

Abstract
Protease inhibitors are the most potent antivirals against HIV-1, but they still lose efficacy against resistant variants. Improving the resistance profile is key to developing more robust inhibitors, which may be promising candidates for simplified next-generation antiretroviral therapies. In this study, we explored analogs of darunavir with a P1 phosphonate modification in combination with increasing size of the P1′ hydrophobic group and various P2′ moieties to improve potency against resistant variants. The phosphonate moiety substantially improved potency against highly mutated and resistant HIV-1 protease variants, but only when combined with more hydrophobic moieties at the P1′ and P2′ positions. Phosphonate analogs with a larger hydrophobic P1′ moiety maintained excellent antiviral potency against a panel of highly resistant HIV-1 variants, with significantly improved resistance profiles. The cocrystal structures indicate that the phosphonate moiety makes extensive hydrophobic interactions with the protease, especially with the flap residues. Many residues involved in these protease-inhibitor interactions are conserved, enabling the inhibitors to maintain potency against highly resistant variants. These results highlight the need to balance inhibitor physicochemical properties by simultaneous modification of chemical groups to further improve resistance profiles.
Keywords: HIV-1 protease, protease inhibitors, drug resistance, SAR studies, X-ray structure
Graphical Abstract

INTRODUCTION
An estimated 38 million people are currently infected with the Human Immunodeficiency Virus (HIV), and over 700,000 people die from HIV/AIDS every year [1]. Despite considerable effort, an HIV vaccine remains elusive. Fortunately, multiple small-molecule inhibitors that target critical viral enzymes (reverse transcriptase, integrase, and protease) are currently available for treating HIV infection [2, 3]. These direct-acting antivirals when used in combination antiretroviral therapy (ART) greatly suppress viral load and reduce rates of resistance in adherent patients [4, 5]. Over time, pill burden and significant side effects reduce drug adherence and increase opportunities for drug resistance, leading to therapy failure [4, 6]. Next-generation therapies could improve patient well-being through simplified ART regimens, via prodrug and long-acting formulation approaches, and improved safety and resistance profiles.
HIV-1 protease is an aspartyl protease that catalyzes the cleavage of viral polyproteins into structural and functional proteins essential for viral maturation [7]. HIV-1 protease is therefore a major target for developing anti-HIV-1 therapies, with nine competitive active site inhibitors approved by the FDA [8]. Protease inhibitors (PIs) are among the most potent antivirals against HIV-1 and key to developing next-generation therapies [9]. Of the nine FDA-approved PIs, only three–lopinavir, atazanavir, and darunavir–are currently in clinical use; darunavir (DRV) is the most potent with an excellent resistance profile [10, 11]. The potential efficacy of DRV and its high genetic barrier to resistance has spurred efforts to evaluate this drug as a potential monotherapy in virologically suppressed patients as an alternative to standard triple-drug ART, but with limited success [12]. However, DRV-based dual-drug regimens have been successful as simplified ART, showing efficacy superior to that of standard triple-drug regimens [13–15]. These precedents suggest that novel, exceptionally potent next-generation HIV-1 PIs could become viable maintenance therapy for patients with suppressed viral replication. To this end, identification of antiviral lead compounds with improved safety and resistance profiles is a high priority.
In recent years, research efforts to develop next-generation PIs have mainly focused on structure-based optimization of the DRV scaffold to further enhance potency, resistance profile, and pharmacokinetic (PK) properties [8, 16, 17]. Numerous analogs of DRV with modifications at all four peptidomimetic moieties have been explored, leading to the discovery of several next-generation PIs, including clinical candidates brecanavir and GS-8374 [18–27]. Both compounds exhibited enhanced antiviral potency against clinically relevant PI-resistant HIV-1 variants compared to DRV. These inhibitors contain large ether-linked moieties at the P1 position, a thiazole group in brecanavir, and a phosphonate group in GS-8374 (Figure 1). Interestingly, both the nonpolar thiazole and the polar phosphonate groups extend outside the protease active site but bind in the same region, making extensive van der Waals (vdW) interactions with the protease (Figure S1), which likely contribute to the improved resistance profile of these PIs [18, 21].
Figure 1.

Structures of HIV-1 protease inhibitors. (A) Darunavir (DRV), TMC-126, and the corresponding P1 phosphonate analog GS-8374. (B) DRV and GS-8374 bound to wild-type HIV-1 protease (PDB: 6DGX and 2I4W, respectively). The protease is depicted as a gray surface and a cartoon representation, with chain A in teal and chain B in magenta. (C) DRV analogs with variations at the P1′ and P2′ positions and the corresponding P1 phosphonate analogs analyzed in this study.
While phosphonate modification at the P1 position can improve the resistance profile of DRV analogs [20], the increased overall polarity of the inhibitor requires a less polar moiety at the P2′ position–such as 4-methoxybenzene–to facilitate membrane permeability, as in GS-8374 [22]. Previous structure-activity relationship (SAR) studies of DRV analogs have also highlighted that the overall hydrophobicity of HIV-1 PIs, as determined by water-octanol partition coefficient (cLogP) and topological polar surface area, is crucial for antiviral activity against resistant variants [18]. We previously designed DRV analogs with combinations of P1′ and P2′ modifications, which we termed UMass1–10 (U1–U10) [28]. We have shown that increasing the size and hydrophobicity of the P1′ group improved inhibitor potency against a panel of drug-resistant HIV-1 variants, even when combined with polar substituents at the P2′ position [28, 29]. Therefore, exploring polar phosphonate modifications at the P1 position in combination with increasingly larger, hydrophobic P1′ groups and various P2′ moieties, could result in PIs with improved physicochemical properties and resistance profiles.
In this study, we incorporated the P1 phosphonate modification into our panel of U1–U10 inhibitors (Figure 1) to identify the optimal combination of modifications to improve potency and resistance profiles. Our extensive enzyme inhibition assays revealed that the phosphonate moiety significantly improved potency against a panel of highly mutated, highly resistant HIV-1 protease variants, compared to the parent compounds as well as DRV. Several phosphonate compounds were more potent than the clinical candidate GS-8374. The cocrystal structures indicate that rather than ‘solvent anchoring’ [20], the P1 phosphonate moiety makes extensive vdW interactions with the protease, especially the flap residues. Many residues involved in these protease-inhibitor interactions are invariant, leading to more robust inhibitors that can maintain potency against highly resistant HIV-1 variants. The resulting PU1–PU10 inhibitors are exceptionally potent with improved resistance profiles and are potential candidates for developing simplified next-generation ART.
RESULTS AND DISCUSSION
Chemistry
The synthesis of HIV-1 protease inhibitors with a P1 phosphonate modification is outlined in Scheme 1. Ring opening of erythro-N-Boc-O-benzyl-L-tyrosine epoxide 1 with primary amines followed by reaction of the resulting amino alcohols 2a–c with sulfonyl chlorides 3–6 in the presence of Na2CO3 as a base under biphasic conditions provided the corresponding (R)-(hydroxyethylamino)sulfonamide intermediates 8–12. The 4-formyl compound 10b was converted to the corresponding hydroxymethyl derivative 11b by reduction with NaBH4 in MeOH. Removal of the Boc group with TFA and subsequent acylation reaction of the resulting amines with the bis-THF carbonate derivative 13 afforded the key O-benzyl-protected intermediates 14–16 and 18. The primary benzyl alcohol in compound 16b was selectively protected as a TBDMS ether to give 17b. Catalytic hydrogenolysis of compounds 14–15 and 17–18 over Pd/C followed by treatment of the resulting phenol derivatives 19–22 with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 and cesium carbonate in anhydrous acetonitrile provided the target phosphonate compounds. For compounds 14b–c, hydrogenolysis also led to simultaneous reduction of the nitro group providing phenols 19b–c. The target compound PU3 with the 4-(hydroxymethyl)benzene P2′ moiety was obtained from 24 by removal of the TDBMS protection by treatment with TBAF.
Scheme 1:

Synthesis of P1 Phosphonate Modified HIV-1 Protease Inhibitors.
Reagents and Conditions: (a) R1-NH2, iPrOH, 80 °C, 3 h, 60–98%; (b) aq. Na2CO3, EtOAc, rt, 12 h, 51–92%; (c) NaBH4, MeOH, 0 °C, 30 min, 81%; (d) TFA, CH2Cl2, rt, 1 h, 100%; (e) DIPEA, CH3CN, 0 °C to rt, 24 h, 64–93%; (f) TBDMS-Cl, DIPEA, DMAP, CH2Cl2, 0 °C to rt, 15 h, 98%; (g) 10 wt% Pd/C, H2 gas, MeOH/EtOAc (1:1), rt, overnight, 55–98%; (h) Cs2CO3, CH3CN, 0 °C, 40 min, rt, 1 h, 62–99%; (i) TBAF, THF, 0 °C, 2 h, 98%.
While the synthetic strategy outlined in Scheme 1 allowed access to P1 phosphonate modified HIV-1 protease inhibitors containing diverse P1′ and P2′ moieties, synthesis of target compounds containing a benzothiazole moiety at the P2′ position proved to be challenging. In these cases, debenzylation of the P1 benzyloxy group was unsuccessful under a variety of conditions including with BBr3. Also, the above strategy required protection/deprotection steps for compounds with a 4-(hydroxymethyl)benzene P2′ moiety. Therefore, we developed an alternative synthetic strategy that allowed late-stage introduction of the P2′ moiety as outlined in Scheme 2. Selective protection of the secondary amine in compounds 2a–c with a Cbz group provided the P1–P1′ intermediates 25a–c with orthogonally protected amines. Boc deprotection with TFA followed by reaction of the resulting amines with bis-THF carbonate derivative 13 gave protected intermediates 26a–c. Removal of both Bn and Cbz protecting groups was achieved simultaneously by catalytic hydrogenolysis, and the resulting free amine was selectively re-protected with a Boc group to afford the phenol derivatives 27a–c. Reaction with 23 in the presence of cesium carbonate afforded the P1 phosphonate derivatives 28a–c. Boc deprotection with TFA followed by treatment of the resulting amine with sulfonyl chlorides 5–7 provided the target phosphonate compounds. This strategy provides a practical alternative route to P1 phosphonate modified HIV-1 protease inhibitors with diverse P2′ moieties (Scheme 2).
Scheme 2:
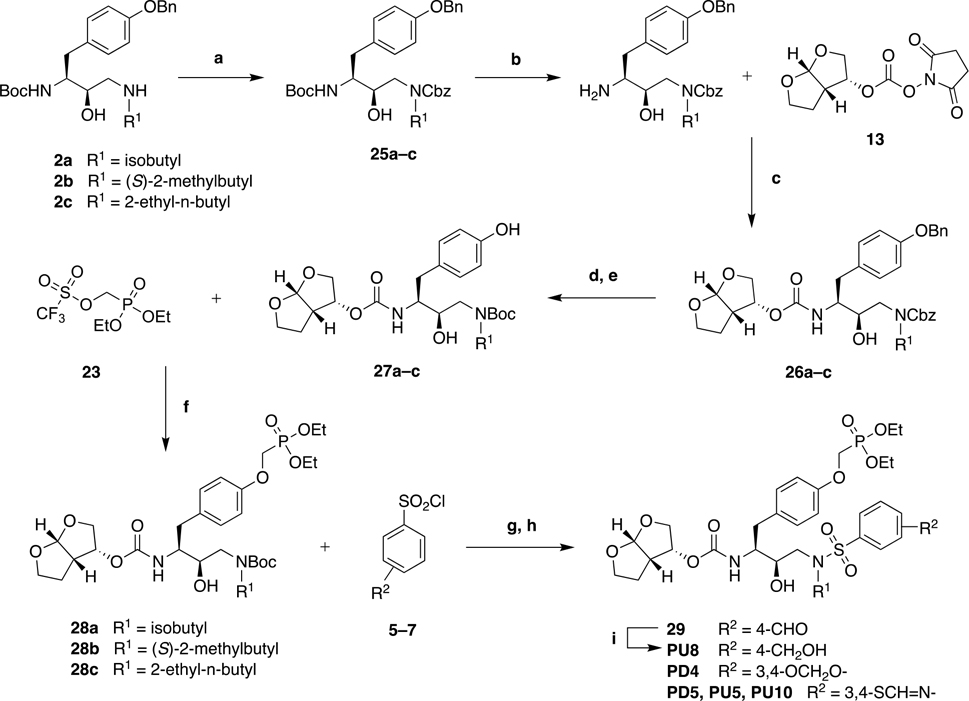
Alternative Synthesis of P1 Phosphonate Modified HIV-1 Protease Inhibitors.
Reagents and Conditions: (a) Benzyl chloroformate, Et3N, CH2Cl2, 0 °C to rt, 15 h, 53–64%; (b) TFA, CH2Cl2, rt, 1 h, 100%; (c) DIPEA, CH3CN, 0 °C to rt, 24 h, 50–75%; (d) 20 wt% Pd(OH)2/C, H2 gas, EtOH/EtOAc (1:1), rt, 3 h; (e) (Boc)2O, Na2CO3, dioxane/H2O (1:1), 0 °C, 30 min, rt, 2 h; 77–91% over two-steps; (f) Cs2CO3, CH3CN, 0 °C, 40 min, rt, 1 h, 82–88%; (g) TFA, CH2Cl2, rt, 1 h, 100% (h) aq. Na2CO3, EtOAc, rt, 12 h, 66–83%; (i) NaBH4, THF, −10 °C, 30 min, 88%.
Enzyme Inhibition and Antiviral Assays
We assessed the potency profiles of P1 phosphonate modified HIV-1 protease inhibitors using biochemical and antiviral assays. The enzyme inhibition constants (Ki) were determined against a panel of six drug-resistant HIV-1 protease variants using a highly sensitive fluorogenic assay [30]. Antiviral potencies (EC50) were determined against wild-type HIV-1 (NL4–3 strain) using a cell-based assay. As we previously reported, DRV and analogs U1–U10 inhibit wild-type HIV-1 protease with a Ki of less than 5 pM (limit of detection) and maintain pM potency against the I84V and I50V/A71V drug-resistant variants [31]. All compounds with a P1 phosphonate modification inhibited the I84V and I50V/A71V protease variants with a potency well below the limit of detection, precluding a comparison between the inhibitors and therefore requiring highly resistant HIV-1 protease variants to assess potency differences and resistance profiles of these PIs (Table 1).
Table 1.
Inhibitory Activity of Phosphonate Compounds against Drug-Resistant Variants of HIV-1 Protease.

| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Inhibitor | R1 | R2 | HIV-1 Protease Ki (nM) |
|||||
| I84V | I50V/A71V | 8Mut | 10Mut | 11Mut | KY24 | |||
|
| ||||||||
| GS-8374 |

|

|
<0.005 | <0.005 | 0.56 ± 0.05 | 2.10 ± 0.10 | 3.60 ± 0.40 | 0.32 ± 0.04 |
| PD4 |

|

|
<0.005 | <0.005 | 2.20 ± 0.50 | 4.50 ± 0.80 | 63.5 ± 15.2 | 0.36 ± 0.05 |
| PD5 |

|

|
<0.005 | <0.005 | 0.93 ± 0.2 | 1.90 ± 0.30 | 15.0 ± 3.5 | 0.23 ± 0.02 |
| PU1 |

|

|
<0.005 | <0.005 | 13.3 ± 3.5 | 146 ± 10 | 259 ± 17 | 2.31 ± 0.09 |
| PU2 |

|

|
<0.005 | <0.005 | 0.78 ± 0.07 | 3.70 ± 0.30 | 10.5 ± 0.9 | 0.16 ± 0.02 |
| PU3 |

|

|
<0.005 | <0.005 | 4.98 ± 0.40 | 52.9 ± 4.2 | 103 ± 3 | 1.89 ± 0.17 |
| PU4 |

|

|
<0.005 | <0.005 | 0.88 ± 0.13 | 4.20 ± 0.30 | 11.0 ± 1.0 | 0.19 ± 0.03 |
| PU5 |

|
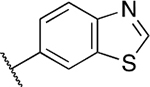
|
<0.005 | <0.005 | 1.80 ± 0.50 | 1.20 ±0.40 | 9.03 ± 4.21 | 0.09 ± 0.01 |
| PU6 |
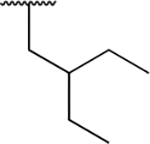
|

|
<0.005 | <0.005 | 4.71 ± 0.73 | 51.6 ± 5.5 | 106 ± 7 | 1.08 ± 0.06 |
| PU7 |
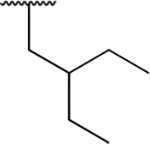
|

|
<0.005 | <0.005 | 0.77 ± 0.06 | 1.26 ± 0.08 | 2.20 ± 0.20 | 0.16 ± 0.04 |
| PU8 |
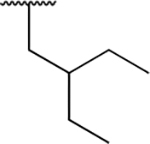
|

|
<0.005 | <0.005 | 2.20 ± 0.40 | 32.1 ± 4.3 | 81.2 ± 10.8 | 0.60 ± 0.10 |
| PU9 |

|

|
<0.005 | <0.005 | 0.37 ± 0.03 | 1.60 ± 0.06 | 6.90 ± 0.50 | 0.08 ± 0.03 |
| PU10 |

|

|
<0.005 | <0.005 | 0.25 ± 0.08 | 0.24 ± 0.09 | 3.20 ± 0.50 | 0.02 ± 0.01 |
| DRV | 0.026 ± 0.006 | 0.075 ± 0.005 | 12.8 ± 0.4 | 156 ± 4 | 759 ± 57 | 6.95 ± 0.12 | ||
The drug-resistant HIV-1 protease variants 8Mut, 10Mut, 11Mut, and KY24 contain the following amino acid substitutions compared to the wild-type NL4-3 protease: 8Mut (I13V, G16E, V32I, L33F, K45I, M46I, V82F, I84V); 10Mut (I13V, G16E, V32I, L33F, K45I, M46I, A71V, L76V, V82F, I84V); 11Mut (I13V, G16E, V32I, L33F, K45I, M46I, I54L, A71V, L76V, V82F, I84V); KY24 (L10V, T12V, I13V, I15V, K20M, V32I, L33F, K43T, M46I, I47V, I54M, D60E, Q61N, I62V, L63P, C67Y, H69K, A71I, I72L, G73S, V77I, V82A, L89V, L90M). NT: not tested.
For a rigorous evaluation of our PIs, we chose three representative highly mutated drug-resistant HIV-1 protease variants, 8Mut, 10Mut, and 11Mut, with overlapping and increasing number of mutations. These variants were identified in viral passaging experiments with increasing concentrations of DRV and thus exhibit increasingly high-level resistance to DRV in accordance with the number of mutations [31, 32]. For example, the 10Mut protease variant exhibited high-level resistance to DRV, with four orders of magnitude weaker potency, yet maintained catalytic efficiency (5.3 s−1μM−1) only about three-fold lower than wild-type protease (17.1 s–1μM–1) [31]. In addition, we included a patient-derived, highly mutated, multidrug-resistant protease variant KY24, which shows high-level resistance to all FDA-approved PIs including DRV [33].
DRV showed significantly reduced potency against all protease variants tested with Ki values ranging from low nM against KY24 (Ki = 6.95 nM) and 8Mut (Ki = 12.8 nM) to high nM against the 10Mut (Ki = 156 nM) and 11Mut variants (Ki = 759 nM), representing three to five orders of magnitude loss in potency compared to wild-type protease (WT Ki = <0.005 nM), as previously reported [31]. For our DRV analogs U1–U10, potency profiles varied significantly depending on the P1′ and P2′ moieties (Table S1). Compounds with the 4-aminobenzene (U1/U5) and 4-(hydroxymethyl)benzene (U3/U8) P2′ moieties exhibited potency profiles similar to that of DRV, except for compounds with the 2-ethyl-n-butyl P1′ moiety which showed slightly improved potencies. In contrast, compounds containing the 4-methoxybenzene (U2/U7), 1,3-benzodioxole (U4/U9), and benzothiazole (U5/U10) moieties at the P2′ position maintained better potency against all variants tested. This trend was particularly pronounced for compounds containing the larger 2-ethyl-n-butyl moiety at the P1′ position. The observed potency profiles of DRV analogs indicate that, while both P1′ and P2′ moieties contribute to potency, the P2′ moiety has a greater impact on potency against resistant variants.
To further improve potency against highly resistant protease variants, we evaluated a series of DRV analogs with a P1 phosphonate modification in combination with diverse P1′ and P2′ moieties. In general, the introduction of a polar phosphonate moiety at the P1 position provided compounds with enhanced potency against highly resistant variants (Table 1, Figure S2). However, the extent of potency improvement varied depending on the P1′ and P2′ moieties present (Figure S3). Firstly, we compared compounds with an isobutyl group at P1′ position as in DRV. The phosphonate compound GS-8374 maintained excellent potency against all resistant variants showing Ki values of 0.32 nM, 0.56 nM, 2.10 nM, and 3.6 nM against KY24, 8Mut, 10Mut, and 11Mut, respectively. Interestingly, the parent compound TMC-126 was equipotent to GS-8374 against the variants tested (Table S1). The phosphonate compounds PD4 and PD5, which contain the 1,3-benzodioxole and benzothiazole moiety at P2′, respectively, also exhibited potency profiles comparable to those of the corresponding parent compounds D4 [10, 34] and D5 [35]. Thus, for compounds with an isobutyl group at P1′, the addition of a phosphonate moiety at P1 did not improve the overall potency profile against resistant variants.
Phosphonate compounds PU1–PU5 incorporate diverse P2′ moieties and increased hydrophobicity at the P1′ position, replacing the isobutyl group in DRV and GS-8374 with the isopentyl group, (S)-2-methylbutyl. Compound PU1 containing the 4-aminobenzene at P2′ was equipotent to DRV against the 10Mut variant but was slightly more active against the 11Mut variant. Whereas PU3 containing the 4-(hydroxymethyl)benzene at P2′ was two to seven-fold more active than DRV against all resistant variants. However, both compounds bearing polar P2′ substituents were significantly less active than GS-8374, which contains a less polar 4-methoxybenzene moiety at the P2′ position. The phosphonate compounds PU2, PU4, and PU5 containing relatively less polar P2′ moieties, 4-methoxybenzene, 1,3-benzodioxole, and benzothiazole, respectively, displayed excellent potency profiles, with Ki values against all resistant variants in the same range as GS-8374. Overall, phosphonate compounds with the isopentyl at the P1′ position were either equipotent or more active than the corresponding inhibitors with the isobutyl group at P1′, indicating that increasing hydrophobic bulk at P1′ is beneficial for maintaining potency against resistant variants. Moreover, the observed potencies of phosphonates PU2, PU4, and PU5 in most cases were substantially better than the corresponding parent compounds (Figures S1, S2).
Phosphonate compounds PU6–PU10 contain a symmetric P1′ isohexyl group, 2-ethyl-n-butyl, with increased size and hydrophobicity compared with the isobutyl in GS-8374 and isopentyl in PU1–PU5. This series of inhibitors showed improved inhibitory potencies against resistant variants compared with PU1–PU5 (Table 1). In this group, PU6 and PU8, containing the polar 4-aminobenzene and 4-(hydroxymethyl)benzene P2′ moieties, inhibited the 10Mut and 11Mut variants less potently than GS-8374, but significantly better than DRV. In addition to showing improved potency against highly resistant HIV-1 protease variants, PU6 was recently identified as a potent inhibitor of the human T-cell lymphotropic virus type 1 (HTLV-1) protease, an aspartyl protease similar to HIV-1 protease [36].
As in the P1′ isobutyl and isopentyl series, phosphonate compounds with the larger P1′ isohexyl group and relatively less polar P2′ moieties, 4-methoxybenzene (PU7), 1,3-benzodioxole (PU9), and benzothiazole (PU10), displayed exceptional potency profiles with Ki values in the pM to low nM range against the resistant variants tested. These inhibitors were more potent than GS-8374 against the 8Mut, 10Mut, and KY24 variants while maintaining similar activity against the 11Mut variant. Overall, PU7, PU9 and PU10 showed improved potency profiles compared to GS-8374.
The observed potency profiles of P1 phosphonate modified HIV-1 protease inhibitors indicate that balancing the polarity of phosphonate group with increased hydrophobicity at the P1′ and P2′ positions is key to maintain potency against highly resistant variants. In general, phosphonate compounds with the less polar P2′ moieties, 4-methoxybenzene, 1,3-benzodioxole, and benzothiazole, inhibited the resistant variants more potently than compounds with the polar 4-aminobenzene and 4-(hydroxymethyl)benzene P2′ moieties, presumably due to enhanced hydrophobic interactions with the protease. The size and shape of the hydrophobic moiety at the P1′ position also impacted potency against the resistant variants. Compounds with the larger and more hydrophobic isohexyl P1′ moiety inhibited the resistant variants more potently than the corresponding analogs with a smaller isobutyl P1′ group. The corresponding analogs with the isopentyl P1′ moiety exhibited either comparable or slightly improved potency than their P1′ isobutyl counterparts. These results indicate that the polar phosphonate modification at P1 requires incorporating more hydrophobic groups at the P1′ and P2′ positions, as increasing hydrophobicity at the P1′ position was beneficial for maintaining potency against highly resistant protease variants. Overall, our approach to explore combination of modifications by exploiting the interdependence between the protease subsites was critical to identify compounds with improved resistance profiles.
Next, we determined antiviral activity of phosphonate analogs against wild-type HIV-1 (NL4–3 strain) in a cell culture-based viral inhibition assay (Table 2). The EC50 values for parent compounds (U1–U10) range from 2.4 to 9.1 nM and are either similar to or 2–3-fold more potent than DRV (EC50 = 7.7 nM), as previously reported (Table S2) [32]. The phosphonate compound GS-8374 was reported to be significantly less active against wild-type HIV-1 than the parent TMC-126 but exhibited an improved resistance profile, superior to that of DRV, as well as improved PK properties [21, 22]. In our assays, GS-8374 showed an EC50 value of 11.8 nM. The corresponding P2′ 1,3-benzodioxole derivative (PD4) was two-fold more active (EC50 = 5.67 nM), while the benzothiazole analog (PD5) was equipotent to GS-8374. Thus, among these three phosphonate compounds with the smaller isobutyl P1′ moiety, the 1,3-benzodioxole derivative PD4 showed enhanced antiviral potency.
Table 2.
Antiviral Activity of Phosphonate Compounds against Wild-Type HIV-1.
| Inhibitor | cLogP | Antiviral EC50 (nM) |
Cytotoxicity CC50 (μM) |
Selectivity Index (SI) | |
|---|---|---|---|---|---|
| 293T cells | TZMbl cells | ||||
|
| |||||
| GS-8374 | 3.10 | 11.8 ± 1.6 | 50 ± 9 | >100 | 4237 |
| PD4 | 2.98 | 5.67 ± 2.3 | 68 ± 4 | >100 | 11993 |
| PD5 | 2.93 | 10.4 ± 2.2 | 45 ± 5 | >100 | 4327 |
| PU1 | 2.49 | 22.9 ± 2.4 | 92 ± 12 | >100 | 4017 |
| PU2 | 3.63 | 9.56 ± 2.5 | 33 ± 2 | 78 ± 4 | 3452 |
| PU3 | 2.68 | 27.3 ± 8.9 | >100 | >100 | >3663 |
| PU4 | 3.51 | 4.16 ± 0.6 | 31 ± 3 | >100 | 7452 |
| PU5 | 3.46 | 9.93 ± 2.8 | 23 ± 3 | 53 ± 8 | 2316 |
| PU6 | 3.02 | 16.0 ± 5.4 | 44 ± 3 | 96 ± 5 | 2750 |
| PU7 | 4.16 | 5.15 ± 1.2 | 22 ± 2 | 59 ± 3 | 4272 |
| PU8 | 3.21 | 17.0 ± 1.3 | 69 ± 6 | >100 | 4059 |
| PU9 | 4.04 | 3.11 ± 2.2 | 21 ± 2 | >100 | 6752 |
| PU10 | 3.99 | 10.7 ± 1.4 | 22 ± 1 | 43 ± 3 | 2056 |
| DRV | 2.39 | 7.73 ± 1.6 | 61 ± 4 | >100 | 7891 |
The antiviral potencies of our phosphonate analogs PU1–PU10 ranged from 3.11 nM to 27.3 nM, showing a broader range of EC50 values compared to the parent compounds. Among these, compounds with the polar 4-aminobenzene (PU1) and 4-(hydroxymethyl)benzene) (PU3) moieties at the P2′ position and isopentyl group at the P1′ position were among the least active, with EC50 values of 22.9 and 27.3 nM, respectively. In P1′ isopentyl series, other analogs containing the less polar 4-methoxybenzene (PU2), 1,3-benzodioxole (PU4) and benzothiazole (PU5) P2′ moieties were 2–5-fold more active (EC50 4.16–9.93 nM), compared with PU1 and PU3. Again, as for the isobutyl series, the 1,3-benzodioxole derivative PU4 was the most active, displaying an EC50 value of 4.16 nM (Table 2).
The antiviral potencies generally improved with the increased size of the hydrophobic moiety at the P1′ position, with EC50 values ranging from 3.11 to 17.0 nM for PU6–PU10 with the larger isohexyl P1′ moiety. The phosphonate compounds PU6, PU7 and PU8 followed this trend. However, the antiviral potencies were largely similar regardless of the size of the P1′ moiety for compounds with a fused bicyclic moiety at the P2′ position. Compounds containing the 1,3-benzodioxole moiety (PD4, PU4, PU9) at the P2′ position but increasingly larger P1′ moieties all showed similar antiviral potencies with EC50 values of 5.67, 4.16, and 3.11 nM, respectively. Similarly, all three compounds with a benzothiazole moiety (PD5, PU5, PU10) also showed comparable potencies. As in enzyme inhibition assays, overall trends were similar with compounds containing polar substituents at the P2′ showing lower antiviral potencies than the corresponding analogs with less polar substituents at that position.
These results agree with previous SAR studies on the DRV scaffold indicating that overall compound hydrophobicity, as estimated by the hydrophobicity descriptor cLogP, impacts cellular antiviral potency against both wild-type HIV-1 and resistant variants. Since incorporating a polar phosphonate moiety at the P1′ position decreased overall lipophilicity, compounds with polar substituents at the P2′ resulted in lower activity in antiviral assays, particularly when combined with a smaller isobutyl group at the P1′ position. Our data shows that the antiviral potency of phosphonate compounds can be enhanced by incorporating less polar substituents at the P2′ position as well as by increasing hydrophobic bulk at the P1′ position.
Among the phosphonate compounds, PU7 and PU9 showed excellent potency profiles in both biochemical and antiviral assays. Therefore, we further evaluated these compounds against a panel of highly resistant HIV-1 variants (Table 3). Antiviral activity of PU7, PU9 and the corresponding parent compounds U7 and U9 was determined against three resistant recombinant HIV-1 clones with 10–19 amino acid substitutions in the protease. In addition, these compounds were also tested against viruses selected to high-level resistance against DRV and different parent compounds [32]. The HIV-1 variant SLK is a multi-PI resistant recombinant virus that shows low-level resistance to DRV. The HIV-1 variants Gag-PR and RT-PR both contain the 10Mut protease sequence, with additional mutations in the Gag and RT regions.
Table 3.
Antiviral activity against resistant HIV-1 variants.
| Virus speciesa | Antiviral EC50 (μM) (fold change) | ||||
|---|---|---|---|---|---|
| DRV | U7 | PU7 | U9 | PU9 | |
| HIV-1-WT | 0.008 ± 0.002 | 0.003 ± 0.001 | 0.005 ± 0.001 | 0.006 ± 0.001 | 0.003 ± 0.002 |
| HIV-1-SLK | 0.056 ± 0.01 (7) | NT | <0.010 (<2) | NT | <0.010 (<3) |
| HIV-1-Gag-PR | 0.77 ± 0.13 (96) | 0.14 ± 0.02 (47) | 0.038 ± 0.005 (6) | NT | NT |
| HIV-1-RT-PR | 1.46 ± 0.13 (183) | 0.25 ± 0.03 (83) | 0.085 ± 0.006 (17) | NT | NT |
| HIV-1-V3 (virus culture) | 1.06 ± 0.52 (133) | 0.80 ± 0.18 (267) | 5.33 ± 1.34 (1066) | 11.1 ± 13.2 (1850) | 1.17 ± 2.03 (390) |
| HIV-1-V4 (virus culture) | 15.8 ± 4.3 (1975) | 5.38 ± 1.38 (1793) | 1.06 ± 0.41 (212) | 5.77 ± 1.44 (962) | 3.90 ± 1.50 (1300) |
| HIV-1-V5 (virus culture) | 1.87 ± 2.22 (234) | 21.8 ± 11.1 (7267) | 0.37 ± 0.11 (74) | 6.44 ± 4.69 (1073) | 13.4 ± 11.0 (4467) |
| HIV-1-V9 (virus culture) | 12.9 ± 9.5 (1613) | 16.7 ± 2.0 (5567) | 0.54 ± 0.18 (108) | >100 (>16667) | 2.85 ± 0.71 (950) |
| HIV-1-DRV (virus culture) | >100 (>12500) | 1.60 ± 0.49 (533) | 0.28 ± 0.12 (56) | 0.78 ± 0.19 (130) | 0.22 ± 0.11 (73) |
NT: not tested.
The resistant HIV-1 variants contain the following amino acid substitutions in HIV-1 protease compared to the wild-type NL4-3 strain.
HIV-1-SLK: L10I, V11I, I13V, K14R, A22V, E35D, M36I, N37D, G48M, F53L, I54V, I62V, L63P, A71V, T74S, V82A, I84V, T91S, Q92K
HIV-1-Gag-PR/RT-PR: I13V, G16E, V32I, L33F, K45I, M46I, A71V, L76V, V82F, I84V (additional mutations in the Gag and RT regions)
HIV-1-V3: L10F, L33F, M46I, I47V, I50V, F53L, L63P, A71V, L76S, V82I, I85V, L89I (81%)
HIV-1-V4: L10F, V11I, I13V, V32I, L33F, K43T, M46L, I54L, A71V, V82I, I84V, L89M, T91S, Q92R (93%)
HIV-1-V5: L10F, I15V, M46I, I47V, I50V, F53L, A71V, V82I, I84V, L89T (89%)
HIV-1-V9: L10F, I13V, L33F, K45R, M46I, I47V, I50V, F53L, I54L, I66F, A71V, T74A, L76S, V82I (95%)
As expected, the HIV-1 variants SLK, Gag-PR, and RT-PR showed increasingly high-level resistance to DRV. Compared to wild-type HIV-1, DRV was 7-fold less potent against SLK, 96-fold less potent against Gag-PR, and 183-fold less potent against RT-PR. The parent compound U7 also showed significantly reduced potency against the Gag-PR and RT-PR variants (EC50 = 0.14 μM and 0.25 μM, respectively). However, PU7 maintained nM potency against SLK, Gag-PR, and RT-PR (EC50 = <10 nM, 38 nM, and 85 nM, respectively), indicating that the phosphonate modification significantly improved potency against highly resistant HIV-1 variants, resulting in an improved resistance profile.
Next, we determined antiviral activity against the single inhibitor-selected virus pools, with viruses selected to high-level resistance against the parent compounds U3, U4, U5, and U9, and DRV (Table 3). In each culture, the most abundant HIV-1 variants (abundance range from 78 to 95%) contained 10–14 mutations in the protease [32]. The viruses from selected HIV-1 cultures V3–V5 and V9 (the culture name indicates a virus pool) were significantly less susceptible to DRV, with EC50 values ranging from 1.06 μM to 15.8 μM, representing over 100 to 2000-fold loss in DRV potency. In several cases, only partial inhibition of virus replication was observed at the highest inhibitor concentration tested (100 μM) (Figure S4). The HIV-1-DRV virus pool selected against DRV was completely resistant to DRV. The parent compounds U7 and U9 also showed substantially reduced potency against these viruses. Interestingly, the V9 virus, which was selected against U9, was completely resistant to U9, but was still susceptible to the corresponding phosphonate analog PU9. Notably, the HIV-1-DRV remained susceptible to both PU7 and PU9 (EC50 = 0.28 μM and 0.28 μM, respectively, compared to >100 μM for DRV). However, only the phosphonate compounds PU7 maintained decent potency against all viruses. Thus, compared to the parent compound U7 and DRV, PU7 maintained antiviral potency against highly resistant HIV-1 variants, resulting in a significantly improved resistance profile.
The antiviral activity data against resistant HIV-1 variants further reinforces the results observed in enzyme inhibition assays, indicating that phosphonate modification improves potency against resistant variants, but only when combined with hydrophobic moieties at the P1′ P2′ positions. Notably, the combination of a larger isohexyl group at P1′ and a less polar 4-methoxybenzene moiety at P2′ proved to be optimal, with the resulting inhibitor PU7 showing an excellent potency and resistance profiles.
Structural Analysis of Protease-Inhibitor Complexes
To investigate the binding and molecular interactions of our new PIs, we determined high-resolution (1.64–2.00 Å) cocrystal structures of all phosphonate compounds with the wild-type HIV-1 protease of the NL4–3 strain. The crystallographic data collection and refinement statistics are summarized in Table S3. The cocrystal structures were all solved in the P212121 space group with one protease homodimer in the asymmetric unit and only one orientation of the bound inhibitor in the protease active site. Following convention, we assigned the protease chains as chain A (non-prime) or chain B (prime) depending on the interactions between the central hydroxyl and the catalytic aspartates Asp25/Asp25′, which orients the inhibitors’ P2 bis–THF moiety to interact with Asp29 and Asp30 of chain A. The cocrystal structures of the parent compounds (U1–U10) were previously determined with the wild-type protease of the SF-2 sequence [28]. To enable direct comparisons of protease-inhibitor interactions, we also determined cocrystal structures of the parent compounds bound to the same wild-type HIV-1 protease (NL4–3 strain) as the phosphonate analogs (Table S4).
The crystal structures of the protease-inhibitor complexes were aligned to compare the inhibitor binding modes (Figure 2). When all the complexes were superimposed, the DRV scaffold of each inhibitor aligned very well. Across the 13 structures of phosphonate compounds, the P1 phosphonate moiety was observed to preferentially adopt one major binding orientation (10 of 13 structures), tucked between the tips of the flaps of the two monomers (Figure 2). However, this moiety is more dynamic than rest of the inhibitor – the phosphonate moiety had higher B-factors and adopted two, likely stochastic, minor binding modes in some structures (Figure 3 and Figure S5). We carefully modeled the phosphonate moiety in each complex based on electron density after simulated annealing to remove model bias. The major binding orientation of the phosphonate moiety indicates that one of the diethyl groups tucks under the side chain of the Phe53 residue and packs against the chain A flap, perpendicular to the Gly-Gly peptide bonds in that region (Figures 2B–C). In the majority of the cocrystal structures, the other phosphonate ethyl group interacts with the side chain of Pro81′, with proximity to the side chain of Val82′. Therefore, the phosphonate oxide was preferentially solvent-exposed, directed at the space between the chain A flap and Arg8′. This major binding orientation enables the phosphonate oxide to utilize the crystallographic water network surrounding the protease active site for stability. Only this oxide participates in solvent anchoring, as previously proposed [20], with most of the remaining atoms of the phosphonate moiety extensively involved in protease-inhibitor van vdW interactions.
Figure 2.

Binding of inhibitors with the P1 phosphonate modification at the HIV-1 protease active site in cocrystal structures. (A) Superposition of all phosphonate compounds shown as sticks, with protease in surface representation. (B) Same as panel A, with protease in cartoon representation. (C) The compounds that share the main biding pose of the phosphonate moiety (10 of 13 inhibitors) are superimposed, with flap (47–53) and 80’s loop (81′–82′) residues interacting with the phosphonate moiety shown as sticks. (D) Inhibitor and residues F53, P81′, and V82′ shown with vdW spheres. (E) All phosphonate compounds from cocrystal structures superimposed and shown as sticks, with protease residues that make vdW contacts labeled. Comparing the phosphonate with the analogous parent compounds, residues with similar (black), more (~0.4–1.0 kcal/mol; orange), and substantially increased (~2–3 kcal/mol; red) vdW contacts.
Figure 3.

Comparison of the binding modes of the (A) parent compounds and the (B) P1 phosphonate analogs in cocrystal structures bound to HIV-1 protease. Each inhibitor is indicated by a different color. Crystallographic B-factors of the (C) parent compounds and the (D) phosphonate analogs mapped onto the inhibitor structure. B-factor values were normalized between all inhibitors to enable a direct comparison. Warmer colors show higher B-factors, indicating relative flexibility.
We used the protease structures to quantify these protease-inhibitor vdW interactions. In all complexes, the phosphonate modification enhanced vdW contacts by an average of 13 kcal/mol relative to the corresponding parent compound (Figure 4, Table S5, Table S6). The additional vdW contacts of the phosphonate PIs compared with the parent compounds were localized to the chain A flaps (residues Ile47, Gly48, Gly49, Gly52, and Phe53) and the chain B 80’s loop (residues Pro81′ and Val82′) (Figure 2C–E, Table S6). The major vdW interactions (~2–3 kcal/mol) were observed at residues Gly48, Gly49, Phe53, and Pro81′; whereas minor improvements (~0.5–1.0 kcal/mol) were observed at residues Ile47, Ile50, Gly52, and Val82′ (Figure 4). While residues Ile47, Gly48, Ile50, and Val82 are known drug-resistance sites, Gly49, Gly52, and Pro81 are typically invariant; increased interactions with these residues can enhance inhibitor potency against resistant variants.
Figure 4.

Protease-inhibitor vdW contacts for phosphonate compounds. (A) Protease residues that have increased vdW contacts upon phosphonate addition relative to the corresponding parent compounds. (B) Mapping vdW contacts of phosphonate compounds onto the protease structure. The protease is in surface representation, with residues colored from blue to red for increasing vdW contacts (gray indicates no contact). (C) Increase in average vdW contacts compared with the parent compounds. Residues in panel A shown on the structure and are colored yellow (I47, G52, I50, and V82′) or red (G48, G49, F53, and P81′) to indicate the most increase in vdW contacts.
Although the inhibitor cocrystal structures were determined with the wild-type protease, they still provide insights into the improved potencies against the highly mutated protease variants. We have previously shown that the primary drug resistance mutations expand the active site at the S1/S1′ and S2/S2′ subsites and reduce vdW contacts with DRV [31]. Therefore, compounds with larger, more hydrophobic P1′ and P2′ moieties would more strongly inhibit the resistant variants by optimally filling the active site of mutant proteases. This analysis is in agreement with the observations here that compounds with the larger isohexyl P1′ inhibited the resistant variant more potently than the corresponding analogs with the isobutyl or isopentyl group at the P1′ position. Also, compounds with a nonpolar 4-methoxybenzene and bicyclic P2′ moieties that provide additional hydrophobic interactions in the S2′ subsite are significantly more active against the resistant variants. For compounds with small, polar substituents at P2′ such as 4-NH2, larger P1′ moieties were needed to improve potency upon phosphonate modification, indicating an interdependency between the subsites.
While the aligned structures show no observed differences in the hydrogen bonding network (Figure S6), our previous research demonstrates that the presence of crystallographic hydrogen bonds is not indicative of the frequency at which those hydrogen bonds are retained in a dynamic system. Molecular dynamics simulations showed that the accumulation of distal mutations, including the I54L flap mutation, significantly reduced correlation motion between DRV and the protease, thereby reducing hydrogen bond frequency [31]. This explains why compounds with small, polar substituents at P2′, which utilize mainly polar interactions, inhibit resistant variants with weaker potency than compounds with both polar and hydrophobic interactions in the S2′ subsite. Inhibitors with the P2′ 4-methoxybenzene, 1,3-benzodioxole, and benzothiazole moieties are also able to participate in hydrogen bonding with the backbone nitrogen of Asp30′ yet have enhanced hydrophobic interactions with the hydrophobic residues that make up the S2′ subsite.
SUMMARY AND CONCLUSIONS
We explored combinations of modifications to the DRV scaffold, including a phosphonate modification at the P1 position, to enhance potency against resistant HIV-1 protease variants. We compared a series of compounds with three hydrophobic moieties of increasing size at the P1′ position and five diverse P2′ moieties with and without the phosphonate addition. We identified combinations of P1, P1′ and P2′ modifications that significantly improved potency against resistant variants and maintained low nM cellular potency. In contrast to a previous suggestion [20, 22], our cocrystal structures revealed that the phosphonate group makes extensive interactions with the protease. We propose that these inhibitors exhibit improved potency against resistant variants due to increased vdW interactions with mostly invariant protease residues.
The inhibitor cocrystal structures revealed that the phosphonate moiety largely interacts with the protease flaps that close over the active site. The flaps open and close to facilitate substrate binding and processing but must remain closed in the inhibited state. We hypothesize that the phosphonate moiety improves potency by interacting with the flaps and shifting the equilibrium toward the closed conformation. Most of the flap residues are invariant, especially the glycine residues that are required for flap flexibility. This residue conservation enables retaining potency against resistant variants.
Our results indicate interdependency between P1 phosphonate and the P1′ and P2′ positions, and thus the need to concomitantly optimize these groups. As expected, the phosphonate modification improved potency but only in combination with relatively less polar P2′ moieties and larger, more hydrophobic P1′ groups. These compounds retained excellent cellular potency despite the polarity of the phosphonate moiety, especially in combination with less polar P2′ moieties. The phosphonate compounds with the larger isohexyl P1′ group maintained potency against resistant variants even when combined with more polar moieties at the P2′ position.
Furthermore, although the phosphonate moiety binds outside the substrate envelope [17, 37], it makes contacts mostly with invariant residues and has the flexibility to tolerate known protease mutations. This beneficial effect of ligand flexibility for improving resistance profile has been seen in HCV NS3/4A protease inhibitors [38]. The improved potency against resistant variants, and interactions mostly with invariant flap residues, suggest an improved barrier to resistance. Such compounds are promising candidates toward developing next-generation drugs that can be used as monotherapy to reduce pill burden, perhaps through long-acting formulations.
EXPERIMENTAL SECTION
Chemistry
General.
All reactions were performed in oven-dried round bottomed flasks fitted with rubber septa under an argon atmosphere unless otherwise noted. Reagents obtained from commercial sources were used as received. Flash column chromatography was performed on a Teledyne ISCO CombiFlash Rf+ system equipped with a UV–vis detector. Thin-layer chromatography (TLC) was performed using silica gel (60 F254) coated aluminum plates (EMD Millipore), and spots were visualized by exposure to ultraviolet light (UV), exposure to iodine adsorbed on silica gel, and/ or exposure to an acidic solution of p-anisaldehyde (anisaldehyde) followed by brief heating. 1H NMR and 13C NMR spectra were acquired on a Bruker Avance III HD 500 MHz NMR instrument. Chemical shifts are reported in ppm (δ scale) with the residual solvent signal used as reference and coupling constant (J) values are reported in hertz (Hz). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, dd = doublet of doublets, m = multiplet, br s = broad singlet), coupling constant in Hz, and number of protons. High-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Orbitrap Velos Pro mass spectrometer coupled with a Thermo Scientific Accela 1250 UPLC and an autosampler using electrospray ionization (ESI) in the positive mode. Analytical HPLC was performed on an Agilent 1200 system equipped with a multiple wavelength detector under the following conditions: column, Phenomenex Hypersil-BDS-5u-C18 (5 μm, 4.6 mm × 250 mm, 130 Å); solvent A, H2O containing 0.1% TFA; solvent B, CH3CN containing 0.1% TFA; gradient, 20% B to 100% B over 15 min followed by 100% B over 5 min; injection volume, 20 μL; flow rate, 1 mL/min. The wavelengths of detection were 254 nm and 280 nm.
Synthesis of P1 Phosphonate Modified HIV-1 Protease Inhibitors
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-(isobutylamino)butan-2-yl)carbamate (2a).
A solution of tert-butyl ((S)-2-(4-(benzyloxy)phenyl)-1-((S)-oxiran-2-yl)ethyl)carbamate 1 (2.00 g, 5.41 mmol) in iPrOH (40 mL) was treated at room temperature with isobutyl amine (1.28 g, 16.2 mmol). The reaction mixture was stirred at 80 °C for 3 h, cooled to room temperature, and concentrated under reduced pressure. The residue was purified by crystallization from a mixture of EtOAc/hexanes (1:4) to provide the amino alcohol 2a (2.30 g, 96%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.34–7.30 (m, 1 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.92–6.89 (m, 2 H), 5.04 (s, 2 H), 4.68 (d, J = 9.0 Hz, 1 H), 3.82–3.70 (m, 1 H), 3.44 (q, J = 6.0 Hz, 1 H), 2.91 (dd, J = 14.0, 4.5 Hz, 1 H), 2.82 (dd, J = 13.5, 7.5 Hz, 1 H), 2.67 (d, J = 4.5 Hz, 2 H), 2.40 (d, J = 7.0 Hz, 2 H), 1.71 (sept, J = 7.0 Hz, 1 H), 1.37 (s, 9 H), 0.92 (d, J = 3.0 Hz, 3 H), 0.90 (d, J = 3.0 Hz, 3 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.58, 156.07, 137.29, 130.67, 130.29, 128.71, 128.05, 127.59, 114.94, 79.45, 70.68, 70.17, 58.12, 54.33, 51.53, 35.91, 28.57, 28.46, 20.70, 20.66 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C26H39N2O4, 443.2905; found 443.2889.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-(((S)-2-methylbutyl)amino)butan-2-yl)carbamate (2b).
The same procedure was used as described above for compound 2a. A solution of tert-butyl ((S)-2-(4-(benzyloxy)phenyl)-1-((S)-oxiran-2-yl)ethyl)carbamate 1 (5.0 g, 13.5 mmol) in iPrOH (100 ml) was treated with (S)-2-methylbutan-1-amine (1.41 g, 16.2 mmol) to give 2b (4.57 g, 74%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.42 (m, 2 H), 7.39–7.36 (m, 2 H), 7.33–7.30 (m, 1 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.91 (d, J = 8.5 Hz, 2 H), 5.03 (s, 2 H), 4.78 (d, J = 9.0 Hz, 1 H), 3.82–3.71 (m, 1 H), 3.46 (q, J = 6.5 Hz, 1 H), 2.90 (dd, J = 14.5, 5.0 Hz, 1 H), 2.82 (dd, J = 14.0, 7.5 Hz, 1 H), 2.68 (d, J = 5.0 Hz, 2 H), 2.51 (dd, J = 11.5, 6.0 Hz, 1 H), 2.39 (dd, J = 11.5, 7.5 Hz, 1 H), 1.51–1.34 (m, 2 H), 1.37 (s, 9 H, overlapping), 1.19–1.09 (m, 1 H), 0.91–0.87 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.67, 156.58, 137.23, 130.71, 130.23, 128.70, 128.06, 127.58, 115.04, 80.08, 70.23, 70.16, 55.63, 53.88, 52.07, 35.77, 33.84, 28.41, 17.52, 11.19 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H41N2O4, 457.3061; found 457.3049.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((2-ethylbutyl)amino)-3-hydroxybutan-2-yl)carbamate (2c).
The same procedure was used as described above for compound 2a. A solution of tert-butyl ((S)-2-(4-(benzyloxy)phenyl)-1-((S)-oxiran-2-yl)ethyl)carbamate 1 (5.0 g, 19.0 mmol) in iPrOH (100 mL) was treated with 2-ethyl-n-butylamine (2.0 g, 20.0 mmol) to give 2c (5.50 g, 79%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.41 (m, 2 H), 7.50–7.36 (m, 2 H), 7.34–7.29 (m, 1 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.91 (d, J = 8.5 Hz, 2 H), 5.04 (s, 2 H), 4.70 (d, J = 9.0 Hz, 1 H), 3.82–3.73 (m, 1 H), 3.44 (app q, J = 5.0 Hz, 1 H), 2.92 (dd, J = 14.5, 5.0 Hz, 1 H), 2.84 (dd, J = 13.5, 7.5 Hz, 1 H), 2.75–2.66 (m, 2 H), 2.56–2.48 (m, 2 H), 1.43–1.23 (m, 5 H), 1.37 (s, 9 H, overlapping), 0.87 (t, J = 7.0 Hz, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.61, 156.21, 137.28, 130.70, 130.16, 128.71, 128.06, 127.59, 114.97, 79.59, 70.55, 70.18, 54.21, 52.68, 51.82, 40.95, 35.93, 28.45, 24.01, 11.06, 11.03 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C28H43N2O4, 471.3218; found 471.3203.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((N-((S)-2-methylbutyl)-4-nitrophenyl)sulfonamido)butan-2-yl)carbamate (8b).
A solution of amino alcohol 2b (1.26 g, 2.76 mmol) in EtOAc (40 mL) was treated at room temperature with a solution of Na2CO3 (0.58 g, 5.52 mmol) in water (40 mL) and 4-nitrobenzenesulfonyl chloride 3 (0.73 g, 3.31 mmol). The resulting reaction mixture was stirred at room temperature overnight. The layers were separated, and the aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with saturated aqueous NaCl solution (50 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–80% EtOAc/hexanes) to give compound 8b (1.63 g, 92%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.33 (d, J = 9.0 Hz, 2 H), 7.95 (d, J = 9.0 Hz, 2 H), 7.44–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.35–7.30 (m, 1 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.93 (d, J = 9.0 Hz, 2 H), 5.05 (s, 2 H), 4.61 (d, J = 6.5 Hz, 1 H), 3.82–3.68 (m, 3 H), 3.24–3.12 (m, 2 H), 3.06 (dd, J = 13.5, 7.0 Hz, 1 H), 2.96 (dd, J = 13.5, 8.0 Hz, 1 H), 2.91–2.81 (m, 2 H), 1.68–1.60 (m, 1 H), 1.46–1.34 (m, 1 H), 1.38 (s, 9 H, overlapping), 1.12–1.02 (m, 1 H), 0.88–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.81, 156.52, 150.10, 145.00, 137.07, 130.53, 129.64, 128.73, 128.67, 128.13, 127.60, 124.40, 115.17, 80.28, 72.07, 70.20, 56.18, 55.33, 52.47, 34.93, 33.21, 28.39, 26.65, 16.92, 11.22 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H44N3O8S, 642.2844; found 642.2837.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((N-(2-ethylbutyl)-4-nitrophenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (8c).
The same procedure was used as described above for compound 8b. Amino alcohol 2c (0.64 g, 1.36 mmol) was treated with Na2CO3 (0.29 g, 2.72 mmol) and 4-nitrobenzenesulfonyl chloride 3 (0.39 g, 1.77 mmol) to give compound 8c (0.72 g, 81%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.34 (d, J = 9.0 Hz, 2 H), 7.95 (d, J = 9.0 Hz, 2 H), 7.45–7.42 (m, 2 H), 7.41–7.36 (m, 2 H), 7.35–7.31 (m, 1 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.93 (d, J = 9.0 Hz, 2 H), 5.05 (s, 2 H), 4.57 (d, J = 6.0 Hz, 1 H), 3.81 (s, 1 H), 3.77–3.68 (m, 2 H), 3.22–3.11 (m, 2 H), 3.10–3.00 (m, 2 H), 2.91–2.80 (m, 2 H), 1.53–1.44 (m, 1 H), 1.40–1.23 (m, 4 H), 1.37 (s, 9 H, overlapping), 0.84 (t, J = 7.4 Hz, 3 H), 0.82 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 157.84, 156.52, 150.14, 144.91, 137.09, 130.55, 129.61, 128.75, 128.70, 128.15, 127.61, 124.44, 115.19, 80.30, 72.33, 70.22, 55.32, 53.85, 52.53, 38.77, 35.07, 28.40, 23.06, 22.90, 10.62, 10.49 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H46N3O8S, 656.3000; found 656.2993.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((4-methoxy-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (9b).
The same procedure was used as described above for compound 8b. Amino alcohol 2b (0.30 g, 0.68 mmol) was treated with Na2CO3 (0.14 g, 1.31 mmol) and 4-methoxybenzenesulfonyl chloride 4 (0.14 g, 0.66 mmol) to give compound 9b (0.29 g, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 9.0 Hz, 2 H), 7.44–7.42 (m, 2 H), 7.40–7.37 (m, 2 H), 7.34–7.31 (m, 1 H), 7.17 (d, J = 8.5 Hz, 2 H), 6.97 (d, J = 9.0 Hz, 2 H), 6.92 (d, J = 8.6 Hz, 2 H), 5.04 (s, 2 H), 4.61 (d, J = 8.5 Hz, 1 H), 3.89 (br s, 1 H), 3.85 (s, 3 H), 3.79–3.66 (m, 2 H), 3.10–2.87 (m, 5 H), 2.76 (dd, J = 14.0, 7.5 Hz, 1 H), 1.66–1.54 (m, 1 H), 1.53–1.44 (m, 1 H), 1.36 (s, 9 H), 1.12–1.00 (m, 1 H), 0.86–0.82 (m, 6 H) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H47N2O7S, 627.3099; found 627.3082.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((N-(2-ethylbutyl)-4-methoxyphenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (9c).
The same procedure was used as described above for compound 8b. Amino alcohol 2c (0.63 g, 1.34 mmol) was treated with Na2CO3 (0.28 g, 2.68 mmol) and 4-methoxybenzenesulfonyl chloride 4 (0.33 g, 1.61 mmol) to give compound 9c (0.73 g, 85%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 9.0 Hz, 2 H), 7.45–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.34–7.30 (m, 1 H), 7.16 (d, J = 8.5 Hz, 2 H), 6.97 (d, J = 9.0 Hz, 2 H), 6.92 (d, J = 8.5 Hz, 2 H), 5.04 (s, 2 H), 4.58 (d, J = 6.5 Hz, 1 H), 3.92 (s, 1 H), 3.85 (s, 3 H), 3.77–3.67 (m, 2 H), 3.11–2.96 (m, 3 H), 2.93 (dd, J = 14.0, 4.5 Hz, 1 H), 2.89–2.79 (m, 2 H, overlapping), 1.49–1.36 (m, 2 H), 1.35 (s, 9 H), 1.32–1.24 (m, 3 H), 0.84 (t, J = 7.4 Hz, 3 H), 0.82 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ163.15, 157.67, 156.13, 137.24, 130.74, 130.10, 129.91, 129.67, 128.73, 128.09, 127.59, 115.04, 114.47, 79.79, 72.97, 70.19, 55.75, 54.93, 54.67, 53.87, 39.12, 34.86, 28.43, 23.22, 22.89, 10.72, 10.45 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H49N2O7S, 641.3255; found 641.3258.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((4-formyl-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (10b).
The same procedure was used as described above for compound 8b. Amino alcohol 2b (1.24 g, 2.71 mmol) was treated with Na2CO3 (0.57 g, 5.41 mmol) and 4-formylbenzenesulfonyl chloride 5 (0.72 g, 3.52 mmol) to give compound 10b (1.50 g, 89%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 10.07 (s, 1 H), 8.01 (d, J = 8.0 Hz, 2 H), 7.93 (d, J = 8.0 Hz, 2 H), 7.44–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.35–7.30 (m, 1 H), 7.15 (d, J = 8.0 Hz, 2 H), 6.92 (d, J = 8.0 Hz, 2 H), 5.04 (s, 2 H), 4.61 (d, J = 5.5 Hz, 1 H), 3.85–3.66 (m, 3 H), 3.19–3.09 (m, 2 H), 3.06 (dd, J = 13.5, 7.5 Hz, 1 H), 2.96–2.82 (m, 3 H), 1.70–1.58 (m, 1 H), 1.47–1.34 (m, 1 H), 1.37 (s, 9 H, overlapping), 1.10–1.01 (m, 1 H), 0.87–0.80 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 190.90, 157.75, 156.36, 144.03, 138.93, 137.13, 130.62, 130.29, 128.73, 128.69, 128.12, 127.59, 115.11, 80.10, 72.39, 70.17, 56.70, 55.08, 53.09, 34.82, 33.33, 28.40, 26.60, 16.95, 11.19 ppm.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((4-(hydroxymethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (11b).
A solution of aldehyde 10b (0.99 g, 1.58 mmol) in MeOH (20 mL) was treated at 0 °C with NaBH4 (0.07 g, 1.90 mmol). The reaction mixture was stirred at 0 °C for 30 min, quenched with saturated aqueous NH4Cl solution (20 mL), partially concentrated under reduced pressure to remove methanol, and extracted with EtOAc (3 × 25 mL). The combined organic fraction was washed with saturated aqueous NaCl solution (25 mL), dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 20–100% EtOAc/hexanes), to give compound 11b (0.80 g, 81%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 2 H), 7.50 (d, J = 8.0 Hz, 2 H), 7.45–7.41 (m, 2 H), 7.40–7.35 (m, 2 H), 7.34–7.30 (m, 1 H), 7.16 (d, J = 8.5 Hz, 2 H), 6.92 (d, J = 8.5 Hz, 2 H), 5.04 (s, 2 H), 4.77 (s, 2 H), 4.62 (d, J = 7.0 Hz, 1 H), 3.80–3.65 (m, 2 H), 3.12–2.98 (m, 3 H), 2.94–2.84 (m, 2 H), 2.80 (dd, J = 13.0, 7.0 Hz, 1 H), 1.60–1.43 (m, 2 H), 1.36 (s, 9 H), 1.11–1.01 (m, 1 H), 0.86–0.77 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.67, 156.21, 151.29, 146.18, 137.21, 130.71, 130.07, 128.74, 128.11, 127.77, 127.60, 127.25, 115.08, 79.92, 70.20, 64.41, 57.34, 54.81, 53.75, 34.69, 33.48, 28.44, 26.57, 17.03, 11.19 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H47N2O7S, 627.3099; found 627.3083.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)butan-2-yl)carbamate (12b).
The same procedure was used as described above for compound 8b. Amino alcohol 2b (1.24 g, 2.71 mmol) was treated with Na2CO3 (0.57 g, 5.41 mmol) and 1,3-benzodioxole-5-sulfonyl chloride 6 (0.72 g, 3.25 mmol) to give compound 12b (1.24 g, 72%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.34–7.30 (m, 2 H), 7.19–7.14 (m, 3 H), 6.92 (d, J = 8.5 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 1 H), 6.06 (s, 2 H), 5.04 (s, 2 H), 4.63 (d, J = 7.5 Hz, 1 H), 3.84 (s, 1 H), 3.79–3.68 (m, 2 H), 3.11–2.97 (m, 3 H), 2.95–2.85 (m, 2 H), 2.80 (dd, J = 13.0, 7.5 Hz, 1 H), 1.64–1.56 (m, 1 H), 1.53–1.44 (m, 1 H), 1.36 (s, 9 H), 1.12–1.01 (m, 1 H), 0.88–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.63, 156.15, 151.54, 148.38, 137.20, 131.69, 130.66, 130.04, 128.67, 128.04, 127.54, 123.20, 114.99, 108.43, 107.68, 102.42, 79.80, 72.73, 70.11, 57.37, 54.75, 53.76, 34.69, 33.45, 28.39, 26.55, 17.01, 11.16 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H45N2O8S, 641.2891; found 641.2877.
tert-Butyl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-(N-(2-ethylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)-3-hydroxybutan-2-yl)carbamate (12c).
The same procedure was used as described above for compound 8b. Amino alcohol 2c (1.52 g, 3.22 mmol) was treated with Na2CO3 (0.68 g, 6.44 mmol) and 1,3-benzodioxole-5-sulfonyl chloride 6 (0.85 g, 3.87 mmol) to give compound 12c (1.04 g, 51%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.41 (m, 2 H), 7.40–7.36 (m, 2 H), 7.35–7.30 (m, 2 H), 7.19–7.15 (m, 3 H), 6.92 (d, J = 8.5 Hz, 2 H), 6.88 (d, J = 8.0 Hz, 1 H), 6.06 (s, 2 H), 5.04 (s, 2 H), 4.61 (d, J = 7.0 Hz, 1 H), 3.93 (s, 1 H), 3.77–3.68 (m, 2 H), 3.11–2.98 (m, 3 H), 2.93 (dd, J = 14.0, 4.5 Hz, 1 H), 2.86 (dd, J = 13.0, 6.0 Hz, 2 H), 1.49–1.37 (m, 2 H), 1.36 (s, 9 H), 1.34–1.24 (m, 3 H), 0.85 (t, J = 7.4 Hz, 3 H), 0.83 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 157.61, 156.12, 151.56, 148.39, 137.19, 131.51, 130.66, 130.00, 128.67, 128.03, 127.54, 123.23, 114.96, 108.45, 107.69, 102.43, 79.77, 72.97, 70.09, 54.91, 54.67, 53.78, 39.03, 34.83, 28.38, 23.14, 22.82, 10.68, 10.41 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H47N2O8S, 655.3048; found 655.3035.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((N-((S)-2-methylbutyl)-4-nitrophenyl)sulfonamido)butan-2-yl)carbamate (14b).
A solution of compound 8b (1.0 g, 1.56 mmol) in anhydrous CH2Cl2 (10 mL) was treated at room temperature with TFA (6 mL). After stirring the mixture at room temperature for 2 h, the solvent was evaporated under reduced pressure. The residue was dissolved in toluene (10 mL), and the solvent was evaporated under reduced pressure. The resulting amine was dissolved in anhydrous CH3CN (20 mL), cooled to 0 °C, and treated with DIPEA (0.61 g, 4.68 mmol) and bis-THF carbonate derivative 13 (0.51 g, 1.87 mmol). After 15 min, the reaction mixture was allowed to warm to room temperature and stirred for 24 h. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% MeOH/CH2Cl2) to give compound 14b (0.99 g, 91%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J = 9.0 Hz, 2 H), 7.96 (d, J = 9.0 Hz, 2 H), 7.44–7.36 (m, 4 H), 7.35–7.31 (m, 1 H), 7.12 (d, J = 8.5 Hz, 2 H), 6.91 (d, J = 8.5 Hz, 2 H), 5.66 (d, J = 5.5 Hz, 1 H), 5.07–5.01 (m, 1 H), 5.03 (s, 2 H, overlapping), 4.91 (d, J = 8.0 Hz, 1 H), 3.96 (dd, J = 9.5, 7.0 Hz, 1 H), 3.88–3.80 (m, 3 H), 3.75–3.67 (m, 2 H), 3.37 (br s, 1 H), 3.21 (dd, J = 15.0, 8.0 Hz, 1 H), 3.12–3.05 (m, 2 H), 2.99 (dd, J = 14.0, 3.5 Hz, 1 H), 2.95–2.88 (m, 2 H), 2.76 (dd, J = 13.5, 8.5 Hz, 1 H), 1.72–1.60 (m, 2 H), 1.58–1.51 (m, 1 H), 1.50–1.40 (m, 1 H), 1.14–1.03 (m, 1 H), 0.88–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.88, 155.80, 150.27, 144.53, 136.95, 130.43, 129.49, 128.77, 128.70, 128.21, 127.57, 124.55, 115.18, 109.44, 73.81, 72.42,70.97, 70.19, 69.71, 56.77, 55.52, 53.14, 45.53, 34.81, 33.41, 26.56, 25.99, 16.97, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H44N3O10S, 698.2742; found 698.2744.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((N-(2-ethylbutyl)-4-nitrophenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (14c).
The same procedure was used as described above for compound 14b. Compound 8c (0.72 g, 1.10 mmol) was treated with TFA (5 mL), and the resulting deprotected amine was treated with DIPEA (0.43 g, 3.30 mmol) and bis-THF carbonate 13 (0.36 g, 1.33 mmol) to give compound 14c (0.73 g, 93%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 8.5 Hz, 2 H), 7.96 (d, J = 9.0 Hz, 2 H), 7.43–7.37 (m, 4 H), 7.35–7.31 (m, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.91 (d, J = 8.5 Hz, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.06–5.01 (m, 1 H), 5.03 (s, 2 H, overlapping), 4.84 (d, J = 8.0 Hz, 1 H), 3.95 (dd, J = 9.5, 6.5 Hz, 1 H). 3.88–3.79 (m, 3 H), 3.74–3.67 (m, 2 H), 3.45 (s, 1 H), 3.19 (dd, J = 15.5, 8.5 Hz, 1 H), 3.14–3.04 (m, 2 H), 3.00 (dd, J = 14.0, 3.5 Hz, 1 H), 2.98–2.89 (m, 2 H), 2.76 (dd, J = 14.5, 9.0 Hz, 1 H), 1.71–1.63 (m, 1 H), 1.59–1.51 (m, 1 H), 1.50–1.45 (m, 1 H), 1.44–1.36 (m, 1 H), 1.33–1.24 (m, 3 H), 0.83 (t, J = 7.4, Hz, 3 H), 0.82 (t, J = 7.4, Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 157.90, 155.81, 150.31, 144.35, 136.96, 130.44, 129.46, 128.79, 128.73, 128.22, 127.58, 124.59, 115.21, 109.44, 73.84, 72.80, 70.89, 70.21, 69.71, 55.48, 54.47, 53.33, 45.52, 39.07, 34.95, 25.99, 23.10, 22.86, 10.69, 10.42 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H46N3O10S, 712.2899; found 712.2915.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((4-methoxy-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (15b).
The same procedure was used as described above for compound 14b. Compound 9b (0.35 g, 0.56 mmol) was treated with TFA (3 mL), and the resulting deprotected amine was treated with DIPEA (0.22 g, 1.67 mmol) and bis-THF carbonate 13 (0.17 g, 0.62 mmol) to give compound 15b (0.30 g, 79%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 8.5 Hz, 2 H), 7.44–7.36 (m, 4 H), 7.35–7.31 (m, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.98 (d, J = 9.0 Hz, 2 H), 6.90 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.06–4.99 (m, 1 H), 5.02 (s, 2 H, overlapping), 4.91 (d, J = 9.0 Hz, 1 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89–3.78 (m, 3 H), 3.85 (s, 3 H, overlapping), 3.74–3.68 (m, 2 H), 3.63 (s, 1 H), 3.12 (dd, J = 15.0, 8.5 Hz, 1 H), 3.07–2.88 (m, 4 H), 2.82–2.73 (m, 2 H), 1.70–1.46 (m, 4 H), 1.13–1.03 (m, 1 H), 0.88–0.83 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.25, 157.74, 155.61, 137.04, 130.53, 129.80, 129.67, 129.64, 128.75, 128.17, 127.56, 115.05, 114.52, 109.43, 73.58, 72.76, 70.93, 70.14, 69.75, 57.55, 55.78, 55.22, 53.88, 45.47, 34.89, 33.61, 26.52, 25.97, 17.07, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H47N2O9S, 683.2997; found 683.2994.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((N-(2-ethylbutyl)-4-methoxyphenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (15c).
The same procedure was used as described above for compound 14b. Compound 9c (0.72 g, 1.12 mmol) was treated with TFA (6 mL), and the resulting deprotected amine was treated with DIPEA (0.48 g, 3.69 mmol) and bis-THF carbonate 13 (0.37 g, 1.35 mmol) to give compound 15c (0.63 g, 81%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 9.0 Hz, 2 H), 7.44–7.36 (m, 4 H), 7.35–7.30 (m, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.99 (d, J = 9.0 Hz, 2 H), 6.90 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.05–5.00 (m, 1 H), 5.02 (s, 2 H, overlapping), 4.86 (d, J = 9.0 Hz, 1 H), 3.95 (dd, J = 9.5, 6.5 Hz, 1 H), 3.88–3.77 (m, 3 H), 3.86 (s, 3 H, overlapping), 3.74–3.67 (m, 3 H), 3.12 (dd, J = 15.5, 8.5 Hz, 1 H), 3.08–2.98 (m, 2 H), 2.96–2.87 (m, 2 H), 2.84–2.74 (m, 2 H), 1.69–1.58 (m, 1 H), 1.56–1.50 (m, 1 H), 1.48–1.37 (m, 2 H), 1.35–1.24 (m, 3 H), 0.84 (t, J = 7.3 Hz, 3 H), 0.82 (t, J = 7.3 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 163.30, 157.77, 155.62, 137.08, 130.54, 129.69, 128.77, 128.18, 127.57, 115.09, 114.56, 109.44, 73.60, 73.14, 70.86, 70.17, 69.74, 55.80, 55.20, 55.09, 53.96, 45.48, 39.32, 35.03, 25.96, 23.23, 22.90, 10.78, 10.43 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C37H49N2O9S, 697.3154; found 697.3158.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-((4-(hydroxymethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (16b).
The same procedure was used as described above for compound 14b. Compound 11b (0.80 g, 1.28 mmol) was treated with TFA (6 mL), and the resulting deprotected amine was treated with DIPEA (0.50 g, 3.83 mmol) and bis-THF carbonate 13 (0.42 g, 1.53 mmol) to give compound 16b (0.79 g, 91%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.0 Hz, 2 H), 7.51 (d, J = 8.0 Hz, 2 H), 7.44–7.35 (m, 4 H), 7.35–7.30 (m, 1 H), 7.12 (d, J = 8.0 Hz, 2 H), 6.89 (d, J = 8.0 Hz, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.05–4.98 (m, 1 H), 5.02 (s, 2 H, overlapping), 4.94 (d, J = 8.0 Hz, 1 H), 4.77 (s, 2 H), 3.95 (app q, J = 11.0, Hz, 1 H), 3.88–3.78 (m, 3 H), 3.73–3.64 (m, 2 H), 3.10 (dd, J = 15.0, 8.0 Hz, 1 H), 3.06–2.86 (m, 4 H), 2.82 (dd, J = 13.0, 7.0 Hz, 1 H), 2.77 (dd, J = 13.5, 8.0 Hz, 1 H), 1.71–1.43 (m, 4 H), 1.09 (sept, J = 7.5 Hz, 1 H), 0.89–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.73, 155.62, 146.55, 137.04, 136.95, 130.50, 129.77, 128.75, 128.16, 127.71, 127.55, 127.29, 115.08, 109.44, 73.59, 72.70, 71.05, 70.15, 69.74, 64.24, 57.54, 55.27, 53.86, 45.46, 34.93, 33.58, 26.57, 25.99, 17.05, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H47N2O9S, 683.2997; found 683.2983.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-((4-(((tert-butyldimethylsilyl)oxy)methyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (17b).
A solution of 16b (0.40 g, 0.59 mmol) in anhydrous CH2Cl2 (15 mL) was cooled to 0 °C and treated with DIPEA (0.15 g, 1.17 mmol), DMAP (0.14 g, 1.17 mmol), and TBDMS-Cl (0.18 g, 1.17 mmol). The reaction was stirred at room temperature for 15 h, and then concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% MeOH/CH2Cl2) to give compound 17b (0.46 g, 98%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.5 Hz, 2 H), 7.48 (d, J = 8.5 Hz, 2 H), 7.44–7.36 (m, 4 H), 7.35–7.30 (m, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.90 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.05–5.00 (m, 1 H), 5.03 (s, 2 H, overlapping), 4.87 (d, J = 9.0 Hz, 1 H), 4.79 (s, 2 H), 3.96 (dd, J = 9.5 Hz, 6.5 Hz, 1 H), 3.88–3.78 (m, 3 H), 3.74–3.67 (m, 2 H), 3.60 (br s, 1 H), 3.15 (dd, J = 15.0 Hz, 8.0 Hz, 1 H), 3.06 (dd, J = 13.5 Hz, 8.0 Hz, 1 H), 3.02–2.95 (m, 2 H), 2.94–2.88 (m, 1 H), 2.83–2.75 (m, 2 H), 1.70–1.46 (m, 4 H), 1.12–1.02 (m, H), 0.95 (s, 9 H), 0.88–0.83 (m, 6 H), 0.12 (s, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.77, 155.63, 147.29, 137.08, 136.44, 130.53, 128.76, 128.16, 127.55, 126.52, 115.08, 109.43, 73.60, 72.76, 70.89, 70.15, 69.74, 64.24, 57.61, 55.27, 53.92, 45.48, 34.88, 33.63, 26.52, 26.03, 25.96, 18.52, 17.06, 11.17, −5.19 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C42H61N2O9SSi, 797.3862; found 797.3860.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)butan-2-yl)carbamate (18b).
The same procedure was used as described above for compound 14b. Compound 12b (1.24 g, 1.94 mmol) was treated with TFA (8 mL), and the resulting deprotected amine was treated with DIPEA (0.75 g, 5.81 mmol) and bis-THF carbonate 13 (0.58 g, 2.13 mmol) to give compound 18b (0.95 g, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.43–7.36 (m, 4 H), 7.35–7.30 (m, 2 H), 7.17 (d, J = 1.5 Hz, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.92–6.88 (m, 3 H), 6.07 (s, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.06–5.00 (m, 1 H), 5.03 (s, 2 H, overlapping), 4.89 (d, J = 9.0 Hz, 1 H), 3.97 (dd, J = 9.5, 6.5 Hz, 1 H), 3.88–3.79 (m, 3 H), 3.75–3.68 (m, 2 H), 3.55 (br s, 1 H), 3.11 (dd, J = 15.0, 8.5 Hz, 1 H), 3.06–2.88 (m, 4 H), 2.82–2.74 (m, 2 H), 1.71–1.46 (m, 4 H), ), 1.14–1.04 (m, 1 H), 0.89–0.84 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.76, 155.63, 151.71, 148.49, 137.07, 131.38, 130.51, 129.78, 128.75, 128.15, 127.55, 123.29, 115.06, 109.42, 108.54, 107.67, 102.53, 73.59, 72.84, 70.89, 70.14, 69.73, 57.61, 55.27, 53.93, 45.47, 34.93, 33.61, 26.54, 25.95, 17.07, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H45N2O10S, 697.2790; found 697.2786.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-(benzyloxy)phenyl)-4-(N-(2-ethylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)-3-hydroxybutan-2-yl)carbamate (18c).
The same procedure was used as described above for compound 14b. Compound 12c (1.04 g, 1.59 mmol) was treated with TFA (7 mL), and the resulting deprotected amine was treated with DIPEA (0.62 g, 4.77 mmol) and bis-THF carbonate 13 (0.47 g, 1.75 mmol) to give compound 18c (0.72 g, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.43–7.36 (m, 4 H), 7.35–7.31 (m, 2 H), 7.17 (d, J = 1.5 Hz, 1 H), 7.13 (d, J = 8.5 Hz, 2 H), 6.92–6.88 (m, 3 H), 6.08 (s, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.05–5.00 (m, 1 H), 5.02 (s, 2 H, overlapping), 4.85 (d, J = 8.5 Hz, 1 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.88–3.76 (m, 3 H), 3.74–3.64 (m, 3 H), 3.11 (dd, J = 15.5, 9.0 Hz, 1 H), 3.07–2.99 (m, 2 H), 2.98–2.88 (m, 2 H), 2.83 (dd, J = 13.5, 6.0 Hz, 1 H), 2.76 (dd, J = 14.5, 9.0 Hz, 1 H), 1.69–1.60 (m, 1 H), 1.55–1.49 (m, 1 H), 1.48–1.39 (m, 2 H), 1.35–1.24 (m, 3 H), 0.86–0.80 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.78, 155.63, 151.76, 148.53, 137.08, 131.20, 130.52, 129.77, 128.77, 128.18, 127.56, 123.34, 115.09, 109.43, 108.58, 107.71, 102.55, 73.62, 73.21, 70.82, 70.16, 69.74, 55.22, 55.17, 54.02, 45.47, 39.33, 35.07, 25.94, 23.22, 22.91, 10.78, 10.44 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C37H47N2O10S, 711.2946; found 711.2936.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-amino-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxy-1-(4-hydroxyphenyl)butan-2-yl)carbamate (19b).
A solution of compound 14b (0.80 g, 1.15 mmol) in a mixture of MeOH and EtOAc (1:1, 20 mL) at room temperature was treated with 10% Pd/C (0.20 g). The resulting mixture was stirred at room temperature under an atmosphere of hydrogen (balloons) for overnight. The reaction mixture was filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% MeOH/CH2Cl2 to give compound 19b (0.46 g, 69%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 9.0 Hz, 2 H), 7.07 (d, J = 8.0 Hz, 2 H), 6.74 (d, J = 8.5 Hz, 2 H), 6.68 (d, J = 8.5 Hz, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.13 (br s, 1 H), 5.03 (dd, J = 6.5 Hz, 1 H), 4.95 (d, J = 9.0 Hz, 1 H), 4.17 (br s, 2 H), 3.97 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89 (td, J = 8.0, 2.0 Hz, 1 H), 3.86–3.78 (m, 2 H), 3.75–3.69 (m, 2 H), 3.65 (br s, 1 H), 3.09 (dd, J = 15.0, 8.5 Hz, 1 H), 3.03–2.90 (m, 4 H), 2.80–2.72 (m, 2 H), 1.75–1.65 (m, 1 H), 1.63–1.55 (m, 2 H), 1.54–1.45 (m, 1 H), 1.13–1.03 (m, 1 H), 0.89–0.83 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 155.67, 154.59, 150.89, 135.28, 130.71, 129.69, 126.10, 115.55, 114.29, 109.47, 73.63, 72.79, 71.02, 69.78, 57.62, 55.29, 53.93, 45.51, 34.90, 33.65, 26.59, 26.00, 17.10, 11.19 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C28H40N3O8S, 578.2531; found 578.2514.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-amino-N-(2-ethylbutyl)phenyl)sulfonamido)-3-hydroxy-1-(4-hydroxyphenyl)butan-2-yl)carbamate (19c).
The same procedure was used as described above for compound 19b. Compound 14c (0.73 g, 1.03 mmol) was treated with 10% Pd/C (0.22 g) to give compound 19c (0.60 g, 98%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 8.5 Hz, 2 H), 7.07 (d, J = 8.5 Hz, 2 H), 6.73 (d, J = 8.5 Hz, 2 H), 6.69 (d, J = 9.0 Hz, 2 H), 5.66 (d, J = 5.5 Hz, 1 H), 5.14 (br s, 1 H), 5.03 (q, J = 6.0 Hz, 1 H), 4.92 (d, J = 8.5 Hz, 1 H), 4.17 (br s, 2 H), 3.95 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89 (td, J = 8.5, 2.5 Hz, 1 H), 3.85–3.77 (m, 2 H), 3.77–3.68 (m, 3 H), 3.09 (dd, J = 15.5, 8.5 Hz, 1 H), 3.05–2.96 (m, 2 H), 2.95–2.89 (m, 2 H), 2.81 (dd, J = 13.0, 6.0 Hz, 1 H), 2.75 (dd, J = 14.0, 8.0 Hz, 1 H), 1.75–1.65 (m, 1 H), 1.64–1.52 (m, 1 H), 1.48–1.38 (m, 2 H), 1.35–1.24 (m, 3 H), 0.83 (t, J = 7.3 Hz, 3 H), 0.82 (t, J = 7.3 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 155.66, 154.59, 150.89, 130.70, 129.72, 129.60, 125.95, 115.55, 114.30, 109.47, 73.62, 73.14, 70.96, 69.77, 55.26, 55.09, 53.95, 45.51, 39.32, 35.01, 25.98, 23.23, 22.92, 10.76, 10.44 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C29H42N3O8S, 592.2687; found 592.2700.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-1-(4-hydroxyphenyl)-4-((4-methoxy-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (20b).
The same procedure was used as described above for compound 19b. Compound 15b (0.60 g, 0.88 mmol) was treated with 10% Pd/C (0.12 g) to give compound 20b (0.32 g, 61%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 9.0, 2 H), 7.07 (d, J = 8.5 Hz, 2 H), 6.99 (d, J = 9.0 Hz, 2 H), 6.74 (d, J = 8.0 Hz, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.28 (s, 1 H), 5.03 (q, J = 6.0 Hz, 1 H), 4.97 (d, J = 8.5 Hz, 1 H), 3.96 (dd, J = 9.5, 6.0 Hz, 1 H), 3.89 (dd, J = 8.0, 2.0 Hz, 1 H), 3.87 (s, 3 H, overlapping), 3.86–3.80 (m, 2 H), 3.75–3.69 (m, 2 H), 3.63 (br s, 1 H), 3.12 (dd, J = 15.5, 8.5 Hz, 1 H), 3.06–2.90 (m, 4 H), 2.81–2.73 (m, 2 H), 1.75–1.65 (m, 1 H), 1.64–1.45 (m, 3 H), 1.13–1.04 (m, 1 H), 0.89–0.83 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.28, 155.72, 154.68, 130.67, 129.65, 129.50, 115.58, 114.55, 109.46, 73.68, 72.79, 71.02, 69.76, 57.54, 55.81, 55.35, 53.84, 45.54, 34.84, 33.62, 26.57, 26.00, 17.07, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C29H41N2O9S, 593.2528; found 593.2524.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((N-(2-ethylbutyl)-4-methoxyphenyl)sulfonamido)-3-hydroxy-1-(4-hydroxyphenyl)butan-2-yl)carbamate (20c).
The same procedure was used as described above for compound 19b. Compound 15c (0.63 g, 0.90 mmol) was treated with 10% Pd/C (0.12 g) to give compound 20c (0.52 g, 95%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 8.5 Hz, 2 H), 7.08 (d, J = 8.0 Hz, 2 H), 7.00 (d, J = 9.0 Hz, 2 H), 6.74 (d, J = 8.5 Hz, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.07 (br s, 1 H), 5.03 (q, J = 6.5 Hz, 1 H), 4.91 (d, J = 8.5 Hz, 1 H), 3.95 (dd, J = 10.0, 6.5 Hz, 1 H), 3.88 (td, J = 8.5, 2.0 Hz, 1 H), 3.88 (s, 3 H, overlapping), 3.84–3.78 (m, 2 H), 3.75–3.68 (m, 3 H), 3.11 (dd, J = 15.0, 8.0 Hz, 1 H), 3.07–2.89 (m, 4 H), 2.83 (dd, J = 13.0, 6.0 Hz, 1 H), 2.75 (dd, J = 14.0, 8.5 Hz, 1 H), 1.74–1.64 (m, 1 H), 1.57–1.51 (m, 1 H), 1.48–1.38 (m, 2 H), 1.35–1.24 (m, 3 H), 0.83 (t, J = 7.4 Hz, 3 H), 0.82 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 163.30, 155.67, 154.55, 130.70, 129.68, 129.61, 129.52, 115.57, 114.57, 109.46, 73.65, 73.13, 70.91, 69.75, 55.81, 55.26, 55.08, 53.93, 45.51, 39.31, 34.98, 25.97, 23.22, 22.90, 10.77, 10.42 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C30H43N2O9S, 607.2684; found 607.2698.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-1-(4-hydroxyphenyl)-4-(N-((S)-2-methylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)butan-2-yl)carbamate (22b).
The same procedure was used as described above for compound 19b. Compound 18b (0.92 g, 1.32 mmol) was treated with 10% Pd/C (0.10 g) to give compound 22b (0.69 g, 86%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 8.5, 2.0 Hz, 1 H), 7.17 (d, J = 1.5 Hz, 1 H), 7.09 (d, J = 8.0 Hz, 2 H), 6.90 (d, J = 8.0 Hz, 1 H), 6.75 (d, J = 8.5 Hz, 2 H), 6.09 (s, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.04 (q, J = 6.0 Hz, 1 H), 4.91 (d, J = 8.5 Hz, 1 H), 4.85 (br s, 1 H), 3.98 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89 (td, J = 8.0, 2.5 Hz, 1 H), 3.86–3.79 (m, 2 H), 3.76–3.69 (m, 2 H), 3.55 (br s, 1 H), 3.10 (dd, J = 15.5, 9.0 Hz, 1 H), 3.06–2.90 (m, 4 H), 2.81–2.73 (m, 2 H), 1.76–1.66 (m, 1 H), 1.64–1.46 (m, 3 H), 1.14–1.04 (m, 1 H), 0.90–0.83 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 155.69, 154.57, 151.74, 148.50, 131.36, 130.69, 129.60, 123.31, 115.61, 109.44, 108.58, 107.71, 102.55, 73.68, 72.88, 70.94, 69.75, 57.66, 55.32, 53.95, 45.50, 34.90, 33.64, 26.55, 25.98, 17.08, 11.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C29H39N2O10S, 607.2320; found 607.2319.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-(N-(2-ethylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)-3-hydroxy-1-(4-hydroxyphenyl)butan-2-yl)carbamate (22c).
The same procedure was used as described above for compound 19b. Compound 18c (0.72 g, 1.01 mmol) was treated with 10% Pd/C (0.07 g) to give compound 22c (0.56 g, 89%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.33 (dd, J = 8.0, 1.5 Hz, 1 H), 7.17 (d, J = 1.5 Hz, 1 H), 7.04 (d, J = 8.5 Hz, 2 H), 6.89 (d, J = 8.5 Hz, 1 H), 6.72 (d, J = 8.5 Hz, 2 H), 6.27 (br s, 1 H), 6.08 (s, 2 H), 5.64 (d, J = 5.0 Hz, 1 H), 5.14 (d, J = 8.5 Hz, 1 H), 5.02 (q, J = 6.0 Hz, 1 H), 3.93 (dd, J = 9.5, 6.0 Hz, 1 H), 3.88–3.77 (m, 3 H), 3.75–3.64 (m, 3 H), 3.11–2.96 (m, 4 H), 2.94–2.85 (m, 2 H), 2.69 (dd, J = 14.0, 9.0 Hz, 1 H), 1.70–1.60 (m, 1 H), 1.51–1.35 (m, 3 H), 1.34–1.26 (m, 3 H), 0.82 (t, J = 7.4 Hz, 3 H), 0.81 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 155.83, 154.96, 151.72, 148.48, 131.14, 130.55, 129.19, 123.31, 115.57, 109.46, 108.55, 107.66, 102.53, 73.69, 73.27, 71.00, 69.76, 55.47, 54.97, 53.79, 45.62, 39.14, 34.94, 25.98, 23.11, 22.87, 10.66, 10.41 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C30H41N2O10S, 621.2477; found 621.2473.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-amino-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxybutan-2-yl)carbamate (PU1).
A solution of compound 19b (0.17 g, 0.29 mmol) in anhydrous CH3CN (10 mL) at 0 °C was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.12 g, 0.38 mmol). Cesium carbonate (0.14 g, 0.44 mmol) was added portion-wise (4 portions over 20 mins). The resulting reaction mixture was stirred at 0 °C for 40 min, warmed to room temperature, stirred for 1 h, and then concentrated under reduced pressure. The residue was dissolved in EtOAc (25 mL) and washed with saturated aqueous NaCl solution (25 mL). The organic fraction was dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% MeOH/CH2Cl2), to give the target compound PU1 (0.18 g, 86%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 8.5 Hz, 2 H), 7.18 (d, J = 8.5 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 6.68 (d, J = 8.5 Hz, 2 H), 5.67 (d, J = 5.5 Hz, 1 H), 5.09–5.03 (m, 2 H), 4.48 (br s, 2 H), 4.30–4.17 (m, 6 H), 4.00 (dd, J = 9.5, 6.0 Hz, 1 H), 3.94–3.88 (m, 1 H), 3.87–3.74 (m, 3 H), 3.71 (dd, J = 9.5, 6.5 Hz, 1 H), 3.66 (br s, 1 H), 3.05–2.93 (m, 4 H), 2.81 (dd, J = 14.0, 7.5 Hz, 1 H), 2.68 (d, J = 13.5 Hz, 1 H), 2.61 (dd, J = 13.5, 6.5 Hz, 1 H), 1.80–1.69 (m, 2 H), 1.62–1.47 (m, 2 H), 1.40–1.35 (m, 6 H), 1.13–1.02 (m, 1 H), 0.88–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.62, 157.50, 155.46, 151.10, 131.17, 130.64, 129.56, 125.61, 114.68, 114.20, 109.40, 73.54, 73.02, 70.97, 69.72, 63.26, 63.21, 63.08, 63.03, 61.67, 57.70, 55.13, 54.20, 45.40, 34.90, 33.65, 26.48, 26.02, 17.12, 16.66, 16.61, 11.14 ppm; 31P NMR (202 MHz, CDCl3) δ 19.27 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H51N3O11PS, 728.2977; found 728.2984; Anal. HPLC: tR 10.50 min, purity 96%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-((4-methoxy-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (PU2).
The same procedure was used as described above for compound PU1. Compound 20b (0.33 g, 0.56 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.22 g, 0.72 mmol) and cesium carbonate (0.27 g, 0.84 mmol) to give the target compound PU2 (0.41 g, 99%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 9.0 Hz, 2 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.98 (d, J = 9.0 Hz, 2 H), 6.87 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.02 (q, J = 6.5 Hz, 1 H), 4.93 (d, J = 9.0 Hz, 1 H), 4.27–4.19 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.90–3.78 (m, 3 H), 3.87 (s, 3 H, overlapping), 3.75–3.67 (m, 2 H), 3.64 (br s, 1 H), 3.11 (dd, J = 15.0, 8.5 Hz, 1 H), 3.05–2.89 (m, 4 H), 2.83–2.75 (m, 2 H), 1.74–1.64 (m, 1 H), 1.63–1.54 (m, 2 H), 1.53–1.44 (m, 1 H), 1.36 (t, J = 7.0 Hz, 6 H), 1.13–1.03 (m, 1 H), 0.88–0.82 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 163.27, 157.73, 157.61, 155.59, 130.92, 130.62, 129.70, 129.64, 114.75, 114.52, 109.41, 73.60, 72.72, 70.94, 69.68, 63.12, 63.07, 63.01, 61.76, 57.51, 55.80, 55.21, 53.83, 45.45, 34.81, 33.61, 26.56, 25.98, 17.06, 16.63, 16.59, 11.17 ppm; 31P NMR (202 MHz, CDCl3) δ 19.22 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H52N2O12PS, 743.2973; found 743.2989; Anal. HPLC: tR 11.89 min, purity 95%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-(((tert-butyldimethylsilyl)oxy)methyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxybutan-2-yl)carbamate (24).
The same procedure was used as described above for compound PU1. Compound 21b (0.29 g, 0.41 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.16 g, 0.55 mmol) and cesium carbonate (0.20 g, 0.62 mmol) to give compound 24 (0.22 g, 62%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 8.5 Hz, 2 H), 7.47 (d, J = 8.0 Hz, 2 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.86 (d, J = 8.5 Hz, 2 H), 5.64 (d, J = 5.0 Hz, 1 H), 5.06–4.98 (m, 2 H), 4.79 (s, 2 H), 4.26–4.18 (m, 6 H), 3.94 (dd, J = 9.5, 6.0 Hz, 1 H), 3.89–3.78 (m, 3 H), 3.74–3.66 (m, 3 H), 3.12 (dd, J = 15.0, 8.0 Hz, 1 H), 3.07–2.95 (m, 3 H), 2.94–2.88 (m, 1 H), 2.84–2.74 (m, 2 H), 1.70–1.42 (m, 4 H), 1.38–1.32 (m, 6 H), 1.12–1.02 (m, 1 H), 0.94 (s, 9 H), 0.87–0.82 (m, 6 H), 0.11 (s, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.61, 157.49, 155.44, 147.36, 136.53, 131.21, 130.64, 127.53, 127.22, 114.71, 109.39, 73.55, 73.06, 71.01, 69.71, 63.98, 63.35, 63.29, 63.21, 63.16, 63.05, 61.68, 57.60, 55.28, 54.10, 45.37, 34.89, 33.58, 26.41, 26.01, 25.82, 25.77, 17.04, 16.62, 16.57, 11.11, −3.46 ppm; 31P NMR (202 MHz, CDCl3) δ 19.28 ppm.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-((4-(hydroxymethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)butan-2-yl)carbamate (PU3).
A solution of compound 24 (0.20 g, 0.23 mmol) in anhydrous THF (10 mL) was cooled to 0 °C and treated dropwise with a solution of TBAF (1M in THF) (0.28 mL, 0.28 mmol). After stirring at 0 °C for 2 h, the reaction was quenched with aqueous NH4Cl solution and extracted with EtOAc (25 mL). The organic layer was dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–20% MeOH/CH2Cl2), to give the target compound PU3 (0.16 g, 96%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 2 H), 7.48 (d, J = 8.0 Hz, 2 H), 7.17 (d, J = 8.5 Hz, 2 H), 6.86 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.21 (d, J = 8.5 Hz, 1 H), 5.04 (q, J = 6.0 Hz, 1 H), 4.73 (d, J = 6.0 Hz, 2 H), 4.27–4.17 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.91–3.79 (m, 3 H), 3.78–3.70 (m, 2 H), 3.69–3.63 (m, 2 H), 3.06–2.91 (m, H), 2.78 (dd, J = 14.0, 7.5 Hz, 1 H), 2.73–2.64 (m, 2 H), 1.77–1.67 (m, 2 H), 1.64–1.54 (m, 1 H), 1.53–1.43 (m, 1 H), 1.39–1.32 (m, 6 H), 1.11–1.01 (m, 1 H), 0.87–0.81 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.56, 157.45, 155.44, 147.34, 136.53, 131.21, 130.60, 127.50, 127.18, 114.67, 109.37, 73.50, 72.99, 71.00, 69.68, 63.91, 63.29, 63.24, 63.18, 63.13, 63.01, 61.64, 57.51, 55.28, 53.99, 45.38, 34.83, 33.53, 26.44, 26.01, 17.01, 16.59, 16.54, 11.10 ppm; 31P NMR (202 MHz, CDCl3) δ 19.34 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H52N2O12PS, 743.2973; found 743.2987; Anal. HPLC: tR 10.16 min, purity 98%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)butan-2-yl)carbamate (PU4).
The same procedure was used as described above for compound PU1. Compound 22b (0.69 g, 1.14 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.44 g, 1.48 mmol) and cesium carbonate (0.56 g, 1.71 mmol) to give the target compound PU4 (0.84 g, 98%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 8.0, 1.5 Hz, 1 H), 7.18–7.13 (m, 3 H), 6.91–6.86 (m, 3 H), 6.09 (s, 2 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.04 (q, J = 6.5 Hz, 1 H), 4.91 (d, J = 8.5 Hz, 1 H), 4.27–4.20 (m, 6 H), 3.98 (dd, J = 9.5, 6.5 Hz, 1 H), 3.88 (td, J = 8.5, 2.5 Hz, 1 H), 3.86–3.78 (m, 2 H), 3.76–3.68 (m, 2 H), 3.57 (br s, 1 H), 3.10 (dd, J = 15.0, 8.5 Hz, H), 3.05–2.90 (m, 4 H), 2.83–2.75 (m, 2 H), 1.75–1.65 (m, 1 H), 1.64–1.56 (m, 2 H), 1.54–1.45 (m, 1 H), 1.37 (t, J = 7.0 Hz, 6 H), 1.14–1.04 (m, 1 H), 0.90–0.83 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.77, 157.65, 155.61, 151.75, 148.52, 131.35, 130.88, 130.61, 123.30, 114.78, 109.41, 108.56, 107.66, 102.57, 73.64, 72.82, 70.90, 69.69, 63.12, 63.07, 63.02, 61.76, 57.64, 55.23, 53.96, 45.44, 34.86, 33.64, 26.55, 25.98, 17.08, 16.65, 16.61, 11.19 ppm; 31P NMR (202 MHz, CDCl3) δ 19.23 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H50N2O13PS, 757.2766; found 757.2778; Anal. HPLC: tR 11.69 min, purity 96%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-amino-N-(2-ethylbutyl)phenyl)sulfonamido)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxybutan-2-yl)carbamate (PU6).
The same procedure was used as described above for compound PU1. Compound 19c (0.60 g, 1.02 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.40 g, 1.32 mmol) and cesium carbonate (0.50 g, 1.53 mmol) to give the target compound PU6 (0.58 g, 77%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.5 Hz, 2 H), 7.18 (d, J = 8.5 Hz, 2 H), 6.89 (d, J = 9.0 Hz, 2 H), 6.70 (d, J = 8.5 Hz, 2 H), 5.67 (d, J = 5.0 Hz, 1 H), 5.05 (q, J = 6.5 Hz, 1 H), 5.01 (d, J = 9.0 Hz, 1 H), 4.51 (br s, 2 H), 4.30–4.17 (m, 6 H), 3.99 (dd, J = 9.5, 6.5 Hz, 1 H), 3.91 (td, J = 8.0, 2.5 Hz, 1 H), 3.88–3.72 (m, 4 H), 3.71 (dd, J = 9.5, 7.0 Hz, 1 H), 3.07–2.93 (m, 4 H), 2.81 (dd, J = 14.0, 7.5 Hz, 1 H), 2.70–2.63 (m, 2 H), 1.80–1.67 (m, 2 H), 1.50–1.33 (m, 8 H), 1.32–1.22 (m, 3 H), 0.83 (t, J = 7.3 Hz, 3 H), 0.82 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 157.64, 157.53, 155.46, 151.23, 131.15, 130.65, 129.61, 125.42, 114.71, 114.13, 109.41, 73.55, 73.31, 70.92, 69.72, 63.25, 63.20, 63.07, 63.02, 61.69, 55.21, 55.09, 54.21, 45.40, 39.32, 34.99, 26.01, 23.29, 22.82, 16.66, 16.62, 10.80, 10.36 ppm; 31P NMR (202 MHz, CDCl3) δ 19.27 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H53N3O11PS, 742.3133; found 742.3139; Anal. HPLC: tR 11.02 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-4-((N-(2-ethylbutyl)-4-methoxyphenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (PU7).
The same procedure was used as described above for compound PU1. Compound 20c (0.52 g, 0.86 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.34 g, 1.11 mmol) and cesium carbonate (0.42 g, 1.29 mmol) to give the target compound PU7 (0.60 g, 92%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 9.0 Hz, 2 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.99 (d, J = 9.0 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.02 (q, J = 6.5 Hz, 1 H), 4.85 (d, J = 9.0 Hz, 1 H), 4.27–4.20 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.90–3.85 (m, 1 H), 3.88 (s, 3 H, overlapping), 3.85–3.76 (m, 2 H), 3.75–3.67 (m, 3 H), 3.11 (dd, J = 15.0, 8.0 Hz, 1 H), 3.07–2.90 (m, 4 H), 2.85–2.76 (m, 2 H), 1.74–1.64 (m, 1 H), 1.60–1.54 (m, 1 H), 1.47–1.39 (m, 2 H), 1.37 (t, J = 7.0 Hz, 6 H), 1.33–1.25 (m, 3 H), 0.83 (t, J = 7.3 Hz, 3 H), 0.82 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 163.32, 157.76, 157.65, 155.59, 130.89, 130.63, 129.68, 129.52, 114.78, 114.57, 109.42, 73.63, 73.07, 70.88, 69.69, 63.14, 63.07, 63.02, 61.78, 55.82, 55.17, 55.08, 53.93, 45.45, 39.32, 34.94, 25.98, 23.21, 22.91, 16.65, 16.61, 10.77, 10.44 ppm; 31P NMR (202 MHz, CDCl3) δ 19.22 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H54N2O12PS, 757.3130; found 757.3140; Anal. HPLC: tR 12.47 min, purity 95%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-4-(N-(2-ethylbutyl)benzo[d][1,3]dioxole-5-sulfonamido)-3-hydroxybutan-2-yl)carbamate (PU9).
The same procedure was used as described above for compound PU1. Compound 22c (0.56 g, 0.90 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.35 g, 1.17 mmol) and cesium carbonate (0.44 g, 1.35 mmol) to give the target compound PU9 (0.69 g, 99%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 8.0, 2.0 Hz, 1 H), 7.18–7.13 (m, 3 H), 6.92–6.86 (m, 3 H), 6.09 (s, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.03 (q, J = 6.0 Hz, 1 H), 4.90 (d, J = 8.5 Hz, 1 H), 4.27–4.20 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.87 (td, J = 8.0, 2.5 Hz, 1 H), 3.85–3.76 (m, 2 H), 3.75–3.68 (m, 2 H), 3.68 (br s, 1 H, overlapping), 3.09 (dd, J = 15.5, 8.5 Hz, 1 H), 3.05–2.90 (m, 4 H), 2.84 (dd, J = 13.5, 6.0 Hz, 1 H), 2.78 (dd, J = 14.0, 8.5 Hz, 1 H), 1.73–1.64 (m, 1 H), 1.60–1.53 (m, 1 H), 1.48–1.38 (m, 2 H), 1.36 (t, J = 7.0 Hz, 6 H), 1.33–1.26 (m, 3 H), 0.83 (t, J = 7.5 Hz, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.75, 157.63, 155.60, 151.77, 148.52, 131.18, 130.90, 130.60, 123.32, 114.77, 109.40, 108.57, 107.68, 102.57, 73.62, 73.15, 70.83, 69.68, 63.11, 63.07, 63.02, 61.75, 55.19, 55.13, 53.98, 45.44, 39.30, 34.97, 25.96, 23.19. 22.90, 16.64, 16.60, 10.75, 10.44 ppm; 31P NMR (202 MHz, CDCl3) δ 19.22 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H52N2O13PS, 771.2922; found 771.2928; Anal. HPLC: tR 12.27 min, purity 96%.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-((tert-butoxycarbonyl)amino)-2-hydroxybutyl)(isobutyl)carbamate (25a).
A solution of amino alcohol 2a (5.04 g, 11.38 mmol) in anhydrous CH2Cl2 (50 mL) was treated at 0 °C with Et3N (2.30 g, 22.76 mmol) followed by dropwise addition of benzyl chloroformate (2.52 g, 14.77 mmol). The reaction mixture was warmed to room temperature and stirred for 15 h. The reaction was diluted with CH2Cl2 (150 mL) and washed with saturated aqueous NaHCO3 solution, water and saturated aqueous NaCl solution (100 mL each). The organic fraction was dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–100% EtOAc/Hexane) to give compound 25a (4.00 g, 61%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.45–7.41 (m, 2 H), 7.40–7.29 (m, 8 H), 7.15 (d, J = 7.5 Hz, 2 H), 6.91 (d, J = 8.0 Hz, 2 H), 5.15 (s, 2 H), 5.04 (s, 2 H), 4.65 (br s, 1 H), 4.47 (br s, 1 H), 3.81–3.68 (m, 2 H), 3.58–3.47 (m, 1 H), 3.35–3.06 (m, 3 H), 2.95–2.68 (m, 2 H), 1.95–1.81 (m, 1 H), 1.37 (s, 9 H), 0.91–0.81 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.85, 157.59, 156.08, 137.26, 136.51, 130.69, 130.22, 128.68, 128.64, 128.20, 128.03, 127.96, 127.56, 114.94, 79.60, 73.96, 70.14, 67.71, 56.01, 54.96, 52.31, 34.96, 28.40, 27.60, 20.15, 20.06 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H45N2O6, 577.3272; found 577.3261.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-((tert-butoxycarbonyl)amino)-2-hydroxybutyl)((S)-2-methylbutyl)carbamate (25b).
The same procedure was used as described above for compound 25a. Amino alcohol 2b (2.28 g, 5.00 mmol) was treated with Et3N (1.01 g, 10.0 mmol) and benzyl chloroformate (1.11 g, 6.50 mmol) to give compound 25b (1.9 g, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.41 (m, 2 H), 7.40–7.29 (m, 8 H), 7.15 (d, J = 7.5 Hz, 2 H), 6.91 (d, J = 8.0 Hz, 2 H), 5.14 (s, 2 H), 5.04 (s, 2 H), 4.62 (br s, 1 H), 4.48 (br s, 1 H), 3.81–3.66 (m, 2 H), 3.57–3.47 (m, 1 H), 3.29 (d, J = 14.0 Hz, 1 H), 3.18 (dd, J = 13.5, 6.5 Hz, 1 H), 3.09 (dd, J = 13.5, 7.5 Hz, 1 H), 2.94–2.68 (m, 2 H), 1.71–1.59 (m, 1 H), 1.42–1.28 (m, 1 H), 1.36 (s, 9 H, overlapping), 1.13–0.98 (m, 1 H), 0.92–0.76 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.92, 157.62, 156.11, 137.26, 136.50, 130.71, 130.19, 128.70, 128.64, 128.22, 128.05, 127.58, 127.11, 114.96, 79.62, 73.94, 70.16, 67.74, 65.50, 54.93, 54.62, 52.28, 34.98, 33.98, 28.42, 26.87, 16.95, 11.39 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H47N2O6, 591.3429; found 591.3419.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-((tert-butoxycarbonyl)amino)-2-hydroxybutyl)(2-ethylbutyl)carbamate (25c).
The same procedure was used as described above for compound 25a. Amino alcohol 2c (4.22 g, 8.98) was treated with Et3N (1.82 g, 18.0 mmol) and benzyl chloroformate (1.99 g, 11.68 mmol) to give compound 25c (2.87 g, 53%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.45–7.41 (m, 2 H), 7.41–7.29 (m, 8 H), 7.15 (d, J = 7.5 Hz, 2 H), 6.91 (d, J = 8.0 Hz, 2 H), 5.15 (s, 2 H), 5.04 (s, 2 H), 4.63 (br s, 1 H), 4.54 (br s, 1 H), 3.82–3.68 (m, 2 H), 3.56–3.46 (m, 1 H), 3.36–3.14 (m, 3 H), 2.96–2.76 (m, 2 H), 1.54–1.44 (m, 1 H), 1.36 (s, 9 H), 1.31–1.17 (m, 4 H), 0.90–0.75 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.89, 157.60, 156.09, 137.25, 136.43, 130.70, 130.19, 128.61, 128.23, 128.12, 128.03, 127.71, 127.56, 127.07, 114.94, 79.61, 73.98, 67.76, 65.41, 54.96, 52.26, 52.01, 39.65, 35.05, 28.40, 23.16, 23.12, 10.69 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H49N2O6, 605.3585; found 605.3573.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl)(isobutyl)carbamate (26a).
A solution of compound 25a (5.50 g, 9.53 mmol) in anhydrous CH2Cl2 (40 mL) was treated at room temperature with TFA (20 mL). After stirring the mixture at room temperature for 2 h, the solvent was evaporated under reduced pressure. The residue was dissolved in toluene (35 mL), and the solution was concentrated under reduced pressure. The resulting amine was dissolved in anhydrous CH3CN (40 mL) and treated at 0 °C with DIPEA (4.92 g, 38.12 mmol) and bis-THF carbonate derivative 13 (3.10 g, 11.44 mmol). After 15 min, the reaction mixture was warmed to room temperature and stirred for 24 h. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography using a silica gel column (RediSep Gold, 80 g, gradient elution with 20–100% EtOAc/hexanes) to give compound 14b (3.00 g, 50%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.30 (m, 10 H), 7.11 (d, J = 7.0 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.15 (s, 2 H), 5.06–4.97 (m, 2 H), 5.02 (s, 2 H, overlapping), 4.24 (br s, 1 H), 3.96 (dd, J = 9.0, 6.5 Hz, 1 H), 3.91–3.81 (m, 3 H), 3.74–3.66 (m, 2 H), 3.60 (dd, J = 14.5, 9.5 Hz, 1 H), 3.21 (d, J = 14.5 Hz, 1 H), 3.16–3.05 (m, 2 H), 2.98–2.87 (m, 2 H), 2.79–2.70 (m, 1 H), 1.94–1.82 (m, 1 H), 1.69–1.58 (m, 1 H), 1.57–1.49 (m, 1 H), 0.86 (d, J = 6.0 Hz, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 159.02, 157.69, 155.60, 137.10, 136.37, 130.47, 130.00, 128.74, 128.70, 128.31, 128.14, 128.02, 127.55, 114.98, 109.43, 74.01, 73.47, 70.93, 70.14, 69.73, 67.88, 56.14, 55.61, 52.45, 45.51, 34.71, 27.68, 25.96, 20.15, 20.07 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H45N2O8, 633.3171; found 633.3165.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl)((S)-2-methylbutyl)carbamate (26b).
The same procedure was used as described above for compound 26a. Compound 25b (1.09 g, 3.22 mmol) was treated with TFA (8 mL), and the resulting deprotected amine was treated with DIPEA (1.66 g, 12.86 mmol) and bis-THF carbonate 13 (1.05 g, 3.86 mmol) to give compound 26b (1.55 g, 75%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.30 (m, 10 H), 7.11 (d, J = 7.0 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.15 (s, 2 H), 5.10–5.00 (m, 2 H), 5.02 (s, 2 H, overlapping), 4.27 (br s, 1 H), 3.95 (dd, J = 9.5, 6.5 Hz, 1 H), 3.91–3.81 (m, 3 H), 3.74–3.66 (m, 2 H), 3.57 (dd, J = 14.0, 8.5 Hz, 1 H), 3.30–3.15 (m, 2 H), 3.11 (dd, J = 13.5, 7.5 Hz, 1 H), 2.99–2.86 (m, 2 H), 2.78–2.70 (m, 1 H), 1.69–1.57 (m, 2 H), 1.56–1.46 (m, 1 H), 1.40–1.29 (m, 1 H), 1.13–1.00 (m, 1 H), 0.89–0.80 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.93, 157.66, 155.58, 137.10, 136.36, 130.47, 130.01, 128.72, 128.66, 128.29, 128.12, 128.03, 127.53, 114.95, 109.43, 73.89, 73.44, 70.92, 70.12, 69.71, 67.85, 55.60, 54.63, 52.27, 45.52, 34.73, 33.99, 26.83, 25.95, 16.96, 11.36 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C37H47N2O8, 647.3327; found 647.3326.
Benzyl ((2R,3S)-4-(4-(benzyloxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl)(2-ethylbutyl)carbamate (26c).
The same procedure was used as described above for compound 26a. Compound 25c (2.87 g, 4.75 mmol) was treated with TFA (12 mL), and the resulting deprotected amine was treated with DIPEA (2.45 g, 19.0 mmol) and bis-THF carbonate 13 (1.54 g, 5.69 mmol) to give compound 26c (2.1 g, 67%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.44–7.30 (m, 10 H), 7.17 (d, J = 8.5 Hz, 2 H), 6.89 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.15 (s, 2 H), 5.06–4.99 (m, 1 H), 5.02 (s, 2 H, overlapping), 4.98–4.89 (m, 1 H), 4.34 (br s, 1 H), 3.99–3.92 (m, 1 H), 3.90–3.77 (m, 3 H), 3.74–3.66 (m, 2 H), 3.63–3.53 (m, 1 H), 3.25–3.11 (m, 3 H), 3.01–2.87 (m, 2 H), 2.79–2.68 (m, 1 H), 1.69–1.43 (m, 3 H), 1.33–1.16 (m, 4 H), 0.86–0.78 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 159.02, 157.66, 155.56, 137.08, 136.28, 130.47, 128.71, 128.64, 128.32, 128.15, 128.11, 127.52, 114.95, 109.41, 74.08, 73.44, 70.87, 70.11, 69.70, 67.90, 55.52, 52.37, 52.05, 45.50, 45.01, 39.71, 34.80, 25.93, 23.17, 23.08, 10.70, 10.63 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C38H49N2O8, 661.3484; found 661.3474.
tert-Butyl ((2R,3S)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-(4-hydroxyphenyl)butyl)(isobutyl)carbamate (27a).
A solution of compound 26a (1.00 g, 1.58 mmol) in a mixture of EtOH and EtOAc (1:1, 10 mL) was treated at room temperature with Pd(OH)2 (20% Pd on activated charcoal) (0.25 g). The reaction mixture was stirred at room temperature under an atmosphere of hydrogen (balloons) for 3 h, filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The resulting amino phenol was dissolved in a mixture of 1,4-dioxane and water (1:1, 10 mL), cooled to 0 °C, and treated with Na2CO3 (0.20 g, 1.90 mmol) followed by di-tert-butyl dicarbonate (0.41 g, 1.90 mmol). The reaction was stirred at 0 °C for 30 min, and then warmed to room temperature and stirred for 2 h. The reaction was diluted with EtOAc (50 mL) and washed with water (25 mL) and saturated aqueous NaCl solution (25 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–100% EtOAc/hexanes) to give compound 27a (0.90 g, 77%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.04 (d, J = 8.5 Hz, 2 H), 6.71 (d, J = 8.0 Hz, 2 H), 6.01 (br s, 1 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.19 (d, J = 6.5 Hz, 1 H), 5.03 (q, J = 7.5 Hz, 1 H), 4.56 (br s, 1 H), 3.95 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89–3.80 (m, 3 H), 3.75–3.66 (m, 2 H), 3.63–3.53 (m, 1 H), 3.14 (d, J = 14.0 Hz, 1 H), 3.04 (d, J = 6.5 Hz, 2 H), 2.95–2.87 (m, 2 H), 2.70 (dd, J = 13.5, 9.5 Hz, 1 H), 1.93–1.80 (m, 1 H), 1.71–1.61 (m, 1 H), 1.55–1.43 (m, 1 H), 1.47 (s, 9 H, overlapping), 0.88 (d, J = 6.7 Hz, 3 H), 0.87 (d, J = 6.5 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 158.70, 155.69, 154.81, 130.56, 129.53, 115.47, 109.47, 80.99, 74.08, 73.53, 71.04, 69.76, 56.50, 55.82, 51.93, 45.59, 34.62, 28.50, 27.84, 26.01, 20.23, 20.15 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C26H41N2O8, 509.2858; found 509.2841.
tert-Butyl ((2R,3S)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-(4-hydroxyphenyl)butyl)((S)-2-methylbutyl)carbamate (27b).
The same procedure was used as described above for compound 27a. Compound 26b (0.95 g, 1.47 mmol) was treated with Pd(OH)2 (0.24 g) and hydrogen gas, and the resulting deprotected amine was treated with Na2CO3 (0.19 g, 1.76 mmol) and di-tert-butyl dicarbonate (0.38 g, 1.76 mmol) to give compound 27b (0.70 g, 91%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.05 (d, J = 8.0 Hz, 2 H), 6.72 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.09 (d, J = 6.5 Hz, 1 H), 5.04 (q, J = 6.5 Hz, 1 H), 3.96 (dd, J = 10.0, 6.5 Hz, 1 H), 3.90–3.79 (m, 3 H), 3.75–3.67 (m, 2 H), 3.62–3.53 (m, 1 H), 3.17–3.08 (m, 2 H), 3.06–2.98 (m, 1 H), 2.95–2.88 (m, 2 H), 2.72 (dd, J = 13.5, 9.5 Hz, 1 H), 1.73–1.58 (m, 2 H), 1.57–1.50 (m, 1 H), 1.47 (s, 9 H), 1.41–1.32 (m, 1 H), 1.14–1.04 (m, 1 H), 0.89 (t, J = 7.5 Hz, 3 H), 0.84 (d, J = 6.5 Hz, 3 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.80, 155.63, 154.63, 130.61, 129.74, 115.46, 109.47, 80.98, 74.10, 73.52, 71.00, 69.76, 55.73, 54.98, 51.92, 45.56, 34.66, 34.15, 28.52, 26.91, 26.00, 17.05, 11.43 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H43N2O8, 523.3014; found 523.3006.
tert-Butyl (2-ethylbutyl)((2R,3S)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-(4-hydroxyphenyl)butyl)carbamate (27c).
The same procedure was used as described above for compound 27a. Compound 26c (2.10 g, 3.18 mmol) was treated with Pd(OH)2 (0.40 g) and hydrogen gas, and the resulting deprotected amine was treated with Na2CO3 (0.34 g, 3.81 mmol) and di-tert-butyl dicarbonate (0.83 g, 3.81 mmol) to give compound 27c (1.40 g, 82%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.02 (d, J = 8.5 Hz, 2 H), 6.70 (d, J = 8.0 Hz, 2 H), 5.63 (d, J = 5.0 Hz, 1 H), 5.24 (d, J = 7.5 Hz, 1 H), 5.02 (q, J = 6.5 Hz, 1 H), 3.93 (dd, J = 10.0, 6.5 Hz, 1 H), 3.88–3.78 (m, 3 H), 3.74–3.64 (m, 2 H), 3.58–3.47 (m, 1 H), 3.29–3.07 (m, 3 H), 2.97–2.86 (m, 2 H), 2.67 (dd, J = 14.0, 9.5 Hz, 1 H), 1.70–1.58 (m, 1 H), 1.55–1.40 (m, 2 H), 1.47 (s, 9 H, overlapping), 1.34–1.20 (m, 4 H), 0.85 (t, J = 7.4 Hz, 3 H), 0.84 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 158.66, 155.73, 155.00, 130.52, 129.30, 115.47, 109.46, 81.04, 74.07, 73.54, 71.01, 69.75, 55.79, 52.31, 51.82, 45.62, 39.78, 34.75, 28.51, 26.01, 23.18, 23.09, 10.81, 10.69 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C28H45N2O8, 537.3171; found 537.3166.
tert-Butyl ((2R,3S)-4-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl)(isobutyl)carbamate (28a).
A solution of compound 27a (0.50 g, 0.98 mmol) in anhydrous CH3CN (15 mL) at 0 °C was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.38 g, 1.28 mmol). Cesium carbonate (0.48 g, 1.47 mmol) was added portion-wise (4 portions over 20 mins). The resulting reaction mixture was stirred at 0 °C for 40 min, warmed to room temperature and stirred for 1 h, and then concentrated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and washed with saturated aqueous NaCl solution (50 mL). The organic fraction was dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% MeOH/CH2Cl2), to give compound 28a (0.53 g, 82%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.13 (d, J = 8.5 Hz, 2 H), 6.86 (d, J = 8.5 Hz, 2 H), 5.64 (d, J = 5.0 Hz, 1 H), 5.09–4.98 (m, 2 H), 4.60 (br s, 1 H), 4.26–4.18 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.89–3.78 (m, 3 H), 3.75–3.66 (m, 2 H), 3.63–3.54 (m, 1 H), 3.12–2.98 (m, 3 H), 2.96–2.88 (m, 2 H), 2.74 (dd, J = 13.5, 9.5 Hz, 1 H), 1.91–1.78 (m, 1 H), 1.71–1.61 (m, 1 H), 1.59–1.51 (m, 1 H), 1.46 (s, 9 H), 1.36 (t, J = 7.0 Hz, 6 H), 0.87 (d, J = 6.5 Hz, 3 H), 0.86 (d, J = 6.5 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 158.70. 157.61, 157.50, 155.48, 131.30, 130.54, 114.62, 109.40, 80.89, 74.18, 73.40, 70.93, 69.67, 63.08, 63.05, 63.00, 61.72, 56.48, 55.60, 53.55, 51.97, 45.46, 34.55, 28.48, 27.84, 25.96, 20.21, 20.14, 16.62, 16.57 ppm; 31P NMR (202 MHz, CDCl3) δ 19.28 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C31H52N2O11P, 659.3303; found 659.3304.
tert-Butyl ((2R,3S)-4-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl)((S)-2-methylbutyl)carbamate (28b).
The same procedure was used as described above for compound 28a. Compound 27b (0.28 g, 0.53 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.21 g, 0.70 mmol) and cesium carbonate (0.26 g, 0.80 mmol) to give compound 28b (0.30 g, 83%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.12 (d, J = 8.5 Hz, 2 H), 6.85 (d, J = 8.5 Hz, 2 H), 5.63 (d, J = 5.5 Hz, 1 H), 5.08–4.98 (m, 2 H), 4.25–4.17 (m, 6 H), 3.94 (dd, J = 9.5, 6.0 Hz, 1 H), 3.88–3.80 (m, 2 H), 3.79–3.73 (m, 1 H), 3.72–3.65 (m, 2 H), 3.60–3.50 (m, 1 H), 3.15–3.03 (m, 3 H), 2.97–2.87 (m, 2 H), 2.72 (dd, J = 14.0, 9.5 Hz, 1 H), 1.70–1.60 (m, 1 H), 1.57–1.50 (m, 1 H), 1.45 (s, 9 H), 1.35 (t, J = 7.0 Hz, 6 H), 1.34–1.18 (m, 3 H), 0.86–0.81 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.70, 157.57, 157.46, 155.45, 131.28, 130.54, 114.58, 109.37, 80.90, 74.20, 73.36, 70.87, 69.64, 63.04, 63.02, 62.97, 61.69, 55.51, 52.24, 51.91, 47.81, 45.44, 39.78, 34.63, 28.48, 25.93, 23.14, 23.07, 16.59, 16.54, 10.78, 10.67 ppm; 31P NMR (202 MHz, CDCl3) δ 19.26 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C32H54N2O11P, 673.3460; found 673.3454.
tert-Butyl ((2R,3S)-4-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxybutyl) (2-ethylbutyl)carbamate (28c).
The same procedure was used as described above for compound 28a. Compound 27c (0.50 g, 0.93 mmol) was treated with (diethoxyphosphoryl)methyl trifluoromethanesulfonate 23 (0.36 g, 1.21 mmol) and cesium carbonate (0.46 g, 1.40 mmol) to give compound 28c (0.56 g, 88%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.14 (d, J = 8.5 Hz, 2 H), 6.87 (d, J = 9.0 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.03 (q, J = 6.5 Hz, 1 H), 4.98 (d, J = 8.0 Hz, 1 H), 4.68 (br s, 1 H), 4.27–4.19 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.90–3.81 (m, 2 H), 3.80–3.75 (m, 1 H), 3.74–3.66 (m, 2 H), 3.63–3.54 (m, 1 H), 3.17–3.03 (m, 3 H), 2.98–2.89 (m, 2 H), 2.75 (dd, J = 14.0, 9.5 Hz, 1 H), 1.72–1.62 (m, 1 H), 1.60–1.52 (m, 1 H), 1.51–1.43 (m, 1 H), 1.47 (s, 9 H, overlapping), 1.36 (t, J = 7.0 Hz, 6 H), 1.32–1.21 (m, 4 H), 0.82 (t, J = 7.4 Hz, 3 H), 0.81 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 158.48, 157.46, 157.34, 155.39, 131.33, 130.47, 114.47, 109.30, 80.72, 74.01, 73.25, 70.80, 69.55, 62.96, 62.91, 61.59, 55.47, 52.08, 51.69, 45.41, 39.70, 34.55, 28.40, 25.86, 23.09, 22.99, 16.51, 16.46, 10.71, 10.60 ppm; 31P NMR (202 MHz, CDCl3) δ 19.24 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H56N2O11P, 687.3616; found 687.3600.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-(N-isobutylbenzo[d][1,3]dioxole-5-sulfonamido)butan-2-yl)carbamate (PD4).
A solution of compound 28a (0.15 g, 0.23 mmol) in anhydrous CH2Cl2 (4 mL) was treated at room temperature with TFA (2 mL). After stirring for 2 h, the reaction was concentrated under reduced pressure. The residue was dissolved in toluene (5 mL) and concentrated under reduced pressure. The resulting deprotected amine was dissolved in EtOAc (5 mL) and treated with a solution of Na2CO3 (0.05 g, 0.46 mmol) in water (5 mL) followed by 1,3-benzodioxole-5-sulfonyl chloride 6 (0.065 g, 0.30 mmol). The biphasic reaction mixture was stirred at room temperature overnight. The layers were separated, and the aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with saturated aqueous NaCl solution (20 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 15 g, gradient elution with 0–10% MeOH/d CH2Cl2), to give the target compound PD4 (0.14 g, 83%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 8.5, 2.0 Hz, 1 H), 7.17–7.12 (m, 3 H), 6.91–6.86 (m, 3 H), 6.09 (s, 2 H), 5.65 (d, J = 5.0 Hz, 1 H), 5.03 (q, J = 6.5 Hz, 1 H), 4.99 (d, J = 9.0 Hz, 1 H, overlapping), 4.27–4.19 (m, 6 H), 3.96 (dd, J = 9.5, 6.5 Hz, 1 H), 3.88 (dd, J = 8.5, 2.5 Hz, 1 H), 3.86–3.78 (m, 2 H), 3.75–3.67 (m, 2 H), 3.64 (br s, 1 H), 3.11 (dd, J = 15.5, 8.5 Hz, 1 H), 3.04–2.89 (m, 4 H), 2.83–2.73 (m, 2 H), 1.83 (sept, J = 6.5 Hz, 1 H), 1.73–1.63 (m, 1 H), 1.60–1.53 (m, 1 H), 1.36 (t, J = 7.5 Hz, 6 H), 0.92 (d, J = 6.5 Hz, 3 H), 0.88 (d, J = 6.5 Hz, 3 H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.70, 157.59, 155.60, 151.72, 148.49, 131.46, 130.98, 130.57, 123.24, 114.73, 109.39, 108.53, 107.61, 102.55, 73.59, 72.91, 70.90, 69.68, 63.06, 63.01, 61.72, 59.03, 55.28, 53.95, 45.44, 34.82, 27.41, 25.96, 20.27, 20.02, 16.62, 16.58 ppm; 31P NMR (202 MHz, CDCl3) δ 19.24 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H48N2O13PS, 743.2609; found 743.2623; Anal. HPLC: tR 11.13 min, purity 98%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-(N-isobutylbenzo[d]thiazole-6-sulfonamido)butan-2-yl)carbamate (PD5).
The same procedure was used as described above for compound PD4. Compound 28a (0.15 g, 0.23 mmol) was treated with TFA (2 mL), and the resulting deprotected amine was treated with Na2CO3 (0.064 g, 0.60 mmol) and 1,3-benzothiazole-6-sulfonyl chloride 7 (0.07 g, 0.30 mmol) to give the target compound PD5 (0.14 g, 81%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.20 (s, 1 H), 8.45 (d, J = 1.5 Hz, 1 H), 8.23 (d, J = 8.5 Hz, 1 H), 7.88 (dd, J = 8.5, 1.5 Hz, 1 H), 7.12 (d, J = 8.5 Hz, 2 H), 6.84 (d, J = 8.5 Hz, 2 H), 5.62 (d, J = 5.5 Hz, 1 H), 5.15 (d, J = 9.0 Hz, H), 4.99 (q, J = 6.0 Hz, 1 H), 4.25–4.17 (m, 6 H), 3.91 (dd, J = 10.0, 6.5 Hz, 1 H), 3.89–3.80 (m, 3 H, overlapping), 3.76 (d, J = 3.0 Hz, 1 H), 3.70–3.64 (m, 2 H), 3.19 (dd, J = 15.0, 8.0 Hz, 1 H), 3.13 (dd, J = 14.5, 2.0 Hz, 1 H, overlapping), 3.04–2.97 (m, 2 H), 2.95–2.85 (m, 2 H), 2.74 (dd, J = 14.5, 9.5 Hz, 1 H), 1.86 (sept, J = 7.0 Hz, 1 H), 1.68–1.58 (m, 1 H), 1.52–1.45 (m, 1 H), 1.34 (t, J = 7.0 Hz, 6 H), 0.89 (d, J = 6.5 Hz, 3 H), 0.87 (d, J = 6.5 Hz, 3 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.21, 157.61, 157.50, 155.67, 155.62, 135.79, 134.48, 131.00, 130.50, 124.90, 124.47, 122.34, 114.64, 109.36, 73.54, 72.75, 70.92, 69.61, 63.05, 63.00, 61.66, 58.65, 55.39, 53.55, 45.48, 34.65, 27.28, 25.92, 20.19, 20.00, 16.57, 16.53 ppm; 31P NMR (202 MHz, CDCl3) δ 19.20 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H47N3O11PS2, 756.2384; found 756.2392; Anal. HPLC: tR 10.55 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-3-hydroxy-4-(N-((S)-2-methylbutyl)benzo[d]thiazole-6-sulfonamido)butan-2-yl)carbamate (PU5).
The same procedure was used as described above for compound PD4. Compound 28b (0.30 g, 0.45 mmol) was treated with TFA (3 mL), and the resulting deprotected amine was treated with Na2CO3 (0.096 g, 0.90 mmol) and 1,3-benzothiazole-6-sulfonyl chloride 7 (0.135 g, 0.58 mmol) to give the target compound PU5 (0.26 g, 75%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.20 (s, 1 H), 8.45 (d, J = 1.5 Hz, 1 H), 8.22 (d, J = 8.5 Hz, 1 H), 7.87 (dd, J = 8.5, 1.5 Hz, 1 H), 7.12 (d, J = 8.5 Hz, 2 H), 6.83 (d, J = 8.5 Hz, 2 H), 5.60 (d, J = 5.0 Hz, 1 H), 5.17 (d, J = 8.5 Hz, 1 H), 4.98 (q, J = 6.5 Hz, 1 H), 4.24–4.16 (m, 6 H), 3.89 (dd, J = 9.5, 6.0 Hz, 1 H), 3.86–3.78 (m, 4 H), 3.69–3.62 (m, 2 H), 3.18–3.09 (m, 2 H), 3.08–2.93 (m, 3 H), 2.91–2.84 (m, 1 H), 2.73 (dd, J = 13.5, 9.0 Hz, 1 H), 1.67–1.56 (m, 1 H), 1.52–1.42 (m, 2 H), 1.33 (t, J = 7.0 Hz, 6 H), 1.31–1.23 (m, 2 H), 0.80–0.76 (m, 6 H) ppm; 13C NMR (126 MHz, CDCl3) δ 158.21, 157.78, 157.67, 155.83, 155.66, 135.45, 134.61, 130.58, 124.98, 124.63, 122.46, 114.80, 109.40, 73.70, 73.02, 70.84, 69.67, 63.12, 63.08, 63.03, 61.76, 55.29, 55.01, 53.84, 45.46, 39.29, 34.89, 25.96, 23.16, 22.89, 16.65, 16.60, 10.74, 10.42 ppm; 31P NMR (202 MHz, CDCl3) δ 19.18 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H49N3O11PS2, 770.2541; found 770.2563; Anal. HPLC: tR 11.78 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-4-((N-(2-ethylbutyl)-4-formylphenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (29).
The same procedure was used as described above for compound PD4. Compound 28c (0.25 g, 0.36 mmol) was treated with TFA (3 mL), and the resulting deprotected amine was treated with Na2CO3 (0.077 g, 0.73 mmol) and 4-formylbenzenesulfonyl chloride 5 (0.10 g, 0.47 mmol) to give compound 29 (0.18 g, 66%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 10.11 (s, 1 H), 8.03 (d, J = 8.5 Hz, 2 H), 7.94 (d, J = 8.5 Hz, 2 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 5.64 (d, J = 5.0 Hz, 1 H), 5.01 (q, J = 6.5 Hz, 1 H), 4.97 (d, J = 8.0 Hz, 1 H), 4.26–4.19 (m, 6 H), 3.93 (dd, J = 9.5, 6.5 Hz, 1 H), 3.86 (td, J = 8.0, 2.0 Hz, 1 H), 3.84–3.78 (m, 2 H, overlapping), 3.73–3.66 (m, 2 H), 3.63 (br s, 1 H), 3.15 (dd, J = 15.5, 8.0 Hz, 1 H), 3.11–3.04 (m, 2 H), 3.01 (dd, J = 14.0, 2.5 Hz, 1 H), 2.98–2.89 (m, 2 H), 2.77 (dd, J = 14.0, 8.5 Hz, 1 H), 1.72–1.62 (m, 1 H), 1.57–1.51 (m, 1 H), 1.50–1.41 (m, 1 H), 1.36 (t, J = 7.0 Hz, 6 H), 1.32–1.22 (m, 4 H), 0.83 (t, J = 7.5 Hz, 3 H), 0.82 (t, J = 7.5 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 190.88, 157.76, 157.64, 155.68, 143.46, 139.10, 130.78, 130.55, 130.38, 128.14, 114.79, 109.40, 73.71, 72.91, 70.91, 69.65, 63.11, 63.09, 63.04, 61.75, 55.32, 54.61, 53.49, 45.48, 39.07, 34.81, 25.97, 23.06, 22.84, 16.62, 16.58, 10.64, 10.40 ppm; 31P NMR (202 MHz, CDCl3) δ 19.17 ppm.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-4-((N-(2-ethylbutyl)-4-(hydroxymethyl)phenyl)sulfonamido)-3-hydroxybutan-2-yl)carbamate (PU8).
A solution of compound 29 (0.18 g, 0.23 mmol) in anhydrous THF (7 mL) was cooled to −10 °C and NaBH4 (0.012 g, 0.28 mmol) was added. The reaction was stirred at −10 °C for 30 min, quenched with saturated aqueous NH4Cl solution (5 mL), and extracted with EtOAc (2 × 15 mL). The combined organic portion was washed with saturated aqueous NaCl solution, dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a silica gel column (RediSep Gold, 12 g, gradient elution with 0–20% MeOH/CH2Cl2), to give the target compound PU8 (0.16 g, 88%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 2 H), 7.51 (d, J = 8.0 Hz, 2 H), 7.20 (d, J = 8.0 Hz, 2 H), 6.89 (d, J = 9.0 Hz, 2 H), 5.68 (d, J = 5.0 Hz, 1 H), 5.10 (d, J = 9.0 Hz, 1 H), 5.07 (q, J = 6.5 Hz, 1 H), 4.81– 4.72 (m, 2 H), 4.30–4.19 (m, 6 H), 4.00 (dd, J = 9.5, 6.5 Hz, 1 H), 3.95–3.90 (m, 1 H), 3.87–3.76 (m, 3 H), 3.69 (dd, J = 9.5, 6.5 Hz, 1 H), 3.67 (d, J = 2.0 Hz, 1 H, overlapping), 3.51 (t, J = 6.0 Hz, 1 H), 3.11–3.02 (m, 2 H), 3.01–2.91 (m, 2 H), 2.81 (dd, J = 14.0, 6.5 Hz, 1 H), 2.67 (dd, J = 13.5, 5.5 Hz, 1 H), 2.56 (d, J = 14.5 Hz, 1 H), 1.81–1.75 (m, 2 H), 1.50–1.41 (m, 2 H), 1.38 (t, J = 7.0 Hz, 3 H), 1.36 (t, J = 7.0 Hz, 3 H, overlapping), 1.32–1.21 (m, 3 H), 0.83 (t, J = 7.4 Hz, 3 H), 0.81 (t, J = 7.4 Hz, 3 H, overlapping), ppm; 13C NMR (126 MHz, CDCl3) δ 157.65, 157.53, 155.43, 147.36, 136.42, 131.20, 130.66, 127.58, 127.27, 114.75, 109.40, 73.59, 73.37, 70.98, 69.73, 64.06, 63.36, 63.31, 63.20, 63.15, 63.09, 61.72, 55.25, 55.21, 54.20, 45.36, 39.25, 34.99, 26.05, 23.25, 22.73, 16.64, 16.59, 10.77, 10.31 ppm; 31P NMR (202 MHz, CDCl3) δ 19.36 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H54N2O12PS, 757.3130; found 757.3137; Anal. HPLC: tR 10.78 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-1-(4-((diethoxyphosphoryl)methoxy)phenyl)-4-(N-(2-ethylbutyl)benzo[d]thiazole-6-sulfonamido)-3-hydroxybutan-2-yl)carbamate (PU10).
The same procedure was used as described above for compound PD4. Compound 28c (0.23 g, 0.34 mmol) was treated with TFA (2 mL), and the resulting deprotected amine was treated with Na2CO3 (0.075 g, 0.68 mmol) and 1,3-benzothiazole-6-sulfonyl chloride 7 (0.102 g, 0.44 mmol) to give the target compound PU10 (0.18 g, 68%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.22 (s, 1 H), 8.47 (d, J = 1.5 Hz, 1 H), 8.27 (d, J = 8.5 Hz, 1 H), 7.90 (dd, J = 8.5, 2.0 Hz, 1 H), 7.15 (d, J = 8.5 Hz, 2 H), 6.88 (d, J = 8.5 Hz, 2 H), 5.65 (d, J = 5.5 Hz, 1 H), 5.01 (q, J = 6.5 Hz, 1 H), 4.87 (d, J = 8.0 Hz, 1 H), 4.27–4.20 (m, 6 H), 3.94 (dd, J = 9.5, 6.5 Hz, 1 H), 3.87 (td, J = 8.5, 2.0 Hz, 1 H), 3.85–3.80 (m, 2 H, overlapping), 3.74–3.66 (m, 3 H), 3.20 (dd, J = 15.0, 8.0 Hz, 1 H), 3.13 (dd, J = 13.0, 8.0 Hz, 1 H), 3.08–3.00 (m, 2 H), 2.96–2.89 (m, 2 H), 2.79 (dd, J = 14.0, 8.5 Hz, 1 H), 1.73–1.63 (m, 1 H), 1.58–1.51 (m, 1 H), 1.50–1.39 (m, 2 H), 1.36 (t, J = 7.0 Hz, 6 H), 1.34–1.26 (m, 3 H), 0.83 (t, J = 7.4 Hz, 3 H), 0.81 (t, J = 7.4 Hz, 3 H, overlapping) ppm; 13C NMR (126 MHz, CDCl3) δ 158.21, 157.78, 157.67, 155.83, 155.66, 135.44, 134.62, 130.77, 130.58, 124.98, 124.63, 122.46, 114.81, 109.40, 73.70, 73.02, 70.84, 69.67, 63.12, 63.08, 63.03, 61.76, 55.29, 55.02, 53.85, 45.46, 39.30, 34.90, 25.96, 23.17, 22.89, 16.65, 16.60, 10.75, 10.42 ppm; 31P NMR (202 MHz, CDCl3) δ 19.19 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C35H51N3O11PS2, 784.2697; found 784.2707; Anal. HPLC: tR 11.75 min, purity 99%.
Biology
Protease Gene Construction
Protease gene construction was carried out as previously described [39, 40]. Briefly, the protease variant genes (I84V, I50V/A71V) were constructed using QuikChange site-directed mutagenesis (Genewiz) onto the NL4–3 wild-type protease on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. The 8Mut and 11Mut variants were constructed by QuikChange site-directed mutagenesis (Genewiz) onto the purchased 10Mut variant on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. The KY24 variant was purchased on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. A Q7K mutation was included to prevent autoproteolysis [41].
The 8Mut variant contains these mutations: I13V, G16E, V32I, L33F, K45I, M46I, V82F, and I84V. The 10Mut includes additional A71V and L76V mutations, and the 11Mut additionally contains the I54L mutation. The KY24 variant contains the following mutations: L10V, T12V, I13V, I15V, K20M, V32I, L33F, K43T, M46I, I47V, I54M, D60E, Q61N, I62V, L63P, C67Y, H69K, A71I, I72L, G73S, V77I, V82A, L89V, L90M.
Protein Expression and Purification
The expression, isolation, and purification of drug resistant HIV-1 proteases used for the enzyme inhibition assays and the wild-tyep HIV-1 protease used for crystallization were carried out as previously described [39, 40]. Briefly, the genes encoding the HIV protease were subcloned into the heat-inducible pXC35 expression vector (ATCC) and transformed into E. coli TAP-106 cells. Cells grown in 6 L of Terrific Broth were lysed with a cell disruptor and the protein was purified from inclusion bodies [42]. The inclusion body centrifugation pellet was dissolved in 50% acetic acid followed by another round of centrifugation to remove impurities. Size-exclusion chromatography was used to separate high molecular weight proteins from the desired protease. This was carried out on a 2.1 L Sephadex G-75 superfine column (Millipore Sigma) equilibrated with 50% acetic acid. The cleanest fractions of HIV protease were refolded into a 10-fold dilution of 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Folded protein was concentrated down to 1–2 mg/mL and stored. This stored protease was used in Ki assays. For crystallography, a final purification was performed with a Pharmacia Superdex 75 FPLC column equilibrated with 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Protease fractions purified from the size exclusion column were concentrated to 1–2 mg/mL using an Amicon Ultra-15 10-kDa device (Millipore) for crystallization.
Enzyme Inhibition Assays
The enzyme inhibition assays were carried out as previously described [29–31, 43]. To determine the enzyme inhibition constant (Ki), in a 96-well plate, each inhibitor was serially diluted, including a no drug control, and incubated with 0.35 nM protein for 1 hour. A 10-amino acid substrate containing an optimized protease cleavage site with an EDANS/DABCYL FRET pair was dissolved in 4% DMSO at 120 μM. Using the Envision plate reader, 5 μL of the 120 μM substrate was added to the 96-well plate to a final concentration of 10 μM. The fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts, for approximately 60 min. Data was analyzed with Prism7. DRV was used as a control in all assays.
Protein Crystallization
The condition reliably producing cocrystals of NL4–3 WT protease bound to PIs was discovered and optimized as previously described [29, 43, 44]. Briefly, all cocrystals were grown at room temperature by hanging drop vapor diffusion method in a 24-well VDX hanging-drop trays (Hampton Research) with a protease concentration of 0.9–2.0 mg/mL with 3- to 5-fold molar excess of inhibitors (0.3–0.5% DMSO) and mixed with the precipitant solution at a 1:2 ratio. The reservoir solution was 23% to 24% (w/v) ammonium sulfate with 0.1 M bis-Tris-methane buffer at pH 5.5, and the crystallization drops were set with 2 μL of well solution and 1 μL of protein–inhibitor solution and micro-seeded (at a 1:100 to 1:1000 dilution) with a cat whisker. Diffraction quality crystals were obtained within 1 wk. As data were collected at 100 K, cryogenic conditions contained the precipitant solution were supplemented with 25% glycerol.
X-ray Data Collection and Structure Solution
X-ray diffraction data were collected and solved as previously described [29, 39, 44]. Diffraction quality crystals were flash frozen under a cryostream when mounting the crystals either at our in-house Rigaku_Saturn944 X-ray system or the Chicago APS Synchrotron Beamline 23–1D-D. The cocrystal diffraction intensities from the Rigaku system were indexed, integrated, and scaled using HKL3000 [45]. Structures shot at the APS were indexed, integrated, and scaled using XDS [46]. All structures were solved using molecular replacement with PHASER [47]. Model building and refinement were performed using Coot [48] and Phenix [49]. Ligands were designed in Maestro and the output sdf files were used in the Phenix program eLBOW [50] to generate cif files containing atomic positions and constraints necessary for ligand refinement. Iterative rounds of crystallographic refinement were carried out until convergence was achieved. To limit bias throughout the refinement process, five percent of the data were reserved for the free R-value calculation [51]. MolProbity [52] was applied to evaluate the final structures before deposition in the PDB. Structure analysis, superposition and figure generation was done using PyMOL [53]. X-ray data collection and crystallographic refinement statistics are presented in the Supporting Information (Table S7).
Intermolecular van der Waals Contact Analysis
To calculate the intermolecular vdW interaction energies the crystal structures were prepared using the Schrödinger Protein Preparation Wizard [54]. Hydrogen atoms were added, protonation states were determined, and the structures were minimized. The protease active site was monoprotonated at Asp25. Subsequently, force field parameters were assigned using the OPLS3 force field [55]. Interaction energies between the inhibitor and protease were estimated using a simplified Lennard–Jones potential, as previously described in detail [56]. Briefly, the vdW energy was calculated for pairwise interactions depending on the types of atoms interacting and the distance between them. For each protease residue, the change in vdW interactions relative to a reference complex in the same space group was also calculated for each variant structure.
Antiviral Assays
293T and TZM-BL [57] cells (NIH AIDS Research and Reference Reagent Program) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum in the presence of penicillin and streptomycin at 37 °C with 5% CO2. To determine the concentration of drugs achieving 50% inhibition of infection compared with the drug-free control, 4.5×106 293T cells were seeded onto a 10-cm plate 24 h before transfection. Cells were transfected with 8 μg of the wild-type plasmid, infectious molecular clone pNL-CH derived from the pNL4–3 clone of HIV-1 using FuGENE 6 transfection reagent (Roche). The culture supernatant of 293T cells transfected with wild-type or PI-resistant HIV-1 variant was removed 18 h after transfection and the cells were washed with 1 × PBS. The 293T cells were collected and transferred to wells of a 24-well plate. Briefly, each drug was serially diluted in the culture medium and the dilutions were added to the wells of a 24-well plate. The 293T cells (0.5 × 106 per well) collected from the transfection were added to wells containing various concentrations of drug. The culture supernatant containing virus particles was harvested 18 h after the 293T cells were reseeded in the presence of drug. This supernatant was filtered through a 0.45-μm-pore-size membrane (Millipore) to remove cell debris then used to infect 2 × 104 TZM-BL cells in a 96-well plate following a procedure previously described [58]. The culture supernatant was removed from each well 48 h post-infection, and the cells were washed with 1 × PBS. For the luciferase assay, infected TZM-BL cells were lysed in 1× reporter lysis buffer (Promega) and the cells were kept at −80 °C. After one freeze-thaw cycle, the cell lysates were transferred into a 96-well assay plate (Costar), and luciferase activity was measured using a luminometer (Promega). The culture supernatant harvested from 293T cells reseeded in the absence of drugs was used as a drug-free control. EC50 was determined based on a dose-response curve generated using GraphPad Prism (version 7.0).
Antiviral Assays with Resistant HIV-1 Variants
Protease inhibitor dilutions were prepared by taking 10 mM stock and performing a five-fold serial dilution using RPMI media (final drug concentration was 100 μM). One dilution of drug was added to each well of a 24-well plate and repeated so each virus would have a full set of dilutions. Viruses for the assay were made by seeding 3 × 106 CEM cells in a 24-well plate and incubating with 250 μL of virus at 37 °C for 2–3 h before bringing the culture to 10 mL with RPMI media. After 48 h, the medium was changed and repeated every 48 h after until the culture had undergone CPE. Infected CEM cells were collected and diluted so that 1 mL of cells could be plated in each well containing a unique drug dilution. Then 24 h later the virus supernatant was collected from each well followed by filtering through a 0.45 μM filter then placed in −80 °C. Viruses were thawed and added to 96-well plates in triplicate. TZM-BL cells were collected and diluted to a concentration of 2 × 105 cells/mL, 100 μL were added on top of the pre-plated viruses. Plates were kept in 37 °C, 5% CO2 in an incubator for 48 h. After 48 h, the cells in the plates were lysed by removing the medium, washing two times with 100 μL PBS, and then lysed with 1 × lysis buffer (made from 5 × Promega Firefly Lysis Buffer). Plates were frozen for at least 24 h and then thawed for 2 h before analyzing with Promega Firefly Luciferase Kit on a luminometer. Data was analyzed with Prism 7 to fit sigmoidal dose–response curves.
Cytotoxicity Assay
The cytotoxicity of phosphonate compounds was assessed using the Cell Cytotoxicity Assay Kit (Abcam, ab112118) according to the manufacturer’s protocol. Briefly, 293T or TZMbl cells were seeded in 96-well plates at a density of 20,000 cells/well and treated with 2-fold serial dilutions of the tested compounds, ranging from 100 μM to 0.78 μM. Cells were incubated in a 37 °C, 5% CO2 incubator for 48 h. Assay solution (Component A from the assay kit) (20 μL) was added into each well, mixed, and the cells were incubated at 37 °C for 2 h. Absorbance was measured at 570 nm and 605 nm. The ratio of OD570/OD605 was used to determine cell viability in each well using no drug and cell-free controls as background. All experiments were performed in triplicate. The concentration of each drug that reduced the cell viability by 50% (CC50) was determined by regression analysis using Prism (version 7.0).
Protein Data Bank accession codes
The PDB accession codes for X-ray cocrystal structures of HIV-1 protease with phosphonate inhibitors: GS-8374, 7LE7; PD4, 7LE8; PD5, 7LE9; PU1, 7LEA; PU2, 7LEB; PU3, 7LEC; PU4, 7LED; PU5, 7LEE; PU6, 6W6T; PU7, 7LEF; PU8, 7LEG; PU9, 7LEH; PU10, 7LEI.
Supplementary Material
Highlights.
SAR studies of HIV-1 protease inhibitors with a P1 phosphonate modification
Simultaneous modification of chemical groups improves potency and resistance profiles
The P1 phosphonate moiety makes extensive hydrophobic interactions with the protease
Targeting resistant variants requires balancing inhibitor physicochemical properties
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences of the NIH (P01 GM109767 and R01 GM135919). This research used resources of the Structural Biology Center (SBC) at the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the beamline specialists at 19-ID for their help in data collection. We thank the AIDS Research and Reference Reagent Program (NIAD, NIH) for reference protease inhibitors, and Drs. William Royer, Brian Kelch and members of the Schiffer and Miller laboratories for helpful discussions.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.World Health Organization (WHO). HIV/AIDS, Fact Sheet (Updated November 2022). https://www.who.int/news-room/fact-sheets/detail/hiv-aids. 2022.
- 2.Zhan P, Pannecouque C, De Clercq E, Liu X Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem, 59 (2016), pp. 2849–2878. [DOI] [PubMed] [Google Scholar]
- 3.Gandhi RT, Bedimo R, Hoy JF, Landovitz RJ, Smith DM, Eaton EF, Lehmann C, Springer SA, Sax PE, Thompson MA, Benson CA, Buchbinder SP, Del Rio C, Eron JJ Jr., Gunthard HF, Molina JM, Jacobsen DM, Saag MS Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2022 recommendations of the International Antiviral Society-USA Panel. JAMA, 329 (2023), pp. 63–84. [DOI] [PubMed] [Google Scholar]
- 4.Cihlar T, Fordyce M Current status and prospects of HIV treatment. Curr. Opin. Virol, 18 (2016), pp. 50–56. [DOI] [PubMed] [Google Scholar]
- 5.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv. (Accessed January 15, 2022).
- 6.World Health Organization (WHO). HIV Drug Resistance, Fact Sheet (Updated November 2022). https://www.who.int/news-room/fact-sheets/detail/hiv-drug-resistance. 2022.
- 7.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U. S. A, 85 (1988), pp. 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh AK, Osswald HL, Prato G Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem, 59 (2016), pp. 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med, 14 (2008), pp. 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem, 48 (2005), pp. 1813–1822. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res, 41 (2008), pp. 78–86. [DOI] [PubMed] [Google Scholar]
- 12.Girard PM, Antinori A, Arribas JR, Ripamonti D, Bicer C, Netzle-Sveine B, Hadacek B, Moecklinghoff C Week 96 efficacy and safety of darunavir/ritonavir monotherapy vs. darunavir/ritonavir with two nucleoside reverse transcriptase inhibitors in the PROTEA trial. HIV Med., 18 (2017), pp. 5–12. [DOI] [PubMed] [Google Scholar]
- 13.Di Cristo V, Adorni F, Maserati R, Annovazzi Lodi M, Bruno G, Maggi P, Volpe A, Vitiello P, Abeli C, Bonora S, Ferrara M, Cossu MV, Oreni ML, Colella E, Rusconi S 96-week results of a dual therapy with darunavir/ritonavir plus rilpivirine once a day vs triple therapy in patients with suppressed viraemia: virological success and non-HIV related morbidity evaluation. HIV Res. Clin. Pract, 21 (2020), pp. 34–43. [DOI] [PubMed] [Google Scholar]
- 14.Casado JL, Vizcarra P, Blanco JL, Montejano R, Negredo E, Espinosa N, Montero M, Mena A, Palacios R, Lopez JC, Vergas J, Galindo MJ, Cabello A, Deltoro MG, Diaz De Santiago A, group BI s. Maintenance of virologic suppression and improvement in comorbidities after simplification to raltegravir plus boosted darunavir among treatment-experienced HIV-infected patients. Int. J. STD AIDS, 31 (2020), pp. 467–473. [DOI] [PubMed] [Google Scholar]
- 15.Hui L, Xiaoxu H, Yuqi W, Peng W, Xin W, Yunyun Y, Xin L Effectiveness and safety analysis of PIs/r based dual therapy in treatment-naive, HIV/AIDS patients: a network meta analysis of randomized controlled trials. Front. Pharmacol, 13 (2022), pp. 811357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Weber IT, Mitsuya H Beyond darunavir: recent development of next generation HIV-1 protease inhibitors to combat drug resistance. Chem. Commun, 58 (2022), pp. 11762–11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthew AN, Leidner F, Lockbaum GJ, Henes M, Zephyr J, Hou S, Rao DN, Timm J, Rusere LN, Ragland DA, Paulsen JL, Prachanronarong K, Soumana DI, Nalivaika EA, Kurt Yilmaz N, Ali A, Schiffer CA Drug design strategies to avoid resistance in direct-acting antivirals and beyond. Chem. Rev, 121 (2021), pp. 3238–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JF, Andrews CW, Brieger M, Furfine ES, Hale MR, Hanlon MH, Hazen RJ, Kaldor I, McLean EW, Reynolds D, Sammond DM, Spaltenstein A, Tung R, Turner EM, Xu RX, Sherrill RG Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett, 16 (2006), pp. 1788–1794. [DOI] [PubMed] [Google Scholar]
- 19.Hazen R, Harvey R, Ferris R, Craig C, Yates P, Griffin P, Miller J, Kaldor I, Ray J, Samano V, Furfine E, Spaltenstein A, Hale M, Tung R, St Clair M, Hanlon M, Boone L In vitro antiviral activity of the novel, tyrosyl-based human immunodeficiency virus (HIV) type 1 protease inhibitor brecanavir (GW640385) in combination with other antiretrovirals and against a panel of protease inhibitor-resistant HIV. Antimicrob. Agents Chemother, 51 (2007), pp. 3147–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cihlar T, He GX, Liu X, Chen JM, Hatada M, Swaminathan S, McDermott MJ, Yang ZY, Mulato AS, Chen X, Leavitt SA, Stray KM, Lee WA Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol, 363 (2006), pp. 635–647. [DOI] [PubMed] [Google Scholar]
- 21.Callebaut C, Stray K, Tsai L, Williams M, Yang ZY, Cannizzaro C, Leavitt SA, Liu X, Wang K, Murray BP, Mulato A, Hatada M, Priskich T, Parkin N, Swaminathan S, Lee W, He GX, Xu L, Cihlar T In vitro characterization of GS-8374, a novel phosphonate-containing inhibitor of HIV-1 protease with a favorable resistance profile. Antimicrob. Agents Chemother, 55 (2011), pp. 1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He G-X, Yang Z-Y, Williams M, Callebaut C, Cihlar T, Murray BP, Yang C, Mitchell ML, Liu H, Wang J, Arimilli M, Eisenberg E, Stray KM, Tsai LK, Hatada M, Chen X, Chen JM, Wang Y, Lee MS, Strickley RG, Iwata Q, Zheng X, Kim CU, Swaminathan S, Desai MC, Lee WA Xu L. Discovery of GS-8374, a potent human immunodeficiency virus type 1 protease inhibitor with a superior resistance profile. MedChemComm, 2 (2011), pp. 1093–1098. [Google Scholar]
- 23.Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori SI, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK Mitsuya H. A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife, 6 (2017), pp. e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK, Rao KV, Nyalapatla PR, Osswald HL, Martyr CD, Aoki M, Hayashi H, Agniswamy J, Wang YF, Bulut H, Das D, Weber IT, Mitsuya H Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants. J. Med. Chem, 60 (2017), pp. 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, P RN., Kovela S., Rao KV., Brindisi M., Osswald HL., Amano M., Aoki M., Agniswamy J., Wang YF., Weber IT., Mitsuya H. Design and synthesis of highly potent HIV-1 protease inhibitors containing tricyclic fused ring systems as novel P2 ligands: structure-activity studies, biological and X-ray structural analysis. J. Med. Chem, 61 (2018), pp. 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh AK, Rao KV, Nyalapatla PR, Kovela S, Brindisi M, Osswald HL, Sekhara Reddy B, Agniswamy J, Wang YF, Aoki M, Hattori SI, Weber IT, Mitsuya H Design of highly potent, dual-acting and central-nervous-system-penetrating HIV-1 protease inhibitors with excellent potency against multidrug-resistant HIV-1 variants. ChemMedChem, 13 (2018), pp. 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh AK, Kovela S, Osswald HL, Amano M, Aoki M, Agniswamy J, Wang YF, Weber IT, Mitsuya H Structure-Based Design of Highly Potent HIV-1 Protease Inhibitors Containing New Tricyclic Ring P2-Ligands: Design, Synthesis, Biological, and X-ray Structural Studies. J Med Chem, 63 (2020), pp. 4867–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nalam MN, Ali A, Reddy GS, Cao H, Anjum SG, Altman MD, Yilmaz NK, Tidor B, Rana TM, Schiffer CA Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem. Biol, 20 (2013), pp. 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rusere LN, Lockbaum GJ, Lee SK, Henes M, Kosovrasti K, Spielvogel E, Nalivaika EA, Swanstrom R, Yilmaz NK, Schiffer CA, Ali A HIV-1 protease inhibitors incorporating stereochemically defined P2’ ligands to optimize hydrogen bonding in the substrate envelope. J. Med. Chem, 62 (2019), pp. 8062–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Windsor IW, Raines RT Fluorogenic Assay for Inhibitors of HIV-1 Protease with Sub-picomolar Affinity. Sci. Rep, 5 (2015), pp. 11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henes M, Lockbaum GJ, Kosovrasti K, Leidner F, Nachum GS, Nalivaika EA, Lee SK, Spielvogel E, Zhou S, Swanstrom R, Bolon DNA, Kurt Yilmaz N, Schiffer CA Picomolar to micromolar: elucidating the role of distal mutations in HIV-1 protease in conferring drug resistance. ACS Chem. Biol, 14 (2019), pp. 2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spielvogel E, Lee SK, Zhou S, Lockbaum GJ, Henes M, Sondgeroth A, Kosovrasti K, Nalivaika EA, Ali A, Yilmaz NK, Schiffer CA, Swanstrom R Selection of HIV-1 for resistance to fifth-generation protease inhibitors reveals two independent pathways to high-level resistance. Elife, 12 (2023), pp. doi: 10.7554/eLife.80328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragland DA, Whitfield TW, Lee SK, Swanstrom R, Zeldovich KB, Kurt-Yilmaz N, Schiffer CA Elucidating the Interdependence of Drug Resistance from Combinations of Mutations. J Chem Theory Comput, 13 (2017), pp. 5671–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother, 51 (2007), pp. 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Surleraux DL, de Kock HA, Verschueren WG, Pille GM, Maes LJ, Peeters A, Vendeville S, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J. Med. Chem, 48 (2005), pp. 1965–1973. [DOI] [PubMed] [Google Scholar]
- 36.Lockbaum GJ, Henes M, Talledge N, Rusere LN, Kosovrasti K, Nalivaika EA, Somasundaran M, Ali A, Mansky LM, Kurt Yilmaz N, Schiffer CA Inhibiting HTLV-1 Protease: A Viable Antiviral Target. ACS Chem Biol, 16 (2021), pp. 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem. Biol, 11 (2004), pp. 1333–1338. [DOI] [PubMed] [Google Scholar]
- 38.Matthew AN, Zephyr J, Hill CJ, Jahangir M, Newton A, Petropoulos CJ, Huang W, Kurt-Yilmaz N, Schiffer CA, Ali A Hepatitis C virus NS3/4A protease inhibitors incorporating flexible P2 quinoxalines target drug resistant viral variants. J. Med. Chem, 60 (2017), pp. 5699–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozen A, Lin KH, Kurt Yilmaz N, Schiffer CA Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A, 111 (2014), pp. 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King NM, Melnick L, Prabu-Jeyabalan M, Nalivaika EA, Yang SS, Gao Y, Nie X, Zepp C, Heefner DL, Schiffer CA Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci, 11 (2002), pp. 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose JR, Salto R, Craik CS Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem, 268 (1993), pp. 11939–11945. [PubMed] [Google Scholar]
- 42.Hui JO, Tomasselli AG, Reardon IM, Lull JM, Brunner DP, Tomich CS, Heinrikson RL Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J. Protein Chem, 12 (1993), pp. 323–327. [DOI] [PubMed] [Google Scholar]
- 43.Rusere LN, Lockbaum GJ, Henes M, Lee SK, Spielvogel E, Rao DN, Kosovrasti K, Nalivaika EA, Swanstrom R, Kurt Yilmaz N, Schiffer CA, Ali A Structural analysis of potent hybrid HIV-1 protease inhibitors containing bis-tetrahydrofuran in a pseudosymmetric dipeptide isostere. J. Med. Chem, 63 (2020), pp. 8296–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lockbaum GJ, Leidner F, Rusere LN, Henes M, Kosovrasti K, Nachum GS, Nalivaika EA, Ali A, Yilmaz NK, Schiffer CA Structural adaptation of darunavir analogues against primary mutations in HIV-1 protease. ACS Infect. Dis, 5 (2019), pp. 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol, 276 (1997), pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch W XDS. Acta Crystallogr D Biol Crystallogr, 66 (2010), pp. 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ Phaser crystallographic software. J. Appl. Crystallogr, 40 (2007), pp. 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr, 60 (2004), pp. 2126–2132. [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr, 66 (2010), pp. 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moriarty NW, Grosse-Kunstleve RW, Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., Sect. D: Biol. Crystallogr, 65 (2009), pp. 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brunger AT Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355 (1992), pp. 472–475. [DOI] [PubMed] [Google Scholar]
- 52.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, Richardson DC MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res, 35 (2007), pp. W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.PyMOL, The PyMOL Molecular Graphics System, Version 2.3. 2020, Schrödinger, LLC. [Google Scholar]
- 54.Schrödinger, Schrödinger Release 2021–3. 2021, Schrödinger, LLC, New York, NY, United States. [Google Scholar]
- 55.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput, 12 (2016), pp. 281–296. [DOI] [PubMed] [Google Scholar]
- 56.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J. Virol, 84 (2010), pp. 5368–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother, 46 (2002), pp. 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SK, Harris J, Swanstrom R A strongly transdominant mutation in the human immunodeficiency virus type 1 gag gene defines an Achilles heel in the virus life cycle. J. Virol, 83 (2009), pp. 8536–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.