

Key Points
Question
Do abatacept, cenicriviroc, or infliximab improve time to recovery for patients hospitalized with COVID-19 pneumonia compared with standard care?
Findings
A randomized, double-masked, placebo-controlled master protocol clinical trial found that treatment with abatacept, cenicriviroc, or infliximab showed no statistically significant difference for the primary end point of time to recovery in patients with COVID-19 pneumonia vs standard care.
Meaning
Abatacept, cenicriviroc, or infliximab did not decrease time to recovery for hospitalized patients with COVID-19 pneumonia.
Abstract
Importance
Immune dysregulation contributes to poorer outcomes in COVID-19.
Objective
To investigate whether abatacept, cenicriviroc, or infliximab provides benefit when added to standard care for COVID-19 pneumonia.
Design, Setting, and Participants
Randomized, double-masked, placebo-controlled clinical trial using a master protocol to investigate immunomodulators added to standard care for treatment of participants hospitalized with COVID-19 pneumonia. The results of 3 substudies are reported from 95 hospitals at 85 clinical research sites in the US and Latin America. Hospitalized patients 18 years or older with confirmed SARS-CoV-2 infection within 14 days and evidence of pulmonary involvement underwent randomization between October 2020 and December 2021.
Interventions
Single infusion of abatacept (10 mg/kg; maximum dose, 1000 mg) or infliximab (5 mg/kg) or a 28-day oral course of cenicriviroc (300-mg loading dose followed by 150 mg twice per day).
Main Outcomes and Measures
The primary outcome was time to recovery by day 28 evaluated using an 8-point ordinal scale (higher scores indicate better health). Recovery was defined as the first day the participant scored at least 6 on the ordinal scale.
Results
Of the 1971 participants randomized across the 3 substudies, the mean (SD) age was 54.8 (14.6) years and 1218 (61.8%) were men. The primary end point of time to recovery from COVID-19 pneumonia was not significantly different for abatacept (recovery rate ratio [RRR], 1.12 [95% CI, 0.98-1.28]; P = .09), cenicriviroc (RRR, 1.01 [95% CI, 0.86-1.18]; P = .94), or infliximab (RRR, 1.12 [95% CI, 0.99-1.28]; P = .08) compared with placebo. All-cause 28-day mortality was 11.0% for abatacept vs 15.1% for placebo (odds ratio [OR], 0.62 [95% CI, 0.41-0.94]), 13.8% for cenicriviroc vs 11.9% for placebo (OR, 1.18 [95% CI 0.72-1.94]), and 10.1% for infliximab vs 14.5% for placebo (OR, 0.59 [95% CI, 0.39-0.90]). Safety outcomes were comparable between active treatment and placebo, including secondary infections, in all 3 substudies.
Conclusions and Relevance
Time to recovery from COVID-19 pneumonia among hospitalized participants was not significantly different for abatacept, cenicriviroc, or infliximab vs placebo.
Trial Registration
ClinicalTrials.gov Identifier: NCT04593940
This study reports the results of 3 randomized, double-masked, placebo-controlled substudies of abatacept, cenicriviroc, and infliximab plus standard care for COVID-19 pneumonia.
Introduction
The immunopathology of COVID-19 involves aberrant immune cell function and dysregulation of cytokine and chemokine networks.1,2,3 The RECOVERY trial demonstrated a mortality benefit for dexamethasone for hospitalized participants receiving supplemental oxygen, making it a mainstay of anti-inflammatory treatment for COVID-19.4 Despite therapeutic advances, considerable morbidity and mortality remain, spurring continued evaluation of therapeutic approaches. Medications disrupting IL-6 signaling (tocilizumab) and the Janus kinase pathway (baricitinib) show benefit in the most severely ill patients with COVID-19.5,6,7,8,9 In April 2020, the US National Institutes of Health (NIH) launched a public-private partnership, Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV), to develop a coordinated research response to COVID-19. The ACTIV-1 Immune Modulator (IM) master protocol evaluated immunomodulatory agents in hospitalized participants with moderate/severe COVID-19. Three agents were selected for study based on their novel mechanisms of action and in vitro data against SARS-CoV-2. These included abatacept (T-cell costimulatory modulator that mitigates T-cell responses), cenicriviroc (CCR2/CCR5 antagonist that reduces monocyte and macrophage functions while sparing neutrophil and T-cell function), and infliximab (tumor necrosis factor α [TNF] inhibitor that binds and neutralizes soluble and transmembrane TNF).3,10 Considerable safety data exist for abatacept and infliximab for other inflammatory disorders. This study reports the results of 3 randomized, double-masked, placebo-controlled substudies of abatacept, cenicriviroc, and infliximab plus standard care for COVID-19 pneumonia.
Methods
Study Design
ACTIV-1 IM is a master protocol designed to evaluate multiple immune modulators plus standard care in participants hospitalized with COVID-19 through randomized, double-masked comparisons with placebo.11 Data from participants receiving placebo were shared among the comparisons to minimize the number of participants receiving placebo, thereby reducing the sample size needed for adequate power for each comparison. The protocol and statistical analysis plan are available in Supplement 1.
Eligibility
The protocol was approved by relevant review boards; all participants provided written informed consent. Participants 18 years or older with confirmed SARS-CoV-2 infection within 14 days, anticipated hospital stay of 72 hours or more, and evidence of pulmonary involvement were eligible. Candidates with pregnancy, liver enzymes more than 10 times the normal value, chronic liver disease, acute kidney injury with glomerular filtration rate less than 30 mL/min (stable chronic kidney insufficiency permitted), severe heart failure, severe neutropenia, lymphopenia, or known or suspected untreated infection including tuberculosis and those who received cytotoxic or biologic-targeted immunomodulators within 4 weeks or 5 half-lives before screening were excluded. Substudy-specific exclusion criteria are outlined in Supplement 1.
Procedures
Demographics and clinical status were extracted from the medical records when available or verified with participants. This included race and ethnicity, which were collected to allow for assessment of study participant representativeness. Study drug or placebo was administered according to substudy randomization. Abatacept (10 mg/kg; maximum dose, 1000 mg) and infliximab (5 mg/kg) were given as a single infusion; cenicriviroc (300-mg loading dose followed by 150 mg twice daily) was an oral medication administered for 28 days (Supplement 1). Participants received standard care at site hospitals, including remdesivir (study provided [Gilead Sciences]) and corticosteroids. Clinical status and safety data were captured daily during hospitalization through day 28 and postdischarge days 8, 11, 15, 29, and 60.
Eligible participants were randomized in a 2-stage process (Figure 1). First, each participant was assigned with equal probability to receive 1 of the immunomodulatory agents (substudy) using an open-label design. Second, each participant was randomized in a masked fashion to the test agent or its matching placebo in an n:1 ratio, where n equals the number of agents for which the participant was eligible.
Figure 1. Flow of Participants in a Master Protocol Assessing Use of Abatacept, Cenicriviroc, and Infliximab to Treat COVID-19 Pneumonia.
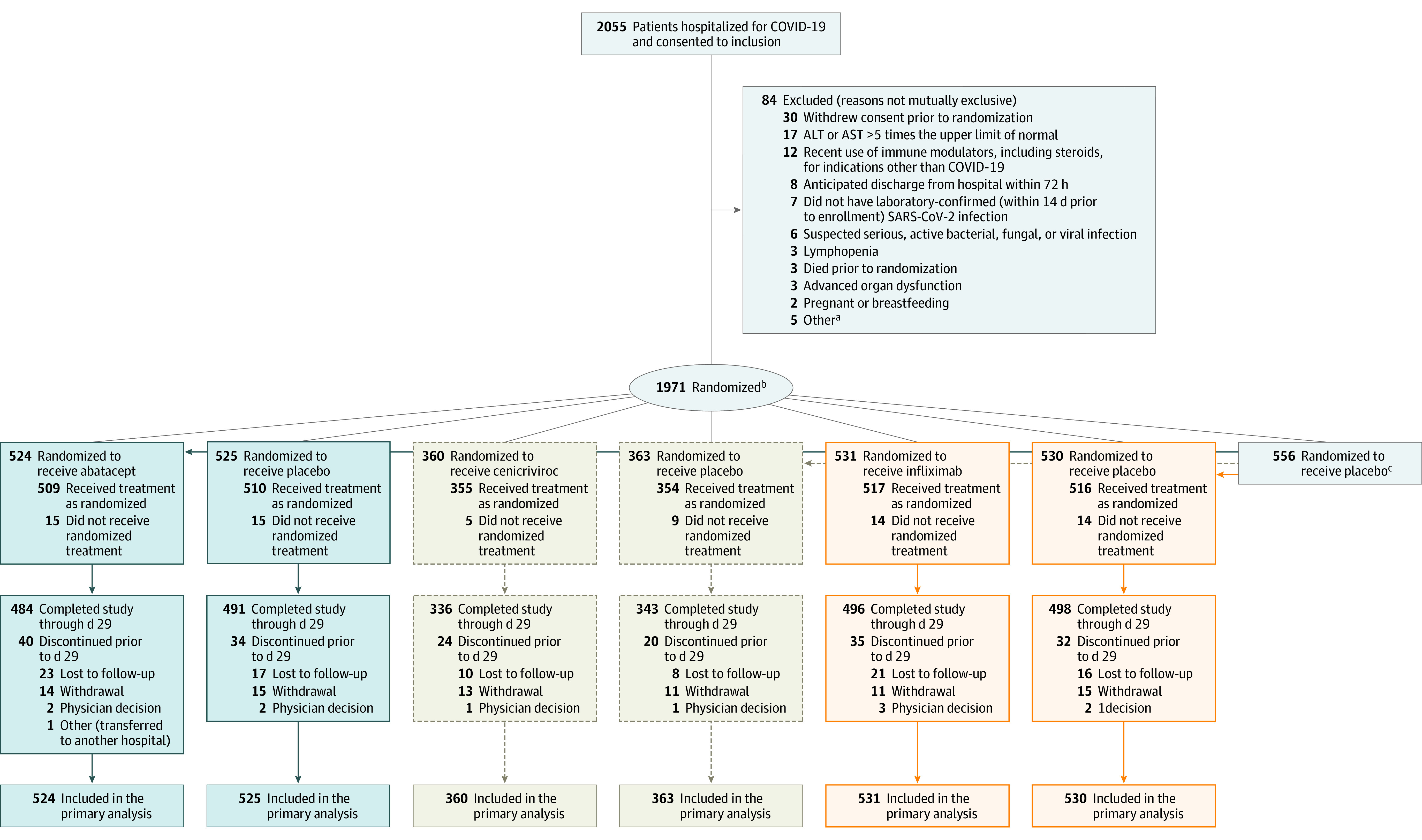
aOne participant with estimated glomerular filtration rate <30 mL/min, known or suspected history of untreated tuberculosis, male or pregnant female <18 years at enrollment, neutropenia, no ongoing illness and radiographic infiltrates by imaging, oxygen saturation as measured by pulse oximetry ≤ 94% on room air, requiring supplemental oxygen, or requiring mechanical ventilation/extracorporeal membrane oxygenation.
bEach participant was randomized (open label) with equal probability to one of the agents available at the time of enrollment after applying agent-specific safety exclusions. Then each participant was assigned in a masked manner to the test agent or its matching placebo in an n:1 ratio, where n equals the number of agents for which that the participant was eligible. Randomization was stratified by geographic location and disease severity.
cOverall, 346 participants were eligible to receive all 3 interventions, 156 were eligible to receive abatacept and infliximab, 10 were eligible for cenicriviroc and infliximab, 4 were eligible for abatacept and cenicriviroc, 19 were eligible for abatacept, 3 were eligible for cenicriviroc, and 18 were eligible for only infliximab.
Data from participants in the placebo group were shared for analysis of each agent once agent-specific eligibility criteria were applied (Supplement 1). Several elements were key to preserving the statistical integrity of shared placebos.11 To avoid introduction of bias related to evolution of disease or standard care over time, the shared placebo group for each substudy analysis only included participants enrolled at a site when that substudy was available at the time of enrollment. The shared placebo group used for substudy analysis was limited to participants who met enrollment criteria for that substudy; participants could be included in the placebo group for 1, 2, or 3 substudies. However, if, for example, the participant did not meet agent-specific criteria for infliximab, they were not included in the placebo group for the infliximab substudy, but could be included in the placebo group for other agents. As a result, the placebo groups for each substudy were similar but not identical. Further, we did not use precision approaches to participant selection, such as biomarkers, that might affect agent-specific randomization.
End Points
The primary outcome of median time to recovery by day 28 was evaluated using an 8-point ordinal scale (higher scores indicate better health; Supplement 2).7 Recovery was identified as the first day a participant attained a score of 6, 7, or 8. Key secondary end points were prespecified in the statistical analysis plan (Supplement 1) as clinical status at day 14, assessed by ordinal scale improvement from baseline, and 28-day mortality. Other prespecified secondary end points were 14-day mortality, 60-day mortality, and clinical status at day 28.
Statistical Analysis
Sample size requirements were based on the ability to detect moderate improvement in time to recovery. For each substudy, an estimated 788 recoveries were required to provide approximately 85% power to detect a recovery rate ratio (RRR) of 1.25 for active study drug vs placebo.7 The primary efficacy analysis was based on the Fine-Gray model.12 Efficacy monitoring used the Lan-DeMets13 alpha spending function analog to the O’Brien-Fleming14 boundary. Futility monitoring used the moderately aggressive, Hwang-Shih-DeCani beta spending function with gamma parameter 2. A gatekeeping approach required the boundary for the primary end point to be crossed to declare a statistically significant result for the 2 key secondary end points (eTables 2, 8, and 14 in Supplement 2). The statistical analysis plan describes the type I error protection for the primary and the 2 key secondary end points (28-day mortality and day 14 clinical status; Supplement 1). Day 28 mortality was analyzed as a binary end point with an indicator variable for treatment group using a logistic regression model. Clinical status at day 14 was analyzed using a proportional odds model on the 8-point ordinal scale. A multiple-imputation approach was used to account for uncertainty of outcomes for participants lost to follow-up (Supplement 1).
The intention-to-treat (ITT) population was used for primary end point analysis as specified in the statistical analysis plan (Supplement 1). All secondary end points and safety analyses were reported for the “treated-as-assigned” population consisting of all randomized participants who received at least 1 dose of the assigned study drug.15,16 All P values presented are 2-sided. All ITT and treated-as-assigned data for the primary and secondary end points were consistent and are presented.
Safety Assessments
Safety assessments included a composite end point of death, serious adverse events (SAEs), or grade 3 (severe) and 4 (potentially life-threatening) AEs through day 60. Secondary infections were collected as AEs of special interest through day 60 and were adjudicated by an independent safety officer.
Results
Participants
Between October 16, 2020, and December 31, 2021, a total of 1971 participants underwent randomization at 95 hospitals (85 clinical research sites) across the ACTIV-1 IM platform (Figure 1). Baseline characteristics for each substudy are presented in Table 1 (treated-as-assigned population is shown in eTable 1 in Supplement 2). Characteristics by region are presented in eTables 3, 4, 9, 10, 15, and 16 in Supplement 2. The abatacept and infliximab substudies completed enrollment when they met the prespecified number of recoveries. On September 2, 2021, at the second interim analysis, the data and safety monitoring board recommended discontinuation of randomization to the cenicriviroc substudy due to futility. Treatment and subsequent follow-up were continued for participants already enrolled in this substudy and receiving study medication. No unmasking of individual treatment assignments was deemed necessary for cenicriviroc.
Table 1. Demographics and Baseline Characteristics for Abatacept, Cenicriviroc, and Infliximab Substudiesa.
| Demographics and baseline characteristics | No. (%) | |||||
|---|---|---|---|---|---|---|
| Abatacept trial | Cenicriviroc trial | Infliximab trial | ||||
| Abatacept (n = 524) | Placebo (n = 525)b | Cenicriviroc (n = 360) | Placebo (n = 363)b | Infliximab (n = 531) | Placebo (n = 530)b | |
| Age, mean (SD), y | 54.8 (14.65) | 55.0 (14.66) | 54.7 (14.32) | 54.1 (14.47) | 54.7 (14.87) | 54.9 (14.60) |
| Median (IQR) | 55.0 (44.0 to 65.0) | 55.0 (45.0-65.0) | 55.0 (46.0-64.0) | 54.0 (44.0-64.0) | 55.0 (44.0-66.0) | 55.0 (45.0-65.0) |
| Sex | ||||||
| Women | 197 (37.6) | 222 (42.3) | 123 (34.2) | 159 (43.8) | 198 (37.3) | 226 (42.6) |
| Men | 327 (62.4) | 303 (57.7) | 237 (65.8) | 204 (56.2) | 333 (62.7) | 304 (57.4) |
| Racec | ||||||
| American Indian or Alaska Native | 5/495 (1.0) | 5/499 (1.0) | 4/337 (1.2) | 3/345 (0.9) | 5/504 (1.0) | 5/507 (1.0) |
| Asian | 16/495 (3.2) | 17/499 (3.4) | 11/337 (3.3) | 12/345 (3.5) | 12/504 (2.4) | 17/507 (3.4) |
| Black or African American | 74/495 (14.9) | 76/499 (15.2) | 44/337 (13.1) | 57/345 (16.5) | 80/504 (15.9) | 73/507 (14.4) |
| Native Hawaiian or Other Pacific Islander | 1/495 (0.2) | 0/499 | 0/337 | 0/345 | 1/504 (0.2) | 0/507 |
| White | 325/495 (65.7) | 327/499 (65.5) | 226/337 (67.1) | 221/345 (64.1) | 328/504 (65.1) | 339/507 (66.9) |
| Multiracial | 0/495 | 3/499 (0.6) | 2/337 (0.6) | 2/345 (0.6) | 2/504 (0.4) | 2/507 (0.4) |
| Other | 74/495 (14.9) | 71/499 (14.2) | 50/337 (14.8) | 50/345 (14.5) | 76/504 (15.1) | 71/507 (14.0) |
| Hispanic or Latino ethnicity | 219 (41.8) | 242 (46.1) | 159 (44.2) | 172 (47.4) | 257 (48.4) | 256 (48.3) |
| Geographic region | ||||||
| US | ||||||
| Northeast | 130 (24.8) | 130 (24.8) | 84 (23.3) | 82 (22.6) | 127 (23.9) | 121 (22.8) |
| South | 100 (19.1) | 99 (18.9) | 68 (18.9) | 68 (18.7) | 98 (18.5) | 97 (18.3) |
| Midwest | 83 (15.8) | 80 (15.2) | 61 (16.9) | 60 (16.5) | 81 (15.3) | 80 (15.1) |
| West | 39 (7.4) | 43 (8.2) | 28 (7.8) | 28 (7.7) | 38 (7.2) | 42 (7.9) |
| Peru | 56 (10.7) | 55 (10.5) | 40 (11.1) | 41 (11.3) | 62 (11.7) | 57 (10.8) |
| Argentina | 52 (9.9) | 57 (10.9) | 33 (9.2) | 38 (10.5) | 56 (10.5) | 62 (11.7) |
| Brazil | 46 (8.8) | 45 (8.6) | 38 (10.6) | 37 (10.2) | 54 (10.2) | 55 (10.4) |
| Mexico | 18 (3.4) | 16 (3.0) | 8 (2.2) | 9 (2.5) | 15 (2.8) | 16 (3.0) |
| Disease severity at baselined | ||||||
| Severe disease | 225 (42.9) | 223 (42.5) | 157 (43.6) | 158 (43.5) | 229 (43.1) | 229 (43.2) |
| Moderate disease | 299 (57.1) | 302 (57.5) | 203 (56.4) | 205 (56.5) | 302 (56.9) | 301 (56.8) |
| Clinical status (8-point ordinal scale) at baselinee | ||||||
| 2: Hospitalized, receiving invasive ventilation or ECMO | 48 (9.2) | 51 (9.7) | 29 (8.1) | 31 (8.5) | 58 (10.9) | 53 (10.0) |
| 3: Hospitalized, receiving noninvasive ventilation or high-flow oxygen | 175 (33.4) | 175 (33.3) | 130 (36.1) | 128 (35.3) | 174 (32.8) | 178 (33.6) |
| 4: Hospitalized, requiring supplemental oxygen | 276 (52.7) | 279 (53.1) | 185 (51.4) | 191 (52.6) | 275 (51.8) | 279 (52.6) |
| 5: Hospitalized, not requiring supplement oxygen, requiring ongoing medical care | 25 (4.8) | 20 (3.8) | 16 (4.4) | 13 (3.6) | 24 (4.5) | 20 (3.8) |
| Time from symptom onset, mean (SD), d | −9.3 (4.33) | −9.9 (5.53) | −9.6 (3.91) | −10.0 (5.98) | −9.9 (4.38) | −9.9 (5.56) |
| Median (IQR) | −9.0 (−12.0 to −7.0) | −9.0 (−12.0 to −7.0) | −9.0 (−12.0 to −7.0) | −10.0 (−12.0 to −7.0) | −10.0 (−13.0 to −7.0) | −9.0 (−12.0 to −7.0) |
| BMI, mean (SD) | 32.6 (8.23) | 32.7 (8.16) | 33.4 (8.42) | 32.9 (7.86) | 32.1 (7.93) | 32.7 (8.11) |
| Median (IQR) | 31.1 (27.5 to 36.1) | 31.3 (27.3 to 36.4) | 31.7 (27.9 to 37.5) | 31.2 (27.4 to 36.9) | 30.5 (26.8 to 35.1) | 31.6 (27.3 to 36.5) |
| Comorbiditiesf | ||||||
| Obesity (BMI ≥30) | 297/518 (57.3) | 308/512 (60.2) | 212/353 (60.1) | 213/355 (60.0) | 279/521 (53.6) | 309/516 (59.9) |
| Hypertension | 219 (41.8) | 218 (41.5) | 154 (42.8) | 151 (41.6) | 211 (39.7) | 215 (40.6) |
| Diabetes | 149 (28.4) | 146 (27.8) | 96 (26.7) | 94 (25.9) | 142 (26.7) | 148 (27.9) |
| Asthma | 38 (7.3) | 55 (10.5) | 49 (13.6) | 34 (9.4) | 37 (7.0) | 55 (10.4) |
| Coronary artery disease | 34 (6.5) | 30 (5.7) | 26 (7.2) | 21 (5.8) | 38 (7.2) | 27 (5.1) |
| History of cancer | 31 (5.9) | 36 (6.9) | 23 (6.4) | 19 (5.2) | 35 (6.6) | 34 (6.4) |
| Chronic obstructive pulmonary disease | 20 (3.8) | 24 (4.6) | 22 (6.1) | 15 (4.1) | 24 (4.5) | 26 (4.9) |
| History of heart failure | 16 (3.1) | 15 (2.9) | 15 (4.2) | 8 (2.2) | 16 (3.0) | 15 (2.8) |
| Severe kidney disease | 4 (0.8) | 9 (1.7) | 2 (0.6) | 5 (1.4) | 3 (0.6) | 7 (1.3) |
| Tuberculosis | 3 (0.6) | 4 (0.8) | 2 (0.6) | 3 (0.8) | 3 (0.6) | 4 (0.8) |
| HIV/AIDS | 3 (0.6) | 5 (1.0) | 0 | 3 (0.8) | 2 (0.4) | 4 (0.8) |
| Severe liver disease | 1 (0.2) | 2 (0.4) | 2 (0.6) | 1 (0.3) | 3 (0.6) | 2 (0.4) |
| Concomitant medicationg | ||||||
| Remdesivir (day 1-5) | 488 (93.1) | 493 (93.9) | 346 (96.1) | 344 (94.8) | 496 (93.4) | 499 (94.2) |
| Corticosteroids (day 1-5) | 468 (89.3) | 488 (93.0) | 324 (90.0) | 328 (90.4) | 480 (90.4) | 492 (92.8) |
| Tocilizumab (any time after randomization) | 14 (2.7) | 15 (2.9) | 14 (3.9) | 9 (2.5) | 9 (1.7) | 13 (2.5) |
| Baricitinib (any time after randomization) | 6 (1.1) | 14 (2.7) | 1 (0.3) | 4 (1.1) | 6 (1.1) | 12 (2.3) |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); ECMO, extracorporeal membrane oxygenation.
Eligible participants were 18 years or older with SARS-CoV-2 infection confirmed no more than 14 days before randomization, anticipated hospital stay of at least 72 hours, and evidence of pulmonary involvement who did not meet any of the study- or drug-specific exclusion criteria listed in Supplement 2.
Placebo participants may be included in 1, 2, or 3 of the placebo columns in this table, depending on whether they were eligible for the agent.
Race was self-reported by participants or extracted from the medical record using the following categories: American Indian or Alaska Native, Asian, Black or African American, Native Hawaiian or Other Pacific Islander White, Other, and Unknown. Multiracial corresponds to participants who identified being more than 1 race.
Disease severity at baseline was calculated as moderate disease (hospitalized participants requiring supplemental oxygen [ordinal scale 4] and hospitalized participants not requiring supplement oxygen but requiring ongoing medical care [ordinal scale 5]) and severe disease (hospitalized participants receiving invasive ventilation or ECMO [ordinal scale 2] and hospitalized participants receiving noninvasive ventilation or high-flow oxygen [ordinal scale 3]).
Clinical status categories 6 (hospitalized, not requiring supplement oxygen, not requiring ongoing medical care), 7 (not hospitalized, limitations on activity, and/or requiring home oxygen), and 8 (not hospitalized, no limitations on activities) were exclusions from the study.
Medical history was collected as free text and categorized using Medical Dictionary for Regulatory Activity. Comorbidities were categorized by clinical review.
SARS-CoV-2 vaccines were not available at the start of the trial and were not tracked consistently once they became available and therefore are not included in the table.
Abatacept Substudy
Primary Outcome
Abatacept did not meet the primary end point of time to recovery (RRR, 1.12 [95% CI, 0.98-1.28]; P = .09) after achieving 811 recoveries in the ITT population (Table 2, Figure 2, and eFigure 1 in Supplement 2). Median time to recovery was the same for abatacept compared with placebo (9 days [95% CI, 8-10]). Across ordinal scale subgroups, the P value for interaction was .36 (Table 2; eTable 2, eFigure 1 in Supplement).
Table 2. Primary and Secondary End Points for Abatacept, Cenicriviroc, and Infliximab Substudies.
| End point | Abatacept trial | Cenicriviroc trial | Infliximab trial | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No./total No. (%) | Estimate (95% CI)a | No./total No. (%) | Estimate (95% CI)a | No./total No. (%) | Estimate (95% CI)a | ||||
| Abatacept | Placebo | Cenicriviroc | Placebo | Infliximab | Placebo | ||||
| Primary outcome | |||||||||
| Recovery through day 28b | |||||||||
| Overall | 414/524 (79.0) | 397/525 (75.6) | 1.12 (0.98-1.28)c | 273/360 (75.8) | 287/363 (79.1) | 1.01 (0.86-1.18)d | 421/531 (79.3) | 405/530 (76.4) | 1.12 (0.99-1.28)e |
| Baseline 8-point ordinal scale | |||||||||
| 2: Hospitalized, receiving invasive ventilation or ECMO | 18/48 (37.5) | 20/51 (39.2) | 1.04 (0.54-2.00) | 7/29 (24.1) | 15/31 (48.4) | 0.40 (0.15-1.06) | 24/58 (41.4) | 20/53 (37.7) | 1.12 (0.61-2.04) |
| 3: Hospitalized, receiving noninvasive ventilation or high-flow oxygen devices | 124/175 (70.9) | 106/175 (60.6) | 1.23 (0.96-1.58) | 84/130 (64.6) | 83/128 (64.8) | 1.04 (0.78-1.40) | 126/174 (72.4) | 112/178 (62.9) | 1.33 (1.04-1.69) |
| 4: Hospitalized, requiring supplemental oxygen | 249/276 (90.2) | 252/279 (90.3) | 1.12 (0.96-1.32) | 166/185 (89.7) | 177/191 (92.7) | 0.98 (0.81-1.19) | 249/275 (90.5) | 254/279 (91.0) | 1.07 (0.91-1.26) |
| 5: Hospitalized, not requiring supplement oxygen, requiring ongoing medical care | 23/25 (92.0) | 19/20 (95.0) | 0.78 (0.42-1.44) | 16/16 (100.0) | 12/13 (92.3) | 1.05 (0.49-2.26) | 22/24 (91.7) | 19/20 (95.0) | 0.80 (0.42-1.50) |
| Secondary outcomesf,g | |||||||||
| Mortality at day 28h | |||||||||
| Overall | 56/509 (11.0) | 77/510 (15.1) | 0.62 (0.41-0.94) | 49/355 (13.8) | 42/354 (11.9) | 1.18 (0.72-1.94) | 52/517 (10.1) | 75/516 (14.5) | 0.59 (0.39-0.90) |
| Baseline 8-point ordinal scale | |||||||||
| 2: Hospitalized, receiving invasive ventilation or ECMO | 19/47 (40.4) | 16/51 (31.4) | 1.63 (0.66-4.05) | 12/27 (44.4) | 8/31 (25.8) | 3.22 (0.92-11.25) | 18/58 (31.0) | 16/53 (30.2) | 1.11 (0.45-2.72) |
| 3. Hospitalized, receiving noninvasive ventilation or high-flow oxygen devices | 29/173 (16.8) | 44/171 (25.7) | 0.48 (0.28-0.84) | 27/130 (20.8) | 27/125 (21.6) | 0.82 (0.43-1.58) | 26/170 (15.3) | 43/174 (24.7) | 0.52 (0.29-0.91) |
| 4 or 5: Hospitalized, with or without supplemental oxygen, requiring ongoing medical care | 8/289 (2.8) | 17/288 (5.9) | 0.43 (0.18-1.03) | 10/198 (5.1) | 7/198 (3.5) | 1.45 (0.52-3.99) | 8/289 (2.8) | 16/289 (5.5) | 0.47 (0.20-1.13) |
| Clinical status at day 14 (No.)i | |||||||||
| Overall | 489 | 494 | 1.19 (0.94-1.50) | 348 | 343 | 0.93 (0.71-1.23) | 502 | 501 | 1.32 (1.05-1.66) |
| Baseline 8-point ordinal scale | |||||||||
| 2: Hospitalized, receiving invasive ventilation or ECMO | 46 | 51 | 1.28 (0.63-2.62) | 27 | 31 | 0.76 (0.29-1.94) | 58 | 53 | 1.28 (0.66-2.52) |
| 3: Hospitalized, receiving noninvasive ventilation or high-flow oxygen devices | 168 | 166 | 1.42 (0.97-2.08) | 129 | 122 | 0.99 (0.63-1.54) | 165 | 169 | 1.48 (1.01-2.17) |
| 4: Hospitalized, requiring supplemental oxygen | 255 | 260 | 1.15 (0.83-1.59) | 176 | 179 | 0.92 (0.62-1.35) | 258 | 261 | 1.26 (0.91-1.74) |
| 5: Hospitalized, not requiring supplement oxygen, requiring ongoing medical care | 20 | 17 | 0.20 (0.06-0.74) | 16 | 11 | 0.71 (0.15-3.46) | 21 | 18 | 0.97 (0.37-1.09) |
| Mortality at day 60 | 74/509 (14.5) | 87/510 (17.1) | 0.74 (0.50-1.08) | 64/355 (18.0) | 49/354 (13.8) | 1.39 (0.89-2.18) | 65/517 (12.6) | 85/516 (16.5) | 0.68 (0.46-1.00) |
Abbreviation: ECMO, extracorporeal membrane oxygenation.
For time to recovery, the estimate is the recovery rate ratio (RRR), which is similar to a hazard ratio. For mortality at day 28 and 60 and clinical status at day 14, the estimate is odds ratio (OR).
The RRR and P value were calculated using a stratified Fine-Gray model with death as a competing risk. Stratification was according to geographic region and baseline disease severity on the 8-point ordinal scale. Age and sex were included as covariates in the model. A number greater than 1 favors abatacept, cenicriviroc, or infliximab.
P = .09.
P = .94.
P = .08.
Treated-as-assigned population.
Mortality at day 14 and clinical status at day 28 are reported in eTables 5, 7, 11, 13, 17, and 19 in Supplement 2.
Mortality at day 28 was analyzed as binary end points using a logistic regression model with an indicator variable for treatment group, geographic region, baseline disease severity on the 8-point ordinal scale, age, and sex. Baseline ordinal scale values of 4 and 5 were combined to address model convergence issues. The OR is based on imputed data per imputation rules as described in the statistical analysis plan. A number less than 1 favors abatacept, cenicriviroc, or infliximab.
Clinical status on day 14 was calculated using a proportional odds logistic regression model and compares the odds of improvement between the treatment and placebo groups on the 8-point ordinal scale at day 14. Age and sex were included as covariates in the model. The OR is based on imputed data per imputation rules as described in the statistical analysis plan. A number greater than 1 favors abatacept, cenicriviroc, or infliximab.
Figure 2. Cumulative Incidence of Time to Recovery (Primary Outcome) and Time to Death (Secondary Outcome).
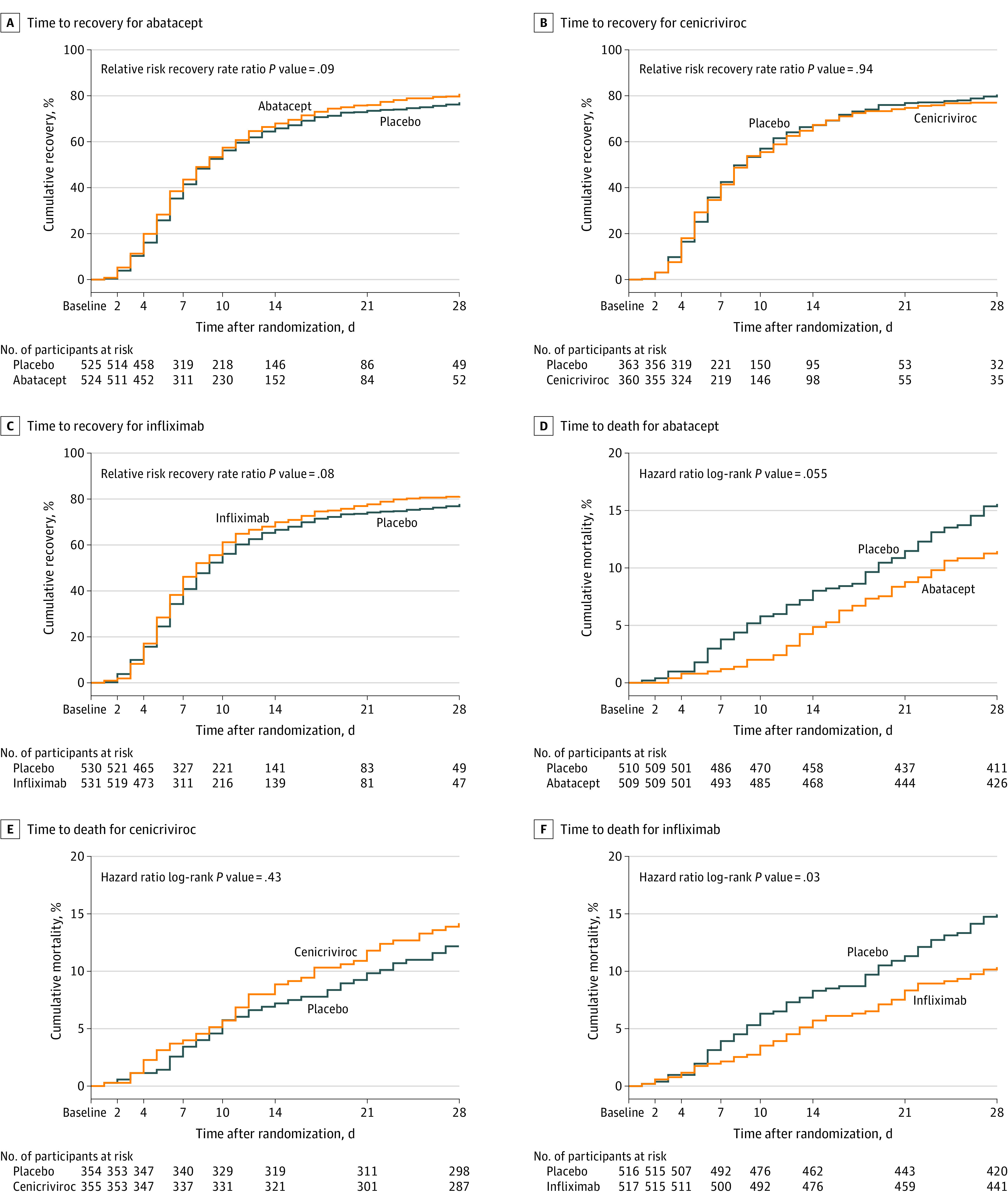
Recovery was defined as the first day the participant scored ≥6 on the 8-point ordinal scale. The median (IQR) observation time for time to recovery and time to death for abatacept, cenicriviroc, and infliximab was 28 (28-28) days.
Secondary Outcomes
Clinical Status
Clinical status was assessed by proportional odds of improvement at day 14 (odds ratio [OR], 1.19 [95% CI, 0.94-1.50]) (Table 2 and eFigure 2 in Supplement 2) and day 28 (OR, 1.35 [95% CI, 1.06-1.73]) (eTable 5 in Supplement 2) in the treated-as-assigned population.
Mortality
All-cause 28-day mortality for participants receiving abatacept was 11.0% compared with 15.1% for those receiving placebo (OR, 0.62 [95% CI, 0.41-0.94]) (Table 2, Figure 2, and eFigures 3-6 in Supplement 2) in the treated-as-assigned population. All-cause 14-day mortality for abatacept was 4.7% compared with 7.8% for placebo (OR, 0.55 [95% CI, 0.32-0.96]), and all-cause 60-day mortality rates were 14.5% for abatacept vs 17.1% for placebo (OR, 0.74 [95% CI, 0.50-1.08]) (eTable 5 and eFigure 7 in Supplement 2).
Safety Assessments
There was no statistically significant difference in the composite safety end point for participants who received abatacept (33.2%) vs placebo (34.9%) (risk difference, −1.7% [95% CI, −7.5% to 4.1%]). SAEs and grade 3 or 4 AEs were not different between the 2 groups. Having any SAE was reported for 128 participants (25.1%) in the abatacept group and 136 (26.7%) in the placebo group. Nine participants (1.8%) in the abatacept group and 7 (1.4%) in the placebo group had an SAE related to the study drug; none were deemed by the sponsor to be suspected unexpected serious adverse reactions related to abatacept administration. Three serious infusion reactions were reported for abatacept and formally adjudicated. One grade 4 SAE met the case definition for anaphylaxis and 2 grade 3 SAEs met criteria for infusion reactions.17 Safety assessment results are presented in Table 3.
Table 3. Safety Composite and Adverse Events for Abatacept, Cenicriviroc, and Infliximab Substudies.
| Adverse event | No. (%) | |||||
|---|---|---|---|---|---|---|
| Abatacept trial | Cenicriviroc trial | Infliximab trial | ||||
| Abatacept (n = 509) | Placebo (n = 510) | Cenicriviroc (n = 355) | Placebo (n = 354) | Infliximab (n = 517) | Placebo (n = 516) | |
| Safety composite and adverse events | ||||||
| Safety compositea | 169 (33.2) | 178 (34.9) | 131 (36.9) | 120 (33.9) | 170 (32.9) | 174 (33.7) |
| SAEb | 128 (25.1) | 136 (26.7) | 102 (28.7) | 84 (23.7) | 125 (24.2) | 130 (25.2) |
| SAE related to study drugc | 9 (1.8) | 7 (1.4) | 3 (0.8) | 3 (0.8) | 6 (1.2) | 7 (1.4) |
| Grade 3 or 4 AE | 129 (25.3) | 136 (26.7) | 105 (29.6) | 97 (27.4) | 146 (28.2) | 131 (25.4) |
| Grade 4 AEd | 55 (10.8) | 66 (12.9) | 51 (14.4) | 46 (13.0) | 63 (12.2) | 64 (12.4) |
| Grade 3 AEe | 109 (21.4) | 102 (20.0) | 92 (25.9) | 76 (21.5) | 124 (24.0) | 97 (18.8) |
| AE related to study drug | 32 (6.3) | 20 (3.9) | 21 (5.9) | 15 (4.2) | 19 (3.7) | 22 (4.3) |
| Secondary infectionsf | ||||||
| Anyg | 82 (16.1) | 73 (14.3) | 53 (14.9) | 52 (14.7) | 79 (15.3) | 72 (14.0) |
| Confirmed | 30 (5.9) | 27 (5.3) | 12 (3.4) | 21 (5.9) | 24 (4.6) | 26 (5.0) |
| Probable | 52 (10.2) | 46 (9.0) | 41 (11.5) | 31 (8.8) | 55 (10.6) | 46 (8.9) |
Abbreviation: SAE, serious adverse event.
Safety composite end point includes any of the following events: deaths, SAEs and grade 3 or 4 AEs through day 60.
SAEs are events that, in the view of either the investigator or the sponsor, result in death, a life-threatening AE, hospitalization or prolongation of existing hospitalization, persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions, or a congenital anomaly/birth defect.
Relatedness to study drug was determined by site principal investigators.
Grade 4 AEs are events that are potentially life threatening.
Grade 3 AEs are events that interrupt usual activities of daily living, significantly affect clinical status, or may require intensive therapeutic intervention.
Secondary infections are new or worsening (after randomization) intercurrent, at least probable, documented serious disease caused by an infection other than SARS-CoV-2, requiring antimicrobial administration and care.
Secondary infections by pathogen are presented in eTables 6, 12, and 18 in Supplement 2.
Secondary infections by day 60 were reported for 82 participants (16.1%) in the abatacept group and 73 (14.3%) in the placebo group, with the types of infections being similar to those in the infliximab substudy (Table 3 and eTable 6 in Supplement).
Cenicriviroc Substudy
Adherence to Study Drug
Overall, 98.4% of participants in the cenicriviroc substudy received a loading dose of the study drug or placebo, with 96.7% receiving at least 1 subsequent maintenance dose. The median (IQR) duration of treatment was 28 (13-29) days. A total of 36.5% of participants discontinued maintenance doses prior to day 29.
Primary Outcome
There was no difference in time to recovery for cenicriviroc compared with placebo (RRR, 1.01 [95% CI, 0.86-1.18]; P = .94) with a median of 8 days (95% CI, 8-9 vs 8-10) for the ITT population (Table 2, Figure 2; eFigure 8 in Supplement 2).
Secondary Outcomes
Clinical Status
There was no difference in the odds of improvement in clinical status at day 14 or day 28 assessed by the ordinal scale (day 14: OR, 0.93 [95% CI, 0.71-1.23]; day 28: OR, 0.89 [95% CI, 0.66-1.19]) (Table 2 and eTable 11 and eFigure 10 in Supplement 2) in the treated-as-assigned population.
Mortality
Mortality through day 28 for the treated-as-assigned population was 13.8% for cenicriviroc and 11.9% for placebo (OR, 1.18 [95% CI, 0.72-1.94]) (Table 2, Figure 2, and eFigure 9 in Supplement 2). Day 14 mortality rates were 8.7% for participants receiving cenicriviroc vs 7.1% for those receiving placebo (OR, 1.31 [95% CI, 0.71-2.40]) and day 60 mortality rates were 18.0% for cenicriviroc and 13.8% for placebo (OR, 1.39 [95% CI, 0.89-2.18]) (eTable 11 and eFigure 11 in Supplement 2).
Safety Assessment
There was no clinically significant difference in the composite safety end point at day 60 between the groups (incidence of 36.9% for cenicriviroc vs 33.9% for placebo; risk difference, 3.0% [95% CI, −4% to 10%]). Grade 3 or 4 AEs occurred with similar frequencies (105 [29.6%] in the cenicriviroc group and 97 [27.4%] in the placebo group) (Table 3). SAEs occurred in 102 participants (28.7%) in the cenicriviroc group and 84 (23.7%) in the placebo group. The percentage of participants who had any secondary infection was similar at day 60 for cenicriviroc and placebo (53 [14.9%] vs 52 [14.7%]) (Table 3 and eTable 12 in Supplement 2).
Infliximab Substudy
Primary Outcome
Infliximab did not meet the primary end point of time to recovery in the ITT population, with an estimated RRR for infliximab vs placebo of 1.12 (95% CI, 0.99-1.28; P = .08) after achieving 826 recoveries (Table 2; eFigure 12 in Supplement 2). Median time to recovery for infliximab was similar to placebo (median of 8 [95% CI, 7-9] vs 9 [95% CI, 8-10] days). Results by ordinal scale are shown in Table 2 and eFigure 12 in Supplement 2. Across ordinal scale subgroups, the P value for interaction was .36, indicating no clear evidence of heterogeneity (eTable 14 in Supplement 2).
Secondary Outcomes
Clinical Status
Clinical status was assessed as a proportional odds of improvement at day 14 (OR, 1.32 [95% CI, 1.05-1.66]) (Table 2 and eFigure 14 in Supplement 2) and day 28 (OR, 1.45 [95% CI, 1.14–1.85]) (eTable 17 in Supplement 2) for the treated-as-assigned population.
Mortality
Mortality through day 28 in participants receiving at least 1 dose of the study medication (treated-as-assigned population) was 10.1% for infliximab and 14.5% for placebo (OR, 0.59 [95% CI, 0.39-0.90]) (Table 2, Figure 2, and eFigures 13 and 15-17 in Supplement 2). Fourteen-day mortality rates were 5.6% for infliximab vs 8.1% for placebo (OR, 0.63 [95% CI, 0.36-1.08]) and 60-day mortality rates were 12.6% for infliximab compared with 16.5% for placebo (OR, 0.68 [95% CI, 0.46-1.00]) (eTable 19 and eFigure 18 in Supplement 2).
Safety Assessments
No difference was observed in the composite safety end point at day 60 (incidence, 32.9% vs 33.7%; risk difference, −0.8% [95% CI, −6.6% to 4.9%]). SAEs occurred in 125 participants (24.2%) in the infliximab group, with 7 events in 6 participants (1.2%) determined by investigators to be related to infliximab. For placebo, SAEs occurred in 130 participants (25.2%) and the events were attributed to trial product in 7 of these participants (1.4%). One participant in the infliximab group experienced a grade 1 (mild) infusion reaction. Safety assessment is presented in Table 3.
The percentage of participants who had any secondary infection was similar at day 60 with infliximab and placebo (79 [15.3%] vs 72 [14.0%]) (Table 3). The most common secondary infections were bacterial pneumonia, bloodstream, and urinary tract infections (eTable 18 in Supplement 2).
Discussion
The ACTIV-1 IM master protocol evaluated the benefit of an additional immunomodulator added to standard care in hospitalized participants with moderate to severe COVID-19 pneumonia. Within this platform, 3 agents with distinct mechanisms of action—abatacept, cenicriviroc, and infliximab—were evaluated. The ACTIV-1 study found that none of the substudies reached a statistically significant primary end point of time to recovery. Thus, 28-day mortality and day 14 clinical status cannot be considered statistically significant based on the predefined gatekeeping approach, despite nominally significant CIs, although these end points are clinically important. For abatacept and infliximab substudies, the primary and 2 key secondary end points were all in the beneficial direction, with the mortality benefit showing remarkable consistency across subgroups (eFigures 3 and 13 in Supplement 2). Additionally, the consistent lack of benefit among all 3 outcomes for cenicriviroc within the same master protocol further supports the validity of the findings for abatacept and infliximab because it illustrates differences in outcomes between the 3 drugs despite each drug being compared with placebo groups that were similar (although not identical).
There is a growing body of evidence for use of a second immunomodulatory agent in addition to corticosteroids. Positive results from several trials prompted the NIH treatment guidelines to include baricitinib and tocilizumab5,6,9,18 as a second immunomodulator in addition to corticosteroids for patients with progressive respiratory failure and evidence of systemic inflammation.19 The current results offer additional information on the efficacy and safety of abatacept and infliximab. Although severe disease is less common in the postvaccination era and SARS-CoV-2 variants become less deadly, immunomodulatory therapy is still critical. The US Centers for Disease Control and Prevention continued to report more than 1000 COVID-19–associated deaths per week in the US as of April 2023.20 Data on the benefit of additional immunomodulators is particularly important in the setting of previous drug shortages within these categories during periods of the pandemic. Additionally, options that are safer for patients with kidney, liver, or clotting disorders fill important gaps.
Similar mortality benefits have been demonstrated for a number of immunomodulatory agents with varying mechanisms (IL-6 inhibition, Janus kinase-1/2 inhibition, TNF inhibition, and select T-cell costimulatory modulation), although further exploration of the role of inflammation in the pathogenesis of SARS-CoV-2 is needed because modulation of other pathways, such as those impacted by cenicriviroc, does not show benefit. The term cytokine storm describes the marked increase in cytokines that triggers a cascade of synergistic inflammatory signaling in severe disease.2 Aberrant T-cell function in severe COVID-19 is complex, involving hyperactivation of CD4+ T cells.1 Circulating monocytes and macrophages also play a key role in inflammatory signaling and fibrosis.21 Subtleties in ACTIV-1 substudies hint at small differences in therapeutic roles. For example, subgroup analysis suggested a larger effect for abatacept among older adults (≥65 years old) and those with diabetes than observed for other subgroups in the abatacept substudy (eFigures 1 and 3 in Supplement 2) or the effects found for the infliximab substudy (eFigures 12 and 13 in Supplement 2). Deeper exploration of dissimilarities among mechanisms and effects of these drugs on the COVID-19 inflammatory cascade could improve insight into the pathogenesis of severe disease.
A key clinical question involves how to treat hospitalized patients receiving low-flow oxygen. Current NIH guidelines do not recommend addition of a second immunomodulator for these patients in the absence of rapidly increasing oxygen needs. The ACTIV-1 IM substudies begin to address this critical question, showing reduced mortality for participants receiving abatacept or infliximab in a general population of participants of all ordinal scales independent of inflammatory markers. Subgroup analysis suggests that the benefits in mortality occur for participants receiving both low-flow and high-flow oxygen/noninvasive ventilation (eFigures 3 and 13 in Supplement 2).
The ACTIV-1 IM master protocol provides a helpful blueprint for a high-quality trial design that allows for concurrent evaluation of multiple agents while simultaneously providing a shared placebo group. Other COVID-19 platform trials conducted provide shared controls, but often do not offer concurrently enrolled controls or a placebo group.4,5,6,22 Use of a shared placebo represents a patient-centered approach to reduce the number of patients receiving placebo, allowing adequate power to evaluate multiple agents with a smaller number of participants enrolled.11 The findings that active and shared placebo groups had similar baseline characteristics, and that there was no benefit seen when cenicriviroc was compared with the shared placebo while benefits were observed for abatacept and infliximab, reinforce the integrity of the shared placebo group in this study.
This study highlights a major challenge in the selection of a primary end point for this and other COVID-19 studies in the setting of a rapidly changing clinical arena.23 A mortality primary end point is considered definitive and free from subjectivity, but can require large participant enrollment potentially resulting in prolonged study recruitment with delayed results. In addition, mortality does not encompass other patient-centered outcomes, which are a greater focus in end points such as time to recovery or clinical status. As research moves forward for COVID-19, the selection of primary end points will be important to encompass evaluable patient-centered outcomes, but take steps to do so with lower enrollment targets.
As SARS-CoV-2 moves from being pandemic to endemic, and while new variants continue to emerge, ongoing morbidity and mortality from this disease is likely. Variants may evade specific antiviral strategies, but treatments that augment host responses are likely to be more durable. Expanding treatment armament available globally and developing optimized treatment strategies remains paramount.
Limitations
The ACTIV-1 study has several limitations. First, ACTIV-1 took place prior to the Omicron era, raising the question of whether the results are limited to the variants represented at that time. However, despite changes in the predominant circulating variant over time and decreased disease severity overall, patients continue to be admitted to the hospital with respiratory failure and evidence of immune dysregulation, highlighting the ongoing need for optimized treatment strategies. Second, although vaccinations became available during the course of the study, data were not available on participant vaccination status. Third, unlike abatacept and infliximab, which were conveniently dosed as 1-time intravenous infusion, cenicriviroc is an oral medication that was administered in this study twice daily for 28 days. This led to challenges with adherence, which may have contributed to the negative results observed in this substudy, although similar adherence issues would also exist in a clinical setting.
Conclusions
Time to recovery from COVID-19 pneumonia among hospitalized participants was not significantly different for abatacept, cenicriviroc, or infliximab vs placebo.
Trial protocol
eTables and eFigures
Nonauthor collaborators
Data sharing statement
References
- 1.Kalfaoglu B, Almeida-Santos J, Tye CA, Satou Y, Ono M. T-cell hyperactivation and paralysis in severe COVID-19 infection revealed by single-cell analysis. Front Immunol. 2020;11:589380. doi: 10.3389/fimmu.2020.589380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Otsuka R, Seino KI. Macrophage activation syndrome and COVID-19. Inflamm Regen. 2020;40:19. doi: 10.1186/s41232-020-00131-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Files DC, Tacke F, O’Sullivan A, Dorr P, Ferguson WG, Powderly WG. Rationale of using the dual chemokine receptor CCR2/CCR5 inhibitor cenicriviroc for the treatment of COVID-19. PLoS Pathog. 2022;18(6):e1010547. doi: 10.1371/journal.ppat.1010547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horby P, Lim WS, Emberson JR, et al. ; RECOVERY Collaborative Group . Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384(8):693-704. doi: 10.1056/NEJMoa2021436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.RECOVERY Collaborative Group . Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10285):1637-1645. doi: 10.1016/S0140-6736(21)00676-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon AC, Mouncey PR, Al-Beidh F, et al. ; REMAP-CAP Investigators . Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med. 2021;384(16):1491-1502. doi: 10.1056/NEJMoa2100433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beigel JH, Tomashek KM, Dodd LE, et al. ; ACTT-1 Study Group Members . Remdesivir for the treatment of Covid-19: final report. N Engl J Med. 2020;383(19):1813-1826. doi: 10.1056/NEJMoa2007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ely EW, Ramanan AV, Kartman CE, et al. ; COV-BARRIER Study Group . Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir Med. 2022;10(4):327-336. doi: 10.1016/S2213-2600(22)00006-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalil AC, Patterson TF, Mehta AK, et al. ; ACTT-2 Study Group Members . Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. 2021;384(9):795-807. doi: 10.1056/NEJMoa2031994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tacke F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin Investig Drugs. 2018;27(3):301-311. doi: 10.1080/13543784.2018.1442436 [DOI] [PubMed] [Google Scholar]
- 11.LaVange L, Adam SJ, Currier JS, et al. ; ACTIV Therapeutics-Clinical Working Group . Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV): designing master protocols for evaluation of candidate COVID-19 therapeutics. Ann Intern Med. 2021;174(9):1293-1300. doi: 10.7326/M21-1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496-509. doi: 10.1080/01621459.1999.10474144 [DOI] [Google Scholar]
- 13.Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70(3):659-663. doi: 10.2307/2336502 [DOI] [Google Scholar]
- 14.O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35(3):549-556. doi: 10.2307/2530245 [DOI] [PubMed] [Google Scholar]
- 15.Gillings D, Koch G. The application of the principle of intention-to-treat to the analysis of clinical trials. Drug Inf J. 1991;25(3):411-424. doi: 10.1177/009286159102500311 [DOI] [Google Scholar]
- 16.US Food and Drug Administration . E9 Statistical Principles for Clinical Trials; 1998. Accessed June 28, 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e9-statistical-principles-clinical-trials
- 17.Rüggeberg JU, Gold MS, Bayas JM, et al. ; Brighton Collaboration Anaphylaxis Working Group . Anaphylaxis: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. 2007;25(31):5675-5684. doi: 10.1016/j.vaccine.2007.02.064 [DOI] [PubMed] [Google Scholar]
- 18.Marconi VC, Ramanan AV, de Bono S, et al. ; COV-BARRIER Study Group . Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. 2021;9(12):1407-1418. doi: 10.1016/S2213-2600(21)00331-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.COVID-19 Treatment Guidelines Panel . Coronavirus disease 2019. (COVID-19) treatment guidelines. National Institutes of Health. Updated May 2, 2023. Accessed June 28, 2023.https://www.covid19treatmentguidelines.nih.gov/
- 20.Centers for Disease Control and Prevention . COVID data tracker. Accessed March 20, 2023. https://covid.cdc.gov/covid-data-tracker/#datatracker-home
- 21.Diaz Soto MP, Lim JK. Evaluating the therapeutic potential of cenicriviroc in the treatment of nonalcoholic steatohepatitis with fibrosis: a brief report on emerging data. Hepat Med. 2020;12:115-123. doi: 10.2147/HMER.S230613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan H, Peto R, Henao-Restrepo AM, et al. ; WHO Solidarity Trial Consortium . Repurposed antiviral drugs for Covid-19: interim WHO solidarity trial results. N Engl J Med. 2021;384(6):497-511. doi: 10.1056/NEJMoa2023184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Douin DJ, Siegel L, Grandits G, et al. Evaluating primary endpoints for COVID-19 therapeutic trials to assess recovery. Am J Respir Crit Care Med. 2022;206(6):730-739. doi: 10.1164/rccm.202112-2836OC [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol
eTables and eFigures
Nonauthor collaborators
Data sharing statement