced, % of CD45 | 71.6 | 68.5 | NA | ≥49.9% |
| PHA, induced, % of CD3 | 73.9 | 79.4 | NA | ≥58.5% |
| B-cell phenotype | ||||
| CD19+ B cells (% of lymphocytes) | 1.1 (L) | 39.0 | 30.1 | 3.8%−18% |
| CD27− IgD+ naïve B cell (% of CD19) | 51.6 | 95.3 | NA | 57.5%−80.0% |
| CD27− IgD+ CD24hi CD38hi transitional B cell (% of CD19) | 0.6 | 3.0 | NA | 0.8%−4.9% |
| CD27− IgD+ CD24mid CD38mid mature naive B cell (% of CD19) | 20.7 (L) | 89.4 (H) | NA | 48.6%−70.7% |
| CD27− IgD+ CD38lo “atypical” naive B cell (% of CD19) | 30.1 (H) | 2.2 | N/A | 1.7%−9.1% |
| CD27+ memory B cells (% of CD19) | 43.5 (H) | 2.1 (L) | 0.6 (L) | 11.5%−37.1% |
| CD27+ IgM− IgD− class switched memory B cell (% of CD19) | 1.5 (L) | 1.7 (L) | 0.2 (L) | 4.7%−19.0% |
| CD27+ IgM+ marginal zone-like memory B cell (% of CD19) | 41.1 (H) | 0 (L) | NA | 6.6%−19.0% |
| CD19hi CD21lo autoreactive-prone B cell (% of CD19) | 30.9 (H) | 1.4 | NA | <6.9% |
| T-cell phenotype | ||||
| CD25hi CD127lo regulatory T cell (% of CD4) | 4.8 (L) | 5 (L) | NA | 5.4%−7.9% |
| CXCR5+ CD45RA− T follicular helper cell (% of CD4) | 16.7 (H) | 6.7 | NA | 5.9%−13.1% |
| PD-1+ CD57− exhausted CD4 T cell (% of CD4) | 11.3 | 6.31 | NA | 0%−19.7% |
| PD-1+ CD57− exhausted CD8 T cell (% of CD8) | 36.2 (H) | 40.4 (H) | NA | 3.0%−13.5% |
| CD4− CD8− TCRab+ (% of CD3) | 0.9 | 1.2 (H) | NA | 0.6%−1.1% |
| Alpha-1-antitrypsin | ||||
| Alpha-1-antitrypsin | 0.3 | NA | NA | 0.2–0.3 g/dL |
| Autoantibody screen | ||||
| Antinuclear antibody screen | Negative | Negative | NA | Negative |
| Anti—double-stranded DNA | 1 IU/mL | NA | NA | ≤ 4 IU/mL |
| Anti—cyclic citrullinated peptide | <16 IU/mL | NA | NA | <16 IU/mL |
| Rheumatoid factor | <14 IU/mL | NA | NA | <14 IU/mL |
| Anti—IFN-α | Positive | Positive | Positive | Nonreactive |
| Anti—IFN-ω | Positive | Positive | Positive | Nonreactive |
| Anti—IL-17A | Positive | Positive | Positive | Nonreactive |
NA, Not applicable/available.
B- and T-cell phenotyping reference ranges reflect mean ± 1 SD of healthy donors measured in-house. Post-HCT, testing results of additional anticytokine antibodies including anti–IFN-β, anti–IL-17F, and anti–IL-22 were found to be negative. Abnormal values are bolded.
IgE.
On IgG replacement therapy.
Off IgG replacement therapy.
In light of frequent ear and sinus infections, “walking” pneumonias in her twenties, and the recent history of severe, acute episode of multilobar pneumonia, T-cell–independent and T-cell–dependent vaccine-specific antibody responses were measured. The patient displayed poor antibody response to polysaccharide pneumococcal vaccine, despite receiving a booster, but normal antibody response to diphtheria and tetanus vaccines (Table I).
On the basis of these findings, the patient was diagnosed with IgG subclass deficiency and specific antibody deficiency and she was subsequently placed on intravenous immunoglobulin replacement therapy. Despite the diagnosis of specific antibody deficiency, given the absence of hypogammaglobulinemia, her immunoglobulin replacement therapy (IgRT) coverage was denied by commercial insurance repeatedly.
After careful review of laboratory data, the patient was noted with prolonged intermittent leukopenia and lymphopenia during the previous year, even after recovery from pneumonia and being weaned from systemic steroids.
Her clinical presentation of frequent and severe infections, persistent lymphopenia (decreased B-cell and T-cell counts), and immune dysregulatory features (eg, alopecia, vitiligo, and premature ovarian failure) led the second allergist to expand on her immune phenotyping. Because there was limited availability of extensive immune phenotyping (naïve/memory T-cell subsets, B-cell phenotyping, lymphocyte proliferation to mitogen and antigens) in the setting of a private practice, a primary immunodeficiency next-generation sequencing panel was pursued by her allergist-immunologist as a next step.
Genetic testing revealed compound heterozygosity of 2 pathogenic RAG1 variants (in trans): c.2924G>A p.Arg975Glu, inherited from her mother, and c.1566G>T p.Trp522Cys, inherited from her father, resulting in a restricted RAG1 recombinase activity (predicted 58% and 42% of residual functional capacity of wild-type protein, respectively, for each variant).3–6 Of note, because next-generation sequencing revealed 2 pathogenic variants of an autosomal-recessive gene, parental Sanger sequencing was clinically indicated to confirm the presence of the described variants in trans.
The patient was then referred to an academic center, where additional detailed history revealed progressive arthralgia and recurrent flares of genital herpes. Further immune phenotyping confirmed persistent lymphopenia, decreased naïve T-cell fraction, normal switched memory B-cell, expansion of marginal zone-like B cells, increased CD19hiCD21lo B cells, and low transitional B cells. The patient displayed normal lymphocyte proliferative responses to phytohemagglutinin and pokeweed mitogens, and decreased lymphocyte proliferative response to tetanus and tuberculin, but normal response to candida antigens. Standard autoantibody testing as part of her rheumatologic evaluation revealed the presence of antithyroglobulin and antithyroperoxidase antibodies (albeit on IgRT), and the result of testing was negative for anti-cyclic citrullinated peptide, antinuclear antibody, rheumatoid factor, and anti–double-stranded DNA antibodies. In addition, the patient had detectable antibodies against IFN-α, IFN-ω, and IL-17A (Table I).
The patient was placed on alpha herpesvirus prophylaxis with daily valacyclovir, along with monthly facilitated subcutaneous IgRT, due to worsening headaches and fatigue with intravenous immunoglobulin and weekly subcutaneous IgRT. There was no need for fungal prophylaxis. On optimization of her IgRT, the frequency of upper respiratory tract infections decreased but did not subside, with a repeated need for short antibiotic courses for sinusitis flares.
Lung disease (Figure 1) was treated with the combination of fluticasone propionate 250 μg/salmeterol 50 μg 2 puffs daily as daily controller, apace with a slow taper of her systemic steroids, as well as increasing her airway clearance with hypertonic saline nebulizations, and vest and chest physical therapy, which showed significant improvement (FEV1 73% and FEV1/forced vital capacity 62.9% of predicted).
Despite improvement in lung function, the patient often complained about her poor quality of life, with chronic cough, recurrent sinus infections, arthralgias, and progression of alopecia and vitiligo. In light of previous reports of high morbidity in patients with pRD and inflammatory lung disease, along with reports of vasculitis and noninfectious complications and pRD,5,7,8 a conversation started between the patient and the immunologist for definitive therapy. The patient was referred to a national referral center specializing in HCT for primary immune deficiency for additional input and was enrolled in protocol NCT03394053, approved by the National Institutes of Health’s Institutional Review Board. In the midst of the 2020–2021 pandemic, coronavirus disease 2019 (COVID-19) precautions were closely followed given the presence of anti–IFN-α antibodies recently found to be associated with severe forms of COVID-19.9 The patient was immunized with mRNA severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccine and mounted anti–SARS-CoV-2 Spike IgG within 2 months of completing her immunization as established on clinical and research grounds.
Donor search revealed no 10/10 HLA-matched related or unrelated donor options, and the patient was offered an HLA-haploidentical HCT under protocol NCT02579967 (ClinicalTrials.gov )10 using her brother, who did not carry either of the RAG1 variants, as donor. The patient’s pre-HCT bone marrow evaluation revealed interstitial T-cell aggregates (predominantly CD8+) and B-cell maturation arrest. The patient received reduced-intensity conditioning with pentostatin, low-dose cyclophosphamide, and busulfan (cumulative exposure 37.8 mg × h/L), a fresh T-cell–replete bone marrow graft, and graft-versus-host disease prophylaxis with high-dose, post-transplantation cyclophosphamide, mycophenolate mofetil, and sirolimus.
At the 1-year time point, the patient is fully engrafted and clinically well without any signs of graft-versus-host disease or other major HCT-related complications. Significant improvement was observed in pigmentation of the neck, elbows, wrists, and thighs regarding vitiligo (Figure 2), along with improvement of alopecia in the scalp and eyebrows following HCT (Figure 3). Following HCT, the patient did not need to restart IgRT and received immunizations with good response (Table I).
FIGURE 3.
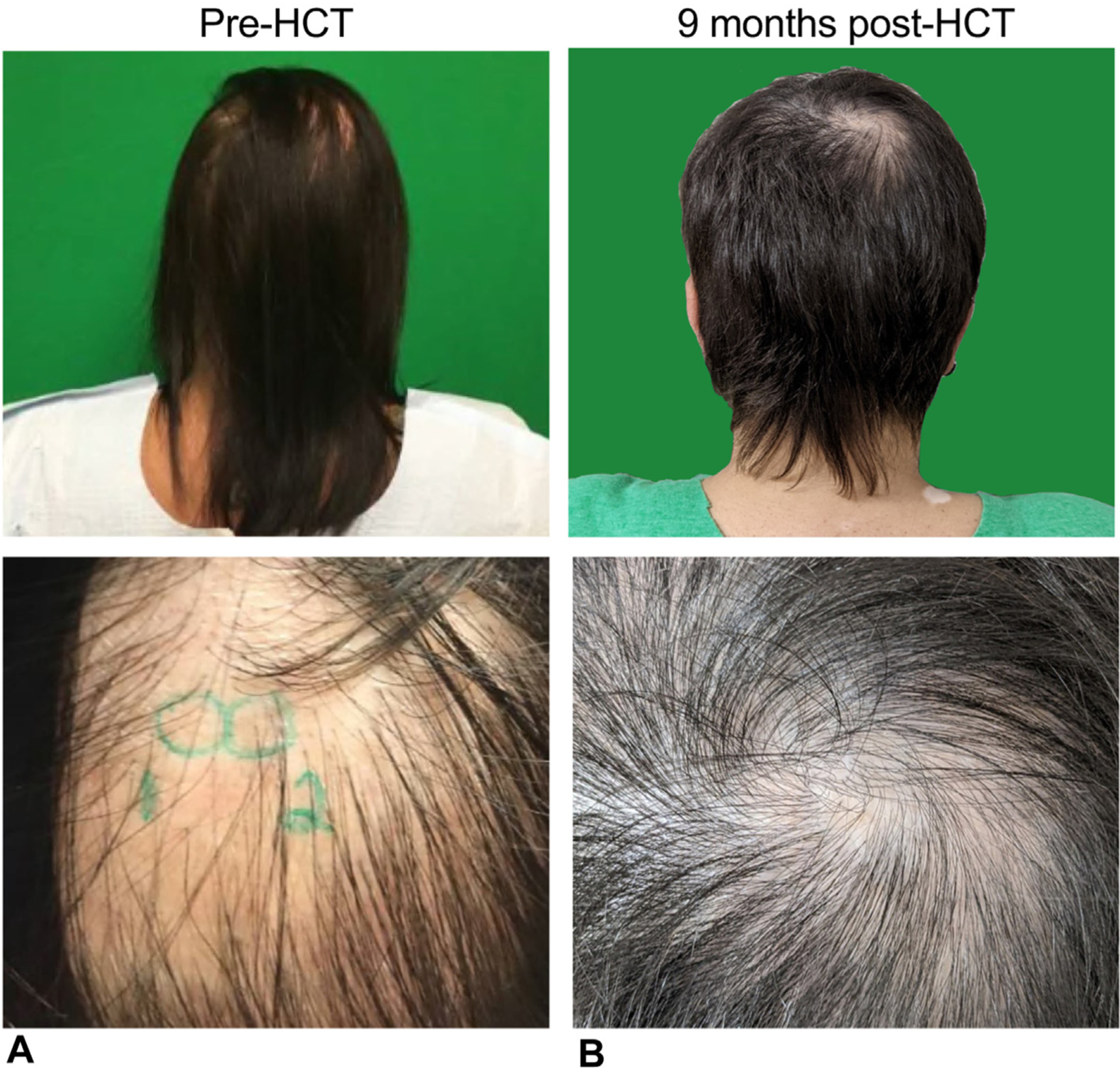
Changes in presentation of alopecia. Alopecia presentation (A) before HCT and (B) 9 months after HCT.
At 18 months post-HCT, the patient had 100% chimerism in T, B, natural killer, and myeloid cells. As a unique aspect of immune reconstitution, we compared the patient’s bone marrow immune subsets before and after HCT. The original block in development of the B-cells form pre-B to large pre–B cell II stage was normalized (Figure 4). This was also reflected by the normal B-cell count in the periphery. At the 12 to 18 month post-HCT period, the naïve B cell compartment was normalized with increased transitional anIure naïve B cells (Table I). Furthermore, a significant decrease in autoreactive-prone B-cell subset (CD19hi CD21lo) was observed. Normalization of total CD4-, toID8-, and naïve CD4- and CD8-cell counts was noted in the T-cell compartment. The skewing in T follicular helper cell compartment before the HCT was no longer detected after HCT. In addition, lymphocyte proliferation to antigens and mitogens was all normal when tested at the 1-year post-HCT time point.
FIGURE 4.
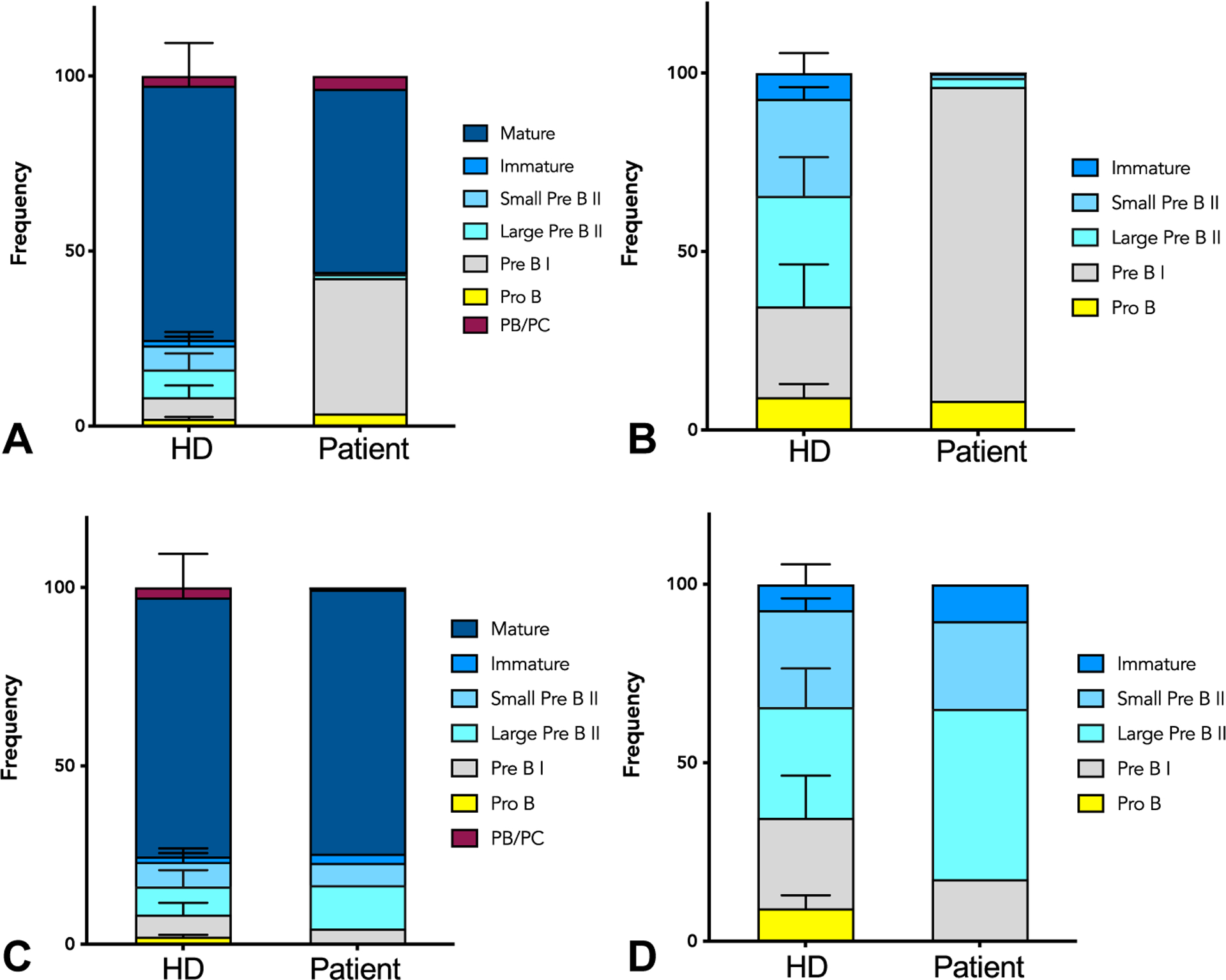
Bone marrow immune subsets. (A) Immune cell composition of the bone marrow of healthy controls (HD) and patient measured by flow cytometry before HCT. (B) B-cell composition at different developmental stages in the bone marrow of healthy controls (HD) and patient, showing block in pre–B I to large pre–B II stage before HCT. (C) Immune cell composition of the bone marrow of healthy controls (HD) and patient measured by flow cytometry after HCT. (D) B-cell composition at different developmental stages in the bone marrow of healthy controls (HD) and patient, showing resolution of the block in pre–B I to large pre–B II stage 1 year after HCT.
Interestingly, the patient had an active COVID-19 infection in summer of 2022, 15 months after HCT and 3 rounds of immunization in 2021 while she remained off IgRT. Even though the presence of anti–IFN-α antibodies is a risk factor in severe COVID-19 infections9 and the patient remained positive for anti–IFN-α antibodies during this time period, her clinical course was mild. This may be explained by the excellent T-cell immune reconstitution and overall function as measured by proliferation studies following HCT. Presumably, the patient built SARS-CoV-2–specific T-cell immunity.
Post-HCT clinical course was complicated only by vulvar herpes simplex virus (HSV) flares (Figure 5). Because the lesions persisted on valacyclovir and remained swab positive for HSV-2 PCR, she was subsequently trialed on various antiviral regimens. Topical imiquimod and later cidofovir were used initially without resolution. Because of concern for resistance, the patient was started on intravenous foscarnet, but logistical barriers prevented optimal dosing, and painful HSV-2 swab–positive lesions spread to her hands. She improved rapidly on treatment with intravenous acyclovir. HSV resistance testing later demonstrated strain susceptibility to both foscarnet and acyclovir. In addition, preacyclovir vulvar biopsy showed HSV-negative granulation tissue with chronic inflammation, indicating that a noninfectious component likely contributed to prior apparent refractoriness. On discharge, she tolerated a gradual decrease from thrice-daily treatment to once-daily prophylactic dosing of valacyclovir over the course of 6 months before flaring again at 1.5 years post-HCT and reverting again to treatment dosing of valacyclovir.
FIGURE 5.
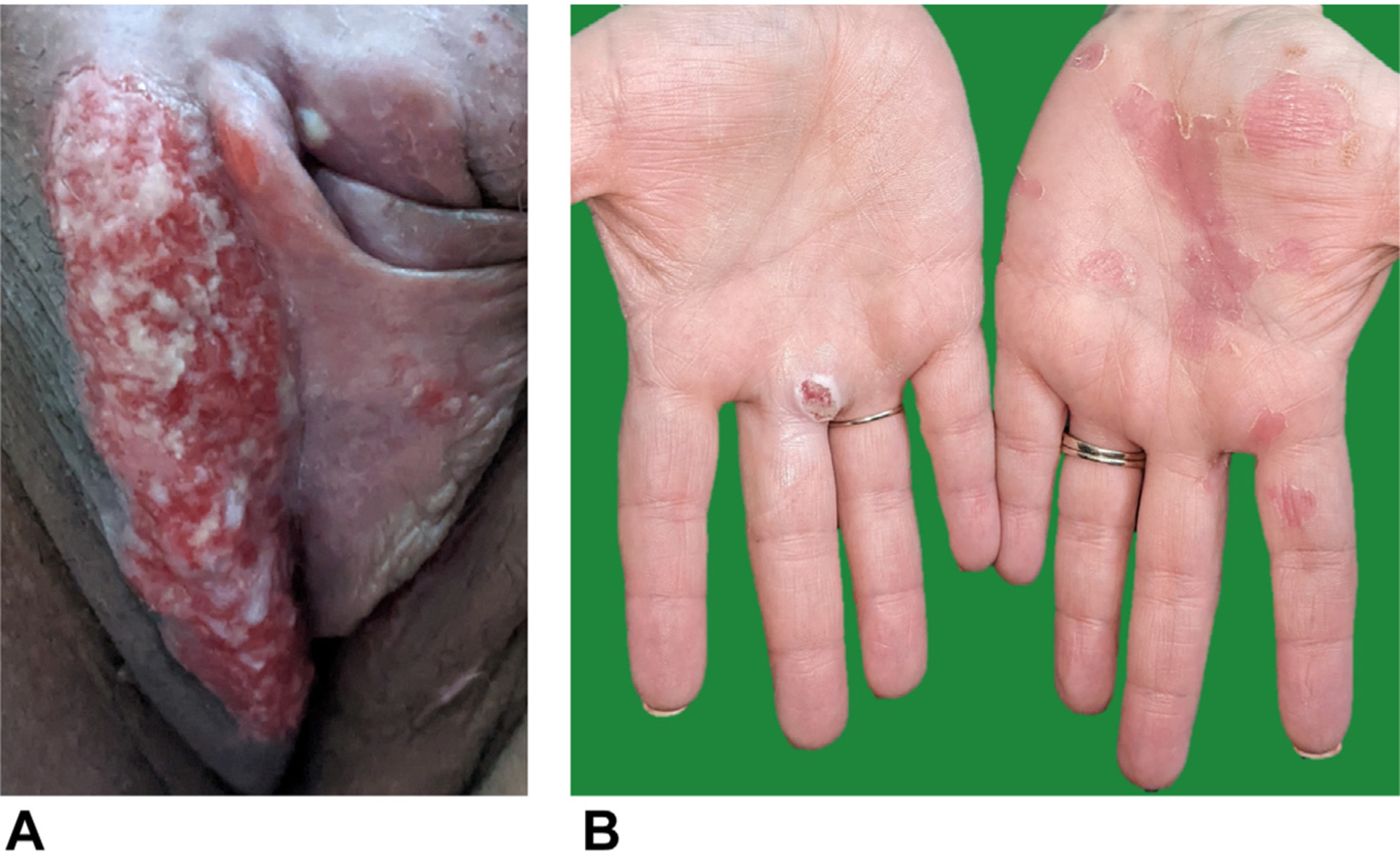
HSV flares post-HCT. (A) HSV-2 swab–positive vulvar lesion at 9 months post-HCT. (B) Spread of HSV lesions to hands at 11 months post-HCT.
In summary, within 2 years from the first inpatient episode and diagnosis of bronchiectasis at age 32 years, our patient with primary diagnosis of asthma, but also infectious and noninfectious complications, received definitive therapy with HCT after discovery of pathogenic RAG1 variants and clinical course that predicted increased risk for mortality and morbidity. The patient has shown complete immune reconstitution with normalization of her lymphocyte counts including her naïve T-cell fractions and appropriate post-HCT antibody response to vaccines. Currently she is undergoing reimmunization per institutional HCT protocol, which will include 23 valent pneumococcal poly-saccharides and SARS-CoV-2 mRNA vaccines.
CONCLUSION
The recombination activating genes (RAG1 and RAG2) are involved in the early phase of the V(D)J recombination, essential for generation of B- and T-cell receptor diversity and development.11,12 Null pathogenic variants in RAG1/2 with undetectable recombinase activity often present with severe combined immunodeficiency with a T−B−NK+ immunophenotype.13 With preserved recombinase activity, partial RAG1/2 deficiencies have a broad clinical spectrum, which includes atypical severe combined immunodeficiency, Omenn syndrome, combined immune deficiency with granuloma and/or autoimmunity(CID-G/AI), and even milder antibody deficiency syndromes.14–18 In the latter 2, residual level of recombinase activity of the mutant protein can range from 20% to 50% of wild-type protein, similar to our case.15–21 However, it is important to note that the average of the estimated recombinase activity of individual alleles may not approximate the overall recombinase activity, as we have previously shown in biallelic systems.5 Variable clinical phenotype has been described in patients with the same hypomorphic RAG1 variant(s), even among members of the same kindred,22 supporting that factors such as environment, infection history, and microbiome can play a role in modifying the B-cell repertoire.
This case exemplifies the pivotal role of clinicians in the systematic evaluation of immune dysregulatory complications and the need for multidisciplinary efforts in the management of pRD noninfectious complications. Furthermore, in adults, as seen in this study, pRD may present with progressive lung disease that can worsen long-term outcome.5,8 Obtaining thorough history for noninfectious complications is of high importance. Among others, evaluation for history of autoimmune cytopenias, autoimmune thyroid disease, vasculitis, vitiligo, alopecia, psoriasis, and myasthenia gravis should be considered, because our patient exhibited several of these features.7,16,21,23,24 Diagnosis can be expedited by immune phenotyping for decreased naïve T-cell compartment, increased autoreactive-prone B cells (CD19hi CD21lo), and antibodies to self-antigens, such as cytokines and blood cells.4,23
Several monogenic causes of combined immunodeficiencies share overlapping clinical history of recurrent infections, especially herpesviruses, signs of immune dysregulation with variable level of antibody deficiency, and laboratory feature of severe lymphopenia, with the present case. While sharing these features, activated phosphoinositide 3-kinase δ syndrome 1 and activated phosphoinositide 3-kinase δ syndrome 2 due to dominant mutations in the PIK3CD or PIK3R1 gene differ by early transitional to follicular B-cell block, whereas patients with cytotoxic T-lymphocyte–associated antigen 4/lipopolysaccharide (LPS)-responsive and beige-like anchor protein deficiency may show more evidence of lymphoproliferation. Pathogenicity of genetic variants in these genes can be confirmed by cytotoxic T-lymphocyte–associated antigen 4 expression and/or transendocytosis assays. Lack of lymphoproliferation may be a dominant factor in differentiating pRD from other CIDs. In these cases, prompt genetic testing is recommended; however, functional assays may be needed for definitive diagnosis.
Special attention is needed to differentiate partial RAG1/2 deficiency with CID versus other antibody syndromes such as common variable immunodeficiency, given their significant clinical and laboratory overlap (eg, frequent infections, autoimmunity, variable lymphopenia, and antibody deficiency). Sole antibody deficiency syndromes are more commonly prone to bacterial but not systemic viral infections. Susceptibility to viral infections, such as recurrent herpes infection in our case, is a possible clue for pRD with CID.25 Another feature that should be kept in mind is that RAG CID patients present with lymphopenia, decreased naïve CD4+ T-cell count, low switched memory B cells, and/or increased fraction of autoreactive-prone (CD19+CD21lo) B cells.26
Allogenic HCT represents a favorable approach to achieve immune reconstitution in patients with RAG1/2 deficiency. Overall survival surpasses 80% in RAG-deficient patients with severe combined immunodeficiency undergoing transplantation from an HLA-matched related donor.27 HCT outcomes for RAG CID-G/AI are limited.28,29 A recent literature review showed that 3 of the 13 transplanted patients died (1 death due to an accident and 2 due to transplant complications). The patients in this review had variable cytoreduction protocols and donor recipient characteristics.30 Five other RAG CID-G/AI patients have been transplanted on the same protocol as the current patient, with favorable outcomes, apart from 1 death due to late sepsis.10,31 Lastly, gene therapy represents an alternative in the path to definitive therapy, currently under an investigatory phase, but with promising results in Rag2−/− mice.32
Our case report highlights several key concepts: patients with history suspicious of IEIs with infectious and noninfectious complications should be evaluated for genetic causes of CID even in the case of mild antibody deficiency syndrome such as specific antibody deficiency and/or IgG subclass deficiency. Lymphopenia with low naïve/memory CD4+ T-cell ratio is a hallmark for CID and should be pursued if feasible. Interestingly, after allogeneic stem cell transplantation, anti–IFN-α, anti–IFN-ω, and anti–IL-17A autoantibodies persisted in our patient, likely due to the presence of patient-derived plasma cells despite the pre-transplant conditioning. Because the patient has displayed SARS-CoV-2–protective titers 6 months after HCT, these will be longitudinally followed to ensure they continue to be protective. Patients with partial RAG deficiency may present with normal quantities of B cells at birth,14 whereas in adulthood progressive B-cell lymphopenia, decline in specific antibody response, and emerging autoantibodies may reflect disease progression. Overall, early diagnosis and assessment of clinical status of pRD can lead to a multidisciplinary approach and definitive therapies, such as HCT.
Conflicts of interest:
M. C. Lopez is an employee and shareholder at Rocket Pharmaceuticals. L. D. Notarangelo is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH). J. E. Walter is a consultant and has research funding from the NIH (R01AI153830), Jeffrey Modell Foundation, and USF Robert A. Good Endowment and serves as a consultant/advisory board member for Takeda, X4-Pharmaceuticals, CSL-Behring, Grifols, ADMA Biologics, Enzyvant, and Regeneron. All other authors do not report any conflicts of interest.
REFERENCES
- 1.Baumann U, Routes JM, Soler-Palacin P, Jolles S. The lung in primary immunodeficiencies: new concepts in infection and inflammation. Front Immunol 2018;9:1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger M, Geng B, Cameron DW, Murphy LM, Schulman ES. Primary immune deficiency diseases as unrecognized causes of chronic respiratory disease. Respir Med 2017;132:181–8. [DOI] [PubMed] [Google Scholar]
- 3.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016;16:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulkhi AA, Dasso JF, Schuetz C, Walter JE. Approaches to patients with variants in RAG genes: from diagnosis to timely treatment. Expert Rev Clin Immunol 2019;15:1033–46. [DOI] [PubMed] [Google Scholar]
- 5.Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol 2018;141:2303–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol 2014;133:1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract 2019;7:1970–1985. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geier CB, Farmer JR, Foldvari Z, Ujhazi B, Steininger J, Sleasman JW, et al. Vasculitis as a major morbidity factor in patients with partial RAG deficiency. Front Immunol 2020;11:574738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020;370:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrova D, Gea-Banacloche J, Steinberg SM, Sadler JL, Hicks SN, Carroll E, et al. Prospective study of a novel, radiation-free, reduced-intensity bone marrow transplantation platform for primary immunodeficiency diseases. Biol Blood Marrow Transplant 2020;26:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 1990;248: 1517–23. [DOI] [PubMed] [Google Scholar]
- 12.Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell 1989;59:1035–48. [DOI] [PubMed] [Google Scholar]
- 13.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014;133: 1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chitty-Lopez M, Westermann-Clark E, Dawson I, Ujhazi B, Csomos K, Dobbs K, et al. Asymptomatic infant with atypical SCID and novel hypomorphic RAG variant identified by newborn screening: a diagnostic and treatment dilemma. Front Immunol 2020;11:1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008;358:2030–8. [DOI] [PubMed] [Google Scholar]
- 16.De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood 2010;116:1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol 2014;134:1375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol 2015;35:280–8. [DOI] [PubMed] [Google Scholar]
- 19.Kutukculer N, Gulez N, Karaca NE, Aksu G, Berdeli A. Novel mutations and diverse clinical phenotypes in recombinase-activating gene 1 deficiency. Ital J Pediatr 2012;38:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharapova SO, Migas A, Guryanova I, Aleshkevich S, Kletski S, Durandy A, et al. Late-onset combined immune deficiency associated to skin granuloma due to heterozygous compound mutations in RAG1 gene in a 14 years old male. Hum Immunol 2013;74:18–22. [DOI] [PubMed] [Google Scholar]
- 21.Reiff A, Bassuk AG, Church JA, Campbell E, Bing X, Ferguson PJ. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J Clin Immunol 2013;33:1289–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuetz C, Pannicke U, Jacobsen EM, Burggraf S, Albert MH, Hönig M, et al. Lesson from hypomorphic recombination-activating gene (RAG) mutations: why asymptomatic siblings should also be tested. J Allergy Clin Immunol 2014; 133:1211–5. [DOI] [PubMed] [Google Scholar]
- 23.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest 2015;125:4135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.H IJ, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol 2014;133:1124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract 2018;6:1769–1771.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosticardo M, Pala F, Notarangelo LD. RAG deficiencies: recent advances in disease pathogenesis and novel therapeutic approaches. Eur J Immunol 2021;51: 1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014;123:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.John T, Walter JE, Schuetz C, Chen K, Abraham RS, Bonfim C, et al. Unrelated hematopoietic cell transplantation in a patient with combined immunodeficiency with granulomatous disease and autoimmunity secondary to RAG deficiency. J Clin Immunol 2016;36:725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuetz C, Gerke J, Ege MJ, Walter JE, Kusters M, Worth AJJ, et al. Hypomorphic RAG deficiency: impact of disease burden on survival and thymic recovery argues for early diagnosis and HSCT. Blood 2023;141:713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan AY, Leiding JW, Liu X, Logan BR, Burroughs LM, Allenspach EJ, et al. Hematopoietic cell transplantation in patients with primary immune regulatory disorders (PIRD): a Primary Immune Deficiency Treatment Consortium (PIDTC) survey. Front Immunol 2020;11:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henrickson SE, Walter JE, Quinn C, Kanakry JA, Bardakjian T, Dimitrova D, et al. Adult-onset myopathy in a patient with hypomorphic RAG2 mutations and combined immune deficiency. J Clin Immunol 2018;38:642–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Til NP, de Boer H, Mashamba N, Wabik A, Huston M, Visser TP, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther 2012;20:1968–80. [DOI] [PMC free article] [PubMed] [Google Scholar]