

Significance
Dysregulated neuroinflammatory responses in the central nervous system (CNS) are known to cause neuronal death in amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases. In this study, we uncover MOK (MAPK/MAK/MRK overlapping kinase) as a signaling kinase that controls immune responses by microglial cells, the main immunocompetent cells in the CNS. Furthermore, we identify an MOK-regulated protein, the epigenetic reader and transcriptional supporter Brd4 (bromodomain-containing protein 4), and demonstrate that MOK controls the inflammatory response by regulating Brd4 chromatin-binding functions on inflammatory genes. Remarkably, we report that MOK is increased in CNS samples from ALS patients and animal models and that administration of a specific MOK inhibitor to ALS mice is able to modify the natural course of disease.
Keywords: amyotrophic lateral sclerosis (ALS), glia, neuroinflammation, signaling kinase, neurodegenerative disease
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal and incurable neurodegenerative disease affecting motor neurons and characterized by microglia-mediated neurotoxic inflammation whose underlying mechanisms remain incompletely understood. In this work, we reveal that MAPK/MAK/MRK overlapping kinase (MOK), with an unknown physiological substrate, displays an immune function by controlling inflammatory and type-I interferon (IFN) responses in microglia which are detrimental to primary motor neurons. Moreover, we uncover the epigenetic reader bromodomain-containing protein 4 (Brd4) as an effector protein regulated by MOK, by promoting Ser492-phospho-Brd4 levels. We further demonstrate that MOK regulates Brd4 functions by supporting its binding to cytokine gene promoters, therefore enabling innate immune responses. Remarkably, we show that MOK levels are increased in the ALS spinal cord, particularly in microglial cells, and that administration of a chemical MOK inhibitor to ALS model mice can modulate Ser492-phospho-Brd4 levels, suppress microglial activation, and modify the disease course, indicating a pathophysiological role of MOK kinase in ALS and neuroinflammation.
Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset neurodegenerative disease in which motor neurons in the cortex, brainstem, and spinal cord gradually degenerate and die off (1, 2). A typical characteristic of ALS and most other neurodegenerative diseases is neuroinflammation, which is associated with disease progression and spreading (1). In particular, several studies have shown that microglia, the main immunocompetent cells in the central nervous system (CNS), become activated and neurotoxic and thereby contribute to motor neuron loss and disease progression (3). Still, the mechanisms driving microglia activation and neurotoxicity in ALS remain incompletely understood, and key molecular mediators along the activated signaling pathways need to be identified (4–6). A powerful emerging view poses that dysregulated neuroinflammatory responses in these disorders may be caused by disruption at the signaling-epigenetics-transcriptional axis level (7, 8).
A hallmark of ALS and other related disorders is the intracellular accumulation of protein inclusions containing misfolded TDP-43 (transactive response DNA-binding protein-43) and in certain cases, RNA-binding protein FUS or SOD1 proteins. TDP-43—a multifunctional protein with RNA/DNA binding activities and physiological localization in the cell nucleus—has been found to display functional abnormalities, including its identification as a major component of cytoplasmic intraneuronal and glial inclusions, in virtually all ALS patients (9, 10) and various ALS animal models (11, 12). Thus, TDP-43 is currently thought to represent a common molecular hub in these disorders, where different pathogenic mechanisms may converge and finally result in neurotoxic cellular outcomes (13).
Recently, we reported that MAPK/MAK/MRK overlapping kinase (MOK), a Ser/Thr kinase that belongs to the mitogen-activated protein kinase (MAPK) superfamily (14), interacts and colocalizes with cytoplasmic TDP-43 inclusions in microglia resulting from exposure of cells to exogenous TDP-43 aggregates (15). MOK is an atypical Ser/Thr signaling kinase, found both in the cytosol and nucleus of different cell types and with a demonstrated function in cilium growth regulation in renal epithelium (16). Up to now, no physiological substrate or downstream-regulated target has been identified for this kinase, and no function or molecular mechanism in relation to immunity or the CNS has been identified.
In this study, we uncover a role of MOK kinase in controlling inflammatory and type-I interferon (IFN) responses in microglia via a Brd4 (bromodomain-containing protein 4)-dependent mechanism and report that MOK-mediated immune functions are dysregulated, and actively participate, in ALS pathophysiology. Thus, we disclose a signaling pathway in neuroinflammation and unveil a mechanism of ALS-associated immune dysregulation.
Results
MOK-Mediated Mechanisms Are Mobilized in Microglial Cells under TDP-43 Aggregation.
To evaluate whether MOK is engaged in cellular processes activated in the context of TDP-43 aggregation, we started by carrying out SLAM-Seq analysis to report on immediate changes in RNA synthesis (17) from SIM-A9 microglial cells exposed to TDP-43 aggregates (TDP43) or sham controls (Sham), with or without pretreatment with AG2P145D/Comp13 (C13) MOK inhibitor (18) (SI Appendix, Fig. S1 A and B). C13 is an unusually specific inhibitor based on the structural, virtual uniqueness of a cysteine gatekeeper in MOK’s active site (18). The results showed a clear difference in the transcriptional changes caused by MOK inhibition for a number of genes. Remarkably, pretreatment with C13 revealed a dramatically different impact in the differentially expressed genes (DEGs) (Fig. 1A, SI Appendix, Fig. S1A, and Dataset S1) and their predicted interaction networks (19) under TDP-43 aggregation compared to control (sham)-treated cells (Fig. 1A). Interestingly, 45% of the DEGs resulting from exposure to TDP-43 aggregates were counterregulated by C13 pretreatment (Fig. 1B and SI Appendix, Fig. S1B), suggesting a possible participation of MOK in microglial responses elicited by TDP-43 aggregates.
Fig. 1.
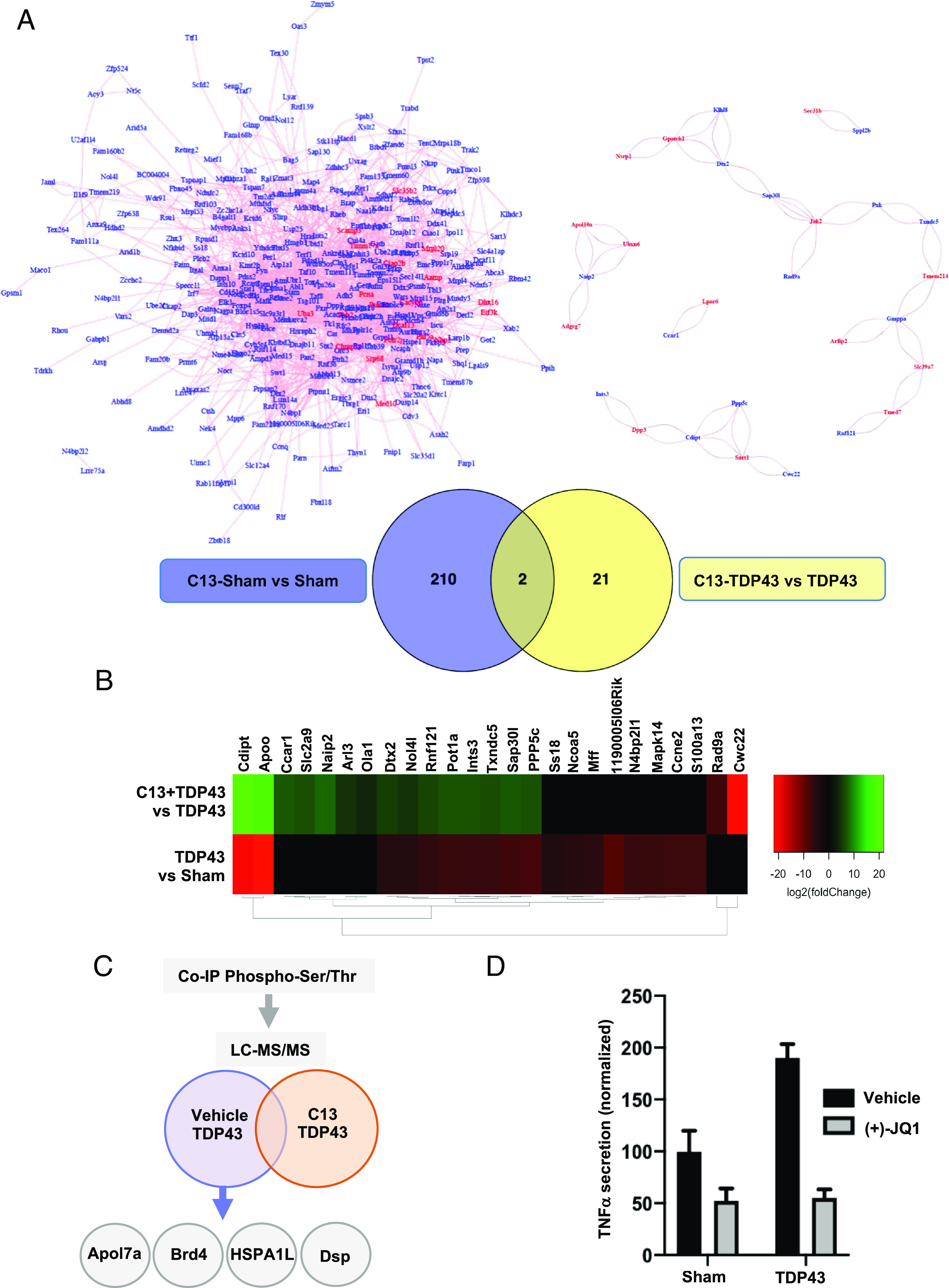
MOK-mediated mechanisms occur in microglial cells upon exposure to TDP-43 aggregates. (A, Bottom) Venn diagrams showing the number of DEGs from analyzed SLAM-Seq data obtained with SIM-A9 cells exposed to 5 µg/mL TDP-43 aggregates (TDP43) or sham aggregates (Sham) for 4 h, pretreated for 1 h with 10 µM C13 [or DMSO (dimethylsulfoxide) as vehicle]. Results are from three independent experiments (N = 3). Indicated are the number of DEGs for “C13 Sham vs. Sham” and “C13-TDP43 vs. TDP43” comparisons (PAdj. < 0.05 and PAdj. < 0.1, respectively). (A, Top) Predicted network analysis based on the two sets of identified DEGs by using GeneMANIA (19). Blue labels correspond to top DEGs and red labels, to inferred genes. (B) Heatmap representing the relative expression profiles (DEGs with PAdj. < 0.05) comparing “TDP43 vs. Sham” and “C13-TDP43 vs. TDP43” treatments. The differential regulation in gene expression changes between both comparisons indicates an effect of MOK inhibitor C13 in the transcriptional profile of microglial cells upon exposure to TDP-43 aggregates. (C) Schematic representation of LC–MS/MS analysis of eluates from antiphospho-Ser/Thr immunoprecipitation assays of lysates from primary microglial cells exposed to 5 µg/mL TDP-43 aggregates, pretreated for 1 h with 10 µM C13 (or DMSO). (D) Determination of TNFα by ELISA from primary microglial cells exposed to 5 µg/mL TDP-43 aggregates (TDP43) or sham aggregates (Sham) overnight, pretreated for 1 h with 10 µM (+)-JQ1 or DMSO (vehicle). Shown values were normalized to Sham control. Data are mean ± SD (N = 2).
Next, to identify proteins whose phosphorylation state could be regulated by MOK kinase in the ALS cellular model, we used immunoprecipitation against phospho-Ser/Thr-containing proteins from primary microglial cells exposed to TDP-43 aggregates, pretreated or not with C13. By subsequent liquid chromatography with tandem mass spectrometry (LC–MS/MS) analyses of both eluates, we identified several proteins in both samples, a few of them exclusively in the absence of MOK inhibitor pretreatment, including desmoplakin, heat-shock protein 1-like protein, Brd4, and apolipoprotein L 7a (Fig. 1C and Datasets S2 and S3). Interestingly, Brd4 has been recently involved in innate immune functions, including microglial responses (20–22). To verify the participation of Brd4 in TDP-43-induced microglial activation, we assessed the impact of the Brd4 inhibitor (+)-JQ1 in TNFα secretion by ELISA (Fig. 1D). Remarkably, the aggregate-elicited cytokine levels were largely suppressed by pretreatment with (+)-JQ1. Taken together, these results indicate that MOK is functionally mobilized in microglial cells under TDP-43 aggregation and may regulate downstream phosphorylation of proteins, including Brd4, involved in microglial activation and immune responses.
MOK Controls Microglial Responses by Mediating Inflammatory and Type-I IFN Pathways.
Given that exposure of microglial cells to pathology-associated TDP-43 species had been found to activate the NLRP3 inflammasome by our group and by others (15, 23, 24), we sought to assess the impact of MOK inhibition in inflammasome activation by using microglial cells exposed to TDP-43 or sham aggregates. Remarkably, the higher levels of interleukin (IL)-1β and IL-18 inflammasome-dependent cytokines induced by TDP-43 aggregates were significantly suppressed by pretreatment with the MOK-inhibitor C13 (SI Appendix, Fig. S1 C and D).
Next, in order to investigate a potential role of MOK in mediating the general inflammatory response in microglia, we assessed TNFα, IL-6, and IL-1β cytokine secretion levels from lipopolysaccharide (LPS)-stimulated primary microglial cells (5 h or 16 h poststimulation), either pretreated or not with C13 (Fig. 2A). Remarkably, C13 pretreatment significantly reduced secretion of all three proinflammatory cytokines otherwise induced by LPS alone. This result was also observed for SIM-A9 cells, in which pretreatment with C13 led to lower TNFα secreted levels after LPS stimulation (SI Appendix, Fig. S2A). Furthermore, western blot analyses evidenced lower Ser536 phosphorylation of p65 signal upon LPS stimulation for C13 pretreated cells, pointing to a role of MOK in the NF-κB pathway in the inflammatory response (SI Appendix, Fig. S2B). Next, to elucidate the effect of C13 treatment on LPS-stimulated microglia at a transcriptional level, RNA-Seq was carried out with RNA isolated from primary microglial cells after stimulation for 5 h with LPS, subjected or not to pretreatment with C13 (Dataset S4). Based on the 158 DEGs identified, we used Ingenuity Pathway Analysis (IPA) software enabling to predict canonical pathways, upstream regulators, functions, and networks (Fig. 2B and Dataset S5). Notably, IPA revealed a number of significantly inhibited (IRF3/7, PML, and STAT1) and activated (CCAAT/enhancer-binding protein beta/CEBPB, NFE2L2/Nrf2, and STAT3) upstream regulators as a result of C13 pretreatment.
Fig. 2.

MOK positively regulates microglial responses, including inflammatory and type-I IFN pathways. (A) Quantification of proinflammatory cytokines by ELISA in supernatants from primary microglial cells stimulated with 1 µg/mL LPS for 5 h (TNFα) or 16 h (IL-6, IL-1β) after pretreatment with 10 µM C13 (or DMSO) for 1 h (N = 4). One-way ANOVA followed by the Tukey post hoc test. (B) Top predicted upstream regulators by IPA from 158 DEGs (C13-treated vs. untreated, PAdj < 0.05) identified by RNA-Seq studies from primary microglial cells stimulated with 1 µg/mL LPS for 5 h. The P-values of overlap are shown in parentheses. (C) Assessment of gene expression levels for a set of key cytokines by qRT-PCR from WT and MOK-KO SIM-A9 cells stimulated with 1 µg/mL LPS for 5 h (N = 3). Data represent fold changes relative to unstimulated WT cells. #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. unstimulated controls. One-way ANOVA followed by the Tukey post hoc test. (D) Quantification of TNFα, IL-6, IFNβ and IL-1α levels by ELISA in supernatants from WT and MOK-KO SIM-A9 cells stimulated with 1 µg/mL LPS for 5 h or overnight for IL-1α (N = 5 for IL-6, TNFα and N = 4 for IFNβ, IL-1α). One-way ANOVA followed by the Tukey post hoc test. (E) Flow cytometry quantification of IL-6+ cells in WT and MOK-KO SIM-A9 cells, previously stimulated with 1 µg/mL LPS for 5 h (Left). Rescue of phenotype in MOK-KO cells resulting from cell transfection with FLAG-MOK-WT or FLAG-MOK-KD constructs and stimulation with 1 µg/mL LPS for 5 h (Right) (N = 3). Student’s t test performed between shown groups, paired, one-tailed. (F) Fold changes of primary motor neurons survival (relative to each control) after exposure to conditioned media from LPS-stimulated WT or MOK-KO cells overnight (N = 3). Student’s t test between shown groups, unpaired, two-tailed. Data in A and C–F are the mean ± SEM. from “N” independent experiments, and *P < 0.05, **P < 0.01, and ***P < 0.001. NS: not significant.
To confirm the participation of MOK in the inflammatory response in microglia, CRISPR/Cas9-generated MOK-KO (MOK-knockout) and WT (wild-type) SIM-A9 cells (SI Appendix, Fig. S2 C and D) were stimulated with LPS for 5 h or 16 h and analyzed by qRT-PCR and/or ELISA for IL-6, TNFα, IFNβ and IL-1α (Fig. 2 C and D and SI Appendix, Fig. S2 E and F), the results clearly demonstrating reduced levels of these key cytokines in LPS-stimulated MOK-KO cells. As expected, this profile of cytokine expression was mirrored by flow cytometry analysis (Fig. 2 E, Left). Importantly, we assessed the functional restoration of MOK activity in MOK-KO cells transfected with a FLAG-MOK construct carrying or not a “kinase-dead” (MOK-KD) mutation (25), by IL-6 expression after LPS stimulation (Fig. 2 E, Right). Remarkably, quantification of IL-6+ cells of (FLAG+) transfected cells demonstrated the rescuing of cytokine expression in MOK-KO cells by recombinant MOK-WT, but not by MOK-KD (Fig. 2E and SI Appendix, Fig. S3). Put together, these results demonstrate that MOK controls the inflammatory response in microglia, and this function is dependent on its kinase activity. To assess the impact in neuronal viability of MOK-regulated microglial responses, we exposed primary motor neurons in culture to conditioned media from LPS-stimulated WT or MOK-KO cells and quantified dead cells by using a fluorescence-based assay (Fig. 2F). As expected, medium from LPS-stimulated WT cells caused a reduction in neuronal cell survival compared to control conditions. Notably, the opposite effect was seen with medium from LPS-stimulated MOK-KO cells, indicating that MOK-mediated microglial responses can be neurotoxic.
We then carried out RNA-Seq analysis by comparing MOK-KO vs. WT SIM-A9 cells, stimulated or not with 1 µg/mL LPS for 5 h (Fig. 3 A and B, SI Appendix, Fig. S4, and Datasets S6–S9). A relatively large number of protein-coding and noncoding DEGs were identified, showing a partial overlap between both cell lines and treatments and including 730 total DEGs (Fig. 3 A, Top) and 424 coding DEGs (Fig. 3 A, Bottom and 3B) found when comparing MOK-KO vs. WT cells stimulated with LPS. Gene ontology (GO) clustering analysis revealed significantly enriched biological processes related to microglial activation and innate immunity and, interestingly, IFNα/IFNβ (type-I IFN) regulation and antiviral responses (Fig. 3 C and D, SI Appendix, Fig. S5A, and Dataset S10). In particular, analysis of coding DEGs that differentially changed for MOK-KO or WT cells upon LPS-stimulation revealed enriched terms related to “innate immunity,” “positive NF-κB regulation,” and “antiviral response” (SI Appendix, Fig. S5B and Dataset S11). Finally, IPA prediction between MOK-KO vs. WT cells upon LPS stimulation unleashed a number of activated (CITED2 and TRIM24) and inhibited (e.g., IRF7 (interferon regulatory factor 7)/IRF3, STAT1, and BHLHE40) upstream regulators (Fig. 3E and Dataset S12) as well as differentially regulated “neuroinflammation,” “pyroptosis,” and “TRIM1” canonical pathways, among others (SI Appendix, Fig. S5C and Dataset S12). The expression of IRF7, a principal type-I IFN master regulator and also a DEG revealed in our RNA-Seq study, was assessed by qRT-PCR and confirmed to be suppressed in the MOK-KO cells upon LPS stimulation (Fig. 3F). Collectively, our results demonstrate a role of MOK in microglia activation and inflammatory response mediated by NF-κB and STAT1 pathways and critically involving induction of IRF7/3/type-I IFN and antiviral cellular responses.
Fig. 3.
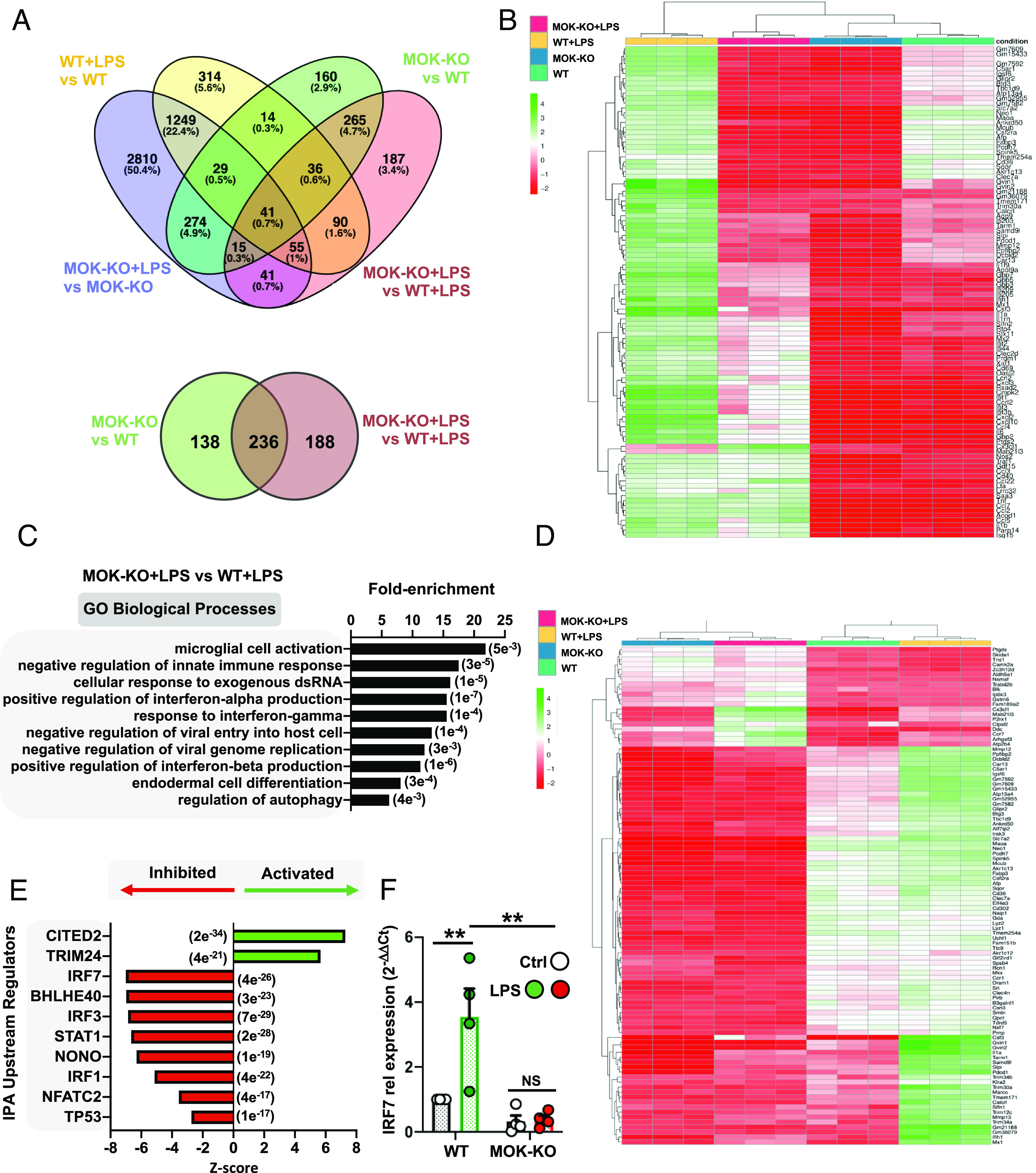
Transcriptomic analysis from RNA-Seq studies of MOK-KO cells and response to LPS stimulation. Data in A–E are from 3 independent experiments (N = 3). (A) Venn diagrams depicting the number of total (Top) and protein-coding (Bottom) DEGs between MOK-KO and WT SIM-A9 cells under basal conditions and/or after 1 µg/mL LPS stimulation for 5 h (PAdj. < 0.05; log2 fold change >2 and <−2); (B) Heatmap showing unsupervised clustering analysis for the top 100 protein-coding genes based on relative expression levels in the four samples; (C) GO “biological process” term enrichment analysis of total DEGs for MOK-KO+LPS vs. WT+LPS. Shown are the top hits based on the P-value (indicated in parentheses) with at least three up-/down-regulated genes; (D) Heatmap depicting clustering analysis for 100 coding DEGs identified for the MOK-KO+LPS vs. WT+LPS comparison (consisting of the top 80% down-regulated and top 20% up-regulated, genes to maintain proportionality of all significant DEGs found); (E) Top 10 IPA-predicted upstream expression regulators from coding DEGs comparing MOK-KO vs. WT cells upon LPS stimulation, indicating positive (activated) or negative (inhibited) z-scores. The results are from all genes that were found to be differentially regulated in the RNA-Seq deseq2 analysis in any comparison with PAdj of <e-5. The P value of overlap is indicated in parentheses; (F) Assessment of IRF7 gene expression by qRT-PCR from WT and MOK-KO SIM-A9 cells stimulated with 1 µg/mL LPS for 5 h. Data represent fold changes relative to unstimulated WT cells (WT Ctrl) (N = 4). One-way ANOVA followed by the Tukey post hoc test. Data are the mean ± SEM. from N independent experiments, and *P < 0.05 and **P < 0.01.
MOK Mediates Immune Responses by Regulating Phospho-Brd4 Levels and Brd4 Chromatin Binding in Microglia.
One of the proteins whose phosphorylated state levels we found to be putatively regulated by MOK (Fig. 1C) corresponds to Brd4 which belongs to the bromodomain and extraterminal domain (BET) bromodomain family, controlling the transcription of genes by binding to acetylated histones in chromatin (26). Brd family members have been linked to cancer and autoimmune disease mechanisms that are still not clear. In particular, Brd4 has been recently involved in innate immune responses, including inflammatory responses (20, 22, 27). To verify that Brd4 is mediating the inflammatory response in our cell line as previously shown in microglia (21, 28), we stimulated SIM-A9 cells in culture with LPS for 5 h after pretreatment with the Brd4 inhibitor (+)-JQ1 (or (−)-JQ1 negative control) and quantified IL-6 and TNFα secretion levels, confirming that (+)-JQ1 effectively suppresses LPS-induced inflammatory responses in both primary microglia and SIM-A9 cells (SI Appendix, Fig. S6 A and B).
Next, to validate our screening result and test whether MOK kinase is indeed regulating phosphorylated Brd4 levels in inflammatory microglia, we quantified Ser492 phosphorylated Brd4 (pBrd4)—previously shown to enable Brd4 chromatin-binding functions (29, 30)—in samples from C13-pretreated SIM-A9 cells stimulated with LPS by western blot, showing a clear attenuation of the pBrd4 band (ca. 250 kDa) in LPS-stimulated cells (SI Appendix, Fig. S6C). Moreover, confocal immunofluorescence (IF) analyses with primary microglia showed that contrary to LPS-stimulated cells displaying higher pBrd4 nuclear levels compared to basal control, C13-pretreatment led to lower pBrd4 nuclear levels following LPS stimulation (Fig. 4 A and B). Importantly, these results were mirrored by LPS-stimulated MOK-KO cells compared to WT cells both by western blot (Fig. 4 C and D and SI Appendix, Fig. S2G) and confocal IF analysis (Fig. 4 E and F), where a significant reduction of nuclear pBrd4 was seen in MOK-KO compared to WT cells upon LPS stimulation. These results clearly demonstrate that MOK kinase regulates nuclear pBrd4 levels in microglia under inflammatory conditions.
Fig. 4.
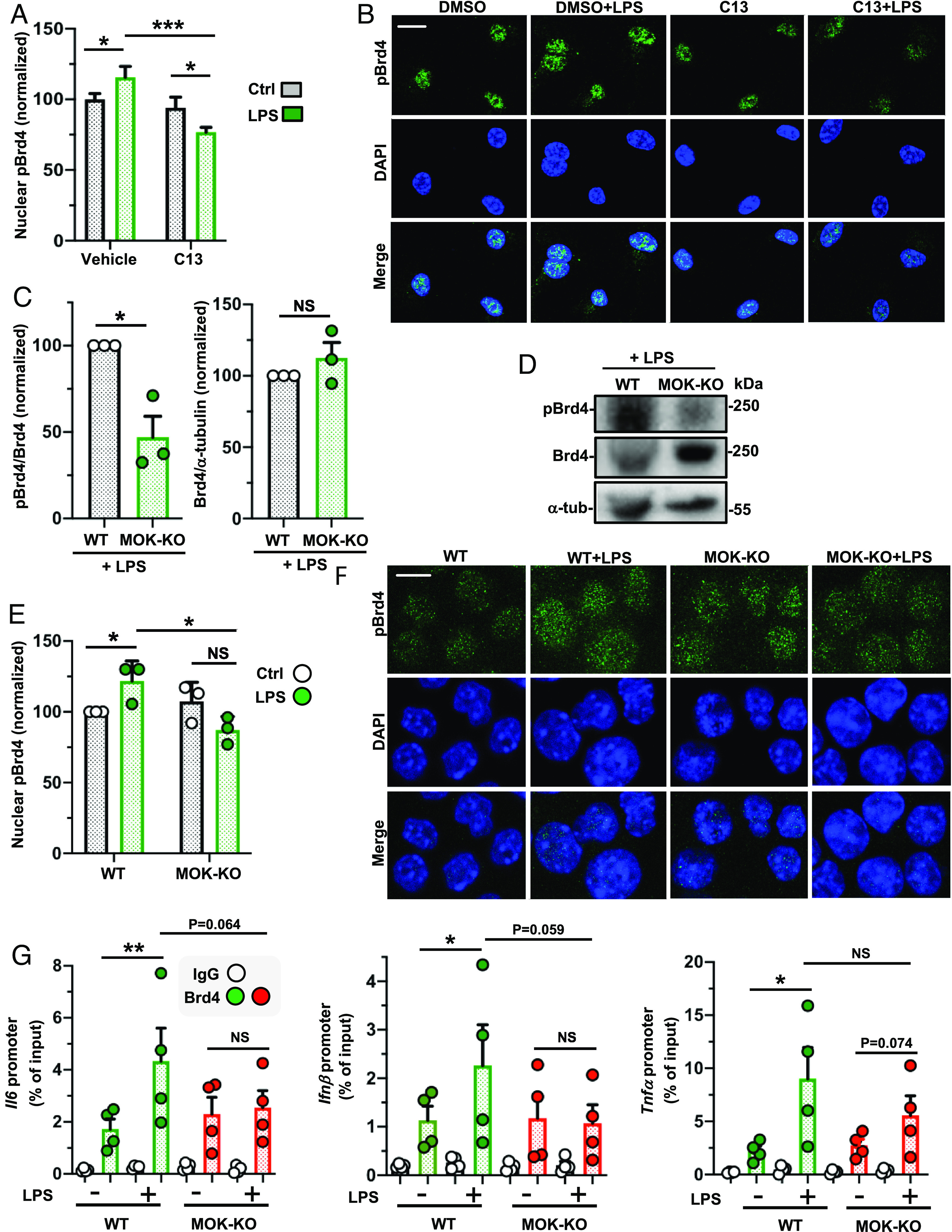
MOK regulates phospho-Ser492-Brd4 levels under neuroinflammatory conditions. (A and B) Quantification of nuclear pBrd4 levels by confocal IF with anti-pBrd4 (green) and DAPI staining of primary microglial cells pretreated with 10 µM C13 (or DMSO) for 1 h and stimulated or not with 1 µg/mL LPS for 4 h. The data and images shown correspond to one experiment (N = 20 analyzed images per condition) and are representative of two independent experiments. (Scale bar: 10 µm.) (C and D) Quantification and representative image of western blot analyses with anti-pBrd4 and anti-Brd4 (normalized to Brd4 or a-tubulin signal, respectively) in lysates of WT and MOK-KO SIM-A9 cells stimulated with 1 µg/mL LPS for 1 h. Data are from three independent experiments (N = 3). Student’s t test, unpaired, one-tailed. (E and F) Quantification and representative images of confocal IF analyses of nuclear pBrd4 levels with anti-pBrd4 (green) and DAPI staining of WT and MOK-KO SIM-A9 cells stimulated or not with 1 µg/mL LPS for 1 h. The signal was enhanced by 50% in all four images in the first row for better visualization. Images are from one out of three independent experiments (N = 3). (Scale bar: 10 µm.) Student’s t test, unpaired, two-tailed. (G) ChIP-PCR assessment of Brd4 binding to specific cytokine promoters. Represented data are relative values (percentage of input) of PCR products for Il6, Ifnβ1, and Tnfα promoters after ChIP assay with either anti-Brd4 or IgG control antibodies with chromatin isolated from WT or MOK-KO cells treated or not with 1 µg/mL of LPS for 1 h (N = 4). Student’s t test between shown groups, ratio-paired, one-tailed. Data are the mean ± SEM. from N independent experiments, and *P < 0.05, **P < 0.01 and ***P < 0.001. NS: not significant.
To assess a possible role of MOK in Brd4 binding to key cytokine gene promoters previously linked to Brd4 recruitment (31–33), we carried out ChIP (chromatin immunoprecipitation)-qPCR assays from SIM-A9 cells after 1 h of LPS stimulation (Fig. 4G). Remarkably, we found that whereas WT cells showed incremented Brd4 binding to Il6, Ifnb1, and Tnfα gene promoters in response to LPS compared to unstimulated cells, such increase was abrogated in MOK-KO cells for Il6, Ifnβ1, and, to a lesser extent, Tnfα promoters. These results demonstrate a role of MOK in positive regulation of Brd4 binding to proinflammatory and type-I IFN gene promoters under inflammatory conditions. Of note, pretreatment with the Brd4-inhibitor (+)-JQ1 resulted in significant reduction of the ChIP-qPCR signal increments, confirming the specificity of Brd4 binding assessment in our assays (SI Appendix, Fig. S6D). Collectively, these results reveal a central role for MOK in Brd4-mediated regulation of cytokine expression upon inflammatory stimulation. On the other hand, the lack of a significant decrease in Brd4 binding to TNFα promoters in LPS-stimulated MOK-KO suggests that Brd4-independent, MOK-mediated mechanisms are also contributing to the upregulation of certain cytokines during microglial responses.
MOK Is Altered in the Spinal Cord from ALS Patients and Mouse Models, and C13 Administration to SOD1G93A Mice Modifies the Disease Course.
Given that MOK was found to be engaged in microglia under TDP-43 aggregation and based on our results showing a role of MOK in the inflammatory response, we wondered whether MOK would be altered in ALS individuals. To address this question, we first assessed phospho-Ser159/Tyr161 MOK (bpMOK), considered to correspond to the fully active form of the kinase (14). Western blot analysis using murine spinal cord tissue homogenates showed a band of ca. 65 kDa, in agreement with previously reported MW for CNS tissue (34) (SI Appendix, Fig. S7A). Next, we carried out immunohistochemistry (IHC) assays to detect bpMOK in spinal cord slices from a TDP-43-based mouse model (TgTDP43) (35) at the late-stage and from sporadic ALS patients. Considering the significant differences in cell size measured between WT and TgTDP43 samples (SI Appendix, Fig. S7B), we normalized the quantitated bpMOK levels in these samples to the cell area. Remarkably, the results revealed significantly higher levels of bpMOK in the mouse model compared to age-matched mice (Fig. 5A). Furthermore, assessment of bpMOK cellular signal in tissue from human subjects also showed increased bpMOK levels in ALS samples compared to age-matched controls (Fig. 5B).
Fig. 5.

MOK is altered in CNS cells in the context of ALS. Quantification of bpMOK levels by IHC analysis in spinal cord tissue samples (Left) and representative IHC images (Right) for spinal tissue samples from (A) TgTDP43 and WT mice (late stage; N = 3) and (B) sporadic ALS patients and age-matched control subjects (N = 4). Student’s t test, unpaired, one-tailed. (Scale bar: 25 µm.) The white arrow indicates one analyzed cell as an example. (C and D, Left) quantification of nuclear bpMOK (C) or pBrd4 (D) levels (green) and DAPI staining by confocal IF of spinal cord organotypic cultures, stimulated or not with 1 µg/mL LPS overnight, from WT and SOD1G93A mice (5 wk old, N = 3). Represented data are the fold change of nuclear bpMOK signal upon LPS stimulation normalized to the corresponding unstimulated controls. Student’s t test, unpaired, two-tailed. (Right) representative images of both IF assays corresponding to each condition. (Scale bar: 25 µm.) (E) Flow cytometry analysis of the bpMOKhi-expressing population in CD11b+ cells isolated from early-onset SOD1G93A or WT mouse spinal cords (14 wk old; N = 9). Student’s t test, unpaired, two-tailed. (F) Time-course body weight monitorization of SOD1G93A mice injected (i.p.) with C13 compound (20 µg per dose) or vehicle (N = 6) starting at 11 wk of age, every other day along 3 wk (gray window). Body weight values are normalized to each mouse’s maximum reached weight (% of maximum). One-way ANOVA followed by the Tukey post hoc test. (G) Flow cytometry analysis of the CD11bhi-expressing live cells isolated from the spinal cord from WT or C13- or vehicle-administered SOD1G93A mice (14 wk old; N = 6 for WT, N = 4 for C13/vehicle-treated SOD1G93A mice). The CD11bhi signal threshold corresponded to the top 5% (on average) for the WT mice. Student’s t test, paired, one-tailed. Data are the mean ± SEM. from N independent experiments, and *P < 0.05, **P < 0.01, and ***P < 0.001. NS: not significant.
To get an insight into a possible engagement of MOK in a pathophysiological context of ALS neuroinflammation, we chose the well-established and neuroinflammation-recapitulating SOD1G93A animal model (24, 36–38), which has additionally been found to display TDP-43 functional abnormalities (39–43). We investigated the relative changes in bpMOK levels induced by LPS-stimulation in spinal cord organotypic cultures from presymptomatic (5 wk old) SOD1G93A mice vs. WT controls, by IF confocal analysis (Fig. 5C). As opposed to WT mice, in which no significant changes were observed when stimulated with LPS, a significant increase in nuclear bpMOK was seen in LPS-stimulated spinal cultures from ALS mice. Remarkably, by performing an analogous study with pBrd4 immunolabeling (Fig. 5D) we observed that whereas LPS stimulation resulted in up-regulated nuclear pBrd4 in spinal cord tissue from healthy mice, no upregulation—but rather, a reduction—in pBrd4 was induced for ALS animals. These results suggest that the MOK-pBrd4 axis activated in the inflammatory response is dysregulated in ALS.
To determine whether the levels of functional MOK kinase could be specifically altered in microglia in the context of ALS, we isolated microglial cells from the spinal cord of WT or SOD1G93A mice at postonset (14 wk old) and quantified the cells displaying high levels of bpMOK among the cell population expressing the microglial surface marker CD11b by flow cytometry (Fig. 5E and SI Appendix, Fig. S7 C and D). Remarkably, bpMOKhigh microglial cells were significantly more abundant in cells isolated from SOD1G93A mice compared to controls, showing ca. fivefold increase in the ALS model.
Finally, to investigate a possible role of MOK in the pathophysiology of ALS in vivo and its potential as a therapeutic target, we peripherally administered the MOK inhibitor C13, or vehicle, to SOD1G93A mice by intraperitoneal injection every other day along 3 wk, starting at the preonset stage (11 wk of age). Assessment of motor performance by the rotarod test did not show significant differences between both groups (SI Appendix, Fig. S7E). Notably, however, C13 treatment resulted in protection during a time window at the postonset stage and reaching statistical significance 1 wk after the end of treatment, according to relative body weight assessment, another standard parameter used to monitor the disease course in this ALS model (Fig. 5F). Finally, to investigate whether the protective effects of C13 administration to SOD1G93A mice might reflect suppressed microgliosis, we analyzed the content of CD11bhigh cells in the spinal cord from treated or nontreated mice past week 14, i.e., just after the end of treatment (Fig. 5G). As expected, flow cytometry analysis showed increased CD11bhigh cell content in SOD1G93A compared to WT mice, indicating increased microglia activation in the ALS spinal cord (44). Remarkably, the analysis also revealed suppressed levels of CD11bhigh in SOD1G93A mice spinal cord as a result of C13 administration, suggesting that MOK is indeed involved in microglial activation and neuroinflammation in the context of ALS. Of note, analysis of brain motor cortex samples from these mice showed overtly lower pBrd4 levels by IF in C13-treated compared to vehicle-treated, ALS mice (SI Appendix, Fig. S7F). All together, these results demonstrate that MOK kinase and its unleashed regulated pathways are altered in ALS spinal cord cells, including microglia, and they indicate that MOK is involved in the pathophysiology of ALS.
Discussion
Previously, we identified the Ser/Thr kinase MOK as a protein that strongly interacts with abnormal cytoplasmic TDP-43 aggregates in microglial cells (15). Until now, no regulated signaling pathway or downstream molecular target has been identified for MOK, and its potential function in immune cells has not been explored. In this study, we obtained a differential profile of phosphorylated protein candidates under TDP-43 aggregation conditions for C13 pretreated cells, suggesting that MOK is regulating downstream phosphorylation events in ALS microglia, including the epigenetic reader Brd4. Furthermore, pretreatment of microglial cells with MOK inhibitor C13 revealed a number of genes whose early expression upon exposure to TDP-43 aggregates would be regulated by MOK, including Dtx2 (regulator of Notch signaling), Ppp5c (Ser/Thr phosphatase involved in various signaling pathways), and Sap30I (transcription repressor via interaction with histone–deacetylase complexes). Together, these data demonstrate that MOK kinase is functionally mobilized in microglia under TDP-43 aggregation conditions, suggesting a role of this kinase in the processes triggered which include proinflammatory responses.
Indeed, by using a uniquely specific inhibitor of MOK kinase (18) and MOK-deleted cells, we showed that MOK kinase controls the inflammatory response induced by the general stimulator LPS in microglia. Moreover, by exogenous expression of recombinant WT or KD MOK protein in MOK-KO cells, we demonstrated that MOK regulates proinflammatory cytokine expression in a kinase-dependent manner. By supporting IL-6, TNFα and IL-1α/β along with the activation of the p65/RelA NF-κB pathway, MOK regulates the canonical inflammatory response, while also supporting IFNβ secretion, which has been lately related to neuroinflammatory processes and disease progression in ALS and other neurodegenerative disorders (45–47). Integrated transcriptomics analysis of enriched pathways indicated that MOK-dependent control of such responses in microglia is mediated by regulation of microglial activation, the innate immune response, IFNα/β (type-I IFN) production, and antiviral responses. IPA prediction of upstream regulators supports that MOK mediates the “neuroinflammation signaling pathway” in LPS-stimulated microglia by stirring the inhibition of CITED2 and TRIM24—a critical negative regulator of inflammatory gene programs in myeloid cells (48–50) and a suppressor of M2 macrophage polarization (51), respectively. In the case of C13-treatment, the top predicted MOK-inhibited regulators were CEBPB, NFE2L2, and STAT3. Consistent with an NF-κB-mediated anti-inflammatory effect of C13 under LPS stimulation, CEBPB was found to be driven by the NF-kB pathway in dendritic cells in the context of inflammation (52) and has been reported to mediate macrophage M2-phenotype polarization (53, 54) and microglial Ab phagocytosis (55). Recently, STAT3 was shown to act coordinately with CEBPB (56) to activate the NFKBIZ/IκB-ζ protein involved in the regulation of inflammatory responses (57), whereas NFLE2L2/Nrf2 is known to elicit anti-inflammatory responses in microglia and other immune cells (58). In addition, as revealed in both transcriptional studies with MOK-KO cells and C13-treated microglia, MOK favors the activation of STAT1 and IRF7/3 upstream regulators, which are well-known IL-6- and type-I IFN-promoting transcription factors, respectively (59). Indeed, the suppression of IFNβ secretion and the transcriptional signature in MOK-deficient inflammatory microglia indicates that MOK regulates the type-I IFN response, which has been lately related to neuroinflammatory processes in ALS and a variety of other neurodegenerative disorders (60–67). BHLHE40, another predicted MOK-activated regulator, has been reported to act as a central transcription factor for proinflammatory gene expression in macrophages and other immune cells (68, 69). Moreover, it has been recently found to be involved in the regulation of the MGnD or disease-associated microglia (DAM) expression signature (38).
Our study uncovered a protein whose phosphorylation state levels are physiologically regulated by MOK, the epigenetic reader Brd4. Brd4 was previously demonstrated to be phosphorylated at Ser492 by casein kinase II/CK2 (30). Therefore, our results indicate that MOK could be acting on pBrd4 levels directly via its Ser/Thr kinase activity or indirectly, possibly by regulating CK2 activity.
Notably, Brd4 has been shown to play crucial roles in innate immune responses by activating transcription of a number of immune genes (26, 70), and BET protein dysfunction has been lately involved in various inflammation-related disorders, including cancer, neurodegenerative disease, and CNS injury (26, 27). In this study, we showed that MOK supports nuclear levels of the critical phospho-Ser492-Brd4 (pBrd4) state, and furthermore, we demonstrated that MOK controls Brd4 binding to cytokine gene promoters in proinflammatory microglia. This unleashed regulation of Brd4 promoter binding and concomitant cytokine expression by MOK upon acute or “nonpathological” inflammatory stimulation could be of utmost relevance as evidence toward an impact of epigenetic factors in ALS and other neurodegenerative disorders has already started to emerge (71, 72). Intriguingly, the MOK-dependent regulation of TNFα secretion we found not to rely on Brd4’s promoter-binding function may still be Brd4-dependent, as Brd4 was reported to control the NF-κB-inhibitor IkBα at the translation stage, in inflammatory macrophage responses (20).
Finally, we showed that the levels of (Ser159Tyr161)-phospho-MOK (bpMOK) are significantly altered in the spinal cord of ALS patients or animal models, indicating that MOK’s physiological functions are dysregulated in the context of ALS. In particular, we reveal a dramatic increase of bpMOKhi microglia in SOD1G93A mice. Moreover, we observed that inflammatory stimulation of spinal cord samples from preonset SOD1G93A mice, as opposed to WT mice, fails to up-regulate nuclear pBrd4 levels, indicating dysfunctional MOK-Brd4 signaling in the ALS model. Remarkably, we observed that peripheral administration of C13 MOK-inhibitor to late preonset ALS mice results in abrogated pBrd4 levels and reduced CD11bhigh-microglia levels in CNS, while significant protection from disease progression was seen along a short period after treatment was discontinued. These results strongly indicate that MOK is involved in ALS pathophysiology and suggest that targeting MOK with a specific inhibitor in a continued manner could be beneficial throughout the disease course. The broad relevance of our findings is further evidenced by two recent studies reporting associations between MOK gene expression levels in peripheral leukocytes with blood pressure (73) and with type 1 diabetes (74), which supports a possible role for MOK in immune dysregulation in the context of other pathological scenarios. Indeed, protein kinases, including immune-related kinases, have been shown to play crucial roles in the physiopathology of ALS and other neurodegenerative proteinopathies and have recently emerged as attractive therapeutic targets in various neurodegenerative disorders (75, 76). Therefore, the identification of MOK-regulated proteins and downstream signaling pathways in immune cells, particularly in the context of neuroinflammation, could set the bases for therapeutic targeting of key pathogenic pathways in ALS and other inflammation-related disorders.
Overall, we unleash a role of MOK kinase related to immunity and the CNS in controlling the inflammatory response in microglia via Brd4-dependent and independent mechanisms and report that MOK microglial levels and its immune-related functions are dysregulated in ALS, with strong implications in disease pathophysiology. Finally, this study unmasks a signaling pathway in neuroinflammation and discloses potential intervention targets in ALS and other neurodegenerative and inflammatory diseases.
Methods
Summarized methods are below. A detailed description can be found in SI Appendix.
General Reagents and DNA Constructs.
LPS from Escherichia coli (Sigma-Aldrich), (−)-JQ1/(+)-JQ1 (Sigma-Aldrich), polymixin B solution (Merck Life Science S.L.), and dimethyl sulfoxide (DMSO, Sigma-Aldrich) were used. Compound AG2P145D/Comp13 (C13) was synthesized and characterized as previously reported (18). Plasmids encoding FLAG-tagged MOK (murine, WT and KD) were described previously (25).
Human Samples.
Spinal cord samples were obtained after voluntary donation to the Brain Bank of the Region of Murcia by patients diagnosed as sporadic ALS and control subjects with no history of neurological diseases. The sample collection fulfilled the ethical standards of our institutions: IMIB-Arrixaca and ISCIII National Biobank Network Review Boards approved the protocol, which was required for Clinical Trial approval (NCT00124539, EudraCT 2006-00309612). Transverse serial segments (1.0 cm) were embedded in paraffin and cut at 7 µm for IHC immunolabeling.
Animal Models.
For IHC studies, spinal cord tissue slices (lumbar, sacral) were from late-stage Prp-TDP43A315T mice (TgTDP43), an ALS/FTD model described previously (35), and WT, age-matched controls. For all the remaining studies, transgenic B6SJL-TgN(SOD1-G93A) 1Gur/J (Jackson Laboratory, USA) male mice carrying the human SOD1 gene with the G93A point mutation (SOD1G93A) (77) and WT counterpart mice were used.
Administration of C13 to SOD1G93A Mice and Phenotype Monitorization.
Mice were injected intraperitoneally every other day with a total of 100 µl solution consisting of either 20 µg of C13 compound (18) in PBS or vehicle control containing 2% DMSO, for a total of 3 wk starting at 11 wk of age. Mice were weighed and tested in rotarod regularly. At the end of treatment, a number of mice from each group were killed, and their spinal cord and brain were removed for microglial cell isolation followed by CD11b assessment by flow cytometry and for pBrd4 IF analysis, respectively.
Cell and Organotypic Culture Preparation and Culture Treatment.
Mixed glial cultures were prepared from cerebral cortices of 1 to 3-d-old C57BL/6 male mice (University of Seville Animal Core Facility), and microglia were isolated as previously described (15). SIM-A9 microglial cell line (CRL-3265) was acquired from American Type Culture Collection (ATCC). Stimulation with LPS (1 µg/mL) was for 1, 5, or 16 h. Pretreatment with 10 µM C13 or 10 µM (−)-JQ1/(+)-JQ1 (or DMSO) was done by preincubating cells for 1 h. TDP-43 and sham aggregates were obtained as previously described (15, 78, 79). SIM-A9 or primary microglial cells were pretreated with 10 µM C13 or DMSO for 1 h and then with 5 µg/mL TDP-43 or sham aggregates in medium containing polymyxin B (10 µg/mL) for 5 h or overnight. Organotypic cultures were prepared as described (15) from lumbar spinal cords of 5-wk-old male SOD1G93A or WT mice. After 1 wk, treatments were performed as described.
MOK-KO Cells.
MOK-KO SIM-A9 cell clones were generated by CRISPR/Cas9 technology (Synthego). Quality control of MOK-KO clones was done by Synthego. The expected mutations were confirmed by Sanger DNA sequencing, and clones were validated by MOK-specific qRT-PCR and western blot by us, as shown in SI Appendix, Fig. S2 C and D.
Primary Spinal Cord Motor Neuron Isolation.
Dissection of the spinal cord from embryonic mice (E12.5) was followed by separation of neurons through mechanical dissociation and enzymatic cleavage followed by density gradient motor neuron selection. Collected neurons were cultured in supplemented neurobasal medium, and culture treatment was carried out at day 5 of culture.
Acute Microglial Cell Isolation from Adult Mice.
Microglial cells from adult mice were highly enriched by Percoll gradient centrifugation, from spinal cord tissues of 14-wk-old mSOD1G93A Tg mice and their WT littermates.
Immunolabeling and Flow Cytometry.
Immunolabeling of acutely isolated cells from mouse spinal cord was done with rat anti-Cd11b-APC (BD Biosciences) and/or anti-bpMOK antibodies (pThr159+pTyr161, StressMarq) followed by antirabbit IgG-Alexa Fluor 488 (Invitrogen) (SI Appendix, Tables S1 and S2). Immunolabeling of transfected SIM-A9 cells was done with anti-FLAG-APC (Abcam) and anti-IL-6-PE (BD Biosciences). Samples were analyzed using the FACSCalibur or LSR Fortessa X-20 cytometer (BD Biosciences), respectively.
IHC.
IHC assays and quantification were double blinded. Fixed spinal cord segments from TgTDP-43 and control mice and controls or human sporadic ALS patients were used for immunolabeling and quantification of bpMOK.
ChIP and ChIP-qPCR.
SIM-A9 cells were cross-linked, washed, and resuspended in PBS containing protease inhibitors. Cell lysates were sonicated to obtain chromatin fragments of 200 to 500 bp. Samples were incubated with anti-BRD4 antibody (Bethyl) and retrieved with Protein G-Dynabeads (Thermo Fisher). Chromatin fragments were treated with proteinase K and purified for ChIP-qPCR (primers in SI Appendix, Table S3).
IF.
IF of cultured cells and spinal organotypic cultures was done by immunolabeling for pBrd4 (Phospho-Brd4, Millipore) and/or bpMOK (pThr159+pTyr161, StressMarq) and subsequent incubation with secondary antibodies, followed by confocal fluorescence analyses. Immunolabeling for IF of brain slices from WT or SOD1G93A mice was for pBrd4 as above, and images were acquired by an automated inverted microscope.
ELISA.
Cytokine quantification by ELISA was done for IL-6, TNF-α, IFN-β and IL-1α from harvested cell culture supernatants. ELISA kits (BD Biosciences or R&D Systems) were used.
Transfection of SIM-A9 Cells.
For the overexpression of FLAG-tagged WT-MOK and KD-MOK constructs, Glial-Mag transfection reagent (OzBiosciences) was used.
Cell Lysate Preparation and Western Blot.
Cells were lysed in lysis buffer containing complete ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail and PhosSTOP (Roche) under denaturing (for western blot) or nondenaturing (for immunoprecipitation) conditions. Samples were subjected to 10/12% SDS-PAGE and semidry or wet transfer (for Brd4) to 0.2 µm nitrocellulose or polyvinylidene fluoride (PVDF) membrane, respectively.
Immunoprecipitation and LC–MS/MS.
IP from cell lysates was done by using the Protein A Dynabeads (Invitrogen) system and rabbit antiphospho-Ser/Thr antibody (Abcam). LC–MS/MS analyses of IP eluates were done in a TOF (5600 Plus, Sciex), and proteins were identified with ProteinPilot v5.0.1 (Sciex) software.
RNA Isolation.
For qRT-PCR purposes, RNA was isolated by using Primezol (Canvax). For mRNA-Seq and RNA-Seq studies, RNA was extracted with the RNeasy Mini Kit (Qiagen). For SLAM-Seq, RNA was obtained by using easy-BLUE (iNtRON Biotechnology) and protected from light during the procedure.
RNA-Seq, mRNA-Seq, and Gene Set Enrichment Analyses.
RNA-Seq data were generated from three biological replicates. RNA-Seq was carried out by the CABIMER Genomics Unit (Seville, Spain) from total RNA or from mRNA isolated with poly-dT-coated beads. GO, pathway enrichment, and IPA were applied on all DEGs obtained for log2 fold change values >2 or <−2 along with a P value < 0.05.
S4U Labeling and SLAM-Seq.
After cell treatment, 4-thiouridine (S4U, Cayman Chemical) was added to 100 µM and incubated for a further 90 min in the dark. After RNA extraction as described, iodoacetamide (Sigma) was conjugated to S4U, and libraries were prepared for analysis.
Statistical Analyses.
Data are presented as mean ± SEM of N independent experiments, unless indicated otherwise. Statistical analyses were performed using either one-way ANOVA followed by the Tukey post hoc test or Student’s t test, two-/one-tailed as indicated in the corresponding figure legend. P values less than or equal to 0.05 were considered statistically significant. Statistical analyses were performed using Prism v.8 software (GraphPad).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Acknowledgments
We are extremely grateful to Prof. Kevan Shokat and Dr. Flora Rutaganira, University of California, San Francisco (San Francisco, USA) for kindly supplying us with C13 compound. We thank patients’ associations (Saca la lengua a la ELA, Juntos contra la ELA, and Reto Todos Unidos/Miquel Valls Fundació Catalana D’ELA) for supporting the PAIDI Research Group (CTS-0160) global mission. Financial support to C.R. was provided by the Spanish Ministry of Economy (RTI2018-098432-B-I00), Fundación Ramón Areces (CIVP19A5938), the Andalusian Regional Government-FEDER (US-1265227), and I+D PAIDI (PY20-01097). Funding to D.P. was given by the PAIDI research group (CTS-0160) and Regional Ministry of Health (PI-0232-2022). V.C-G. is supported by the Institute of Health Carlos III, Spain, cofunded by the European Social Fund (CP19/00046). S.M. was supported by Generalitat Valenciana (GVA: Prometeo/2018/041 and CIPROM/2021/018); the Spanish Ministry of Economy, Industry and Competitiveness (MINECO), the Spanish State Research Agency (AEI) cofunded by European Regional Development Fund (ERDF), European Union (PID2020-118171RB-100); and Instituto de Salud Carlos III (RD16/001/0010, cofunded by ERDF).
Author contributions
J.A.P.-C., L.S.-C., J.M.F., R.S., D.P., and C.R. designed research; J.A.P.-C., L.S.-C., J.M.F., V.C.-G., A.A., M.G.-L., R.G.-G., M.L.-L., D.P., and C.R. performed research; D.L., R.H.B., S.M., Y.M., G.G.T., R.S., and M.G.-D. contributed new reagents/analytic tools; J.A.P.-C., L.S.-C., J.M.F., A.A., M.G.-L., R.G.-G., X.W.Y., M.L.-L., D.L., G.G.T., R.S., M.G.-D., D.P., and C.R. analyzed data; and Y.M., R.S., M.G.-D., D.P., and C.R. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Preprint Servers: BioRxiv. The copyright holder for this preprint is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission.
Data, Materials, and Software Availability
RNA-Seq raw data have been deposited at the Gene Expression Omnibus (GEO) database: 1) Dataset S1: GSE233569 (80), 2) Dataset S4: GSE233719 (81), and 3) Datasets S6–S9: GSE234160 (82).
Supporting Information
References
- 1.Caballero-Hernandez D., et al. , The ‘omics’ of amyotrophic lateral sclerosis. Trends Mol. Med. 22, 53–67 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Feldman E. L., et al. , Amyotrophic lateral sclerosis. Lancet 400, 1363–1380 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ransohoff R. M., How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Van Harten A. C. M., Phatnani H., Przedborski S., Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 44, 658–668 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke B. E., Patani R., The microglial component of amyotrophic lateral sclerosis. Brain 143, 3526–3539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCauley M. E., Baloh R. H., Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 137, 715–730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Troutman T. D., Kofman E., Glass C. K., Exploiting dynamic enhancer landscapes to decode macrophage and microglia phenotypes in health and disease. Mol. Cell 81, 3888–3903 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeh H., Ikezu T., Transcriptional and epigenetic regulation of microglia in health and disease. Trends Mol. Med. 25, 96–111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arai T., et al. , TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Neumann M., et al. , Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Torres P., et al. , A motor neuron disease mouse model reveals a non-canonical profile of senescence biomarkers. Dis. Model. Mech. 15, dmm049059 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood A., Gurfinkel Y., Polain N., Lamont W., Lyn Rea S., Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 22, 4705 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao Y.-Z., Ma J., Dou J.-Z., The role of TDP-43 in neurodegenerative disease. Mol. Neurobiol. 59, 4223–4241 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Miyata Y., Akashi M., Nishida E., Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 4, 299–309 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Leal-Lasarte M. M., Franco J. M., Labrador-Garrido A., Pozo D., Roodveldt C., Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 31, 2797–2816 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Broekhuis J. R., Verhey K. J., Jansen G., Regulation of cilium length and intraflagellar transport by the RCK-kinases ICK and MOK in renal epithelial cells. PLoS One 9, e108470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aprile-Garcia F., Tomar P., Hummel B., Khavaran A., Sawarkar R., Nascent-protein ubiquitination is required for heat shock–induced gene downregulation in human cells. Nat. Struct. Mol. Biol. 26, 137–146 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Garske A. L., Peters U., Cortesi A. T., Perez J. L., Shokat K. M., Chemical genetic strategy for targeting protein kinases based on covalent complementarity. Proc. Natl. Acad. Sci. U.S.A. 108, 15046–15052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warde-Farley D., et al. , The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao Y., et al. , Brd4 modulates the innate immune response through Mnk2–eIF4E pathway-dependent translational control of IκBα. Proc. Natl. Acad. Sci. U.S.A. 114, E3993–E4001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., et al. , (+)-JQ1 attenuated LPS-induced microglial inflammation via MAPK/NFκB signaling. Cell Biosci 8, 60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeMars K. M., Yang C., Castro-Rivera C. I., Candelario-Jalil E., Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochem. Biophys. Res. Commun. 497, 410–415 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhao W., et al. , TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 273, 24–35 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Deora V., et al. , The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68, 407–421 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Miyata Y., Ikawa Y., Shibuya M., Nishida E., Specific association of a set of molecular chaperones including HSP90 and Cdc37 with MOK, a member of the mitogen-activated protein kinase superfamily. J. Biol. Chem. 276, 21841–21848 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Liu L., Yang C., Candelario-Jalil E., Role of BET proteins in inflammation and CNS diseases. Front. Mol. Biosci. 16, 748449 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang N., Wu R., Tang D., Kang R., The BET family in immunity and disease. Signal Transduct. Target. Ther. 6, 23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung K. H., et al. , RNA sequencing reveals distinct mechanisms underlying BET inhibitor JQ1-mediated modulation of the LPS-induced activation of BV-2 microglial cells. J. Neuroinflamm. 12, 12–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korb E., Herre M., Zucker-Scharff I., Darnell R. B., Allis C. D., BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci. 18, 1464–1473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu S. Y., Lee A. Y., Lai H. T., Zhang H., Chiang C. M., Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 49, 843–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belkina B. S., Nikolajczyk G. V., Dennis A. C., Macrophage inflammatory responses BET inhibitor JQ1 impair mouse inflammation: Brd2 genetic disruption and BET protein function is required for. J. Immunol. 190, 3670–3678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan Y. M., Kirkham P., Barnes P. J., Adcock I. M., Brd4 Is essential for IL-1b-induced inflammation in human airway epithelial cells. PLoS One 9, e95051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik N., et al. , Suppression of interferon β gene transcription by inhibitors of bromodomain and extra-terminal (BET) family members. Biochem. J. 468, 363–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen T., Wu D., Moskaluk C. A., Fu Z., Distinct expression patterns of ICK/MAK/MOK protein kinases in the intestine implicate functional diversity. PLoS One 8, e79359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wegorzewska I., Bell S., Cairns N. J., Miller T. M., Baloh R. H., TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 18809–18814 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gois A. M., Mendonça D. M. F., Freire M. A. M., Santos J. R., In vitro and in vivo models of amyotrophic lateral sclerosis: An updated overview. Brain Res. Bull. 159, 32–43 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Gerbino V. V, et al. , The Loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106, 789–805.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Krasemann S., et al. , The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan X., Vocadlo D., Krieger C., Mislocalization of TDP-43 in the G93A mutant SOD1 transgenic mouse model of ALS. Neurosci. Lett. 458, 70–74 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Cai M., Lee K.-W., Choi S.-M., Yang E. J., TDP-43 modification in the hSOD1G93A amyotrophic lateral sclerosis mouse model. Neurol. Res. 37, 253–262 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Marino M., et al. , Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 36, 492–504 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Pasetto L., et al. , Targeting extracellular cyclophilin A reduces neuroinflammation and extends survival in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 37, 1413–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeon S.-G., et al. , Pathological Modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 mutations. Mol. Neurobiol. 56, 2007–2021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rock R. B., et al. , Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 17, 942–964 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrett J. P., et al. , Interferon-β plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that frives chronic neurodegeneration. Neurobiol. Dis. 40, 2357–2370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu C.-H., et al. , TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan P.-H., Ji J., Hsing C.-H., Tan R., Ji R.-R., Emerging roles of type-I interferons in neuroinflammation, neurological diseases, and long-haul COVID. Int. J. Mol. Sci. 23, 14394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pong Ng H., et al. , CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB J. 34, 12100–12113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X., et al. , Action by CITED2 in the nucleus B κ negative feedback regulation of NF. J. Immunol. 186, 539–548 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Gun-Dong K., et al. , CITED2 restrains proinflammatory macrophage activation and response. Mol. Cell. Biol. 38, e00452-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu T., et al. , Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 10, 4353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanmarco L. M., et al. , Identification of environmental factors that promote intestinal inflammation. Nature 611, 801–809 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruffell D., et al. , A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. U.S.A. 106, 17475–17480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamkin D. M., et al. , C/EBPβ regulates the M2 transcriptome in β-adrenergic-stimulated macrophages. Brain. Behav. Immun. 80, 839–848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu J., et al. , Peli1 impairs microglial Aβ phagocytosis through promoting C/EBPβ degradation. PLOS Biol. 18, e3000837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muromoto R., et al. , Regulation of NFKBIZ gene promoter activity by STAT3, C/EBPβ, and STAT1. Biochem. Biophys. Res. Commun. 613, 61–66 (2022). [DOI] [PubMed] [Google Scholar]
- 57.Annemann M., et al. , Atypical IκB proteins in immune cell differentiation and function. Immunol. Lett. 171, 26–35 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Cuadrado A., Brain-protective mechanisms of transcription factor NRF2: Toward a common strategy for neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 62, 255–277 (2022). [DOI] [PubMed] [Google Scholar]
- 59.Mogensen T. H., IRF and STAT transcription factors–From basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front. Immunol. 1, 3047 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roy E. R., et al. , Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 55, 879–894.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue F., Tian J., Yu C., Du H., Guo L., Type I interferon response-related microglial Mef2c deregulation at the onset of Alzheimer’s pathology in 5×FAD mice. Neurobiol. Dis. 152, 105272 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez S., et al. , Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Sci. Transl. Med. 13, eaaz4699 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barrett J. P., et al. , Traumatic brain injury induces cGAS activation and type I interferon signaling in aged mice. Front. Immunol. 12, 710608 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nazmi A., et al. , Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67, 1254–1276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Friedman B. A., et al. , Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Main B. S., et al. , Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia 64, 1590–604 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Wang R., Yang B. O., Zhang D., Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59, 946–58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zafar A., Ng H. P., Kim G. D., Chan E. R., Mahabeleshwar G. H., BHLHE40 promotes macrophage pro-inflammatory gene expression and functions. FASEB J. 35, 21940 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cook M. E., Jarjour N. N., Lin C. C., Edelson B. T., Transcription factor Bhlhe40 in immunity and autoimmunity. Trends Immunol. 41, 1023–1036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu X., et al. , Bromodomain containing protein 4 (BRD4) regulates expression of its interacting coactivators in the innate response to respiratory syncytial virus expression of host defense mechanisms and remodeling genes through changes in promoter accessibility. Front. Mol. Biosci. 8, 728661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berson A., Nativio R., Berger S. L., Bonini N. M., Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jimenez-Pacheco A., et al. , Epigenetic mechanisms of gene regulation in amyotrophic lateral sclerosis. Adv. Exp. Med. Biol. 978, 255–275 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Huang Y., et al. , Genetic and environmental effects on gene expression signatures of blood pressure: A transcriptome-wide twin study. Hypertension 71, 457–464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sopic M., et al. , Downregulation of mapk/mak/mrk overlapping kinase 1 in peripheral blood mononuclear cells of pediatric patients with type 1 diabetes mellitus. J. Med. Biochem. 41, 282–289 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.García-García R., Martín-Herrero L., Blanca-Pariente L., Pérez-Cabello J., Roodveldt C., Immune signaling kinases in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int. J. Mol. Sci. 22, 13280 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo W., Vandoorne T., Steyaert J., Staats K. A., Van Den Bosch L., The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain 143, 1651–1673 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gurney M. E., et al. , Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Capitini C., Conti S., Perni M., Guidi F., Cascella R., TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One 9, 86720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cascella R., et al. , Quantification of the relative contributions of loss-of-function and gain-of-function mechanisms in TAR DNA-binding protein 43 (TDP-43) proteinopathies. J. Biol. Chem. 291, 19437–19448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roodveldt C., Yap X. W., Sawarkar R., SLAM-Seq analysis of early transcriptional changes by C13 MOK-inhibitor in microglial cells exposed to TDP-43-aggregates. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE233569. Deposited 26 May 2023.
- 81.Roodveldt C., Franco J. M., Gómez-Lima M., Effect of C13 MOK-inhibitor on LPS-induced responses in primary microglial cells. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE233719. Deposited 30 May 2023.
- 82.Roodveldt C., Pozo D., Effect of MOK in LPS-induced responses in microglial cells. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE234160. Deposited 5 June 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Data Availability Statement
RNA-Seq raw data have been deposited at the Gene Expression Omnibus (GEO) database: 1) Dataset S1: GSE233569 (80), 2) Dataset S4: GSE233719 (81), and 3) Datasets S6–S9: GSE234160 (82).