

Abstract
Background:
The terminal complement C5 inhibitor eculizumab is approved in Japan for relapse prevention in aquaporin-4 antibody-positive (AQP4+) neuromyelitis optica spectrum disorder (NMOSD) and is undergoing mandatory post-marketing surveillance (PMS) of clinical use.
Objectives:
The objective of the study is to assess the real-world, long-term safety and effectiveness of eculizumab in Japanese patients with AQP4+ NMOSD.
Design:
Regulatory-mandated PMS analysis implemented as an all-case surveillance of all patients with AQP4+ NMOSD who have been treated with eculizumab in Japan since its approval in November 2019.
Methods:
This PMS interim analysis assessed the safety and effectiveness of eculizumab in Japanese patients with AQP4+ NMOSD from November 2019 to April 2022.
Results:
Of 147 patients treated with eculizumab who consented to publication, 71 had at least one case report form collected and locked at the interim analysis data cut-off, constituting the safety analysis set; three patients from PREVENT (NCT01892345) were excluded from the effectiveness analysis set. Twelve and 10 patients in the safety and effectiveness analysis sets discontinued, respectively. In the safety analysis set, 67/71 patients (94.4%) were female, mean illness duration was 6.8 [standard deviation (SD): 6.2] years, mean age at eculizumab initiation was 50.7 (SD: 13.3) years, and mean eculizumab treatment duration was 44.6 (SD: 23.7) weeks. At diagnosis of NMOSD, 34/71 patients (47.9%) and 35/71 patients (49.3%) in the safety analysis set had symptoms of optic neuritis and transverse myelitis, respectively. In the safety analysis set, 19/71 patients (26.8%) reported adverse events, 10/71 (14.1%) reported adverse drug reactions (ADRs), and 7/71 (9.9%) reported serious ADRs; no meningococcal infections were observed. In the effectiveness analysis set, 64/68 patients (94.1%) were female, mean disease duration was 6.9 (SD: 6.3) years, mean age at eculizumab initiation was 50.6 (SD: 13.2) years, and 27/68 (39.7%) were tested for C5 genetic polymorphism (all negative). In the 2 years before eculizumab, 51/68 patients (75.0%) experienced relapse. Relapse rate was 0.02/patient-year after eculizumab initiation versus 0.74/patient-year in the 2 years before eculizumab. Overall, 37/68 patients (54.4%) were prescribed immunosuppressants in the 6 months before and 19/40 (47.5%) in the 6–12 months after starting eculizumab treatment. The proportion of patients taking >10 mg/day of prednisolone decreased from 45.6% at 24–20 weeks before to 23.1% and 0% at 48–52 and 100–104 weeks after eculizumab, respectively.
Conclusion:
This article reports interim PMS data for Japanese patients and provides updated real-world evidence for the safety of eculizumab and its effectiveness at preventing relapses in patients with AQP4+ NMOSD. Safety and effectiveness results are consistent with those from PREVENT.
Keywords: aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder, eculizumab, post-marketing surveillance, real-world evidence, relapse prevention
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a disabling inflammatory disorder that predominantly affects the optic nerves and spinal cord. 1 It is characterized by debilitating and unpredictable relapses, and relapse-related tissue damage leads to the accumulation of substantial, and mostly irreversible, disability.1,2 Evidence suggests that the pathogenesis of NMOSD directly involves serum antibodies (immunoglobulin G [IgG]) targeting the water channel aquaporin-4 (AQP4), which is expressed mainly in astrocyte foot processes in the central nervous system. 3 Preclinical data indicate that anti-AQP4 IgG binding to the AQP4 water channel triggers the complement cascade, ultimately leading to uncontrolled terminal complement activation, inflammation, and membrane attack complex formation. This results in complement-mediated astrocyte injury and destruction, which promotes immune-mediated neuronal demyelination.4–6
The prevalence of NMOSD in Japan is estimated to be 3.42 per 100,000 people. 7 The prevalence of NMOSD in East Asian populations appears to be higher than in White populations. 8 More than 90% of patients with aquaporin-4 antibody-positive (AQP4+) NMOSD in Japan are treated using oral prednisolone. 9 Immunosuppressive therapies such as azathioprine and tacrolimus may be used, often in combination with prednisolone. Conversely, the anti-CD20 monoclonal antibody rituximab – which acts by depleting B cells – is rarely used for the treatment of patients with AQP4+ NMOSD in Japan.
The humanized monoclonal antibody eculizumab (Soliris®; Alexion Pharma GK, Tokyo, Japan) acts by inhibiting cleavage of the terminal complement protein C5 into C5a, which is pro-inflammatory, and C5b, which coordinates the formation of the membrane attack complex. 10 It is approved for the treatment of NMOSD in 41 countries and was approved in Japan in November 2019 for the prevention of relapses in adults with AQP4+ NMOSD. The drug was also approved in 2019 for the treatment of adults with AQP4+ NMOSD in the United States (June) and the European Union (August). Approval was based on data from a phase 3, randomized, double-blind, placebo-controlled, time-to-event trial (PREVENT; NCT01892345), in which eculizumab demonstrated a significant effect on the time to first adjudicated relapse compared with placebo (relative risk reduction: 94%; hazard ratio: 0.06; p < 0.001).11,12 Results of the PREVENT open-label extension (OLE; NCT02003144) revealed persistently low adjudicated annualized relapse rates for eculizumab versus the PREVENT placebo group [0.025 (95% confidence interval [CI]: 0.013–0.048) and 0.350 (95% CI: 0.199–0.616), respectively] after a median follow-up of 133 weeks. 13 With longer follow-up (192 weeks), 96% of patients were found to be free from adjudicated relapses, and more than 90% of patients receiving eculizumab monotherapy did not experience any worsening of disability. 10
The entry and inclusion criteria for PREVENT restricted the patient population by disease severity, prior treatment status, and ongoing immunosuppressive therapy use. This contrasts with clinical practice, in which the patient population is more diverse. Therefore, the efficacy and safety of eculizumab in a broader group of patients have not been fully established. The Japanese Pharmaceuticals and Medical Devices Agency (PMDA) requires the manufacturer to conduct mandatory post-marketing surveillance (PMS) of patients treated with any newly approved drug to understand the characteristics of the patients treated and to obtain additional safety and effectiveness data in clinical practice. This article reports the real-world safety and effectiveness data from the latest interim analysis of the PMS, for all patients in Japan diagnosed with AQP4+ NMOSD who were treated with eculizumab between November 2019 and April 2022. It provides the first robust real-world evidence of the safety and effectiveness of eculizumab in this indication, which should help to inform the treatment of patients with this rare disease in a clinical practice setting.
Methods
Study design
This regulatory-mandated PMS is being implemented as an all-case surveillance, based on the conditions of approval by Japanese regulatory authorities. As such, it includes all patients with AQP4+ NMOSD treated with eculizumab in Japan since its approval in November 2019. Patients are being enrolled over a planned 5-year period, from November 2019 to November 2024, and surveillance will continue for up to 7 years, from November 2019 to November 2026. In cases of treatment discontinuation before November 2026, surveillance will continue for 8 weeks after the last treatment dose. The results reported here are for the period from the date of eculizumab approval (November 2019) through the interim analysis data cut-off on 1 April 2022. The protocol for the PMS analysis was confirmed by the PMDA and the study is being conducted in accordance with Good Post-Marketing Study Practice ordinance. 14
Treatment
Patients received the approved dose and schedule of eculizumab (intravenous infusion), 900 mg once weekly for 4 weeks, followed by 1200 mg 1 week later, and 1200 mg every 2 weeks thereafter (fixed dose irrespective of body weight). 15 Patients who were receiving eculizumab during the OLE of the PREVENT study were switched to treatment with commercially available eculizumab as soon as possible following approval. Meningococcal vaccination is required ⩾2 weeks before the first dose of eculizumab.
Data collection
Data are extracted from the medical chart of each patient and documented in a case report form (CRF), collected at 26 weeks and 1 year, then yearly thereafter. Following CRF collection, CRFs are locked using the following process: collected CRFs are checked for any pharmacovigilance events; any pharmacovigilance events are re-examined (attending physician provides additional details); CRFs are re-collected with detailed information about any pharmacovigilance events; CRFs are re-checked; and CRFs are locked. Patients who received eculizumab as an investigational product before enrollment had relevant data collected retrospectively from the time of first administration.
Outcomes
The following data are being collected: patient demographics and disease characteristics at entry into the PMS, results of testing for the terminal complement protein C5 c.2654G>A polymorphism (which prevents C5 binding and inhibition by eculizumab and is associated with poor treatment response), duration of eculizumab therapy, number of doses received, reasons for eculizumab discontinuation (if applicable), and number and type of other NMOSD therapies [including oral steroids, immunosuppressive therapies, intravenous immunoglobulin (IVIg), plasma exchange, and biologics] received during the 6 months before, up to 6 months after, and at 6–12, 12–18, and 18–24 months after eculizumab initiation.
Adverse events and adverse drug reactions (ADRs) were classified according to the Japanese version of the Medical Dictionary for Regulatory Activities. 16 ADRs were defined as those adverse events for which a relationship to eculizumab treatment was not excluded in the opinion of the patient’s physician and was confirmed by the sponsor based on information provided by the physician. Serious ADRs were defined as ADRs that result in death, life-threatening events, hospitalization or prolonged hospitalization for treatment, persistent or significant disability or dysfunction, congenital anomalies, or any other important medical events. Diagnosis of relapse was at the attending physician’s discretion and physician-reported relapses were not subject to adjudication by an independent committee, as with PREVENT. Therefore, the way relapses were reported in this study reflected real-world clinical practice.
Data/analysis sets
All analyses were descriptive. Continuous variables are summarized descriptively using means, standard deviations (SDs), 95% CIs, medians, and ranges. Missing data were not imputed. Categorical variables are based on observed data and are presented as counts and percentages.
Results
Demographics and disease characteristics
In total, 147 patients were enrolled in this PMS study (Figure 1). All patients provided consent for publication. At the 1 April 2022 interim analysis data cut-off, CRFs had been collected and locked for 71 patients, all of whom were included in the safety analysis set; three patients were excluded from the effectiveness analysis set (n = 68) owing to prior participation in the phase 3 PREVENT study.
Figure 1.

Patient disposition.
CRF, case report form.
In the safety analysis set, 67/71 patients (94.4%) were female, and the mean duration of illness was 6.8 (SD: 6.2; range: 0.1–25.0) years (Table 1). The mean age at eculizumab initiation was 50.7 (SD: 13.3; range: 19–84) years. At diagnosis of NMOSD, half of patients in the safety analysis set had symptoms of optic neuritis (34/71; 47.9%) and half had symptoms of transverse myelitis (35/71; 49.3%).
Table 1.
Baseline demographics and disease characteristics (data for the safety analysis set, n = 71 and for the effectiveness analysis set, n = 68).
| Safety analysis set (n = 71) | Effectiveness analysis set (n = 68) | |
|---|---|---|
| Age, years, mean (SD) | 50.7 (13.3) | 50.6 (13.2) |
| Female, n (%) | 67 (94.4) | 64 (94.1) |
| Disease duration, years, mean (SD) | 6.8 (6.2) | 6.9 (6.3) |
| <2 years, n (%) | 20 (28.2) | 20 (29.4) |
| ⩾2 years, n (%) | 51 (71.8) | 48 (70.6) |
| Symptoms at NMOSD diagnosis, n (%) | ||
| Optic neuritis | 34 (47.9) | 32 (47.1) |
| Transverse myelitis | 35 (49.3) | 34 (50.0) |
| Brain stem symptoms | 15 (21.1) | 14 (20.6) |
| Cerebral symptoms | 5 (7.0) | 5 (7.4) |
| Other | 2 (2.8) | 2 (2.9) |
| Any relapse in the 2 years before eculizumab administration, n (%) | 51 (71.8) | 51 (75.0) |
| Relapse count in the 2 years before eculizumab administration, n (%) | ||
| 1 | 22 (43.1) | 22 (43.1) |
| 2 | 14 (27.5) | 14 (27.5) |
| 3 | 9 (17.6) | 9 (17.6) |
| ⩾4 | 6 (11.8) | 6 (11.8) |
| Terminal complement protein C5 genetic polymorphism, n (%) | ||
| Positive | 0 (0.0) | 0 (0.0) |
| Negative | 27 (38.0) | 27 (39.7) |
| Not measured | 42 (59.2) | 39 (57.4) |
| Blank | 0 (0.0) | 0 (0.0) |
| Meningococcal vaccination received, n (%) | 70 (98.6) | 67 (98.5) |
| Prior NMOSD treatment, n (%) | ||
| Steroids | 69 (97.2) | 66 (97.1) |
| Immunosuppressive therapy | 40 (56.3) | 40 (58.8) |
| Plasma exchange | 28 (39.4) | 27 (39.7) |
| Intravenous immunoglobulin | 5 (7.0) | 5 (7.4) |
| Biologics | 3 (4.2) | 3 (4.4) |
| Other | 17 (23.9) | 16 (23.5) |
NMOSD, neuromyelitis optica spectrum disorder; SD, standard deviation.
In the effectiveness analysis set, 64/68 patients (94.1%) were female, the mean disease duration was 6.9 (SD: 6.3; range: 0.1–25.0) years, the mean age at eculizumab initiation was 50.6 (SD: 13.2; range: 19–84) years, and 27/68 (39.7%) were tested for C5 genetic polymorphism (all negative; C5 genetic polymorphism testing was at the discretion of the attending physician) (Table 1).
Treatment with eculizumab
At the interim cut-off date, the mean duration of eculizumab treatment was 44.6 (SD: 23.7) weeks, up to a maximum of 104 weeks in the safety analysis set (Table 2). At the interim cut-off date, 59/71 patients (83.1%) were receiving eculizumab treatment, with 12 discontinuations. Reasons given for discontinuation were adverse events (n = 3; two pyrexia and one pulmonary hypertension), physician decision (n = 6), patient decision (n = 5), and other (n = 1).
Table 2.
Duration of eculizumab treatment and eculizumab treatment status (data for the safety analysis set, n = 71).
| Patients who received eculizumab, n (%) (Safety analysis set, n = 71) | |
|---|---|
| Overall eculizumab treatment duration, weeks | |
| Mean (SD) | 44.6 (23.7) |
| Median (range) | 52.1 (0.1–104.4) |
| Total eculizumab cumulative dose, mg | |
| Mean (SD) | 26,126 (13,393) |
| Median (range) | 31,114 (900–63,086) |
| Eculizumab treatment status, ongoing/discontinued, n (%) | |
| At week 26 | 62 (87.3)/9 (12.7) |
| At year 1 | 40 (56.3)/1 (1.4) |
| At year 2 | 4 (5.6)/2 (2.8) |
| At interim analysis data cut-off (1 April 2022) | 59 (83.1)/12 (16.9) |
| Reason for discontinuation | |
| Adverse event | 3 (4.2) |
| Death | 0 (0.0) |
| Lost to follow-up | 0 (0.0) |
| Physician decision | 6 (8.5) |
| Patient decision | 5 (7.0) |
| Relapse | 0 (0.0) |
| Symptomatic improvement | 0 (0.0) |
| Other | 1 (1.4) |
| Blank | 0 (0.0) |
SD, standard deviation.
Safety
Overall, the safety results among these Japanese patients (Table 3) were consistent with those from PREVENT and its OLE.11,13 In this PMS study, 31.69 adverse events per 100 patient-years (PY) were reported, compared with 183.50 adverse events per 100 PY in PREVENT and its OLE combined for eculizumab-treated patients. 13 Overall, 19/71 patients (26.8%) experienced an adverse event. Of the 29 different adverse or serious adverse events reported, no single event was reported in more than 5% of patients in the safety analysis population.
Table 3.
Summary of adverse events for all patients (data for the safety analysis set, n = 71).
| Patients who received eculizumab (safety analysis set, n = 71) | ||
|---|---|---|
| n (%) | n per 100 patient-years | |
| Any adverse event | 19 (26.8) | 31.69 |
| Any ADR | 10 (14.1) | 16.68 |
| Any serious adverse event | 11 (15.5) | 18.35 |
| Any serious ADR | 7 (9.9) | 11.68 |
| Relapse | 1 (1.4) | 1.67 |
| Relapse (ADR) | 0 (0.0) | 0.00 |
| Meningococcal infection | 0 (0.0) | 0.00 |
| Meningococcal infection (ADR) | 0 (0.0) | 0.00 |
| Nonmeningococcal infection | 8 (11.3) | 13.34 |
| Cellulitis | 1 (1.4) | 1.67 |
| Meningitis bacterial | 1 (1.4) | 1.67 |
| Meningitis herpes | 1 (1.4) | 1.67 |
| Pneumonia | 1 (1.4) | 1.67 |
| Pneumonia pneumococcal | 1 (1.4) | 1.67 |
| Pyelonephritis | 1 (1.4) | 1.67 |
| Urinary tract infection | 1 (1.4) | 1.67 |
| Viral infection | 1 (1.4) | 1.67 |
| Escherichia pyelonephritis | 1 (1.4) | 1.67 |
| Pyrexia | 2 (2.8) | 3.34 |
| Nonmeningococcal infection (ADR) | 4 (5.6) | 6.67 |
| Noninfection adverse event | 12 (16.9) | 20.02 |
| Citrobacter infection | 1 (1.4) | 1.67 |
| Device-related infection | 1 (1.4) | 1.67 |
| Abnormal sensation in eye | 1 (1.4) | 1.67 |
| Eyelid edema | 1 (1.4) | 1.67 |
| Hypertension | 1 (1.4) | 1.67 |
| Pulmonary hypertension | 1 (1.4) | 1.67 |
| Constipation | 1 (1.4) | 1.67 |
| Diarrhea | 1 (1.4) | 1.67 |
| Hepatic function abnormal | 1 (1.4) | 1.67 |
| Alopecia | 1 (1.4) | 1.67 |
| Erythema | 1 (1.4) | 1.67 |
| Myalgia | 1 (1.4) | 1.67 |
| Systemic lupus erythematosus | 1 (1.4) | 1.67 |
| Nephrolithiasis | 1 (1.4) | 1.67 |
| Renal impairment | 1 (1.4) | 1.67 |
| Condition aggravated | 1 (1.4) | 1.67 |
| Malaise | 1 (1.4) | 1.67 |
| Pyrexia | 1 (1.4) | 1.67 |
| C-reactive protein increased | 1 (1.4) | 1.67 |
| Noninfection adverse event (ADR) | 7 (9.9) | 11.68 |
| Infusion site reaction | 1 (1.4) | 1.67 |
| Death | 0 (0.0) | 0.00 |
ADR, adverse drug reaction.
Ten patients reported ADRs. These ADRs were five infections (cellulitis, bacterial meningitis, herpes meningitis, pneumonia, and a streptococcal catheter-related infection), two skin and subcutaneous tissue disorders (alopecia and erythema), two musculoskeletal and connective tissues disorders [myalgia and systemic lupus erythematosus (SLE)], two pyrexia cases, and a single case each of eyelid edema, pulmonary hypertension, diarrhea, renal impairment, and increased C-reactive protein.
There were 10 serious ADRs, experienced by seven patients: two cases of pyrexia and one case each of cellulitis, bacterial meningitis (blood culture: negative), herpes meningitis, pneumonia, a streptococcal catheter-related infection, pulmonary hypertension, SLE, and renal impairment. No meningococcal infections were reported during this PMS analysis up to the interim cut-off date. These results are consistent with those observed in PREVENT and its OLE. 13
No patients discontinued eculizumab treatment owing to death. However, one patient who discontinued owing to an adverse event (pulmonary hypertension) subsequently died. This death was recorded as the outcome of SLE and thrombotic microangiopathy. The SLE was recorded as occurring on the same day as the pulmonary hypertension and was also judged to be related to eculizumab use. The death occurred more than 7 months after the date of the final eculizumab dose.
Effectiveness: NMOSD relapses
In the effectiveness analysis set, 51/68 patients (75.0%) experienced at least one relapse in the 2 years before the start of eculizumab treatment. Of these, 29 experienced at least two relapses (Table 1). Among patients receiving eculizumab up to the interim cut-off date, one relapse was reported in the effectiveness analysis set, with a relapse rate of 0.02/PY (one relapse in 57.0 PY of treatment; Figure 2). This compared favorably with a relapse rate of 0.74/PY in the 2 years before treatment (101 relapses in 136.0 PY of treatment). The single relapse experienced during eculizumab treatment was transverse myelitis. The physician judged it to be nonserious, but the final classification of the relapse was serious. The patient who experienced the relapse continued eculizumab treatment. This patient had experienced one previous relapse in the 2 years before eculizumab treatment and had tested negative for the terminal complement protein C5 genetic polymorphism.
Figure 2.
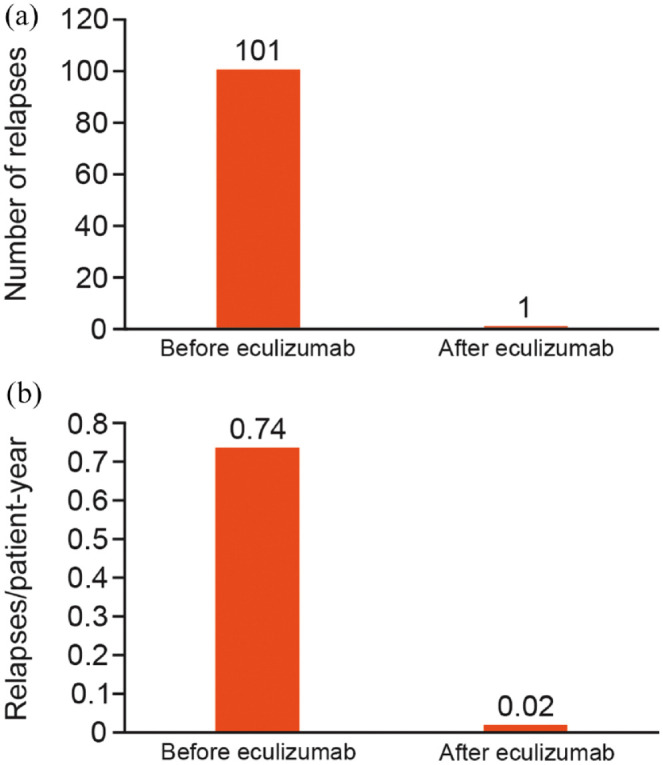
(a) Number of relapses and (b) relapse rates per patient-year before and after eculizumab (data for the effectiveness analysis set, n = 68).
Concomitant use of NMOSD medication
Changes in treatment with the oral steroid prednisolone and any concomitant immunosuppressive therapies were at the treating physician’s discretion. The mean daily dose of prednisolone decreased after the initiation of eculizumab, from 16.0 mg/day (SD: 11.1 mg/day) at 24–20 weeks before eculizumab to 8.6 mg/day (SD: 4.8 mg/day) at 48–52 weeks after eculizumab and 5.3 mg/day (SD: 1.0 mg/day) at 100–104 weeks after eculizumab (Figure 3(a)). This corresponds to a 67.1% decrease in the mean daily dose of prednisolone treatments over the study period. The proportion of patients taking a prednisolone dose >10 mg/day decreased from 45.6% at 24–20 weeks before eculizumab treatment to 23.1% at 48–52 weeks after initiation of eculizumab (Figure 3(b)). The overall proportion of patients prescribed concomitant prednisolone in the effectiveness analysis set was reduced from 67/68 (98.5%) within the 6 months before eculizumab treatment to 37/40 (92.5%) within the 6–12 months after eculizumab initiation. The patient who experienced relapse during eculizumab treatment received a decrease in concomitant prednisolone dose after eculizumab initiation, followed by an increase. The dose increase took place following the relapse.
Figure 3.

Use of the oral steroid prednisolone before and after initiation of eculizumab (data for the effectiveness analysis set, n = 68). (a) Average daily dose of prednisolone in 4-week ranges, in which crosses represent means, horizontal lines represent medians, colored boxes represent first and third quartiles, whiskers represent minimum and maximum values, and circles represent outliers (defined as a value that is larger than 1.5 times the first to third quartile range). (b) Percentage of patients treated with prednisolone by average daily dose in 4-week ranges.
The proportion of patients who were prescribed concomitant immunosuppressive therapies decreased slightly from 37/68 (54.4%) to 19/40 (47.5%) over the same period. Concomitant immunosuppressants included azathioprine, cyclosporine, cyclophosphamide, fingolimod, mycophenolate mofetil, and tacrolimus. At eculizumab initiation, 16 patients were taking azathioprine, three were taking cyclosporine, one was taking mycophenolate mofetil, 13 were taking tacrolimus, and no patients were taking cyclophosphamide or fingolimod. No patients were taking multiple immunosuppressants at eculizumab initiation. Doses of concomitant immunosuppressive therapies were not increased in any patients.
Intravenous methylprednisolone (IVMP) treatment was required in 38/68 patients (55.9%) in the 6 months before eculizumab; 17 of these patients required IVMP therapy once, while 21 required it twice or more. Conversely, IVMP treatment was required in only 2/68 patients (2.9%) in the 6 months after eculizumab initiation. The patient who experienced relapse during eculizumab treatment did not receive IVMP treatment. The mean dose of IVMP was 985 mg/day in the 6 months before eculizumab initiation and 1000 mg/day in the 6 months after. In the 6 months before eculizumab initiation, most patients received 1000 mg/day; one patient received 1000 mg/day and 500 mg/day alternately for each cycle. The standard dose of IVMP is 1000 mg/day.
IVIg treatment was required in 5/68 patients (7.4%) and plasma exchange was required in 15/68 patients (22.1%) in the 6 months before eculizumab administration. In the 6 months after eculizumab initiation, no usage of IVIg treatment was reported and only one of the 68 patients (1.5%) required plasma exchange. No usage of either treatment was reported 6 months or later after eculizumab initiation. The patient who experienced relapse during eculizumab treatment did not receive plasma exchange.
Discussion
The results of this interim PMS analysis of Japanese patients provide an update to the current real-world evidence for the long-term safety and effectiveness of eculizumab as a treatment for patients with AQP4+ NMOSD. In these patients, eculizumab was well tolerated and no cases of meningococcal infections were reported among the treated cohort by the interim analysis data cut-off on 1 April 2022. Menactra® (Sanofi Pasteur Inc., Swiftwater, PA, 18370, USA) was used as the meningococcal vaccine for most patients; this vaccine is covered by medical insurance in Japan and is effective against meningococcal serotypes A, C, Y, and W-135. No vaccine against meningococcal serotype B is approved for use in Japan.
By the interim data cut-off, one relapse (transverse myelitis) had been reported. The relapse rate was 0.02 relapses/PY, compared with a rate of 0.74 relapses/PY in the 2 years before initiating eculizumab. No formal definition of relapse was used, unlike in PREVENT, in which relapses were adjudicated by an independent committee. These findings are consistent with the safety and efficacy results of eculizumab in PREVENT and its OLE, including the evidence for eculizumab as monotherapy.10,11,13 However, it should be noted that the number of Japanese patients who received eculizumab in previous eculizumab clinical studies was limited. Among patients receiving eculizumab in PREVENT, nine patients were Japanese, 36.5% (n = 35/96) identified as being from the Asia-Pacific region (Australia, Hong Kong, Japan, South Korea, Malaysia, Taiwan, and Thailand), and 38.5% (n = 37/96) identified as being of Asian race.11,17 Pharmacokinetic and pharmacodynamic studies involving patients from PREVENT showed no differences between Japanese and non-Japanese patients. 18 Adverse events were lower in this PMS analysis than in PREVENT and its OLE, possibly due to under-reporting in the PMS compared with the clinical trial.
A heterozygous variation in the terminal complement protein C5 (c.2654G>A) prevents C5 binding and inhibition by eculizumab and is associated with a poor treatment response. This C5 variant was first identified in Japanese patients with paroxysmal nocturnal hemoglobinuria. The overall prevalence of this C5 variant in patients with NMOSD is unknown, but low rates of occurrence have been reported in healthy Japanese (3.5%) and Chinese (0.8%) populations. 19 In the present study, none of the 27 patients who were tested for the C5 genetic polymorphism harbored the genetic variation.
The reduction in the use of prescribed concomitant NMOSD treatments observed in this PMS study suggests that patients with NMOSD at risk of adverse events or intolerant to immunosuppressive therapies may benefit from a reduction in dose of concomitant oral steroids and may experience a decrease in treatment burden. A recent subanalysis of PREVENT and its OLE revealed that 96% of patients receiving eculizumab monotherapy were free from adjudicated relapses after approximately 4 years, with no worsening of disability during PREVENT. 10 Given the high clinical and quality of life burden of NMOSD, this requires further exploration.
Steroids are frequently used to help manage NMOSD, particularly in Japan, where oral prednisolone is the most commonly used drug to treat NMOSD (use in 92.6% of cases, compared with 15.8% in Germany). 9 Their continued use is despite limited clinical trial evidence and relatively poor rates of relapse prevention. 20 As shown in this PMS study, treatment with eculizumab can reduce or abolish the need for steroid use in a number of patients. This is in line with data from the PREVENT OLE, in which over a third of eculizumab-treated patients (44/119; 37.0%) were able to stop or decrease the use of background immunosuppressive therapy. 13 IVMP treatment and plasma exchange, both of which are used to manage acute-phase NMOSD,21–23 could be avoided by most patients in this PMS study.
In Japan, eculizumab is currently indicated for the prevention of relapses in NMOSD in adult patients with AQP4+ NMOSD. 15 In a retrospective cohort study, patients with NMOSD who had a relapse incurred more outpatient hospital encounters, longer lengths of stay, and greater mean annualized costs compared with patients who did not relapse. 24 Therefore, the use of eculizumab within this indication offers both health and economic benefits owing to its favorable benefit–risk profile, its potential to improve outcomes, and its potential to lower healthcare resource use by reducing the rate of relapses. In addition, in the present study, concomitant immunosuppresive therapy and high-dose corticosteroid use decreased following eculizumab treatment. In a post hoc analysis of PREVENT and its OLE, eculizumab monotherapy (with no concomitant immunosuppressive therapy) was associated with a lower rate of serious infections than placebo. 10 Therefore, eculizumab use may further decrease healthcare resource utilization by reducing the rate of potential adverse events associated with immunosuppressive therapy use.
Study limitations
This regulatory-mandated all-case surveillance PMS study is an observational study and is therefore subject to certain limitations inherent to any observational study, such as potential reporting bias, lack of a control group (only patients with AQP4+ NMOSD who were treated with eculizumab were assessed), data originating from daily medical practice (data were extracted from patient medical charts and relapses and adverse events were defined by the attending physician), and some missing data (missing data were not imputed). Given that this was an interim analysis of PMS data, the duration of observation was relatively short, but this will increase as the PMS data collection continues.
Conclusion
This interim analysis of a PMS study in Japan offers the first demonstration in a real-world setting that eculizumab is effective in preventing relapses in patients with AQP4+ NMOSD. Treatment with eculizumab was also associated with a reduction in the concomitant use of immunosuppressive therapies. These findings in Japanese patients are consistent with the efficacy and safety results from the global phase 3 PREVENT study of eculizumab versus placebo in AQP4+ NMOSD and its OLE, in which Japanese participants were low in number.10,11 Further data will become available over the course of the 7-year surveillance period to help understand the long-term impact of eculizumab in Japanese patients with AQP4+ NMOSD. In the meantime, these interim results provide important real-world evidence about the safety and effectiveness of eculizumab.
Acknowledgments
Medical writing support was provided by Corin Wing, PhD, and Caitlin Edgell, PhD, of Oxford PharmaGenesis, Oxford, UK, and was funded by Alexion Pharma GK, AstraZeneca Rare Disease.
Footnotes
ORCID iD: Ichiro Nakashima  https://orcid.org/0000-0002-2612-8948
https://orcid.org/0000-0002-2612-8948
Contributor Information
Ichiro Nakashima, Division of Neurology, Tohoku Medical and Pharmaceutical University, 1-12-1 Fukumuro, Miyagino-ku, Sendai, Miyagi 983-8512, Japan.
Jin Nakahara, Department of Neurology, Keio University School of Medicine, Tokyo, Japan.
Hiroaki Yokote, Department of Neurology, Nitobe Memorial Nakano General Hospital, Tokyo, Japan.
Yasuhiro Manabe, Department of Neurology, National Hospital Organization Okayama Medical Center, Okayama, Japan.
Kazumi Okamura, Alexion Pharma GK, Tokyo, Japan.
Kou Hasegawa, Alexion Pharma GK, Tokyo, Japan.
Kazuo Fujihara, Department of Multiple Sclerosis Therapeutics, Fukushima Medical University School of Medicine, Fukushima, Japan; Multiple Sclerosis and Neuromyelitis Optica Center, Southern TOHOKU Research Institute for Neuroscience, Koriyama, Japan.
Declarations
Ethics approval and consent to participate: Ethics approval was not required for this post-marketing surveillance analysis.
Consent for publication: All patients included in this post-marketing surveillance analysis provided written consent for publication.
Author contributions: Ichiro Nakashima: Conceptualization; Writing – original draft; Writing – review & editing.
Jin Nakahara: Conceptualization; Writing – original draft; Writing – review & editing.
Hiroaki Yokote: Conceptualization; Writing – original draft; Writing – review & editing.
Yasuhiro Manabe: Conceptualization; Writing – original draft; Writing – review & editing.
Kazumi Okamura: Conceptualization; Writing – original draft; Writing – review & editing.
Kou Hasegawa: Conceptualization; Writing – original draft; Writing – review & editing.
Kazuo Fujihara: Conceptualization; Writing – original draft; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research reported, the development of this manuscript, and the open access publishing fee were funded by Alexion Pharma GK, AstraZeneca Rare Disease, which is the manufacturer of eculizumab. Data were collected by the investigators, analyzed by statisticians employed by the funder, and interpreted by the authors (both investigators and employees of the funder).
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Ichiro Nakashima reports personal fees from Alexion Pharma GK, Chugai, Biogen Japan, Mitsubishi Tanabe Pharma, and Novartis, and grants from LSI Medience, the Ministry of Education, Science and Technology of Japan, and the Ministry of Health, Labor and Welfare of Japan (MHLW).
Jin Nakahara reports personal fees from AbbVie, Alexion Pharma GK, Asahi Kasei Medical, Biogen, Bristol Myers Squibb, Chugai, CSL Behring, Daiichi Sankyo, Eisai, Kyorin, Mitsubishi Tanabe Pharma, Novartis, Otsuka, Roche, Takeda, and Teijin Pharma; research scholarships from AbbVie, Boehringer Ingelheim, Chugai, Daiichi Sankyo, EA Pharma, Eisai, JB, Mitsubishi Tanabe Pharma, Otsuka, Shionogi, Sumitomo Pharma, Teijin Pharma, and Tsumura; and grants from the Ministry of Education, Science and Technology of Japan, the MHLW, and Biogen.
Hiroaki Yokote reports personal fees from Biogen Japan, Mitsubishi Tanabe Pharma, Novartis, Chugai Pharma, and Alexion Pharma GK; and grants from the MHLW.
Yasuhiro Manabe declared no potential conflicts of interest with respect to this work.
Kazumi Okamura and Kou Hasegawa are employees of, and hold stock in, Alexion Pharma GK, AstraZeneca Rare Disease.
Kazuo Fujihara has received personal fees and other support from AbbVie, Asahi Kasei Medical, Biogen, Chugai, Eisai, Merck Group, Mitsubishi Tanabe Pharma, Novartis, Ono, Roche, Sumitomo Dainippon, Takeda, Teijin Pharma, UCB, and Viela Bio (formerly MedImmune); and grants from the Ministry of Education, Science and Technology of Japan and the MHLW.
Availability of data and materials: Not applicable.
References
- 1.Wingerchuk DM, Hogancamp WF, O’Brien PC, et al. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999; 53: 1107–1114. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6: 805–815. [DOI] [PubMed] [Google Scholar]
- 3.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012; 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujihara K, Misu T, Nakashima I, et al. Neuromyelitis optica should be classified as an astrocytopathic disease rather than a demyelinating disease. Clin Exp Neuroimmunol 2012; 3: 58–73. [Google Scholar]
- 5.Fujihara K.Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol 2019; 32: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutra BG, Da Rocha AJ, Nunes RH, et al. Neuromyelitis optica spectrum disorders: spectrum of MR imaging findings and their differential diagnosis. Radiographics 2018; 38: 169–193. [DOI] [PubMed] [Google Scholar]
- 7.Jarius S, Paul F, Weinshenker BG, et al. Neuromyelitis optica. Nat Rev Dis Primers 2020; 6: 85. [DOI] [PubMed] [Google Scholar]
- 8.Hor JY, Asgari N, Nakashima I, et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol 2020; 11: 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asseyer S, Masuda H, Mori M, et al. AQP4-IgG autoimmunity in Japan and Germany: differences in clinical profiles and prognosis in seropositive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin 2021; 7: 20552173211006862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pittock SJ, Fujihara K, Palace J, et al. Eculizumab monotherapy for NMOSD: data from PREVENT and its open-label extension. Mult Scler 2022; 28: 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 2019; 381: 614–625. [DOI] [PubMed] [Google Scholar]
- 12.Alexion Europe SAS. Summary of product characteristics, Soliris (eculizumab), https://www.ema.europa.eu/en/medicines/human/EPAR/soliris (accessed 17 August 2022).
- 13.Wingerchuk DM, Fujihara K, Palace J, et al. Long-term safety and efficacy of eculizumab in aquaporin-4 IgG-positive NMOSD. Ann Neurol 2021; 89: 1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ministry of Health Labour Welfare. MHLW ordinance related to standards for conducting post-marketing surveys and studies on drugs (MHLW ordinance no. 171) [in Japanese], https://www.pmda.go.jp/files/000161574.pdf (accessed 5 October 2022).
- 15.Soliris for intravenous infusion 300 and mg (package insert) [in Japanese]. Tokyo, Japan: Alexion Pharma GK, 2022. [Google Scholar]
- 16.Brown EG, Wood L, Wood S.The medical dictionary for regulatory activities (MedDRA). Drug Saf 1999; 20: 109–117. [DOI] [PubMed] [Google Scholar]
- 17.Pharmaceuticals Medical Devices Agency. Soliris intravenous infusion 300 mg review report [in Japanese], https://www.pmda.go.jp/drugs/2019/P20191205001/870056000_22200AMX00316_A100_1.pdf (accessed 1 September 2022).
- 18.Singh P, Gao X, Kleijn HJ, et al. Eculizumab pharmacokinetics and pharmacodynamics in patients with neuromyelitis optica spectrum disorder. Front Neurol 2021; 12: 696387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishimura J, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med 2014; 370: 632–639. [DOI] [PubMed] [Google Scholar]
- 20.Takai Y, Kuroda H, Misu T, et al. Optimal management of neuromyelitis optica spectrum disorder with aquaporin-4 antibody by oral prednisolone maintenance therapy. Mult Scler Relat Disord 2021; 49: 102750. [DOI] [PubMed] [Google Scholar]
- 21.Kosiyakul P, Songwisit S, Ungprasert P, et al. Effect of plasma exchange in neuromyelitis optica spectrum disorder: a systematic review and meta-analysis. Ann Clin Transl Neurol 2020; 7: 2094–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 2016; 79: 206–216. [DOI] [PubMed] [Google Scholar]
- 23.Kleiter I, Gahlen A, Borisow N, et al. Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 2018; 5: e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foley D, Williams T, Polson M.Costs of illness associated with relapses in neuromyelitis optica spectrum disorder: an administrative claims database analysis. J Manag Care Spec Pharm 2022; 28: S45. [Google Scholar]