

Significance
KRAS(G12D) is a prevalent activating mutation associated with devastating cancers. Although recent breakthroughs have demonstrated that KRAS(G12D) is no longer undruggable, there are still few direct inhibitors selective to KRAS(G12D). We describe a series of synthetic binding proteins, monobodies, that were exquisitely selective to KRAS(G12D). When used as genetically encoded intracellular biologics, they inhibited signaling and tumorigenesis mediated by KRAS(G12D). Structural and systematic mutational analyses revealed a conformation of KRAS(G12D) distinct from those reported previously and key features for the high selectivity of the monobodies, which will guide the development of next-generation inhibitors against KRAS(G12D). The relative ease of generating these monobody inhibitors and gaining mechanistic insights suggests their utility as tool reagents for interrogating challenging therapeutic targets.
Keywords: drug discovery, protein engineering, intracellular biologics, conformational plasticity, protein-protein interaction
Abstract
The G12D mutation is among the most common KRAS mutations associated with cancer, in particular, pancreatic cancer. Here, we have developed monobodies, small synthetic binding proteins, that are selective to KRAS(G12D) over KRAS(wild type) and other oncogenic KRAS mutations, as well as over the G12D mutation in HRAS and NRAS. Crystallographic studies revealed that, similar to other KRAS mutant-selective inhibitors, the initial monobody bound to the S-II pocket, the groove between switch II and α3 helix, and captured this pocket in the most widely open form reported to date. Unlike other G12D-selective polypeptides reported to date, the monobody used its backbone NH group to directly recognize the side chain of KRAS Asp12, a feature that closely resembles that of a small-molecule inhibitor, MTRX1133. The monobody also directly interacted with H95, a residue not conserved in RAS isoforms. These features rationalize the high selectivity toward the G12D mutant and the KRAS isoform. Structure-guided affinity maturation resulted in monobodies with low nM KD values. Deep mutational scanning of a monobody generated hundreds of functional and nonfunctional single-point mutants, which identified crucial residues for binding and those that contributed to the selectivity toward the GTP- and GDP-bound states. When expressed in cells as genetically encoded reagents, these monobodies engaged selectively with KRAS(G12D) and inhibited KRAS(G12D)-mediated signaling and tumorigenesis. These results further illustrate the plasticity of the S-II pocket, which may be exploited for the design of next-generation KRAS(G12D)-selective inhibitors.
Activating, missense mutations of RAS genes (KRAS, HRAS, and NRAS) are frequently associated with human cancers and play important roles in oncogenic transformation (1, 2). Oncogenic RAS proteins have long been considered undruggable, due to an apparent lack of binding pockets suitable for small-molecule inhibitors and the minute differences of RAS mutants from the wild-type proteins. However, the discovery of covalent inhibitors targeting KRAS(G12C) in the GDP-bound state (3, 4) has revolutionized the field on two fronts: the revelation of a dynamic and druggable pocket under the switch II region, termed S-II pocket, and the effectiveness of mutant-selective targeting with G12C inhibitors (5, 6), followed by recent FDA approval of sotorasib and adagrasib. These discoveries have revised the notion that RAS is undruggable and sparked renewed enthusiasm for drug discovery against other oncogenic RAS mutant proteins.
RAS mutants other than G12C for which the covalent inhibitor approach cannot be readily applied still present formidable challenges for inhibitor discovery. Among these mutants, KRAS(G12D) is prevalent in particularly challenging cancers, including pancreatic and colorectal cancers (7). Until recently, there were no reports of small-molecule inhibitors selective to KRAS(G12D). Consequently, there have been intense efforts to develop KRAS(G12D)-selective inhibitors using polypeptides. There are two families of cyclic peptides that target KRAS(G12D) (8, 9). Both classes of compounds bind in the S-II pocket. Sakamoto et al. have developed cyclic peptides that have ~fourfold selectivity for KRAS(G12D) over the wild type (8). These peptides can be derivatized into a cell-permeable form, which was shown to inhibit RAS-mediated signaling in cells and tumor growth in a mouse xenograft model. However, the low selectivity of these peptides and the presence of wild-type RAS proteins in cells (KRAS4A, KRAS4B, HRAS, and NRAS) make it difficult to attribute the observed effects of these peptides solely to selective inhibition of the KRAS(G12D) allele in these cancer cells. Zhang et al. have developed cyclic peptides that bind dominantly to the GTP-bound state of KRAS(G12D) as well as those bind to both GTP- and GDP-bound states (9). However, these peptides have low affinity and minimal cell permeability, and their efficacy in a cellular context has not been reported.
Recently, a small-molecule inhibitor that selectively targets KRAS(G12D), MRTX1133, has been developed (10, 11). The molecule appears effective at inhibiting the proliferation of cells harboring G12D mutation but not cells harboring wild-type KRAS. Unlike the G12C-selective drugs, the inhibitor targeting KRAS(G12D) is noncovalent and its efficacy in the clinic will depend on several factors such as its pharmacokinetic profile in humans and therapeutic window for safe dosage. This impressive success has established the feasibility of selectively targeting KRAS(G12D) in a noncovalent manner. To comprehensively define how one can selectively target this prevalent KRAS mutation, it is still important to examine a diverse array of drug modalities against KRAS(G12D) and define the extent of plasticity of the S-II pocket.
Our group has utilized the monobody system, synthetic binding proteins, to generate intracellular biologics targeting RAS and other proteins (12). We recently reported monobody inhibitors that are highly selective to KRAS(G12V) and KRAS(G12C), demonstrating the feasibility of achieving selective and noncovalent inhibition and degradation of these KRAS mutants (13). Although intracellular biologics in the protein form are difficult to deliver into cells, similar to peptides, they can be readily delivered into cells as genetically encoded reagents using viruses, expression vectors, and mRNA constructs. Furthermore, monobodies and other intracellular biologics can be used as the RAS-targeting moiety as a degrader fusion protein with a subunit of E3 ubiquitin ligases, such as VHL and SPOP (13–16). Thus, intracellular biologics such as monobodies are powerful tools for establishing the feasibility of selective targeting, elucidating the structural basis of specificity and characterizing the effects of selective inhibition (and degradation) in cellular and organismal contexts.
Here, we report monobodies that are highly selective toward KRAS(G12D) and potently inhibit KRAS(G12D)-mediated signaling and tumorigenesis as intracellular biologics. Three crystal structures of monobody–KRAS(G12D) complexes revealed a conformation of the S-II pocket that is distinct from those previously reported, and, coupled with deep mutational scanning, defined the basis for the selectivity toward KRAS(G12D) and different nucleotide-bound states.
Results
Development of the Initial Monobody Selective to KRAS(G12D), 12D1.
To develop monobodies that demonstrate high selectivity for KRAS(G12D), we used established methods that combine phage display and yeast display technologies. Initial selection was performed using naïve phage-display libraries with KRAS(G12D)·GTPγS as the target for positive selection and KRAS(WT)·GDP for negative selection. Throughout this work, we used the KRAS4B isoform exclusively, and we refer to KRAS4B as KRAS for brevity. After rounds of phage-display library sorting, the enriched pool of monobody clones was transferred to a yeast display library. Using fluorescence-activated cell sorting, we screened for clones that bound the KRAS(G12D)·GTPγS and bound negligibly to KRAS(WT)·GDP or KRAS(WT)·GTPγS. We identified clone 12D1 that showed high selectivity toward KRAS(G12D)·GTPγS, weaker binding to KRAS(G12D)·GDP, and negligible binding to KRAS(WT) and other KRAS mutants tested in either GDP- or GTPγS-bound state (Fig. 1 A and B). Biolayer interferometry (BLI) using purified protein samples further demonstrated that 12D1 bound at least 25-fold more tightly to KRAS(G12D)·GTPγS than to KRAS(WT)·GTPγS and KRAS(WT)·GDP (Fig. 1C). This monobody bound to KRAS(G12D)·GTPγS ~ninefold more tightly than to KRAS(G12D)·GDP (Fig. 1C). It inhibited the binding of RAF RBD to KRAS(G12D)·GTPγS with an IC50 value of 54 nM (Fig. 1D), indicating that the interaction of KRAS(G12D) with RAF RBD and 12D1 was mutually exclusive. These results demonstrate the feasibility of developing monobody inhibitors highly selective to KRAS(G12D).
Fig. 1.
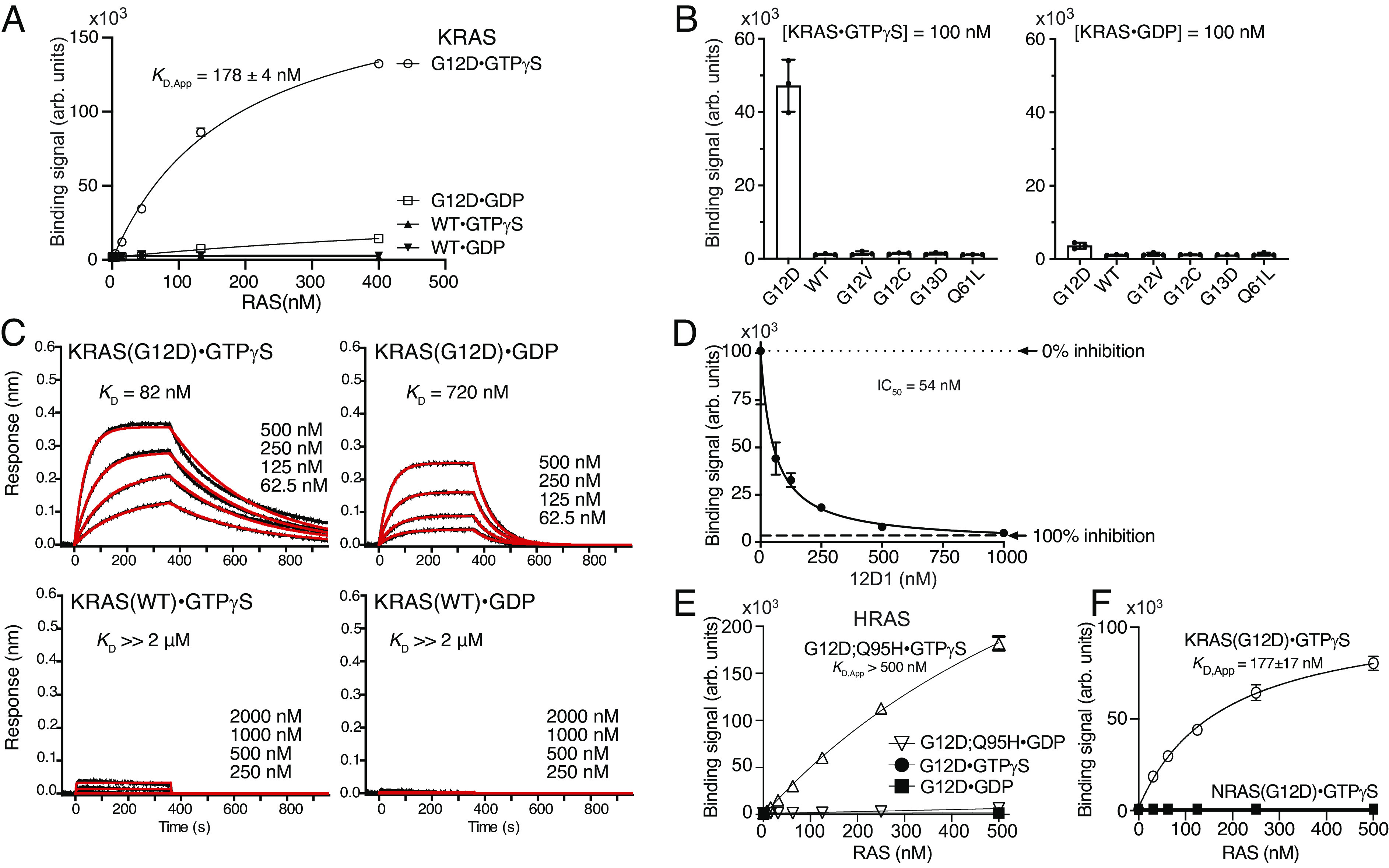
KRAS(G12D)-selective monobody, 12D1. (A) Binding titration of 12D1 displayed on the yeast cell surface to the indicated KRAS proteins. The mean (n = 3; technical replicates) of the median fluorescence intensity is shown. (B) Specificity of 12D1 to different KRAS mutants bound to GTPγS (Left) and GDP (Right) at 100 nM. The bars indicate the means (n = 3, technical replicates). (C) BLI sensorgrams of the interaction between 12D1 and KRAS. The indicated KRAS proteins were immobilized and binding of a soluble 12D1 sample was measured. The KD values are from the global fit of a 1:1 binding model to the data. (D) Inhibition by 12D1 of the binding of KRAS(G12D)•GTPγS to RAF RBD. Binding of 150 nM biotinylated KRAS(G12D)•GTPγS in complex with streptavidin-Dylight650 to RAF RBD immobilized on M270 Dynabeads in the presence of 0 nM to 1,000 nM 12D1. Complete inhibition (100% inhibition) is defined as the signal intensity in the absence of KRAS(G12D). (E) 12D1 does not bind to HRAS(G12D) in the absence of the KRAS-mimicking mutation, Q95H. (F) 12D1 does not bind to NRAS (G12D). Binding titration of the indicated HRAS and NRAS mutant samples to 12D1 displayed on the yeast cell surface is shown.
12D1 did not bind to HRAS(G12D)·GTPγS (Fig. 1E) or to NRAS(G12D)·GTPγS (Fig. 1F). Residue 95, located in the S-II pocket, is not conserved among the RAS isoforms. It is Gln in HRAS and Leu in NRAS. We found that the introduction of the Q95H mutation in HRAS to mimic KRAS promoted the binding of 12D1 to HRAS (Fig. 1E), indicating that 12D1 specifically recognizes three features of KRAS(G12D), i.e., G12D mutation, the GTP-bound state, and His95. A KRAS(G12C) inhibitor, sotorasib (AMG510), similarly recognizes H95, one of three residues that form a pocket for drug binding (17).
Structural Basis for the Selectivity of 12D1 toward KRAS(G12D).
To elucidate the mechanism underlying the specific recognition of KRAS(G12D) by monobody 12D1, we crystallized a surface mutant version of 12D1, 12D1(K63S), in complex with the “Cys-light” version of KRAS(G12D), and determined the structure at 2.52 Å (SI Appendix, Table S1 and Fig. S1). For brevity, we will refer to 12D1(K63S) as 12D1 and the Cys-light version of KRAS simply as KRAS, hereafter. None of these mutations are located near the monobody–KRAS interface.
The monobody and KRAS molecules retained their overall folds, as expected (Fig. 2A). The monobody framework excluding the diversified regions takes on a conformation close to the parental FN3 domain structure (PBD: 1FNA, RMSD for Ca = 0.63 Å). Similarly, the RAS molecule excluding the switch regions and the nucleotide is similar to a previous KRAS(G12D)·GppNHp structure (PDB: 5USJ, RMSD for Ca = 0.69 Å). The interface area between the two molecules is 914 Å2, a typical interface size for monobody–target complexes (18). Interestingly, we found that the nucleotide in the crystal was more consistent with GDP than GTPγS that was utilized for crystallization (SI Appendix, Fig. S1 A and B). We speculate that GTPγS was slowly hydrolyzed to GDP during the crystallization process, as reported previously for other GTPases (19, 20). The affinity of 12D1 to KRAS(G12D)·GDP is high enough to maintain the complex at high protein concentrations used for crystallization. Thus, the structure represents the complex of monobody 12D1 and KRAS(G12D)·GDP. We will describe the structure of an affinity-matured monobody bound to KRAS(G12D)·GTPγS later in the paper.
Fig. 2.
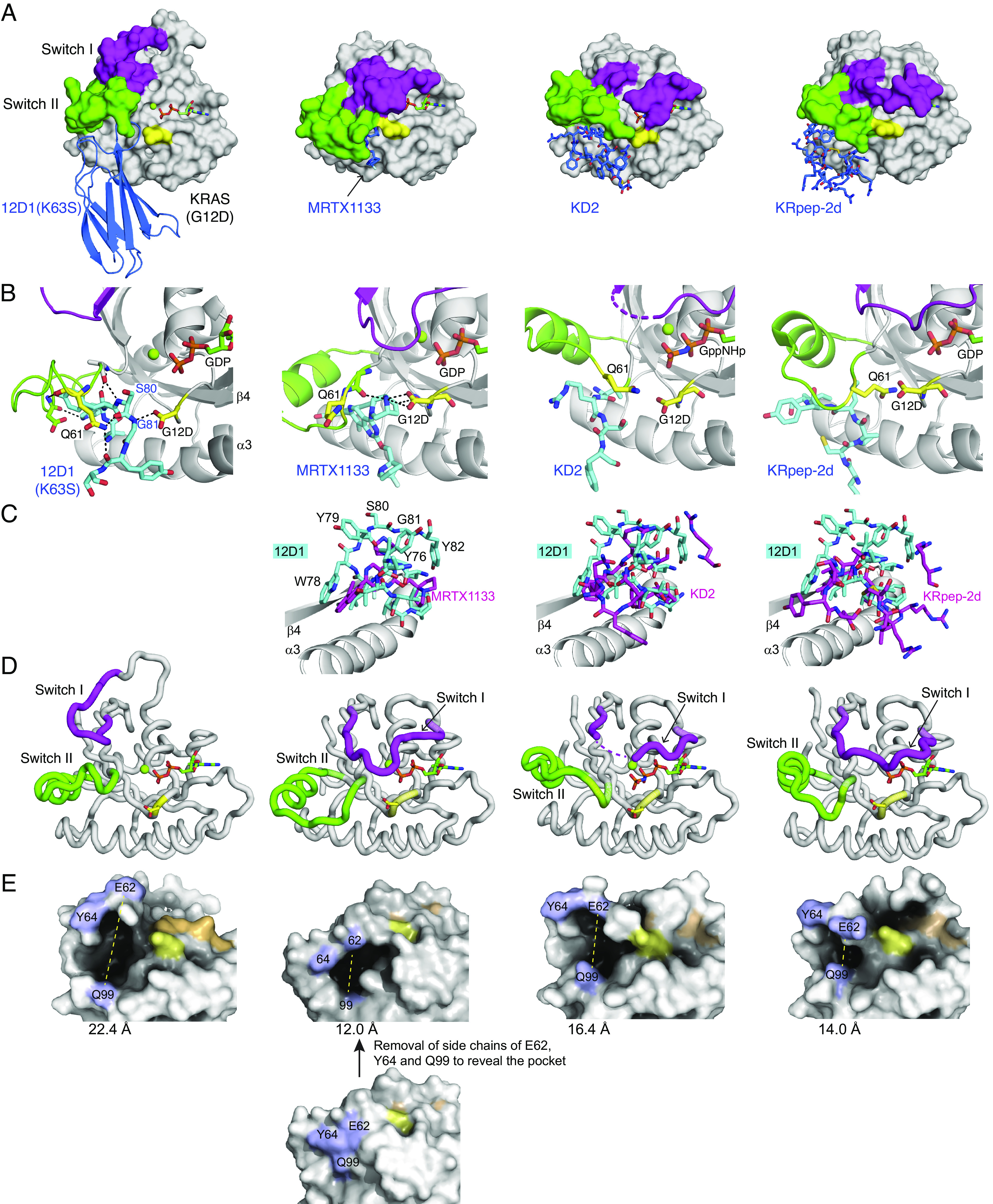
The crystal structure of 12D1(K63S) in complex with KRAS(G12D)•GDP, and comparisons with the structures of other KRAS(G12D) inhibitors. (A) Comparison of 12D1(K63S)–KRAS(G12D)•GDP with MRTX1133–KRAS(G12D)•GDP (PBD: 7RPZ), KD2–KRAS(G12D)•GppNHp (PDB: 6WGN), and KRpep-2d–KRAS(G12D)•GDP (PDB: 5XCO). 12D1(K63S) and other inhibitors are depicted in blue. The switch I and switch II regions and the G12D residue are shown in magenta, green, and yellow, respectively. MRTX1133 is only marginally visible in this mode of presentation. (B) Close-up views of the G12D and Q61 residues (yellow) and surrounding residues of inhibitors (cyan) in the four structures. The hydrogen bonds are indicated as dashed lines. 12D1(K63S) directly interacts with the side chain of the G12D mutation, which resembles interaction between MRTX1133 and the G12D mutation. (C) The superpositions of other inhibitors (magenta) on 12D1 residues (cyan) in the S-II pocket. The structures were superimposed using residues 76–162 of KRAS as the reference. The β4 and α3 regions of KRAS are shown to orient the reader. (D) A comparison of the backbone conformations of the switch I (magenta) and switch II (green) regions of KRAS in the four structures. The inhibitors are not shown for clarity. (E) A comparison of the S-II pocket in the four structures. E62RAS, Y64RAS, and Q99RAS resides are shown in light blue, the nucleotides are shown in orange, and Asp12 in yellow. The distance between E62RAS and Q99RAS (yellow dotted line) is indicated under each structure. For MRTX1133–KRAS(G12D)•GDP, the side chains of E62RAS, Y64RAS, and Q99RAS are removed in order to expose the pocket.
The crystal structure revealed that 12D1 bound primarily to the S-II pocket, the cleft between the switch II and the α3 helix (Fig. 2B and SI Appendix, Fig. S1C). This binding surface differs from another RAS mutant–specific monobody, 12VC1, that binds to both switch I and II regions and selectively recognizes the G12C and G12V mutants (13) (SI Appendix, Fig. S1D). 12D1 mainly interacts with the P-loop and the switch II region via the FG loop (residues 78–82), and with the α3 helix via the C and D stands and the DE-loop (SI Appendix, Fig. S1 C and D). This mode of recognition of the S-II pocket is similar to other inhibitors specific to KRAS(G12D), i.e., the KD2 peptide, the KRpep-2d peptide, and MRTX1133 (8–10) (Fig. 2 A and B), confirming that S-II pocket is a “druggable site” for achieving selectivity to KRAS(G12D).
Although 12D1 and the other three inhibitors of KRAS(G12D) bind to the S-II pocket, 12D1 presents substantially more bulk in the pocket (Fig. 2C). W78, Y79, S80, G81, and Y82 of 12D1 occupy the space that is occupied by residues of the RAS switch II region in the other structures, and consequently capture an enlarged S-II pocket conformation (see the next section).
Closer inspection of the interface revealed that 12D1 directly interacts with the side chain of Asp12RAS using a hydrogen bond with the backbone NH group of Gly81Mb (Fig. 2B and SI Appendix, Fig. S1C). Hereafter, residue numbers for RAS and 12D1 will be indicated with RAS and Mb in superscript, respectively, for clarity. This interaction bears striking resemblance with that between the nitrogen atom of MRTX1133 and the Asp12RAS side chain (Fig. 2B) (10). In contrast, other KRAS(G12D)-directed peptide inhibitors with moderate selectivity do not form direct contacts with the Asp12RAS side chain (Fig. 2B, the right two panels). Taken together, the observed direct interactions with the Asp12RAS side chain rationalize the high selectivity of 12D1 and MRTX1133 to the G12D mutant.
In addition to Gly81Mb, the backbone of Ser80Mb forms multiple hydrogen bonds with Ala59RAS and Gln61RAS, suggesting that Ser80Mb is another key residue for the interaction between 12D1 and KRAS(G12D) (Fig. 2B and SI Appendix, Fig. S1C). The side chains of Thr49Mb and Phe31Mb interact with the side chain of His95RAS in the α3 helix via a hydrogen bond and π–π stacking interaction (SI Appendix, Fig. S1E), respectively, suggesting its contribution to selective binding of 12D1 to KRAS(G12D) over HRAS(G12D) and NRAS(G12D) (Fig. 1E). These structural features rationalize the specific recognition of KRAS(G12D) by 12D1.
12D1 Captures an Enlarged S-II Pocket Conformation.
Similar to other G12D-selective inhibitors reported to date, 12D1 binds to the switch II pocket. However, the conformations of the switch I and II regions of KRAS(G12D) in the 12D1 complex are substantially different from those of the regions in previously reported RAS structures (SI Appendix, Fig. S2A). The greater volume of 12D1 moieties that occupy the S-II pocket displaces switch II toward switch I. The loop region between the β3 strand and the α2 helix, including the switch II region, in the 12D1-bound KRAS(G12D) is shifted toward the switch I region (“up” in our depiction) compared with the region in other RAS structures (Fig. 2D and SI Appendix, Fig. S2B). Switch I region in the 12D1–KRAS(G12D) complex also takes on a conformation distinct from that in KRAS(G12D) bound to other inhibitors as well as in RAS structures in the absence of a bound inhibitor (Fig. 2D and SI Appendix, Fig. S2A). Unfortunately, we cannot unambiguously define factors contributing to the stabilization of the observed conformation of switch I, because the switch I region is involved in crystal packing (SI Appendix, Fig. S2C). These differences are likely to be caused by 12D1 presenting larger bulk in the S-II pocket. Consistent with this view, the pocket formed between switch II loop and the α3 helix is substantially larger in the 12D1 complex than that of the other complexes with G12D inhibitors (Fig. 2E). To quantify these changes, we measured the distance between the Cα positions of E62 in switch II and Q99 in α3 in each structure (Fig. 2E). The distance in the 12D1 complex (22.4 Å) is much greater than that in the MRTX1133 complex (12.0 Å) and the cyclic peptide complexes (16.4 and 14.0 Å, respectively).
Together, our structure revealed yet another conformation of the switch I and II regions of RAS(G12D), further defining the plasticity of the switch regions in the RAS mutant. It also revealed the most open form of the switch II groove to date, which may be useful for structure-guided drug discovery.
Affinity Maturation of 12D1 Monobody.
To improve the potency of 12D1 affinity, we subjected its gene to error-prone PCR and screened for higher-affinity clones, which yielded 12D2 that contained two mutations, I20V and P51S, both located outside the diversified positions in the starting library (Fig. 3A). 12D2 had approximately fourfold higher affinity to KRAS(G12D)·GTPγS than that of 12D1 and also showed less discrimination of the GTPγS- and GDP-bound states (Fig. 3B and SI Appendix, Fig. S3A). In the crystal structure, P51Mb is located near the interface with RAS(G12D), suggesting that the P51S mutation improved the binding interface. Ile20Mb is located away from the interface and its side chain is a part of the hydrophobic core of the monobody. As such, the role of the I20V mutation is unclear.
Fig. 3.
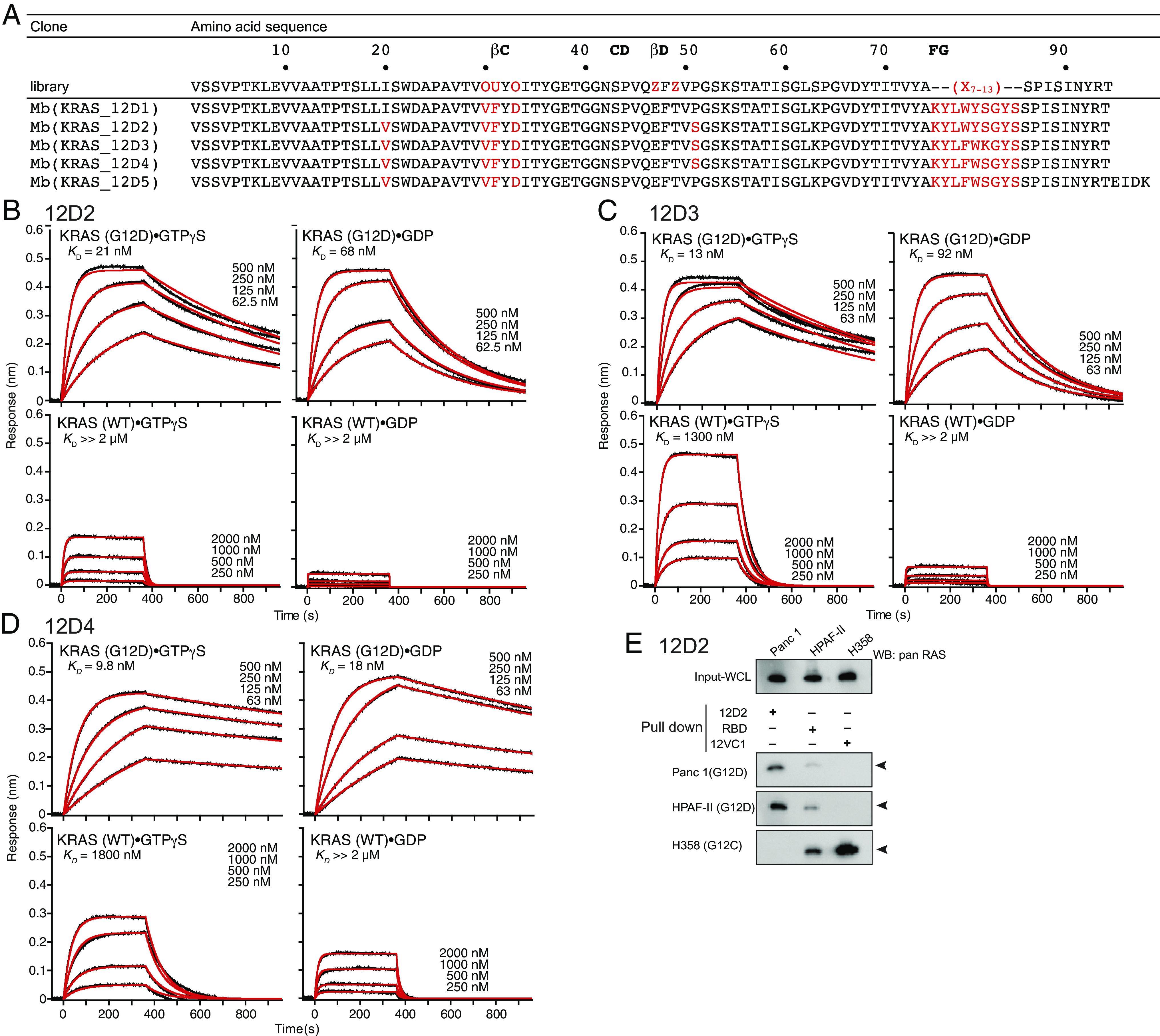
Affinity maturation of KRAS(G12D)-selective monobodies. (A) Amino acid sequences of monobody clones. Residues in red indicate mutations with respect to the parental scaffold. (B–D) BLI sensorgrams of 12D2 (B), 12D3 (C), and 12D4 (D) interacting with immobilized KRAS. The KD value was determined from the global fit of the data with a 1:1 binding model. (E) The capture of endogenous RAS proteins with 12D2. The “pull down” reaction was done with the indicated monobodies (12D2 and 12VC1) and RAF RBD, followed by immunoblotting using an anti-pan RAS antibody. 12VC1 is selective to KRAS(G12C) and KRAS(G12V) and used as a control.
We next introduced directed mutations to residues in the FG loop of 12D2, the segment containing most residues that form extensive contacts with KRAS(G12D) in the crystal structure of 12D1 (Fig. 2). We introduced three adjacent randomized positions in the 9-residue sequence of the FG loop, KYLWYSGYS (Fig. 3A), to generate three sublibraries (XXXWYSGYS, KYLXXXGYS, and KYLWYSXXX, where X denotes a randomized position with all 20 genetically encoded amino acids), followed by library sorting with yeast display. These experiments resulted in clones 12D3 and 12D4 with further improved affinity to KRAS(G12D)·GTPγS (Fig. 3 C and D and SI Appendix, Fig. S3 D and G). 12D4 contained two mutations W78F and Y79W, and 12D3 contained an additional mutation, S80K (Fig. 3A), originating from the library in which the middle three residues (positions 78–80) were randomized. 12D3 maintained the preference toward KRAS(G12D)·GTPγS over KRAS(G12D)·GDP (Fig. 3C) as seen in 12D1 and 12D2. In contrast, 12D4 bound to both GTPγS- and GDP-bound forms of KRAS(G12D) with similar affinity (Fig. 3D). These monobodies exhibited high selectivity for KRAS(G12D), with up to a 180-fold difference in affinity between KRAS(G12D) and KRAS(WT) (Fig. 3 B–D). All the monobodies bound only very weakly to other KRAS mutants in either nucleotide-bound form as tested in the yeast display format (SI Appendix, Fig. S3 B, E, and H), and discriminated KRAS(G12D) and HRAS(G12D) by recognizing the difference at position 95 (SI Appendix, Fig. S3 C, F, and I). A pull-down assay with clone 12D2 further confirmed its selectivity toward the KRAS(G12D) mutant. 12D2 selectively captured endogenous RAS from cancer cell lines Panc-1 and HPAF-II that contain KRAS(G12D), but not from H358 cells containing KRAS(G12C) (Fig. 3E). Together, we successfully improved the affinity of the initial monobody while maintaining its high selectivity to KRAS(G12D).
Crystal Structures of 12D5 Monobody in Complex with KRAS(G12D)·GTPγS and with KRAS(G12D)·GDP.
We attempted to crystallize 12D4, but its limited solubility made it difficult to prepare crystals. Reverting Ser to Pro at position 51 and extending the C terminus with the EIDK sequence that is present in human fibronectin improved the solubility. This monobody, termed 12D5 (Fig. 3A), had fourfold weaker affinity to KRAS(G12D)·GTPγS than that of 12D4, but still maintained high specificity to KRAS(G12D) over KRAS(WT) and slight preference to KRAS(G12D)·GTPγS over KRAS(G12D)·GDP (SI Appendix, Fig. S4A). We determined the crystal structure of 12D5 in complex with KRAS(G12D). We observed two copies of the complex in the asymmetric unit, but, intriguingly, with different states of the nucleotide bound to KRAS(G12D), one copy with GTPγS and the other with GDP. The conformations and the Mg2+ coordination of GTPγS and GDP are consistent with those in other KRAS structures (SI Appendix, Fig. S4B). In the 12D5–KRAS(G12D)·GTPγS complex, the N-terminal region of the symmetry-related KRAS(G12D) molecule is engaged in coordinating Mg2+ (SI Appendix, Fig. S4C). The presence of GDP in the other copy is likely due to the hydrolysis of GTPγS used for crystallization, as in the 12D1–KRAS(G12D)·GDP structure.
The overall conformations of the two 12D5–KRAS complexes in the asymmetric unit as well as that of the 12D1–KRAS complex are nearly identical except for the switch I region (Fig. 4 A and B and SI Appendix, Fig. S4C), consistent with the ability of 12D5 to bind to both nucleotide states of KRAS(G12D) and the small differences in the sequences between 12D1 and 12D5 (Fig. 3A).
Fig. 4.

Crystal structures of 12D5(K63S) in complex with KRAS(G12D)•GTPγS and with KRAS(G12D)•GDP. (A) A comparison of 12D5(K63S)–KRAS(G12D)•GTPγS (Left) and 12D5(K63S)–KRAS(G12D)•GDP (Center) with 12D1(K63S)–KRAS(G12D)•GDP (Right). The color scheme is the same as in Fig. 2. (B) Superpositions of 12D5(K63S)–KRAS(G12D)•GTPγS (Left) and 12D1(K63S)–KRAS(G12D)•GDP (Right) on 12D5(K63S)–KRAS(G12D)•GDP. (C) Close-up views of the G12DRAS and Q61RAS residues (yellow) and surrounding residues of 12D1 or 12D5 (cyan). The hydrogen bonds are indicated as dashed lines. (D) Close-up views of the S-II pocket and surrounding residues of 12D1 or 12D5 (cyan).
A conspicuous difference between the interfaces in the GTPγS- and GDP-bound complexes is the interactions involving S80Mb. It directly interacts with GTPγS, whereas it has no interaction with GDP (Fig. 4C), rationalizing the preferential binding of 12D5 toward KRAS(G12D)·GTP over KRAS(G12D)·GDP. In addition, switch I displayed distinct conformations, including partial disorder, between the GTPγS- and GDP-bound complexes (Fig. 4A and SI Appendix, Fig. S4C), strongly suggesting that the switch I region is inherently flexible.
Although two residues in the FG loop are mutated in 12D5 (W78F and Y79W) with respect to 12D1, these mutations do not alter the FG loop conformation, and key interactions involving the common residues between 12D1 and 12D5, including that between G81Mb and G12DRAS, are maintained (Fig. 4C). A mutated position in 12D5, W79Mb, presents its side chain more deeply into the S-II pocket than that of Y79Mb in 12D1 (Fig. 4D). The interface area between 12D5 and KRAS(G12D) is indeed larger than that between 12D1 and KRAS(G12D) (943 Å2 vs. 914 Å2, SI Appendix, Table S1). Thus, we can reason that the W78F and Y79W mutations in 12D3, 12D4, and 12D5 increased the binding surface area while keeping the FG loop conformation, contributing to improved affinity compared with 12D1.
The conformations of the switch I and II regions in KRAS(G12D)·GTPγS in complex with 12D5 also substantially differ from that in the RAF RBD–bound RAS (SI Appendix, Fig. S5A). Given the high similarity between 12D5- and 12D1-bound KRAS(G12D) molecules (except for the switch I region), this observation rationalizes the inhibition by 12D1 of the interaction between the RAF RBD and KRAS(G12D) (Fig. 1D). MRTX1133 causes similar but smaller-scale distortion of the RBD-binding surface of KRAS(G12D)·GppCp and disrupts the RBD–KRAS(G12D) interaction (11). We found similar incompatibility between the conformations of 12D5-bound KRAS(G12D)·GTPγS and KRAS bound to the RBD of PI3K (SI Appendix, Fig. S5B), suggesting that these monobodies also inhibit KRAS(G12D)-mediated activation of PI3K. In addition, structural overlay showed that 12D5 binding to RAS would interfere with the interaction of RAS and SOS (SI Appendix, Fig. S5C), suggesting that 12D1 and its variants inhibit allosteric activation of SOS by KRAS(G12D)·GTP.
Deep Mutational Scanning.
To more comprehensively define residues of 12D4 important for binding to KRAS(G12D), we performed deep mutational scanning (21). Using the crystal structures as a guide, we selected a total of 19 residues in 12D4 that were expected to be located in or near the binding interface and generated a yeast-display library in which one of these positions was diversified to all 20 amino acids at a time (Fig. 5A). The library was sorted into three classes: first, clones that exhibited a binding profile similar to the wild-type 12D4 when measured with 10 nM KRAS(G12D) (“high binders”); second, those similar to the wild type when measured with 100 nM KRAS(G12D) (“binders”); and third, those that did not show binding when measured with 100 nM KRAS(G12D) (“nonbinders”) (Fig. 5A). The experiment was performed with KRAS(G12D)·GTPγS and KRAS(G12D)·GDP, resulting in a total of six pools. These pools were subjected to deep sequencing and the numbers of reads for individual mutations were determined. We identified 181 and 160 unique mutants for binders and nonbinders to KRAS(G12D)·GTPγS and 149 and 186 unique mutants for binders and nonbinders to KRAS(G12D)·GDP, respectively. In total, we generated 362 unique sequences. As expected, there were high degrees of overlap between the high binder and binder pools, and there was little overlap between the nonbinder pool and the high binder and binder pools, indicating successful separation of clones with distinct binding properties (Fig. 5 B and C).
Fig. 5.
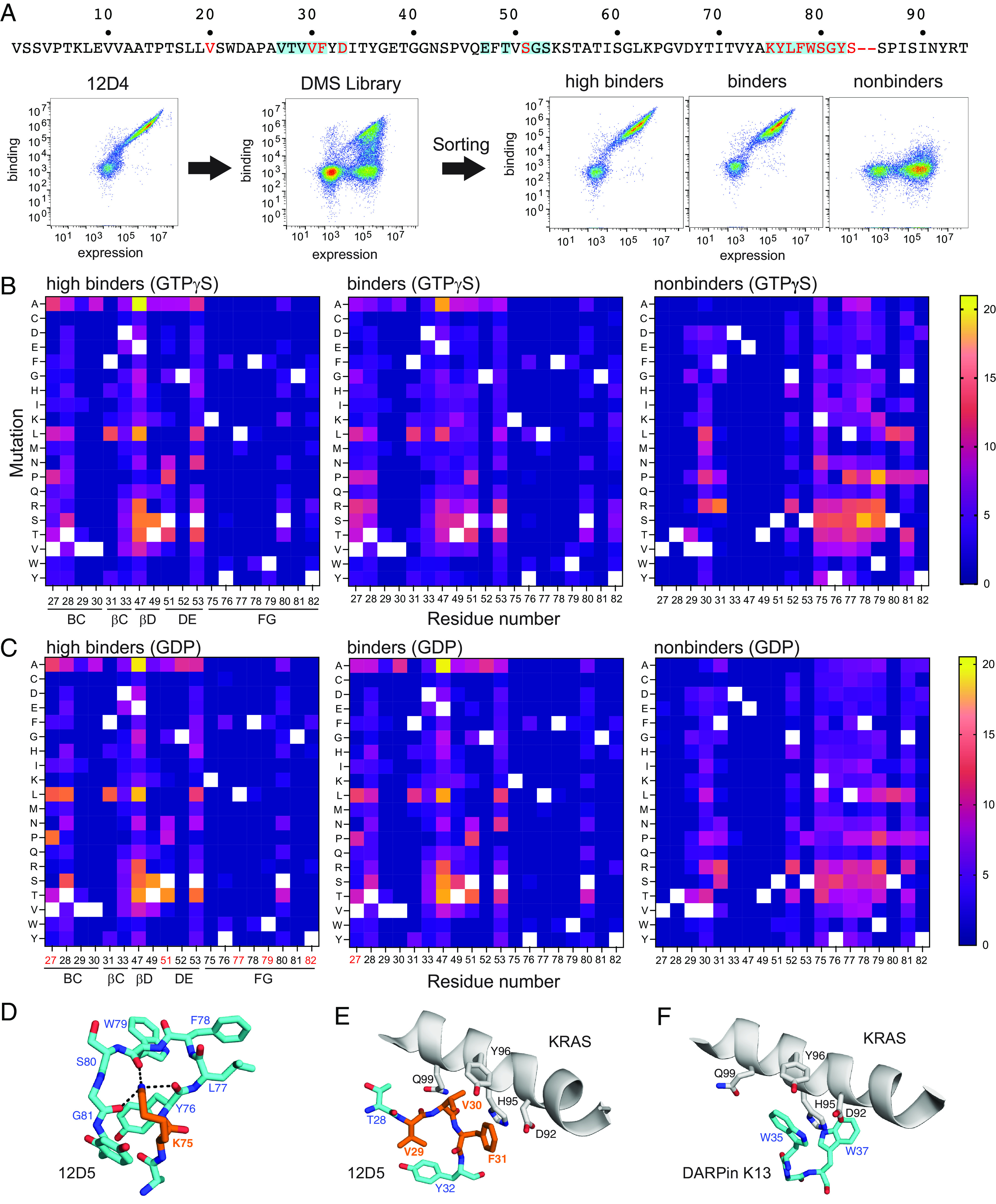
Deep mutational scanning analysis of 12D4 interface residues. (A) Flow cytometry profiles monitoring the binding of KRAS(G12D)•GTPγS (100 nM) to yeast cells displaying 12D4 (Left), the deep mutational scanning library (Middle), and sorted pools (Right). The mutated positions in the 12D4 sequence are highlighted with the cyan boxes. (B) Heat map representation of the numbers of reads for substitutions recovered in the three pools from library sorting with KRAS(G12D)•GTPγS. The number of reads for each substitution was normalized to the total number of reads for each entire pool and multiplied by 1,000. The white squares indicate the wild-type residues. The locations of the residues in the monobody secondary structure elements are indicated on the bottom of the Left panel. (C) Heat map representation of the numbers of reads for substitutions recovered in the three pools from library sorting with KRAS(G12D)•GDP. The data were analyzed in the same manner as in panel B. The residue numbers in red indicate positions whose amino acid profile differs between the pools sorted with KRAS(G12D)•GTPγS and KRAS(G12D)•GDP. (D and E) Close-up views of K75Mb and adjacent residues (D), and of V29Mb, V30Mb, F31Mb, and adjacent residues (E) in the 12D5–KRAS(G12D)•GTPγS complex. Nε of K75Mb interacts with three backbone carbonyl groups of residues in the FG loop of 12D5, depicted with the dashed lines. (F) The region corresponding to that depicted in E of DARPin K13 in complex with KRAS(WT) (PDB:6H46). The π–π stacking interaction between W35 of DARPin K13 and H95 of KRAS(WT) resembles the interaction between F31Mb of monobody 12D5 and H95 of KRAS(G12D) depicted in E.
In both high binder and binder pools, only two of the 19 positions, K75Mb and G81Mb, did not permit any substitution. As described above, the backbone NH group of G81Mb is directly engaged with the Asp12RAS side chain in the crystal structure of 12D1. The inability to mutate G81Mb strongly suggests the importance of this interaction, as the side chain alterations are likely to perturb the position and orientation of the interaction between G81Mb NH and Asp12RAS. The side chain of K75Mb also points toward the side chain carboxyl of Asp12RAS. Intriguingly, the positively charged Nε of K75Mb does not directly interact with the Asp12RAS carboxyl. Instead, it is cradled by three backbone carbonyl groups of monobody residues (residues 76Mb, 79Mb, and 81Mb) (Fig. 5D), suggesting that this feature is important for maintaining the ring-like structure of the FG loop that supports most of the monobody residues interacting with the S-II pocket (Figs. 2B and 4C).
An additional eight positions allowed limited numbers of substitutions in the high binder and binder pools. Residues 76–79 and 82 in the FG loop fall in this class. This segment is tightly packed in the S-II pocket (Fig. 4 C and D), rationalizing these characteristics. High conservation of the remaining residues in this class can also be rationalized by the crystal structure. V30Mb and F31Mb form direct hydrophobic contacts with the α3 helix (Fig. 5E) and V29Mb is part of the hydrophobic core of the monobody scaffold and important for its stability (22). G52 Mb forms a tight turn with a positive phi angle, an energetically reasonable conformation only for Gly. F31 can be replaced with His, Lys, Leu, and Try, substitutions that may maintain close ring stacking or hydrophobic packing against H95 (Fig. 5E). Similar ring stacking is observed in a KRAS-specific DARPin, K13 (Fig. 5F) (23). The remaining positions allowed most substitutions. Together, the deep mutational scanning data are consistent with the 12D1 and 12D5 crystal structures, further validating them.
A few positions show different patterns between the corresponding pools recovered with KRAS(G12D)·GTPγS and KRAS(G12D)·GDP (residues 27Mb, 51Mb, 77Mb, 79Mb, and 82Mb; Fig. 5 B and C), suggesting that these residues sense differences between the GTP- and GDP-bound states. However, we noted minimal conformational differences of these residues and their adjacent residues in the 12D5 crystal structures (Fig. 4 B and C), indicating that additional information is required for rationalizing these data. Still, these data guide future efforts in expanding the sequences of KRAS(G12D)-binding monobodies and tuning their nucleotide-state specificity.
Intracellularly Expressed Monobodies Selectively Engage and Inhibit KRAS(G12D).
We reformatted 12D2, 12D3, and 12D4 in the form of mCherry fusion proteins into genetically encoded reagents and expressed them intracellularly. These monobodies colocalized with overexpressed KRAS(G12D) fused to EGFP, but not with EGFP-KRAS(WT) (Fig. 6A), indicating that these monobodies selectively engaged with KRAS(G12D) in the cellular context. Similarly, CFP-fused monobodies 12D2, 12D3, and 12D4, but not CFP alone, specifically captured KRAS(G12D) when coexpressed in HEK293T cells (Fig. 6B). By contrast, they did not capture KRAS(G13D) or KRAS(G12R). These results confirmed the selectivity of these monobodies to KRAS(G12D) in cells.
Fig. 6.
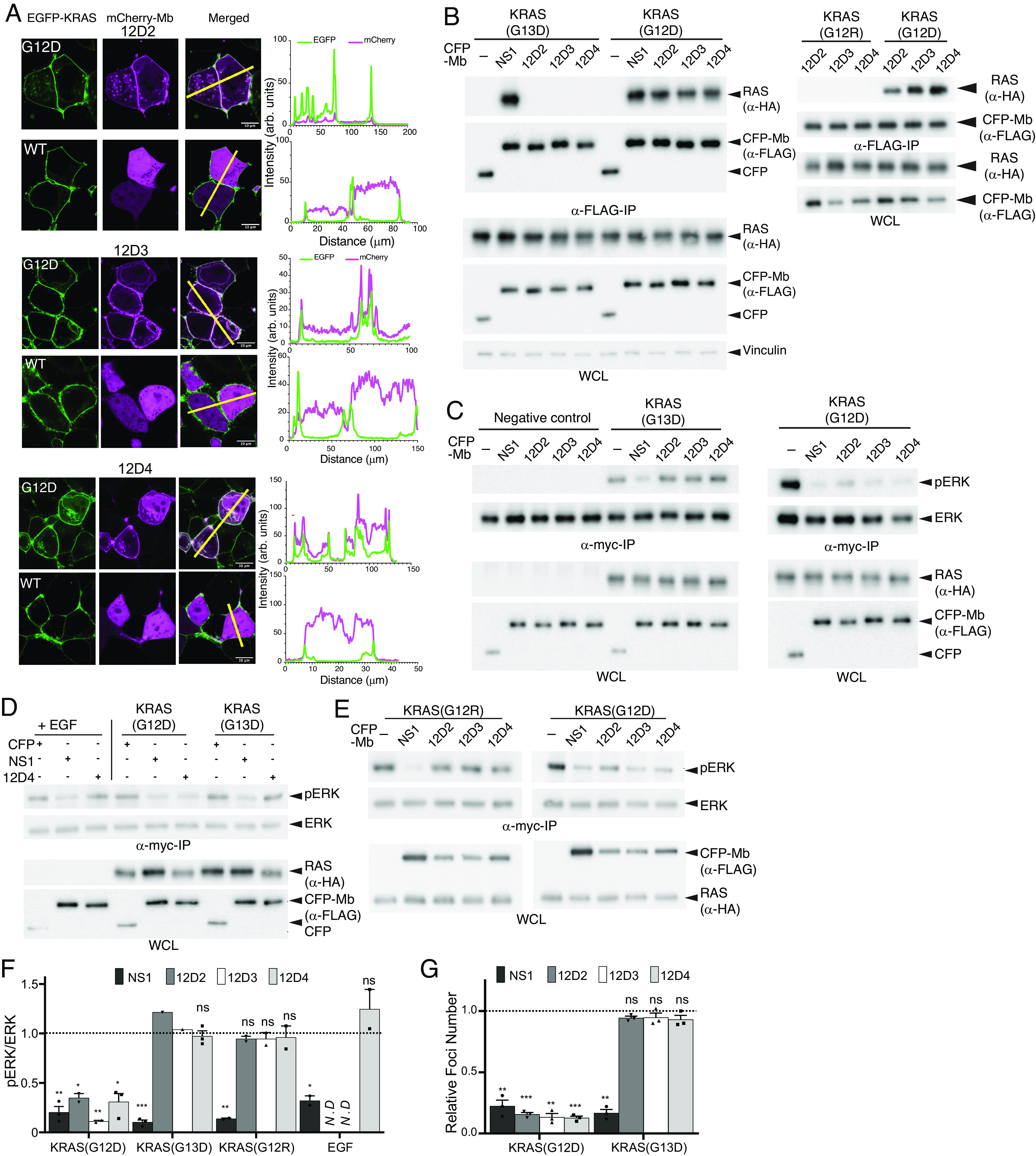
Selective engagement and inhibition of KRAS(G12D) by monobodies. (A) Colocalization of mCherry-fused 12D2, 12D3, and 12D4 (pseudo-color magenta) with overexpressed EGFP-fused KRAS(G12D) and KRAS(WT) (pseudo-color green) in HEK293T cells. The scale bar denotes 10 μm. The graphs on the right show the fluorescence intensity profiles under the yellow lines in the microscopy images. (B) Coimmunoprecipitation of HA-tagged KRAS mutants and FLAG- and CFP-tagged monobodies. The proteins were coexpressed in HEK-293 cells and the capture of KRAS proteins was probed. The NS1 monobody, which is agnostic to KRAS mutations, was used as a control. (C–E) Effects of monobodies on ERK–MAPK activation mediated by KRAS mutants and by EGF stimulation. CFP-tagged monobodies and MYC-tagged ERK were coexpressed in HEK293 cells, and phosphorylation of MYC-tagged ERK was detected following MYC IP and western blotting. CFP and CFP-NS1 were used as controls. The negative control lanes in C show results with cells without the expression of a KRAS mutant. (F) Quantification of pERK levels for data in panels C–E. The pERK level was first normalized to the total ERK level. The resulting value in the presence of the indicated monobody was divided by the value for CFP alone. The dotted line at 1 represents the normalized pERK level by CFP alone without inhibition. The P values were determined using a Student’s t test between CFP-monobody and CFP for each condition. (G) Inhibition of KRAS(G12D)-mediated transformation of NIH/3T3 cells with monobodies, as measured by quantification of foci numbers. The raw data are provided in SI Appendix, Fig. S7A. Each bar represents the ratio of foci number with CFP-monobody to that with CFP alone (n = 3; mean ± SD). The P values were determined using a Student’s t test between CFP-monobody and CFP for each condition. *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.001.
We next tested the ability of these monobodies to selectively inhibit the activity of oncogenic KRAS mutants in cells. Transient expression of 12D2, 12D3, and 12D4 potently inhibited KRAS(G12D)-mediated activation of ERK–MAPK, but was ineffective at inhibiting ERK activation by KRAS(G13D) and KRAS(G12R), as well as epidermal growth factor (EGF)-induced ERK activation mediated by wild-type RAS (Fig. 6 C–F). In contrast, the NS1 monobody, which inhibits KRAS and HRAS and is not selective for a particular RAS mutant (24), inhibited ERK–MAPK activation mediated by both KRAS mutants and EGF treatment. Further, 12D2, 12D3, and 12D4 inhibited the oncogenic transformation of NIH/3T3 cells by KRAS(G12D) but not KRAS(G13D) (Fig. 6G).
Next, we tested the effect of expressing 12D4 monobody in human PDAC tumor cells. Pa14C PDAC cells, harboring KRAS(G12D), and PSN-1 PDAC cells, harboring KRAS(G12R), were stably transduced with a doxycycline (DOX)-inducible expression vector encoding CFP-12D4. DOX-induced expression of CFP-tagged 12D4 inhibited ERK–MAPK activation and anchorage-independent growth of Pa14C cells but not PSN-1 cells, as expected from the selectivity of 12D4 (SI Appendix, Fig. S6). Furthermore, DOX-induced 12D4 expression reduced the growth of Pa14C tumors, but not PSN1 tumors, when injected into athymic nude mice (Fig. 7 A and B). Western blot analysis of tumors revealed reduced pERK levels in Pa14C12D4 tumors but not PSN112D4 tumors as anticipated (Fig. 7 C and D). Further, Ki-67 and cleaved caspase staining of cell suspensions isolated from tumor tissues revealed decreased proliferation and increased apoptosis in Pa14C12D4 vs. PSN112D4 tumors (Fig. 7 E and F), again consistent with selective inhibition of KRAS(G12D) functions by 12D4.
Fig. 7.
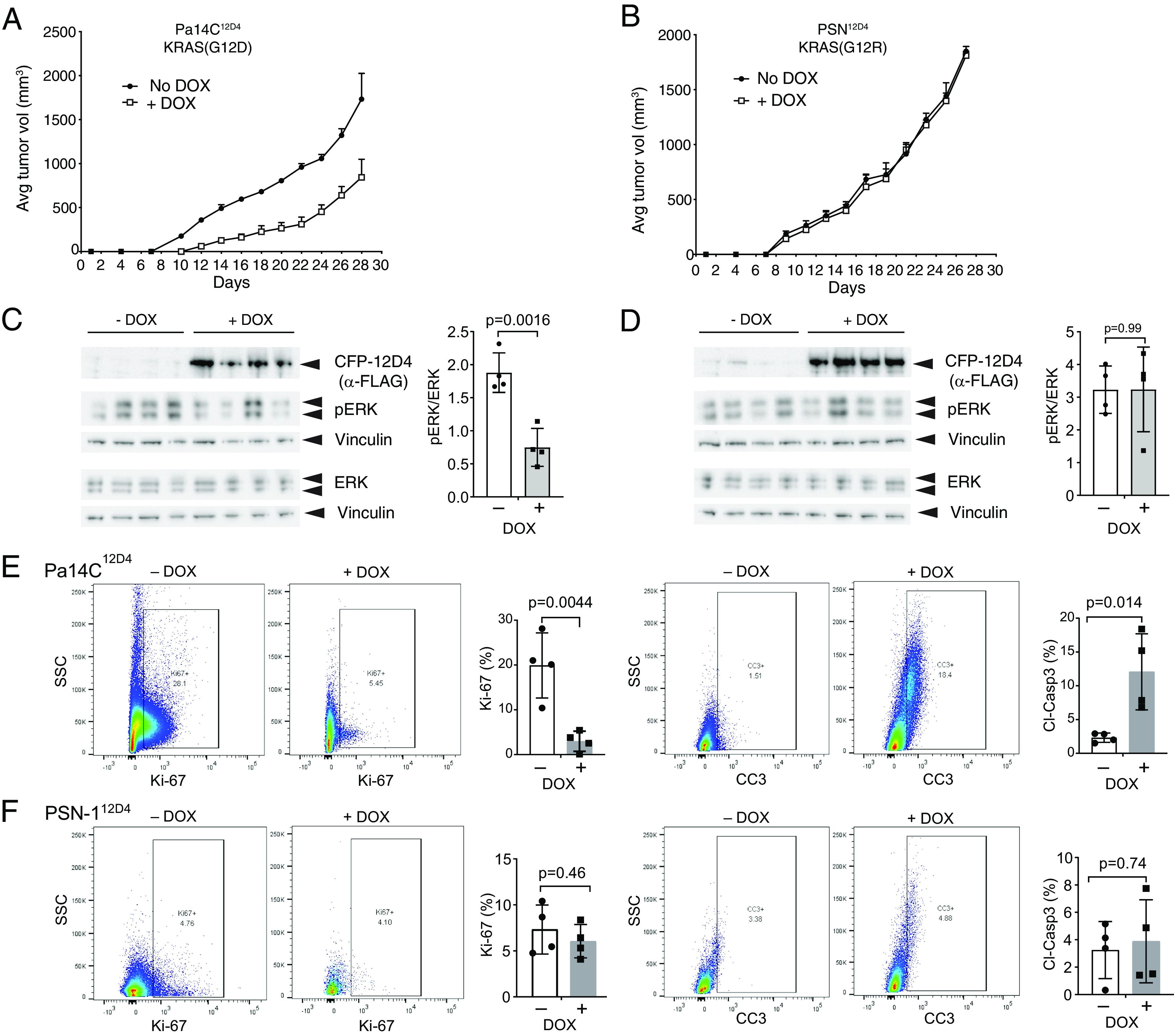
Selective inhibition of KRAS(G12D)-driven tumorigenesis by monobody 12D4. (A and B) The effects of 12D4 expression on tumor growth in mouse xenograft models of Pa14C cells harboring KRAS(G12D) (A) and of PSN1 cells harboring KRAS(G12R) (B). The engineered PDAC cells were injected subcutaneously in the flanks of athymic nude mice. The mice were treated without (−) or with (+) DOX beginning on day 2 following injection, and tumor development was monitored. The mean tumor volumes are shown (n = 4 per condition). The error bars indicate SD. (C and D) Effect of DOX-induced CFP-12D4 expression on ERK–MAPK activation. Tumor lysates were probed for the indicated proteins by immunoblotting. Vinculin was used as a loading control. The graphs show the normalized pERK/ERK ratio for each set of tumor lysates. The P values were calculated using an unpaired Student’s t test. (E and F) Flow cytometric analysis for Ki-67 and cleaved caspase-3 for tumor cells from various cohorts. The gating strategy is shown in SI Appendix, Fig. S7 B–F. Cells from the dox-induced conditions had fewer cells in the analysis because they had low viability. The P values were calculated using an unpaired Student’s t test.
Discussion
Monobodies developed in this work further expand the toolbox of biologic reagents for controlling oncogenic RAS mutants and elucidating RAS biology. Clearly, the success of MRTX1133 in achieving extraordinarily high specificity and potency toward KRAS(G12D) overshadows our success in developing a biologics reagent with the highest level of specificity to date, to our knowledge, toward KRAS(G12D). However, favorable attributes of monobody reagents, including the ease of genetic encoding and intracellular delivery, flexibility in constructing fusion proteins including those with E3 ligase subunits, e.g., VHL and SPOP, and with fluorescent proteins, and the ability to control their expression in a temporal and tissue-specific manner, will complement small-molecule inhibitors in advancing RAS research (12).
The crystal structures of 12D1 and 12D5 in complex with KRAS(G12D) show remarkable similarity with the structure of MRTX1133 bound to KRAS(G12D); in particular, the placement of a nitrogen atom that interacts with the side chain of Asp12RAS, a feature that appears to be crucial for achieving high specificity toward the G12D mutation. At the same time, the 12D1 and 12D5 structures add snapshots of the S-II pocket to the growing body of atomic structures of RAS bound to noncovalent S-II-pocket ligands (8–10), which collectively define a highly malleable nature of this pocket. The deep mutational scanning data consisting of hundreds of 12D4 variants substantially expand the knowledge of the structure–activity relationship of ligands binding to the S-II pocket. Together, these data inform further design of inhibitors for KRAS(G12D).
We were able to develop KRAS(G12D)-specific monobodies by applying our standard protein-engineering technologies. This relatively straightforward pathway is in direct contrast to the challenge that small-molecule inhibitor discovery for KRAS(G12D) has presented over the past four decades. The discovery of the S-II pocket with the covalent inhibitors for KRAS(G12C) was crucial for revising the notion that RAS has no pockets suitable for small molecules and for achieving the “initial traction” toward the ultimate development of MRTX1133 (10). By contrast, we were able to develop the 12D1 monobody by the combination of positive and negative sorting of monobody libraries without prior knowledge of a targetable pocket. These results further demonstrate the maturity of synthetic binding protein technologies to achieve exquisite specificity and potency even for the most challenging targets. Clearly, the strategy employed for KRAS(G12D) in this work is readily applicable to other challenging targets that are currently considered undruggable, for example, Q61 mutations of RAS. Furthermore, advances in technologies for cellular delivery of proteins and nucleic acids may enable therapeutic use of monobodies targeting intracellular molecules.
Materials and Methods
Protein preparation, monobody development, biophysical characterization, and crystal structure determination were performed essentially as described previously (13, 25). Deep mutational scanning was performed following published methods (21, 26). Colocalization, immunoprecipitation, signaling assays, and mouse xenograft experiments were performed essentially as described previously (27, 28). Further details on methods used in this study are described in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Jerzy Osipiuk and Kemin Tan for assistance with data collection at the Advanced Photon Source and for advice on structural determination; Dhirendra Simanshu for advice on RAS crystallization and data analysis; Joseph Taft for advice on DNA extraction from yeast for deep mutational scanning; Koki Makabe for advice on refinement of the structure; Mark Philips for the pEGFP-RAS(WT) vector; and Genome Technology Center and Microscopy Laboratory of New York University Grossman School of Medicine for technical assistance. This work was supported by the NIH grants R21 CA246457 (T.H.), R01 CA212608 (J.P.O. and S.K.), and R01 CA194864 (S.K.). J.P.O. was supported by grants from the Department of Veterans Affairs Biomedical Laboratory Research and Development Service MERIT Award (1I01BX002095) and the NCI (P30 CA138313). K.W.T. was supported in part by an NIH fellowship (F32 CA225131) and an American Cancer Society fellowship (PF-18-180-01-TBE). The core facilities of NYU School of Medicine were partially supported by the Cancer Center Support Grant P30CA016087. Results shown in this report are derived from work performed at the beamline 19-ID at Argonne National Laboratory, Structural Biology Center (SBC), at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Author contributions
P.A., T.H., I.K., E.G., A.K., G.K., K.W.T., M.C.O., J.P.O., and S.K. designed research; P.A., T.H., I.K., E.G., A.K., G.K., M.W., M.Z., K.W.T., J.L., and S.K. performed research; L.M. and I.B. contributed new reagents/analytic tools; J.P.O. analyzed data; and P.A., T.H., I.K., J.P.O., and S.K. wrote the paper.
Competing interests
S.K. is a co-founder and holds equity in Aethon Therapeutics and Revalia Bio. P.A., T.H., A.K., K.W.T., and S.K. are listed as inventors of pending patents on technology described in this paper filed by New York University. A.K. and S.K. are listed as inventors on issued and pending patents on the monobody technology filed by the University of Chicago and Novartis. S.K. has received research funding from Aethon Therapeutics, Argenx BVBA, Black Diamond Therapeutics, and Puretech Health. S.K. received consulting fees from Aethon Therapeutics and Black Diamond Therapeutics.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
John P. O’Bryan, Email: obryanjo@musc.edu.
Shohei Koide, Email: Shohei.Koide@nyulangone.org.
Data, Materials, and Software Availability
Structural coordinates data have been deposited in Protein Data Bank (8EZG (29) and 8F0M (30)). All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., Der C. J., Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug. Discov. 13, 828–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore A. R., Rosenberg S. C., McCormick F., Malek S., RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug. Discov. 19, 533–552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M., K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janes M. R., et al. , Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Canon J., et al. , The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Hallin J., et al. , The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prior I. A., Hood F. E., Hartley J. L., The frequency of Ras mutations in cancer. Cancer Res. 80, 2969–2974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakamoto K., Masutani T., Hirokawa T., Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 10, 21671 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z., et al. , GTP-state-selective cyclic peptide ligands of K-Ras(G12D) block its interaction with Raf. ACS Cent. Sci. 6, 1753–1761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X., et al. , Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J. Med. Chem. 65, 3123–3133 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Hallin J., et al. , Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 28, 2171–2182 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Akkapeddi P., Teng K. W., Koide S., Monobodies as tool biologics for accelerating target validation and druggable site discovery. RSC Med. Chem. 12, 1839–1853 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng K. W., et al. , Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 12, 2656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson L. M., et al. , Inducible degradation of target proteins through a tractable affinity-directed protein missile system. Cell Chem. Biol. 27, 1164–1180.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bery N., Miller A., Rabbitts T., A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 11, 3233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim S., et al. , Exquisitely specific anti-KRAS biodegraders inform on the cellular prevalence of nucleotide-loaded states. ACS Cent. Sci. 7, 274–291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanman B. A., et al. , Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J. Med. Chem. 63, 52–65 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Gilbreth R. N., Koide S., Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr. Opin. Struct. Biol. 22, 413–420 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Donnell J. P., et al. , Timing and reset mechanism of GTP hydrolysis-driven conformational changes of Atlastin. Structure 25, 997–1010.e1014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berghuis A. M., Lee E., Raw A. S., Gilman A. G., Sprang S. R., Structure of the GDP-Pi complex of Gly203–>Ala gialpha1: A mimic of the ternary product complex of galpha-catalyzed GTP hydrolysis. Structure 4, 1277–1290 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Fowler D. M., et al. , High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koide A., Gilbreth R. N., Esaki K., Tereshko V., Koide S., High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. U.S.A. 104, 6632–6637 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bery N., et al. , KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 10, 2607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer-Smith R., et al. , Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 13, 62–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teng K. W., Koide A., Koide S., Engineering binders with exceptional selectivity. Methods Mol. Biol. 2491, 143–154 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Hattori T., et al. , Creating MHC-restricted neoantigens with covalent inhibitors that can be targeted by immune therapy. Cancer Discov. 13, 132–145 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan I., et al. , Identification of the nucleotide-free state as a therapeutic vulnerability for inhibition of selected oncogenic RAS mutants. Cell Rep. 38, 110322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan I., Spencer-Smith R., O’Bryan J. P., Targeting the alpha4-alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 38, 2984–2993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hattori T., et al. , Monobody 12D1 bound to KRAS(G12D). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/8EZG. Deposited 31 October 2022.
- 30.Hattori T., et al. , Monobody 12D5 bound to KRAS(G12D). Worldwide Protein Data Bank (wwPDB). https://www.rcsb.org/structure/8F0M. Deposited 03 November 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Structural coordinates data have been deposited in Protein Data Bank (8EZG (29) and 8F0M (30)). All study data are included in the article and/or SI Appendix.