

Abstract
The COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 is a global health emergency. The main protease is an important drug target in coronaviruses. It plays an important role in the processing of viral RNA-translated polyproteins and is highly conserved in the amino acid sequence and three-dimensional structure, making it a good drug target for which several small molecule inhibitors are available. This paper describes the various anti-severe acute respiratory syndrome coronavirus 2 inhibitor drugs targeting Mpro discovered since the severe acute respiratory syndrome coronavirus 2 outbreak at the end of 2019, with all these compounds inhibiting severe acute respiratory syndrome coronavirus 2 Mpro activity in vitro. This provides a reference for the development of severe acute respiratory syndrome coronavirus 2 Mpro-targeted inhibitors and the design of therapeutic approaches to address newly emerged severe acute respiratory syndrome coronavirus 2 mutant strains with immune evasion capabilities.
Keywords: covalent inhibitor, Mpro, non-covalent inhibitor, severe acute respiratory syndrome coronavirus 2, targeted inhibitor
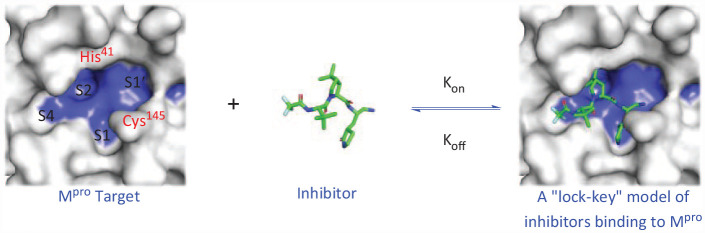
Introduction
Background
Since the outbreak of novel coronavirus pneumonia (COVID-19) in December 2019, the resulting global pandemic posed a huge threat to human life and health, and caused significant losses to the global economy. Therefore, it is urgent to develop effective prevention and treatment strategies.1–3 The genome sequence of the pathogen of COVID-19 was first published on 11 January 2020. The pathogen was identified as a novel coronavirus (2019-nCoV), which was later named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Virus Taxonomy (ICTV). 4
At present, the world is still fighting SARS-CoV-2 and has rapidly deployed effective vaccines to combat the pandemic. 5 Although many vaccines have been approved for the prevention of COVID-19, most COVID-19 vaccines were developed for the early pandemic strains, due to the rapid and frequent variation of the virus, including the Alpha (B.1.1.7) variant, the Beta (B.1.351) variant, the Gamma (P.1) variant, the Delta (B.1.617.2) variant, and the Omicron (B.1.1.529) variant. 6 The low titer of the antibody leads to a shortening of the protection time of the vaccine, limited protection of the naturally acquired and vaccine-induced antibodies against infection by the Omicron variant strain, and the resurgence of SARS-CoV-2 infection in the highly vaccinated population, which impairs the efficacy of the vaccine. 7 Therefore, it is crucial to develop more effective specific drugs and treatment methods for more conservative viral targets to curb the spread of COVID-19 and reduce mortality. 8
Biological basis of targeted SARS-CoV-2 Mpro treatment
SARS-CoV-2 is a positive single-stranded RNA virus with a genome of about 30,000 nucleotides. Genome sequencing shows that the genome sequence of this pathogenic coronavirus has 96% homology with that of bat coronavirus and belongs to the same genus as the pathogen SARS-CoV that caused the outbreak of SARS in 2003 β-coronavirus, with 89.1% nucleotide similarity and 79% homology. 9 The RNA genome of SARS-CoV-2 contains at least six open reading frames (ORFs), but the first ORF (ORF1a/b) accounts for about two-thirds of the length of the genome. It directly translates into two large overlapping precursor proteins pp1a and pp1ab (Figure 1). They must be cut and processed into functional subunits for replication and transcription activities. 10 The cleavage process is completed by two viral proteases: the main protease Mpro (also known as 3CLpro) and the papain-like protease PLpro, both of which are encoded by ORF1a, and these polyproteins are processed into 16 non-structural proteins (NSPs). 11 These NSPs are involved in the production of subgenomic RNA, encoding four main structural proteins (envelope (E) protein, membrane (M) protein, spike (S) protein, and nucleocapsid (N) protein), and other auxiliary proteins. 12 SARS-CoV-2 mainly infects host cells by binding with angiotensin-converting enzyme 2 (ACE2) on the surface of host cells through the receptor binding domain (RBD) of the S protein. Therefore, the hydrolysis of pp1a/pp1ab by proteolytic enzymes is an indispensable key step in the process of viral RNA replication and transcription, and plays a crucial role in the life cycle of COVID-19.
Figure 1.

(a) Structure and (b) genomic composition of SARS-CoV-2. 2
One of the most characteristic drug targets of coronavirus is the main protease Mpro, which is a chymotrypsin-like cysteine protease that exists in the non-structural protein 5 (NSP5) of the virus. 13 Like papain, this enzyme is essential for processing the multi-protein translated from the virus RNA. 14 Mpro is a cysteine protease with three domains (Domains I–III) (Figure 2), which is involved in most mature cleavage events in the precursor protein. Mpro is encoded by ORF1 as an NSP5. It cleaves the poly-protein pp1ab of the virus at 11 different positions, and the core cleavage motif is Leu-Gln↓(Ser/Ala/Gly).15,16 The active site of Mpro contains a catalytic dimer composed of Cys145 and His 41 . The catalytic dimer consists of four main pockets, which are marked according to their position relative to the shear bond of the substrate. The active site is located in the gap between the two N-terminal domains of the three domains of the monomer, while the C-terminal helical domain is involved in the regulation and dimerization of the enzyme. 10 Inhibiting Mpro activity will prevent virus replication, so Mpro protease is an important antiviral target.13,17,18 Although there are many potential drug targets on SARS-CoV-2,1,19–22 such as RNA-dependent RNA polymerase RdRp, spinous RBD, and glucose regulatory protein GRP78, Mpro only cleaves peptides after glutamine (Gln) residues, and the human body lacks its homologous protein enzyme. 23 Its structure has been verified by X-ray crystallography, and its active form is a homodimer. 10 The active center with less plasticity is mainly composed of His 41 and Cys145, which is relatively conservative in pathogenic β-coronaviruses and not easy to mutate,24,25 so it becomes an ideal target for drug development against COVID-19.
Figure 2.

Three-dimensional structure of SARS-CoV-2 Mpro from two different perspectives. 15
Targeted inhibitors of SARS-CoV-2 main protease
From the perspective of the interaction mechanism of targeting Mpro targets, the reported SARS-CoV-2 Mpro inhibitors can be divided into two categories: One type is a covalent inhibitor that can bind to the active pocket of Mpro, and the groups on the compound react with the target site to form covalent bonds, thereby inhibiting its biological function. The other type is non-covalent inhibitors that use non-covalent interactions with binding pockets to reversibly bind to enzyme targets, while the inhibitors themselves do not react. The Mpro inhibitors reported in this paper were tested for their cytotoxicity against Vero E6, HPAEpiC, and A549 cells. Both in vivo and in vitro activity tests showed low/submicromolar activity, and examination of the pharmacokinetics also showed good oral absorption effects. This provides a reference for the rational design of more inhibitors and the discovery of candidate drugs for the treatment of COVID-19.
Covalent inhibitors
Mpro covalent inhibitors contain electrophilic warheads or connect electrophilic groups to modify known non-covalent ligand fragments. Cys145 is used as the anchor point of the electrophilic warhead for covalent binding, resulting in changes in protein conformation and playing an inhibitory role. The covalent bond formed with the target allows the inhibitor to have a high potential energy and a high coordination efficiency. In addition, it can prolong the duration of pharmacological action and lower the frequency of administration, thus potentially reducing the drug dosage to improve safety. According to the classification of electrophilic warheads, covalent inhibitors mainly include ketoamides, nitriles, sulfonates, Michael acceptors, aldehydes, and organic selenium compounds.
α-Ketoamides
The team of Rolf Hilgenfeld at the University of Lübeck designed, synthesized, and evaluated the inhibitory activity of several α-ketoamide peptidomimetic inhibitors against SARS-CoV-2 Mpro(compounds 1–4) (Figure 3(a)). 15 To improve the half-life of 1 in plasma, the P3–P2 amide bond is hidden in a pyridone ring, preventing cellular proteases from entering the bond and lysing it, while replacing the hydrophobic cinnamoyl group with a slightly less hydrophobic Boc group which increases the solubility of this compound in plasma and reduces its binding to plasma proteins, leading to compound 2. Compound 2 was optimized, and Compound 3 was obtained by replacing the P2 cyclohexyl of 2 with cyclopropyl and improved the antiviral activity against SARS-CoV-2. However, Compound 4, obtained by removing the Boc group, showed a loss of activity and weakened inhibition, indicating that the Boc and hydrophobic groups were necessary for the interaction between Mpro and the cell membrane. Thus, the crystal structure of the SARS-CoV-2 Mpro complex with the α-ketoamide inhibitor 3 was reported (Figure 3(b)) and its lung tissue level was monitored (IC50 = 0.67 μM, EC50 = 1.75 μM). 15 The pharmacokinetic profiles demonstrate significant pulmonary tropism and suitability for administration via the inhalation route, 11 providing a useful framework for the development of anti-coronavirus drugs containing ketoamide inhibitors.
Figure 3.

(a) Chemical structures of the inhibitors 1–4; (b) crystal structure of 3 bound to the Mpro active site. 15
In May 2022, the team of Professor Lei Jian and Yang Shengyong of West China Hospital of Sichuan University reported a series of potent Mpro inhibitors 5 (Y180) (Figure 4(a)) containing an α-ketoamide and obtained using the Ugi four-component reaction (Ugi-4CR) (IC50 = 8.1 nM). 26 An acetyl group was introduced at the terminal acyl of the amide, which allowed the formation of an α-ketoamide warhead in the S1′ pocket. Furthermore, to reduce the epimerization of the most active epimer ((R)-epimer) to its less active epimer ((S)-configuration), deuterium was used to replace the exchangeable hydrogen attached to the chiral carbon linking the two amides (Figure 4(b)). Compound 5 is effective against wild-type SARS-CoV-2, B.1.1.7 (Alpha), B.1.617.1 (Kappa), and P.3 (Theta), with EC50 values of 11.4, 20.3, 34.4, and 23.7 nM, respectively. Oral treatment with 5 showed significant antiviral efficacy. It was also very effective against the B.1.1.529 (Omicron) variant in vitro and in vivo, significantly reducing the viral load, reducing tissue damage, and improving the survival rate of infected animals. The covalent bond formed with the target gives the inhibitor high potential energy and ligand efficiency, and can prolong the time of efficacy, lower the frequency of administration, and possibly reduce the drug dose, thus improving safety.
Figure 4.

(a) Chemical structure of compound 5 and (b) detailed interactions of Mpro and compound 5. 26
Compound 6 (Boceprevir; Figure 5(a)) is an FDA-approved serine protease inhibitor for the treatment of hepatitis C virus (HCV). 27 Its ketoamide group can covalently bind with Ser139 of HCV NS3 protease. 28 The drug can also inhibit Mpro enzyme activity with an IC50 value of 4.13 μM and an EC50 value of 1.90 μM for SARS-CoV-2. 29 In the Mpro–Boceprevir composite structure (PDB: 6ZRU and 7C6S), the nucleophilic Cys145 in Mpro forms a C–S covalent bond with the ketone carbon of 6, the oxygen of the α-ketoamide in 6 forms H bonds with the Cys145 and Gly143 backbone chain amides, occupying oxygen anion holes, and the hydroxy group covalently added by the α-ketoamide forms hydrogen bonds with His 41 of the side chain, both of which are stable conformations. The backbone of His164 and Glu166 forms an H bond with the amide bond on the backbone of compound 6. The dimethylcyclopropyl moiety is inserted deep into the S2 pocket and forms extensive hydrophobic contacts with His 41 , Met 49 , Met165, Asp187, Arg188, and Gln189. The tert-butyl group is directed into the S4 pocket and is stabilized by several H bonds to the backbone oxygen of Glu166 and by hydrophobic interactions with the side chains of Met165, Gln192, Leu167, and Pro168 (Figure 5(b)). Due to 6 being an approved drug for the treatment of HCV, there are a large number of data in terms of dose, toxicity, and pharmacokinetics, which can accelerate the research and development process of its use in SARS-CoV-2 treatment.
Figure 5.

(a) Chemical structure of 6 and (b) the interaction diagram of Mpro and 6. 30
Nitriles
In September 2020, the Pfizer company reported compound 8 (PF-00835231) as a potent inhibitor of SARS-CoV-2 Mpro. 31 This compound had been previously identified for the treatment of SARS-CoV, and given that SARS-CoV Mpro and SARS-CoV-2 Mpro have homology, it was repurposed. It is based on the phosphate prodrug 7 (PF-07304814) designed by Pfizer, which hydrolyzes into the active form 8 when administered in the body. The phosphate prodrug 7 was also described to increase the potential for the intravenous treatment of COVID-19. 32 However, 7 has to be administered intravenously, making it less appealing for massive distribution and relegating its use to hospital settings. Meanwhile, due to the polarity of 8, it cannot be absorbed by the intestine through oral administration. Therefore, based on the modification of compound 7, an oral Mpro inhibitor 9 (PF-07321332) was prepared (Figure 6(a)). 33
Figure 6.

(a) Chemical structures of the inhibitors 7, 8, and 9; (b) SARS-CoV-2 Mpro-bound crystal structure of 9; and (c) a reversible covalent Cys145 adduct is formed with the nitrile substituent in 9. 33
Compound 9 (also called Nirmatrelvir) is a pseudo-peptide covalent inhibitor with a nitrile warhead. It can directly bind to the cysteine catalytic residue of the SARS-CoV-2 Mpro protein, and selectively and reversibly inhibit SARS-CoV-2 Mpro activity (IC50 = 19.5 nM, EC50 = 74.5 nM, Ki = 3.11 nM).33,34 Compound 9 has similarities with the dimethylcyclopropylproline and tert-leucine groups of Boceprevir. The molecular simulation also shows that the interaction of inhibitor 9 is similar to that of Boceprevir, and the interaction of the pyrrolidone group with the H bond between Glu166 is similar to that of 8. The eutectic structure of 9 (PDB: 7RFW) shows that the P1′ nitrile group of the inhibitor and the mercaptan group of Cys145 form a reversible covalent thioimidate and form two hydrogen bonds with Cys145 and Gly143. 18 The bicyclic tetrahydropyrrole part and trifluoroacetyl group occupy the S2 and S4 pockets, respectively. The (S)-γ-lactam at the P1 site and the amide backbone at the P3–P4 site established multiple hydrogen bonds with His163, Glu166, and Gln189, respectively (Figure 6(b)). The antiviral activity in mice showed that 9 could effectively reduce the viral load of SARS-CoV-2 in the lungs of mice and slow down the progression of the disease. In clinical trials involving healthy human participants, oral plasma concentrations exceeded in vitro antiviral cell efficacy. 33 A pharmacokinetic property study showed that the CYP3A4 enzyme oxidatively metabolizes multiple sites of 9 (e.g. tert-butyl, azabicyclic, and pyrrolidone), resulting in rapid clearance.
Ritonavir is a human immunodeficiency virus type 1 (HIV-1) protease inhibitor, which has no activity against SARS-CoV-2 Mpro. However, ritonavir can bind to the CYP3A4 enzyme to inhibit the metabolism of 9. This increases the concentration of 9 in plasma and prevents its inactivation by liver metabolism. In December 2021, the US FDA approved compound 9 and the low-dose HIV antiviral drug ritonavir (CYP3A4 inhibitor) combined inhibitor PAXLOVID as the first oral antiviral drug for the treatment of COVID-19, which is intended to be administered in combination to treat and prevent COVID-19. 35
Meanwhile, Pfizer is also working on the second-generation COVID-19 oral administration of compound 10 (PF-07817883). A phase I clinical trial (NCT05580003) was released in clinical trials to evaluate the safety and blood levels of 10 in healthy people. Currently, it has entered the phase II clinical trial (NCT05799495) to understand the effect and safety of the study medicine of 10 in adults. The new drug still selects the Mpro target, but its oral absorption effect is better than Paxlovid. Though similar to Paxlovid, the drug does not need to be administered in combination with ritonavir, as Paxlovid currently is. Compound 10 no longer requires ritonavir to prolong blood concentration, which could reduce the risk of interactions with other drugs. Pfizer has not spoken extensively about the program, and it did not come up at the company’s J.P. Morgan presentation, though it was highlighted at a recent Pfizer investor event on the high-value pipeline of the company.
Sulfonates
Vuong et al. have described two types of peptide-like covalent inhibitors, namely parent 11 (GC-373; Figure 7(a)) and its prodrug 12 (GC-376; Figure 7(b)). These two drugs were previously used to treat feline infectious peritonitis (FIP) and showed good antiviral activity against the SARS-CoV-2 virus. The IC50 values are 0.40 and 0.19 μM, and the EC50 values are 1.5 and 0.92 μM, respectively.36,37 The crystal structures and NMR analyses of SARS-CoV2 Mpro using 11 and 12 showed that the prodrug 12 was converted into the parent drug 11, resulting in the covalent connection of the drug to Cys145 as a hemi-thioacetal, indicating that the bisulfite group had left 12. The glutamine surrogate in the P1 position of 11 forms a hydrogen bond with the side chain of His163 and Glu166, and a hydrophobic interaction with His172. The carbonyl in P3 forms a hydrogen bond with the backbone amide of Glu166 (Figure 7(c)). The (S)-γ-lactam ring of the P1 part of 12 forms a hydrogen bond with the His163 and Glu166 side chains and the main chain of Phe140, combined with the S1 site of Mpro. 38 The carbamate bond of 12 forms hydrogen bonds with the main chain of Glu166 and the side chain of Gln189, connecting the isobutyl of P2 to the phenylmethyl ester interacting with the aliphatic S4 site, and the carbonyl group in P3 forms a hydrogen bond with the backbone amide of Glu166, further stabilizing the binding of inhibitors (Figure 7(d)). Compared with the previous inhibitors, the phenylmethyl ester of 12 is highly complementary for the S4 site, and extensive non-polar interactions may significantly improve the effectiveness of this compound. Compound 12 forms an extensive hydrogen bond network with active sites, making 12 one of the most effective SARS-CoV-2 Mpro inhibitors in vitro. Compound 12 has been proven to have more potent inhibition efficiency than Boceprevir. 30 In cell cultures, 11 and 12 can block virus replication and are non-toxic, which makes them more advantageous than Boceprevir in clinical practice. However, the side effects demonstrated by 12 in the tests may limit its use.39,40
Figure 7.

Chemical structures of the inhibitors (a) 11 and (b) 12 and the interaction diagrams of (c) 11 and (d) 12 with the active site complex of Mpro.36,37
Michael acceptor
The team of Yang Haitao and Rao Zihe and that of Jiang Hualiang used computer-aided drug design to determine a potential inhibitor 13 (N3) (Figure 1(a)) for the main protease Mpro of SARS-CoV-2, 41 which contains a Michael acceptor as a warhead and can effectively fight against a variety of coronaviruses, including SARS-CoV and the Middle East respiratory syndrome coronavirus (MERS-CoV). 42 Compound 13 has antiviral effects in SARS-CoV-2-infected Vero cells (EC50 = 16.77 μM). The crystal structure of the complex of SARS-CoV-2 Mpro and inhibitor 13 was analyzed (PDB 6LU7). The β–C atom on the α,β-unsaturated ester of 13 forms a covalent bond (1.8 Å C–S distance) with Cys145 (Figure 8(b)), indicating that a Michael addition reaction has occurred, confirming that compound 13 is a Michael-type inhibitor. In addition, the lactam occupies the S1 pocket and forms a hydrogen bond with His163, and the isobutyl of the Leu side chain is inserted into the S2 pocket. The side chain of Val of P3 is exposed, indicating that these parts may not be the key to inhibiting the activity. The structure of these parts can be optimized to improve the efficacy and pharmacokinetic characteristics. The Ala side chain on the P4 side is inserted into the S4 pocket to form a small hydrophobic pocket together with the surrounding amino acids, which also helps to lock the inhibitor in the substrate-binding pocket.
Figure 8.

(a) The covalent inhibitor 13 containing the Michael acceptor and (b) its co-crystallization with Mpro. 41
Aldehydes
The Chinese Academy of Sciences and Shanghai University of Science and Technology jointly designed and synthesized two SARS-CoV-2 Mpro peptide covalent inhibitors 14 and 15 with an aldehyde group as the warhead. 11 Both compounds have good antiviral activity. The IC50 values measured by the fluorescence resonance energy transfer (FRET) method are 53 and 40 nM. In Vero-E6 cells, the EC50 values of SARS-CoV-2 are 0.53 and 0.72 μM, respectively. The eutectic structures of 14 and 15 with SARS-CoV-2 Mpro (Figure 9) show that the aldehyde group as an electrophilic group forms a covalent bond with the mercaptan group of Cys145. The (S)-γ-lactam ring and indole-2-formyl group occupy the S1 and S4 pockets, respectively, and establish multiple hydrogen bonds with His163 and Glu166. The cyclohexyl group of 14 and the 3-fluorophenyl group of 15 occupy the S2 pocket and form important hydrophobic interactions, in which the fluorine atom of the phenyl further establishes a hydrogen bond with Gln189. Due to its good pharmacokinetic characteristics and low toxicity, 14 has now entered clinical research (Clinical trial No. NCT04766931). Compound 14 was later transferred to Frontier Biotechnologies Inc. and named FB2001. It is administered through atomization inhalation and now has entered into phase II/III clinical trials (NCT05675072).
Figure 9.

Chemical structures of inhibitors (a) 14 and (b) 15; close-up view of (c) 14 and (d) 15 combined with Mpro pocket. 11
Qiao et al. 23 synthesized 32 new small molecular compounds containing bicyclic proline fragments through rational drug design, which can effectively inhibit the activity of SARS-CoV-2 Mproin vitro (IC50 7.6–748.5 nM). The eutectic structure of the most active compound 16 (MI-23, IC50 = 7.6 nM) and Mpro reveal the interaction mode (Figure 10(a) and (b)). The carbon of the warhead aldehyde interacts with the sulfur atom of the catalytic residue Cys145 to form a 1.8 Å covalent bond, and the oxygen of the aldehyde interacts with the main chain amide of Cys145 and Gly143 to form two hydrogen bonds. The (S)-γ-lactam ring of P1 is deeply inserted into the S1 pocket, forming two hydrogen bonds with the side chain of His163 (2.8 Å) and the main chain of Phe140 (3.3 Å). The rigid P2 bicyclic proline group points to a hydrophobic S2 pocket and forms hydrophobic interactions with Met165, Gln189, His 41 , Met 49 , Asp187, and Arg188. The main chain oxygen of P3 interacts with the main chain amide of Glu166 through a 2.9 Å hydrogen bond, and the 1-ethyl-3,5-difluorobenzene part assumes an extended conformation and occupies the S4 site, and forms a multiple hydrophobic interaction with the residue of the catalytic pocket. Two compounds 17 (MI-09) and 18 (MI-30) with high activity, good pharmacokinetic properties, and safety showed excellent antiviral activity in transgenic mice infected with SARS-CoV-2 (Figure 10(c) and (d)). Oral or intraperitoneal injection can significantly reduce the lung viral load and lung pathological damage, and has the potential to develop anti-SARS-CoV-2 drugs.
Figure 10.

(a) Chemical structure of 16; (b) interaction between Mpro and 16; chemical structures of (c) 17 and (d) 18.23.
Organic selenium compounds
Compound 19 (Ebselen) (Figure 11(a)) is an organoselenium molecule that can function as a glutathione peroxidase and peroxiredoxin mimic. 43 It has been shown to form a seleno-sulfide bond with thiol groups of cysteine (Cys) in a number of proteins which results in anti-inflammatory, antimicrobial, and neuroprotective effects. 44 Compound 19 has extremely low cytotoxicity (when administered orally; the median lethal dose of rats is more than 4600 mg kg−1), and its safety to humans has been evaluated in many clinical trials.45,46 Compound 19 was identified as a SARS-CoV-2 Mpro inhibitor in high-throughput screening with an IC50 value of 0.67 µM. 36 Amporndanai et al. 44 evaluated the inhibitory effect of Ebselen derivatives on SARS-CoV-2 Mpro, two of which showed a greater inhibitory effect on SARS-CoV-2 replication. From the eutectic study of Mpro-Ebselen and another effective compound (MR6-31-2), it was found that these compounds combine at Mpro-catalytic sites, form covalent bonds by providing selenium atoms, and block the His–Cys catalytic binary to inhibit Mpro-activity and virus replication (Figure 11(b)). Compound 19 and its derivatives have been proved to be effective in inhibiting papain-like protease (PLpro), which is another key protease in inhibiting SARS-CoV-2 replication. 47 This dual inhibition of the two key enzymes of virus replication may be the reason for the effective antiviral effect of Ebselen, which has an important guiding role in the development of virus double-target inhibitors.
Figure 11.
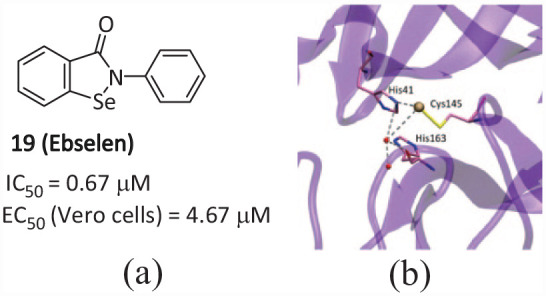
(a) Chemical structure of the inhibitor 19 and (b) details of the crystal structure of the selenium-Mpro complex. 48
Non-covalent inhibitors
Although the covalent bond formed by an electrophilic warhead in a covalent inhibitor and the amino acid residues in the active pocket of the target protein can help to prolong the action time of a drug in vivo, it usually has potential toxic side effects and targeting problems. Non-covalent inhibitors that competitively bind to target proteins through weak reversible binding can greatly reduce these risks. Therefore, compared with covalent compounds, non-covalent inhibitors may have a higher selectivity for Mpro.49,50
S-217622
Compound 20 (S-217622) (Figure 2(a)) is the first non-covalent, non-peptide SARS-CoV-2 Mpro inhibitor with oral activity (IC50 = 13 nM). In April 2022, Shionogi Pharmaceutical Research Center released part of the phase II/III clinical trial data of its oral SARS-CoV-2 Mpro inhibitor 20, which was used to treat COVID-19, at the ECCMID conference. 51 The data showed that it was well tolerated and effective against the Omicron strain. On 22 November 2022, Japan’s Shionogi Pharmaceutical announced that the emergency use authorization of 20 had been approved by the Japanese Pharmaceutical and Medical Devices Agency (PMDA). This approval was based on the clinical phase II/III test results of the drug and was used for COVID-19 infected people above 12 years old. The IC50 value of the biochemical activity of 20 is 0.013 μM. The antiviral activity EC50 value is 0.37 μM. Human and rat liver microsomes have high metabolic stabilities of 96% and 88%, a high oral absorption rate of 97%, and a low clearance rate.
Figure 12(b) shows the X-ray co-crystal structure (PDB: 7VU6) of Mpro complexed with 20. In the S1 site, the 1-methyl-1H-1,2,4-triazole unit fits into the S1 pocket, forming a hydrogen bond with the sidechain NH of His163. The distinctive His 41 flip was maintained in the complex, and the 2,4,5-trifluorobenzylic moiety occupied the hydrophobic S2 pocket and stacked with the side chain of His 41 . The P1′ ligand, 6-chloro-2-methyl-2H-indazole moiety, entered into hydrogen bonding with the Thr 26 mainchain NH and a hydrophobic contact with Met 49 . A variety of interactions and a high degree of fit with the catalytic pocket result in 20 having excellent inhibitory activity against Mpro. In addition, compound 20 showed a high level of inhibition against different virus strains (Alpha–Omicron), which dose-dependently inhibited the replication of SARS-CoV-2 in the lungs of mice. It can quickly reduce the viral load and shorten the time for viral titer to turn negative, with good tolerance. 52 Compared with 9, compound 20 is expected to remove the dependence on P450 enzyme inhibitors and achieve single-drug treatment of COVID-19.
Figure 12.

(a) Chemical structure of the inhibitor 20; (b) X-ray eutectic structure of 20 with Mpro. 51
MCULE-5948770040 and HL-3-68
Clyde of the National Virtual Biotechnology Laboratory (NVBL) of the United States and others discovered a new non-covalent small molecule inhibitor 21 (MCULE-5948770040) (Figure 13(a)) that acts on the main protease of SARS-CoV-2, via high-throughput virtual screening (HTVS) and a targeted compound library (IC50 = 0.68 µM, Ki = 2.9 µM). 53 By resolving its room-temperature X-ray crystal structure at 1.8 Å (Figure 13(c)), compound 21 was found to be bound to a cleft in the active site of Mpro. It occupies subsites S1 and S2, forming stable hydrogen bonds and hydrophobic interactions. The uracil P1 group of the ligand is located on the S1 substituent driven by polarity, with the dichlorobenzene substituent of P2 occupying most of the hydrophobic S2 substituent. Furthermore, the position of the P2-dichlorobenzene group is stabilized by π–π stacking interactions with Gln189 and the imidazole side chain of catalytic His 41 , with an interatomic distance of ∼3.8 Å. The binding of 21 to the Mpro active site crack led to the overturn of the His 41 side chain and the rotation of the χ2 angle, creating a favorable geometric structure for π–π stacking with P2-dichlorobenzene in the complex structure. Molecular dynamics simulations illustrate that the interaction between ligands and Mpro changes the conformation state of Mpro, and points toward a mechanism of complementary interaction and inter-domain movement, including the proximal and distal movement of binding sites.
Figure 13.

The chemical structures of (a) 21 and (b) 22; X-ray crystal structures of Mpro complexed with (c) 21 and (d) 22.54.
Kneller et al. 54 optimized the structure of 21 and obtained 22 (HL-3-68) (Figure 13(b)), which has a stronger inhibitory effect on SARS-CoV-2 Mpro than 21, with an IC50 value of 0.29 μM. Through structure–activity relationship (SAR) studies, it was found that 22 stabilized the main interaction by “locking” the S1 and S2 sites, thus greatly reducing the flexibility of the loop around the main binding sites. The eutectic of 22 and Mpro further shows that the S2 pocket is very sensitive to minor changes in ligand properties (Figure 13(d)). A chlorine atom is introduced at the fifth position of the phenyl group, which makes Met 49 rotate to accommodate additional chlorine atoms. It has a Van der Waals interaction with Cys 44 , which enhances the combination with Mpro. Molecular dynamics (MD) simulations of the Mpro-HL-3-68 complex shows that the ligand 22 can stably bind to Mpro, forming a stronger interaction than 21. SAR research analyzed the binding of non-covalent ligands and elucidated new details of Mpro as a drug target. This process provides a scalable framework for the further design of Mpro non-covalent inhibitors and the rapid discovery of feasible lead molecules targeting SARS-CoV-2.
Masitinib
Professor Savas Tay’s team at the University of Chicago screened a library of 1900 clinically safe drugs against OC43, a human beta coronavirus that causes the common cold, and evaluated the top hits against SARS-CoV-2. They discovered that oral bioavailable tyrosine kinase inhibitor 23 (Masitinib) (Figure 14(a)) can effectively inhibit the activity of SARS-CoV-2 Mpro (IC50 = 2.5 μM, IC50 = 2.5 μM). 55 When compound 23 was used to treat mice infected with SARS-CoV-2, the virus titer in the lung and nasal cavity decreased by more than 200 times, the lung inflammation was reduced, and it was also effective for all variant strains tested in vitro (including B.1.1.7, B.1.351, and P.1). The co-crystal of 23 and SARS-CoV-2 Mpro(Figure 14(b)) shows that 23 is non-covalently bound to the catalytic pocket between the domains I and II of Mpro and blocks the key catalytic residues of the two active sites in the dimer. The pyridine ring has hydrophobic and Van der Waals interactions with the residue around the pocket, and is inserted into the S1 pocket to form a hydrogen bond with His163. The aminothiazole ring forms two hydrogen bonds with His164 and the key residue Cys145. The hydrophobic toluene ring occupies the S2 pocket and forms an important π–π stacking interaction with His 41 . The N-methylpiperazine group is located outside the catalytic pocket, and the absence of 23 is observed in the S1′ and S4 pockets, indicating that structural optimization can be carried out in these places to further improve the inhibitory activity of Mpro while reducing the inhibitory activity of tyrosine kinase and reducing the related side effects. 42
Figure 14.

(a) Chemical structure of 23; (b) band diagram of 23 interacting with Mpro at the active site, with the two catalytic residues marked with red stars. 55
ML188 and 23R
Compound 24 (ML188) (Figure 15(a)) is a non-covalent inhibitor aimed at SARS-COV-1 Mpro (IC50 = 1.5 μM, EC50 = 12.9 μM). 56 Lockbaum et al. 57 described the characteristics of the complex of SARS-CoV-2 Mpro and non-covalent inhibitor 24, and solved the eutectic structure of the complex of 24 and SARS2-Mpro (Figure 15(c)) with a resolution of 2.39 Å. The interaction between protease and the inhibitor is through Van der Waals forces, and the SARS2-Mpro-ML188 force is −62.0 kcal mol−1. Compared with SARS1-Mpro, non-covalent inhibitor 24 has a stronger binding effect on SARS2-Mpro. The complex of these two proteases is very similar, but the slight difference may lead to the higher efficacy of 24 against SARS2-Mpro, which indicates that 24 may provide a scaffold for a powerful non-covalent pan-coronavirus inhibitor.
Figure 15.

The chemical structures of (a) 24 and (b) 25; co-crystal structure of Mpro in complex with (c) 24 and (d) 25.57,49
Kitamura et al. 49 discovered a non-covalent small molecule inhibitor 25 (23R) (Figure 15(b)) based on the co-crystallization of GC376, calpain inhibitors XII, and ML188 (R) with Mpro using structural drug design and an Ugi-4CR methodology. It has high selectivity for host proteases, effective enzymatic inhibition, and antiviral activity in vitro. The IC50 value of SARS-CoV-2 Mpro in Vero E6 cells is 0.20 μM and the EC50 value is 1.27 μM. The X-ray crystal structure of SARS-CoV-2 Mpro in complex with 25 (Figure 15(d)) revealed a ligand-induced binding pocket between the S2 and S4 pockets, in which the benzyl ring from the terminal α-methylbenzyl fits in a pocket. This conformation is strengthened by π-stacking interactions with the first phenyl group of the biphenyl substituent. The benzyl ring is inserted into this binding pocket, strengthening binding with Mpro. This new binding mode of 25 was characterized by continuous intramolecular π-stacking, in which the phenyl group was clamped by furanyl and benzyl groups, contributing to its potent inhibition of Mpro with a favorable mechanism of action and robust cellular antiviral activity.
CCF0058981
Han et al. 58 from Cleveland University in the United States conducted a series of structural optimizations on the Mpro inhibitor ML300 of SARS-CoV-1. The non-covalent small molecule inhibitor 26 (CCF0058981) (Figure 16(a)) (IC50 = 68 nM) of SARS-CoV-2 was ultimately determined, making significant progress compared to the original SARS-CoV-1 ML300-derived inhibitors. 59 Investigation of the 3-chlorophenyl P2sp analogue of 27 (Figure 16(b)) afforded 26. Substituting the 3-thienyl group of 27 with a 3-chlorophenyl moiety resulted of a significantly increased inhibitory effect. Compound 26 exhibited potent antiviral activity with EC50 values of 0.497 and 0.558 μM against SARS-CoV-2-infected Vero E6 cells in the cytopathic effect (CPE) inhibition assay and plaque reduction assay, respectively, being submicromolar in both assays, and thus achieving strong nanomolar biochemical inhibition of SARS-CoV-2 Mpro, submicromolar antiviral efficacy against SARS-CoV-2 live virus, and inhibition of plaque formation. The EC50/IC50 ratio of 26 is 7.4, which is a high ratio among current Mpro non-covalent inhibitors. Its curative effect is equivalent to that of the clinical RNA polymerase inhibitor Remdesivir. The optimization of this series of compounds is currently underway, focusing on improving the Drug Metabolism and Pharmacokinetics (DMPK) profile and strong inhibition of CYP enzymes, with the aim of further improving the biochemical and cellular efficacy, and providing a direction for optimizing the structure of future inhibitors.
Figure 16.

Chemical structures of (a) 26 and (b) 27; (c) binding diagram of 26 in the Mpro activity pocket. 58
WU-04 (Mprosevir)
Recently, a non-covalent small molecule SARS-CoV-2 Mpro inhibitor 28 (WU-04) (Figure 17(a)), with an IC50 value of 72 nM and an EC50 value of 12 nM against SARS-CoV-2 in A549-hACE2 cells was discovered by the research team at West Lake University through DNA-encoded Library (DEL) technology screening. 60 The co-crystalline display of 28 with Mpro (Figure 17(b)) shows that the isoquinoline rings are well inserted into the S1 pocket and form hydrogen bonds with His163. The carbonyl group at the 3-position of the isoquinoline ring and the methylcarbamoyl group form two hydrogen bonds with Asn142 and Glu166. The phenyl ring and Gln189 form an amino–π interaction, and the 4-Br of the phenyl ring extends into the S4 pocket and establishes a halogen bond with Thr190. The presence of a strongly electron-withdrawing 6-nitro group that reaches the S2 pocket is significant as it boosts the potency by strengthening both amino–π and the halogen bond interactions. Therefore, replacing the nitro group with other electron-withdrawing groups or hydrogen results in a decline or absence of inhibitory activity, which can be attributed to this key factor. The ability of 28 to inhibit SARS-CoV-2 replication was evaluated by the mouse model, and the efficacy of 28in vivo was verified. In vivo antiviral activity showed that 28 exhibited anti-SARS-CoV-2 activity similar to that of PF-07321332 in K18-hACE2 mice when the same dose was administered orally. At present, compound 28 has completed preclinical studies on pharmacokinetics, efficacy, pharmacology, and safety evaluation, and was officially approved by the Center for Drug Evaluation (NMPA) to enter clinical practice in China.
Figure 17.

Chemical structure of (a) 28; (b) interaction of 28 with Mpro. 60
Conclusion and Outlook
SARS-CoV-2 is still spreading and mutating around the world, damaging public health. The highly conserved main protease-targeting sequence has attracted more and more attention from researchers. In recent years, many types of SARS-CoV-2 Mpro inhibitors have been reported. The most common are covalent inhibitors that contain electrophilic warheads and take part in covalent binding with Cys145. Due to the existence of highly reactive electrophilic warheads, the covalent inhibitors have potential safety problems of targeting and the disadvantages of poor oral bioavailability. Non-covalent small molecule inhibitors show inhibitory activity mainly through a weak reversible interaction with the amino acid residues in the Mpro-catalytic pocket, but the lack of a strong interaction with the Mpro-catalytic pocket may lead to the need to extend the administration time or increase the dosage, which may lead to drug resistance. Therefore, non-covalent inhibitors that combine the characteristics of small molecules and the advantages of covalent inhibitor warheads may have a better potential for drug formation, but the currently reported inhibitors are few or have not entered the clinical trial stage.
Although many drugs have shown promising results in the fight against SARS-CoV-2, further validation is needed to determine whether these drugs pose risks due to the relatively short research periods. In addition, some examples have poor oral absorption effects, 61 high prices, and have side effects caused by drug–drug interactions. 62 As the clinical trials of ordinary COVID-19 patients did not meet expectations, 63 more clinical trials are needed to verify the safety and effectiveness of these drugs. Therefore, researchers still need to make continuous efforts to develop more powerful anti-SARS-CoV-2 inhibitors to cope with the emerging SARS-CoV-2 variants with immune escape function.
Since no known cysteine protease in the human body has the same cleavage site as Mpro and lacks its homologous protease, this indicates that in addition to Mpro-targeted inhibitors, it will be a promising direction to use protein degradation targeted chimerism (PROTAC) technology to develop degradation agents targeting the Mpro catalytic pocket as anti-coronavirus drugs. It is expected that in the near future, there will be more efficient Mpro inhibitors reasonably designed based on existing structures and using established technical means, and the development of new and powerful anti-coronavirus drugs will also benefit the discovery of other drug targets.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Jiangsu Province (Grant No. BK20211001), the Young and Middle-aged Academic Leaders of Jiangsu Province Youth and Blue Project (2021), and the National Research Project Incubation Fund of Chengxian College, Southeast University (Grant No. 2022NCF001). We are grateful to the High Performance Computing Center of Nanjing Tech University for supporting the computational resources.
ORCID iDs: Zhuxin She  https://orcid.org/0009-0000-5186-4202
https://orcid.org/0009-0000-5186-4202
References
- 1.Mhatre S, Srivastava T, Naik S, et al. Phytomedicine 2021; 85, 153286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perico L, Benigni A, Casiraghi F, et al. Nat Rev Nephrol 2021; 17: 46–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu RG, WU J, Bai X, et al. Chin J Virol 2020; 36: 927–935. [Google Scholar]
- 4.Wang XB, Wang A, Qu LL, et al. J Agric Food Chem 2020; 68: 14426–14437. [DOI] [PubMed] [Google Scholar]
- 5.Tregoning JS, Flight KE, Higham SL, et al. Nat Rev Immunol 2021; 21: 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao WH, Chen XR, Zheng CF, et al. Emerg Microbes Infec 2022; 11: 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keehner J, Horton LE, Binkin NJ, et al. New Engl J Med 2021; 385: 1330–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Fang C, Zhang Q, et al. Protein Cell 2022; 13: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu RJ, Zhao X, Li J, et al. Lancet 2020; 395: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunther S, Reinke PYA, Fernandez-Garcia Y, et al. Science 2021; 372: 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai WH, Zhang B, Jiang XM, et al. Science 2020; 368: 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramajayam R, Tan KP, Liang PH. Biochem Soc T 2011; 39: 1371–1375. [DOI] [PubMed] [Google Scholar]
- 13.Anand K, Ziebuhr J, Wadhwani P, et al. Science 2003; 300: 1763–1767. [DOI] [PubMed] [Google Scholar]
- 14.Hilgenfeld R. Febs J 2014; 281: 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang LL, Lin DZ, Sun XYY, et al. Science 2020; 368: 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoops K, Kikkert M, van den Worm SHE, et al. Plos Biol 2008; 6: 1957–1974. [Google Scholar]
- 17.Zhang LL, Lin DZ, Kusov Y, et al. J Med Chem 2020; 63: 4562–4578. [DOI] [PubMed] [Google Scholar]
- 18.Pillaiyar T, Manickam M, Namasivayam V, et al. J Med Chem 2016; 59: 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibrahim IM, Abdelmalek DH, Elshahat ME, et al. J Infection 2020; 80: 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia S, Liu MQ, Wang C, et al. Cell Res 2020; 30: 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li TT, Han XJ, Gu CJ, et al. Nat Commun 2021; 12: 1–11.33397941 [Google Scholar]
- 22.Yan RH, Zhang YY, Li YN, et al. Science 2020; 367: 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao JX, Li YS, Zeng R, et al. Science 2021; 371: 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang DK, Chen J, Deng LB, et al. Infect Genet Evol 2010; 10: 84–88. [DOI] [PubMed] [Google Scholar]
- 25.Goyal B, Goyal D. ACS Comb Sci 2020; 22: 297–305. [DOI] [PubMed] [Google Scholar]
- 26.Quan BX, Shuai HP, Xia AJ, et al. Nat Microbiol 2022; 7: 716–725. [DOI] [PubMed] [Google Scholar]
- 27.Tomei L, Failla C, Santolini E, et al. J Virol 1993; 67: 4017–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malcolm BA, Liu R, Lahser F, et al. Antimicrob Agents Ch 2006; 50: 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oerlemans R, Ruiz-Moreno AJ, Cong YY, et al. RSC Med Chem 2021; 12: 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu LF, Ye F, Feng Y, et al. Nat Commun 2020; 11: 4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman RL, Kania RS, Brothers MA, et al. J Med Chem 2020; 63: 12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boras B, Jones RM, Anson BJ, et al. Nat Commun 2021; 12: 6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Owen DR, Allerton CMN, Anderson AS, et al. Science 2021; 374: 1586–1593. [DOI] [PubMed] [Google Scholar]
- 34.Lamb YN. Drugs 2022; 82: 585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Catlin NR, Bowman CJ, Campion SN, et al. Reprod Toxicol 2022; 108: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma CL, Sacco MD, Hurst B, et al. Cell Res 2020; 30: 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vuong W, Khan MB, Fischer C, et al. Nat Commun 2020; 11, 5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He XY, Zhen HY, Wu PY, et al. Chin J Med Chem 2021; 31: 301–311. [Google Scholar]
- 39.Pedersen NC, Kim Y, Liu HW, et al. J Feline Med Surg 2018; 20: 378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim Y, Liu HW, Kankanamalage ACG, et al. Plos Pathogens 2016; 12: e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin ZM, Du XY, Xu YC, et al. Nature 2020; 582: 289–293. [DOI] [PubMed] [Google Scholar]
- 42.Chen RF, Gao YL, Liu H, et al. RSC Med Chem 2023; 14: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nogueira CW, Barbosa NV, Rocha JBT. Arch Toxicol 2021; 95: 1179–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amporndanai K, Meng XL, Shang WJ, et al. Nat Commun 2021; 12: 3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masaki C, Sharpley AL, Cooper CM, et al. Psychopharmacology 2016; 233: 2655–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kil J, Lobarinas E, Spankovich C, et al. Lancet 2017; 390: 969–979. [DOI] [PubMed] [Google Scholar]
- 47.Weglarz-Tomczak E, Tomczak JM, Talma M, et al. Sci Rep-UK 2021; 11: 3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agost-Beltran L, de la Hoz-Rodriguez S, Bou-Iserte L, et al. Molecules 2022; 27: 2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitamura N, Sacco MD, Ma CL, et al. J Med Chem 2022; 65: 2848–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang CH, Stone EA, Deshmukh M, et al. ACS Central Sci 2021; 7: 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Unoh Y, Uehara S, Nakahara K, et al. J Med Chem 2022; 65: 6499–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen G, Guo H, Zhang F. Chin J Pharm 2022; 53: 763–774. [Google Scholar]
- 53.Clyde A, Galanie S, Kneller DW, et al. J Chem Inf Model 2022; 62: 116–128. [DOI] [PubMed] [Google Scholar]
- 54.Kneller DW, Li H, Galanie S, et al. J Med Chem 2021; 64: 17366–17383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drayman N, DeMarco JK, Jones KA, et al. Science 2021; 373: 931–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacobs J, Grum-Tokars V, Zhou Y, et al. J Med Chem 2013; 56: 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lockbaum GJ, Reyes AC, Lee JM, et al. Viruses-Basel 2021; 13, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han SH, Goins CM, Arya T, et al. J Med Chem 2022; 65: 2880–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turlington M, Chun A, Tomar S, et al. Bioorg Med Chem Lett 2013; 23: 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou NK, Shuai L, Zhang LJ, et al. ACS Cent Sci 2023; 9: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sendi P, Razonable RR, Nelson SB, et al. Clin Microbiol Infec 2022; 28: 1230–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marzolini C, Kuritzkes DR, Marra F, et al. Ann Intern Med 2022; 175: 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang R, Mylonakis E. Ann Intern Med 2021; 174: JC17–JC17. [DOI] [PubMed] [Google Scholar]