

Abstract
Bruton’s tyrosine kinase (BTK) is a target for treating B-cell malignancies and autoimmune diseases, and several BTK inhibitors are already approved for use in humans. Heterobivalent BTK protein degraders are also in development, based on the premise that proteolysis targeting chimeras (PROTACs) may provide additional therapeutic benefits. However, most BTK PROTACs are based on the BTK inhibitor ibrutinib raising concerns about their selectivity profiles, given the known off-target effects of ibrutinib. Here we disclose the discovery and in vitro characterization of BTK PROTACs based on the selective BTK inhibitor GDC-0853 and the cereblon recruitment ligand pomalidomide. PTD10 is a highly potent BTK degrader (DC50 0.5 nM) that inhibited cell growth and induced apoptosis at lower concentrations than the two parent molecules, as well as three previously reported BTK PROTACs, and had improved selectivity compared to ibrutinib-based BTK PROTACs.
Keywords: Bruton’s tyrosine kinase, BTK, PROTAC, apoptosis, selectivity
Graphical Abstract

INTRODUCTION
Bruton’s tyrosine kinase (BTK) is a nonreceptor tyrosine kinase that belongs to the hepatocellular carcinoma (Tec) family of kinases and is an essential component of the B-cell receptor (BCR) signaling pathway. The BCR pathway regulates the survival, activation, proliferation, and differentiation of B cells. Therefore, BTK has been recognized as a valid therapeutic target for B-cell malignances, including chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and Waldenstrom macroglobulinemia (WM).1 In addition, BTK inhibitors are also in development for the treatment of autoimmune diseases such as rheumatoid arthritis and multiple sclerosis.2
BTK is comprised of a kinase domain, an amino terminal pleckstrin homology (PH) domain, a proline-rich Tec homology (TH) domain, and two SRC homology (SH) domains, SH2 and SH3.1,3 The majority of BTK inhibitors target the kinase domain and approved drugs as well as those in development include compounds that bind both covalently and non-covalently to the enzyme. Ibrutinib is a first-in-class BTK inhibitor that binds irreversibly to the BTK kinase domain and has been approved by the FDA for the treatment of relapsed/refractory MCL, CLL, and WM.4,5 Ibrutinib suffers from off-target effects and inhibits other kinases including B-lymphoid tyrosine kinase (BLK), bone marrow tyrosine kinase gene in chromosome X (BMX), hematopoietic cell kinase (HCK), Erb-b2 receptor tyrosine kinase 2 (ERBB2), Janus kinase 3 (JAK3), IL2 inducible T cell kinase (ITK) and epidermal growth factor receptor (EGFR).4 This has driven the development of BTK inhibitors with increased selectivity, and four additional irreversible BTK inhibitors, acalabrutinib, zanubrutinib, tirabrutinib and orelabrutinib, have so far been approved.6–9 Ibrutinib and as well as the other irreversible inhibitors covalently modify C481 in the BTK active site, and drug resistance caused by the C481S mutation is a problem faced by all these compounds.10 Thus there are also programs to discover selective, reversible inhibitors of BTK that bind equally well to the C481S mutant,11 as well as the application of targeted protein degradation as a therapeutic modality.12
Targeted protein degradation is an emerging therapeutic strategy in which the ubiquitin-proteasome house-keeping system is hijacked by heterobifunctional proteolysis targeting chimeras (PROTACs) to catalyze the removal of the protein of interest from the cell. Targeted protein degradation differs fundamentally from the small molecule inhibition, and this therapeutic modality is being applied extensively across disease space.13 PROTACs are comprised of a flexible linker that connects two ligands, one that binds to the protein of interest (POI), and a second that binds to and recruits an E3 ligase into a ternary complex where the POI is then ubiquitinated. To date there are several reports of BTK PROTACs, most derived from ibrutinib in which the acrylamide group is replaced with a linker to the E3 ligase ligand.14–16 While the ibrutinib-based PROTACs efficiently degrade BTK with nanomolar DC50 values,17,18 these heterobivalent compounds also catalyze the degradation of off-target proteins including BLK, C-terminal Src kinase (CSK), HCK and LYN,17–19 and thus there remains a need for BTK PROTACs with improved selectivity.
Here we report the design and synthesis of a small library of BTK PROTACs based on GDC-0853 (fenebrutinib).20 GDC-0853 is a potent, reversible inhibitor of BTK and thus inhibits enzymes that also have mutations to C481, the most commonly mutated residue in cases of acquired resistance to irreversible inhibitors such as ibrutinib. GDC-0853 is also more selective than ibrutinib, and so PROTACs based on GDC-0853 are expected to exhibit higher selectivity than the corresponding ibrutinib-based PROTACs. We investigated the effect of varying linker length, exit vector, and the point of attachment to ligands for the cereblon E3 ligase on the ability of the PROTACs to degrade BTK and cause phenotypic effects including inhibition of cell growth and the induction of apoptosis. Most of the compounds demonstrated low nanomolar DC50 values and PTD10 is the most potent compound with an optimized DC50 value of 0.5 nM (Figure 1). Two compounds, PTD13 and PTD15, exhibited low degradation efficiency with DC50 values > 100 nM and > 3000 nM, respectively, and cell-based experiments demonstrated that these compounds did not catalyze ternary complex formation efficiently. Using label-free proteomics we demonstrated that PTD10 and PTD14 had improved degradation selectivity compared to the ibrutinib-based PROTAC P13I. In addition, PTD10 showed enhanced phenotypic effects including the inhibition of cell growth and induction of cell apoptosis compared to the two parent molecules GDC-0853 and pomalidomide, as well as three previously reported BTK PROTACs DD-03-171, MT-802, and P13I (Figure 1).14,15,21 We also examined the impact of the PROTACs on BCR signaling and demonstrated that PTD10 inhibited phosphorylation of downstream proteins at lower concentrations than the parent molecule GDC-0853. Collectively, we have developed a series of novel BTK PROTACs with enhanced selectivity and phenotypic effects that will be useful for chemical tools and serve as potential drug leads for treating diseases caused by B-cell dysregulation.
Figure 1:
Structures of BTK PROTACs
RESULTS
PROTAC Design
Heterobivalent PROTACs targeted at BTK were designed based on GDC-0853, a reversible BTK inhibitor with high potency and selectivity. The crystal structure of GDC-0853 bound to BTK shows that the oxetane group of the inhibitor points out of the binding site and is exposed to the solvent, providing a feasible site for linker attachment (Figure 2).20
Figure 2: GDC-0853 in complex with BTK.

(A) The structure of GDC-0853 bound to BTK from PDB ID: 5VFI. The figure was made with Pymol.22 (B) The structure of GDC-0853.
Subsequently, we synthesized a series of heterobivalent compounds in which the warheads were tethered via the piperazine ring to different E3 ligase ligands using a variety of linkers, including alkyl, ethylene glycol (PEG) and mixed linkers. The majority of the compounds were designed to recruit the E3 ligase cereblon (CRBN) and were based on pomalidomide. However, we also synthesized analogs in which the pomalidomide amino group was replaced with an oxygen, moved the point of attachment to the phthalimide ring from C4 to C5, replaced pomalidomide with thalidomide and lenalidomide, and synthesized an analog that incorporated a ligand to recruit the von Hippel–Lindau (VHL) E3 ligase. We also synthesized analogs lacking a methyl group on the GDC-0853 piperazine ring based on the knowledge that this group modulates the residence time of the inhibitor on BTK.20
Chemistry
The synthesis of GDC-0853 warhead 1 is shown in Scheme 1 and is based on a method described by Genentech.20
Scheme 1. Synthesis of GDC-0853 warhead 1.

aReagents and conditions: (a) Pd(OAc)2, BINAP, K3PO4, dioxane, 110 °C; (b) H2, Pd/C, EtOAc/EtOH; (c) Pd2(dba)3, Xantphos, Cs2CO3, dioxane, 100 °C; (d) Pd(OAc)2, Xphos, KOAc, B2pin2, dioxane, 100 °C; (e) Pd(dppf)Cl2, K3PO4, THF/H2O, 50 °C; (f) NaBH4, MeOH, rt; (g) TFA, DCM, rt.
Intermediate 1 was prepared from 5-bromo-2-nitropyridine and Boc-protected 3-methyl piperazine using palladium-catalyzed cross-coupling reaction. Following nitro reduction, Buchwald-Hartwig coupling with 3,5-dibromo-1-methylpyridin-2(1H)-one resulted in intermediate 3. Intermediate 4 was then synthesized from 3 by borylation with bis(pinacolato)diboron, and then reacted with aromatic chloride to afford intermediate 5. GDC-0853 warhead 1 was subsequently obtained by reduction of the aldehyde and removal of the Boc group.
The synthesis of GDC-0853 warhead 2, which lacks a methyl group on the piperazine ring, followed the same synthetic route as GDC-0853 warhead 1 except that the Buchwald-Hartwig coupling reaction with 3,5-dibromo-1-methylpyridin-2(1H)-one used 1-Boc-4-(6-amino-3-pyridyl)piperazine and (Scheme 2).
Scheme 2. Synthesis of GDC-0853 warhead 2.

aReagents and conditions: (a) Pd2(dba)3, Xantphos, Cs2CO3, dioxane, 100 °C; (b) Pd(OAc)2, Xphos, KOAc, B2pin2, dioxane, 100 °C; (c) Pd(dppf)Cl2, K3PO4, THF/H2O, 50 °C; (d) NaBH4, MeOH, rt; (e) TFA, DCM, rt.
The E3 ligase ligands attached to linkers were either obtained from Sigma-Aldrich, or synthesized as shown in Scheme 3. Compound 12 and 14 were synthesized as described,23,24 while Compound 15 was synthesized by a nucleophilic aromatic substitution reaction (SNAr) using 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione and 12-aminododecanoic acid.
Scheme 3. Synthesis of E3 ligase ligand conjugates 12, 14, and 15.

aReagents and conditions: (a) DIPEA, NMP, 110 °C; (b) HCl in dioxane, rt; (c) DIPEA, NMP, 110 °C, Microwave, 2 h; (d) TFA, DCM, rt; (e) DIPEA, DMF, 90 °C.
The PROTACs were subsequently prepared either by alkylation of the GDC-0853 warheads with the alkyl iodide of the E3 ligase ligand or by coupling the warhead to the carboxylic acids on the linker (Scheme 4).
Scheme 4. Two typical approaches for synthesizing PTD PROTACs.

aReagents and conditions: (a) HATU, DIPEA, DMF, rt; (b) DIPEA, DMF, rt.
Biological Activity
The ability of the PROTACs to degrade BTK was first evaluated in Ramos and JeKo-1 cell lines by western blot analysis after 17 h incubation. Initially, we synthesized PTD3 by replacing pomalidomide in PTD2 with a VHL ligand. However, no degradation was observed with PTD3 at concentrations lower than 3000 nM (Figure S1). Therefore, we decided to use ligands for the CRBN E3 ligase. At first, we aimed to optimize the linker length and linker composition that connected the GDC-0853 warhead to pomalidomide. Most of the resulting PROTACs (PTD1, PTD2, PTD4 and PTD8-PTD11) degraded BTK efficiently at nanomolar concentrations (Table 1). The degradation potency improved as the linker length decreased, and PTD10 was found to be the most potent degrader with a DC50 of 0.5 nM, suggesting that the optimized geometry for ternary complex formation and ubiquitin transfer was achieved with the shortest linker between the two warheads (Figure 3). As was the case for most degraders, the classical Hook effect was observed for PTD10 at concentrations > 333 nM (Figure 3).
Table 1.
Structures and DC50 Values for PTD PROTACs Based on Pomalidomide

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compounds | Linker | R | DC50 (nM) Ramos cellsa |
Dmax (%) Ramos cellsa |
DC50 (nM) JeKo-1 cellsa |
Dmax (%) JeKo-1 cellsa |
| PTD1 |
|
Me | 31.2 ± 16.4 | 94 | 32.1 ± 1.9 | 97.1 |
| PTD2 |
|
Me | 6.6 ± 2.5 | 97.1 | 4.1 ± 1.8 | 97.7 |
| PTD4 |
|
Me | 7.0 ± 2.3 | 95.6 | 9.7 ± 4.6 | 96.9 |
| PTD5 |
|
H | 4.0 ± 1.0 | 98.3 | 7.2 ± 3.4 | 98.8 |
| PTD6 |
|
H | 5.3 ± 2.4 | 97.2 | 6.8 ± 2.7 | 97.7 |
| PTD7 |

|
H | 2.0 ± 0.8 | 96.9 | 3.5 ± 1.2 | 98 |
| PTD8 |

|
Me | 3.2 ± 0.8 | 95 | 3.3 ± 1.4 | 97 |
| PTD9 |

|
Me | 0.9 ± 0.2 | 95.7 | 1.0 ± 0.3 | 97.4 |
| PTD10 |

|
Me | 0.5 ± 0.2 | 97.1 | 0.6 ± 0.2 | 95.7 |
| PTD11 |

|
Me | 1.3 ± 0.3 | 96.1 | 4.1 ± 0.9 | 95.1 |
| PTD15 |
|
Me | 113 ± 20 | 89 | 348 ± 135 | 89.8 |
Cells were treated with PROTACs at concentrations from 3000 nM to 0.15 nM (3-fold dilutions) for 17 h. Western blots were used to quantify BTK levels by normalizing to β-actin. DC50 values are reported as the mean ± standard deviation from two independent experiments.
Figure 3. BTK degradation in Ramos and JeKo-1 cells.
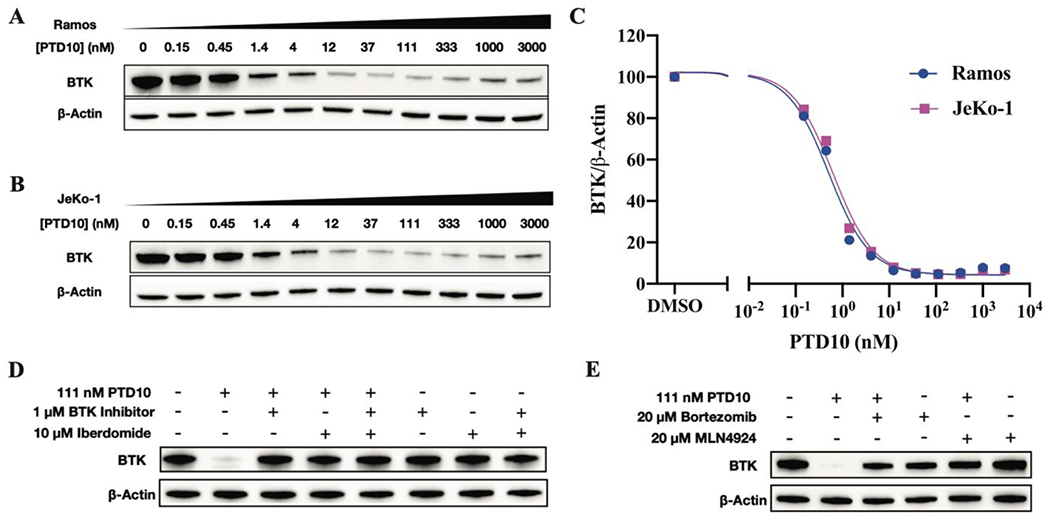
Western blots of BTK degradation by PTD10 in (A) Ramos and (B) JeKo-1cell lines. Cells were treated with different concentrations of PTD10 from 3000 nM to 0.15 nM (3-fold dilution) for 17 h. (C) Dose-dependent BTK degradation in Ramos and JeKo-1 cell lines. For Ramos and JeKo-1 cells, the compound concentration inducing 50% of protein degradation (DC50) was 0.5 ± 0.2 nM and 0.6 ± 0.2 nM, respectively, and the maximal degradation was >95%. (D) Ramos cells were incubated with PTD10 for 17 h in the presence or absence of the BTK inhibitors GDC-0853 or iberdomide (CC-220). (E) Ramos cells were pre-treated with the proteasome inhibitor bortezomib or the NEDD8-activating enzyme E1 inhibitor MLN-4924 for 2 h, and then treated with PTD10 for 4 h. Western blots are representative of two independent experiments.
To extend the SAR, we synthesized analogs in which the methyl group on the piperazine ring had been removed. The resulting compounds, PTD5, PTD6, and PTD7, degraded BTK with similar DC50 values compared to the same compounds bearing the methyl group, suggesting that any alteration in the kinetics of BTK engagement caused by removing the methyl group did not alter the ability of the PROTACs to catalyze degradation (Table 1). We also replaced pomalidomide in PTD10 with lenalidomide (PTD12) which resulted in a ~8-fold decrease in degradation activity (DC50 0.5 nM compared to 4 nM), consistent with the respective binding affinities of the two E3 ligase ligands for CRBN (Table 2).25 Interestingly, PTD13, in which the thalidomide E3 ligase ligand was directly connected to the GDC-0853 warhead, demonstrated poor degradation activity with DC50 > 3000 nM and Dmax 30% (Table 2). This suggests that the alteration in the orientation of the warheads impairs the ability of the compound to stabilize ternary complex formation and/or produce a ternary complex that efficiently ubiquitylates BTK.
Table 2.
Structures and DC50 Values for PTD10 Analogs with Modifications to the E3 Ligase Ligand and Linker Attachment Site

| |||||
|---|---|---|---|---|---|
|
| |||||
| Compounds | CRBN E3 Ligase Ligand | DC50 (nM) Ramos cellsa |
Dmax (%) Ramos cellsa |
DC50 (nM) JeKo-1 cellsa |
Dmax (%) JeKo-1 cellsa |
| PTD12 |

|
3.6 ± 1.2 | 89.8 | 4.6 ± 1.4 | 89.2 |
| PTD13 |

|
>3000 | 30.1 | >3000 | 30 |
| PTD14 |

|
1.7 ± 0.4 | 96.2 | 3.1 ± 0.6 | 97 |
| PTD16 |

|
0.9 ± 0.2 | 97.3 | 0.8 ± 0.2 | 97.6 |
Cells were treated with PROTACs at concentrations from 3000 nM to 0.15 nM (3-fold dilutions) for 17 h. Western blots were used to quantify BTK levels by normalizing to β-actin. DC50 values are reported as the mean ± standard deviation from two independent experiments.
The Gray group has reported that a nitrogen-to-oxygen modification on the pomalidomide aryl attachment point can reduce the ability of immunodulatory imide drugs (IMiDs) to recruit Ikaros family zinc finger 1/3 (IKZF1/3).21 Consequently, we synthesized the PTD10 analog PTD14 with this modification. The DC50 of PTD14 for degrading BTK was 1.7 nM, which is similar to PTD10 (Table 2). Finally, we investigated the impact of altering the linker attachment site and found that moving the linker from the C4 (PTD10) to the C5 (PTD16) position on the pomalidomide phthalimide ring has little effect on degradation (Table 2),14 suggesting that both attachment sites produce PROTACs that can catalyze productive ternary complex formation.
In order to verify the mechanism of action of the BTK degraders, we co-treated Ramos cells with 111 nM PTD10 and either 1 μM GDC-0853 or 20 μM iberdomide (CC-220) for 17 h (Figure 3). BTK degradation was completely blocked, which suggests that PTD10 binds to the active site of BTK and that degradation is dependent on CRBN. In addition, no BTK degradation was observed in Ramos cells treated with GDC-0853 and/or pomalidomide at concentrations from 3000 nM to 0.15 nM, (Figure S2), indicating that the parent molecules cannot catalyze degradation. Finally, in order to substantiate that PTD10 degrades BTK through the ubiquitin-proteasome pathway, Ramos cells were pre-treated with the proteasome inhibitor bortezomib or the neddylation inhibitor MLN-4924 for 2 h before treatment with 111 nM PTD10 for 4 h (Figure 3). The inhibition of BTK degradation by bortezomib or MLN-4924 indicates that the degradation is ubiquitin-proteasome dependent.
Binding affinity, live-cell target occupancy and ternary complex formation
We were interested in the low degradation efficiency of PTD13 and PTD15. Consequently, a time-resolved fluorescence resonance energy transfer (TR-FRET) assay was performed to measure the binding affinity of PTD10, PTD13, PTD15, and GDC-0853 to BTK. The Kd value for PTD10 (2.28 nM) was increased about ~10-fold compared to that for GDC-0853 (0.35 nM) while the value for PTD13 (0.86 nM) was ~3-fold larger than that for GDC-0853 (Table 3). In contrast, the binding affinity of PTD15 was reduced ~300-fold to 107 nM compared to GDC-0853. Thus, changes in affinity for the target cannot be used alone to explain the SAR for degradation efficiency in our system.
Table 3.
BTK Binding and Target Engagement of BTK and CRBN in HEK293 Cells
| Compound | DC50 (nM) Ramos cellsa |
BTK Kd (nM)b | BTK IC50 (nM)c | CRBN IC50 (nM)c | Ternary Complex Formationd |
|---|---|---|---|---|---|
| GDC-0853 | 0.35 | 12.1 ± 1 (1.1) |
NDe | ||
| Pomalidomide | NDe | NDe | 83 ± 10 (0.8) |
||
| PTD10 | 0.5 ± 0.2 | 2.28 | 97 ± 11 (1.9) |
107 ± 19 (1.0) |
100% |
| PTD13 | >3,000 | 0.86 | 320 ± 30 (1.4) |
7000 ± 700 (1.0) |
0 |
| PTD15 | 113 ± 20 | 107 | 3212 ± 208 (1.3) |
5000 ± 300 (0.5) |
~75% |
To gain more insight into the mechanism of action of the PROTACs, we measured the target engagement of BTK and CRBN by PTD10, PTD13, and PTD15 in HEK293 cell lines using NanoBRET. The Nano-Luciferase (Nano-Luc) based bioluminescence resonance energy transfer (NanoBRET) target engagement assay is an intracellular competitive displacement assay, where the unlabeled compound competes a BRET-acceptor tracer from the target resulting in reduced BRET.26 Since we intended to determine the rank-order affinity of the compounds, a single concentration of the tracer was used.
We compared the intracellular engagement of the parent small molecule warheads with that observed for PTD10, PTD13, and PTD15 (Figure 4). While PTD10 was 8-fold less potent than GDC-0853 at engaging BTK, this PROTAC had a similar affinity for CRBN compared to pomalidomide (Table 3). In addition, the interaction of PTD10 with BTK displayed positive cooperativity (Hill slope ~ 2) in contrast to either of the two individual warheads. Furthermore, PTD13 was 30-fold less potent than GDC-0853, and PTD15 was 250-fold less potent than GDC-0853 at engaging BTK. At the same time, PTD13 and PTD15 showed low affinity for CRBN with IC50 values higher than 5,000 nM.
Figure 4. Intracellular target engagement and ternary complex formation.

(A) and (B) NanoBRET was used compare the ability of the warheads (GDC-0853 and pomalidomide) and the PROTACs (PTD10, PTD13, PTD15) to bind to (A) BTK or (B) CRBN in HEK293 cells. The cells were transiently transfected with the BTK-NanoLuc® Fusion vector or the NanoLuc®-CRBN Fusion vector. After 24 h, the NanoBRET™ tracer reagent (1 μM K5 tracer or 0.5 μM CRBN tracer) and a serial dilution of test compounds were added and the cells were incubated at 37 °C for 2 h. Data are presented as the mean value ± SD (n= 2 technical replicates). Two independent biological replicates were performed with similar results. IC50 values are given in Table 3. (C) Ternary complex formation catalyzed by PTD10, PTD13, and PTD15. HEK293 cells were transiently transfected with BTK-NanoLuc® Fusion vector HaloTag-CRBN Fusion Vector overnight. The transfected HEK293 cells were treated with a serial dilution of test compounds. Data are represented as the mean value ± SD (n= 2 technical replicates). Two independent biological replicates were performed with similar results.
The binary target engagement assays cannot account for the differential ability of PTD13 and PTD15 to degrade BTK since PTD15 has lower affinity for BTK both in vitro and in cells than PTD13 while neither PROTAC binds CRBN with affinities similar to either PTD10 or pomalidomide. We therefore hypothesized that the low degradation activity of PTD13 might be due to the inability of this compound to promote ternary complex formation. We therefore used a live-cell NanoBRET assay to investigate the ability of PTD10, PTD13, and PTD15 to promote ternary complex formation in HEK293 cells (Figure 4). HEK293 cells were transiently transfected with BTK-NanoLuc® Fusion vector and HaloTag®-CRBN Fusion Vector, and the BRET signal was monitored at various concentrations of each compound following addition of the NanoBRET 618 ligand and the NanoBRET NanoGlo substrate. While PTD10 demonstrated prominent ternary complex formation, no ternary complex was observed in the presence of PTD13. In contrast, PTD15 generated a maximum BRET signal that was ~75% of that resulting from PTD10, suggesting that PTD15 could stimulate ternary complex formation despite apparently low affinity for CRBN in the binary target engagement assay. These results correlate with the ability of each compound to degrade BTK where DC50 values were PTD10 < PTD15 < PTD13. PTD13 differs from PTD10 by the removal of a CH2 group and replacement of NH with CH2 which we assume has altered the relative orientation of the two warheads such that PTD13 is unable to stabilize ternary complex between BTK and CRBN.
PTD10 targets BTK with improved selectivity
To evaluate the selectivity of the BTK PROTACs, we treated TMD8 cells with 111 nM PTD10, PTD14, and P13I, and performed a label-free mass-spectrometry proteomics study (Figure 5). We chose to evaluate changes in protein levels 6 h after treatment since we speculated that changes in protein abundance at this time would mainly originate directly from protein degradation rather than from downstream effects such as transcriptional or translational changes resulting from BTK degradation. Our results demonstrate that PTD10 and PTD14 are highly selective BTK degraders and did not affect the abundance of other kinases, including CSK, spleen tyrosine kinase (SYK), FRK, HCK, LYN, BAZ1B, tyrosine kinase 2 (TYK2), and Fes/Fps. In contrast, P13I, which is based on ibrutinib, showed significant degradation of some off-targets, specifically, CSK and FRK. It should be noted that Src, bone marrow tyrosine kinase gene in chromosome X protein (Bmx), and Fgr, the reported off-targets of GDC-0853 were not detected in the proteomics study.
Figure 5. PTD10 and PTD14 are potent and selective degraders of BTK.

TMD8 cells were treated with PTD10 (A), PTD14 (B), and P13I (C) at 111 nM for 6 h, respectively. Protein levels were quantified using label-free proteomics analysis. Data are plotted as fold change (Log2) of proteins versus −Log10(P-value) in the treated samples compared to the DMSO controls.
We were also interested in determining the impact of the PROTACs on the abundance of neo-substrates, and we found that both PTD10 and P13I degrade IKZF1and IKZF3. However, PTD14 demonstrated no degradation of these neo-substrates, indicating the impact of using an aryl ether as the exit vector from the phthalimide ring, although all three compounds degraded the less well-studied neo-substrate of ZNF280D. In addition, PTD10, PTD14, and P13I did not affect the levels of the cell cycle protein G1 to S phase transition 1 (GSPT1) which is essential for the G1 to S phase transition 1. We further validated the effects of PTD10 and PTD14 on IKZF1/3 and GSPT1 using western blot assays (Figure S1).
Cell-Based Phenotypic Studies
PTD10 inhibits cell growth with enhanced efficacy
We investigated the effect of PTD10 on cell growth in TMD8 and Mino cells, two cell lines that are sensitive to BTK inhibition and degradation. PTD10 was found to be a highly potent at inhibiting cell growth, with IC50 values of 1.4 nM and 2.2 nM for TMD8 and Mino cells, respectively (Figure 6, Table 4). For TMD8 cells, PTD10 was 10-fold more potent than GDC-0853, and over 100-fold more potent than pomalidomide, while in Mino cells, PTD10 was 3-fold more potent than GDC-0853, and 10-fold more potent than pomalidomide (Figure 6). These results suggest that BTK degradation has a more profound effect on the inhibition of cell growth than BTK inhibition. In addition, PTD10 was a more potent inhibitor of cell growth in both cell lines compared to the previously reported PROTACs, P13I, DD-03-171, and MT-802 (Figure 6). Taken together, these results demonstrate that PTD10 is a highly potent inhibitor of TMD8 and Mino cell growth.
Figure 6. Inhibition of cell growth by PTD10.

Cells were treated with compound for 48 h after which cellular viability was quantified using the CellTiter-Glo 2.0 assay. (A) and (B) Comparison of PTD10 with GDC-0853, pomalidomide, and the BTK PROTACs DD-03-171, MT-802, and P13I in TMD8 cells. (C) and (D) Comparison of PTD10 with GDC-0853, pomalidomide, and the BTK PROTACs DD-03-171, MT-802, and P13I in Mino cells. Data were fit to an IC50 equation in GraphPad Prism where the Hill slope was allowed to float and are represented as the mean value ± SD from two technical replicates in one representative experiment. Similar results were obtained in two additional independent experiments. IC50 values are given in Table 4.
Table 4.
Effect of BTK Degradation on Cell Viability
| Compound | TMD8 Cells IC50 (nM)a |
Mino Cells IC50 (nM)a |
|---|---|---|
| PTD10 | 1.4 ± 0.2 (1.6) | 2.2 ± 0.8 (1.0) |
| GDC-0853 | 12.6 ± 1.4 (2.0) | 7.8 ± 2.2 (1.4)b |
| Pomalidomide | 187 ± 54 (0.8) | 20.8 ± 2.5 (2.0) |
| DD-03-171 | 5.1 ± 0.9 (1.5) | 10.9 ± 4.6 (0.6) |
| MT-802 | 12.0 ± 1.4 (1.6) | 100c |
| P13I | 15.0 ± 2.8 (1.2) | 10.4 ± 2.4 (3.7) |
Numbers in parentheses are the Hill slopes.
Cell viability was ~20% at the highest concentration of compound.
Cell viability was ~40% at the highest concentration of compound.
PTD10 induces cell apoptosis
After showing that PTD10 potently affected cell viability, we next used flow cytometry to explore the induction of apoptosis by PTD10 in TMD8 and Mino cell lines. PTD10 was found to induce apoptosis in a dose-dependent manner with robust apoptosis detected at 333 nM (Figure 7). The hook effect was observed at higher concentrations, consistent with the degradation studies. We also performed similar experiments with GDC-0853 and pomalidomide using the same doses (Figure S3). Whereas GDC-0853 did not cause apoptosis in Mino cells, a dose-dependent effect was observed in TMD8 cells. In contrast, pomalidomide, acting as an immunomodulatory imide drug, induced apoptosis in Mino cells in a dose-dependent manner but showed little effect on TMD8 cells. It should be noted that PTD10 induced apoptosis in both cell lines at lower concentrations than either GDC-0853 or pomalidomide. In addition, we also found that PTD10 was equal to or better than P13I, DD-03-171, and MT-802 at inducing apoptosis in TMD8 and Mino cells lines (Figure 7).
Figure 7. Induction of apoptosis resulting from degradation of BTK.

(A) Induction of apoptosis by PTD10 in TMD8 cells and in (B) Mino cells. Cells were treated with PTD10 for 48 h at concentrations ranging from 3000 nM to 0.15 nM (3-fold dilutions) and with 1% DMSO as a control. (C) Induction of apoptosis by PTD10, P13I, DD-03-171, and MT-802 in TMD8 and (D) Mino cells treated with test compounds at 1.4, 12, 111, and 1000 nM for 48 h. Apoptosis was examined using Alexa Fluor 488 Annexin-V/ propidium iodide (PI) staining. Data were collected on a CytoFLEX flow cytometry instrument. Percentages of live, apoptotic, and necrotic cells were analyzed in Prism 8.0. Data are the mean of two independent experiments and errors are the standard deviation of the mean.
To determine the mechanism of cell apoptosis, TMD8 and Mino cells were treated with PTD10 for 48 h and the expression levels of cleaved caspase-3, poly ADP-ribose polymerase (PARP), and mitochondrial apoptotic pathway proteins (Bcl-2 family) were quantified by western blot (Figure 8). A dose-dependent decrease in the levels of PARP and caspase-3 as well as an increase in the level of cleaved-PARP and cleaved caspase-3 were observed. In addition, a significant down-regulation was observed in the levels of B-cell lymphoma-2 (Bcl-2) and myeloid cell leukemia-1 (Mcl-1) over a range of concentrations. Taken together, these results indicate that PTD10 induces apoptosis via activation of the caspase-dependent pathway and mitochondrial pathway in TMD8 and Mino cell lines.
Figure 8. PTD10 induces cell apoptosis via activation of the caspase-dependent pathway and mitochondrial pathway.

(A) The effect of PTD10 on the expression levels of capase-3, cleaved-caspase-3, PARP, cleaved-PARP, Bcl-2, and MCL-1 in TMD8 cells and (B) in Mino cells. The cells were treated for 48 h with concentrations of PTD10 ranging from 3000 nM to 0.15 nM (3-fold dilution) and with 1% DMSO as a control. Protein expression levels were determined via western blot, and the data are representative of two independent experiments.
PTD10 has higher potency than GDC-0853 at inhibiting BCR signaling pathway
To gain insight into the molecular mechanism underlying the inhibition of cell growth, we next investigated the influence of the compounds on the BCR signaling pathway. TMD8 and Mino cells were treated with PTD10 or GDC-0853, and then activated with goat F(ab’) 2 anti-human IgM. Cells that were not treated or stimulated served as controls. For PTD10, degradation of BTK leads to a series of cellular responses in downstream signaling pathways (Figure 9). Robust downregulation was observed after 48 h incubation not only for the phospho-protein levels of p-PLCγ2, p-BTK, p-AKT, and p-NF-κB, but also for total protein of phospholipase Cγ2 (PLCγ2), AKT, and nuclear factor κB (NF-κB). In addition, while phosphorylation of extracellular signal-regulated kinase (ERK) was not affected in the Mino cell line, activation of ERK was observed in TMD8 cells, which suggests some compensatory effects in the MAPK/ERK pathway for this specific cell line. Importantly, the inhibitory effect of GDC-0853 on BCR signaling pathway was less prominent compared to that of PTD10 in both cell lines (Figure 9), consistent with observation that PTD10 was a more potent inhibitor of cell growth than GDC-0853.
Figure 9. PTD10 exhibits more prominent effects on downstream cell signaling pathways than GDC-0853 in TMD8 and Mino cell lines.

(A) TMD8 cells were serum starved and then treated with serial dilutions of PTD10 or (B) GDC-0853 from 3000 nM to 0.15 nM (3-fold dilutions) for 48 h in media. Mino cells were treated in a similar fashion with either (C) PTD10 or (D) GDC-0853. Cells were stimulated with 10 μg/mL goat F(ab’) 2 anti-human IgM for 10 mins before harvesting. Cells that were not treated or stimulated served as controls. The level of p-PLCγ2(Y1217), PLCγ2, p-BTK, BTK, p-AKT, AKT, p-NF-κB, NF-κB, p-ERK, ERK, as wells as two housekeeping proteins, vinculin and GAPDH, were quantified using western blots. All western blotting data are representative of three independent experiments.
DISCUSSION
Targeted protein degradation is a rapidly developing therapeutic approach that is being applied to multiple oncology targets including BTK. Four BTK PROTACs are in phase 1 clinical trials and there are a number of publications describing BTK degraders. However, despite this progress, the structural diversity of existing BTK PROTACs is limited, and most are based on the non-selective BTK inhibitor ibrutinib. Here, we describe the synthesis and cellular characterization of BTK PROTACs based on GDC-0853, a potent and reversible BTK inhibitor with improved selectivity.
PROTAC catalyzed targeted protein degradation occurs via the formation of a ternary complex between the target protein and an E3 ligase that results in ubiquitination and degradation of the target protein. Linkerology is a critical factor in productive ternary complex formation,27,28 and in our studies we found that PTD10, the compound with the shortest linker, was the most potent BTK degrader with a sub-nanomolar DC50 value. This is interesting since short linkers are not commonly observed in PROTAC designs.28 In addition, while PTD13 was more potent than PTD15 at engaging BTK and had similar potency for CRBN target engagement, PTD13 demonstrated much lower degradation potency than PTD15. This result suggests that a certain amount of target engagement is sufficient but not necessary to form the ternary complex, while the conformation and geometry of the PROTAC molecules are clearly of great importance for degradation activity.
Quantitative proteomics is a useful tool for evaluating PROTAC selectivity.29 Off-target activities can arise from interaction of the target warhead with weakly binding proteins, while immunomodulatory drugs (IMiDs), which function as CRBN E3 ligase ligands, degrade neo-substrates, including the transcription factors IKZF1 and IKZF3,30 and translation regulator GSPT1,31 by acting as molecular glues. It should be noted that despite degrading neo-substrates, this activity increases the anti-tumor effects especially for the treatment of B-cell malignances.21,32 To date, most of the published BTK PROTACs based on ibrutinib have been reported to degrade some common kinases other than BTK, including CSK, LYN and BLK.17–19 In our studies, we demonstrated that our PROTACs show improved selectivity for degrading BTK without affecting other kinases including CSK, SYK, FRK, HCK, LYN, BAZ1B, TYK2, and Fes/Fps. Unfortunately, we did not detect the reported off-targets of GDC-0853 (Src, Bmx, and Fgr) in our studies, and thus we cannot compare the selectivity of PTD10 with GDC-0853. We also observed the degradation of neo-substrates IKZF1 and IKZF3 by PTD10 but not by PTD14, indicating that E3 ligase ligand selectivity can be tuned by altering the exit vector. It is also worth emphasizing that the levels of GSPT1 was not affected by the GDC-0853-based PROTACs.
Since PTD10 had high potency and selectivity for degrading BTK, we further explored the phenotypic effects caused by this PROTAC including inhibition of cell growth and induction of cell apoptosis. While PTD10 showed potent degradation effects in all seven B-cell malignant cell lines (Figure S4), it only exhibited complete cell growth inhibition in TMD8 and Mino cell lines (Figure S5), suggesting that BTK is important for the proliferation of these two cell lines. We found that PTD10 exhibited superior potency for inhibiting cell growth and inducing apoptosis in comparison to GDC-0853, pomalidomide, as well other reported BTK PROTACs. The cleavage of PARP and caspase 3 and the downregulation of Bcl-2 and Mcl-1 indicates that PTD10 induces apoptosis via activation of the caspase-dependent pathway and mitochondrial pathway.
BTK is an essential component of the BCR signaling pathway regulating the survival, activation, proliferation, and differentiation of B cells. Inhibition of BTK and down-stream BCR signaling pathway has been shown to be the mechanism for the therapeutic effect of ibrutinib.33 In addition, GDC-0853 has been reported to inhibit BCR signaling by deactivation of BTK, AKT(S473), PLCγ2(Y1217), and ERK(T202/Y204).34 In our studies, we compared the inhibition effect of GDC-0853 and PTD10 on BCR, as well as PI3K/AKT, MAPK/ERK, and NF-κB pathways. Our results suggest that PTD10 is more potent than GDC-0853 for inhibiting the phosphorylation of BTK, leading to the downregulation and deactivation of downstream effectors including PLCγ2, AKT, and NF-κB. It should be noted that the activation of ERK in TMD8 cell line indicates some compensatory effects in the MAPK/ERK pathway for this specific cell line.
CONCLUSION
In summary, we have discovered a novel series of BTK PROTAC based on GDC-0853, a best-in-class BTK inhibitor. We found that the conformation and geometry of PROTAC molecules was critical for the degradation activity, and that alteration of the exit vector from the CRBN ligand modulated the degradation of neo-substrates. Significantly, the most potent compound PTD10 demonstrated enhanced degradation activity with a sub-nanomolar DC50 value, while proteomic analysis revealed that PTD10 had higher selectivity than other ibrutinib-based BTK PROTACs. Furthermore, PTD10 exhibited improved phenotypic effects including cell growth inhibition and induction of cell apoptosis compared to the warheads, while mechanism of action studies revealed that PTD10 induces apoptosis via activation of the caspase-dependent and mitochondrial pathways. Consistent with these results, PTD10 was found to have more prominent effects on the BCR signaling pathway than GDC-0853. Therefore, these results indicate that PTD10 is a promising pharmaceutical research tool and is a promising starting point for the development of therapeutics against B-cell malignances and autoimmune diseases.
EXPERIMENTAL SECTION
Chemistry
All chemical reactions were performed under nitrogen using anhydrous conditions unless otherwise noted. All chemicals were purchased from commercial suppliers and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) using pre-coated 220 μm silica gel 60 F254 TLC plates that were visualized by fluorescence quenching under UV light. A CombiFlash NextGen 300+ instrument equipped with a variable wavelength UV detector and a fraction collector was used to perform flash column chromatography. All target compounds were purified on a Shimadzu Prominence high-performance liquid chromatography (HPLC) instrument equipped with a semi-preparative 250 × 10 mm Luna 5 μm 100 Å C18 column and a multi-wavelength UV detector. The flow rate was set at 4 mL/min, and a gradient program from 50 to 80% MeCN in water was used for elution. All final compounds were purified to >95% as assessed by HPLC with detection at 254 and 353 nm. High-resolution mass spectra (HRMS) and low-resolution mass spectra were obtained using a Bruker Impact II QTOF mass spectrometer equipped with an Agilent 1290 Infinity II UHPLC and an Agilent 6110 single quadrupole mass spectrometer equipped with a 1260 HPLC, respectively. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker Ascend 700, Bruker 500 Advance, or Bruker 400 Nanobay spectrometers. Chemical shifts are expressed in δ ppm referenced to an internal standard, tetramethylsilane (δ = 0 ppm). Abbreviations used in describing peak signals are br=broad, s=singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet. Microwave irradiation reaction was performed on the Discover S microwave synthesizer.
General Chemistry Procedure
BTK PROTAC DD-03-171 was purchased from TOCRIS (cat#7160), and MT-802 was purchased from MedChemExpress (cat#HY-122562). The synthesis of P13I has been described previously.15
Synthesis of GDC-0853 warhead 1.
Tert-butyl (S)-3-methyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate (1).
A 50 mL three-neck round bottle flask equipped with a magnetic stirrer and a reflux condenser was charged with 5-bromo-2-nitropyridine (3.1 g, 15.28 mmol), (S)-tert-butyl 3- methylpiperazine-1-carboxylate (3 g, 14.98 mmol), K3PO4 (8.1 g, 38.2 mol), Pd(OAc)2 (50.45 mg, 0.22 mmol), and BINAP (139.8 mg, 0.22 mmol). 1,4-Dioxane (30 mL) was added to the flask under nitrogen, and the mixture was stirred at 110 °C overnight. After cooling to room temperature, the mixture was filtered and extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/ DCM) to give 1 as a yellow solid (2.3 g, 48% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 9.1 Hz, 1H), 8.08 (d, J = 3.0 Hz, 1H), 7.15 (dd, J = 9.2, 3.1 Hz, 1H), 4.05 (d, J = 58.3 Hz, 3H), 3.57 (dt, J = 12.5, 3.6 Hz, 1H), 3.28 (td, J = 12.4, 11.7, 3.7 Hz, 2H), 3.13 (s, 1H), 1.48 (s, 9H), 1.20 (d, J = 6.6 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 154.89, 148.87, 147.62, 133.37, 120.34, 119.89, 80.49, 49.88, 48.21, 46.73, 43.29, 42.24, 41.09, 28.37, 13.78, 13.45. [ESI-MS] (ESI+): m/z calcd for C15H23N4O4, [M + H]+ 323.2, found 323.2
Tert-butyl (S)-4-(6-aminopyridin-3-yl)-3-methylpiperazine-1-carboxylate (2).
A 100 mL single round bottom flask was charged with 1 (1.12 g, 3.10 mmol), 10% palladium on carbon (50% water, 180 mg), EtOAc (5 mL), and EtOH (30 mL). The mixture was then stirred at room temperature under nitrogen for 6 h. After all of the starting material has been consumed, hydrogen was replaced with nitrogen. The catalyst was removed by filtration through a pad of Celite, and the filtrate was concentrated to give 2 as an orange solid (1 g, 99% yield). The product was used in the next step without purification. [ESI-MS] (ESI+): m/z calcd for C15H25N4O4, [M + H]+ 293.2, found 293.2.
Tert-butyl (S)-4-(6-((5-bromo-1-methyl-2-oxo-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)-3-methylpiperazine-1-carboxylate (3).
A 50 mL three-neck round bottom flask equipped with a magnetic stirrer and a reflux condenser was charged with 3,5-dibromo-1-methylpyridin-2(1H)-one (761 mg, 2.85 mmol), Cs2CO3 (1.3g, 3.99 mmol), Xantphos (131.9 mg, 0.228 mmol), and Pd2(dba)3 (104.4 mg, 0.114 mmol). Then, tert-butyl (S)-4-(6-aminopyridin-3-yl)-3-methylpiperazine-1-carboxylate 2 (1 g, 3.42 mmol) dissolved in 1,4-dioxane (24 mL) was added to the flask under nitrogen, and the mixture was stirred at 100 °C overnight. After cooling to room temperature, the mixture was filtered and extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: hexane/ethyl acetate) to give 3 as a dark orange solid (773.4 mg, 56.9% yield). 1H NMR (700 MHz, CDCl3) δ 8.58 (d, J = 2.4 Hz, 1H), 7.98 (s, 1H), 7.79 (s, 1H), 7.26 – 7.22 (m, 1H), 6.93 (d, J = 2.5 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 3.86 – 3.62 (m, 1H), 3.58 (s, 3H), 3.49 (d, J = 59.5 Hz, 4H), 3.00 (d, J = 33.0 Hz, 2H), 1.47 (s, 10H), 0.93 (d, J = 6.2 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 156.86, 154.89, 149.11, 140.23, 139.43, 138.70, 132.27, 130.35, 129.73, 125.64, 117.72, 112.62, 100.28, 79.83, 52.94, 49.78, 48.72, 47.60, 46.24, 44.33, 43.08, 40.98, 37.78, 28.43, 13.66, 13.01. [ESI-MS] (ESI+): m/z calcd for C21H29BrN5O3, [M + H]+ 478.1, found 478.1
Tert-butyl (S)-3-methyl-4-(6-((1-methyl-2-oxo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate (4).
A 25 mL two-neck round bottom flask was charged with, Pd(OAc)2 (35.93 mg, 0.16 mmol), and Xphos (152.55 mg, 0.32 mmol), KOAc (471.1 mg, 4.8 mol), B2pin2 (487.6 mg, 1.92 mol). Then, tert-butyl (S)-4-(6-((5-bromo-1-methyl-2-oxo-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)-3-methylpiperazine-1-carboxylate 3 (763.7 mg, 1.6 mmol) dissolved in dioxane (10 mL) was added to the flask under nitrogen, and the mixture was stirred at 100 °C for 3 h. After cooling to room temperature, the mixture was filtered through a celite pad, and the filtrate was concentrated to yield the crude product which was purified using flash column chromatography (SiO2: Et2O/ EtOAc) to give the title compound 4 as a yellow solid (590 mg, 70.2% yield). [ESI-MS] (ESI+): m/z calcd for C27H41BN5O5, [M + H]+ 526.3, found 526.3.
Tert-butyl (S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-formyl-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridine-3-yl)-3-methylpiperazine-1-carboxylate (5).
A 25 mL Schlenk tube was charged with tert-butyl (S)-3-methyl-4-(6-((1-methyl-2-oxo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate 4 (590 mg, 1.12 mmol), 4-chloro-2-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)nicotinaldehyde (579 mg, 1.68 mmol), Pd(dppf)Cl2 (164 mg, 0.22 mmol), and K3PO4 (715.1 mg, 3.37 mmol). THF/H2O (4.8 mL/1.2 mL) was added under nitrogen, and the mixture was stirred at 50 °C overnight. After cooling to room temperature, the mixture was extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/DCM) to give 5 as a yellow solid (360 mg, 45.4% yield). 1H NMR (700 MHz, CDCl3) δ 9.95 (s, 1H), 8.62 – 8.59 (m, 1H), 8.51 (d, J = 5.1 Hz, 1H), 7.94 (s, 1H), 7.85 (s, 1H), 7.24 (dd, J = 16.1, 6.8 Hz, 2H), 6.94 – 6.90 (m, 1H), 6.81 – 6.75 (m, 2H), 4.38 (t, J = 5.6 Hz, 2H), 4.17 (t, J = 5.6 Hz, 2H), 3.76 (dd, J = 31.1, 18.1 Hz, 1H), 3.66 (s, 3H), 3.59 – 3.49 (m, 1H), 3.40 (d, J = 51.8 Hz, 3H), 2.99 (d, J = 35.8 Hz, 2H), 2.54 (s, 2H), 2.46 (s, 2H), 1.47 (s, 9H), 1.24 (s, 6H), 0.92 (d, J = 6.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 189.51, 158.90, 157.52, 154.91, 151.54, 149.82,149.59, 147.34, 141.89, 140.08, 139.37, 138.95, 131.73, 130.48, 129.87, 127.64, 127.61, 125.86, 125.85, 122.29, 115.96, 115.44, 112.52, 111.18, 79.83, 53.03, 49.90, 48.79, 47.82, 46.95, 46.95, 46.50, 45.88, 45.88, 44.28, 43.21, 42.53, 40.93, 39.51, 38.22, 30.31, 28.44, 13.74, 13.14. [ESI-MS] (ESI+): m/z calcd for C39H47N8O5, [M + H]+ 707.4, found 707.3.
Tert-butyl (S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridine-3-yl)-3-methylpiperazine-1-carboxylate (6).
A 10 mL single-neck round bottom flask was charged with tert-butyl (S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-formyl-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridine-3-yl)-3-methylpiperazine-1-carboxylate 5 (27.4 mg, 0.0388 mmol) and MeOH (2 mL). NaBH4 (1.9 mg, 0.05 mmol) was added to the flask at 0 °C on an ice bath and the mixture was stirred at 0 °C for 5 min, and then stirred at room temperature for 1 h after which it was filtered and concentrated to give 6 as a yellow solid (27 mg, 98%). The crude product was used for next step without further purification. [ESI-MS] (ESI+): m/z calcd for C39H49N8O5, [M + H]+ 709.4, found 709.4.
(S)-2-(3’-(hydroxymethyl)-1-methyl-5-((5-(2-methylpiperazin-1-yl)pyridine-2-yl)amino)-6-oxo-1,6-dihydro-[3,4’-bipyridin]-2’-yl)-7,7-dimethyl-3,4,7,8-tetrahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one (GDC-0853 warhead 1).
To a solution of 6 (27 mg, 0.0381mmol) in DCM (3 mL) was added TFA (0.5 mL) dropwise. The mixture was stirred at room temperature for 2 h. After all of the starting material had been consumed, the mixture was concentrated in vacuo to give (1) as a brown liquid (22.7 mg, 98% yield). The crude product was used for the next step without purification.
Synthesis of GDC-0853 warhead 2.
Tert-butyl 4-(6-((5-bromo-1-methyl-2-oxo-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate (7).
A 250 mL three-neck round bottom flask equipped with a magnetic stirrer and a reflux condenser was charged with 1-Boc-4-(6-amino-3-pyridyl)piperazine (2 g, 7.185 mmol), 3,5-dibromo-1-methylpyridin-2(1H)-one (1.74 g, 6.53 mmol), Cs2CO3 (2.98 g, 9.14 mmol), Xantphos (302.3 mg, 0.522 mmol), and Pd2(dba)3 (239.2 mg, 0.261 mmol). Then 1,4-dioxane (30 mL) was added under nitrogen, and the mixture was stirred at 100 °C overnight. After cooling to room temperature, the mixture was filtered and extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: hexane/EtOAc) to give 7 as a white solid (1.283 g, 42.4% yield). 1H NMR (700 MHz, CDCl3) δ 8.57 (d, J = 2.5 Hz, 1H), 7.99 (d, J = 2.9 Hz, 1H), 7.81 (s, 1H), 7.26 (dd, J = 8.9, 3.0 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 8.9 Hz, 1H), 3.60 (d, J = 7.3 Hz, 8H), 3.06 (t, J = 5.0 Hz, 3H), 1.50 (s, 9H). 13C NMR (176 MHz, CDCl3) δ 156.89, 154.66, 148.72, 141.52, 135.95, 132.31, 128.39, 125.63, 117.68, 112.67, 100.33, 80.02, 50.26, 44.09, 42.94, 37.80, 28.44. [ESI-MS] (ESI+): m/z calcd for C20H27BrN5O3, [M + H]+ 464.1, found 464.1.
Tert-butyl 4-(6-((1-methyl-2-oxo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate (8).
A 25 mL three-neck round bottom flask was charged with tert-butyl 4-(6-((5-bromo-1-methyl-2-oxo-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate 7 (463.1 mg, 1 mmol), Pd(OAc)2 (22.45 mg, 0.1 mmol), Xphos (95.34 mg, 0.2 mmol), KOAc (294.42 mg, 3 mol), and B2pin2 (304.73 mg, 1.2 mol). Dioxane (10 mL) was then added to the flask under nitrogen and the mixture was stirred at 100 °C for 2 h. After cooling to room temperature, the mixture was filtered through a celite pad, and the filtrate was concentrated to yield the crude product which was purified using flash column chromatography (SiO2: Et2O/ EtOAc)) to give the title compound 8 as a yellow solid (254 mg, 49.7% yield). [ESI-MS] (ESI+): m/z calcd for C26H39BN5O5, [M + H]+ 512.3, found 512.3.
Tert-butyl 4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-formyl-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridine-3-yl)piperazine-1-carboxylate (9).
A 10 mL Schlenk tube was charged with tert-butyl 4-(6-((1-methyl-2-oxo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2-dihydropyridin-3-yl)amino)pyridine-3-yl)piperazine-1-carboxylate 8 (254 mg, 0.5 mmol), 4-chloro-2-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)nicotinaldehyde (256.2 mg, 0.74 mmol), Pd(dppf)Cl2 (72.7 mg, 0.1 mmol), and K3PO4 (316.4 mg, 1.5 mmol). THF/H2O (3.2 mL/0.8 mL) was added to the tube under nitrogen, and the mixture was stirred at 50 °C overnight. After cooling to room temperature, the mixture was extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/ DCM) to give the title compound 9 as a yellow solid (75 mg, 21.8% yield). 1H NMR (700 MHz, CDCl3) δ 9.96 (s, 1H), 8.60 (d, J = 2.4 Hz, 1H), 8.52 (d, J = 5.1 Hz, 1H), 7.93 (d, J = 3.0 Hz, 1H), 7.85 (s, 1H), 7.27 – 7.23 (m, 2H), 6.93 (d, J = 2.4 Hz, 1H), 6.83 – 6.77 (m, 2H), 4.41 – 4.36 (m, 2H), 4.18 (t, J = 5.7 Hz, 2H), 3.67 (s, 3H), 3.58– 3.56 (m, 4H), 3.05 – 3.01(m, 4H), 2.55 (s, 2H), 2.48 (s, 2H), 1.48 (s, 9H), 1.25 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 189.52, 158.89, 157.49, 154.66, 151.51, 149.82, 148.95, 147.36, 141.90, 141.41, 136.07, 131.77, 128.36, 127.61, 127.53, 125.82, 122.26, 115.96, 115.24, 112.57, 111.16, 79.96, 74.92, 50.24, 46.94, 45.86, 42.51, 40.92, 39.49, 38.22, 30.31, 28.42, 24.83. [ESI-MS] (ESI+): m/z calcd for C38H45N8O5, [M + H]+ 693.3, found 693.4.
Tert-butyl 4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridine-3-yl)piperazine-1-carboxylate (10).
A 10 mL single-neck round bottom flask was charged with compound 9 (28.4 mg, 0.041 mmol) and MeOH (2 mL). NaBH4 (1.9 mg, 0.05 mmol) was added to the mixture at 0 °C on an ice bath. The mixture was stirred at 0 °C for 5 min, and then stirred at room temperature for 1 h. The mixture was filtered and concentrated to give the title compound 10 as a yellow solid (27.9 mg, 98%). The crude product was used for next step without further purification. [ESI-MS] (ESI+): m/z calcd for C38H47N8O5, [M + H]+ 695.4, found 695.4.
2-(3’-(hydroxymethyl)-1-methyl-6-oxo-5-((5-(piperazin-1-yl)pyridine-2-yl)amino)-1,6-dihydro-[3,4’-bipyridin]-2’-yl)-7,7-dimethyl-3,4,7,8-tetrahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one (GDC-0853 warhead 2).
To a solution of 10 (27.9 mg, 0.04mmol) in DCM (3 mL) was added TFA (0.5 mL) dropwise, and the mixture was stirred at room temperature for 2 h. After all of the starting material had been consumed, the mixture was concentrated in vacuo to give the title compound GDC-0853 warhead 2 as a brown liquid (23.2 mg, 97% yield). The crude product was used for next step without purification.
Synthesis of E3 ligase ligand conjugates.
Tert-butyl (2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)glycinate (11).
Compound 11 was synthesized according to the literature.23 A 10 mL two-neck round bottom flask equipped with a stirrer and a reflux condenser was charged with lenalidomide (259.3 mg, 1 mmol). NMP (5 mL) was then added to the flask, followed by DIPEA (387.7 mg, 3 mmol), and tert-butyl bromoacetate (234. 1 mg, 1.2 mmol), and the mixture was stirred at 110 °C overnight. After cooling to room temperature, the mixture was extracted with EtOAc (3 × 30 mL). The organic layers were combined, washed with brine five times, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/ DCM) to give the title compound 11 as a white solid (122 mg, 32.7% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 7.29 (t, J = 7.7 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.02 (t, J = 6.5 Hz, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.30 – 4.13 (m, 2H), 3.90 (d, J = 5.8 Hz, 2H), 2.93 (ddd, J = 17.2, 13.6, 5.4 Hz, 1H), 2.63 (dt, J = 17.2, 3.7 Hz, 1H), 2.33 (qd, J = 13.2, 4.4 Hz, 1H), 2.04 (dtd, J = 12.0, 4.9, 1.9 Hz, 1H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 173.38, 171.69, 170.62, 169.19, 143.72, 132.62, 129.57, 127.14, 112.83, 111.41, 81.17, 52.01, 46.13, 45.68, 31.73, 28.20, 23.24. [ESI-MS] (ESI+): m/z calcd for C19H24N3O5, [M + H]+ 374.2, found 374.1.
(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)glycine (12).
A 10 mL single-neck round bottom flask was charged with compound 11 (14 mg, 0.038 mmol) and 4M HCl in dioxane (3 mL). The mixture was stirred at room temperature overnight. After all of the starting material had been consumed, the mixture was concentrated in vacuo to give the title compound 12 as a white solid (11.4 mg, 95% yield). The crude product was used for the next step without purification.
Tert-butyl (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)glycinate (13).
Compound 13 was synthesized according to the literature.24 A solution of 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (552.4 mg, 2 mmol), tert-butyl glycinate (314.8 mg, 2.4 mmol), and DIPEA (775.4 mg, 6 mmol) in NMP (5 mL) was heated at 130 °C in a microwave reactor with microwave absorbance at 200 watts for 2 h. After cooling to room temperature, the mixture was extracted with EtOAc (3 × 30 mL). The organic layers were combined, washed with brine five times, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/ DCM) to give the title compound 13 as a yellow solid (84.7 mg, 11% yield). 1H NMR (700 MHz, DMSO-d6) δ 11.08 (s, 1H), 7.60 (d, J = 8.3 Hz, 1H), 7.36 (t, J = 6.3 Hz, 1H), 6.97 (s, 1H), 6.89 (d, J = 8.5 Hz, 1H), 5.05 (dd, J = 12.9, 5.5 Hz, 1H), 4.01 (d, J = 6.2 Hz, 2H), 2.88 (ddd, J = 17.1, 13.9, 5.4 Hz, 1H), 2.61 – 2.51 (m, 2H), 2.01 (dtd, J = 12.9, 5.3, 2.3 Hz, 1H), 1.43 (s, 9H). 13C NMR (176 MHz, DMSO-d6) δ 173.30, 170.62, 169.88, 168.06, 167.60, 154.59, 134.45, 125.42, 117.55, 81.55, 49.13, 45.36, 31.45, 28.19, 22.67. [ESI-MS] (ESI+): m/z calcd for C19H22N3O6, [M + H]+ 388.1, found 388.1.
(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)glycine (14).
To a solution of 13 (14.7 mg, 0.038 mmol) in DCM (3 mL) was added TFA (0.5 mL) dropwise, and the mixture was stirred at room temperature for 3 h. After all of the starting material had been consumed, the mixture was concentrated in vacuo to give the title compound 14 as a yellow solid (11 mg, 87% yield). The crude product was used for next step without purification.
12-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)dodecanoic acid (15).
A 50 mL two-neck round bottom flask equipped with a stirrer was charged with 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (552.44 mg, 2 mmol) and 12-aminododecanoic acid (516 mg, 2.4 mmol). DMF (10 mL) was added to the flask, followed by DIPEA (12.9 g, 100 mmol), and the mixture was stirred at 90 °C overnight. After cooling to room temperature, the mixture was extracted with DCM (3 × 20 mL). The organic layers were combined, washed with brine five times, dried over MgSO4, and concentrated to yield the crude product which was purified using flash column chromatography (SiO2: MeOH/ DCM) to give the title compound 15 as a yellow solid (360 mg, 38.2% yield). 1H NMR (700 MHz, DMSO-d6) δ 11.11 (s, 1H), 7.58 (dd, J = 8.5, 7.1 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 7.0 Hz, 1H), 6.53 (t, J = 5.9 Hz, 1H), 5.06 (dd, J = 12.9, 5.5 Hz, 1H), 3.29 (q, J = 6.7 Hz, 2H), 2.89 – 2.86 (m, 1H), 2.59 (ddd, J = 17.2, 4.4, 2.4 Hz, 1H), 2.18 (t, J = 7.4 Hz, 2H), 2.03 (dtd, J = 13.1, 5.4, 2.4 Hz, 1H), 1.57 (p, J = 7.1 Hz, 2H), 1.48 (p, J = 7.3, 6.7 Hz, 2H), 1.36 – 1.28 (m, 4H), 1.24 (s, 12H). 13C NMR (176 MHz, DMSO-d6) δ 179.74, 178.04, 175.32, 174.17, 172.53, 167.52, 151.65, 141.49, 137.41, 122.39, 115.58, 114.21, 53.75, 47.04, 45.08, 44.97, 44.85, 44.73, 44.61, 44.49, 44.37, 41.00, 38.90, 36.20, 35.98, 34.20, 34.16, 34.12, 33.98, 33.89, 33.78, 31.53, 29.73, 27.38. HRMS (ESI+): m/z calcd for C25H34N3O6, [M + H]+ 472.2442, found 472.2438.
Synthesis of PTD PROTACs
4-((10-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-10-oxodecyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD1).
A 10 mL single-neck round bottom flask equipped with a stirrer was charged with GDC-0853 warhead 1 (13.43 mg, 0.022 mmol), 10-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)decanoic acid (Pomalidomide-C9-CO2H, cat. # 901525 Sigma-Aldrich) (8.16 mg, 0.018 mmol), and HATU (8.4 mg, 0.022 mmol). Then DIPEA (142.2 mg, 1.1 mmol) dissolved in DMF (1 mL) was added to the flask under nitrogen, and the mixture was stirred at room temperature overnight. The mixture was purified by flash column chromatography without any workup (SiO2: MeOH/ DCM), and then reverse-phase prep-HPLC was used to give the title product PTD1 as a yellow solid (6.2 mg, 32.6% yield). 1H NMR (700 MHz, CDCl3) δ 8.62 (s, 1H), 8.49 (d, J = 5.1 Hz, 1H), 8.44 – 8.33 (m, 1H), 8.02 – 7.89 (m, 2H), 7.84 (s, 1H), 7.49 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.30 – 7.24 (m, 1H), 7.08 (d, J = 7.0 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 6.7 Hz, 2H), 6.23 (t, J = 5.7 Hz, 1H), 4.91 (dd, J = 12.5, 5.4 Hz, 1H), 4.64 (d, J = 12.1 Hz, 1H), 4.51 (s, 1H), 4.31 (d, J = 12.0 Hz, 1H), 4.16 (dd, J = 8.8, 5.0 Hz, 2H), 3.97 – 3.77 (m, 3H), 3.71 (s, 3H), 3.64 (d, J = 12.7 Hz, 1H), 3.55 (dd, J = 18.8, 10.1 Hz, 1H), 3.47 – 3.36 (m, 2H), 3.26 (q, J = 6.5 Hz, 2H), 3.08 – 2.95 (m, 2H), 2.91 – 2.69 (m, 4H), 2.57 (d, J = 11.4 Hz, 2H), 2.51 (s, 2H), 2.40 – 2.30 (m, 2H), 2.13 (dt, J = 15.4, 4.8 Hz, 1H), 1.67 – 1.64 (m, 4H), 1.41 (p, J = 7.3 Hz, 2H), 1.38 – 1.30 (m, 8H), 1.27 (s, 6H), 0.93 (dd, J = 27.9, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 172.03, 171.99, 171.07, 171.04, 169.54, 168.42, 168.39, 167.66, 161.74, 157.65, 154.32, 150.19, 148.39, 147.04, 141.97, 139.45, 136.13, 132.51, 129.75, 127.72, 125.76, 123.84, 117.21, 116.67, 112.66, 111.38, 111.30, 109.86, 76.85, 59.44, 53.45, 51.34, 48.89, 48.20, 47.20, 45.96, 45.86, 42.71, 42.61, 41.67, 40.94, 39.56, 38.54, 33.30, 33.04, 31.46, 30.33, 29.72, 29.46, 29.31, 29.30, 29.28, 29.20, 29.14, 26.82, 25.44, 25.28, 22.85, 14.22, 14.15, 14.08, 14.03, 13.69, 13.61. HRMS (ESI+): m/z calcd for C57H68N11O8, [M + H]+ 1034.5247, found 1034.5256. HPLC Purity: 100%.
2-(2-((7-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-7-oxoheptyl)oxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)acetamide (PTD2).
PTD2 was synthesized using the same protocol as that use to synthesize PTD1. GDC-0853 warhead 1 and 7-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-2-oxoethoxy)ethoxy)heptanoic acid (Pomalidomide-PEG2-butylCO2H, cat. # 910449 Sigma-Aldrich) were subjected to the reaction conditions to yield a white solid (8.2 mg, 30.4% yield). 1H NMR (700 MHz, CDCl3) δ 10.49 (s, 1H), 9.11 (s, 1H), 8.88 (d, J = 8.5 Hz, 1H), 8.67 (d, J = 3.3 Hz, 1H), 8.51 (d, J = 5.0 Hz, 1H), 7.95 (d, J = 10.5 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.85 (s, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.60 (d, J = 7.3 Hz, 1H), 7.38 (d, J = 5.0 Hz, 1H), 7.28 (t, J = 8.7 Hz, 1H), 6.88 – 6.82 (m, 2H), 5.08 (s, 1H), 4.98 (dd, J = 12.9, 5.3 Hz, 1H), 4.66 (d, J = 12.1 Hz, 1H), 4.53 (s, 1H), 4.33 (d, J = 11.9 Hz, 1H), 4.23 (d, J = 3.6 Hz, 2H), 4.18 (t, J = 6.0 Hz, 2H), 3.96 – 3.79 (m, 4H), 3.75 (d, J = 4.9 Hz, 2H), 3.73 (s, 3H), 3.62 (dt, J = 38.9, 11.1 Hz, 2H), 3.53 (t, J = 6.8 Hz, 2H), 3.41 (q, J = 22.5, 18.8 Hz, 2H), 3.09 – 2.97 (m, 2H), 2.96 – 2.75 (m, 3H), 2.65 – 2.55 (m, 2H), 2.53 (s, 2H), 2.38 (dt, J = 29.3, 7.6 Hz, 2H), 2.17 (dt, J = 11.9, 5.5 Hz, 1H), 1.64 (dt, J = 31.2, 7.3 Hz, 4H), 1.41 – 1.35 (m, 4H), 1.29 (s, 6H), 0.94 (dd, J = 26.5, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.13, 169.51, 168.42, 168.03, 166.88, 161.75, 157.64, 154.32, 150.26, 148.39, 141.96, 139.50, 136.75, 136.27, 131.41, 129.74, 128.89, 128.86, 127.71, 125.75, 125.28, 123.87, 118.80, 117.25, 116.51, 116.45, 116.22, 112.56, 111.29, 71.73, 71.63, 71.61, 71.07, 70.02, 70.00, 59.44, 51.35, 49.39, 48.85, 48.20, 47.29, 45.95, 45.87, 42.71, 41.76, 40.94, 39.56, 38.53, 33.06, 32.82, 31.59, 30.27, 29.72, 29.44, 29.21, 25.95, 25.92, 25.08, 22.58, 14.11. HRMS (ESI+): m/z calcd for C58H68N11O11, [M + H]+ 1094.5094, found 1094.5100. HPLC Purity: 96%.
(2S,4R)-1-((S)-2-(2-(2-((7-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-7-oxoheptyl)oxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (PTD3).
PTD3 was synthesized according to the protocol of synthesis of PTD1. GDC-0853 warhead 1 and 7-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)ethoxy)heptanoic acid ((S,R,S)-AHPC-PEG2-butylCO2H, cat. # 910600 Sigma-Aldrich) were subjected to the reaction conditions to yield a white solid (3.6 mg, 14.6%). 1H NMR (700 MHz, CDCl3) δ 8.69 – 8.63 (m, 2H), 8.48 (d, J = 5.1 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 4.9 Hz, 1H), 7.82 (s, 1H), 7.46 (q, J = 7.6, 6.9 Hz, 1H), 7.36 (t, J = 6.9 Hz, 4H), 7.30 (dd, J = 8.5, 3.4 Hz, 1H), 7.26 (q, J = 5.5 Hz, 1H), 6.82 (d, J = 13.0 Hz, 2H), 5.06 (t, J = 7.0 Hz, 1H), 4.74 (td, J = 8.0, 3.5 Hz, 1H), 4.66 – 4.61 (m, 1H), 4.57 (dd, J = 15.0, 6.7 Hz, 1H), 4.54 – 4.47 (m, 3H), 4.33 (dt, J = 14.3, 7.1 Hz, 2H), 4.18 – 4.13 (m, 2H), 4.10 (dd, J = 11.8, 5.4 Hz, 1H), 4.01 (d, J = 6.5 Hz, 2H), 3.91 – 3.81 (m, 2H), 3.78 (d, J = 12.9 Hz, 1H), 3.71 (d, J = 1.4 Hz, 3H), 3.69 – 3.56 (m, 7H), 3.55 – 3.48 (m, 1H), 3.46 (t, J = 6.7 Hz, 2H), 3.43 – 3.34 (m, 2H), 3.08 – 2.94 (m, 3H), 2.61 – 2.52 (m, 3H), 2.52 – 2.50 (m, 4H), 2.32 (dtq, J = 30.7, 15.3, 7.2 Hz, 3H), 2.13 (t, J = 11.1 Hz, 1H), 1.58 (q, J = 7.1 Hz, 2H), 1.39 – 1.31 (m, 4H), 1.27 (s, 6H), 0.95 (s, 9H), 0.94 – 0.89 (m, 3H). 13C NMR (176 MHz, CDCl3) δ 171.95, 171.35, 170.76, 161.75, 157.64, 154.33, 150.31, 150.26, 148.47, 148.39, 141.97, 139.51, 138.18, 131.64, 131.34, 130.92, 129.75, 129.52, 128.86, 128.14, 127.72, 125.75, 123.86, 117.24, 112.55, 111.30, 71.47, 71.45, 71.21, 71.19, 70.38, 70.17, 69.94, 69.89, 59.44, 58.35, 57.16, 57.14, 56.69, 48.20, 45.96, 43.25, 42.71, 41.72, 40.94, 39.56, 38.52, 35.82, 34.90, 33.17, 32.94, 29.39, 29.31, 29.29, 26.42, 25.96, 25.94, 25.35, 25.20, 16.09, 14.12, 13.72. HRMS (ESI+): m/z calcd for C67H87N12O10S, [M + H]+ 1251.6383, found 1251.6394. HPLC Purity: 97%.
2-(2-((6-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)hexyl)oxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)acetamide (PTD4).
A 10 mL single-neck round bottom flask equipped with a stirrer was charged with GDC-0853 warhead 1 (18.9 mg, 0.031 mmol), and N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-2-(2-((6-iodohexyl)oxy)ethoxy)acetamide (Pomalidomide-PEG2-butyl iodide, cat. # 904724 Sigma-Aldrich) (23.23 mg, 0.039 mmol). DIPEA (20.1 mg, 0.15 mmol) dissolved in DMF (1 mL) was added to the flask under nitrogen and the mixture was stirred at room temperature overnight. The mixture was purified by flash column chromatography without any workup (SiO2: MeOH/ DCM), and then reverse-phase prep-HPLC to give the title product PTD4 as a white solid (2.2 mg, 6% yield). 1H NMR (700 MHz, CDCl3) δ 10.49 (s, 1H), 9.01 (s, 1H), 8.87 (d, J = 8.4 Hz, 1H), 8.61 (s, 1H), 8.51 (d, J = 5.1 Hz, 1H), 8.08 (s, 1H), 7.94 (s, 1H), 7.73 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 7.3 Hz, 1H), 7.50 (s, 1H), 7.34 (d, J = 5.1 Hz, 1H), 6.84 (d, J = 4.9 Hz, 2H), 5.34 (q, J = 5.5 Hz, 1H), 4.95 (dd, J = 12.8, 5.4 Hz, 1H), 4.65 (d, J = 12.1 Hz, 1H), 4.51 (s, 1H), 4.31 (s, 1H), 4.23 – 4.19 (m, 2H), 4.19 – 4.14 (m, 2H), 3.88 (s, 1H), 3.81 (dd, J = 5.7, 3.3 Hz, 2H), 3.73 (dd, J = 5.8, 3.2 Hz, 2H), 3.72 (s, 3H), 3.66 – 3.53 (m, 2H), 3.50 (t, J = 6.4 Hz, 2H), 3.08 – 2.95 (m, 3H), 2.94 – 2.73 (m, 4H), 2.57 (d, J = 11.0 Hz, 3H), 2.51 (s, 2H), 2.22 (t, J = 7.6 Hz, 1H), 2.16 (ddd, J = 11.1, 5.9, 3.4 Hz, 1H), 2.01 (q, J = 7.1, 6.6 Hz, 1H), 1.40 – 1.30 (m, 8H), 1.27 (s, 6H), 0.89 (dd, J = 16.2, 9.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 170.96, 169.53, 168.41, 168.02, 166.83, 161.74, 157.63, 154.26, 148.47, 142.03, 136.76, 136.33, 131.42, 129.70, 127.74, 125.72, 125.28, 123.78, 118.82, 116.98, 116.20, 111.35, 71.77, 71.21, 71.14, 70.15, 59.43, 57.39, 51.72, 49.38, 48.22, 45.96, 42.72, 40.94, 39.56, 38.62, 35.92, 31.59, 29.71, 29.54, 29.34, 29.25, 29.20, 29.13, 27.24, 26.36, 25.53, 25.48, 25.46, 23.44, 22.57. HRMS (ESI+): m/z calcd for C57H68N11O10, [M + H]+ 1065.5145, found 1065.5148. HPLC Purity: 95%.
2-(2-((7-(4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)piperazin-1-yl)-7-oxoheptyl)oxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)acetamide (PTD5).
PTD5 was synthesized according to the protocol of synthesis of PTD1. GDC-0853 warhead 2and 7-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-2-oxoethoxy)ethoxy)heptanoic acid (Pomalidomide-PEG2-butylCO2H, cat. # 910449 Sigma-Aldrich) were subjected to the reaction conditions to yield a yellow solid (17.7 mg, 48% yield). 1H NMR (700 MHz, CDCl3) δ 10.46 (s, 1H), 9.19 (s, 1H), 8.85 (d, J = 8.5 Hz, 1H), 8.62 (d, J = 2.3 Hz, 1H), 8.47 (d, J = 5.1 Hz, 1H), 7.89 (d, J = 3.0 Hz, 1H), 7.85 (s, 1H), 7.80 (d, J = 2.3 Hz, 1H), 7.71 (t, J = 7.9 Hz, 1H), 7.56 (d, J = 7.3 Hz, 1H), 7.34 (d, J = 5.0 Hz, 1H), 7.24 (dd, J = 9.0, 2.9 Hz, 1H), 6.85 – 6.80 (m, 2H), 5.05 (s, 1H), 4.95 (dd, J = 12.8, 5.4 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H), 4.49 (s, 1H), 4.31 (d, J = 12.1 Hz, 1H), 4.19 (d, J = 3.2 Hz, 2H), 4.15 (dt, J = 8.5, 4.4 Hz, 2H), 3.85 (d, J = 12.0 Hz, 1H), 3.80 (dd, J = 5.7, 3.5 Hz, 2H), 3.75 (t, J = 5.3 Hz, 2H), 3.71 (dd, J = 5.8, 3.4 Hz, 2H), 3.70 (s, 3H), 3.62 – 3.58 (m, 2H), 3.49 (t, J = 6.7 Hz, 2H), 3.03 (dt, J = 17.5, 5.2 Hz, 4H), 2.92 – 2.72 (m, 3H), 2.56 (d, J = 10.9 Hz, 2H), 2.50 (s, 2H), 2.35 (t, J = 7.5 Hz, 2H), 2.14 (dtt, J = 12.7, 5.2, 2.3 Hz, 1H), 1.59 (dq, J = 27.8, 7.0 Hz, 4H), 1.35 (dq, J = 8.5, 4.7 Hz, 4H), 1.26 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 171.81, 171.22, 169.52, 168.42, 168.09, 166.87, 161.74, 157.62, 150.25, 149.55, 148.38, 141.96, 140.87, 136.73, 136.27, 131.40, 129.74, 128.79, 128.64, 127.69, 125.75, 125.26, 123.85, 118.78, 117.29, 116.21,112.66, 111.27, 71.71, 71.59, 71.07, 70.02, 59.42, 50.80, 50.36, 49.39, 48.20, 45.95, 45.52, 42.70, 41.45, 40.94, 39.55, 38.52, 33.06, 31.58, 29.43, 29.17, 25.92, 25.13, 22.58. HRMS (ESI+): m/z calcd for C57H66N11O10, [M + H]+ 1080.4938, found 1080.4941. HPLC Purity: 97 %.
2-(2-((6-(4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)piperazin-1-yl)hexyl)oxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)acetamide (PTD6).
PTD6 was synthesized according to the protocol of synthesis of PTD4. GDC-0853 warhead 2 and N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-2-(2-((6-iodohexyl)oxy)ethoxy)acetamide (Pomalidomide-PEG2-butyl iodide, cat. # 904724 Sigma-Aldrich) were subjected to the reaction conditions to yield a white solid (2.2 mg, 6% yield). 1H NMR (700 MHz, CDCl3) δ 10.42 (s, 1H), 8.87 (d, J = 8.4 Hz, 1H), 8.61 (d, J = 2.3 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.89 (d, J = 3.0 Hz, 1H), 7.83 (d, J = 2.3 Hz, 1H), 7.76 (s, 1H), 7.72 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 7.3 Hz, 1H), 7.36 (d, J = 5.1 Hz, 1H), 7.24 (dd, J = 8.9, 3.0 Hz, 1H), 6.84 (s, 1H), 6.78 (d, J = 8.9 Hz, 1H), 5.08 – 5.03 (m, 1H), 4.93 (dd, J = 13.0, 5.4 Hz, 1H), 4.64 (d, J = 11.9 Hz, 1H), 4.50 (s, 1H), 4.32 (s, 1H), 4.22 (d, J = 15.6 Hz, 1H), 4.18 – 4.14 (m, 3H), 3.88 – 3.73 (m, 5H), 3.71 (s, 3H), 3.54 (d, J = 6.6 Hz, 2H), 3.10 (d, J = 5.5 Hz, 4H), 2.91 – 2.84 (m, 2H), 2.75 – 2.69 (m, 1H), 2.67 (t, J = 5.0 Hz, 4H), 2.57 (d, J = 10.9 Hz, 3H), 2.51 (s, 2H), 2.40 (ddt, J = 19.7, 12.0, 6.0 Hz, 2H), 2.13 (tt, J = 7.8, 2.9 Hz, 1H), 1.53 – 1.50 (m, 2H), 1.39 – 1.31 (m, 6H), 1.27 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 171.71, 169.37, 168.58, 168.45, 166.99, 161.75, 154.29, 150.36, 148.40, 141.90, 141.25, 136.74, 136.20, 135.28, 131.59, 129.70, 128.58, 127.82, 127.68, 125.79, 125.41, 123.92, 118.83, 117.23, 116.29, 115.85, 112.53, 111.27, 71.81, 71.60, 70.99, 69.67, 59.46, 58.16, 52.73, 49.62, 49.56, 48.20, 45.95, 42.72, 40.95, 39.56, 38.50, 31.84, 30.35, 29.30, 27.21, 26.12, 25.88, 22.57. HRMS (ESI+): m/z calcd for C56H66N11O10, [M + H]+ 1051.4989, found 1051.4996. HPLC Purity: 95%.
4-((7-(4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)piperazin-1-yl)-7-oxoheptyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD7).
PTD7 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 2 and 7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptanoic acid (Pomalidomide-C6-CO2H, cat. # 901496 Sigma-Aldrich) were subjected to the reaction conditions to yield a yellow solid (15.2 mg, 60.7% yield). 1H NMR (700 MHz, CDCl3) δ 8.69 (d, J = 15.0 Hz, 1H), 8.65 (d, J = 2.4 Hz, 1H), 8.50 (d, J = 5.0 Hz, 1H), 7.93 (d, J = 3.0 Hz, 1H), 7.87 (s, 1H), 7.82 (d, J = 2.4 Hz, 1H), 7.50 (dd, J = 8.6, 7.1 Hz, 1H), 7.36 (d, J = 5.0 Hz, 1H), 7.27 (dd, J = 8.9, 2.9 Hz, 1H), 7.09 (d, J = 7.1 Hz, 1H), 6.91 – 6.83 (m, 3H), 6.24 (t, J = 5.6 Hz, 1H), 5.07 (s, 1H), 4.92 (dd, J = 12.4, 5.4 Hz, 1H), 4.66 (d, J = 12.0 Hz, 1H), 4.52 (s, 1H), 4.38 – 4.30 (m, 1H), 4.18 (t, J = 6.2 Hz, 2H), 3.87 (d, J = 12.7 Hz, 1H), 3.78 (tq, J = 13.0, 6.4, 4.8 Hz, 2H), 3.72 (s, 3H), 3.65 – 3.62 (m, 2H), 3.28 (p, J = 6.6 Hz, 2H), 3.09 – 3.04 (m, 4H), 2.93 – 2.86 (m, 1H), 2.83 – 2.70 (m, 2H), 2.61 – 2.54 (m, 2H), 2.52 (s, 2H), 2.39 (t, J = 7.5 Hz, 2H), 2.13 (dtd, J = 11.8, 4.7, 4.3, 2.3 Hz, 1H), 1.70 (p, J = 7.3 Hz, 4H), 1.49 – 1.41 (m, 4H), 1.28 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 171.50, 171.23, 169.55, 168.59, 167.65, 161.74, 157.63, 154.31, 150.27, 149.63, 148.38, 146.98, 141.98, 140.85, 136.17, 136.15, 132.49, 131.42, 129.76, 128.73, 128.67, 127.70, 125.73, 123.87, 117.30, 116.66, 116.28, 112.66, 111.42, 111.29, 109.86, 59.42, 50.82, 50.49, 48.89, 48.20, 45.95, 45.51, 42.70, 42.54, 41.43, 40.94, 39.55, 38.52, 33.05, 31.44, 30.28, 29.08, 29.07, 26.74, 25.14, 22.83. HRMS (ESI+): m/z calcd for C53H60N11O8, [M + H]+ 978.4621, found 978.4625. HPLC Purity: 100%.
4-((7-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-7-oxoheptyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD8).
PTD8 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and 7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptanoic acid (Pomalidomide-C6-CO2H, cat. # 901496 Sigma-Aldrich) were subjected to the reaction conditions to yield a yellow solid (7.5 mg, 40% yield). 1H NMR (700 MHz, CDCl3) δ 8.67 (dt, J = 5.0, 2.4 Hz, 1H), 8.50 (d, J = 5.0 Hz, 1H), 7.97 – 7.93 (m, 1H), 7.91 – 7.87 (m, 1H), 7.83 (d, J = 2.4 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 7.37 (d, J = 5.0 Hz, 1H), 7.30 – 7.26 (m, 1H), 7.10 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.84 (d, J = 7.8 Hz, 2H), 6.24 (q, J = 5.6, 4.5 Hz, 1H), 5.07 (dd, J = 9.9, 4.1 Hz, 1H), 4.92 (ddd, J = 12.8, 5.4, 2.2 Hz, 1H), 4.65 (d, J = 12.0 Hz, 1H), 4.52 (s, 1H), 4.34 (d, J = 12.2 Hz, 1H), 4.17 (t, J = 5.9 Hz, 2H), 3.94 – 3.78 (m, 2H), 3.72 (s, 3H), 3.68 – 3.35 (m, 5H), 3.28 (q, J = 6.6 Hz, 2H), 3.08 – 2.95 (m, 2H), 2.89 (dd, J = 17.0, 3.9 Hz, 1H), 2.84 – 2.70 (m, 2H), 2.58 (d, J = 11.5 Hz, 2H), 2.52 (s, 2H), 2.42 – 2.31 (m, 2H), 2.13 (dtd, J = 9.9, 4.9, 2.3 Hz, 1H), 1.70 (t, J = 7.5 Hz, 4H), 1.45 (ddd, J = 22.4, 12.4, 5.1 Hz, 4H), 1.30 – 1.26 (m, 6H), 0.94 (dd, J = 27.6, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.67, 171.14, 171.09, 169.56, 167.65, 161.74, 157.64, 154.31, 150.50, 150.27, 148.39, 146.99, 141.97, 139.47, 136.16, 132.50, 131.35, 129.76, 128.88, 128.86, 127.71, 125.74, 123.87, 117.27, 116.67, 116.57, 116.50, 112.59, 111.44, 111.30, 109.87, 59.43, 51.37, 48.91, 48.89, 48.20, 45.95, 42.71, 42.57, 41.72, 40.94, 39.56, 38.54, 33.09, 32.80, 31.46, 31.44, 29.12, 29.10, 26.78, 26.76, 25.29, 25.25, 25.09, 22.83, 14.14, 13.79, 13.70. HRMS (ESI+): m/z calcd for C54H62N11O8, [M + H]+ 992.4777, found 992.4777. HPLC Purity: 95%.
4-((4-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-4-oxobutyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD9).
PTD9 was synthesized according to the protocol of synthesis of PTD1. GDC-0853 warhead 1 and 4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butanoic acid (Pomalidomide-C3-CO2H, cat. # 901500 Sigma-Aldrich) were subjected to the reaction conditions to yield a yellow solid (7.4 mg, 41.3% yield). 1H NMR (700 MHz, CDCl3) δ 8.68 (q, J = 2.4 Hz, 1H), 8.51 (d, J = 5.0 Hz, 1H), 8.40 – 8.30 (m, 1H), 7.95 (dd, J = 8.5, 2.8 Hz, 1H), 7.89 (d, J = 4.8 Hz, 1H), 7.84 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 8.6, 7.0 Hz, 1H), 7.37 (d, J = 4.8 Hz, 1H), 7.30 – 7.25 (m, 1H), 7.12 (d, J = 7.1 Hz, 1H), 7.02 (dd, J = 8.6, 2.7 Hz, 1H), 6.85 (d, J = 9.8 Hz, 2H), 6.35 (t, J = 5.9 Hz, 1H), 5.08 (d, J = 8.5 Hz, 1H), 4.93 (dd, J = 12.5, 5.4 Hz, 1H), 4.67 (d, J = 12.0 Hz, 1H), 4.53 (s, 1H), 4.34 (s, 1H), 4.21 – 4.16 (m, 2H), 3.94 – 3.83 (m, 2H), 3.73 (s, 3H), 3.67 – 3.50 (m, 3H), 3.46 – 3.37 (m, 4H), 3.08 – 2.98 (m, 2H), 2.90 (dd, J = 17.0, 3.8 Hz, 1H), 2.85 – 2.71 (m, 2H), 2.64 – 2.55 (m, 2H), 2.53 (s, 2H), 2.48 (dq, J = 16.2, 9.4, 8.2 Hz, 1H), 2.14 (qd, J = 6.5, 5.9, 3.3 Hz, 1H), 2.06 (q, J = 7.0 Hz, 2H), 1.29 (s, 6H), 0.94 (t, J = 7.2 Hz, 3H).13C NMR (176 MHz, CDCl3) δ 171.03, 170.57, 169.51, 167.60, 161.75, 157.63, 154.33, 150.31, 150.25, 148.39, 146.95, 141.96, 139.42, 136.25, 132.48, 131.33, 131.31, 129.76, 128.85, 127.71, 125.76, 123.85, 117.27, 116.91, 116.51, 112.55, 112.52, 111.66, 111.29, 110.06, 59.43, 51.26, 48.90, 48.20, 47.56, 47.44, 45.95, 45.72, 42.71, 42.12, 41.88, 40.94, 39.56, 38.53, 31.45, 30.35, 30.29, 29.79, 29.73, 29.62, 24.56, 24.48, 24.46, 22.80, 14.26, 14.19, 13.76. HRMS (ESI+): m/z calcd for C51H56N11O8, [M + H]+ 950.4308, found 950. 4308. HPLC Purity: 99%.
4-((2-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-2-oxoethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD10).
PTD10 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and (2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycine (Thalidomide-NH-CH2-CO2H, cat. # HY-131717 MedChemExpress) were subjected to the reaction conditions to yield a yellow solid (12.5 mg, 51.3% yield). 1H NMR (700 MHz, CDCl3) δ 8.65 (s, 1H), 8.48 (d, J = 5.0 Hz, 1H), 8.16 (s, 1H), 7.93 (s, 1H), 7.82 (d, J = 4.7 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.34 (dd, J = 5.4, 2.2 Hz, 1H), 7.30 (s, 1H), 7.20 – 7.13 (m, 2H), 6.82 (d, J = 13.7 Hz, 3H), 5.07 (s, 1H), 4.93 (dd, J = 12.5, 5.4 Hz, 1H), 4.64 (d, J = 12.1 Hz, 1H), 4.51 (s, 1H), 4.31 (d, J = 11.7 Hz, 1H), 4.16 (q, J = 5.1, 4.2 Hz, 2H), 4.13 – 4.02 (m, 2H), 3.89 (d, J = 48.8 Hz, 2H), 3.71 (s, 3H), 3.69 – 3.32 (m, 4H), 3.15 – 2.98 (m, 2H), 2.90 – 2.79 (m, 2H), 2.73 (ddd, J = 17.6, 13.4, 5.1 Hz, 1H), 2.57 (d, J = 11.2 Hz, 2H), 2.51 (s, 2H), 2.14 – 2.09 (m, 1H), 1.27 (s, 6H), 0.96 (d, J = 53.9 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.03, 168.90, 168.87, 168.29, 167.60, 166.19, 161.75, 157.63, 154.34, 148.41, 145.49, 142.00, 139.18, 136.15, 132.69, 129.76, 127.73, 125.74, 123.81, 117.27, 116.94, 112.11, 111.30, 111.17, 59.43, 50.34, 48.99, 48.96, 48.20, 45.96, 42.71, 42.40, 40.94, 39.56, 38.56, 31.45, 29.72, 29.34, 22.74, 14.15, 13.95. HRMS (ESI+): m/z calcd for C49H52N11O8, [M + H]+ 922.3995, found 922. 3996. HPLC Purity: 99%.
4-((2-(2-(3-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-3-oxopropoxy)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD11).
PTD11 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and 3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)propanoic acid (Pomalidomide-PEG2-CO2H, cat. # 901526 Sigma-Aldrich) were subjected to the reaction conditions to yield a yellow solid (3.9 mg, 19.2% yield). 1H NMR (700 MHz, CDCl3) δ 8.65 (d, J = 2.4 Hz, 1H), 8.57 – 8.44 (m, 2H), 7.94 – 7.86 (m, 2H), 7.83 (dd, J = 4.6, 2.3 Hz, 1H), 7.48 (q, J = 7.5 Hz, 1H), 7.35 (d, J = 4.8 Hz, 1H), 7.27 – 7.22 (m, 1H), 7.12 – 7.07 (m, 1H), 6.91 (t, J = 9.0 Hz, 1H), 6.84 – 6.79 (m, 2H), 6.52 – 6.47 (m, 1H), 5.06 (s, 1H), 4.91 (dt, J = 12.8, 4.7 Hz, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.51 (s, 1H), 4.32 (s, 1H), 4.16 (q, J = 5.1, 4.1 Hz, 2H), 3.94 – 3.75 (m, 4H), 3.74 – 3.62 (m, 9H), 3.60 – 3.35 (m, 5H), 3.01 (dddd, J = 37.5, 16.7, 8.4, 4.1 Hz, 2H), 2.90 – 2.84 (m, 1H), 2.82 – 2.58 (m, 5H), 2.57 (d, J = 11.3 Hz, 2H), 2.51 (s, 2H), 2.16 – 2.10 (m, 1H), 1.27 (s, 6H), 0.92 (dd, J = 11.9, 6.1 Hz, 3H).13C NMR (176 MHz, CDCl3) δ 171.06, 169.90, 169.32, 167.60, 161.74, 157.64, 154.34, 150.25, 148.38, 146.86, 146.83, 141.92, 139.45, 136.08, 132.54, 131.35, 129.75, 128.88, 127.69, 125.79, 123.85, 117.26, 116.81, 116.78, 116.51, 112.58, 111.74, 111.27, 110.39, 70.62, 70.60, 70.51, 70.48, 70.46, 70.44, 69.49, 69.47, 69.39, 67.76, 67.74, 67.53, 59.45, 51.45, 51.40, 48.93, 48.90, 48.67, 48.44, 48.19, 47.27, 47.25, 46.00, 45.95, 42.72, 42.43, 42.40, 41.74, 40.95, 39.56, 38.54, 33.57, 33.35, 31.44, 31.42, 22.87, 14.04, 13.94, 13.78, 13.59. HRMS (ESI+): m/z calcd for C54H62N11O10, [M + H]+ 1024.4676, found 1024.4673. HPLC Purity: 98%.
3-(4-((2-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-2-oxoethyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (PTD12).
PTD12 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and compound 12 were subjected to the reaction conditions to yield a white solid (8.5 mg, 47.3% yield). 1H NMR (700 MHz, CDCl3) δ 8.67 (t, J = 2.2 Hz, 1H), 8.48 (d, J = 5.0 Hz, 1H), 8.36 (t, J = 6.1 Hz, 1H), 7.95 (t, J = 2.7 Hz, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.81 (t, J = 2.7 Hz, 1H), 7.38 – 7.33 (m, 2H), 7.28 (dd, J = 14.7, 5.3 Hz, 2H), 6.84 (d, J = 9.8 Hz, 2H), 6.72 – 6.68 (m, 1H), 5.22 (dt, J = 13.6, 4.1 Hz, 1H), 5.08 – 5.03 (m, 1H), 4.89 (d, J = 4.3 Hz, 1H), 4.64 (d, J = 11.9 Hz, 1H), 4.49 (d, J = 14.3 Hz, 1H), 4.38 (d, J = 15.5 Hz, 1H), 4.33 (d, J = 13.1 Hz, 1H), 4.25 (d, J = 15.5 Hz, 1H), 4.16 (q, J = 5.1, 4.2 Hz, 2H), 4.05 – 3.97 (m, 2H), 3.96 – 3.82 (m, 2H), 3.71 (s, 3H), 3.68 – 3.53 (m, 2H), 3.49 – 3.38 (m, 1H), 3.14 – 3.00 (m, 2H), 2.91 – 2.78 (m, 2H), 2.56 (d, J = 11.5 Hz, 2H), 2.50 (s, 2H), 2.38 – 2.29 (m, 1H), 2.18 (ddd, J = 13.4, 6.0, 3.5 Hz, 1H), 1.26 (s, 6H), 0.96 (dd, J = 41.9, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.22, 169.92, 169.67, 167.18, 167.16, 161.75, 157.63, 154.36, 150.20, 148.38, 142.06, 141.97, 139.16, 132.11, 131.25, 129.77, 129.75, 128.96, 127.72, 126.88, 125.76, 123.82, 117.27, 116.69, 116.66, 113.20, 113.16, 112.56, 111.29, 59.44, 51.83, 48.19, 48.06, 45.96, 45.02, 44.66, 44.64, 44.55, 42.71, 42.47, 40.94, 39.56, 38.53, 31.62, 23.48, 14.30,14.21, 14.07,14.00. HRMS (ESI+): m/z calcd for C49H54N11O7, [M + H]+ 908.4202, found 908.4197. HPLC Purity: 100%.
4-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazine-1-carbonyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD13).
PTD13 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and 2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindoline-4-carboxylic acid (cat. # EN300–27082444 Enamine) were subjected to the reaction conditions to yield a white solid (5.4 mg, 34.5% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.14 (s, 1H), 8.63 (q, J = 3.1, 2.4 Hz, 1H), 8.51 – 8.44 (m, 2H), 8.01 – 7.92 (m, 2H), 7.89 – 7.75 (m, 2H), 7.48 (t, J = 2.8 Hz, 1H), 7.41 – 7.33 (m, 2H), 7.26 (dd, J = 9.1, 3.6 Hz, 1H), 6.56 (d, J = 3.9 Hz, 1H), 5.21 – 5.11 (m, 1H), 4.97 (d, J = 6.9 Hz, 1H), 4.42 (dd, J = 18.8, 12.5 Hz, 3H), 4.22 (dd, J = 22.5, 12.3 Hz, 3H), 4.10 – 3.80 (m, 3H), 3.60 (s, 3H), 3.28 – 3.10 (m, 2H), 3.08 – 2.81 (m, 3H), 2.64 – 2.54 (m, 3H), 2.43 (s, 2H), 2.08 (dd, J = 13.0, 7.9 Hz, 1H), 1.24 – 1.21 (m, 6H), 1.08 (d, J = 6.5 Hz, 1H), 0.96 (d, J = 6.4 Hz, 1H), 0.77 – 0.67 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 173.21, 170.21, 167.14, 166.47, 165.55, 165.43, 159.76, 157.20, 155.32, 149.49, 149.18, 148.39, 141.33, 139.67, 136.00, 135.88, 133.89, 132.22, 131.94, 131.33, 131.23, 130.12, 128.72, 127.89, 127.30, 127.09, 126.28, 126.01, 124.31, 124.21, 116.81, 116.45, 116.31, 113.60, 109.29, 63.56, 57.64, 51.33, 49.55, 49.50, 48.34, 46.85, 46.72, 46.63, 45.79, 42.42, 41.56, 41.20, 38.04, 31.75, 31.41, 31.34, 30.65, 30.53, 29.20, 29.16, 22.37, 14.43, 12.71. HRMS (ESI+): m/z calcd for C48H49N10O8, [M + H]+ 892.3729, found 892.3727. HPLC Purity: 98%.
4-(2-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-2-oxoethoxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD14).
PTD14 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1and 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (Thalidomide-O-CO2H, cat. # HY-103597 MedChemExpress) were subjected to the reaction conditions to yield a light yellow solid (7.1 mg, 37% yield). 1H NMR (700 MHz, CDCl3) δ 8.61 (d, J = 29.5 Hz, 1H), 8.48 (q, J = 5.0, 4.3 Hz, 1H), 8.36 – 8.20 (m, 1H), 7.95 – 7.72 (m, 3H), 7.72 – 7.68 (m, 1H), 7.52 (dd, J = 7.3, 3.5 Hz, 1H), 7.41 – 7.30 (m, 2H), 7.26 (d, J = 16.1 Hz, 1H), 6.87 – 6.81 (m, 2H), 5.08 – 4.92 (m, 4H), 4.65 (s, 1H), 4.50 (s, 1H), 4.32 (s, 1H), 4.19 – 4.13 (m, 2H), 3.86 (d, J = 12.9 Hz, 1H), 3.70 (d, J = 2.5 Hz, 3H), 3.66 – 3.36 (m, 3H), 3.13 – 2.95 (m, 2H), 2.93 – 2.65 (m, 4H), 2.57 (d, J = 9.8 Hz, 2H), 2.51 (s, 2H), 2.16 – 2.10 (m, 1H), 1.27 (s, 6H), 0.96 – 0.73 (m, 3H). 13C NMR (176 MHz, CDCl3) δ 170.90, 170.87, 166.79, 165.61, 165.53, 161.75, 157.63, 155.28, 155.08, 154.39, 154.36, 150.18, 148.40, 141.94, 139.32, 136.80, 133.91, 133.86, 131.24, 129.75, 127.69, 127.68, 125.81, 125.79, 123.81, 123.76, 120.21, 120.14, 119.83, 117.13, 111.27, 68.65, 68.58, 68.46, 59.42, 59.38, 51.09, 49.34, 49.25, 48.18, 47.89, 45.95, 45.77, 42.71, 42.45, 40.96, 39.56, 38.53, 31.41, 30.36, 30.29, 22.58, 22.56, 14.02. HRMS (ESI+): m/z calcd for C49H51N10O9, [M + H]+ 923.3835, found 923.3835. HPLC Purity: 99%.
4-((12-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-12-oxododecyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD15).
PTD15 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and compound 15 were subjected to the reaction conditions to yield a yellow solid (11.7 mg, 36.2% yield). 1H NMR (700 MHz, CDCl3) δ 8.65 (d, J = 2.3 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 8.41 (dd, J = 30.7, 12.7 Hz, 1H), 7.93 (dd, J = 10.9, 2.8 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 2.2 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.28 – 7.24 (m, 1H), 7.08 (d, J = 7.1 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 10.3 Hz, 2H), 6.22 (t, J = 5.6 Hz, 1H), 5.08 – 5.03 (m, 1H), 4.91 (dd, J = 12.5, 5.4 Hz, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.50 (s, 1H), 4.32 (d, J = 11.8 Hz, 1H), 4.16 (q, J = 5.1, 4.3 Hz, 2H), 3.96 – 3.78 (m, 2H), 3.71 (s, 3H), 3.63 (tt, J = 10.3, 5.0 Hz, 1H), 3.59 – 3.43 (m, 2H), 3.42 – 3.37 (m, 1H), 3.25 (q, J = 6.6 Hz, 2H), 3.07 – 2.95 (m, 2H), 2.88 (dt, J = 17.0, 3.4 Hz, 1H), 2.83 – 2.70 (m, 2H), 2.57 (d, J = 11.5 Hz, 2H), 2.50 (s, 2H), 2.39 – 2.30 (m, 2H), 2.12 (dtd, J = 12.3, 5.0, 2.3 Hz, 1H), 1.65 (s, 2H), 1.43 – 1.38 (m, 2H), 1.36 – 1.24 (m, 20H), 0.93 (dd, J = 27.6, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.08, 171.06, 169.53, 168.42, 167.66, 161.74, 157.64, 154.33, 150.25, 148.38, 147.04, 141.93, 139.51, 136.12, 132.51, 131.36, 131.35, 129.75, 128.87, 127.70, 125.78, 123.84, 117.24, 116.65, 116.49, 116.43, 112.55, 111.34, 111.27, 109.83, 59.45, 51.38, 48.89, 48.19, 47.23, 45.95, 45.90, 42.71, 42.66, 41.68, 40.94, 39.56, 38.53, 33.32, 33.06, 31.47, 30.28, 29.48, 29.46, 29.42, 29.39, 29.27, 29.22, 26.92, 25.47, 25.31, 22.83, 13.67. HRMS (ESI+): m/z calcd for C59H72N11O8, [M + H]+ 1062.5560, found 1062.5561. HPLC Purity: 99%.
5-((2-((S)-4-(6-((2’-(7,7-dimethyl-1-oxo-1,3,4,6,7,8-hexahydro-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-2-yl)-3’-(hydroxymethyl)-1-methyl-6-oxo-1,6-dihydro-[3,4’-bipyridin]-5-yl)amino)pyridin-3-yl)-3-methylpiperazin-1-yl)-2-oxoethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PTD16).
PTD16 was synthesized according to the protocol for the synthesis of PTD1. GDC-0853 warhead 1 and compound 14 were subjected to the reaction conditions to yield a yellow solid (11.7 mg, 38.5% yield). 1H NMR (700 MHz, CDCl3) δ 8.68 (d, J = 2.3 Hz, 1H), 8.48 (d, J = 5.0 Hz, 1H), 8.20 (s, 1H), 7.96 (s, 1H), 7.92 – 7.87 (m, 1H), 7.83 (t, J = 2.0 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.29 (dt, J = 8.9, 2.4 Hz, 1H), 6.97 (t, J = 2.7 Hz, 1H), 6.84 (d, J = 9.9 Hz, 3H), 5.90 (q, J = 3.9 Hz, 1H), 5.08 – 5.03 (m, 1H), 4.94 (dd, J = 12.6, 5.4 Hz, 1H), 4.64 (d, J = 11.9 Hz, 1H), 4.51 (s, 1H), 4.32 (t, J = 11.6 Hz, 1H), 4.19 – 4.12 (m, 2H), 4.02 (t, J = 4.7 Hz, 1H), 3.97 (ddd, J = 24.3, 12.4, 3.5 Hz, 1H), 3.91 – 3.77 (m, 2H), 3.71 (s, 3H), 3.68 – 3.31 (m, 4H), 3.16 – 3.00 (m, 2H), 2.91 – 2.70 (m, 3H), 2.57 (d, J = 11.3 Hz, 2H), 2.51 (s, 2H), 2.13 (ddd, J = 11.8, 7.6, 4.4 Hz, 1H), 1.27 (s, 6H), 0.98 (dd, J = 42.1, 6.3 Hz, 3H). 13C NMR (176 MHz, CDCl3) δ 171.03, 168.34, 167.95, 167.85, 167.36, 166.07, 161.75, 157.63, 154.35, 152.20, 150.79, 150.21, 148.38, 141.96, 139.09, 139.07, 134.80, 131.44, 131.24, 129.76, 129.00, 127.72, 125.76, 125.54, 123.82, 119.02, 117.54, 117.42, 117.25, 116.72, 116.70, 112.57, 111.30, 105.97, 105.87, 59.45, 50.29, 49.13, 48.91, 48.19, 48.13, 48.01, 45.96, 44.68, 44.33, 44.23, 42.71, 42.54, 40.94, 39.56, 38.53, 31.47, 22.78, 14.36, 14.14. HRMS (ESI+): m/z calcd for C49H52N11O8, [M + H]+ 922.3995, found 922.3993. HPLC Purity: 100%.
P13I 4-((2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione.
P13I was synthesized according to the literature.15 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.54 – 7.45 (m, 1H), 7.11 (d, J = 7.1 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 4.91 (dd, J = 12.0, 5.3 Hz, 1H), 3.73 (t, J = 5.4 Hz, 2H), 3.68 (d, J = 4.8 Hz, 10H), 3.48 (t, J = 5.5 Hz, 2H), 3.38 (t, J = 5.0 Hz, 2H), 2.94 – 2.67 (m, 4H), 2.12 (dd, J = 11.6, 5.7 Hz, 1H). [ESI-MS] (ESI+): m/z calcd for C21H27N6O7, [M + H]+ 475.2, found 475.1. HPLC Purity: 100%.
Biological Assays
Cell lines
Ramos (cat. # CRL-1596), JeKo-1 (cat. # CRL-3006), NAMALWA (cat. # CRL-1432), Mino (cat. # CRL-3000), and HEK293 (cat. # CRL-1573) cell lines were purchased from American Type Culture Collection (ATCC). TMD8 and HBL-1 cell lines were kind gifts from Dr. Richard E. Davis at MD Anderson Cancer center. Ramos, NAMALWA, TMD8, and HBL-1 cell lines were cultured in RPMI 1640 media (cat. # MT10040CV, ThermoFisher Scientific) supplemented with 10% FBS (cat. # MT35010CV, ThermoFisher Scientific), and 1% penicillin-streptomycin (10,000 U/ml) (cat. # 15140122, ThermoFisher Scientific). JeKo-1 and Mino cell lines were cultured in RPMI 1640 media (cat. # MT10040CV, ThermoFisher Scientific) supplemented with 20% FBS (cat. # MT35010CV, ThermoFisher Scientific), and 1% penicillin-streptomycin (10,000 U/ml) (cat. # 15140122 ThermoFisher Scientific). HEK293 cell line was cultured in DMEM media (cat. # 11965118, ThermoFisher Scientific) supplemented with 10 % FBS (cat. # MT35010CV, ThermoFisher Scientific), and 1% penicillin-streptomycin (10,000 U/ml) (cat. # 15140122 ThermoFisher Scientific). All the cell lines were maintained in a humidified incubator at 37°C and 5% CO2.
Western blots
Cells (2 × 106) were seeded in a 12-well plate and incubated with varying concentrations of compounds for different periods of time. Cells were then harvested and washed twice with cold phosphate-buffered saline (PBS) pH 7.4 (cat. # 10010023 ThermoFisher Scientific), and the cell pellets were lysed with CelLytic M (cat. # C2978 Sigma-Aldrich) supplemented with protease inhibitor (cat. # A32955 ThermoFisher Scientific) and phosphatase inhibitor (cat. # 78420 ThermoFisher Scientific). Lysates were centrifuged at 14,000 x g for 45 min, 4 °C and the total protein content of the supernatant was quantified using the Pierce BCA Protein Assay kit (cat. # 23225 ThermoFisher Scientific). Samples were reduced and denatured at 95 °C in a heating block, and an equal amount of protein (8-30 μg per lane) was loaded onto a precast 4-20% Tris-glycine gel (Mini-PROTEAN TGX, cat. # 4561095 & 4561096 Bio-Rad) for electrophoretic separation, followed by transfer of the resolved proteins onto nitrocellulose membranes (cat. # 1704159 Bio-Rad) using mini trans-blot electrophoretic transfer cell (Bio-Rad). The membranes were blocked with BSA (cat. # A7030 Sigma-Aldrich) (5% wt/vol) in TBS (cat. # 1706435 Bio-Rad) supplemented with 1% Tween 20 (cat. # 1706531 Bio-Rad) for 1 h at room temperature. The membranes were then probed with primary antibodies in TBS-T containing 1% BSA at 4°C overnight. After washing three times with TBST, the membranes were incubated with horse radish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. The membranes were then washed again with TBST, treated with the chemiluminescent HRP substrate (cat. # 34076 ThermoFisher Scientific) and visualized using a Bio-Rad ChemiDoc imaging instrument. All western blots were subsequently processed and quantified using the accompanying Bio-Rad Image Lab software. The following antibodies were purchased from Cell Signaling Technology: anti-BTK (cat. # 8547), anti-α/β tubulin (cat. # 2148), anti-β-actin (cat. # 4970), anti-GAPDH (cat. # 5174), anti-Vinculin (cat. # 13901), anti-PLCγ2 (cat. # 3872), anti-phospho- PLCγ2 (Try1217) (cat. # 3871), anti-caspase-3 (cat. # 9662), anti-cleaved caspase-3 (Asp175) (cat. # 9661), anti-PARP (46D11) (cat. # 9532), anti-Bcl-2 (D17C4) (cat. # 3498), anti-Aiolos (D1C1E) (cat. # 15103), anti-Ikaros (D6N9Y) (cat. # 14859), anti-AKT (Ser473) (cat. # 4691), anti-phospho-AKT (Ser473) (cat. # 4060), anti-NFκB (D14E12) (cat. # 8242), anti-phospho-NFκB (Ser536) (cat. # 3033), anti-p44/42 MAPK(Erk1/2) (137F5) (cat. # 4695), anti-phospho-p44/42 MAPK(Erk1/2) (Thr202/Tyr204) (cat. # 4370), and anti-rabbit IgG HRP-linked (cat. # 7074).
Cell viability
TMD8 cells and Mino cells were used for measuring cell viability. TMD8 cells were seeded in a 96-well plate at a density of 4,000 cells/well while Mino cells were seeded in 96-well plate at a density of 10,000 cells/well. The cells were then treated with test compounds at different concentrations from 3,000 nM to 0.15 nM (3-fold dilution) and DMSO (1%) as the vehicle control. After incubating for 48 h (TMD8 cells) or 72 h (Mino cells), cell viability was determined using the CellTiter-Glo 2.0 Assay (Promega cat. # G9242) according to the manufacturer’s instructions. A four-parameter dose-response equation was used for curve fitting in GraphPad Prism software and IC50 values were calculated as the mean ± SD from triplicates of three independent experiments.
Cell apoptosis
Cells were seeded in 12-well plates at a density of 2 × 106 cells/well and treated with test compounds at concentrations ranging from 3,000 nM to 0.15 nM (3-fold dilution) or DMSO (1%) as the vehicle control. After incubation for 48 h, cells were collected, and washed twice with cold PBS. Cell apoptosis was examined by using the Dead cell apoptosis kit Annexin V Alexa Fluor 488 and propidium iodide (PI) (cat. #V13241, Thermo Fisher) following the manufacturer’s instructions. Apoptosis data were collected on a CytoFLEX flow cytometer. Percentages of live, apoptotic and necrotic cells were determined by analyzing the data in GraphPad Prism 8.0. Data are presented as the mean of two independent experiments.
TR-FRET biochemical binding assays
TR-FRET was used to quantify the binding of the PROTACs to purified full-length unactivated BTK using the KINETICfinder® assay in which the binding and displacement of an Alexa Fluor647-labeled kinase tracer to BTK was quantified by TR-FRET detection with a terbium (Tb) antibody (Life Technologies). The assays were performed in black 384 Well Small Volume HiBase Microplates (Greiner) containing 0.05 nM BTK, 10 nM kinase Tracer and 2 nM Tb antibody in assay buffer (50 mM HEPES, pH 7.5, 10 mM MgCl2, 0.01% Brij-35, 1 mM DTT, and 1% DMSO). For all experiments, 4-point 10-fold serial dilutions of 100X concentrated test compounds were prepared in DMSO. Positive control wells contained no test compound and negative control wells contained no kinase. The kinetic assays were read continuously at 37 °C in a PHERAstar FSX plate reader (BMG LABTECH). After subtracting the background signal, the TR-FRET data were fit to a competitive binding equation described by Motulsky and Mahan to calculate the Kd values.
NanoBRET target engagement assays
Live-cell target engagement assays for BTK and CRBN were performed using an assay kit from Promega (cat. #N2500 & N2910, Promega). All the procedures were performed according to the manufacturer’s instructions. HEK293 cells were transiently transfected with BTK-NanoLuc® Fusion vector or NanoLuc®-CRBN Fusion vector using FuGENE® HD Transfection Reagent (cat. #E2311, Promega). After incubating for 24 h, HEK293 cells were seeded in a 96 well plate (cat. #3600, Corning) at a density of 2 × 105 cells/mL. The NanoBRET™ tracer reagent (1 μM K5 tracer or 0.5 μM CRBN tracer) and serial concentrations of test compounds were added to the cells after which the plates were incubated at 37°C for 2 h. To measure the NanoBRET in live cells, NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor were added to the cells, and luminescence was detected using a BioTek Synergy Neo 2 plate reader equipped with filter cubes EM 450/50, 610LP| DM 550 (part. #1035074, BioTek) and EX540/25 | DM 550 (part. #1035015, BioTek). The NanoBRET ratio is expressed in milliBRET units and calculated according to the equation: BRET = [(Acceptor sample/ Donor sample) – (Acceptor no trace control/ Donor no trace control)] x 1000. The BRET percentage (%) was calculated using the BRET ratio of the sample divided by the BRET ratio of the DMSO control. A four-parameter dose-response equation was used for curve fitting in GraphPad Prism software, and IC50 values are reported as the mean ± SD from duplicates of two independent experiments.
NanoBRET ternary complex formation assays
HEK293 cells were seeded in a six-well plate at a density of 4 × 105 cells/mL in 2 mL of culture media. After recovering for 5 h at 37°C and 5% CO2, the cells in each well were transiently transfected with 0.5 μg BTK-NanoLuc® Fusion vector (cat. #N2441, Promega) and 2 μg HaloTag®-CRBN Fusion Vector (cat. #N2691, Promega) using the FuGENE® HD Transfection Reagent (cat. #E2311, Promega). After 21 h, 40 μL of the transfected cells were replated on a 384-well white plate (cat. #3570, Corning) at a density of 2 × 105 cells/mL in the presence or absence of HaloTag® NanoBRET 618 ligand (cat. #G9801, Promega). After incubating for 18 h, 10 μL of MG-132 proteasome inhibitor in Opti-MEM® I Reduced Serum Media no phenol red was added to each well to give a final concentration of 10 μM. After incubation for 30 min, 10 μL of test compounds in Opti-MEM® I Reduced Serum Media no phenol red (cat. # 11058021 ThermoFisher Scientific) were added to each well to give final concentrations ranging from 3,000 nM to 0.15 nM (3-fold dilutions). Following a further 2.5 h, the NanoBRET NanoGlo Substrate (cat. #N1571, Promega) was added according to the manufacturer’s instructions and the donor and acceptor signal was quantified using a BioTek Synergy Neo 2 plate reader equipped with filter cubes EM 450/50, 610LP| DM 550 (part. #1035074, BioTek) and EX540/25 | DM 550 (part. #1035015, BioTek). The NanoBRET ratio is expressed in milliBRET units and was calculated according to the equation: BRET = [(Acceptor sample/ Donor sample) – (Acceptor no 618 ligand/ Donor no 618 ligand)] x 1000.
Label-free mass spectrometry-based proteomics
TMD8 cells (2 × 106) were treated with 111 nM of the BTK PROTACs or DMSO for 6 h in duplicate. Cells were harvested and washed twice with cold Tris-buffered saline (TBS), and the cell pellets were lysed using a sonicator with a microtip in 100 μL lysis buffer (5% SDS, 50 mM TEAB pH 8.5) supplemented with protease inhibitor (cat. # A32955 ThermoFisher Scientific) and phosphatase inhibitor (cat. # 78420 ThermoFisher Scientific). Lysates were centrifuged at 14,000 x g for 15 min at room temperature and the total protein content of the supernatant was determined using the Pierce BCA Protein Assay kit (cat. # 23225 ThermoFisher Scientific).
Aliquots of the lysis samples (50 μL) each containing 200 μg of protein were reduced with 10 mM DTT at 55°C for 30 min, followed by alkylation with 50 mM iodoacetamide at room temperature in the dark for 30 min. Subsequently, 5 μL of 12% H3PO4 was added to the samples, followed by 350 μL of S-Trap bind/wash buffer (90% methanol/100 mM TEAB) to produce a microprecipitate. Samples were then loaded onto a S-Trap mini cartridge (cat. # K02-mini-10 Protifi), and washed three times according to the manufacturer’s instruction. The samples were digested with trypsin (20 μg) in a humified incubator overnight at 37°C. Peptides were eluted according to the manufacturer’s instructions and the samples were then dried by SpeedVac and resuspended in 20 μL 0.1% formic acid for peptide analysis by nano LC-MS/MS.
Parent peptide mass, collision-induced fragment mass information, and peptide abundance values were obtained by a liquid chromatography-electrospray ionization tandem mass spectrometry (LC-MS/MS) using an orbital trap instrument (Thermo Q-Exactive HF) followed by protein database searching. HPLC C18 columns were prepared using P-2000 CO2 laser puller (Sutter Instruments) and silica tubing (100 μm ID × ∼20 cm) and were self-packed with 3 μm Reprosil C 18 resin. Peptides (~10 μg in a 1 μL injection volume) were separated on the resolving column with a flow rate of rate of 300 nL/min, using a gradient elution step 0–40% with MeCN and 0.1% HCOOH (0.23%/min) over 40 min, followed by a 10 min wash with 90% MeCN and a 10 min wash step with isocratic 90% MeCN. Electrospray ionization was achieved using a spray voltage of 2.2 kV. Data-dependent MS and MS-MS acquisitions were made using a survey scan (m/z 375–1400) with maximum fill of 50 ms followed typically by 20 consecutive product ion scans (m/z 100–1600). Parent ion with charge states of 2+, 3+, and 4+ were selected with a 15 s exclusion period. MS data were collected using Xcaliber (Thermo).
Raw data were analyzed using the Proteome Discover v2.4 software (Thermo) using label free quantitation. Resolution of MS and MS/MS data searches were set to 10 ppm and 0.05 Da, respectively. Two skipped trypsin cleavages and M-oxidation, KR-deamidation, ST-dehydration PTMs were allowed. Peptide and spectral cutoffs were set to <1% false discovery rates (FDR). Protein identifications were binned at <1% and <5% FDR cutoffs. The human UniProt dataset (73,101 entries) was used for data alignment. Fold change ratios of PROTAC versus DMSO vehicle control were obtained by matched peptide-based label free quantitation and p-values were calculated by Benjamini-Hochberg correction for FDR.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health grant R01GM102864 to P.J.T. and the Chemistry-Biology interface training grant T32GM136572 at Stony Brook University. We thank Dr. Richard E. Davis for providing the TMD8 and HBL-1 cells. We thank Yuzhang Wang for assistance making the Table of Contents Graphic.
ABBREVIATIONS
- BTK
Bruton’s tyrosine kinase
- BCR
B-cell receptor
- BLK
B-lymphoid tyrosine kinase
- BMX
bone marrow tyrosine kinase gene in chromosome X
- Bcl-2
B-cell lymphoma 2
- CLL
chronic lymphocytic leukemia
- CSK
C-terminal Src kinase
- CRBN
cereblon
- DLBCL
diffuse large B-cell lymphoma
- DC50
compound concentration inducing 50% of protein degradation
- ERBB2
Erb-b2 receptor tyrosine kinase 2
- GSPT1
G1 to S phase transition 1
- HRP
horse radish peroxidase
- HCK
hematopoietic cell kinase
- ITK
IL2 inducible T cell kinase
- IMiDs
immunodulatory imide drugs
- IKZF1
Ikaros
- IKZF3
Aiolos
- JAK3
Janus kinase 3
- MCL
mantle cell lymphoma
- Mcl-1
myeloid cell leukemia 1
- NF-κB
nuclear factor κB
- Nano-Luc
Nano-Luciferase
- NanoBRET
Nano-Luciferase (Nano-Luc) based bioluminescence resonance energy transfer
- PROTACs
proteolysis targeting chimeras
- PH
pleckstrin homology
- POI
protein of interest
- PI
propidium iodide
- PARP
poly ADP-ribose polymerase
- PLCγ2
phospholipase Cγ2
- SH
SRC homology
- SYK
spleen tyrosine kinase
- TR-FRET
time-resolved fluorescence resonance energy transfer
- TYK2
Tyrosine kinase 2
- TBS
Tris-buffered saline
- TH
Tec homology
- VHL
Von Hippel–Lindau
- WM
Waldenstrom macroglobulinemia
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://.
Supplementary Figures S1–S5 including degradation of neo-substrates, mechanism of action studies, induction of apoptosis, BTK degradation in various cell lines, and inhibition of cell growth. NMR spectra and HPLC traces (PDF)
Molecular formula strings (CSV)
The authors declare the following competing financial interest(s): P.J.T. is the cofounder of Chronus Pharmaceuticals Inc.
REFERENCES
- (1).Hendriks RW; Yuvaraj S; Kil LP Targeting Bruton’s tyrosine kinase in B cell malignancies Nat Rev Cancer, 2014, 14, 219–232. DOI: 10.1038/nrc3702 [DOI] [PubMed] [Google Scholar]
- (2).Ringheim GE; Wampole M; Oberoi K Bruton’s Tyrosine Kinase (BTK) Inhibitors and Autoimmune Diseases: Making Sense of BTK Inhibitor Specificity Profiles and Recent Clinical Trial Successes and Failures Front Immunol, 2021, 12, 662223. DOI: 10.3389/fimmu.2021.662223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hyvonen M; Saraste M Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia EMBO J, 1997, 16, 3396–3404. DOI: 10.1093/emboj/16.12.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Arthur R; Valle-Argos B; Steele AJ; Packham G Development of PROTACs to address clinical limitations associated with BTK-targeted kinase inhibitors Explor Target Antitumor Ther, 2020, 1, 131–152. DOI: 10.37349/etat.2020.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pan Z; Scheerens H; Li SJ; Schultz BE; Sprengeler PA; Burrill LC; Mendonca RV; Sweeney MD; Scott KC; Grothaus PG; Jeffery DA; Spoerke JM; Honigberg LA; Young PR; Dalrymple SA; Palmer JT Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase ChemMedChem, 2007, 2, 58–61. DOI: 10.1002/cmdc.200600221 [DOI] [PubMed] [Google Scholar]
- (6).Wu J; Liu C; Tsui ST; Liu D Second-generation inhibitors of Bruton tyrosine kinase J Hematol Oncol, 2016, 9, 80. DOI: 10.1186/s13045-016-0313-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Guo Y; Liu Y; Hu N; Yu D; Zhou C; Shi G; Zhang B; Wei M; Liu J; Luo L; Tang Z; Song H; Guo Y; Liu X; Su D; Zhang S; Song X; Zhou X; Hong Y; Chen S; Cheng Z; Young S; Wei Q; Wang H; Wang Q; Lv L; Wang F; Xu H; Sun H; Xing H; Li N; Zhang W; Wang Z; Liu G; Sun Z; Zhou D; Li W; Liu L; Wang L; Wang Z Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase J Med Chem, 2019, 62, 7923–7940. DOI: 10.1021/acs.jmedchem.9b00687 [DOI] [PubMed] [Google Scholar]
- (8).Liclican A; Serafini L; Xing W; Czerwieniec G; Steiner B; Wang T; Brendza KM; Lutz JD; Keegan KS; Ray AS; Schultz BE; Sakowicz R; Feng JY Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on - and off - target inhibition Biochim Biophys Acta Gen Subj, 2020, 129531. DOI: 10.1016/j.bbagen.2020.129531 [DOI] [PubMed] [Google Scholar]
- (9).Dhillon S Orelabrutinib: First Approval Drugs, 2021, 81, 503–507. DOI: 10.1007/s40265-021-01482-5 [DOI] [PubMed] [Google Scholar]
- (10).Woyach JA; Furman RR; Liu TM; Ozer HG; Zapatka M; Ruppert AS; Xue L; Li DH; Steggerda SM; Versele M; Dave SS; Zhang J; Yilmaz AS; Jaglowski SM; Blum KA; Lozanski A; Lozanski G; James DF; Barrientos JC; Lichter P; Stilgenbauer S; Buggy JJ; Chang BY; Johnson AJ; Byrd JC Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib N Engl J Med, 2014, 370, 2286–2294. DOI: 10.1056/NEJMoa1400029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gu D; Tang H; Wu J; Li J; Miao Y Targeting Bruton tyrosine kinase using non-covalent inhibitors in B cell malignancies J Hematol Oncol, 2021, 14, 40. DOI: 10.1186/s13045-021-01049-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nalawansha DA; Crews CM PROTACs: An Emerging Therapeutic Modality in Precision Medicine Cell Chem Biol, 2020, 27, 998–1014. DOI: 10.1016/j.chembiol.2020.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation Proc Natl Acad Sci U S A, 2001, 98, 8554–8559. DOI: 10.1073/pnas.141230798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Buhimschi AD; Armstrong HA; Toure M; Jaime-Figueroa S; Chen TL; Lehman AM; Woyach JA; Johnson AJ; Byrd JC; Crews CM Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation Biochemistry, 2018, 57, 3564–3575. DOI: 10.1021/acs.biochem.8b00391 [DOI] [PubMed] [Google Scholar]
- (15).Sun Y; Zhao X; Ding N; Gao H; Wu Y; Yang Y; Zhao M; Hwang J; Song Y; Liu W; Rao Y PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies Cell Res, 2018, 28, 779–781. DOI: 10.1038/s41422-018-0055-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sun Y; Ding N; Song Y; Yang Z; Liu W; Zhu J; Rao Y Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas Leukemia, 2019, 33, 2105–2110. DOI: 10.1038/s41375-019-0440-x [DOI] [PubMed] [Google Scholar]
- (17).Gabizon R; Shraga A; Gehrtz P; Livnah E; Shorer Y; Gurwicz N; Avram L; Unger T; Aharoni H; Albeck S; Brandis A; Shulman Z; Katz BZ; Herishanu Y; London N Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs J Am Chem Soc, 2020, 142, 11734–11742. DOI: 10.1021/jacs.9b13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Guo WH; Qi X; Yu X; Liu Y; Chung CI; Bai F; Lin X; Lu D; Wang L; Chen J; Su LH; Nomie KJ; Li F; Wang MC; Shu X; Onuchic JN; Woyach JA; Wang ML; Wang J Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry Nat Commun, 2020, 11, 4268. DOI: 10.1038/s41467-020-17997-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tinworth CP; Lithgow H; Dittus L; Bassi ZI; Hughes SE; Muelbaier M; Dai H; Smith IED; Kerr WJ; Burley GA; Bantscheff M; Harling JD PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding ACS Chem Biol, 2019, 14, 342–347. DOI: 10.1021/acschembio.8b01094 [DOI] [PubMed] [Google Scholar]
- (20).Crawford JJ; Johnson AR; Misner DL; Belmont LD; Castanedo G; Choy R; Coraggio M; Dong L; Eigenbrot C; Erickson R; Ghilardi N; Hau J; Katewa A; Kohli PB; Lee W; Lubach JW; McKenzie BS; Ortwine DF; Schutt L; Tay S; Wei B; Reif K; Liu L; Wong H; Young WB Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development J Med Chem, 2018, 61, 2227–2245. DOI: 10.1021/acs.jmedchem.7b01712 [DOI] [PubMed] [Google Scholar]
- (21).Dobrovolsky D; Wang ES; Morrow S; Leahy C; Faust T; Nowak RP; Donovan KA; Yang G; Li ZN; Fischer ES; Treon SP; Weinstock DM; Gray NS Bruton tyrosine kinase degradation as a therapeutic strategy for cancer Blood, 2019, 133, 952–961. DOI: 10.1182/blood-2018-07-862953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).PyMOL. The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC, 2015. DOI: [Google Scholar]
- (23).Qiu X; Sun N; Kong Y; Li Y; Yang X; Jiang B Chemoselective Synthesis of Lenalidomide-Based PROTAC Library Using Alkylation Reaction Org Lett, 2019, 21, 3838–3841. DOI: 10.1021/acs.orglett.9b01326 [DOI] [PubMed] [Google Scholar]
- (24).Liu J; Plewe MB; Wang J; Han X; Chen L Tropomyosin Receptor Kinase (Trk) Degradation Compounds And Methods Of Use, WO 2020/038415 Al, February, 27 2020
- (25).Yang K; Zhao Y; Nie X; Wu H; Wang B; Almodovar-Rivera CM; Xie H; Tang W A Cell-Based Target Engagement Assay for the Identification of Cereblon E3 Ubiquitin Ligase Ligands and Their Application in HDAC6 Degraders Cell Chem Biol, 2020, 27, 866–876 e868. DOI: 10.1016/j.chembiol.2020.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Robers MB; Dart ML; Woodroofe CC; Zimprich CA; Kirkland TA; Machleidt T; Kupcho KR; Levin S; Hartnett JR; Zimmerman K; Niles AL; Ohana RF; Daniels DL; Slater M; Wood MG; Cong M; Cheng YQ; Wood KV Target engagement and drug residence time can be observed in living cells with BRET Nat Commun, 2015, 6, 10091. DOI: 10.1038/ncomms10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hendrick CE; Jorgensen JR; Chaudhry C; Strambeanu II; Brazeau JF; Schiffer J; Shi Z; Venable JD; Wolkenberg SE Direct-to-Biology Accelerates PROTAC Synthesis and the Evaluation of Linker Effects on Permeability and Degradation ACS Med Chem Lett, 2022, 13, 1182–1190. DOI: 10.1021/acsmedchemlett.2c00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Troup RI; Fallan C; Baud MGJ Current strategies for the design of PROTAC linkers: a critical review Explor Target Antitumor Ther, 2020, 1, 273–312. DOI: 10.37349/etat.2020.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Burslem GM; Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery Cell, 2020, 181, 102–114. DOI: 10.1016/j.cell.2019.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sievers QL; Petzold G; Bunker RD; Renneville A; Slabicki M; Liddicoat BJ; Abdulrahman W; Mikkelsen T; Ebert BL; Thoma NH Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN Science, 2018, 362, eaat0572. DOI: 10.1126/science.aat0572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ishoey M; Chorn S; Singh N; Jaeger MG; Brand M; Paulk J; Bauer S; Erb MA; Parapatics K; Muller AC; Bennett KL; Ecker GF; Bradner JE; Winter GE Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders ACS Chem Biol, 2018, 13, 553–560. DOI: 10.1021/acschembio.7b00969 [DOI] [PubMed] [Google Scholar]
- (32).Yang Z; Sun Y; Ni Z; Yang C; Tong Y; Liu Y; Li H; Rao Y Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently Cell Res, 2021, 31, 1315–1318. DOI: 10.1038/s41422-021-00533-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cinar M; Hamedani F; Mo Z; Cinar B; Amin HM; Alkan S Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis Leuk Res, 2013, 37, 1271–1277. DOI: 10.1016/j.leukres.2013.07.028 [DOI] [PubMed] [Google Scholar]
- (34).Reiff SD; Muhowski EM; Guinn D; Lehman A; Fabian CA; Cheney C; Mantel R; Smith L; Johnson AJ; Young WB; Johnson AR; Liu L; Byrd JC; Woyach JA Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: a new treatment strategy for ibrutinib-resistant CLL Blood, 2018, 132, 1039–1049. DOI: 10.1182/blood-2017-10-809020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.