

Significance
Altered heart rate variability (HRV), arising from impaired chronotropic response of sinoatrial node (SAN) pacemaker activity to vagal input, has been proposed as an early diagnostic marker in sepsis. However, the underlying pathophysiological changes in the SAN have not been investigated yet. We identified the downstream M2R-GIRK signaling in a lipopolysaccharide-induced proxy septic model is impaired, consequent to reduced expression of GIRK isoforms, leading to compromised chronotropic regulation of SAN pacemaking and HRV by IKACh. Intriguingly, downregulation of GIRK channels was not directly mediated by the common proinflammatory cytokines elevated in sepsis. Our findings provide insights into parasympathetic regulation of SAN and clinical monitoring of altered HRV in sepsis, which potentially help to reduce risk of death in patients.
Keywords: carbachol, Ca2+ transient, heart rate variability, I KACh , Langendorff’s heart
Abstract
Sepsis has emerged as a global health burden associated with multiple organ dysfunction and 20% mortality rate in patients. Numerous clinical studies over the past two decades have correlated the disease severity and mortality in septic patients with impaired heart rate variability (HRV), as a consequence of impaired chronotropic response of sinoatrial node (SAN) pacemaker activity to vagal/parasympathetic stimulation. However, the molecular mechanism(s) downstream to parasympathetic inputs have not been investigated yet in sepsis, particularly in the SAN. Based on electrocardiography, fluorescence Ca2+ imaging, electrophysiology, and protein assays from organ to subcellular level, we report that impaired muscarinic receptor subtype 2-G protein-activated inwardly-rectifying potassium channel (M2R-GIRK) signaling in a lipopolysaccharide-induced proxy septic mouse model plays a critical role in SAN pacemaking and HRV. The parasympathetic responses to a muscarinic agonist, namely IKACh activation in SAN cells, reduction in Ca2+ mobilization of SAN tissues, lowering of heart rate and increase in HRV, were profoundly attenuated upon lipopolysaccharide-induced sepsis. These functional alterations manifested as a direct consequence of reduced expression of key ion-channel components (GIRK1, GIRK4, and M2R) in the mouse SAN tissues and cells, which was further evident in the human right atrial appendages of septic patients and likely not mediated by the common proinflammatory cytokines elevated in sepsis.
Sepsis refers to a life-threatening, dysregulated host response to infection, manifesting into multiple organ dysfunction and ultimately death, unless diagnosed and treated promptly (1). Sepsis is a major health problem that accounted for 11 million deaths out of 49 million reported cases worldwide in 2017 and nearly 40% of fatal outcome were related to cardiovascular complications (2). The precise etiology and mechanism(s) underlying cardiac dysfunction in sepsis are complex and not entirely understood, which hinders timely diagnosis and therapeutic intervention in patients. Various mediators such as proinflammatory cytokines, nitric oxide, and reactive oxygen species have been linked to cardiac dysfunction (3, 4).
A number of clinical studies over the past two decades have correlated the disease severity and mortality in septic patients with altered heart rate variability (HRV), as a consequence of dampened cardiac response to vagal/parasympathetic stimulation (5–8). HRV refers to beat-to-beat variability (BBV) between successive RR intervals on an electrocardiogram (ECG). This variability predominantly arises from adaptive autonomic nervous system modulations of a specialized group of cardiac cells in the sinoatrial node (SAN) that constitute the natural pacemaker of heart. Besides, a certain degree of BBV is intrinsic to the SAN cells due to fluctuations in the spontaneous pacemaking machinery (9, 10). Previous animal studies on isolated SAN cells and tissues showed that spontaneous diastolic depolarization and action potential firing in SAN cells are generated by a functional interplay between two tightly linked intracellular clocks, namely the membrane clock and the calcium clock. The membrane clock involves periodic opening of transmembrane ion channels: hyperpolarization-activated cyclic nucleotide-gated (HCN) channels encoding the funny current (If), voltage-gated Ca2+ channels (L-type Cav1.3 and T-type Cav3.1), Ca2+-activated K+ channels, and transient receptor potential channels (11). The calcium clock involves local Ca2+ release through ryanodine receptors from the sarcoplasmic reticulum (SR), which can be either spontaneous (12) or activated by Cav1.3 channels (13, 14). Local Ca2+ release is coupled to diastolic depolarization via activation of the cardiac Na+/Ca2+ exchanger (NCX1) (10, 15–17). Sympathetic stimulation of beta (β)-adrenergic receptor on isolated SAN cells promotes action potential firing rate (18, 19) and reduces BBV (9) via coupled G-protein alpha stimulatory subunit (Gαs)–mediated cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) pathway, which is counterbalanced by the parasympathetic stimulation of muscarinic receptor subtype 2 (M2R)–inhibitory subunit Gαi leading to lower action potential firing rate and higher BBV (9, 10, 19–21). Moreover, upon M2R stimulation by agonists in isolated SAN cells, the coupled G-protein beta-gamma (Gβγ) subunit directly activates acetylcholine-activated inwardly rectifying potassium current (IKACh). The cardiac IKACh is conducted by G protein-activated inwardly rectifying potassium (GIRK) channels, consisting of GIRK1 and GIRK4 heteromers, which results in K+ efflux and membrane hyperpolarization with consequent reduction in action potential firing rate (19, 20, 22, 23). IKACh plays a major role in the generation of HRV in response to vagal stimulation and in recovering basal heart rate after sympathetic activation (22, 23).
Various animal models of sepsis point out the impact of the parasympathetic nervous system on cardiac dysfunction. In a lipopolysaccharide (LPS)-induced endotoxemic rat model, the observed narrowed BBV of the isolated SAN-atria region was linked to blunted or uncoupled cardiac response to cholinergic stimulation (24). Other studies reported that electrical stimulation of the vagus nerve attenuated the myocardial inflammation and hemodynamic impairment in LPS-induced endotoxemia in vagotomized rats (25, 26). Another rat model of sepsis obtained by cecal ligation and puncture demonstrated altered biphasic redistribution and phosphorylation of M2R receptor in the isolated heart (27). However, the molecular mechanism(s) downstream to vagal/parasympathetic stimulation particularly in the SAN tissue have not been explored in detail in sepsis. Given the fact that GIRK1/4 channels are mainly expressed in SAN and atrial tissues, and are involved in determination of HRV (22, 23), we hypothesized that impaired vagal activity during sepsis might be related to pathological changes in the M2R-GIRK signaling in SAN tissue. Thus, the present study was designed to elucidate whether the responsiveness of SAN cells, tissue, and heart to parasympathetic stimulation is compromised under septic conditions due to modifications in the M2R-GIRK signaling, thereby potentially impairing HRV.
Results
LPS Attenuates Parasympathetic Modulation of Heart Rate and Its Variability.
We first induced sepsis in mice via intraperitoneal (i.p.) injection of LPS (5 mg/kg), an endotoxin from gram-negative bacteria, and after 6 h, recorded ECGs from the isolated hearts under Langendorff’s perfusion in relation to parasympathetic stimulation. We compared the beating rate and HRV measurements (SDNN, RMSSD, and pNN6, defined in the method section) calculated from ECGs on the hearts isolated from septic versus phosphate-buffered saline (PBS)-injected control mice. As illustrated in Fig. 1A, the beating rate of isolated hearts was unaffected by LPS injection in mice (414.22 ± 10.23/min) compared to controls (399.58 ± 13.11/min). Perfusion for 10 min with 0.1 µM carbachol (CCh), a muscarinic receptor agonist, significantly reduced the heart rate in controls by 39% (243.81 ± 12.06/min). In contrast, CCh could marginally, albeit nonsignificant, reduce the beating rate in septic hearts (383.73 ± 14.08/min, 7.4% reduction). LPS altered neither the SDNN (Fig. 1B, 0.81 ± 0.22 ms vs. 0.83 ± 0.12 ms) nor RMSSD (Fig. 1C, 0.69 ± 0.21 ms vs. 0.46 ± 0.09 ms) compared to controls. However, upon CCh perfusion, hearts from control mice exhibited significantly higher SDNN (Fig. 1B, 2.88 ± 0.16 ms, 3.5-fold) and RMSSD (Fig. 1C, 2.46 ± 0.39 ms, 5.4-fold). SDNN of hearts from LPS-injected mice also increased significantly following CCh perfusion (Fig. 1B, 1.85 ± 0.31 ms, 2.3-fold) but to a much lesser extent than in controls, as evident from the significant interaction effect in the mixed two-way ANOVA (P = 0.032). Meanwhile, the increase in RMSSD of hearts from LPS-injected mice did not reach significance (Fig. 1C, 1.19 ± 0.35 ms, 1.7-fold) following CCh perfusion. SDNN and RMSSD data together reflect that LPS attenuated the increase in total autonomic variability as well as short-term variation between successive heart beats in response to CCh. Further, baseline pNN6 data were zero in most of the control and LPS-injected mice. So, the difference or increase in pNN6 data between baseline and CCh response were compared in Fig. 1D. Clearly, the percentage increase in pNN6 following CCh perfusion was significantly higher in the hearts from control mice than those from LPS-injected mice (3.15% vs. 0.3%), which further reflects a lower response of heart rate to parasympathetic stimulation.
Fig. 1.

LPS attenuates parasympathetic modulation of heart rate and its variability. Scatter plots showing mean ± SEM for (A) heart rate, (B) SDNN and (C) RMSSD in the absence (−) and presence (+) of 0.1 µM CCh and median with first and third quartiles for (D) difference in pNN6 following 0.1 µM CCh perfusion in the isolated heart from LPS-injected mice vs. controls. N = 6; **P < 0.01, ***P < 0.001, nsP > 0.05; mixed two-way ANOVA followed by simple effect analysis in A–C; percentile bootstrap–based test of equal medians in D.
LPS Dampens CCh-Mediated Negative Chronotropic Effect on Ca2+ Dynamics in SAN Tissue.
The heartbeat is generated by SAN pacemaker mechanism, which depends on the activity of the membrane and the calcium clocks. Cholinergic stimulation is known to modulate Ca2+ homeostasis during SAN pacemaking and LPS impaired CCh effect on HR and HRV in isolated hearts (Fig. 1). So, we examined the effect of CCh on Ca2+ dynamics in SAN tissues isolated from control and LPS-administered mice. Fig. 2A illustrates pacemaker activity reflected by automatic intracellular Ca2+-transients, expressed as change in fluorescence signal of Cal-520-indicator over time in the SAN tissue of control vs. LPS-injected mouse in response to CCh. CCh perfusion for 10 min significantly reduced the rate of spontaneous Ca2+-transients in control SAN tissues by 25% (Fig. 2B, 284.76 ± 27.21/min to 213.51 ± 32.19/min), which correlates well with the reduced beating rate of isolated hearts as observed in Fig. 1A. Meanwhile, the rate of Ca2+-transients following CCh perfusion was also significantly reduced in SAN tissues from LPS-injected mice by 9.4% (Fig. 2B, 328.99 ± 32.29/min to 298.17 ± 34.26/min). However, the magnitude of reduction in LPS-treated SAN tissues was significantly lower than in control tissues, evident from the significant interaction effect in the mixed two-way ANOVA (P = 0.015). CCh reduced the peak amplitude by 23.6% (Fig. 2C) and prolonged the relaxation time to 90% from peak (RT90) by 15.5% (Fig. 2D) in the control SAN tissues whereas these parameters were unaffected in the SAN tissues of LPS-injected mice. Under basal conditions, there was no difference in these parameters between SAN tissues of LPS-injected mice and control mice (Fig. 2 B–D), supporting the results obtained from isolated hearts (Fig. 1).
Fig. 2.

LPS dampens CCh-mediated negative chronotropic effect on Ca2+ dynamics in SAN tissue. (A) Representative traces of Ca2+-sensitive Cal-520 fluorescence (F/Fb) depicting baseline-corrected intracellular Ca2+-transients over time in the SAN tissue isolated from control (Left) and LPS-injected (Right) mice in the absence (−) and presence (+) of 0.1 µM CCh. Scatter plots showing mean ± SEM for (B) Ca2+-transients/min, (C) peak amplitude and (D) RT90 in the absence (−) and presence (+) of 0.1 µM CCh in the isolated SAN tissues from LPS-injected mice vs. controls. N = 8; *P < 0.05, ***P < 0.001, nsP > 0.05; mixed two-way ANOVA followed by simple effect analysis.
LPS Inhibits CCh-Mediated Increase in Steady-State Current Density and IKACh in Isolated SAN Cells.
Since LPS blunted CCh-induced reduction in heart rate and frequency of spontaneous Ca2+ transients (Figs. 1 and 2), we investigated the impact of CCh on membrane current in SAN cells isolated from control and LPS-injected mice. In whole-cell patch-clamp recordings, a ramp protocol from −100 mV to +40 mV was applied over 1.5 s to elicit quasi-steady-state current before and after 0.1 µM CCh perfusion (Fig. 3 A and B). The major differences between control and LPS groups in response to CCh were noticeable at negative and positive voltages (Fig. 3C and SI Appendix, Fig. S1 A–E). Upon CCh perfusion, control SAN cells showed a significant increase in the inward current density at −100 mV (Fig. 3C), −90 and −80 mV (SI Appendix, Fig. S1 A and B) as well as the outward current density at +20, +30, and +40 mV (SI Appendix, Fig. S1 C–E). In contrast, CCh perfusion failed to alter the current density in inward and outward directions in SAN cells from LPS-injected mice, except for reduction at −80 mV. In SI Appendix, Fig. S1F, plotting the difference between CCh and basal current (ICCh-Ibasal, referred as CCh-induced) revealed that IKACh mediated CCh effect at both negative and positive voltages, and showed inward rectifying properties in control SAN cells. In contrast, LPS suppressed the CCh-induced inward current at negative voltages and abrogated most of the difference current at positive voltages. The inward current was further confirmed to be mediated by IKACh in a separate set of experiments where a GIRK channel inhibitor tertiapin-Q (20, 22) at 300 nM significantly attenuated CCh-mediated increase in current density at −100 mV (Fig. 3D). We next evaluated IKACh at −35 mV holding potential, as reported previously (22). Fig. 3E illustrates representative current recordings, demonstrating prominent activation of IKACh within few seconds upon CCh perfusion in a control cell but negligible activation in a cell from LPS-injected mouse. Averaged data plotted in Fig. 3F show that CCh significantly increased IKACh in control SAN cells (1.24 ± 0.23 pA/pF) compared to cells from LPS-injected mice (0.35 ± 0.11 pA/pF). We further investigated if LPS diminished IKACh activation independent of Gβγ-coupled M2R stimulation by CCh. To do so, we used ML297 and 3hi2one-G4, direct activators of heteromeric GIRK1/4 and homomeric GIRK4 channels respectively (28, 29), for measuring IKACh at −100 mV in ramp recordings. Three micromolar ML297 significantly increased the inward current density at −100 mV in the control SAN cells but not in the cells from LPS-injected mice (SI Appendix, Fig. S1G), suggesting disruption of GIRK1/4 channel function and/or expression. In contrast, 30 µM 3hi2one-G4 failed to elicit IKACh at −100 mV in the cells from both control and LPS-injected mice (SI Appendix, Fig. S1H), suggesting that in SAN cells, GIRK4 forms functional heterotetramers with GIRK1 but not GIRK4 homotetramers.
Fig. 3.
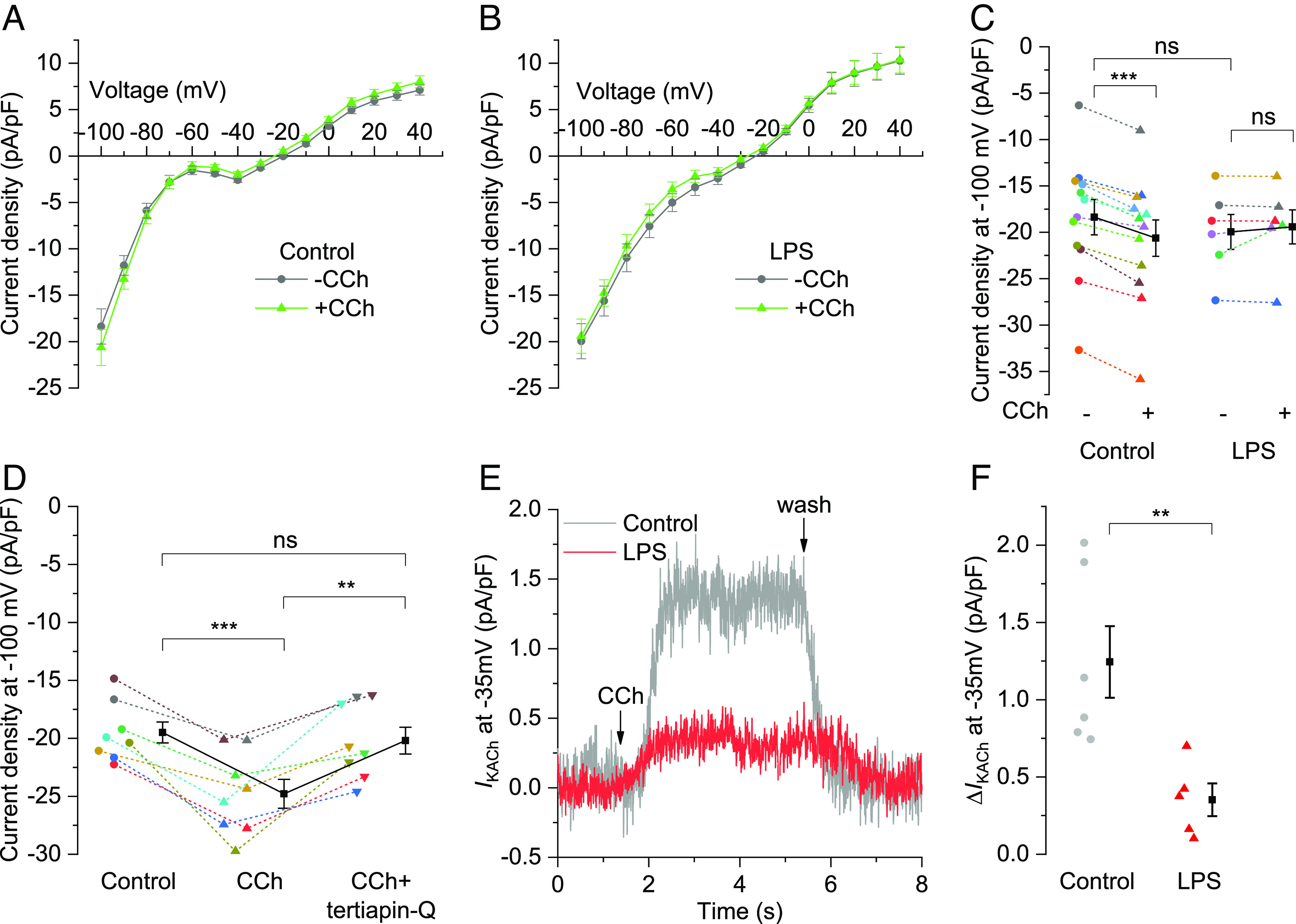
LPS inhibits CCh-mediated increase in steady-state current density and IKACh in isolated SAN cells. (A and B) Traces showing mean ± SEM of current density from −100 to +40 mV ramp over 1.5 s and (C) scatter plot with mean ± SEM for current density at −100 mV in the absence (−) and presence (+) of 0.1 µM CCh in the isolated SAN cells from (B) LPS-injected mice (N = 6) vs. (A) controls (N = 12). (D) Scatter plot with mean ± SEM for current density at −100 mV in isolated control SAN cells (N = 8) prefused with 0.1 µM CCh ± 300 nM tertiapin-Q. (E) Representative traces and (F) scatter plot with mean ± SEM representing change in the IKACh outward current density at −35 mV holding potential following 0.1 µM CCh perfusion in SAN cells isolated from LPS-injected mice (N = 5) vs. control mice (N = 6). **P < 0.01, ***P < 0.001, nsP > 0.05; (C) mixed two-way ANOVA followed by simple effect analysis, (D) repeated measure one-way ANOVA followed by within-group effect analysis with Bonferroni adjustment, (F) independent samples t test.
LPS Reduces Expression of M2R and GIRK Channels Involved in Parasympathetic Regulation of Heart Rate.
GIRK1 and GIRK4 channels are integral components of membrane delimited M2R-induced activation of IKACh (19, 20, 22). So, we investigated whether LPS administration could alter expression of M2R and/or GIRK channel isoforms accounting for loss of IKACh and reduced response to CCh in SAN pacemaker cells, intact SAN tissues and whole hearts isolated from LPS-injected mice in the functional assays described above. Western blots of SAN tissues from LPS-injected mice indeed showed significantly lower expression of GIRK1 (0.33 ± 0.05 fold), GIRK4 (0.51 ± 0.09 fold), and M2R (0.49 ± 0.09 fold) compared to the SAN tissues from control mice (Fig. 4A). Next, we investigated if LPS directly impacts the expression of these receptors and ion channels in intact SAN tissues in situ in addition to the above in vivo effects. In vitro incubation of the isolated SAN tissues with LPS for 6 h also significantly reduced the expression of GIRK1 (0.65 ± 0.04 fold), GIRK4 (0.74 ± 0.05 fold), and M2R (0.69 ± 0.04 fold) compared to the control SAN tissues (Fig. 4B); however, the effect was less pronounced than in the SAN tissues from LPS-injected mice. Next, we evaluated the expression of M2R and GIRK1/4 channels in the human right atrial appendages (RAA) from septic patients and similarly observed reduced expression of GIRK1 (0.43 fold), GIRK4 (0.45-fold), and M2R (0.55-fold) compared to the control human tissues (SI Appendix, Fig. S2), which corroborates the findings in the SAN tissues from mice following in vivo or in vitro LPS treatment.
Fig. 4.
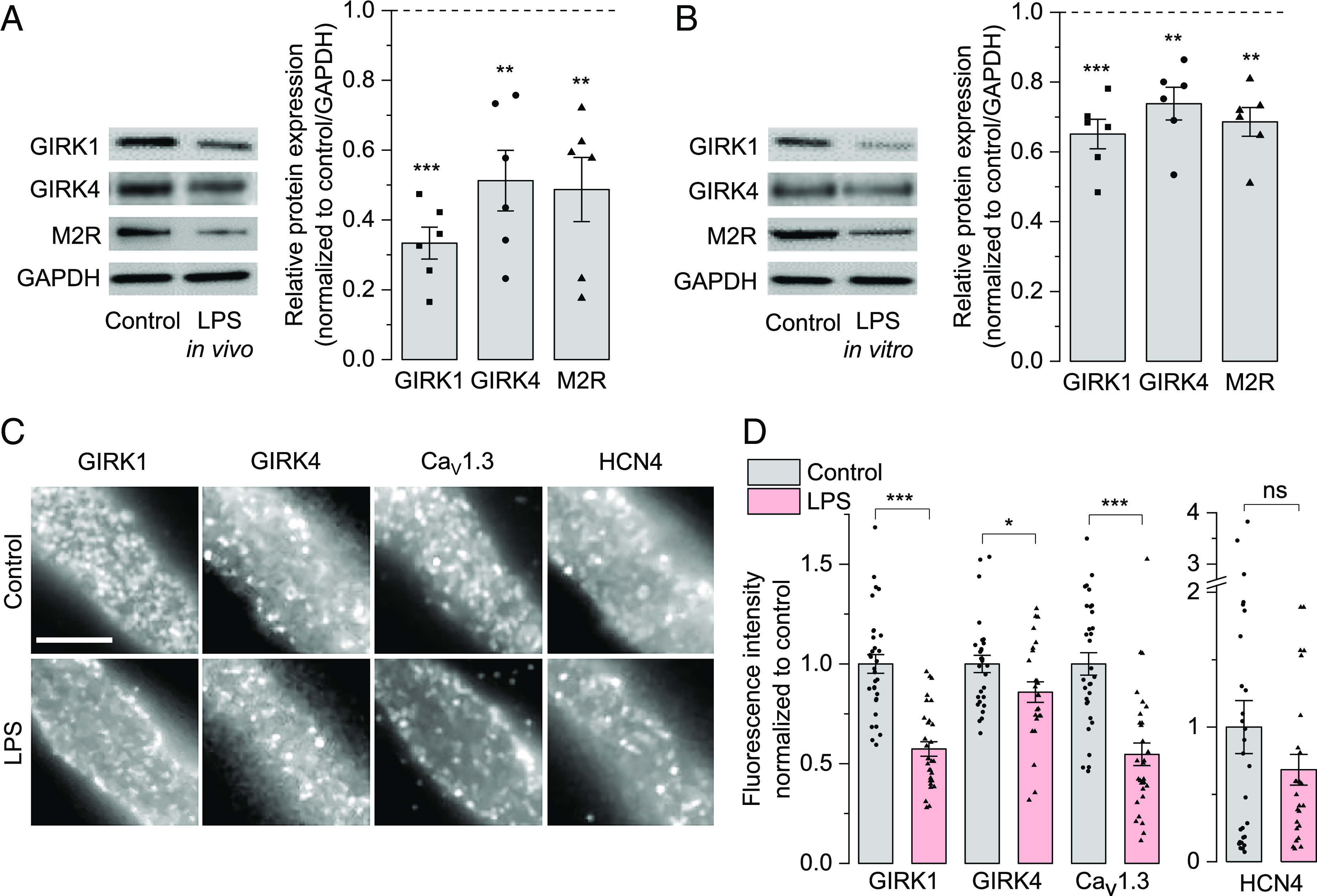
LPS reduces expression of ion-channels involved in parasympathetic modulation. Representative western blots (Left) and bar graphs with mean ± SEM of densitometric evaluation of immunoreactive bands (Right) showing reduced expression of GIRK1, GIRK4, and M2R in mouse SAN tissue after 6 h of (A) LPS i.p. injection in mice (in vivo) or (B) LPS incubation (in vitro). GAPDH is used as a loading control. (C) Representative immunofluorescence images of SAN cells isolated from control (above) and LPS-injected (below) mice showing the distribution of GIRK1, GIRK4, CaV1.3 and HCN4 in discrete clusters at the membrane. (Scale bar = 5 µm.) (D) Bar graphs with mean ± SEM showing the fluorescence intensities of membrane-expressed GIRK1, GIRK4, Cav1.3 and HCN4. Data are expressed as symbols in panels B (N = 6 independent experiments) and D (N = 31 control cells and 30 LPS cells for colabeled pair GIRK1, Cav1.3; 28 control cells and 25 LPS cells for GIRK4, HCN4 pair); *P < 0.05, **P < 0.01, ***P < 0.001; nsP > 0.05; one-sample t test in B; unpaired bootstrap t test in D.
To determine whether LPS-induced reduction of GIRK1/4 expression in the SAN tissues is reflected at the membrane levels, we performed fluorescence immunolabeling of SAN cells isolated from control and LPS-injected mice. We also assessed the membrane expression of other ion channels involved in SAN pacemaking, such as Cav1.3 channels (14) and If-conducting HCN4 (22). Fig. 4C shows the immunolabeled GIRK1, GIRK4, Cav1.3, and HCN4 homogenously localized in discrete clusters at the membrane in both conditions, suggesting that LPS does not alter the overall channel distribution. However, quantitative analysis of fluorescence intensity (Fig. 4D) revealed significant reduction of GIRK1 (0.57 ± 0.04 fold) and GIRK4 (0.86 ± 0.05 fold) membrane levels in SAN cells from LPS-injected mice compared to controls, indicating that LPS diminishes the amount of functional GIRK channels at the surface. The membrane amount of Cav1.3 was also significantly decreased (0.55 ± 0.06 fold) whereas HCN4 showed strong variability with no significant change. Low-magnification images of the SAN cells from which the enlarged regions in Fig. 4C were isolated are provided in SI Appendix, Fig. S3 and display their typical thin and elongated morphology. SI Appendix, Fig. S4 clarifies the membrane localization of the analyzed channels as a rim surrounding the intracellular region. Although some intracellular labeling consistent with organelle localization was observed (arrowheads), this was segregated in restricted regions and clearly distinguishable from the cell membrane rim (arrows). This differs from the homogeneously distributed clusters represented in Fig. 4C, consistent with membrane ion channels segregated in functional domains.
Since the intermediate G-protein subunits, namely Gαi and Gβγ, mediate cAMP-PKA pathway inhibition and IKACh activation, respectively, upon M2R stimulation by CCh (20), we checked if LPS affects the expression of Gαi2 and Gβ too. However, we did not observe any change in expression from western blots of SAN tissues from LPS-injected mice compared to those from control mice (SI Appendix, Fig. S5). We further checked whether the reduced expression in SAN is mediated by common proinflammatory cytokines like tumor necrotic factor-alpha (TNF-α), interleukin-1 beta (IL-1β) or interleukin-6 (IL-6). Elevated level of these cytokines has been previously shown to promote systemic inflammation (25, 26) and is associated with reduced HRV in septic rodents (30). We incubated SAN tissues with TNF-α (50 ng/mL), IL-1β (10 ng/mL), or IL-6 (10 ng/mL) for 6 h. These cytokines did not affect expression of M2R and GIRK1/4 channels compared to the control tissues (SI Appendix, Fig. S6 A and B), suggesting that cytokine elevation in sepsis may not directly affect expression of channels involved in IKACh in the SAN tissues.In addition, we investigated if IKACh activation by CCh in SAN cells was affected upon incubation with 10 ng/mL IL-6 for 2 h. IL-6-incubated cells showed a similar increase in CCh-activated IKACh density at −100 mV compared to control cells (SI Appendix, Fig. S6C), indicating no direct effect of cytokines like IL-6 on GIRK channel activation in addition to expression.
Discussion
Our study presents evidence of impaired M2R-GIRK signaling pathway in the SAN tissues of LPS-injected septic mice. As summarized in Fig. 5, LPS reduced expression of key ion-channels and receptors (GIRK1, GIRK4, and M2R). This diminished expression led to diminished parasympathetic regulation of pacemaker activity at multiple levels namely, i) IKACh activation in SAN cells, ii) reduction in the frequency of spontaneous Ca2+ transients of SAN tissues, and iii) lowering of heart rate and increase in HRV parameters.
Fig. 5.

Schematic diagram illustrating the impact of LPS-induced sepsis on M2R-GIRK signaling in SAN tissue pacemaking and HRV. Upon M2R stimulation by a muscarinic agonist, CCh in SAN cells, the βγ subunit of the coupled G-protein directly activates outward K+ current (IKACh) via inwardly rectifying potassium (GIRK) channels, consisting of GIRK1 and GIRK4 heteromers, which results in K+ efflux and membrane hyperpolarization. Further downstream signaling leads to reduction in Ca2+ mobilization and contractility of SAN tissues and decrease in heart rate and increase in HRV. LPS-induced sepsis reduces expression of M2R, GIRK1, and GIRK4 channels, which diminishes the above-mentioned effects of CCh stimulation.
Our findings are in line with previous studies showing that genetic ablation of GIRK4 channels and hence lack of IKACh in isolated SAN tissue and atrial myocytes attenuated the effect of vagal stimulation, namely lowering of heart rate and increase in HRV, in isolated hearts as well as conscious mice (22, 23). We further revealed substantial reduction in expression of GIRK1, GIRK4, and M2R in RAA of septic patients, which might in turn contribute to the diminished vagal response and impaired HRV observed in these patients (5, 6). However, LPS did not affect the expression of M2R-coupled Gβ subunit that directly activates IKACh in mouse SAN tissues but attenuated Gβγ-independent and direct GIRK1/4 channel activation by ML297 in mouse SAN cells, which indicates that LPS impairs GIRK1/4 channel function and expression independent of regulation of Gβ expression. Our group has previously reported impairment of If in human RAAs following LPS incubation without affecting HCN2/4 expression, which further compromises cardiac pacemaking in response to autonomic inputs (31, 32). Here, we did not observe change in membrane expression of HCN4 channels in the SAN cells from LPS-injected mice, in line with our previous report. In addition, our data show that SAN cells most likely conduct IKACh via functional heterotetramers of GIRK1/4 but not GIRK4 homotetramers since GIRK4-specific activator, 3hi2one-G4, failed to elicit IKACh in the SAN cells from both control and LPS-injected mice. Moreover, this evidence further supports LPS-induced reduction of IKACh in SAN cells is due to concomitant down regulation of GIRK1 and GIRK4.
In previous reports, elevated common proinflammatory cytokines like TNF-α, IL-1β, and IL-6 have been associated with blunted antiinflammatory vagal tone leading to cardiac inflammation and reduced HRV in LPS-induced sepsis in rodents (26, 30). Among the common cytokines, IL-6 was previously found to be highly elevated in plasma of LPS-administered mice within 1 h (30), and IL-6 incubation of atrial-SAN tissues isolated from control mice for 30 min reduced CCh response on beating rate by 4.7-fold (33). However, direct incubation of SAN tissues isolated from control mice with these common cytokines for 6 h in the present study did not affect the expression of M2R and GIRK channels. Moreover, isolated control SAN cells in direct contact with IL-6 for 2 h failed to alter CCh-activated IKACh. These findings together suggest that the common cytokines interfere with the vagal response associated with reduced HRV in sepsis most likely via indirect systemic mechanism(s), rather than directly targeting M2R-GIRK1/4 function and expression. However, we cannot exclude that less-known cytokines may contribute to direct effect of LPS and further mechanistic studies are required to establish the effect of the other cytokines on HRV during sepsis.
Tachycardia and reduced HRV are mostly detected in septic patients (6–8) and reported in LPS-administered rats in vivo though the basal beating rate of SAN-atrial tissue isolated from these rats was unaffected (24). Besides, conscious GIRK4 knockout mice showed no change in basal heart rate but reduction in HRV in telemetry (23). Under our ex vivo experimental conditions, we did not observe significant change in basal beating rate and HRV of isolated hearts and in basal Ca2+ transient rate of isolated SAN tissues from LPS-injected mice compared to controls. The impact of LPS-induced endotoxemia on heart rate and HRV as well as GIRK4 knockout on HRV in previous findings in the whole animal likely requires additional factors like elevated sympathetic inputs and/or eicosanoids (24, 34), which were absent in our experiments designed to investigate parasympathetic inputs on the isolated SAN cell/tissue and heart. The parasympathetic inhibition of chronotropic action on SAN tissues and heart isolated from LPS-injected mice was compromised, as evident from the diminished effect of a muscarinic agonist, CCh, on the rate of spontaneous Ca2+ transients in the SAN and heart rate. This functional impairment of negative chronotropic effect is likely a direct consequence of reduced expression of M2R and GIRK channels in SAN tissues. This outcome is consistent with previous studies that have documented the inhibitory role of M2R-GIRK pathway on action potential firing rate in isolated SAN cells and heart rate (20–23).
In addition to slowing action potential firing rate in isolated SAN cells, Gαi-coupled M2R stimulation attenuates SR-Ca2+ release and reuptake via inhibition of cAMP-PKA-mediated phosphorylation of downstream ion-channels and pumps (20). We observed these effects in SAN tissues isolated from control mice as reduced peak amplitude and longer RT90 of Ca2+-transients following CCh administration. However, these effects were absent in the SAN tissues of LPS-injected mice with lower expression of M2R and Cav1.3 but not Gαi2, suggesting that cAMP-PKA downstream signaling could not be inhibited independent of Gαi2 expression. LPS-induced reduction in membrane expression of Cav1.3 channels in the SAN cells is in line with previous findings of reduced expression of Cav1.3 in the atrial tissues isolated from LPS-injected guinea pigs (35). This warrants investigation of LPS effect on cAMP-PKA cascade and L-type Cav1.3 currents in SAN cells/tissues in future studies.
In summary, our findings demonstrate a pivotal role of downregulation of M2R-GIRK signaling in SAN pacemaking and HRV in a murine sepsis model and likely contribution to the diminished vagal response and reduced HRV observed in septic patients. Furthermore, our data support the emerging practice to monitor HRV as an early prognostic marker for sepsis (5–8, 36). Within 6 h of LPS administration, expression of M2R and GIRK channel is reduced with consequences on vagal responsiveness and HRV. Hence, HRV monitoring at the onset of sepsis, when clinical symptoms are subtle and can easily be overlooked, is a noninvasive and efficient tool for timely diagnosis and therapeutic intervention of this fatal disease in patients.
Materials and Methods
Animals.
The experimental procedure and number of animals used were approved by the ethics committee of the Medical University of Graz and Federal Ministry of Science, Research, and Economy of the Republic of Austria (BMWFW-66.010/0101-WF/V/3b/2016), and the University of Montpellier and the French Ministry of Agriculture (protocol no: 2017010310594939). The experiments were conducted according to the Directive of the European Parliament and of the Council of September 22, 2010 (2010/63/EU). C57BL/6 wild-type mice (both sexes, 14 to 20 wk old) were administered with LPS (5 mg/kg, i.p.) to elicit a septic response for 6 h as reported previously (30) and compared to PBS-injected controls.
Human RAA.
The use of human tissue was approved by the ethics committee of the Medical University of Graz, Austria (19–109 ex 07/08) and all patients gave prior informed consent according to the WMA Declaration of Helsinki, 2013. RAA were obtained from two septic and two nonseptic (control) patients undergoing cardiac surgery.
Reagents.
All reagents used were of molecular biology grade and purchased from Sigma-Aldrich or Carl Roth unless specified otherwise.
Langendorff Perfusion.
Mice were injected with heparin (1,000 U/kg, i.p.) and anesthetized with ketamine (80 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Depth of anesthesia was monitored by toe pinch reflex, and mice were killed by cervical dislocation. Hearts were rapidly excised and arrested in ice-cold Krebs–Henseleit buffer. After cannulation of the aorta with a 20-gauge needle, retrograde perfusion was established at a constant flow of 20 mL/min/g wet weight with a modified Krebs–Henseleit bicarbonate buffer (in mM: 118 NaCl, 25 NaHCO3, 1.2 KH2PO4, 4.8 KCl, 1.2 MgSO4, 1.25 CaCl2, 11 glucose, pH 7.4) using the ISO-HEART perfusion system (Hugo Sachs Elektronik) as described previously with slight modifications (37). The perfusate was filtered through a 5-µm filter before reaching the heart and continuously gassed with carbogen (95% O2, 5% CO2). Heart temperature was maintained at 37 °C throughout the experiments, monitored with a physitemp probe (Physitemp Instruments). After removal of the left auricle, a tiny fluid-filled balloon made of a small square of polyethylene film was inserted into the left ventricle and connected to a pressure transducer via a 4F biluminal monitoring catheter (Vygon). An epicardial ECG was recorded throughout the experimental period with the use of two stainless steel electrodes attached directly to the base of the right atrium and left ventricle, respectively. Hearts were perfused for 30 min to establish stable baseline conditions, followed by 15-min perfusion with 0.1 µM CCh.
RR intervals (1,000 baseline and 1,000 after 10 min of CCh perfusion) were extracted using HRV module of LabChart 7 Pro software (ADInstruments). Each RR interval was plotted as a function of time (tachogram) or as a function of the preceding RR (Poincaré plot) to select the normal RR intervals excluding artifact and ectopic beats, as reported previously (38). Heart rate and time-domain statistical measures of HRV (SDNN, RMSSD, and pNN6) were calculated from RR interval using MATLAB™. SDNN, the SD of all normal RR intervals (in ms), estimates the total HRV arising from autonomic nervous system. RMSSD denotes the square root of the mean of the sum of the squares of differences between adjacent normal RR intervals (in ms), quantifying short-term variation between two successive beats as a measure of parasympathetic regulation. pNN6 is the percentage of consecutive normal RR intervals differing more than 6 ms, and reflects the relatively abrupt and fast changes in HR, attributed to cardiac parasympathetic activity.
Isolation of SAN Cells.
SAN cells were isolated as described previously (22, 39). Briefly, mice were heparinized (250 U, i.p., Gilvasan Pharma), anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.), and killed by cervical dislocation. The beating heart was quickly removed and transferred to the Tyrode’s solution (37 °C) containing (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES-NaOH, and 5.5 d-glucose (adjusted to pH 7.4 with NaOH). The SAN tissue was excised by cutting along the crista terminalis and the interatrial septum, and transferred into a low-Ca2+, low-Mg2+ solution containing (in mM): 140 NaCl, 5.4 KCl, 0.5 MgCl2, 0.2 CaCl2, 1.2 KH2PO4, 50 taurine, 5.5 d-glucose, 5 HEPES, and 1 mg/mL bovine serum albumin (BSA) (adjusted to pH 6.9 with NaOH). The tissue was enzymatically digested by adding 0.15 mg/mL Liberase TH (Roche) and 0.5 mg/mL elastase (Worthington Biochemical Corporation) for ~24 min at 37 °C. The digested tissue was washed and transferred to a modified Kraftbrühe solution containing (in mM): 100 K+-L-glutamate, 10 K+-aspartate, 25 KCl, 10 KH2PO4, 2 MgSO4, 20 taurine, 5 creatine, 0.5 EGTA, 20 glucose, 5 HEPES, and 1 mg/mL BSA (pH 7.2 with KOH). Single cells were manually dissociated in Kraftbrühe solution at 37 °C using a Pasteur pipette. Physiological extracellular Na+ and Ca2+ concentrations were reestablished by adding aliquots of solutions containing (in mM): 10 NaCl, 1.8 CaCl2, and subsequently, Tyrode’s solution containing 1 mg/mL BSA. Cells were used on the day of isolation.
Electrophysiological Recordings.
Electrophysiological recordings were performed in the whole-cell configuration of the patch-clamp technique using Axopatch 200B or Multiclamp amplifiers connected to Digidata 1550B interface (Molecular Devices) and Clampex v11.1 software. For IL-6 group, control SAN cells were incubated with 10 ng/mL IL-6 for 2 h before recordings. SAN cells were harvested on a custom-made glass-bottom dish and mounted on the stage of an inverted microscope (Zeiss Axiovert 135) with a 40×/0.75 objective. Prewarmed (37 °C) Tyrode’s solution was perfused near the cell using a multi-MPRE8 heating pen and cFlow perfusion controller (Cell MicroControls). Only elongated, spontaneously beating cells were patched with patch pipettes filled with intracellular solution containing (in mM): 80 K+-aspartate; 50 KCl, 3 Na+-ATP, 2 CaCl2, 1 MgCl2, 5 HEPES, and 5 EGTA (adjusted to pH 7.2 with KOH), and a tip resistance of 2 to 3 MΩ. The current was recorded from a holding potential of −80 mV using a voltage ramp protocol (1.5 s in duration) from −100 mV to +40 mV (37) before and after perfusion with 0.1 µM CCh, 300 nm tertiapin-Q (Alomone Labs) or 3 µM ML297 in Tyrode’s solution. For measuring IKACh (22), the current was recorded at −35 mV holding potential and after reaching a stable current (baseline), 0.1 µM CCh was added, and the current was recorded till it reached a plateau, usually within a few seconds. IKACh was calculated as the difference between the baseline and the plateau current. Current density was calculated by dividing the current amplitudes with cell membrane capacitance to allow comparison among cells of different size.
Ca2+ Imaging of SAN Tissue.
Intact SAN/atrial tissue was isolated from the excised mouse heart in Tyrode’s solution prewarmed to 37 °C as described previously (17, 40). The right and the left atria were pinned flat to the bottom of a 35-mm cell culture dish coated with Sylgard (Sylgard 184 Silicone elastomer kit; Dow Corning) of ~2-mm thickness. The tissue was loaded with 5 µM Cal-520 AM dye (Biozol) and 0.02% Pluronic F-127 (Thermo Fisher Scientific) in the Tyrode’s solution at 20 to 22 °C for 1 h in dark, washed twice with fresh Tyrode’s solution and kept for 15 min to allow deesterification of Cal-520 AM. The dish with Cal-520-loaded tissue was then mounted on an upright microscope (Olympus BX51WI) with a 5×/0.75 objective and perfused with Tyrode’s solution at 37 °C using dual automatic temperature controller (Warner Instruments). The SAN area was excited with 470-nm light-emitting diode (LED) of Omicron LEDHUB (Omicron-Laserage Laserprodukte GmbH), and the fluorescent images were captured at 100 frames/s in Visiview v5.0 software (Visitron Systems) using a 530/43-nm emission filter (Semrock) with an ORCA-Flash 4.0 v2 complementary metal oxide semiconductor (CMOS) camera (Hamamatsu). The fluorescence intensity (F) proportional to the intracellular Ca2+ concentration in the SAN region was analyzed after background subtraction and normalized to baseline fluorescence Fb (F/Fb). The Ca2+-transient parameters (transients/min, peak amplitude, RT90) were calculated using peak analysis module of LabChart 7 Pro software (ADInstruments).
Western Blot.
Western blot was performed as described previously (37). SAN tissues were used immediately after isolation except when incubated with TNF-α (50 ng/mL), IL-1β (10 ng/mL) or IL-6 (10 ng/mL) (Peprotech) for 6 h. The isolated tissue was homogenized in 50 to 100 µL ice-cold lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, 10% glycerol, pH 7.4 adjusted with NaOH) containing protease inhibitor cocktail tablet (Roche) with Precellys 24 tissue homogenizer (2 × 20 s, 6,500 rpm) and then was centrifuged at 13,000 rpm, 4 °C for 15 min. Protein estimation of solubilized tissue lysates was performed using the bicinchoninic acid (BCA) protein assay kit (Thermo Fischer Scientific). Then, 25 to 30 µg protein was added to 10 µL of 4× NuPAGE lithium dodecyl sulphate (LDS) sample buffer containing 2 µL sample reducing agent (Thermo Fisher Scientific), resolved by electrophoresis in NuPAGE 4 to 12% Bis-Tris gel and transferred to nitrocellulose membranes (Thermo Fisher Scientific). Membranes were blocked with 5% (w/v) nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST, 25 °C, 1 h) and cut till 80 KDa protein ladder since all the detected proteins were below 80 KDa molecular weight, and limited amount of working batch of antibodies could be utilized for incubation. The following primary antibodies (diluted in 5% [w/v] BSA-TBST) were used for incubation overnight at 4 °C: rabbit anti-GIRK1 (1:1,000, Alomone Labs Cat# APC-005, RRID: AB_2040113), rabbit anti-GIRK4 (1:500, Alomone Labs Cat# APC-027, RRID: AB_2039943), rabbit anti-CHRM2 (1:500, Alomone Labs Cat# AMR-002, RRID: AB_2039995), rabbit anti-Gαi2 (1:2,000, GeneTex Cat# GTX101779, RRID: AB_10721709), and mouse anti-Gβ (1:500, Santa Cruz Biotechnology Cat# sc-166123, RRID: AB_2109632). For normalization, membranes were stripped with stripping buffer (58.4 g/L NaCl, 7.5 g/L glycine, pH 2.15 adjusted with HCl) and incubated with anti-GAPDH (1:10,000, Proteintech Cat# 60004-1-Ig, RRID: AB_2107436) as a primary antibody. After washing, the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (1:20,000, Thermo Fisher Scientific Cat# 31460, RRID: AB_228341) or goat anti-mouse IgG (1:10,000, Jackson ImmunoResearch Labs Cat# 115-035-146, RRID: AB_2307392) in 4% [w/v] nonfat milk-TBST (25 °C, 1 h). Immunoreactive bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) in ChemiDoc MP Imaging System (Bio-Rad), and densitometric evaluation was performed using Image Lab 6.0 software (Bio-Rad). The relative expression for each protein was calculated by normalizing to the control samples and GAPDH as a loading control.
Quantitative Fluorescence Immunolabeling.
Fixed cells were immunolabeled for fluorescence imaging as described previously (41). SAN cells were isolated from three control and three LPS-injected mice, as described above and plated on poly-l-lysine-coated glass coverslips with 18-mm diameter at plating density of 25 to 30 cells/100 µL. Two hours later, the cells were fixed with 4% paraformaldehyde, 4% sucrose in PBS at room temperature for 10 min. The fixed cells were incubated in 5% normal goat serum in PBS containing 0.1% BSA and 0.2% Triton X-100 (PBS/BSA/Triton) for 30 min. Cells were then exposed to the primary antibodies in PBS/BSA/Triton at 4 °C overnight, washed, and incubated with the fluorochrome-conjugated secondary antibodies at room temperature for 1 h. The primary antibodies used were rabbit anti-GIRK1 (1:1,000, Alomone Labs Cat# APC-005, RRID: AB_2040113), rabbit anti-GIRK4 (1:1,000, Alomone Labs Cat# APC-027, RRID: AB_2039943), guinea-pig anti-CaV1.3 (1:500, Alomone Labs Cat# ACC-005-GP, RRID: AB_2756614), and guinea-pig anti-HCN4 (1:500, Alomone Labs Cat# APC-052-GP, RRID: AB_2340957). The secondary antibodies goat anti-rabbit Alexa-594 (Thermo Fisher Scientific Cat# A-11012, RRID: AB_2534079) and goat anti-guinea-pig Alexa-647 (Thermo Fisher Scientific Cat# A-21450, RRID: AB_2735091) were used at 1:4,000. Coverslips were mounted in p-phenylenediamine glycerol to retard photobleaching and observed with a conventional inverted epifluorescence IX3 microscope (Olympus) using a 60×/1.42 apochromat oil-immersion objective lens (UPLXAPO60XO, Olympus) and SOLA LED solid-state light engine (Lumencor) light source. Images were acquired with a scientific CMOS Prime back side-illuminated (BSI) camera (Teledyne Photometrics) using the same acquisition time for all pictures. Two images were captured for each cell, one at the level of the cell membrane adhering to the coverslips and another at the longitudinal focal plane passing through the cell showing the membrane as a rim around the cell.
Quantitative fluorescence analysis was performed with the ImageJ-based open-source software package Fiji (ImageJ 1.53t) (42). For each SAN cell, membrane and background regions were selected from the image at the level of the cell membrane adhering to the coverslips using the polygon selection tool, and mean fluorescence values were measured. Since HCN4 or GIRK1 immunostaining displayed distinct membrane localization, they were used as reference to observe the membrane and its regions adhering to the coverslip. The membrane regions with the costained GIRK4 and Cav1.3, respectively, were coimaged and analyzed accordingly. Membrane fluorescence values were background subtracted, obtaining one value for each cell representing the channel levels at the membrane. Absolute fluorescence values were normalized to the average fluorescence of control cells. For data presentation, representative membrane regions were selected and assembled in a composite using Photoshop version 24.1.0.
Statistical Analysis.
Data were analyzed using IBM SPSS Statistics 27 (IBM Corporation), R version 4.1.0 (2021–05–18) and RStudio version 1.4.1717. N represents the number of cells or tissues from at least 6 mice unless specified otherwise. The approximate normal distribution of data was assessed by z-value of skewness and kurtosis, and P-value of Shapiro–Wilk test. If normally distributed and unless specified otherwise, mixed two-way ANOVA was applied to evaluate interaction and main effects followed by simple effects. The between-group factor was treatment (Control vs. LPS) and the within-group, repeated measure factor was baseline (-CCh) vs. CCh response. In case of pNN6 data, the baseline values in both control and LPS groups were mostly zeroes, thus the difference between baseline and CCh response for control and LPS groups was calculated and the medians of the delta values were compared by the R function medpb2 as described by Wilcox in Chapter 7.5.3 (43). This function uses the percentile bootstrap method to account for tied values. Data from two independent groups were analyzed by unpaired bootstrap t test. Data from more than two paired groups were analyzed by repeated measure one-way ANOVA followed by within-group effect analysis with Bonferroni adjustment. Western blot data were analyzed using one-sample t test, with reference value for control samples set to one. All tests were two-tailed, and P values < 0.05 were considered significant. Normally distributed data were plotted using OriginPro 2018 (OriginLab Corporation) as scatter plots along with mean ± SEM, otherwise median with first and third quartiles were plotted.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by research grants from the Austrian Science Funds (Hertha Firnberg T1066 and Zukunftskolleg ZK17 to S.S., stand-alone project P33225 to V.D.B.), and the Fondation Leducq (TNE FANTASY 19CVD03 to M.E.M.). We acknowledge Diomedes E. Logothetis for providing 3hi2one-G4.
Author contributions
N.S., K.Z.-P., B.P., and S.S. designed research; N.S., P.M., C.N.K., G.W., V.D.B., E.T., P.L., A.G., M.E.M., B.P., and S.S. performed research; P.M., V.D.B., W.S., R.A., H.M., B.M., D.v.L., A.G.T., M.E.M., B.P., and S.S. contributed new reagents/analytic tools; N.S., K.Z.-P., P.M., C.N.K., G.W., V.D.B., M.E.M., B.P., and S.S. analyzed data; and N.S., K.Z.-P., P.M., C.N.K., G.W., V.D.B., E.T., P.L., A.G., W.S., R.A., H.M., B.M., D.v.L., A.G.T., M.E.M., B.P., and S.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. P.A.S. is a guest editor invited by the Editorial Board.
Contributor Information
Brigitte Pelzmann, Email: brigitte.pelzmann@medunigraz.at.
Susanne Scheruebel, Email: susanne.scheruebel@medunigraz.at.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Singer M., et al. , The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudd K. E., et al. , Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kakihana Y., Ito T., Nakahara M., Yamaguchi K., Yasuda T., Sepsis-induced myocardial dysfunction: Pathophysiology and management. J. Intensive Care 4, 22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pierrakos C., Velissaris D., Bisdorff M., Marshall J. C., Vincent J. L., Biomarkers of sepsis: Time for a reappraisal. Crit Care 24, 287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt H., et al. , Autonomic dysfunction predicts mortality in patients with multiple organ dysfunction syndrome of different age groups. Crit. Care Med. 33, 1994–2002 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Chen W. L., et al. , Heart rate variability measures as predictors of in-hospital mortality in ED patients with sepsis. Am. J. Emerg. Med. 26, 395–401 (2008). [DOI] [PubMed] [Google Scholar]
- 7.de Castilho F. M., Ribeiro A. L. P., da Silva J. L. P., Nobre V., de Sousa M. R., Heart rate variability as predictor of mortality in sepsis: A prospective cohort study. PLoS One 12, e0180060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pong J. Z., et al. , Combining heart rate variability with disease severity score variables for mortality risk stratification in septic patients presenting at the emergency department. Int. J. Environ. Res. Public Health 16, 1725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yaniv Y., et al. , Synchronization of sinoatrial node pacemaker cell clocks and its autonomic modulation impart complexity to heart beating intervals. Heart Rhythm. 11, 1210–1219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaniboni M., Cacciani F., Lux R. L., Beat-to-beat cycle length variability of spontaneously beating guinea pig sinoatrial cells: Relative contributions of the membrane and calcium clocks. PLoS One 9, e100242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesirca P., et al. , Pharmacologic approach to sinoatrial node dysfunction. Annu. Rev. Pharmacol. Toxicol. 61, 757–778 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vinogradova T. M., et al. , Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94, 802–809 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Torrente A. G., et al. , L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc. Res. 109, 451–461 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Louradour J., et al. , L-type Cav1.3 calcium channels are required for beta-adrenergic triggered automaticity in dormant mouse sinoatrial pacemaker cells. Cells 11, 1114 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogdanov K. Y., et al. , Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99, 979–987 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Yaniv Y., et al. , Stochasticity intrinsic to coupled-clock mechanisms underlies beat-to-beat variability of spontaneous action potential firing in sinoatrial node pacemaker cells. J. Mol. Cell Cardiol. 77, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torrente A. G., et al. , Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc. Natl. Acad. Sci. U.S.A. 112, 9769–9774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yaniv Y., et al. , Real-time relationship between PKA biochemical signal network dynamics and increased action potential firing rate in heart pacemaker cells: Kinetics of PKA activation in heart pacemaker cells. J. Mol. Cell Cardiol. 86, 168–178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behar J., Ganesan A., Zhang J., Yaniv Y., The autonomic nervous system regulates the heart rate through cAMP-PKA dependent and independent coupled-clock pacemaker cell mechanisms. Front. Physiol. 7, 419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyashkov A. E., et al. , Cholinergic receptor signaling modulates spontaneous firing of sinoatrial nodal cells via integrated effects on PKA-dependent Ca(2+) cycling and I(KACh). Am. J. Physiol. Heart. Circ. Physiol. 297, H949–959 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Borren M. M., et al. , Effects of muscarinic receptor stimulation on Ca2+ transient, cAMP production and pacemaker frequency of rabbit sinoatrial node cells. Basic Res. Cardiol. 105, 73–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mesirca P., et al. , The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 142, 113–126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wickman K., Nemec J., Gendler S. J., Clapham D. E., Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Gholami M., et al. , Endotoxemia is associated with partial uncoupling of cardiac pacemaker from cholinergic neural control in rats. Shock 37, 219–227 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Borovikova L. V., et al. , Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Schulte A., Lichtenstern C., Henrich M., Weigand M. A., Uhle F., Loss of vagal tone aggravates systemic inflammation and cardiac impairment in endotoxemic rats. J. Surg. Res. 188, 480–488 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Dong L. W., Tang C., Liu M. S., Biphasic redistribution of muscarinic receptor and the altered receptor phosphorylation and gene transcription are underlying mechanisms in the rat heart during sepsis. Cardiovasc. Res. 45, 925–933 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann K., et al. , ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 4, 1278–1286 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui M., et al. , A novel small-molecule selective activator of homomeric GIRK4 channels. J. Biol. Chem. 298, 102009 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fairchild K. D., et al. , Endotoxin depresses heart rate variability in mice: Cytokine and steroid effects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1019–1027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zorn-Pauly K., et al. , Endotoxin impairs the human pacemaker current If. Shock 28, 655–661 (2007). [PubMed] [Google Scholar]
- 32.Scheruebel S., et al. , I(f) blocking potency of ivabradine is preserved under elevated endotoxin levels in human atrial myocytes. J. Mol. Cell Cardiol. 72, 64–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hajiasgharzadeh K., Mirnajafi-Zadeh J., Mani A. R., Interleukin-6 impairs chronotropic responsiveness to cholinergic stimulation and decreases heart rate variability in mice. Eur. J. Pharmacol. 673, 70–77 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Takayama K., et al. , Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat. Med. 11, 562–566 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Aoki Y., et al. , Role of ion channels in sepsis-induced atrial tachyarrhythmias in guinea pigs. Br. J. Pharmacol. 166, 390–400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinten V. M., van Meurs M., Renes M. H., Ligtenberg J. J. M., Ter Maaten J. C., Protocol of the sepsivit study: A prospective observational study to determine whether continuous heart rate variability measurement during the first 48 hours of hospitalisation provides an early warning for deterioration in patients presenting with infection or sepsis to the emergency department of a Dutch academic teaching hospital. BMJ Open 7, e018259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koyani C. N., et al. , Dipeptidyl peptidase-4 independent cardiac dysfunction links saxagliptin to heart failure. Biochem. Pharmacol. 145, 64–80 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Thireau J., Zhang B. L., Poisson D., Babuty D., Heart rate variability in mice: A theoretical and practical guide. Exp. Physiol. 93, 83–94 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Mangoni M. E., Nargeot J., Properties of the hyperpolarization-activated current (I(f)) in isolated mouse sino-atrial cells. Cardiovasc. Res. 52, 51–64 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Ahammer H., et al. , Sinoatrial beat to beat variability assessed by contraction strength in addition to the interbeat interval. Front. Physiol. 9, 546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Biase V., Flucher B. E., Obermair G. J., Resolving sub-synaptic compartments with double immunofluorescence labeling in hippocampal neurons. J. Neurosci. Methods 176, 78–84 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Schindelin J., et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilcox R., Modern Statistics for the Social and Behavioural Sciences: A Practical Introduction (CRC Press, Taylor & Francis Group, Boca Raton (FL), ed. 2, 2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.