

Abstract
Our understanding of tissue-resident memory (TRM) T cell biology has been largely developed from acute infection models in which antigen is cleared and sterilizing immunity is achieved. Less is known about TRM cells in the context of chronic antigen persistence and inflammation. Here, we investigated factors that underlie TRM maintenance in a kidney transplantation model in which TRM cells drive rejection. In contrast to acute infection, we found that TRM cells declined dramatically in the absence of cognate antigen, antigen presentation, or antigen sensing by the T cells. Depletion of graft-infiltrating dendritic cells or interruption of antigen presentation after TRM cells were established was sufficient to disrupt TRM maintenance and reduce allograft pathology. Likewise, removal of IL-15 transpresentation or of the IL-15 receptor on T cells during TRM maintenance led to a decline of TRM cells, and IL-15 receptor blockade prevented chronic rejection. Therefore, antigen and IL-15 presented by dendritic cells play non-redundant key roles in CD8 TRM cell maintenance in settings of antigen persistence and inflammation. These findings provide insights that could lead to improved treatment of chronic transplant rejection and autoimmunity.
One Sentence Summary:
Maintenance of tissue-resident memory T cells reveals non-redundant, essential roles for cognate antigen and interleukin-15.
INTRODUCTION
Organ transplantation is a life-saving treatment for patients with end-stage organ disease. However, chronic rejection remains a barrier to long-term allograft survival. In mice and humans, chronic allograft rejection is characterized by a persistent T cell infiltrate associated with gradual loss of graft function and eventual graft failure (1–3). CD8 T cells play a key role in graft failure through cytokine production and direct cytolysis of parenchymal cells (4, 5).
Alloreactive effector T cells are generated in secondary lymphoid tissues and migrate to transplanted organs where they re-engage with antigen-presenting cells (6–9). After the initial effector phase, memory T cells form in secondary lymphoid tissues or in the allograft and they either reenter circulation or take up residence in the tissue. Resident memory T (TRM) cells are a non-circulating, long-lived population with the potential to mount rapid, in situ immune responses by coordinating both local innate and adaptive immune cells (10–14). TRM cells are phenotypically and transcriptionally distinct from circulating memory T cells (3, 13–17). TRM cells express markers of activation, retention, and adhesion (CD69, CD103, CD49a, PD-1), produce effector cytokines (IFNγ), and proliferate locally (3, 10, 12, 18–20).
We have previously shown that TRM cells form in murine kidney allografts and mediate chronic rejection (3). This was established by demonstrating that antigen specific and polyclonal CD8 T cells in the graft have a phenotype and transcriptional profile consistent with TRM cells, do not recirculate after parabiosis or retransplantation, and exhibit high functionality in vivo and ex vivo. TRM cells have also been characterized in human kidney, lung, and small bowel allografts by phenotypic and transcriptional analysis (21–23). However, it is unclear what factors are responsible for the long-term maintenance of TRM cells in kidney allografts. In contrast to barrier tissues in which TRM cells have traditionally been studied, such as the skin, small intestine, and the female reproductive tract (13, 24, 25), the kidney is a non-barrier tissue. Moreover, alloantigen persists in the local tissue environment of the graft. Although persistent antigenic stimulation has been shown to lead to progressive loss of cytokine production, as well as loss of cytotoxic and proliferative abilities of T cells in the context of chronic viral infection and cancer (26–28), T cells retain their function and mediate pathology in human autoimmunity and transplantation (29–37). Moreover, it is still debated whether memory T cells are maintained in the absence of antigenic stimulation or require periodic interaction with their cognate antigen to persist (38–41). This same debate extends to TRM cells (42–46).
The observation that functional TRM cells form and persist in settings of chronic antigen and inflammation suggests that additional signals in the tissue microenvironment could participate in the maintenance of these cells. Of particular interest is the role of interleukin-15 (IL-15), a cytokine crucial for the maintenance of memory CD8 T cells, including TRM cells in the native kidney (47), during homeostasis (i.e., non-inflamed conditions). IL-15 appears to contribute to memory T cell maintenance by promoting basal proliferation (48, 49). However, the role of IL-15 during chronic inflammation remains to be elucidated. Previous studies have shown that supplemental IL-15 can enhance tumor-specific CD8 T cell function (50, 51) and that increased IL-15 expression is associated with functional tumor-infiltrating T cells and disease-free survival in colorectal cancer (52). Moreover, IL-15 is elevated in various autoimmune diseases (53–56) as well as in renal allograft rejection (57). Altogether, these observations support a role for IL-15 in TRM cell homeostasis in the setting of persistent antigen and chronic inflammation.
Here, we examined the contributions of antigen and IL-15 to TRM cell maintenance in a mouse model of kidney transplantation. Transplanted kidney tissue represents a unique and important landscape to study TRM cells because: 1) antigen is persistent; 2) the kidney is a non-barrier tissue; 3) inflammation is chronic; and 4) TRM cells form and function. Previous studies delving into the roles of antigen and IL-15 in TRM cell maintenance examined these factors individually in the context of acute viral infection in barrier tissues (46, 58). In contrast to those studies, we report here that antigen and IL-15 play non-redundant key roles in the maintenance of TRM cells in allograft tissue. Importantly, we demonstrate that blocking IL-15 from binding to its receptor on T cells prevents chronic rejection and significantly prolongs allograft survival.
RESULTS
TRM cells form in renal allograft tissue and receive TCR signaling
To study the maintenance of TRM cells in allograft tissue, we transplanted allogeneic (Balb/c × B6.OVA) F1.Act-mOVA (F1.OVA) kidneys, which express chicken ovalbumin (OVA) ubiquitously on cell surfaces, into C57BL/6J (B6) recipients. Two days later, OVA-specific T cell receptor (TCR) transgenic OT-I effector CD8 T cells were transferred to the transplant recipients (Fig. 1A). These OT-I effector CD8 T cells (Fig. S1A) were generated and sorted from a donor mouse immunized with anti-DEC-205/OVA as detailed in Methods. In our model (Fig. 1A), the transferred OT-I cells served as a traceable, donor antigen-specific T cell population that was also required for complete rejection of the allograft (3). As previously reported (3), CD8 T cells with a prototypic cell surface, transcriptional, and non-recirculatory TRM profile form in the graft from recipient-derived polyclonal T cells that recognize Balb/c antigens and from monoclonal OT-I cells that recognize OVA257–265 SIINFEKL peptide in the context of H-2Kb. Together, they mediate chronic rejection. Of note, all transferred effector OT-I cells localized to the graft and adopted a TRM phenotype (3). In the current study, increasing fibrosis and infiltrates containing CD103+ T cells could be appreciated within the renal cortex by days 28–42 (Fig. 1B). Flow cytometry confirmed that after gating on extravascular CD8+ T cells as shown in Fig. S1B, all OT-I (CD90.1/CD90.2+) and a majority of host polyclonal CD8 (CD90.2+) T cells in the graft expressed a surface phenotype consistent with TRM by 28 days post-transplantation (Fig. 1C): CD44+CD62L–CD69+CD103+/–KLRG1–CD49a+. OT-I cells underwent homeostatic proliferation as assessed by BrdU uptake, lacked inhibitory receptor expression (TIM-3), and were functional as measured by production of IFNγ after restimulation. Lack of EdU uptake administered 1 hour prior to harvest indicates that the OT-I cells were not rapidly dividing effector cells (Fig. 1C).
Fig.1. TRM cells form in renal allografts and encounter local cognate antigen.
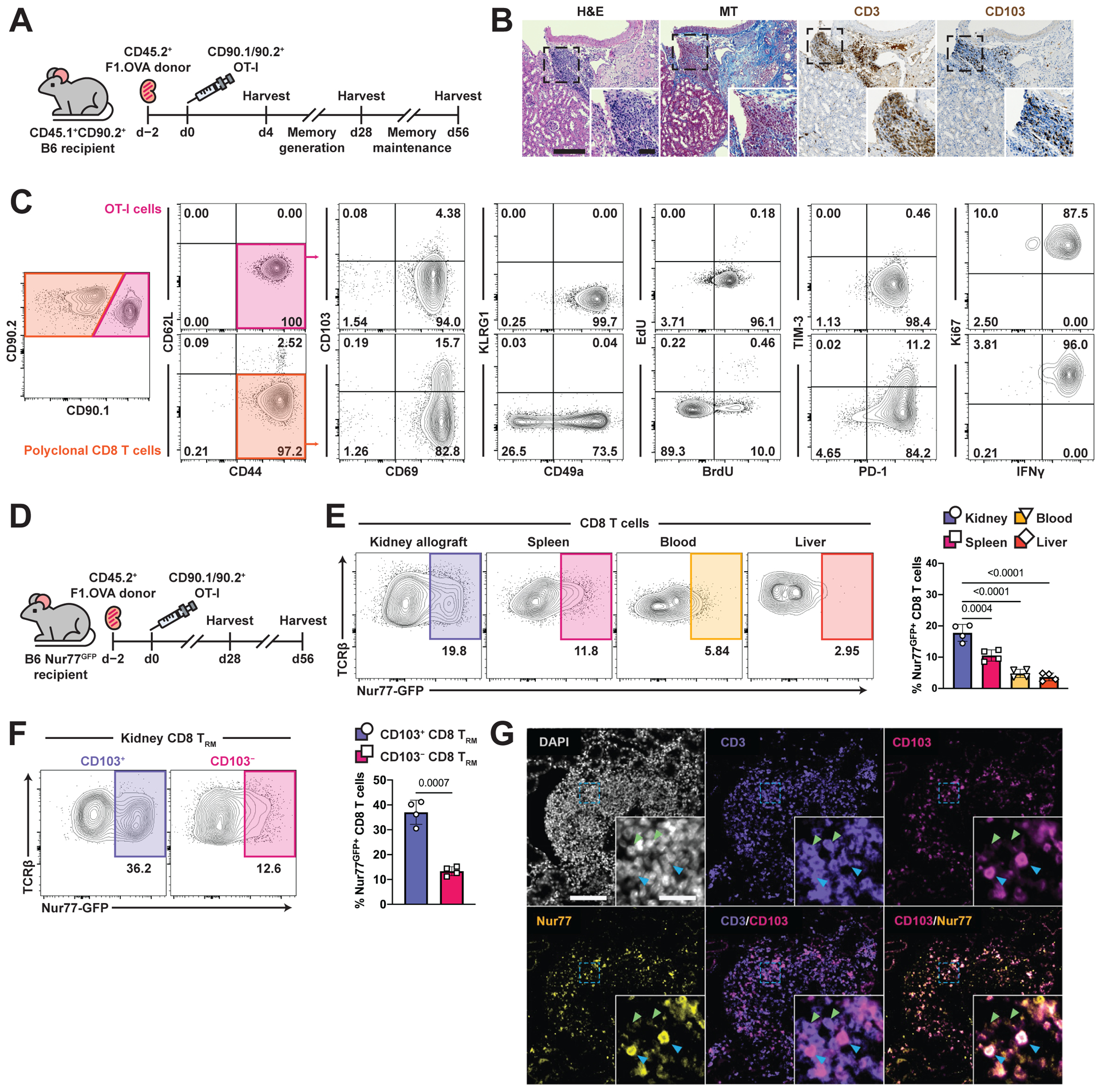
(A) (Balb/c × B6.OVA) F1 (F1.OVA) kidney allografts were transplanted into B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Transplant recipients were harvested at indicated times.
(B) H&E, Masson’s trichrome (MT), and anti-CD3 and anti-CD103 stained sections of renal allograft tissue 56 days post-transplantation. Insets highlight areas of T cell infiltration. Blue in MT staining indicates fibrosis. Whole image scale bar: 200 μm. Inset scale bar: 50 μm. n = 6 mice.
(C) Transferred CD90.1/90.2+ OT-I and recipient-derived polyclonal CD90.2+ CD8 T cells from 28-day post-transplant kidney allografts were analyzed for TRM markers after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. BrdU water was administered 7 days before and EdU injected 1 hour prior to harvest. n = 8 mice.
(D) F1.OVA kidney allografts were transplanted into Nur77-GFP B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Transplant recipients were harvested on days 28 and 56. n = 4 mice per time point.
(E) Representative plots (left) and percentage (graph, right) of Nur77-GFP expression by CD8 T cells in various tissues on day 56.
(F) Representative plots (left) and percentage (graph, right) of Nur77-GFP expression based on CD103 expression.
(G) Immunofluorescence staining of renal allograft tissue on day 56 for DAPI (white), CD3 (purple), CD103 (magenta), and Nur77 (yellow). Blue arrows point to cells co-expressing CD3, CD103 and Nur77. Green arrows point to cells expressing only CD3. Whole image scale bar: 100 μm. Inset scale bar: 20 μm. n = 5 mice.
P values were determined by (E, F) two-tailed unpaired t test.
To assess whether intragraft TRM cells continue to receive cognate interactions through their TCR, we transplanted F1.OVA kidney into Nur77-GFP recipients (Fig. 1D). In Nur77-GFP mice, GFP is coupled to Nur77 expression, which is upregulated specifically by TCR signaling (59) – thus, allowing us to examine which polyclonal T cells had undergone recent cognate antigen stimulation. As shown in Fig. 1E and 1F, a greater proportion of polyclonal CD8 T cells expressed Nur77-GFP within renal allograft tissue compared to other tissues on day 56. Importantly, Nur77 expression was enriched in CD103+ T cells in the graft (Fig. 1F and 1G), suggesting that polyclonal TRM cells had recently encountered cognate antigen. We also found that intragraft OT-I cells on day 56 had upregulated Nur77 expression based on intracellular staining (Fig. S1C), indicating that they too have recently encountered their cognate antigen.
Reduced number of intragraft TRM cells in the absence of cognate antigen
To investigate the role of cognate antigen in TRM cell generation or maintenance, we transplanted either F1.OVA or F1 (Balb/c × B6) kidneys to B6 recipients, followed by adoptive transfer of effector OT-I cells (Fig. 2A). F1 renal allografts lack OVA, and hence, transferred effector OT-I cells do not encounter cognate antigen in the graft of the recipient. Dramatically fewer TRM OT-I cells were present in F1 allografts compared to F1.OVA allografts on day 56 post-transplantation (Fig. 2B). OT-I cells were not found outside of the renal allograft (Fig. S2A), indicating that the reduced number of intragraft OT-I cells in the absence of cognate antigen on day 56 was not due to egress from the graft. Quantification of intragraft OT-I cells over time revealed a sizeable, proliferating population of OT-I cells in both types of allografts on day 4 (Fig. 2C, S2B), which declined over time in F1 grafts but remained stable in F1.OVA grafts (Fig. 2C, S2C). In contrast, polyclonal CD8 T cells, which recognize Balb/c antigens present in F1 allografts, declined much less (Fig. 2D, S2D). The smaller number of OT-I cells in F1 grafts on day 4 (Fig. 2C) could be explained by reduced effector T cell infiltration in the absence of cognate antigen (6). Intragraft OT-I cells harvested on day 4 from either graft were CD44+CD62L–KLRG1– but approximately half were CD49a– (Fig. S2E). In contrast, OT-I cells harvested on days 28 and 56 had acquired the typical TRM phenotype (CD44+CD62L–KLRG1–CD49a+). Moreover, renal allograft pathology (cellular infiltrate and fibrosis) was diminished in the absence of cognate antigen, consistent with the role of TRM OT-I cells in chronic rejection (Fig. 2E) (3). Together, these results indicate that cognate antigen is required for either optimal generation or maintenance of TRM cells in the graft.
Fig. 2. Reduced number of intragraft TRM cells in the absence of cognate antigen.
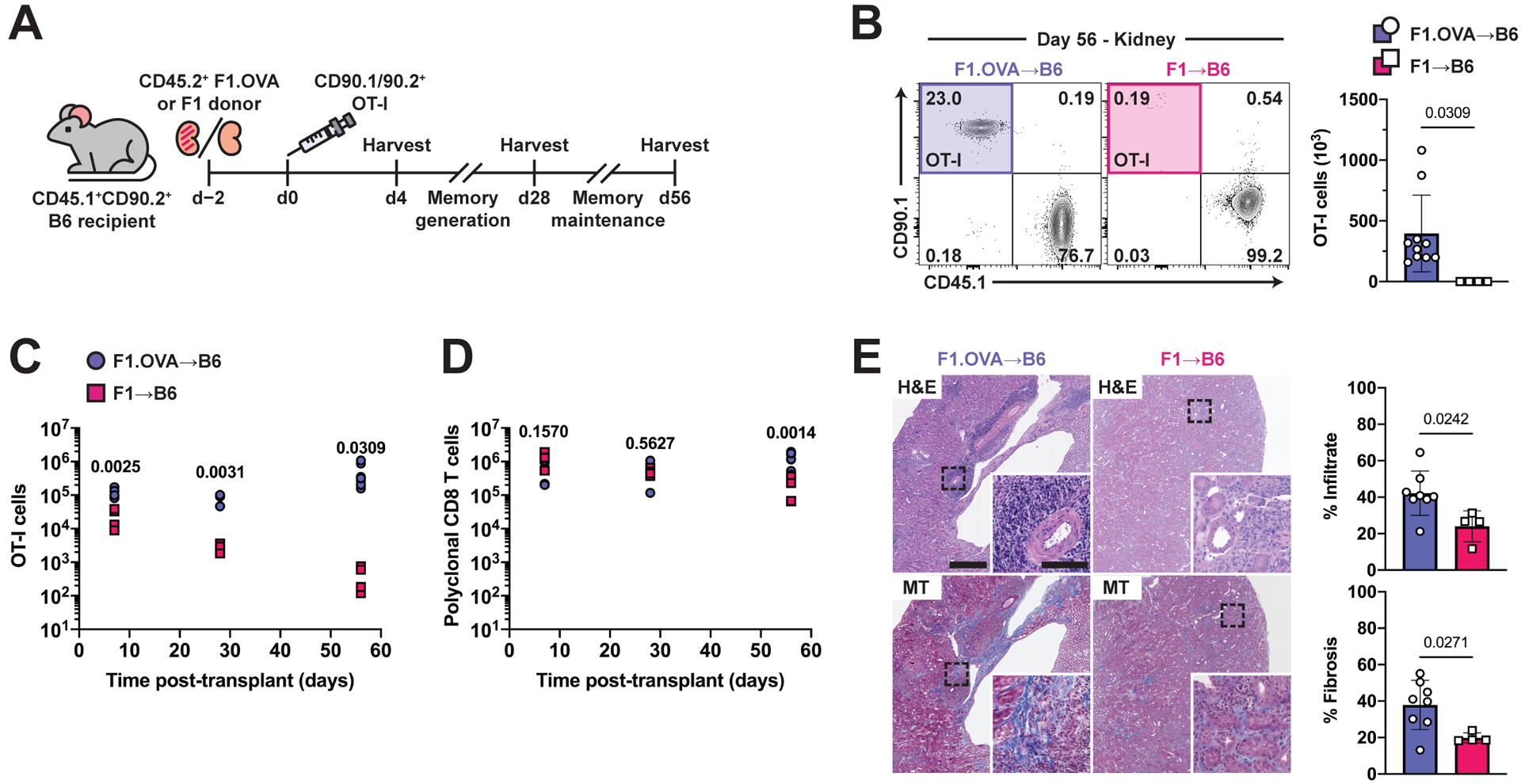
(A) F1.OVA or (Balb/c × B6) F1 kidney allografts were transplanted into B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Transplant recipients were harvested on day 4 to study effector CD8 T cell graft infiltration, and on days 28 and 56 to study maintenance of CD8 TRM cells. Flow analysis was performed after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. n = 4–8 mice per group.
(B) Percentage (representative plots, left) and absolute number (graphs, right) of CD90.1+ OT-I cells on day 56.
(C, D) Enumeration of CD90.1+ OT-I cells (C) and polyclonal CD45.1+CD90.1− CD8 T cells (D) in renal allograft tissue on days 4, 28, and 56. n = 4–8 mice per group per time point.
(E) H&E and Masson’s trichrome-stained sections of F1.OVA renal allograft tissue (representative images, left) and quantification of infiltrate and fibrosis (graphs, right) day 56. Whole image scale bar: 500 μm. Inset scale bar: 100 μm.
P values were determined by (B-E) two-tailed unpaired t test.
TCR affinity modulates TRM cell abundance in the graft
To further investigate the role of cognate antigen, we studied the effect of modulating TCR affinity (i.e., none, intermediate, or high affinity) on the number of TRM cells present in the graft. To do so, we co-adoptively transferred effector OT-I and P14 cells to F1.OVA kidney transplant recipients and harvested grafts 56 days later (Fig. 3A). TCR transgenic P14 cells are LCMV gp33–41 (KAVYNFATC)-specific CD8 T cells that do not recognize either the OVA or Balb/c antigens present in the allograft. Effector P14 cells were generated by infecting donor mice with LCMV as described in Methods. At an early time point (day 6), similar numbers of OT-1 and P14 cells were present in the graft, whereas a much smaller number of P14 cells (10-fold less) was present on day 56 (Fig. 3B, S3A), despite the two cell types encountering the same inflammatory graft environment. Similar to OT-I cells, P14 cells were only found in renal allograft tissue (Fig. S3B) and exhibited a TRM-like phenotype (Fig. S3C). We next co-adoptively transferred effector OT-I and OT-3 cells to transplant recipients to test the effect of reducing TCR affinity to cognate antigen on generation or maintenance of TRM cells (Fig. 3C) (60). TCR transgenic OT-I and OT-3 are both OVA257–265 (SIINFEKL)-specific CD8 T cells but compared to the high affinity TCR of OT-I cells, the TCR of OT-3 cells has lower affinity to the cognate antigen (61). As shown in Fig. 3D & S3A, a significantly (p=0.02 and p=0.03 respectively) smaller number of TRM OT-3 cells (2–3-fold less) was observed in the F1.OVA grafts on day 56 although OT-3 and OT-I cell numbers were similar on day 6. A lower percentage of intragraft OT-3 population expressed CD69 but the vast majority expressed CD103 and all were CD44hiCD62Llo and lacked typical exhaustion markers (Fig. 3E, S3D–E). These results further support a role for cognate antigen in either the generation or maintenance of TRM cells.
Fig. 3. TCR affinity of CD8 T cells is positively associated with TRM maintenance and phenotype.
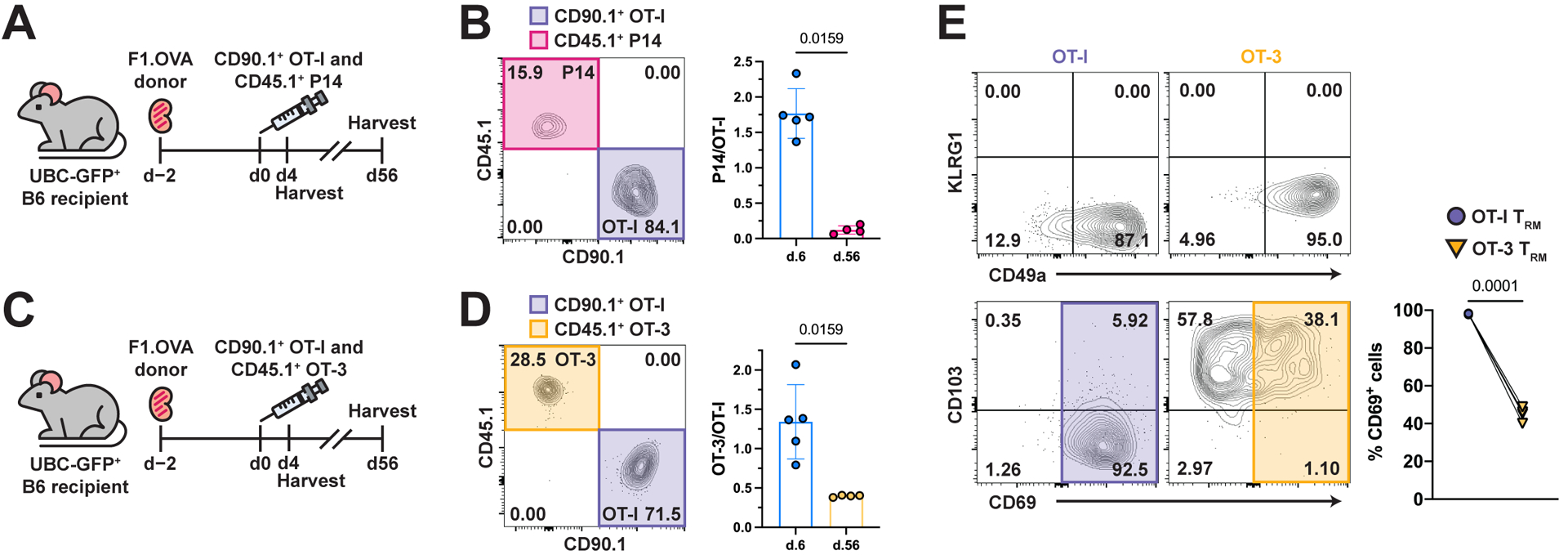
(A) F1.OVA kidney allografts were transplanted into GFP+ B6 recipients followed by co-adoptive transfer of 1 × 106 effector OT-I and P14 cells 2 days post-transplantation. Transplant recipients were harvested on days 6 (n = 5) and 56 (n = 4) to quantify CD8 TRM after gating on extravascular graft CD8+ T cells as shown in Fig. S1B.
(B) Percentage (representative d.56 plot, left) and ratio (graph, right) of CD45.1+ P14 to CD90.1+ OT-I cells on days 6 and 56.
(C) (Balb/c × B6.OVA) F1.OVA kidney allografts were transplanted into GFP+ B6 recipients followed by co-adoptive transfer of 1 × 106 CD90.1/90.2+ effector OT-I cells and CD45.1+ effector OT-3 cells 2 days post-transplantation. Transplant recipients were harvested on days 6 (n = 5) and 56 (n = 4) to quantify CD8 TRM after gating on extravascular graft CD8+ T cells as shown in Fig. S1B.
(D) Percentage (representative d.56 plot, left) and ratio (graph, right) of OT-3 to OT-I cells on days 6 and 56.
(E) Percentage (representative plots, left) of OT-I and OT-3 cells expressing TRM cell markers and percentage (graph, right) of OT-I and OT-3 cells that were CD69+ in the renal allograft on day 56.
P values were determined by (B, D, E) two-tailed paired t test.
Antigen presentation by recipient cells is required for TRM cell maintenance
In addition to the cognate interaction in secondary lymphoid tissues required for naïve T cell activation, effector T cells require a second cognate interaction within graft tissue to cause rejection (7, 8). These cognate interactions can consist of cross-presentation, whereby host DCs present alloantigen on self-MHC, or cross-decoration, whereby host DCs acquire intact, donor MHC-peptide complexes from graft cells. Therefore, we tested the effect of deleting either cross-presentation or cross-decoration on TRM OT-I cell numbers in the graft. To do so, we used combinations of recipient and donor mice lacking the H-2Kb MHC class I molecule (Fig. 4A). Here, tracking effector OT-I cells provides two advantages over tracking polyclonal T cells: 1) OT-I cells are an antigen-specific T cell population for which antigen presentation can be manipulated specifically by removing H-2Kb expression from either donor or recipient; and 2) unlike endogenous polyclonal T cells, effector OT-I cells were activated in wild-type mice prior to transfer, thus bypassing naïve T cell activation defects associated with the absence of H-2Kb. Significantly less TRM OT-I cells were present in allografts on day 56 in the absence of either cross-presentation (p=0.02) or cross-decoration (p=0.006)(Fig. 4B). The decrease of OT-I cells was not due to egress from renal allograft tissue into circulation (Fig. S4A). Moreover, OT-I cells declined more over time in the absence of either cross-presentation or cross-decoration (Fig. S4B and S4C), but remained functional as assessed by IFNγ production, albeit less so than in the presence of both antigen presentation pathways (Fig. 4C). These findings lend further support for the key role of cognate antigen in the generation or maintenance of functional TRM cells in allograft tissue.
Fig. 4. MHC-I presentation of cognate antigen via cross-priming and cross-decoration are required for TRM cell maintenance.
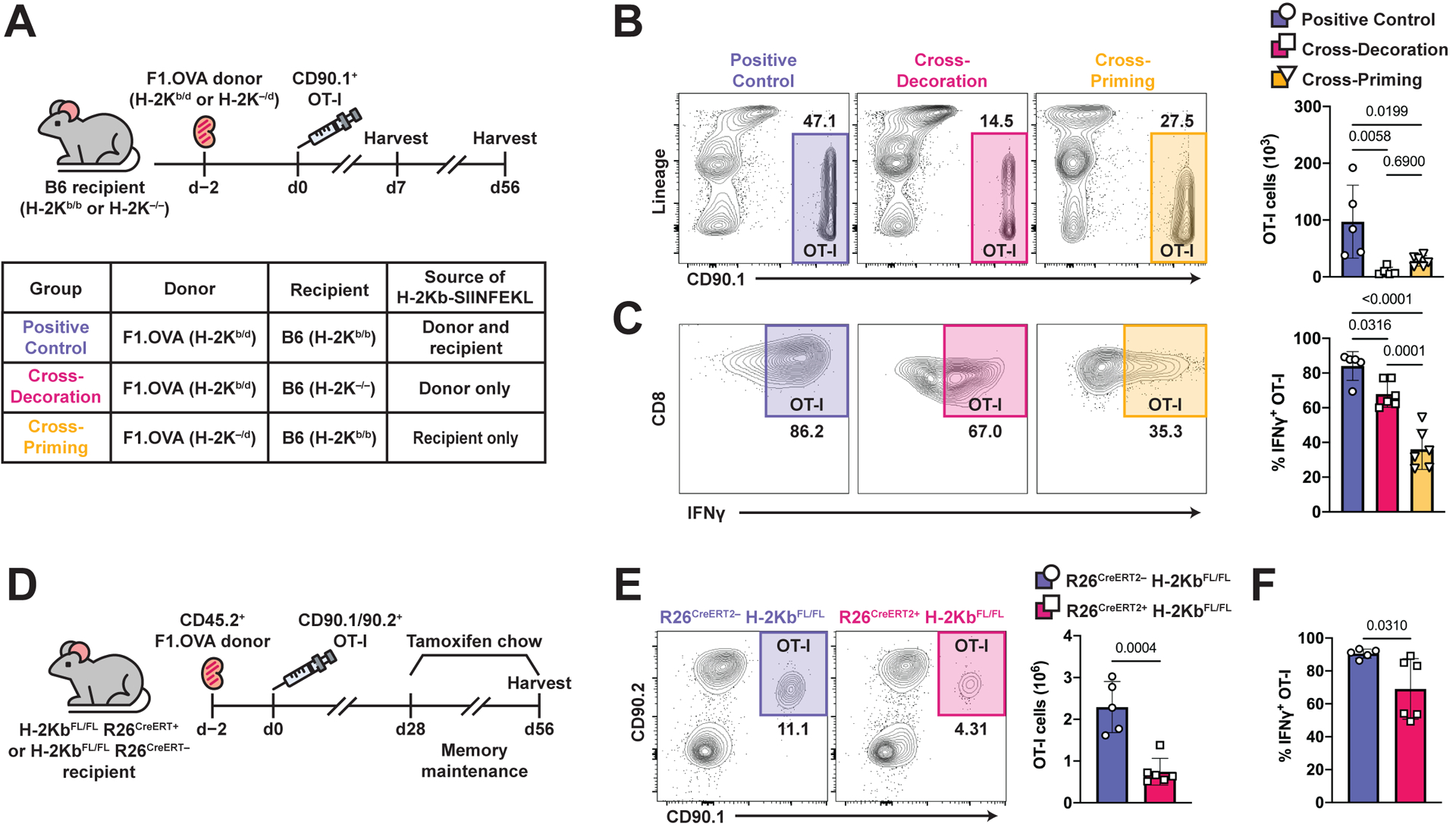
(A) F1.OVA H-2Kb-sufficient (H-2Kb/d) or F1.OVA H-2Kb-deficient (H-2K–/d) kidney allografts were transplanted into B6 H-2Kb-sufficient (H-2Kb/b) or B6 H-2Kb-deficient (H-2Kb–/–) recipients followed by co-adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Transplant recipients were harvested on days 7 and 56 to study maintenance of CD8 TRM cells. Flow analysis was performed after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. n = 5–6 mice per group.
(B) Representative flow plots and absolute number (graph) of OT-I cells from positive control mice (intact cross-priming and cross-decoration), cross-decoration only mice (deficient cross-priming), and cross-priming only mice (deficient cross-decoration) on day 56.
(C) Representative flow plots and frequency (graph) of IFNγ+ OT-I cells from positive control mice, cross-decoration only mice, and cross-priming only mice on day 56.
(D) F1.OVA kidney allografts were transplanted into either H-2KbFL/FL R26CreERT2– or H-2KbFL/FL R26CreERT2+ B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Tamoxifen chow was introduced from days 28–56, then recipients were subsequently harvested. n = 5–6 mice per group.
(E) Representative flow plots (left) and absolute number (graph) of TRM OT-I cells after tamoxifen treatment at 56 days post-transplantation.
(F) Frequency of IFNγ+ OT-I cells after restimulation with F1.OVA donor splenocytes.
P values were determined by (B, C) one-way ANOVA with Tukey’s correction, and (E, F) two-tailed unpaired t test.
To specifically test the role of antigen presentation during maintenance of TRM cells, we crossed H-2KbFL/FL mice with Rosa26Cre-ERT2 mice and used them as recipients to achieve tamoxifen-inducible deletion of H-2Kb (62). Tamoxifen was administered from 28–56 days post-transplantation to delete H-2Kb after TRM cells had already formed in the graft (Fig. 4D). H-2Kb ablation was achieved by day 42 (Fig. S4D) and resulted in reduction of intragraft TRM OT-I cells on day 56 (Fig. 4E). A previous report found that deletion of MHC-I in inflammatory settings results in NK cell-mediated missing-self reactivity towards MHC-I deficient cells (63). In our experiments, OT-I cells have intact MHC-I, and furthermore, we did not observe reduction of circulating CD4 T cells following tamoxifen-induced MHC-I deletion (Fig. S4E). Therefore, reduction of OT-I cell number on day 56 was not due to NK cell-mediated killing. Similar to our findings above (Fig. 4C), OT-I cells from H-2Kb-deficient recipients had decreased IFNγ production (Fig. 4F). Altogether, these findings demonstrate that cognate antigen presentation by recipient cells is essential in the maintenance of functional TRM cells in allograft tissue.
Recipient CD11c+ DCs are required for the maintenance of TRM cells during chronic allograft rejection
After transplantation, CD11c+ DCs derived from recipient monocytes become the principal antigen-presenting cell (APC) in the graft and their depletion prevents and interrupts graft rejection (7). Here, we confirmed that the majority of intragraft myeloid cells originate from recipient monocytes (Fig. S5A–S5D), and that the CD11c+ DCs are the predominant cell population presenting OVA (H-2Kb-SIINFEKL complex) (Fig. S5E). To address the hypothesis that cognate antigen is essential during the maintenance phase of TRM cells, we investigated the effect of temporal CD11c+ cell depletion (days 28–42) on the number of intragraft TRM cells using CD11c-DTR bone marrow chimeras as recipients (Fig. 5A). B6 chimeras (B6 bone marrow to B6 mice) served as controls. DT administration effectively depleted intragraft DCs (Fig. 5B) and significantly reduced intragraft TRM OT-I (p=0.002)(Fig. 5C) and both CD8 and CD4 polyclonal TRM cell populations (p<0.0001 and p=0.01, respectively) (Fig. 5D, Fig. S5F) in CD11c-DTR:B6 chimeric recipients. This reduction was associated with decreased tissue infiltration and fibrosis (Fig. 5E). Altogether, these data demonstrate that elimination of CD11c+ DCs during the maintenance phase reduces TRM cell numbers.
Fig. 5. Graft-infiltrating DCs are required for TRM cell maintenance and graft pathology.
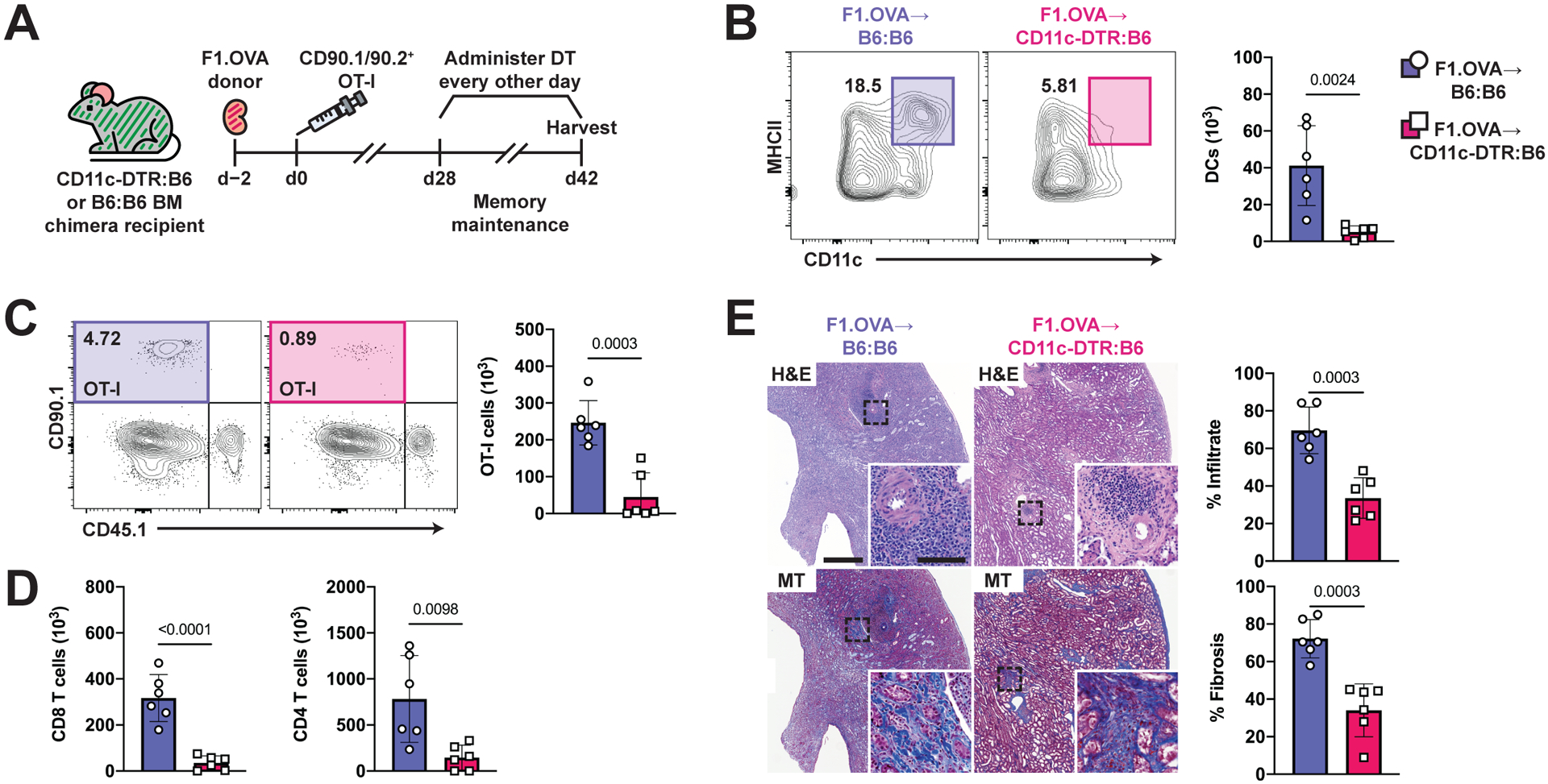
(A) F1.OVA kidney allografts were transplanted into CD11c-DTR:B6 chimeras (B6 mice reconstituted with CD11c-DTR bone marrow) or control B6:B6 chimeras (B6 mice reconstituted B6 bone marrow) followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Diphtheria toxin was administered to both groups every other day from days 28–42. Flow analysis was performed after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. n = 6 mice per group.
(B) Representative flow plots and absolute number (graph) of intragraft CD11c+MHCII+ DCs after DT treatment.
(C) Representative flow plots and absolute number (graphs) of TRM OT-I cells after DT treatment.
(D) Absolute number of recipient-derived polyclonal CD8 and CD4 TRM cells after DT treatment.
(E) H&E and MT-stained sections of F1.OVA renal allograft tissue (representative images) and quantification of infiltrate and fibrosis (graphs) from bone marrow chimera recipients on day 42 following DT treatment . Whole image scale bar: 500 μm. Inset scale bar: 100 μm.
P values were determined by (B-E) two-tailed unpaired t test.
Defective maintenance and function of TRM cells in the absence of IL-15 signaling
In addition to cognate interactions, DCs provide non-cognate signals to T cells that regulate their phenotype, function, and survival. In the setting of transplantation, antigen is persistent and inflammation is chronic; therefore, one would expect T cell dysfunction, yet functional and proliferating TRM cells persist in allograft tissue (Fig. 1C) (3). We considered the possibility that non-cognate interactions, specifically IL-15 signaling, support the maintenance and function of intragraft T cells (50, 51). Previous studies have shown that the cytokine IL-15 is essential for the long-term survival of TRM cells in various tissues (47, 58, 64). However, these studies employed acute viral infection models where inflammation resolves and antigen is cleared. In the context of organ transplantation, inflammation is chronic and antigen is persistent. Signaling by IL-15 requires trans-presentation by CD215 (IL-15Rα) to a responder cell expressing CD122 (IL-2Rβ) (Fig. 6A). Therefore, we developed a multiplex immunofluorescence panel to evaluate DCs expressing CD215 and their interactions with T cells (Fig. S6A), and stained renal allograft tissue 56 days post-transplantation (Fig. 6B). A substantial fraction of intragraft DCs (CD11c+CD11b+) expressed CD215 (36 ± 12%), constituting nearly 100% of all CD215+ cells in the allograft (Fig. 6B). Moreover, CD215+ DCs were found in closer proximity to intragraft T cells than non-DCs (Fig. 6C). Detection of IL-15 protein has been difficult, which may be in part due to the sequestering of IL-15 to the cell surface by CD215, or the inability of antibody to bind to IL-15 complexed to CD215. To overcome this limitation, we stained for IL-15 mRNA and found IL-15 expression throughout renal allograft tissue, especially in areas of high infiltration (Fig. S6B and S6C). We also found high CD122 expression on intragraft T cells on days 7 and 56 post-transplantation (Fig. S6D). These findings indicate that the components of the IL-15 ligand/receptor system are present in abundance in the local environment of the renal allograft.
Fig. 6. TRM cell maintenance and function depends on local IL-15 signaling in renal allograft tissue.
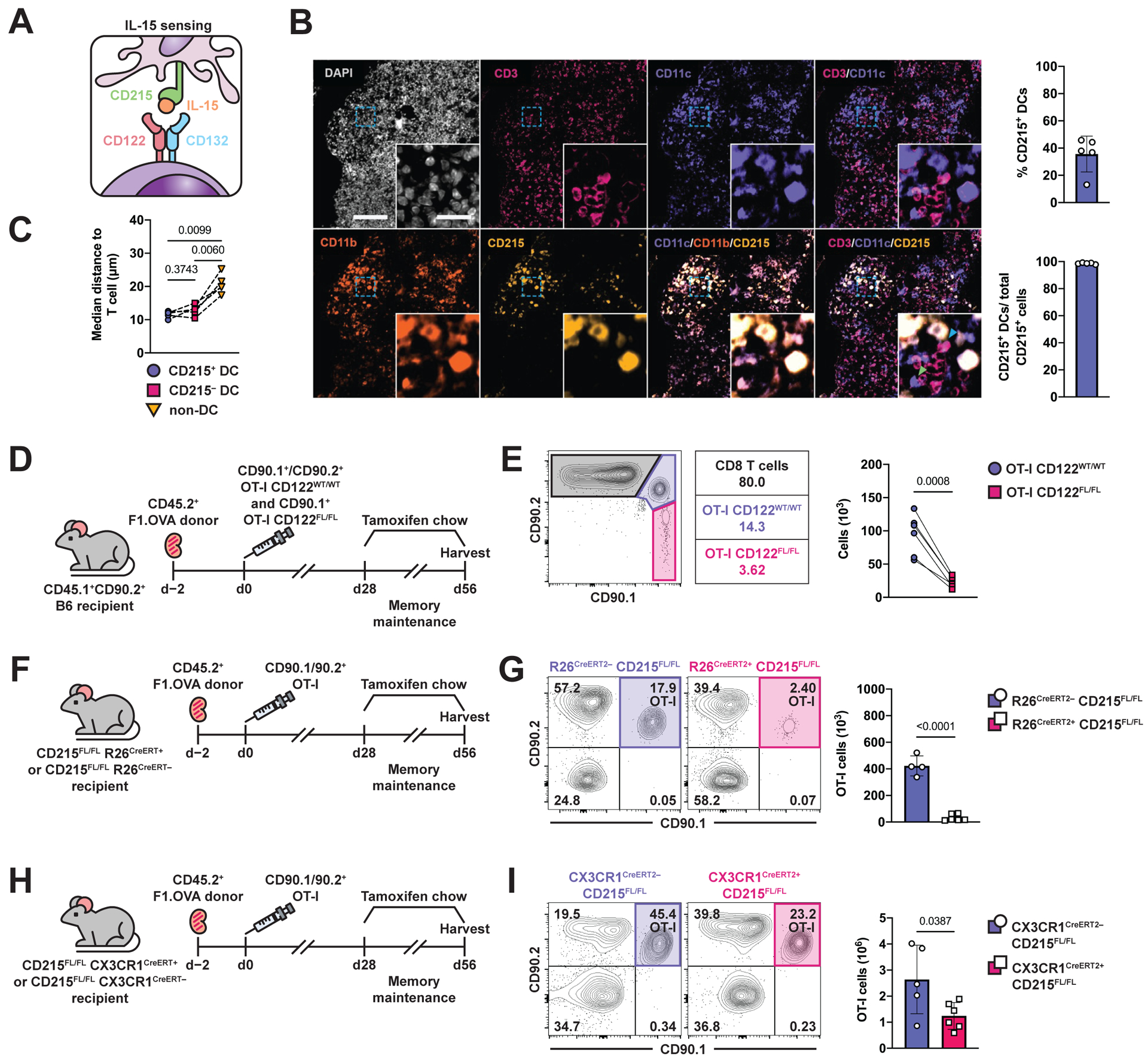
(A) Schematic of IL-15 signaling.
(B) Multiplex immunofluorescence staining of F1.OVA renal allograft on day 56 post-transplantation for nuclei (DAPI, white), T cells (CD3, magenta), dendritic cells (CD11b/CD11c, purple/orange), and CD215 expression (yellow). Close proximity of CD3+ cell with CD11c+CD215+cell indicated by blue arrow. Close proximity of CD3+ cell with CD11c+CD215–cell indicated by green arrow. Percentage of APCs expressing CD215 and percentage of CD215-expressing APCs out of total CD215+ cells shown in graphs. Whole image scale bar: 200 μm. Cropped inset scale bar: 20 μm. n = 5 mice.
(C) Median distance of T cells from CD215+ APCs, CD215– APCs, and non-APCs.
(D) F1.OVA kidney allografts were transplanted into CD45.1+ B6 recipients followed by co-adoptive transfer of 1 × 106 effector OT-I CD122WT/WT and effector OT-I CD122FL/FL R26CreERT2+ cells 2 days post-transplantation. Tamoxifen chow was introduced from days 28–56 post-transplantation, then recipients were subsequently harvested. Flow analysis was performed after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. n = 6 mice.
(E) Representative flow plots and absolute number (graph) of TRM OT-I on d.56 following tamoxifen treatment.
(F) F1.OVA kidney allografts were transplanted into either CD215FL/FL R26CreERT2– or CD215FL/FL R26CreERT2+ B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Tamoxifen chow was introduced from days 28–56. n = 4–6 mice per group.
(G) Representative flow plots and absolute number (graph) of TRM OT-I cells on day 56 following tamoxifen treatment.
(H) F1.OVA kidney allografts were transplanted into either CD215FL/FL CX3CR1CreERT2– or CD215FL/FL CX3CR1CreERT2+ B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Tamoxifen chow was introduced from days 28–56. n = 5–6 mice per group.
(I) Representative flow plots and absolute number (graph) of TRM OT-I cells on day 56 following tamoxifen treatment.
P values were determined by (C) mixed-effects model with Tukey’s correction, (E) two-tailed paired t test, and (G, I) two-tailed unpaired t test.
To investigate the role of IL-15 in TRM cell maintenance, we first deleted CD122 on TRM OT-I cells after they had established in the graft. To do so, we crossed CD122FL/FL mice and Rosa26CreERT2 mice onto the OT-I RAG–/– background (hereafter, referred to as OT-I CD122FL/FL) to achieve tamoxifen-inducible deletion of CD122, and used these mice to generate effector OT-I CD122FL/FL cells. Effector OT-I CD122WT/WT and OT-I CD122FL/FL cells were co-adoptively transferred to B6 recipients of F1.OVA renal allografts (Fig. 6D), and tamoxifen was administered from 28–56 days post-transplantation. CD122 staining confirmed the ablation of CD122 on transferred OT-I CD122FL/FL cells after tamoxifen treatment (Fig. S7A). Relative to OT-I CD122WT/WT cells, OT-I CD122FL/FL cells were present in significantly (p=0.0008) lower numbers in the grafts on day 56 (Fig. 6E). No significant change was observed in expression of CD69 and CD103 (Fig. S7B). These results suggest that IL-15 is important for TRM cell maintenance, but do not rule out a contribution of IL-2, which also binds to the CD122 subunit.
To specifically ablate IL-15 signaling, we next deleted CD215, which is required for IL-15 trans-presentation on recipient cells, by crossing CD215FL/FL mice with Rosa26Cre-ERT2 mice to allow for tamoxifen-inducible deletion of CD215. Tamoxifen was administered from 28–56 days post-transplantation to delete CD215 after TRM OT-I cells had already formed in the graft (Fig. 6F). Induced deletion of CD215 caused a reduction in both intragraft TRM OT-I cells (Fig. 6G) and recipient-derived polyclonal TRM cells (Fig. S7C), without altering the splenic lymphocyte compartment (Fig. S7D–S7F). Upon ex vivo restimulation with donor F1.OVA splenocytes, graft OT-I and polyclonal T cells from CD215FL/FL Rosa26Cre-ERT2+/– recipients produced less IFNγ and expressed less Ki67 and BCL-2 (Fig. S7G and S7H). Given the large overlap between CD11c, CD11b and CD215 expression in our immunofluorescence staining (Fig. 6B), we also asked whether trans-presentation of IL-15 by intragraft DCs is important for TRM maintenance. Since a substantial proportion of intragraft DCs express CX3CR1 (Fig. S5C), we crossed CD215FL/FL mice with CX3CR1Cre-ERT2 mice to allow for tamoxifen-inducible deletion of CD215 on these DCs. Tamoxifen was administered from 28–56 days post-transplantation (Fig. 6H). Induced deletion of CD215 on DCs caused an approximately 50% reduction [(−1.40 ± 0.58) × 106 cells] in intragraft TRM OT-I cells TRM cells (Fig. 6I). Altogether, these data indicate that IL-15 trans-presented by CD215-expressing cells, which include intragraft DCs, is required for the maintenance and function of TRM cells in renal allograft tissue.
Antibody blockade of IL-15 signaling disrupts TRM cell maintenance and prevents chronic allograft rejection
Given the importance of IL-15 signaling for TRM cell maintenance in renal allograft tissue, we hypothesized that antibody blockade of IL-15 sensing would deplete intragraft TRM cells and prevent damage to the graft. To test this hypothesis, we treated murine transplant recipients with chronically rejecting grafts three times weekly between 28 and 42 days post-transplantation with anti-CD122 antibody (Fig. 7A). Anti-CD122 (clone: ChMBC7) is a chimeric rat/mouse antibody that has been engineered to eliminate Fc-mediated effector functions (64–66). An additional group of animals were treated with isotype control antibody. Since CD122 also contributes to IL-2 signaling (Fig. S7I), a group receiving anti-CD25 (clone: 7D4) was included as a control to distinguish between blocking IL-15 and IL-2 signaling. After two weeks of systemic treatment, fewer TRM OT-I and recipient-derived polyclonal T cells were found in renal allografts of mice treated with anti-CD122 compared to isotype controls (Fig. 7B and 7C). In contrast, anti-CD25 treatment did not significantly reduce the number of intragraft TRM OT-I or polyclonal T cells. Anti-CD122 treatment did not have a significant effect on intragraft B, NK, and regulatory T cell number (Fig. S7J). Numbers of splenic lymphocytes, including CD44+ CD8 and CD4 T cells, were also not affected (Fig. S7K–S7M). Importantly, decreased renal allograft infiltration and fibrosis was observed in anti-CD122-treated, but not anti-CD25-treated, recipients compared to isotype controls (Fig. 7D). Altogether, these data indicate that anti-CD122 selectively disrupts TRM cell maintenance in renal allograft tissue and reduces graft pathology.
Fig. 7. Antibody blockade of IL-15 signaling disrupts TRM cell maintenance in renal allograft tissue and prevents chronic rejection.
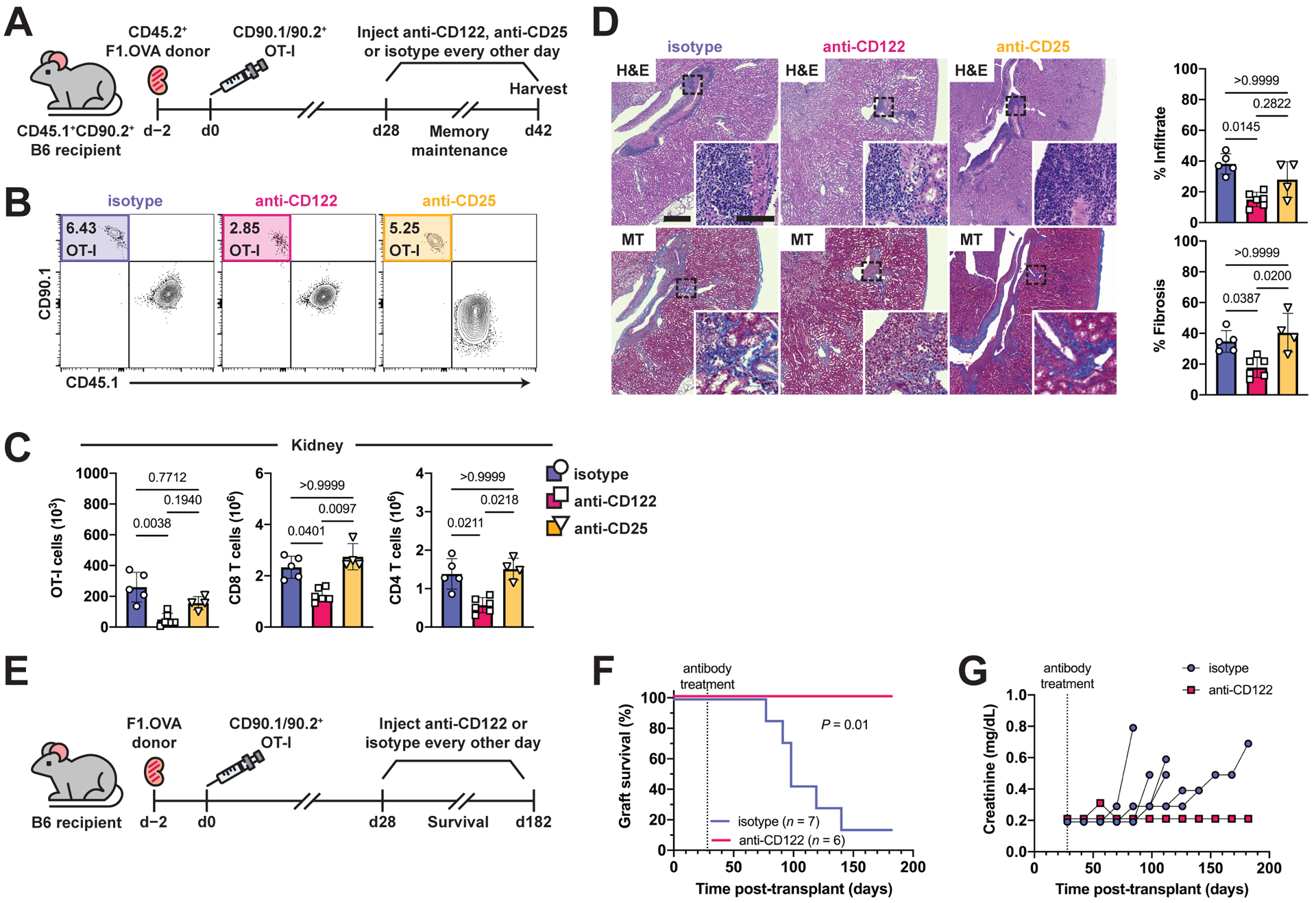
(A) F1.OVA kidney allografts were transplanted into CD45.1+ B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Isotype, anti-CD122, or anti-CD25 antibodies were administered three times weekly from days 28–42 post-transplantation. Flow analysis was performed after gating on extravascular graft CD8+ T cells as shown in Fig. S1B. n = 4–6 mice per group.
(B) Percentage of TRM OT-I cells after antibody treatment.
(C) Absolute numbers of TRM OT-I and CD8 and CD4 recipient-derived polyclonal TRM cells after antibody treatment.
(D) H&E and MT-stained sections of F1.OVA renal allograft tissue and quantification of infiltrate and fibrosis (graphs) from antibody-treated recipients on day 42. Whole image scale bar: 400 μm. Inset scale bar: 100 μm.
(E) F1.OVA kidney allografts were transplanted into CD45.1+ B6 recipients followed by adoptive transfer of 1 × 106 effector OT-I cells 2 days post-transplantation. Isotype or anti-CD122 antibody were administered three times weekly beginning on day 28 post-transplantation. n = 6–7 mice per group.
(F) Kaplan-Meier curve of graft survival after antibody treatment.
(G) Serum creatinine measurements throughout the course of antibody treatment.
P values were determined by (C, D) Kruskal–Wallis one-way ANOVA with Dunn’s correction, and (F) log-rank (Mantel–Cox) test.
Currently, no immunotherapies have been clinically proven to be effective in the prevention or treatment of chronic rejection. Given that IL-15 signaling is required for TRM cell maintenance and TRM cells mediate chronic rejection (3), we tested whether long-term CD122 blockade prevents chronic rejection. Transplant recipients with established intragraft TRM cells (28 days post-transplantation) were treated three times weekly with anti-CD122 antibody or isotype control antibody until graft failure or until the end of the 182-day study period (Fig. 7E). Recipient mice underwent bilateral nephrectomy prior to receiving a renal allograft, thus tying recipient survival to graft survival. Long-term treatment with anti-CD122 prevented graft failure as evidenced by 100% recipient survival at 182 days (Fig. 7F). Analysis of serum creatinine also revealed that anti-CD122 treatment preserved allograft function (serum creatinine remained normal) (Fig. 7G). These data suggest that anti-CD122 blockade could be a potential therapy for prolongation of human transplant survival, given the selective targeting of alloreactive TRM cells and prevention of chronic rejection.
DISCUSSION
Recent studies have sought to identify the factors that underlie TRM cell maintenance in non-lymphoid tissues (44, 45, 47, 58, 67). It is emergent that there is heterogeneity in TRM maintenance depending on the environments in which they reside (68–70). Here, we examined TRM cell maintenance in a setting where TRM cells reside in the kidney, a non-barrier tissue, and where antigen and inflammation are persistent. We found that cognate antigen and IL-15 play important non-redundant roles in the maintenance and functionality of TRM cells. Both were required. Presentation of antigen and IL-15 were mediated by intragraft DCs, and ablation of DCs led to disruption of TRM cell maintenance. Our findings extend our understanding of TRM cell maintenance to settings where antigen is persistent, inflammation is chronic, and TRM cells reside in non-barrier tissue. Such findings are not only relevant to transplantation, but also apply to TRM cell maintenance and function in tumors and autoimmune diseases (18, 71–73).
In the skin, antigen has been shown to be necessary for the optimal generation of TRM cells after acute viral infection (44, 45), but it remained to be defined whether antigen plays a role in TRM maintenance. Several studies have indicated that TRM cells persist independently of antigen (42, 43, 46). These studies focused on TRM cell persistence in skin or mucosal barrier tissues (46, 58) or in RAG–/– hosts where memory-like cell generation is driven by lymphopenia and not cognate antigen (43). Here, we investigated TRM maintenance in a non-barrier tissue free from environmental and commensal antigens where cognate antigen was continually present. Using multiple approaches, which included modulating TCR affinity for cognate antigen and temporally removing antigen or antigen presentation, we demonstrated that antigen is required for the maintenance of TRM cells. Notably, we observed a shift towards a lower proportion of OT-3 TRM expressing CD69 (Fig. 3E). CD69– TRM cells have been previously reported (74), and it is possible that antigen affinity modulates CD69 expression as the OT-3 TCR has lower affinity for the OVA SIINFEKL peptide than the OT-I TCR. We also observed reduced IFNγ expression upon recall of TRM if only cross-presentation was present in the graft (Fig. 4C). This can be potentially explained by higher density of donor MHCI-peptide complexes on cross-decorated DCs, where staining of donor molecules is punctate, versus diffuse distribution of recipient MHCI-peptide complexes along the DC surface during cross-presentation (75).
Similarly, we established a key non-redundant role for IL-15 in TRM cell maintenance by temporally deleting IL-15 trans-presentation or IL-15 receptor, and antibody blockade of IL-15 sensing. The IL-15 findings are consistent with published data in which IL-15 was shown to be important for the maintenance of TRM cells in skin, lungs, and native kidney (47, 58, 64). Our findings that DCs are the predominant cell type presenting cognate antigen and IL-15 in the graft, and that DC depletion interrupts TRM maintenance, suggest that DCs could provide antigen and IL-15 signals simultaneously to TRM cells. This is plausible given that DCs form a dense network in the graft that interacts intimately with infiltrating T cells (7, 76). Although earlier blockade of IL-15 signaling could further decrease fibrosis, we suspect that would not be the case because effector T cells have a stronger reliance on IL-2, whereas memory T cells have a stronger reliance on IL-15 because they lack or have much less IL-2Rα (CD25) expression. It is also possible that other APCs in the kidney such as CD11c– macrophages contribute to IL-15 trans-presentation.
Our observation that TRM cells continue to receive TCR stimulation in the graft tissue raises the possibility of T cell dysfunction or exhaustion under these conditions, as has been observed in tumors and chronic viral infection (77–80). However, we find that antigen is in fact required for the maintenance and function of TRM cells. We speculate that chronic inflammation in the graft may prevent T cell dysfunction, possibly via the actions of survival or proliferation signals. IL-15 could provide such signals (81, 82) because: 1) IL-15 drives the homeostatic proliferation of memory T cells (48); 2) IL-15 is upregulated in rejecting allograft tissue (57); 3) local IL-15 is necessary for intragraft TRM cell maintenance (Fig. 6 and 7); 4) intratumoral IL-15 enhances the function of infiltrating CD8 T cells and is associated with increased patient survival (50, 52); 5) IL-15 administration expanded CD8 effector memory T cells in SIV-infected monkeys with controlled viremia (83); and 6) in the chronic LCMV model, tissue-specific deletion of IL-15 enhanced T cell exhaustion (84). Additional studies are needed to test whether IL-15 or other factors rescue TRM cells continually exposed to antigen from terminal exhaustion or increase the generation of functional TRM cells from progenitor-like precursors in the graft (82, 84).
Limitations of our study include inability to track endogenous allospecific polyclonal T cells and reliance on transferred TCR-transgenic T cells to investigate the roles of antigen and IL-15. Transferred effector T cells, particularly ones with a high affinity transgenic TCR, could potentially behave differently from effectors generated from endogenous naive T cells.
Overall, our study demonstrates that TRM cells persist and function in a non-barrier tissue containing persistent antigen and chronic inflammation as a result of cognate antigen and IL-15 signaling. We thus offer a different lens on TRM cell biology by revealing their maintenance in an environment that emulates chronic diseases afflicting humans, such as autoimmunity and transplant rejection. Furthermore, we have identified actionable targets for clinical translation given that chronic rejection remains a major obstacle to long-term survival of allografts. Manipulating these maintenance signals could ablate pathogenic TRM cells that likely underlie chronic pathologies.
MATERIALS AND METHODS
Study Design
We utilized a mouse kidney transplantation model to study for role of tissue-resident memory T cells in transplantation. Three biological replicates (3 individual transplant recipients) per group were included in each experiment. Experiments were repeated once resulting in a total of up to 6 biological replicates. Both sexes of mice were used, but males were preferred for the transplantation procedure due to size and anatomy. Sample sizes were based on prior observations that three to six biological replicates were sufficient to discern statistically significant differences between groups, with observed effect sizes >0.5. Prospective exclusion criteria were transplant recipient death within the first 7 days after transplantation (technical failure). All other data points were included, and no outliers were excluded. All end points were prospectively selected. It was not possible to blind the study because of the need to identify donors and recipients. Initial flow cytometry data analysis was not blinded, but analysis by a second investigator was blinded.
Animals
B6.CD45.2 (C57BL/6J; Thy1.2, CD45.2), B6.CD45.1 (B6.SJL-PtprcaPepcb/BoyJ, Thy1.2, CD45.1), DsRed [B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J], C57BL/6-Tg(UBC-GFP)30Scha/J, CD11c-DTR (B6.FVB-1700016L21RikTg(Itgax-DTR/EGFP)57Lan/J), Rosa26-CreERT2 (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J), CD215FL/FL (C57BL/6-Il15ratm2.1Ama/J), and CD122FL/FL (B6.129S1-Il2rbtm1Ukl/J) were from Jackson Laboratory (Jax). B6.CD45.1 (B6.SJL-PtprcaPepcb/BoyCrl) were from Charles River Laboratories. B6.Kb–/–(B6.129P2-H2-Kbtm1N12) were from Taconic. B6.Act-OVA.H-2Kb–/– mice were generated by breeding B6.Act-OVA mice with B6.Kb–/– mice. F1.OVA (F1.Act-mOVA) and F1.OVA.Kb–/– (F1.Act-mOVA.H-2Kb–/–) mice were generated by breeding BALB/cJ mice with B6.Act-OVA (C57BL/6-Tg(CAG-OVAL)-916Jen/J) and B6.Act-OVA.H-2Kb–/– mice, respectively. OT-I mice (C57BL/6-Tg[TcraTcrb]1100Mjb/J; CD45.2) were obtained from Jax and maintained on a RAG–/– Thy1.1 or RAG–/– DsRed Thy1.1/Thy1.2 background. P14 mice were provided by L.P.K. (University of Pittsburgh) and crossed with RAG–/– and CD45.1 mice to generate P14 RAG–/– CD45.1 mice. OT-3 TCRα–/– CD45.1 were provided by Dr. Dietmar Zehn (Lausanne University Hospital) (61). H-2KbFL/FL mice were provided by A.J.J. (Mayo Clinic) (62). OT-I CD122FL/FL R26CreERT2 mice were generated by breeding CD122FL/FL and Rosa26-CreERT2 mice onto the OT-I RAG–/– Thy1.1 background. CD215FL/FL R26CreERT2 mice were generated by breeding CD215FL/FL mice with Rosa26-CreERT2 mice. H-2KbFL/FL R26CreERT2 mice were generated by breeding H-2KbFL/FL mice with Rosa26-CreERT2 mice. All mouse work was performed in compliance with ethical regulations and was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Kidney transplantation
Mouse kidney transplants were performed as previously described (3). Recipient native kidneys were removed during the transplantation procedure. Allograft rejection was monitored by visual observation of recipients for signs of uremia (lethargy, decreased mobility, and ruffled hair) or death. Serial serum creatinine measurements were performed using an i-Stat Analyzer (Abbott) at a lower detection limit of 0.2 mg/dL.
Generation and adoptive transfer of effector OT-I, OT-3, and P14 cells
To generate effector OT-I and OT-3 cells, B6.CD45.1 mice were injected i.v. with 25μg anti-DEC-205-OVA hybrid antibody (W.D.S., University of Pittsburgh), 50μg anti-CD40 (clone: FGK4.5), and 5 × 105 splenocytes from either OT-I RAG–/– Thy1.1, OT-I RAG–/– DsRed Thy1.1/Thy1.2+, OT-I RAG–/– Thy1.1 CD122FL/FL Rosa26-CreERT2, or OT-3 TCRα–/– CD45.1+ mice. To generate effector P14 cells, mice were infected with 2 × 105 plaque-forming units of LCMV Armstrong followed by i.v. injection of 1 × 103 splenocytes from P14 RAG–/– CD45.1+ mice. Effector OT-I, OT-3, or P14 cells were high-speed sorted 6 days later using a BD FACS Aria (BD Biosciences) by gating on CD4–Lineage (CD11c, CD11b, CD16/32, Ter119, B220, F4/80, and CD49b)– followed by appropriate congenic markers. Sorted 1 × 106 effector cells (CD44+CD62L– (Fig. S1A)) were injected i.v. into transplanted mice as indicated. Effector OT-3 cell generation was comparable to effector OT-I cells (Fig. S3B).
Preparation of single cell suspensions from tissue
Mice were anesthetized and injected with 3μg anti-CD45 BUV395 (clone: 30-F11, BD Biosciences) i.v. 2 minutes before euthanasia to label intravascular cells (85). Single cell suspensions of kidney, spleen, liver, lung, and blood were prepared as previously described (3). RBC lysis was performed on spleen and blood for 5 min using RBC lysis buffer (R7757, Sigma Aldrich). Kidney, liver, and lung were homogenized (gentleMACS, Miltenyi Biotec), followed by digestion with collagenase IV (350 U/mL), DNAse I (50 U/mL), and 5% FBS in RPMI for 45 minutes at 37°C. Leukocytes were isolated by gradient centrifugation using Lympholyte M (CedarLane Labs).
Flow cytometry
Cells were incubated with Fc Block (clone: 2.4G2, BD Biosciences) for 10 minutes, stained with antibodies (Table S1) for 30 minutes and dead cells labeled with Zombie NIR Fixable Viability Kit (BioLegend). Intracellular staining was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). BrdU and EdU staining were performed with Phase Flow BrdU Kit (BioLegend) and Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Invitrogen), respectively. Anti-BrdU-Alexa Fluor 647 (clone: MoBU-1, BioLegend), which does not cross-react with EdU, was used for BrdU staining. For intracellular cytokine staining, cells from allograft tissue were stimulated ex vivo with CFSE-labelled donor F1.OVA splenocytes at a 1:1 ratio in the presence of GolgiPlug (BD Biosciences) for 16 hours at 37°C, followed by staining for cell surface markers and intracellular staining. Samples were acquired on a 5-laser Cytek Aurora and analyzed with FlowJo V10 software (BD Biosciences).
Bone marrow chimeras
CD11c-DTR:B6 and B6:B6 bone marrow chimeras were generated by irradiating B6.CD45.1 mice with 10 Gy followed by adoptive transfer 10 × 106 BM cells i.v. from CD11c-DTR or B6.CD45.2 mice, respectively. Mice received sulfatrim food for 14 days after irradiation. Reconstitution was confirmed 56 days after bone marrow transplantation by analyzing peripheral blood cell composition via flow cytometry.
In vivo treatment
To deplete CD11c-expressing DCs, CD11c-DTR:B6 and B6:B6 bone marrow chimera recipients were injected with diphtheria toxin i.p. (4 ng per gram of body weight) every 2 days. To block CD122 signaling, 200 μg of anti-CD122 (clone: ChMBC7, J.N. Biosciences), anti-CD25 (clone: 7D4, BioXcell), or isotype (clone: MBP100, J.N. Biosciences) antibody were injected i.v. three times per week for 14 days, beginning 28 days post-transplantation. For allograft survival experiments, antibodies were injected every week until the end of the study (182 days post-transplantation).
Histological analysis
Kidney allograft tissue was fixed in formalin, paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E), Masson’s trichrome (MT), and periodic acid–Schiff (PAS) stain (Magee-Womens Research Institute Histology and Microimaging Core, University of Pittsburgh). For immunofluorescence, formalin-fixed, paraffin-embedded (FFPE) kidney allograft tissue sections were baked for 1 hour at 60°C, followed by deparaffinization and rehydration. Antigen retrieval was performed at pH 6.0 for 45 minutes at 90°C (Agilent), followed by protein block (Agilent) for 20 minutes at room temperature. Slides were then stained with primary antibodies for 16 hours at 4°C. Following avidin/biotin blocking, slides were incubated with biotinylated secondary antibody for 30 minutes at room temperature, and then streptavidin-conjugated quantum dots for 30 minutes at room temperature. Stained sections were mounted in EcoMount (Biocare Medical). RNAscope fluorescent in-situ hybridization for detection of IL-15 was performed by Advanced Cell Diagnostics. DAPI was used to visualize nuclei. Positive and negative control probes were tested. Slides were scanned on a Zeiss Axioscan.Z1 with a 20× objective and analyzed in QuPath (86), ImageJ, and Python.
Statistics and reproducibility
Statistical analysis was performed using Prism v.9 (GraphPad). Parametric and non-parametric tests were used as indicated in figure legends. All p values, regardless of statistical significance, were reported.
Supplementary Material
Acknowledgements:
Our gratitude goes to Heth Turnquist, JoAnne Flynn, Mandy McGeachy, Matthew Nicotra, Simon Watkins, and Faruk Sacirbegovic for helpful discussions of experimental design and interpretation; Dietmar Zehn for providing OT-3 mice; William Shufesky for technical assistance with imaging; Sarah Rosenberger for generating anti-DEC-205/OVA; and the Unified Flow Cytometry Core (Lisa Borghesi, Dewayne Falkner, Aarika MacIntyre, Heidi Gunzelman, Nan Sheng, and Nevil Abraham) for sorting cells and technical assistance.
Funding:
This work was supported by National Institute of Health (NIH) grant R01 AI049466 (F. Lakkis), R01 AI145881 (M. Oberbarnscheidt), R01 HL143349 (W. Shlomchik), R01 GM136148 (L. Kane), R01 NS103212 (A. Johnson), T32 grant GM008208 (R. Tieu), T32 grant AI074490 (R. Tieu), F30 grant DK124925 (R. Tieu), and the American Society of Transplantation Sanofi Basic Science Fellowship Research Grant (N. Feizi). The Unified Flow Cytometry Core was funded by NIH grants 1S10OD011925-01 and 1S10OD019942-01.
Footnotes
Competing interests: Dr. J. Yun Tso is a cofounder and managing partner of JN Biosciences LLC.
Data and materials availability:
All data are available in the main text or the supplementary materials. All materials will be made available upon request.
References and Notes
- 1.Ibrahim S, Dawson DV, Sanfilippo F, Predominant infiltration of rejecting human renal allografts with T cells expressing CD8 and CD45RO. Transplantation 59, 724–728 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Loupy A, Haas M, Roufosse C, Naesens M, Adam B, Afrouzian M, Akalin E, Alachkar N, Bagnasco S, Becker JU, Cornell LD, Clahsen-van Groningen MC, Demetris AJ, Dragun D, Duong van Huyen JP, Farris AB, Fogo AB, Gibson IW, Glotz D, Gueguen J, Kikic Z, Kozakowski N, Kraus E, Lefaucheur C, Liapis H, Mannon RB, Montgomery RA, Nankivell BJ, Nickeleit V, Nickerson P, Rabant M, Racusen L, Randhawa P, Robin B, Rosales IA, Sapir-Pichhadze R, Schinstock CA, Seron D, Singh HK, Smith RN, Stegall MD, Zeevi A, Solez K, Colvin RB, Mengel M, The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. American Journal of Transplantation 20, 2318–2331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abou-Daya KI, Tieu R, Zhao D, Rammal R, Sacirbegovic F, Williams AL, Shlomchik WD, Oberbarnscheidt MH, Lakkis FG, Resident memory T cells form during persistent antigen exposure leading to allograft rejection. Science immunology 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg AS, Singer A, Cellular Basis of Skin Allograft Rejection: An In Vivo Model of Immune-Mediated Tissue Destruction. Annual review of immunology 10, 333–358 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Hancock WW, Gao W, Faia KL, Csizmadia V, Chemokines and their receptors in allograft rejection. Current opinion in immunology 12, 511–516 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Walch JM, Zeng Q, Li Q, Oberbarnscheidt MH, Hoffman RA, Williams AL, Rothstein DM, Shlomchik WD, Kim JV, Camirand G, Lakkis FG, Cognate antigen directs CD8+ T cell migration to vascularized transplants. Journal of Clinical Investigation 123, 2663–2671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhuang Q, Liu Q, Divito SJ, Zeng Q, Yatim KM, Hughes AD, Rojas-Canales DM, Nakao A, Shufesky WJ, Williams AL, Humar R, Hoffman RA, Shlomchik WD, Oberbarnscheidt MH, Lakkis FG, Morelli AE, Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nature communications 7, 12623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes AD, Zhao D, Dai H, Abou-Daya KI, Tieu R, Rammal R, Williams AL, Landsittel DP, Shlomchik WD, Morelli AE, Oberbarnscheidt MH, Lakkis FG, Cross-dressed dendritic cells sustain effector T cell responses in islet and kidney allografts. Journal of Clinical Investigation 130, 287–294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG, Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proceedings of the National Academy of Sciences 99, 6175–6180 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D, Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (New York, N.Y.) 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN, Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science (New York, N.Y.) 346, 101–105 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR, Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nature immunology 10, 524–530 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T, The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nature immunology 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, Inoue T, Kluger Y, Kaech SM, Kurosaki T, Okada T, Flavell RA, KLRG1(+) Effector CD8(+) T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48, 716–729.e718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, Friedman AL, Shen Y, Farber DL, Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell reports 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA, Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Science translational medicine 7, 279ra239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, Bevan MJ, The molecular signature of tissue resident memory CD8 T cells isolated from the brain. Journal of Immunology 189, 3462–3471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, Hoog C, Tjernlund A, Michaelsson J, Folkersen L, Mjosberg J, Blomqvist L, Ehrstrom M, Stahle M, Bryceson YT, Eidsmo L, CD49a Expression Defines Tissue-Resident CD8(+) T Cells Poised for Cytotoxic Function in Human Skin. Immunity 46, 287–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, Mackay LK, Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nature immunology 19, 183–191 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, Masopust D, Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nature immunology 19, 173–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Leur K, Dieterich M, Hesselink DA, Corneth OBJ, Dor F, de Graav GN, Peeters AMA, Mulder A, Kimenai H, Claas FHJ, Clahsen-van Groningen MC, van der Laan LJW, Hendriks RW, Baan CC, Characterization of donor and recipient CD8+ tissue-resident memory T cells in transplant nephrectomies. Scientific reports 9, 5984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E, Carpenter D, Marboe C, Benvenuto L, Shah L, Robbins H, Hook JL, Sykes M, D’Ovidio F, Bacchetta M, Sonett JR, Lederer DJ, Arcasoy S, Sims PA, Farber DL, Generation and persistence of human tissue-resident memory T cells in lung transplantation. Science immunology 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuber J, Shonts B, Lau SP, Obradovic A, Fu J, Yang S, Lambert M, Coley S, Weiner J, Thome J, DeWolf S, Farber DL, Shen Y, Caillat-Zucman S, Bhagat G, Griesemer A, Martinez M, Kato T, Sykes M, Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Science immunology 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R, Dynamic T cell migration program provides resident memory within intestinal epithelium. Journal of Experimental Medicine 207, 553–564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iijima N, Iwasaki A, A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (New York, N.Y.) 346, 93–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R, Viral immune evasion due to persistence of activated T cells without effector function. Journal of Experimental Medicine 188, 2205–2213 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R, Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. Journal of Experimental Medicine 207, 2187–2194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, Vyth-Dreese FA, Luiten RM, Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. Journal of Investigative Dermatology 129, 2220–2232 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, Goronzy JJ, Weyand CM, CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. Journal of Experimental Medicine 195, 1325–1336 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Di Meglio P, Villanova F, Navarini AA, Mylonas A, Tosi I, Nestle FO, Conrad C, Targeting CD8(+) T cells prevents psoriasis development. Journal of Allergy and Clinical Immunology 138, 274–276.e276 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schröder R, Deckert M, Schmidt S, Ravid R, Rajewsky K, Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. Journal of Experimental Medicine 192, 393–404 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF, Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis and Rheumatology 52, 201–211 (2005). [DOI] [PubMed] [Google Scholar]
- 34.O. M. T. S. Group, A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. New England Journal of Medicine 313, 337–342 (1985). [DOI] [PubMed] [Google Scholar]
- 35.Haverty TP, Sanders M, Sheahan M, OKT3 treatment of cardiac allograft rejection. Journal of Heart and Lung Transplantation 12, 591–598 (1993). [PubMed] [Google Scholar]
- 36.Cosimi AB, Jenkins RL, Rohrer RJ, Delmonico FL, Hoffman M, Monaco AP, A randomized clinical trial of prophylactic OKT3 monoclonal antibody in liver allograft recipients. Archives of Surgery 125, 781–784; discussion 785 (1990). [DOI] [PubMed] [Google Scholar]
- 37.Maraninchi D, Gluckman E, Blaise D, Guyotat D, Rio B, Pico JL, Leblond V, Michallet M, Dreyfus F, Ifrah N, et al. , Impact of T-cell depletion on outcome of allogeneic bone-marrow transplantation for standard-risk leukaemias. Lancet (London, England) 2, 175–178 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Lau LL, Jamieson BD, Somasundaram T, Ahmed R, Cytotoxic T-cell memory without antigen. Nature 369, 648–652 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Hou S, Hyland L, Ryan KW, Portner A, Doherty PC, Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature 369, 652–654 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Gray D, Matzinger P, T cell memory is short-lived in the absence of antigen. Journal of Experimental Medicine 174, 969–974 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oehen S, Waldner H, Kündig TM, Hengartner H, Zinkernagel RM, Antivirally protective cytotoxic T cell memory to lymphocytic choriomeningitis virus is governed by persisting antigen. Journal of Experimental Medicine 176, 1273–1281 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T, Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proceedings of the National Academy of Sciences 109, 7037–7042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D, Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. Journal of Immunology 188, 4866–4875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC, Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. Journal of Experimental Medicine 213, 951–966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, König PA, Tao S, Tao R, Heikenwälder M, Busch DH, Korn T, Kastenmüller W, Drexler I, Gasteiger G, Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. Journal of Experimental Medicine 213, 3075–3086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wijeyesinghe S, Beura LK, Pierson MJ, Stolley JM, Adam OA, Ruscher R, Steinert EM, Rosato PC, Vezys V, Masopust D, Expansible residence decentralizes immune homeostasis. Nature 592, 457–462 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, Masopust D, IL-15-Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. Journal of Immunology 196, 3920–3926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A, IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity 9, 669–676 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, Malynn BA, Ma A, Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity 31, 811–822 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA, Restifo NP, IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proceedings of the National Academy of Sciences 101, 1969–1974 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, Tan X, Sutton SE, Cooke MP, Ohlén C, Greenberg PD, Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nature medicine 12, 335–341 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, Lafontaine L, Bilocq AM, Kirilovsky A, Tosolini M, Waldner M, Berger A, Fridman WH, Rafii A, Valge-Archer V, Pagès F, Speicher MR, Galon J, Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Science translational medicine 6, 228ra237 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, Ebert EC, Green PH, Jabri B, Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366 (2004). [DOI] [PubMed] [Google Scholar]
- 54.McInnes IB, al-Mughales J, Field M, Leung BP, Huang FP, Dixon R, Sturrock RD, Wilkinson PC, Liew FY, The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nature medicine 2, 175–182 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, Murase K, Cutler C, Ho VT, Alyea EP, Armand P, Blazar BR, Antin JH, Soiffer RJ, Ritz J, Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Science translational medicine 5, 179ra143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N, Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. Journal of Immunology 185, 5693–5703 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Pavlakis M, Strehlau J, Lipman M, Shapiro M, Maslinski W, Strom TB, Intragraft IL-15 transcripts are increased in human renal allograft rejection. Transplantation 62, 543–545 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR, T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA, T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. Journal of Experimental Medicine 208, 1279–1289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Restifo NP, Gattinoni L, Lineage relationship of effector and memory T cells. Current opinion in immunology 25, 556–563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Enouz S, Carrié L, Merkler D, Bevan MJ, Zehn D, Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. Journal of Experimental Medicine 209, 1769–1779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malo CS, Huggins MA, Goddery EN, Tolcher H, Renner DN, Jin F, Hansen MJ, Pease LR, Pavelko KD, Johnson AJ, Non-equivalent antigen presenting capabilities of dendritic cells and macrophages in generating brain-infiltrating CD8+ T cell responses. Nature communications 9, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bern MD, Parikh BA, Yang L, Beckman DL, Poursine-Laurent J, Yokoyama WM, Inducible down-regulation of MHC class I results in natural killer cell tolerance. Journal of Experimental Medicine 216, 99–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richmond JM, Strassner JP, Zapata L Jr., Garg M, Riding RL, Refat MA, Fan X, Azzolino V, Tovar-Garza A, Tsurushita N, Pandya AG, Tso JY, Harris JE, Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Science translational medicine 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan X, Dong Y, Tsurushita N, Tso JY, Fu W, CD122 blockade restores immunological tolerance in autoimmune type 1 diabetes via multiple mechanisms. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathews DV, Dong Y, Higginbotham LB, Kim SC, Breeden CP, Stobert EA, Jenkins J, Tso JY, Larsen CP, Adams AB, CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. Journal of Clinical Investigation 128, 4557–4572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, Hippen KL, Blazar BR, Masopust D, Kelekar A, Vulchanova L, Hogquist KA, Jameson SC, The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Christo SN, Evrard M, Park SL, Gandolfo LC, Burn TN, Fonseca R, Newman DM, Alexandre YO, Collins N, Zamudio NM, Souza-Fonseca-Guimaraes F, Pellicci DG, Chisanga D, Shi W, Bartholin L, Belz GT, Huntington ND, Lucas A, Lucas M, Mueller SN, Heath WR, Ginhoux F, Speed TP, Carbone FR, Kallies A, Mackay LK, Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nature immunology 22, 1140–1151 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, Jameson SC, The Functional Requirement for CD69 in Establishment of Resident Memory CD8(+) T Cells Varies with Tissue Location. Journal of Immunology 203, 946–955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frizzell H, Fonseca R, Christo SN, Evrard M, Cruz-Gomez S, Zanluqui NG, von Scheidt B, Freestone D, Park SL, McWilliam HEG, Villadangos JA, Carbone FR, Mackay LK, Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Science immunology 5, (2020). [DOI] [PubMed] [Google Scholar]
- 71.Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, Validire P, Besse B, Mami-Chouaib F, CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. Journal of Immunology 194, 3475–3486 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, Wilmott JS, Scolyer RA, Tuting T, Palendira U, Gyorki D, Mueller SN, Huntington ND, Bedoui S, Holzel M, Mackay LK, Waithman J, Gebhardt T, Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Krebs CF, Reimers D, Zhao Y, Paust HJ, Bartsch P, Nuñez S, Rosemblatt MV, Hellmig M, Kilian C, Borchers A, Enk LUB, Zinke M, Becker M, Schmid J, Klinge S, Wong MN, Puelles VG, Schmidt C, Bertram T, Stumpf N, Hoxha E, Meyer-Schwesinger C, Lindenmeyer MT, Cohen CD, Rink M, Kurts C, Franzenburg S, Koch-Nolte F, Turner JE, Riedel JH, Huber S, Gagliani N, Huber TB, Wiech T, Rohde H, Bono MR, Bonn S, Panzer U, Mittrücker HW, Pathogen-induced tissue-resident memory T(H)17 (T(RM)17) cells amplify autoimmune kidney disease. Science immunology 5, (2020). [DOI] [PubMed] [Google Scholar]
- 74.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, Masopust D, Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hughes AD, Zhao D, Dai H, Abou-Daya KI, Tieu R, Rammal R, Williams AL, Landsittel DP, Shlomchik WD, Morelli AE, Oberbarnscheidt MH, Lakkis FG, Cross-dressed dendritic cells sustain effector T cell responses in islet and kidney allografts. J Clin Invest 130, 287–294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yatim KM, Gosto M, Humar R, Williams AL, Oberbarnscheidt MH, Renal dendritic cells sample blood-borne antigen and guide T-cell migration to the kidney by means of intravascular processes. Kidney international 90, 818–827 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Wherry EJ, Blattman JN, Murali-Krishna K, Van Der Most R, Ahmed R, Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of Virology 77, 4911–4927 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mueller SN, Ahmed R, High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proceedings of the National Academy of Sciences 106, 8623–8628 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, Delorenzi M, Pinschewer DD, Zehn D, High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. Journal of Experimental Medicine 213, 1819–1834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jabri B, Abadie V, IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nature Reviews Immunology 15, 771–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hashimoto M, Im SJ, Araki K, Ahmed R, Cytokine-Mediated Regulation of CD8 T-Cell Responses During Acute and Chronic Viral Infection. Cold Spring Harbor perspectives in biology 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Picker LJ, Reed-Inderbitzin EF, Hagen SI, Edgar JB, Hansen SG, Legasse A, Planer S, Piatak M Jr., Lifson JD, Maino VC, Axthelm MK, Villinger F, IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. Journal of Clinical Investigation 116, 1514–1524 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu J, Weisshaar N, Hotz-Wagenblatt A, Madi A, Ma S, Mieg A, Hering M, Mohr K, Schlimbach T, Borgers H, Cui G, Skeletal muscle antagonizes antiviral CD8(+) T cell exhaustion. Science advances 6, eaba3458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D, Intravascular staining for discrimination of vascular and tissue leukocytes. Nature protocols 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW, QuPath: Open source software for digital pathology image analysis. Scientific reports 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. All materials will be made available upon request.