

Abstract
A 72-year-old man was admitted for examination of proteinuria (9.14 g/day) and leg edema. Essential thrombocythemia (ET) was diagnosed because of thrombocytosis (platelet count, 57.9×104/μL), elevated megakaryocytes in bone marrow biopsy, and JAK2 V617 mutation. Kidney biopsy led to a diagnosis of focal segmental glomerulosclerosis (FSGS) cellular variant (characterized by glomerular capillaries filled with swollen endothelial cells containing foam cells) in 6 glomeruli, FSGS tip variant in 5 glomeruli, and additional FSGS variants in other glomeruli. Affected glomeruli had anti-CD61 antibody staining-positive megakaryocyte infiltrations. ET may induce FSGS because megakaryocyte infiltration increases intraglomerular pressure, resulting in hypertension and proteinuria.
Keywords: essential thrombocythemia, nephrotic syndrome, focal segmental glomerulosclerosis
Introduction
Focal segmental glomerulosclerosis (FSGS) is diagnosed when light microscopy shows segmental scarring of the glomerulus, immunofluorescence shows immunoglobulin M (IgM) and C3 staining in the scarred segment, and electron microscopy shows no electron dense deposits but extensive foot process effacement (1). However, there is no common and consistent view on the pathogenesis of FSGS, and attempts have been made to collect information and elucidate the pathogenesis by classifying the disease into 5 histologic types based on morphologic characteristics. Secondary FSGS occurs in patients with underlying diseases, such as hereditary, viral, or drug-induced diseases, and primary FSGS occurs in patients with no apparent underlying disease (2).
Myeloproliferative disorders (or neoplasms), a group of rare blood cancers in which the bone marrow produces an excess of red blood cells, white blood cells, or platelets, are classified into 6 disease groups (3). Three of these groups, polycythemia vera, essential thrombocythemia (ET), and primary myelofibrosis, have recently attracted attention as causes of secondary FSGS.
ET is a rare myeloproliferative neoplasm characterized by clonal overproduction of platelets (thrombocytes) by megakaryocytes (i.e., abnormal platelets) in the bone marrow. Patients with ET are known to be at increased risk of thrombotic disorders, such as cerebrovascular disease, myocardial infarction, and pulmonary embolism, and pregnancy complications, but the association between ET and kidney complications is less well known. Only a few reports have described such complications, i.e., FSGS, IgA nephropathy, and fibrillary glomerulonephritis (4-9).
We experienced a 72-year-old man with characteristic FSGS lesions on kidney biopsy who had concurrent ET. Here, we discuss the relationship of FSGS with ET by referring to the Columbia classification (2). We believe that this case is a valuable example of the diverse FSGS pathology observed in many glomeruli and may further support the association of FSGS with myeloproliferative diseases. We focus on ET and discuss the pathophysiology of secondary FSGS due to ET with the aim to improve understanding of the pathogenesis of FSGS.
Case Report
A 72-year-old Japanese man was admitted for evaluation of proteinuria and leg edema. At the age of 67, he was diagnosed with early stage gastric cancer and underwent endoscopic submucosal dissection and was found to have hypertension (142/88 mmHg); however, he did not wish to be treated with antihypertensive drugs and did not visit a medical institution after discharge. Five months before the current admission, hypertension (234/110 mmHg) was noted during a dental visit, and treatment was initiated with antihypertensive drugs, including calcium antagonists (amlodipine 10 mg) and a diuretic (furosemide 20 mg). Four months after starting treatment, edema of the lower legs appeared and proteinuria was observed, and the patient was admitted to our hospital one month later.
At admission, the patient was 169 cm tall and weighed 57.9 kg. His blood pressure was 128/66 mmHg; heart rate, 80 beats/min; and temperature, 36.6°C. Heart and breath sounds were normal, but edema was present in the lower extremities.
The complete blood count was as follows: erythrocytes, 5.93×106/μL; hemoglobin, 12.8 g/dL; leucocytes, 12,400 /μL; and thrombocytes, 57.9×104/μL. The results of blood chemistry tests were as follows: serum albumin, 1.9 g/dL; serum creatinine, 0.75 mg/dL; estimated glomerular filtration rate, 78.2 mL/min/1.73 m2; lactate dehydrogenase, 371 U/L; C-reactive protein, less than 0.1 mg/dL; iron, 3.3 μg/dL; unsaturated iron binding capacity, 232 μg/dL; ferritin, 7 μg/dL; IgG, 773 mg/dL; IgA, 166.2 mg/dL; IgM, 88.9 mg/dL; total complement activity (assessed as CH50), 57 U/mL; complement 3, 101 mg/dL; complement 4, 28 mg/dL; antinuclear antibody, negative; anti-dsDNA antibody, negative; hepatitis B surface antigen, negative; hepatitis C virus antibody, negative; and syphilis lipid antibody, negative. No findings were suggestive of an autoimmune or infectious disease. Urinary protein excretion was 9.14 g/day, and the urinary sediment contained 1 to 5 erythrocytes per high power field.
Ultrasound showed splenomegaly but no morphological abnormalities in the kidneys or liver. The patient had no history of drug treatment other than the antihypertensive medications started before admission and no history of low birth weight or information suggestive of human immunodeficiency virus (HIV). A bone marrow biopsy was performed to evaluate thrombocytosis.
Bone marrow biopsy
Flow cytometry showed no clear abnormalities. In the bone marrow specimen, megakaryocytes were increased (53 counts/mm2; Fig. 1) and the G-band showed 5/20 46, XY, del(20)(q11). Genetic testing was positive for the JAK2 V617 mutation and negative for the BCR-ABL mutation. ET was diagnosed according to the revised diagnostic criteria because of the high platelet count (>400×103/µL) and JAK2 V617F mutation and because iron deficiency, polycythemia vera, chronic myeloid leukemia, and myelofibrosis myelodysplastic syndrome were ruled out (3). A kidney biopsy was performed to evaluate proteinuria.
Figure 1.
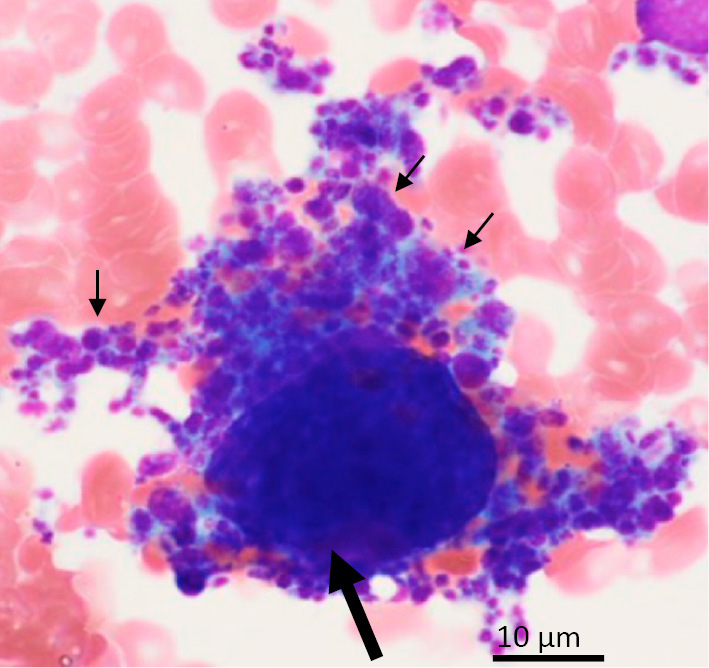
Bone marrow biopsy. Megakaryocytes (arrow) produced a large number of platelets (small arrows). Wright staining; original magnification ×1,000.
Kidney biopsy
Light microscopic examination of the biopsy specimen revealed global sclerosis with luminal hyalinosis in 2 out of 56 glomeruli. In general, glomerular hypertrophy was unremarkable. The FSGS cellular variant characterized by glomerular capillaries filled with swollen endothelial cells containing foam cells was noted in 6 glomeruli. Overlying podocytes showed hypertrophy and hyperplasia. Other adjacent areas of the same glomerulus were slightly collapsed (Fig. 2A). Five glomeruli showed FSGS tip variant, characterized by herniation of the affected segmental sclerosis lesion into the tubular lumen, both with (Fig. 2B) and without foam cells (Fig. 2C). One glomerulus showed FSGS collapsing variant, characterized by global collapse of the glomerular capillary lumina due to wrinkling, collapse of the glomerular basement membranes, and marked hyperplasia of the overlying podocytes (Fig. 2D); another showed FSGS perihilar variant with luminal hyalinosis (Fig. 2E); and another showed FSGS not otherwise specified (NOS) variant, characterized by hyalinosis of the capillary lumina (Fig. 2F). Foam cells were seen among Bowman's epithelial cells (Fig. 2G). Mild tubular atrophy and fibrosis of the interstitium were noted. Interlobular arteries showed moderate fibroelastosis. Immunoglobulins and complement were not seen on immunofluorescence microscopy. Electron microscopy showed no electron-dense deposits but did reveal an increase in the mesangial matrix and foot process effacement in more than 80% of podocytes in observable glomeruli. No detachment of podocytes from the glomerular basement membrane was evident (Fig. 2H). Immunohistological evaluation showed anti-CD61 antibody staining-positive platelet and megakaryocyte infiltrations in glomerular capillaries (Fig. 2I) but no fibrin thrombus formation, indicating positivity with phosphotungstic acid-hematoxylin staining.
Figure 2.
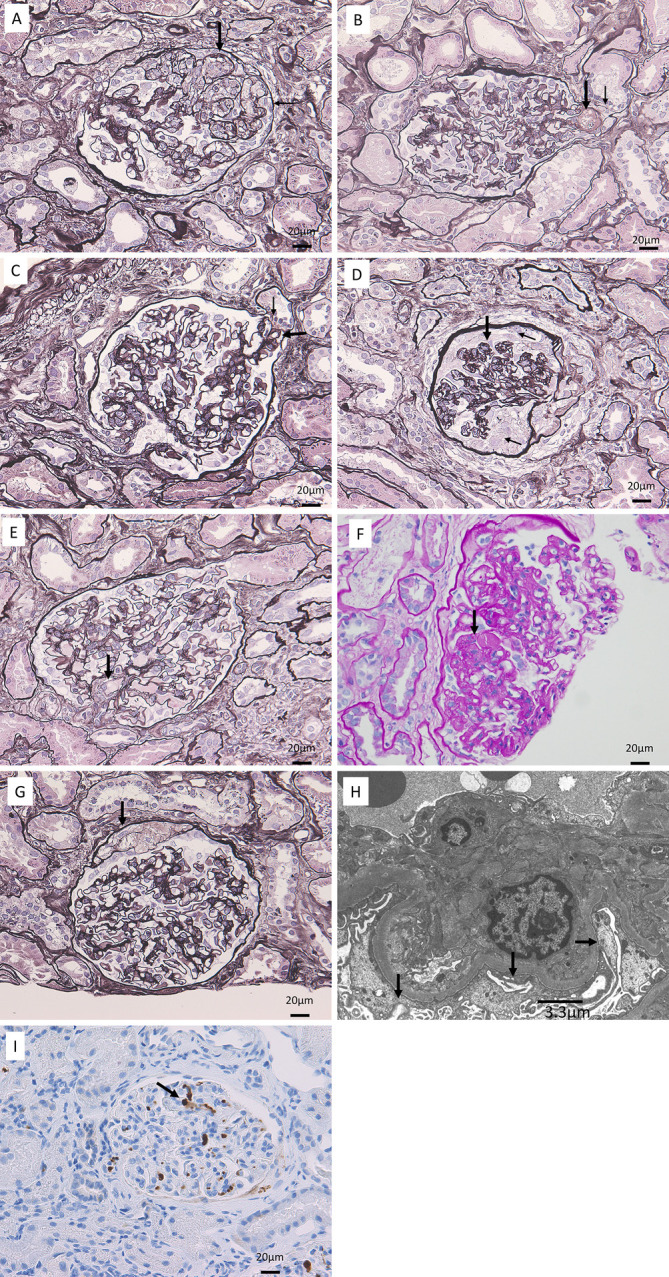
Kidney biopsy findings. A: Prominent focal segmental glomerulosclerosis (FSGS) cellular variant (large arrow), characterized by glomerular capillaries filled with swollen endothelial cells containing foam cells. Overlying podocytes showed hypertrophy and hyperplasia (small arrow). Periodic acid methenamine silver staining; original magnification ×200. B: FSGS tip variant (large arrow), characterized by a segmental sclerosis lesion with foam cells herniating into the tubular lumen. Overlying podocytes showed capping (small arrow). Periodic acid methenamine silver staining; original magnification ×200. C: FSGS tip variant (large arrow), characterized by herniation of the affected segmental sclerosis lesion into the tubular lumen. The overlying podocytes showed capping (small arrow). Periodic acid methenamine silver staining; original magnification ×200. D: FSGS collapsing variant (large arrow) characterized by global collapse of the glomerular capillary lumina due to wrinkling and collapse of glomerular basement membranes, with marked hyperplasia of the overlying podocytes (small arrows). Periodic acid methenamine silver staining; original magnification ×200. E: FSGS perihilar variant with luminal hyalinosis (arrow). F: FSGS not otherwise specified variant with luminal hyalinosis (arrow). Periodic acid-Schiff staining; original magnification ×200. G: Foam cells among Bowman’s epithelial cells (arrow). Periodic acid methenamine silver staining; original magnification ×200 H: Electron microscopy revealed an increase in the mesangial matrix and foot process effacement (arrows) in more than 80% of glomerular capillaries. I: Immunohistological examination showed infiltrations of anti-CD61 antibody staining-positive megakaryocytes (arrow) in glomerular capillaries. Original magnification ×200.
Clinical course
In addition to the continued use of the calcium antagonists and diuretic, post-hospitalization nutritional dietary treatment with restriction of salt intake to 6 g/day and water intake to 1 L/day and additional use of angiotensin II receptor antagonists led to a body weight loss of 5 kg and a reduction in urinary protein to 2.5 g/day. Subsequently, treatment was started with 50 mg/day prednisolone and 100 mg/day aspirin. Three months later, urinary protein had decreased to 1.5 g/day, and the prednisolone dose was gradually reduced; however, the urinary protein level remained elevated (1.0 g/day).
Ten months after hospitalization, hydroxycarbamide was started to treat ET and adjusted in the range of 1,000 to 2,000 mg. Eight months later, thrombocytes decreased to approximately 20.0×104/μL, blood pressure normalized without hypertensive agents, and urinary protein decreased to less than 0.2 g/day (Fig. 3). Urinary occult blood was not evident during follow-up. Sadly, the patient developed acute myeloid leukemia (without ET) after another 8 months and died 3 months later.
Figure 3.

Clinical course. The Figure shows changes in platelets and proteinuria over time and drug treatment; symptoms improved with prednisolone, but aspirin caused femoral subcutaneous hematoma and was discontinued.
Discussion
A few reports have been published on ET and renal impairment, and together they suggest that FSGS lesions are a feature of renal involvement in patients with ET. In these reports, FSGS is a representative renal complication of ET, but IgA nephropathy and fibrillary glomerulonephritis are also present. The reports are described briefly below and summarized in Table.
Table.
Summary of Case Reports on Focal Segmental Glomerulosclerosis Associated with Essential Thrombocythemia.
| Reference No. | Age | Sex | Pathological diagnosis | Proteinuria (g/gCr) | Plt (×104) |
|---|---|---|---|---|---|
| (2) | 25 | M | FSGS | 2.2 | 107 |
| (2) | 39 | F | FSGS | 5 | 12 |
| (3) | 68 | M | Fibrillary glomerulonephritis | 7.5 | 122 |
| (4) | 76 | M | FSGS | 12.7 | 87 |
| (5) | 74 | F | No data | No data | 68 |
| (6) | 76 | F | IgAN | No data | 49 |
| (6) | 62 | F | No data | 1.5 | 98 |
| (7) | 59 | M | IgAN | 0.56 | 42 |
Pathological diagnosis, urine protein, and platelets described in each report. Those not listed are indicated as “no data”.
FSGS: focal segmental glomerulosclerosis, IgAN: IgA nephropathy
Au et al. presented kidney biopsy findings from 2 cases of ET (4). Case 1 was a 25-year-old man diagnosed with ET on the basis of hypertension, cerebral infarction, and proteinuria; kidney biopsy showed diffuse mesangial sclerosis with segmental glomerular capillary thickening. And case 2 was a 39-year-old man diagnosed with ET on the basis of hypertension and proteinuria; kidney biopsy showed FSGS.
Asaba et al. reported on a 68-year-old man who developed proteinuria 28 years after hyperthrombocytosis was noted and was subsequently diagnosed with ET (5). Kidney biopsy showed periodic acid Schiff-positive deposits throughout 5 glomeruli and in the mesangial area and along the capillary wall in 2 glomeruli. Immunofluorescence staining for IgG was positive. Electron microscopy showed randomly disposed, 10- to 20-nm nonbranching fibrils. Fibrillary glomerulonephritis was diagnosed.
Haraguchi et al. described a 68-year-old man with increased proteinuria and ET after splenectomy (6). Kidney biopsy showed segmental sclerosis and hyalinosis in 17 of 26 glomeruli and intracapillary infiltration of foam cells. Tip lesions were also noted. FSGS, probably cellular and tip variants, was suggested. An electron micrograph revealed foot process effacement. The patient had increased serum levels of transforming growth factor-β, basic fibroblast growth factor, and platelet-derived growth factor BB.
Said et al. evaluated glomerular disease in 11 patients with myeloproliferative neoplasms, including 1 patient with ET and FSGS whose kidney biopsy showed segmental sclerosis with prominent luminal hyalinosis and adhesion to Bowman’s capsule (7). CD61 immunohistology showed infiltration of intracapillary megakaryocytes.
Fujita et al. presented the case of a 78-year-old woman with ET (8). Kidney biopsy showed moderate mesangial proliferation with several crescents. Immunohistological staining was positive for IgG and IgA, and electron microscopy showed electron-dense deposits in the mesangium. IgA nephropathy was diagnosed.
Rahimian et al. described a 57-year-old man with ET. Kidney biopsy showed diffuse thickening of capillary membranes and mild mesangial matrix expansion and hypercellularity. Immunofluorescence staining was positive for IgA. IgA nephropathy was diagnosed, although no electron microscopic evaluation was performed (9).
The Columbia classification of FSGS reported by D'Agati et al. defines 5 variants of FSGS based on the morphology of the lesions but does not consider the underlying disease or the implications of the 5 variants (2). We attempted to use this system to classify the morphological features of FSGS in our patient and noted that our case included all 5 variants, as follows: FSGS cellular variant, characterized by segmental endocapillary hypercellularity, including foam cells and some karyorrhectic leukocytes; FSGS tip variant, characterized by segmental lesions involving the tip domain at the origin of the tubular pole; FSGS collapsing variant, characterized by global collapse of the tuft and podocyte hypertrophy and hyperplasia with tubular degenerative changes; FSGS perihilar variant, characterized by both increased matrix (sclerosis) and glassy hyalinosis deposited in the vascular pole segment of the tuft; and FSGS NOS variant, characterized by a discrete segmental lesion of sclerosis with wrinkling and collapse of glomerular basement membranes and hyaline deposits. The prominent FSGS variants were the cellular and tip variants.
In our case, CD61 staining confirmed that the ET-related infiltrations of megakaryocytes in glomeruli were related to FSGS, which is in agreement with the report by Said et al. (7).
Almost 20 years have passed since the Columbia classification of FSGS was published, but there have been no new developments in defining the relationship between each variant and the clinical picture. The identified characteristics can be summarized as follows: collapsing variant is either idiopathic in origin or associated with HIV, is more commonly found in Black patients, and shows poor prognosis; perihilar variant shows secondary adaptive forms stemming from nephron loss or glomerular hypertension (i.e., due to obesity, reflux nephropathy, and sickle cell disease), usually accompanied by glomerular hypertrophy; cellular variant shows early stage lesions and has a poor prognosis; tip variant responds to steroid therapy and has a better prognosis; and FSGS NOS variant has been observed from repeat biopsies. The first 4 variants may evolve into FSGS NOS over time (2,10). Secondary FSGS due to elevated intraglomerular pressure has been reported to be associated with the perihilar variant, but the cellular and tip variants seen in our case have not been mentioned in relation to glomerular hypertension.
The relationship between FSGS and hypertension is unclear, but nephron loss was suggested to cause FSGS because of pressure loading on the residual glomerulus, especially in patients with hypertension. When a kidney biopsy shows signs of FSGS in a patient with hypertension, proteinuria, and reduced renal function, hypertension can be assumed to have induced the FSGS lesions; however, no evidence for this relationship has been published. On the other hand, a few articles reported that FSGS cellular variant-like lesion is related to the development of hypertension (11).
The fact that our case was characterized by FSGS cellular and tip variants can be easily explained by the following inference in relation to intraglomerular pressure: FSGS cellular variant, which is characterized by segmental endocapillary hypercellularity, may indicate the presence of intraglomerular hypertension due to infiltration of megakaryocytes into the glomerulus, and FSGS tip variant may indicate that elevated glomerular hypertension has resulted in glomerular invasion into the ureteric pole.
Usually, even when FSGS is diagnosed, the majority of glomeruli are almost normal, and only a few glomeruli show signs of FSGS. Foot process effacement is seen only in glomeruli with abnormal glomerular lesions, which are present in at most 30% to 40% of glomeruli. In contrast, in the present case, foot process effacement was observed in more than 80% of the glomeruli. We believe that in our case, abnormal platelets associated with ET caused endothelial cell damage in many glomeruli, leading to epithelial cell damage, which in turn led to FSGS lesions (mainly cellular and tip variants) and proteinuria. In addition, hypertension was induced by the development of the glomerular lesions.
In conclusion, this case was characterized by findings corresponding mainly to FSGS cellular and tip variants and by ET-related, CD61-positive cells in the glomeruli. The pathogenesis of proteinuria in FSGS remains unclear. However, consistent with post-treatment effects in ET, blood pressure normalized in our case and proteinuria disappeared, suggesting that ET-related pathologies induce elevated glomerular hypertension, which may be associated with the appearance of proteinuria and the formation of FSGS lesions.
The present study adhered to the Declaration of Helsinki, and the patient gave his consent for the case report to be published.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Colvin RB, Chang A. Podocytopathies. In: Diagnostic Pathlogy: Kidney Disease. 3rd ed. Elsevier, Philadelphia, 2019: 48-89. [Google Scholar]
- 2. D'Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 43: 368-382, 2004. [DOI] [PubMed] [Google Scholar]
- 3. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127: 2391-2405, 2016. [DOI] [PubMed] [Google Scholar]
- 4. Au WY, Chan KW, Lui SL, Lam CC, Kwong YL. Focal segmental glomerulosclerosis and mesangial sclerosis associated with myeloproliferative disorders. Am J Kidney Dis 34: 889-893, 1999. [DOI] [PubMed] [Google Scholar]
- 5. Asaba K, Tojo A, Onozato ML, et al. Fibrillary glomerulonephritis associated with essential thrombocytosis. Clin Exp Nephrol 7: 296-300, 2003. [DOI] [PubMed] [Google Scholar]
- 6. Haraguchi K, Shimura H, Ogata R, et al. Focal segmental glomerulosclerosis associated with essential thrombocythemia. Clin Exp Nephrol 10: 74-77, 2006. [DOI] [PubMed] [Google Scholar]
- 7. Said SM, Leung N, Sethi S, et al. Myeloproliferative neoplasms cause glomerulopathy. Kidney Int 80: 753-759, 2011. [DOI] [PubMed] [Google Scholar]
- 8. Fujita K, Hatta K. Renal biopsy cases in myeloproliferative neoplasms (MPN). CEN Case Rep 2: 215-221, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rahimian S, Johnson T, Herb R. A case of essential thrombocythemia and IgA nephropathy with literature review of the concurrence. Case Rep Oncol Med 2019: 5086963, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han MH, Kim YJ. Practical application of Columbia classification for focal segmental glomerulosclerosis. Biomed Res Int 2016: 9375753, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hasegawa J, Hoshino J, Suwabe T, et al. Characteristics of intravascular large B-cell lymphoma limited to the glomerular capillaries: a case report. Case Rep Nephrol Dial 5: 173-179, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]