

Abstract
Pulmonary fibrotic diseases are characterized by proliferation of lung fibroblasts and myofibroblasts and excessive deposition of extracellular matrix proteins. Depending on the specific form of lung fibrosis, there can be progressive scarring of the lung, leading in some cases to respiratory failure and/or death. Recent and ongoing research has demonstrated that resolution of inflammation is an active process regulated by families of small bioactive lipid mediators terms “specialized pro-resolving mediators” (SPMs). While there are many reports of beneficial effects of SPMs in animal and cell culture models of acute and chronic inflammatory and immune diseases, there have been fewer reports investigating SPMs and fibrosis, especially pulmonary fibrosis. Here, we will review evidence that resolution pathways are impaired in interstitial lung disease, and that SPMs and other similar bioactive lipid mediators can inhibit fibroblast proliferation, myofibroblast differentiation, and accumulation of excess extracellular matrix in cell culture and animal models of pulmonary fibrosis, and we will consider future therapeutic implications of SPMs in fibrosis.
Keywords: pro-resolving mediators, SPMs, interstitial lung disease, idiopathic pulmonary fibrosis, scleroderma, sarcoidosis, fibroblast
1. Introduction
Pulmonary fibrotic diseases are characterized by proliferation of lung fibroblasts and myofibroblasts and excessive deposition of extracellular matrix proteins. In some forms of fibrosis, including idiopathic pulmonary fibrosis (IFP), progressive scarring of the lung can lead to respiratory failure and even death. Several subtypes of pulmonary fibrosis all classified under the umbrella term “interstitial lung disease” (ILD), that can be caused by various insults to the lung including chemical, immunological, physical and radiological, as well as genetic factors and susceptibilities. IPF is an irreversible, progressive disease with no known cause and is one of the most serious forms of lung fibrosis. Other common ILDs include chronic hypersensitivity pneumonitis (HP) and autoimmune connective tissue (CT)-ILD. Although the FDA has approved pirfenidone and nintedanib for IPF, these drugs only slow the progression of disease in some patients and have significant side effects. The global incidence of IPF is rising, with a mean survival time of 3–5 years after diagnosis that is shorter than that for lung cancer, highlighting an urgent need for more effective therapies (Jeganathan, et al., 2021; Marshall, et al., 2018). The FDA has granted orphan drug status to several upcoming IPF therapies entering clinical trials, but more options are urgently needed.
There is a great deal of attention being paid to the field of resolution of inflammation, and the families of bioactive lipids that promote resolution, termed specialized pro-resolving mediators, or SPMs. SPMs have proven to have potent anti-inflammatory and pro-resolving effects in many animal models of disease (reviewed in (Cagnina, et al., 2022; Chiang & Serhan, 2020)). They are exciting candidates for drug development; since they are endogenously produced, they are expected to be well tolerated, and they are easily derivatized to increase their selectivity, potency, and improve their pharmacokinetic properties. SPMs act through G protein coupled receptors (GPCRs), and GPCRs are the largest family of targets for drug development (Park, et al., 2020).
However, it is not immediately obvious if lung fibrosis or other fibrosing diseases should be candidates for SPM therapy. Some of the key effector cells in fibrosis are fibroblasts and myofibroblasts, and the involvement of classical inflammatory or immune processes is less clear. Here, we will review emerging evidence from cell culture, animal models and human patient samples that SPMs have therapeutic potential to treat IPF and other lung fibrosing diseases.
2. Overview of pulmonary fibrosis/interstitial lung disease
Interstitial lung disease (ILD) is a general classification that encompasses a number of diseases that cause scaring or fibrosis of the lungs. ILDs can be chronic and progressive, and can lead to irreversible lung damage and loss of lung function. Depending on the specific disease, therapy can be very challenging (Behr, et al., 2021; Farrand, et al., 2020; Wijsenbeek, et al., 2022). Tissue fibrosis can be understood as an aberrant wound healing process, in which there is epithelial damage, activation of fibroblasts and differentiation of fibroblasts to myofibroblasts. Myofibroblasts are contractile and matrix producing cells with features of both fibroblasts and smooth muscle cells. Fibrosing ILDs are characterized by epithelial injury and repair, proliferation of lung fibroblasts and myofibroblasts, production of excess extracellular matrix proteins including collagens and fibronectin, increased crosslinking of extracellular matrix (ECM), dysregulation of matrix metalloproteinases, and fibroblast resistance to apoptosis. This can lead to increased thickness of alveolar septae, fibrotic destruction of the normal lung architecture, loss of gas exchange, and in severe disease, death due to respiratory failure (Meyer, 2017; Upagupta, et al., 2018) (Figure 1).
Figure 1. Radiologic and histologic features of Idiopathic Pulmonary Fibrosis (IPF).
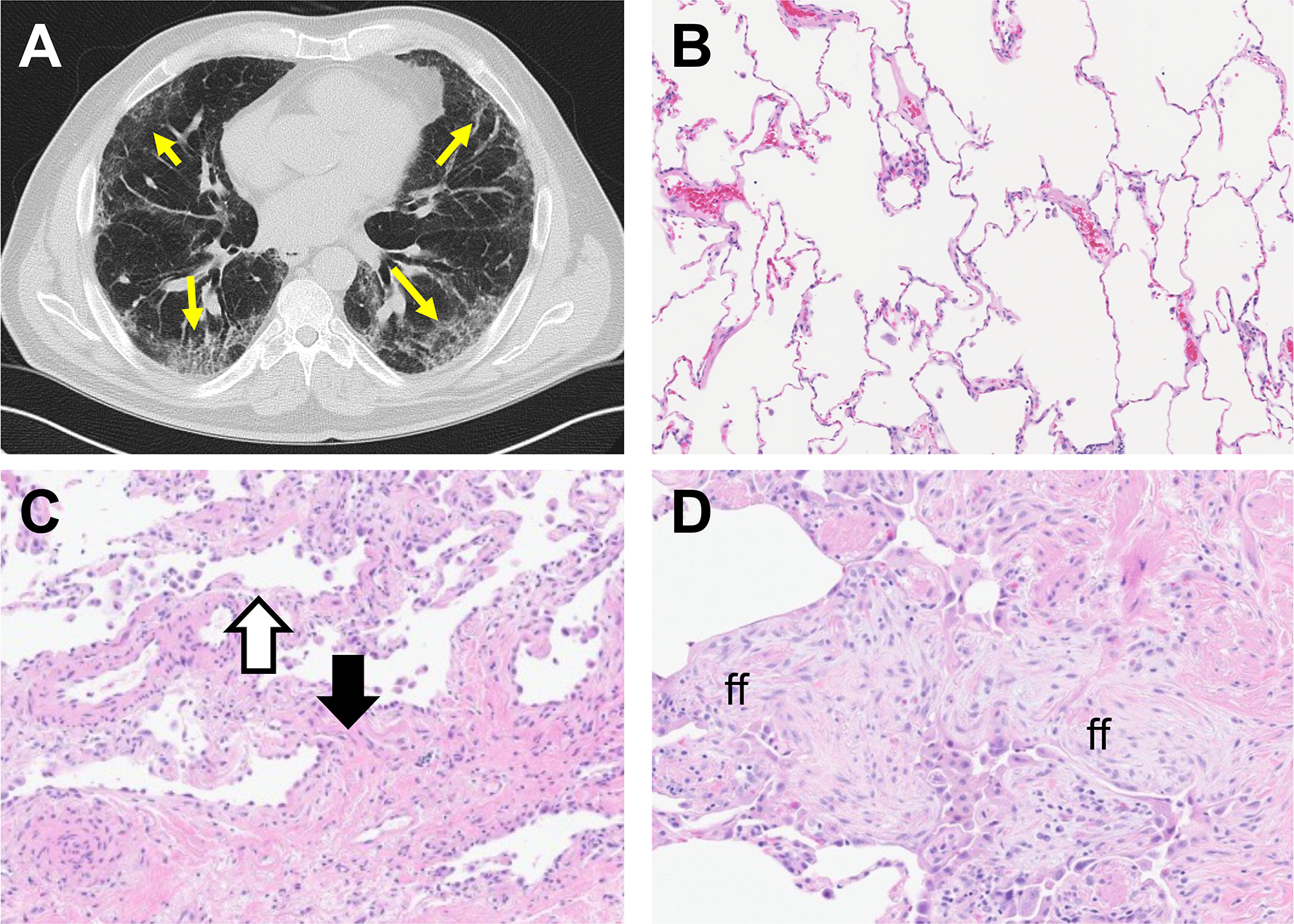
A. High resolution CT scan from patient with IPF. Yellow arrows point to honeycombing and fibrotic features. B-D. H&E stained sections of normal (B) and IPF (C, D) lung tissue. In C, the black arrow indicates fibrotic tissue while the white area indicates adjacent uninvolved parenchyma. In D, ff indicates fibroblastic foci, a hallmark feature of IPF.
Some ILDs have immune or inflammatory involvement. Hypersensitivity pneumonitis (HP), caused by chronic exposure to antigens such as mold or bird dander, can develop into chronic fibrosis and/or chronic inflammatory subtypes. Autoimmune connective tissue (CT) diseases including scleroderma and rheumatoid arthritis can have fibrotic involvement of the lungs. Sarcoidosis is an immune disorder of unknown origin that commonly involves the lungs and is characterized by granulomas and fibrosis. Therapeutic ionizing radiation treatment for breast, esophageal or lung cancer can also cause fibrotic changes in the lung. The risk is proportional to the cumulative radiation dose and the volume irradiated. Radiation-induced fibrosis may be preceded by pneumonitis, but up to 90% of irradiated patients exhibit fibrotic change on CT scans without developing clinically significant pneumonitis (Kalman, et al., 2018). Finally, approximately 10 to 30% of ILDs are diagnosed as idiopathic pulmonary fibrosis (IPF), exhibiting progressive tissue scarring with no known cause (Coultas, et al., 1994; Duchemann, et al., 2017; Hyldgaard, et al., 2014). Two drugs, nintedanib and pifenidone, have been FDA approved to treat IPF, but at best they only slow disease progression in some patients and have significant side effects. The only true cure for IPF at present is lung transplantation, which is not available or appropriate for all patients.
The pathogenesis of IPF is complex and incompletely understood. Evidence suggests that pulmonary fibrosis follows repetitive epithelial injury and aberrant wound healing. Gene defects in familial IPF (encompassing approximately 10–20% of all IPF patients (Borie & Crestani, 2019; Fernandez, et al., 2012; Garcia-Sancho, et al., 2011)) also implicate lung epithelial cell function in IPF, and mouse models of drug-induced or genetically controlled epithelial damage result in fibrogenic pathologies (T. Liu, et al., 2019; Takezaki, et al., 2019; Yasutomo, 2021).
There is strong evidence that the cytokine transforming growth factor-β (TGFβ) is the key pro-fibrotic cytokine. TGFβ promotes activation of fibroblasts and differentiation of fibroblasts to myofibroblasts, and upregulates production and crosslinking of ECM (Biernacka, et al., 2011; Frangogiannis, 2020). TGFβ can be produced by many cell types and is sequestered in the matrix in latent form and can be activated by a variety of stimuli including enzymatic action, mechanical stress, integrins, and low pH (Kottmann, et al., 2012; Munger, et al., 1999; Wipff, et al., 2007). Other pro-fibrotic chemokines include fibroblast growth factor (FGF), platelet-derived growth factor (PDGF) and insulin-like growth factor (IGF).
Most activated fibroblasts and myofibroblasts in IPF lung tissue are believed to arise from proliferation and differentiation of resident lung fibroblasts (Kendall & Feghali-Bostwick, 2014). However, there is some evidence that some fibroblast-like cells arise via differentiation from epithelial cells in a process called epithelial-mesenchymal transition (EMT), driven by TGFβ (Willis & Borok, 2007; Willis, et al., 2005; L. Yao, et al., 2019). While EMT is described in animal models of lung fibrosis, this process is less well described in human lung. Evidence from rodent models also suggests that some circulating bone marrow-derived cells express fibroblast-like markers (fibrocytes) and can migrate to the site of fibrosis and contribute to disease pathology (Ashley, et al., 2017; Xu, et al., 2009). These processes are well-described in vitro and in rodent models of IPF but their contribution to human disease is uncertain.
Pulmonary fibrosis is also characterized by vascular remodeling, encompassing neovascularization at the boundary between areas of normal and fibrotic lung architecture as well as destruction of the capillary bed in fibrotic foci (Barratt & Millar, 2014). Vascular remodeling in pulmonary fibrosis includes muscularization of vessels, vasoconstriction and perivascular fibrosis, mediated in part by vascular smooth muscle cells. Vascular remodeling including vascular pruning has been demonstrated in experimental models of pulmonary fibrosis (Abdalla, et al., 2015; Santos-Ribeiro, et al., 2023). Pulmonary hypertension due to vascular remodeling occurs in 40% of IPF patients and is associated with increased mortality (Shorr, et al., 2007). Drivers of vascular remodeling in pulmonary fibrosis include TGFβ and CTGF (Tsujino, et al., 2017; T. Wang, et al., 2018; Yanagihara, et al., 2022). The cytokine vascular endothelial growth factor (VEGF) aggravates pulmonary fibrosis in experimental animal models, while blockade of VEGF signaling attenuates experimental fibrosis (Chaudhary, et al., 2007; Farkas, et al., 2009; K. S. Lee, et al., 2008). The FDA approved anti-fibrotic drug nintedanib acts in part by inhibiting the VEGF receptor (Hilberg, et al., 2008).
3. The role of inflammation in pulmonary fibrosis
The question of how much inflammatory or immune mechanisms contribute to the pathogenesis of some forms of pulmonary fibrosis is a contentious debate. There are varying levels of evidence of immune mechanism involvement in the ILD subtypes. There is a strong immune and inflammatory component to CT and HP ILDs, and sarcoid, CT and HP patients can respond to anti-inflammatory and immunotherapies (Boerner, et al., 2020; Morisset, et al., 2017; Vorselaars, et al., 2013), implicating inflammation in the progression of disease. However, nonspecific immunotherapy is harmful for IPF patients. The PANTHER-IPF (Prednisone, Azathioprine, and N-acetylcysteine) clinical trial demonstrated nonspecific immunotherapy offers no clinical benefit and even increases death and hospitalization in IPF patients (Idiopathic Pulmonary Fibrosis Clinical Research, et al., 2012). Histologically, IPF does not exhibit many of the features of classical cellular inflammation. There are signs that non-classical inflammation is present in IPF, but it remains unknown if it is an important driver of fibrosis.
The role of immune cells and soluble mediators in pulmonary fibrosis is reviewed in depth elsewhere (Kolahian, et al., 2016). Briefly, both adaptive and innate immune cells can modulate pro-fibrotic behavior of fibroblasts. Fibroblasts and myofibroblasts are highly responsive to growth factors and cytokines produced by immune cells. There is a complicated and nuanced role for macrophages in fibrosis as they produce multiple cytokines, chemokines, and proteases. In general, classically activated (M1) macrophages are thought to be pro-inflammatory, while alternatively activated macrophages (M2) are believed to promote resolution of inflammation and resolve the wound healing process. However, as M2 macrophages produce TGFβ and PDGF, their presence in IPF tissue is viewed with suspicion (Misharin, et al., 2017; Sennello, et al., 2017; Y. Yao, et al., 2016; Zhang, et al., 2018). T cells can provide both anti- and pro-fibrotic effectors based on subtypes and temporal stage of fibrosis. Th1 cytokines (INF-γ, IL-12) are generally considered antifibrotic (Keane, et al., 2001) while Th2 cytokines (IL-17, IL-13) have reported pro-fibrotic activities (Saito, et al., 2003). Early stage Tregs are pro-fibrotic, whereas late stage Tregs dampen fibrosis (Boveda-Ruiz, et al., 2013; F. Wang, et al., 2020). The harmful versus beneficial roles of macrophage and T-cell phenotypes remain under investigation.
4. Specialized pro-resolving mediators (SPMs)
In spite of its bad reputation, inflammation is often a beneficial and necessary process. Inflammation serves to mitigate damage from injury, initiate repair processes, and recruit inflammatory and immune cells to clear debris and to orchestrate immune responses to pathogens. “Resolution of inflammation” refers to the return to homeostasis after an inflammatory response, and includes several distinct hallmarks including; class switching of recruited macrophages from pro-inflammatory M1 phenotype to a pro-resolving M2 phenotype; nonphlogistic (non-inflammatory) phagocytosis of pathogens; increased phagocytosis and clearance of apoptotic neutrophils (efferocytosis); and decreased production of pro-inflammatory cytokines and chemokines (reviewed in (Panigrahy, et al., 2021)). It is now well-established that resolution of inflammation is an active process that is mainly controlled by families of “specialized pro-resolving mediators” (SPMs)—bioactive lipids derived from omega-3 and omega-6 polyunsaturated fatty acids through a series of enzymatic reactions (Figure 2) (Chiang & Serhan, 2017; Duvall & Levy, 2016; Serhan, et al., 2015). The first bioactive eicosanoids to be described were the prostaglandins (PGs) and leukotrienes (LTs), which are derived from arachidonic acid via cyclooxygenases and generally have pro-inflammatory effects. The first pro-resolving lipids to be identified were the lipoxins, which are derived from arachidonic acid via 5- and 15-lipooxygenase (LO) enzymes (Serhan, et al., 1984). As the field expanded, additional classes of SPMs were identified, termed resolvins, protectins and maresins, derived from docosahexaenoic acid (DHA) and eicosapenatenoic acid (EPA) (Serhan, et al., 2000; Serhan, et al., 2012). Although not formally classified as SPMs, other DHA products have pro-resolving or anti-inflammatory effects. The epoxyeicosatrienoic acids (EETs) are derived from DHA via the action of cytochrome P450 epoxygenases, while hydroxyeicosatetraenoic acids (HETEs) are generated by 12- and 15-LOX. HETEs and EETs have independent pro-resolving activity and can also act as indirect precursors for other SPMs (Spector, et al., 2004; Zeldin, 2001).
Figure 2. Diagram of synthetic pathways for the major classes of SPMs.

SPMs are derived from Omega-3 and –6 polyunsaturated fatty acids through a series of enzymatic reactions. Pro-resolving mediators consist of Resolvins (Rv-), Maresins (MaR-), Protectins (PD-), and Lipoxins (LX-). Pro-inflammatory mediators Prostaglandins and Leukotrienes are derived from Arachidonic Acid. Abbreviations are defined in the main text and abbreviations table. Created with Biorender.com.
The newest class of SPMs are known as conjugates of tissue regeneration. It was originally found that maresin-1 can be conjugated to glutathione. Subsequently, the glutathione moiety can be cleaved by gamma-glutamyl transferase to a dipeptide, and further cleaved by dipeptidases to a cysteine monopeptide linked to maresin-1 (Dalli, et al., 2016). It was discovered that these cisteinyl-conjugated SPMs promoted resolution of inflammation but also promoted tissue repair and regeneration, and they were named maresin conjugates of tissue regeneration (MCTR)-1, -2 and -3, respectively. Subsequently, cysteinyl-conjugated forms of D resolvins and protectins were identified and termed resolvin conjugates of tissue regeneration (RCTRs) and protectin conjugates of tissue regeneration (PCTRs) (Dalli, et al., 2015; Jouvene, et al., 2019; Ramon, et al., 2016).
The endogenous sources for SPMs are not well understood. SPMs have been detected in human peripheral blood, immune tissues like spleen and lymph nodes, nervous system tissues including the brain and spinal fluid, in lung tissue, sputum, exhaled breath condensates and bronchoalveolar lavage fluid, and in many other compartments (Claria, et al., 2013; Colas, et al., 2014; Giera, et al., 2012; Levy, et al., 2007; Mas, et al., 2012). SPMs can be produced by B cells, T cells, macrophages, neutrophils, platelets, epithelial cells, endothelial cells and fibroblasts. However, the exact cellular sources of SPMs in specific tissues or specific disease states are incompletely understood. In animal models, treatment with SPMs have been shown to block inflammation and promote resolution in a wide variety of lung diseases including asthma, emphysema, cystic fibrosis, and multiple infection models (Cagnina, et al., 2022).
SPMs act via a family of GPCRs. Eight SPM receptors have been identified to date, including DRV1/GPR32 (D resolvin receptor 1, binds RvD1, RvD3, and RvD5), DRV2/GPR18 (D resolvin receptor 2, binds RVD2), FPR2/ALX (formyl peptide receptor 2/lipoxin A receptor, binds RvD1, RvD3 and LXA4), ERV1/CMKLR1/ChemR23 (E resolvin receptor 1, binds RvE1), BLT1/LTB4R (leukotriene B4 receptor, binds RvE1 and RvE2), GPR37 (binds protectin D1), LGR6 (binds maresin 1), and GPR101 (binds n-3 D-resolvins) (Figure 3) (Chiang & Serhan, 2020). Some receptors can bind multiple SPMs, and this has important implications for the potential therapeutic uses of SPMs. For example, resolvin D1 (RvD1) and lipoxin A4 (LXA4) can both bind either DRV1/GPR32 or FPR2/ALX, and we and others have reported that RvD1 exerts anti-inflammatory and pro-resolving effects via these receptors (Hsiao, et al., 2014). However, FPR2 is also a ligand for serum amyloid A, which provokes pro-inflammatory responses (Tylek, et al., 2021). Similarly, resolvin E1 (RvE1) can bind to either ERV1/ChemR23 or BLT1. As leukotrienes are classical pro-inflammatory mediators, this also highlights the importance of ligand structure and receptor selectivity, as different ligands that bind to the same GPCR can trigger different downstream effects, and understanding ligand structure, binding, and GPCR downstream signaling will be critical to development of SPMs and SPM derivatives as new pharmacotherapeutics (Merlin, et al., 2022; Park, et al., 2020).
Figure 3. SPM receptor network.

Diagram of the GPCR signaling network for SPMs. Multiple SPMs (left) can bind the same receptors (right). Green indicates pro-resolving mediators, and red indicates pro-inflammatory mediators. Abbreviations are defined in the main text and abbreviations table. Created with Biorender.com.
Production of SPMs is spatially and temporally regulated, with pro-inflammatory lipids being produced in the initial phases of an inflammatory response, shifting to production of pro-resolving mediators during the resolution phase. This is accomplished by altering expression levels of the key synthetic enzymes in the pro-inflammatory and pro-resolving eicosanoid pathways. Many SPMs can also be degraded into products with significantly reduced biological activity by enzymes including eicosanoid oxidoreductase (EOR, also known as 15-hydroxyprostaglandin dehydrogenase or 15-PGDH) and soluble epoxide hydrolases (sEHs) (Y. P. Sun, et al., 2007; Wagner, et al., 2017). Production of SPM-degrading enzymes is an endogenous part of the temporal regulation of resolution, but inappropriate expression of these enzymes can prolong chronic inflammation by blocking normal resolution responses.
Another important concept in SPM biology is transcellular biosynthesis, in which one cell type produces an SPM intermediate, which is taken up by the target cell and converted to the final bioactive SPM (Capra, et al., 2015). This contributes to the specific spatial and temporal regulation of SPM production. Finally, it is important to note that SPMs are pro-resolving and anti-inflammatory without being immunosuppressive. Classical anti-inflammatory therapies like corticosteroids can suppress immune responses, limiting inflammation but increasing susceptibility to infection, while SPMs have been shown in multiple animal models to reduce inflammation while improving antibacterial and antiviral immune responses (Basil & Levy, 2016; Bhat, et al., 2021; Codagnone, et al., 2018; Croasdell, et al., 2016; Dalli, et al., 2016; Isopi, et al., 2020; P., et al., 2022; Ramon, et al., 2016; Recchiuti, et al., 2021; Ruiz, et al., 2019).
SPMs are endogenous bioactive lipids that have potent anti-inflammatory and pro-resolving activities against a wide variety of acute and chronic inflammatory and infectious disease models. However, evidence also suggests that SPMs may be able to improve outcomes in pulmonary fibrosis, a group of diseases in which the role of inflammation is much less obvious.
5. The case for SPMs and pulmonary fibrosis
Fibroblasts and myofibroblasts are not classical immune or inflammatory cells, so it is not obvious that SPMs would regulate their function. On the other hand, fibrosis appears to be an aberrant wound healing and repair process, suggesting that SPMs might play a role in promoting resolution and repair in fibrosis. CT and sarcoid ILDs have significant immune system involvement and are amendable to corticosteroid therapy, suggesting that SPMs could promote resolution of these forms of fibrosis via immune cell interactions. However, it remains unclear if we should expect SPMs to have a role in controlling fibrotic diseases that lack a clear underlying immune component.
Data on levels of SPMs in samples from patients with ILDs is extremely limited. A study of 5-LOX derived eicosanoids in bronchoalveolar lavage fluid from scleroderma patients identified elevated levels of LTB4 and PGE2 and a decreased ratio of LXA4:LTB4 (Kowal-Bielecka, et al., 2005), while LC-MS/MS analysis of eicosanoids in human lung tissue found significantly lower levels of 8,9-EET, 11,12-EET and 14,15-EET in lung tissue from IPF patients compared to control tissue (H. S. Kim, et al., 2021). Untargeted metabolomics analysis of lung tissue from patients undergoing transplantation for end-stage silicosis found upregulated arachidonic acid metabolites including PGE2 and several D/J series prostaglandins (Pang, et al., 2021). This is a clearly under-studied area, and the availability of LC-MS/MS methods for lipidomics analysis with sensitivities in the picogram range presents significant opportunities for new discoveries in this area.
In the absence of strong patient data, we must rely on cell culture and animal models. Many of the animal models of pulmonary fibrosis have an inflammatory component, making it difficult to determine if the beneficial effects of SPMs on fibrosis are due to direct effects on pro-fibrotic mechanisms, or are an indirect result of reducing the inflammation that precedes fibrosis in these models. This can be overcome to a certain extent by administering the SPM intervention to the animals after the acute inflammatory phase but before the fibrotic phase, and by using in vitro cultures of lung cells to investigate whether SPMs inhibit pro-fibrotic phenotypes and the specific mechanisms involved.
5.1. Data from animal models
The most widely used animal model of pulmonary fibrosis is the bleomycin model. Bleomycin is a chemotherapeutic drug that was found to cause pulmonary fibrosis as a side-effect in cancer patients, that was developed into an animal model of lung fibrosis (T. Liu, et al., 2017). Bleomycin can be instilled directly into the lungs via intranasal aspiration, oropharyngeal aspiration, or intratracheal delivery, or it can be delivered systemically by osmotic minipump or daily subcutaneous injections. Bleomycin is believed to cause fibrosis via DNA damage and oxidative stress to the lung epithelium and lung resident fibroblasts (Walters & Kleeberger, 2008). Bleomycin is also a strong pro-inflammatory stimulus and the single dose lung delivery models are characterized by an inflammatory phase from day 1 to day 7, transitioning to a fibrotic phase, with most investigators harvesting fibrotic endpoints between day 14 and day 35.
Several studies have reported that pre-treating mice with DHA or a fish oil diet prior to administration of bleomycin resulted in reduced fibrosis, reduced collagen content, and preserved lung mechanical function, suggesting that dietary ingestion or lung administration of SPM precursors inhibits the inflammatory and fibrotic responses by changing the spectrum of bioactive mediators produced in response to injury (Galdino de Souza, et al., 2022; Kennedy, et al., 1989; Zhao, et al., 2014). Mice treated with a LXA4 analog with increased in vivo stability exhibited reduced expression of collagen and α-smooth muscle actin (αSMA), a differentiation marker of myofibroblasts, when the analog was given on day 0 or day 4 after bleomycin administration (Martins, et al., 2009). Subsequently, the same group reported that this analog was also effective at reducing fibrosis, improving the survival of alveolar type II (AT-II) epithelial cells, and reducing expression of TGFβ, when given on days 7 and 10 after bleomycin administration, after the acute inflammatory phase. The analog also restored the balance of pro-resolving M2 macrophages in lung tissue (Guilherme, et al., 2013). Two synthetic FPR2/ALX agonists, BML-111 and Quin-C1, attenuated experimental fibrosis in the bleomycin mouse model, with decreased accumulation of collagen, preservation of lung architecture, and decreased expression of TGFβ (He, et al., 2011; Ji, et al., 2018). LXA4 is also implicated as an anti-fibrotic lipid mediator by a study demonstrating that the pro-inflammatory eicosanoid thromboxane A2 negatively regulates expression of the LXA4 receptor FPR2/ALX, preventing LXA4 signaling. Administration of a thromboxane synthesis inhibitor to mice treated with bleomycin resulted in increased expression of FPR2/ALX and decreased collagen gene expression (Sato, et al., 2004).
A group of related studies examined the effects of MaR1, MCTR1, and protectin DX (PDX) in bleomycin-treated mice. Administration of the SPMs was started late, after the acute inflammatory response. MaR1 was given on days 14–21 with harvest at day 28, while MCTR1 and PDX were given from days 8–21 with harvest on day 21. All three SPMs demonstrated significant anti-fibrotic activity at doses as low as 10 ng/mouse/day, with reductions in expression of ECM proteins including collagen and fibronectin, reduced expression of TGFβ, preserved lung compliance, and improved blood oxygenation. The authors also reported significant preservation of epithelial cell numbers and function (H. Li, et al., 2017; Pan, et al., 2021; Y. Wang, et al., 2015). In another study using late administration of an SPM, mice received bleomycin by continuous infusion via an implanted osmotic minipump for 7 days, with 17(R)-RvD1 administered by i.p. injection on days 21–25 with harvest on day 28. 17(R)-RvD1, sometimes referred to as aspirin-trigged (AT)RvD1 (Y. P. Sun, et al., 2007), is an RvD1 stereoisomer with similar bioactivity but greater resistance to degradation by endogenous EOR enzymes. Even with this brief, late treatment, 17(R)-RvD1 attenuated fibrosis as determined by Masson’s trichrome staining, collagen mRNA, and lung tissue hydroxyproline content, an indirect measure of tissue collagen (Yatomi, et al., 2015). Co-administration of RvD1 with Boc-PLPLP, a peptide antagonist of the RvD1 receptor FPR2/ALX, reversed the effects, demonstrating that the actions of 17(R)-RvD1 in this model were receptor-dependent (Yatomi, et al., 2015). A more recent study also investigated 17(R)-RvD1 in the bleomycin model. The authors reported that administration of 17(R)-RvD1 7 and 10 days after bleomycin treatment significantly reduced inflammation and fibrosis, with reductions in both tissue and brochoalveolar lavage macrophages, neutrophils and inflammatory cytokines, along with reductions in tissue fibrosis, collagen accumulation, and expression of αSMA (Guilherme, et al., 2023). Interestingly, the authors also reported decreased production of extracellular vesicles by macrophages, and inhibition of angiogenesis, suggesting additional routes by which SPMs might inhibit pulmonary fibrosis.
As discussed above, analysis of human lung tissue found significantly lower levels of 8,9-EET, 11,12-EET and 14,15-EET in IPF patients. The EETs are produced from DHA via the action of cytochrome P450 hydrolase enzymes and are metabolized by endogenous soluble epoxide hydrolases (sEH). Soluble epoxide hydrolase inhibitors (sEHIs) have been developed by several pharmaceutical companies as potential therapies for hypertension, inflammation, metabolic syndrome and other indications (Shen & Hammock, 2012). sEHIs raise levels of endogenous EETs by blocking their metabolism, and have shown promise in pre-clinical animal models of lung inflammation (Podolin, et al., 2013; J. Yang, et al., 2015). Kim et al. reported that the sEHI 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) effectively attenuated bleomycin-induced fibrosis when given starting on day 0 (H. S. Kim, et al., 2021), while Tao et al. reported that TPPU inhibited bleomycin fibrosis when given starting on day 7, with reductions in lung collagen and αSMA-positive myofibroblasts (J. H. Tao, et al., 2022). This provides strong evidence that endogenous EETs have anti-fibrotic properties that can be enhanced by inhibiting their degradation.
Another group of eicosanoids with potent anti-fibrotic activity in vitro are the prostaglandins including PGE2 and 15dPGJ2 (reviewed in (K. Li, et al., 2021). PGE2 is normally considered a potent pro-inflammatory prostaglandin, but has been shown to inhibit lung fibroblast differentiation to myofibroblasts and production of ECM (Epa, et al., 2015; Kolodsick, et al., 2003; Mukherjee, et al., 2019). In healthy lung, one source for PGE2 is local production by epithelial cells, which supports the hypothesis that damaged lung epithelium either promotes fibrosis or permits fibrosis to occur via removal of anti-fibrotic checkpoints (Epa, et al., 2015). PGE2 is metabolized to an inactive form by the enzyme 15-PGDH. Bärnthaler et al. reported that lungs from IPF patients exhibited higher expression of 15-PGDH by immunostaining, and that treating human IPF precision cut lung slices with the 15-PGDH inhibitor SW033291 increased levels of PGE2 and decreased expression of collagen. SW033291 protected mice from bleomycin-induced fibrosis, with reductions in lung collagen, reduced apoptosis of AT-II cells, reduced proliferation of fibroblasts, and reduced accumulation of fibrocytes (Barnthaler, et al., 2020). Similar results were obtained in a mouse model of lung fibrosis induced by intravenous, rather than intratracheal, administration of bleomycin. SW033291 reduced fibrosis and collagen accumulation in lung tissue and preserved lung mechanical function (Smith, et al., 2020).
Limited investigations have been carried in out in other mouse models of pulmonary fibrosis. Apolipoprotein A1 (ApoA1) protected mice from an experimental silicosis model, with reductions in fibrotic nodules and TGFβ expression. ApoA1 has anti-inflammatory and anti-oxidative properties and was found to be reduced in lung tissue from IPF patients, but in the silicosis model it appeared to act by increasing LXA4 (E. Lee, et al., 2013). Mice exposed to a high dose of ionizing radiation delivered to the left lung developed fibrosis at 6 weeks, which was attenuated by daily administration of LXA4. The effect of LXA4 was again receptor-dependent, as it could be blocked by an FPR2/ALX antagonist (H. Kim, et al., 2020). Finally, it has been shown that mechanical ventilation can contribute to lung damage, and, when combined with other insults, can sometimes lead to abnormal repair in patients who are placed on mechanical ventilation for acute respiratory distress syndrome (ARDS) and other conditions. Yang et al. reported that using a mouse model of ventilation-induced fibrosis, treatment with RvD1 at 10–1000ng/mouse/day reduced lung fibrosis, lung collagen content, and expression of TGFβ, αSMA and vimentin, and that the effect of RvD1 was dependent on FRP2/ALX (Y. Yang, et al., 2019).
We also highlight here some key results from non-lung disease models that illustrate the therapeutic and translational potential of SPMs in fibrosing diseases. A fish oil diet or treatment with PDX reduced kidney fibrosis in a model of obesity-induced end stage renal disease (Perazza, et al., 2021), while RvD1 mitigated kidney fibrosis induced by unilateral ureteral obstruction (Y. B. Sun, et al., 2013). RvD1 also attenuated right atrial fibrosis in a rat model of pulmonary hypertension, with suppression of collagen and TGFβ expression and increased M2 macrophages (Hiram, et al., 2021). These models are interesting because they exhibit less classical inflammation than the bleomycin mouse model and support the concept that SPMs can promote resolution of fibrosis independent of inflammation.
5.2. SPM effects on fibroblasts and epithelial cells
As mentioned above, most animal models of pulmonary fibrosis exhibit some level of inflammation, and it is difficult to clearly determine whether the anti-fibrotic effects of SPMs are due to direct effects on the mechanisms of fibrosis or are indirect results of decreased inflammation. However, experiments in cultured lung fibroblasts and epithelial cells strongly support that SPMs have direct anti-fibrotic effects.
Several studies have reported that LXA4 inhibited differentiation of lung fibroblasts to myofibroblasts after stimulation with TGFβ or CTGF. These studies were performed in multiple cell types, including primary human lung fibroblasts from healthy donors and IPF patients, a cloned human lung fibroblast cell line, and the NIH3T3 mouse embryonic fibroblast cell line. LXA4 is reported to inhibit fibroblast differentiation, proliferation, migration, and activation of the key TGFβ -responsive transcription factor Smad2/3 via its receptor FPR2 (Herrera, et al., 2015; Roach, et al., 2015; Wu, et al., 2006; Zheng, et al., 2016). Similarly, the FPR2/ALX agonist BML-111 suppressed expression of αSMA in NIH3T3 fibroblasts (Ji, et al., 2018). RvD1 and RvD2 have also been reported to inhibit TGFβ-stimulated fibroblast proliferation and migration via the FPR2/ALX receptor (Herrera, et al., 2015; Zheng, et al., 2016; Zheng, et al., 2018) (Table 1).
Table 1.
Anti-fibrotic effects of SPMs and other bioactive lipids in vitro.
| Treatment (Concentration) | Cell Type | Effects | Reference |
|---|---|---|---|
| Lung fibroblasts | |||
| 11,12-EET 1000nM |
MRC-5 cell line, primary HLFs from IPF patients | Decreased TGFβ-stimulated differentiation, collagen production and Smad signaling. | (Kim, et al., 2021) |
| 14,15-EET 500–1000nM |
Primary murine fibroblasts and NIH3T3 fibroblasts | Inhibited TGFβ-stimulated differentiation, collagen expression, and Smad signaling. | (Tao, et al., 2022) |
| D/J series PGs NA* |
Primary HLFs | Activated HLFs produce D/J series PGs that inhibit myofibroblast differentiation of bystander HLFs. | (Lacy, et al., 2016; Lacy, et al., 2019) |
| LXA4 10nM |
HLF cell line | Inhibited CTGF-stimulated proliferation via STAT and pI3K pathways. | (Wu, et al., 2006) |
| LXA4 1–100nM |
Primary HLFs | Reduced TGFβ driven fibroblast proliferation, collagen production, and myofibroblast differentiation. Mediated via FPR2/ALX. | (Zheng, et al., 2016) |
| LXA4 1–10nM |
Primary HLFs from normal and IPF donors | Inhibited myofibroblast differentiation, collagen expression and stress fiber formation. | (Roach, et al., 2015) |
| PGE2 NA* |
Primary HLFs | Epithelial cells can inhibit HLF differentiation in co-culture via production of PGE2 | (Epa, et al., 2015) |
| PGE2 10–1000nM |
Primary HLFs | Inhibited TGFβ-promoted changes in Ca2+, inhibited expression of collagen and αSMA, inhibited stress fiber formation. | (Mukherjee, et al., 2019),(Kolodsick, et al., 2003) |
| RvD1 10–100nM |
Primary HLFs | Inhibited fibroblast proliferation, collagen production, and myofibroblast differentiation | (Zheng, et al., 2018) |
| Epithelial cells | |||
| LXA4 1–100nM |
Primary Human AT-II cells | Promoted proliferation, inhibited apoptosis, and promoted wound healing. | (Zheng, et al., 2016) |
| MaR1 0.1–100nM |
MLE-12 (immortalized mouse lung epithelial cell line) | Inhibited TGFβ driven EMT | (Wang, et al., 2015) |
| PDX 1–100nM |
Primary rat AT-II | Inhibited TGFβ driven EMT | (Li, et al., 2017) |
| RvD1 10–100nM |
Primary Human AT-II cells | Inhibited EMT in response to TGFβ. Promoted wound repair and proliferation. | (Zheng, et al., 2018) |
| RvD1 1–100nM |
BEAS-2B (immortalized bronchial epithelial cell line) | Reduced markers of EMT in cells exposed to acid and stretch (model of mechanical ventilation-induced fibrosis); dependent on FPR2/ALX. | (Yang, et al., 2019) |
| Non-lung in vitro results | |||
| 12-HETE, 15-HETE | Mouse dermal fibroblasts | Inhibited collagen production and MAP kinase activity | (Kronke, et al., 2012) |
| DHA 0–100μM |
Human dermal fibroblasts | Inhibited expression of αSMA and collagen | (Maeshige, et al., 2019) |
| LXA4 10–100nM |
NIH3T3 fibroblasts | Inhibited TGFβ-stimulated migration but did not alter expression of αSMA or collagen. | (Herrera, et al., 2015) |
| MaR1 1–100nM |
Mouse Glomerular mesangial cells (GMCs) | Reduced activation and expression of TGFβ and fibronectin | (Tang, et al., 2017) |
| RvD1 1–20nM |
HT-29 (immortalized human intestinal epithelial cell line) | Reduced EMT in HT-29 cells. HT-29 cells treated with RvD1 produced anti-fibrotic substances in conditioned medium that could inhibit myofibroblast differentiation of fibroblasts. | (Zeng, et al., 2022) |
| RvD2 10–100nM |
NIH3T3 fibroblasts | Inhibited TGFβ-stimulated migration but did not alter expression of αSMA or collagen | (Herrera, et al., 2015) |
| WKYMVm (FPR2 ligand) 1 mM |
Immortalized rat hepatic stellate cells | Reduced collagen and αSMA expression in TGFβ treated cells | (Jun, et al., 2021) |
NA, not available.
Abbreviations:
αSMA, α-smooth muscle actin
AT-II, alveolar type II epithelial cells
CTGF, connective tissue growth factor
EET, epoxyeicosatrienoic acid
EMT, epithelial-mesenchymal transformation
FPR2/ALX, formyl peptide receptor 2/lipoxin A4 receptor
HETE, hydroxyeicosatetraenoic acid
HLF, human lung fibroblast
IPF, idiopathic pulmonary fibrosis
LXA4, lipoxin A4
PG, prostaglandin
PG, prostaglandins
PGE2, prostaglandin E2
RvD1, resolvin D1
DHA, docosahexaenoic acid
In addition to PGE2 discussed above, prostaglandins in the D/J family, especially 15dPGJ2, have potent anti-fibrotic activity in vitro. While PGE2 acts via the E-prostaglandin receptors (EP1-EP4), 15dPGJ2 and other D/J PGs are ligands for the transcription factor peroxisome proliferator-activated receptor (PPAR)γ. There are several reports that PPARγ ligands block TGFβ-stimulated lung myofibroblast differentiation and function (Burgess, et al., 2005; Lacy, et al., 2016; Milam, et al., 2008). Highlighting the importance of bioactive lipids as autocoid factors, activated human lung fibroblasts (HLFs) first produce pro-inflammatory mediators, but undergo a class-switch to produce D/J series PGs that have potent anti-fibrotic activities on bystander HLFs (Lacy, et al., 2016; Lacy, et al., 2019). More recently, it was reported that 11,12-EET and 14,15-EET inhibited expression of αSMA, collagen, proliferation and Smad phosphorylation in mouse lung fibroblasts, also via PPARγ (H. S. Kim, et al., 2021; J. H. Tao, et al., 2022).
As mentioned, epithelial cells are a source of anti-fibrotic factors including PGE2, and it is hypothesized that epithelial damage can lead to lung fibrosis. The lung epithelium is also subject to epithelial-mesenchymal transition (EMT), in which epithelial cells are differentiated to mesenchymal-like cells that express fibroblast markers, become contractile, and produce excess ECM. This process is well-described in cell culture, although its contribution to fibrosis in vivo is contested. LXA4 and RvD1 were both shown to block EMT and promote proliferation and wound healing in human AT-II cells (Zheng, et al., 2016; Zheng, et al., 2018). RvD1 also inhibited expression of mesenchymal markers in bronchial epithelial cells subjected to cyclic stretching, a model of mechanical ventilation-induced lung fibrosis (Y. Yang, et al., 2019). These effects could be blocked by an FPR2/ALX antagonist, demonstrating the effects were receptor-dependent. Additionally, it was recently reported that MaR1 could inhibit TGFβ-stimulated EMT of the MLE-12 mouse alveolar epithelial cell line, and PDX inhibited EMT in primary rat AT-II cells (H. Li, et al., 2017; Y. Wang, et al., 2015).
Vascular smooth muscle cells (VSMCs) contribute to the pathology of pulmonary fibrosis via vascular remodeling leading to pulmonary hypertension. Although studies of the effect of SPMs on VSMCs have mainly investigated acute vascular injury models, some findings may be extrapolated to suggest that SPMs will have beneficial effects in pulmonary fibrosis that are mediated through VSMCs as well as fibroblasts and epithelial cells. VSMCs can synthesize SPMs including D-series resolvins and protectins and express the FPR2/ALX, DRV1/GPR32 and ERV1/ChemR23 receptors (Chatterjee, et al., 2017; Ho, et al., 2010). EPA, LXA4, RvD1, RvD2, and RvE1 have each been reported to inhibit VSMC proliferation, migration, and contractility in a receptor-dependent manner (Akagi, et al., 2015; Artiach, et al., 2018; Hiram, et al., 2015; Hiram, et al., 2014; Ho, et al., 2010; Kurahara, et al., 2020; G. Liu, et al., 2018; Miyahara, et al., 2013; Mottola, et al., 2017). MaR1 also inhibited migration and proliferation of rat pulmonary arterial smooth muscle cells in vitro in a receptor (LGR6) dependent manner (H. Li, et al., 2022).
Lastly, we will briefly summarize studies on SPMs in fibroblasts from other organs, as they demonstrate that many of the signaling mechanisms that promote or attenuate fibrotic phenotypes are shared among fibroblasts from different sources. RvD1 inhibited activation and proliferation of kidney and periodontal ligament fibroblasts (Y. B. Sun, et al., 2013; Zarrough, et al., 2022), and also blocked EMT of intestinal epithelial cells in a model of intestinal fibrosis (Zeng, et al., 2022). LXA4 inhibited EMT in a human kidney epithelial cell line (Wu, et al., 2010). MaR1 reduced activation and expression of TGFβ and the ECM protein fibronectin in glomerular mesangial cells, a type of mesenchymal stromal cell with properties similar to fibroblasts (Tang, et al., 2017). A novel peptide ligand of FPR2/ALX reduced collagen expression in rat hepatic stellate cells (ECM producing cells in the fibroblast lineage) and attenuated liver fibrosis in a rat model of cirrhosis (Jun, et al., 2021). Similar to the relationship between epithelial cells and fibroblasts, natural killer (NK) cells exhibit anti-fibrotic properties in liver fibrosis and inhibit activation of hepatic stellate cells. Inhibition of E-prostanoid 3 receptor (EP3), the PGE2 receptor, reduced cytotoxic activity of NK cells toward hepatic stellate cells in vitro and aggravated liver fibrosis in a mouse model, strongly supporting an anti-fibrotic role for PGE2 (X. Tao, et al., 2022).
6. Lessons learned from other chronic lung diseases
The interstitial lung diseases are characterized by parenchymal or interstitial fibrosis, that is, scarring, thickening and ECM accumulation in the interstitial spaces between alveoli, eventually resulting in destruction of the gas exchange surface. Several other lung diseases include fibrosis of the small airways, rather than the parenchyma, resulting in airway narrowing that contributes to disease pathology. Here, we will briefly discuss evidence for the use of SPMs in moderating small airways fibrosis.
The pathologies of chronic obstructive pulmonary disease (COPD) include emphysema (airspace enlargement), chronic bronchitis, and small airway fibrosis (Barnes, 2019). Although there has been considerable research into the use of SPMs to promote resolution of chronic inflammation in COPD (reviewed in (Thatcher, et al., 2019), the most common rodent models of COPD do not develop small airways fibrosis, and thus, studies to date are silent on whether SPMs can prevent or resolve small airways fibrosis in COPD, and a new experimental approach is needed to address the question.
Asthma, although predominantly an immune disorder, also has a component of small airways fibrosis, and SPMs have been extensively studied in multiple asthma models (reviewed in (Cagnina, et al., 2022)). However, little work has directly addressed bioactive lipids and small airways fibrosis. The leukotriene receptor inhibitors pranlukast and montelukast inhibited experimental allergic airway disease in a mouse model of asthma, including reducing expression of αSMA and collagen genes, collagen accumulation in lung tissue, and TGFβ signaling (Hur, et al., 2018; Shin, et al., 2013). A study by Abreu et al. examined the effect of mesenchymal stromal cell therapy on a model of house dust mite-induced asthma in mice. Stromal cells delivered to the lungs after induction of asthma were more effective at reducing asthma phenotypes when they were pretreated with EPA. EPA-treated stromal cells produced RvD1 and reduced lung expression of TGFβ and collagen, and reduced airways fibrosis (Abreu, et al., 2018). In a more recent study, administration of LXA4 reduced deposition of collagen and gene expression of collagen and αSMA in the mouse ovalbumin asthma model (Y. Liu, et al., 2021). However, since the LXA4 was administered before the antigenic challenge, this study demonstrates a preventative effect rather than a pro-resolving or treatment effect. Previous studies have reported that arginase-2 expression in human lung fibroblasts is associated with airway remodeling in asthma. Duggirala et al. reported that EPA and a combination of EPA and DHA reduced expression of arginase-2 in primary lung fibroblasts from asthma patients, but DHA alone or RvD1 did not, providing mixed evidence for the role of SPMs in asthma airway remodeling in human patients (Duggirala, et al., 2022).
Cystic fibrosis (CF) is a genetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator gene. The resulting in disruptions in sodium, chloride, and bicarbonate ion transport lead to fluid imbalance in the lung, reduced mucociliary clearance, and chronic pathogen colonization, resulting in chronic epithelial injury and epithelial remodeling. SPMs have been extensively studied in CF in the context of promoting resolution of chronic inflammation and improving bacterial clearance (Briottet, et al., 2020). However, little attention has been paid specifically to airway remodeling. There are reports that lung fibroblasts from CF patients exhibit elevated production of TGFβ, collagen deposition and myofibroblast differentiation, although investigations of SPM effects on CF lung fibroblasts have not been reported (Harris, et al., 2013; Huaux, et al., 2013). It is hypothesized that much of the airway remodeling in CF derives from repeated injury to epithelial cells resulting in EMT (Rout-Pitt, et al., 2018). LXA4 promoted markers of epithelial repair including ion channel activation, proliferation, migration, and wound repair in CF and non-CF human airway epithelial cells in an FPR2/ALX dependent manner (Buchanan, et al., 2013). LXA4 also prevented disruption of epithelial tight junctions by Pseudomonas aeruginosa in CF and non-CF bronchial epithelial cell lines (Higgins, et al., 2016).
7. Conclusion and future directions.
Interstitial lung diseases represent a chronic wound repair process that has failed to resolve and become pathogenic, leading to accumulation of fibrotic tissue in the lung parenchyma. Although some ILDs including sarcoidosis and CT-ILD have an immune/inflammatory pathology, the role of inflammation in IPF is unclear. SPMs have shown clear anti-fibrotic effects in lung fibroblasts, epithelial cells and mouse models. However, the mouse lung fibrosis models used to date have significant inflammatory components, making it difficult to separate anti-inflammatory from anti-fibrotic effects. One way to address this problem would be to use the bleomycin mouse model but focus on late treatment, beginning on day 7 or later. While this does not completely bypass the inflammatory response, it focuses more on the fibrotic response than treatments begun earlier. Another approach would be to use a less inflammatory based mouse model of IPF. One such model is transient overexpression of TGFβ in lung tissue by a recombinant adenovirus (Sime, et al., 1997; Warshamana, et al., 2002). Administration of a recombinant adenovirus expressing constitutively active TGFβ results in transient inflammation and expression of TGFβ lasting about 7–10 days, with progressive fibrosis developing between days 21–42. SPMs could be administered after day 7, bypassing the immune reaction to the adenovirus particles. Genetic mouse models of familial IPF that involve inducible epithelial damage may also have less of a classical inflammatory component (Yasutomo, 2021).
On the other hand, perhaps it is not important if we can’t separate the anti-inflammatory effects of SPMs from their pro-resolving and pro-repair functions. ILDs have complex pathologies and some of them have a clear inflammatory component, so it may be less important to determine if the exact mechanism of action of SPMs in ILD is primarily anti-inflammatory or primarily anti-fibrotic, as long as they work. When fibrosis is preceded or accompanied by an inflammatory or immune response, treating the inflammation or immune condition with SPMs could be expected to also reduce concomitant fibrosis, even if SPMs do not act directly on fibroblasts or myofibroblasts. This approach might be particularly useful in HP, autoimmune CT-ILDs, and small airway fibrosis in asthma.
There are several distinct approaches to translating SPM research into new clinical therapies. First are the SPMs themselves. As they are endogenous produced, SPM-based therapies are expected to be well-tolerated. SPMs are easily derivatized to improve their pharmacokinetics and increase their target specificity, and they are active in the nanomolar range, which is achievable with typical routes of administration. The second approach involves directly targeting the GPCRs that carry out SPM signaling. GPCRs are the largest target for drug development. GPCRs are highly sensitive to the structures of the ligands that bind them, as illustrated by the ability of FPR2/ALX and BLT1 to have pro-inflammatory or pro-resolving effects depending on the ligand and context (Merlin, et al., 2022). The third approach is illustrated by the development of soluble epoxide hydrolase inhibitors (sEHIs) which block the action of endogenous and pathogen-derived sEHs to metabolize and inactivate anti-inflammatory EETs and HETEs (Guan, et al., 2021). sEHIs have anti-inflammatory effects in several cell culture and animal models, and anti-fibrotic effects in the mouse bleomycin model. There are other endogenous enzymes that metabolize SPMs, including eicosanoid oxidoreductases, and the promise offered by sEHIs suggests that developing inhibitors of eicosanoid oxidoreductase could also be a fruitful approach. Specialized pro-resolving mediators and other bioactive lipids represent a target rich environment for novel anti-fibrotic therapies.
Acknowledgements:
The authors wish to thank Drs. Apostolos Perelas and Raghavendra Pillappa for lung images. This work was supported in part by NIH grants R01HL127001 and R01HL133761, and by the Chandler-Pollock-Solimano-Thomas Pulmonary Fibrosis Research Fund. MTF is a recipient of a Pulmonary Fibrosis Foundation Scholar award and is a recipient of an NIH Fellowship F32HL154525.
Abbreviations
- 15-PGDH
15-hydroxyprostaglandin dehydrogenase, also known as eicosanoid oxidoreductase or EOR
- αSMA
α-smooth muscle actin
- AT-II
alveolar type II cells
- BLT1
leukotriene B4 receptor
- CF
cystic fibrosis
- COPD
Chronic Obstructive Pulmonary Disease
- COX2
cyclooxygenase 2
- CT
connective tissue
- CTGF
connective tissue growth factor
- DHA
docosahexaenoic acid
- DRV
D resolvin receptor
- ECM
extracellular matrix
- EET
epoxyeicosatrienoic acid
- EMT
epithelial-mesenchymal transition
- EP3
E-prostanoid 3 receptor
- EPA
eicosapentaenoic acid
- ERV
E resolvin receptor
- FGF
fibroblast growth factor
- FPR2/ALX
formyl peptide receptor 2/lipoxin A4 receptor
- GPCR
G-protein coupled receptor
- HETE
hydroxyeicosatetraenoic acid
- HLF
human lung fibroblast
- HP
hypersensitivity pneumonitis
- IGF
insulin-like growth factor
- ILD
interstitial lung disease
- IPF
Idiopathic pulmonary fibrosis
- LO
lipooxygenase
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- LXA4
lipoxin A4
- MaR1
maresin 1
- MCTR
maresin conjugate of tissue regeneration
- NK
natural killer cell
- PPARγ
Peroxisome proliferator-activated receptor γ
- PCTR
protectin conjugate of tissue regeneration
- PD1
protectin D1
- PDGF
platelet-derived growth factor
- PDX
protectin DX
- PG
prostaglandin
- PGE2
prostaglandin E2
- RCTR
resolvin conjugate of tissue regeneration
- RvD1
resolvin D1
- RvD2
resolvin D2
- RvE1
resolvin E1
- sEH
soluble epoxide hydrolase
- sEHI
soluble epoxide hydrolase inhibitor
- SPM
specialized pro-resolving lipid mediator
- TGFβ
transforming growth factor-β
- VSMC
vascular smooth muscle cell
Footnotes
Conflicts of interest:
Dr. Sime is a paid consultant for Boehringer Ingelheim, UCB and Three Lakes Foundation. She serves on the board member for the American Thoracic Society, the scientific council of the Parker Francis Foundation, an advisory Board for Galecto and the medical scientific advisory committee for the Pulmonary Fibrosis Foundation. She receives grant funding from the NIH. Dr. Sime holds patents related to fibrosis. The other authors have no financial interests to disclose.
References Cited
- Abdalla M, Sabbineni H, Prakash R, Ergul A, Fagan SC, & Somanath PR (2015). The Akt inhibitor, triciribine, ameliorates chronic hypoxia-induced vascular pruning and TGFbeta-induced pulmonary fibrosis. Br J Pharmacol, 172, 4173–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu SC, Lopes-Pacheco M, da Silva AL, Xisto DG, de Oliveira TB, Kitoko JZ, de Castro LL, Amorim NR, Martins V, Silva LHA, Goncalves-de-Albuquerque CF, de Castro Faria-Neto HC, Olsen PC, Weiss DJ, Morales MM, Diaz BL, & Rocco PRM (2018). Eicosapentaenoic Acid Enhances the Effects of Mesenchymal Stromal Cell Therapy in Experimental Allergic Asthma. Front Immunol, 9, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi D, Chen M, Toy R, Chatterjee A, & Conte MS (2015). Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J, 29, 2504–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artiach G, Carracedo M, Claria J, Laguna-Fernandez A, & Back M (2018). Opposing Effects on Vascular Smooth Muscle Cell Proliferation and Macrophage-induced Inflammation Reveal a Protective Role for the Proresolving Lipid Mediator Receptor ChemR23 in Intimal Hyperplasia. Front Pharmacol, 9, 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley SL, Wilke CA, Kim KK, & Moore BB (2017). Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal Immunol, 10, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2019). Small airway fibrosis in COPD. Int J Biochem Cell Biol, 116, 105598. [DOI] [PubMed] [Google Scholar]
- Barnthaler T, Theiler A, Zabini D, Trautmann S, Stacher-Priehse E, Lanz I, Klepetko W, Sinn K, Flick H, Scheidl S, Thomas D, Olschewski H, Kwapiszewska G, Schuligoi R, & Heinemann A (2020). Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J Allergy Clin Immunol, 145, 818–833 e811. [DOI] [PubMed] [Google Scholar]
- Barratt S, & Millar A (2014). Vascular remodelling in the pathogenesis of idiopathic pulmonary fibrosis. QJM, 107, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil MC, & Levy BD (2016). Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol, 16, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, Hammerl P, Koschel D, Blaas S, Wirtz H, Ficker JH, Neumeister W, Schonfeld N, Claussen M, Kneidinger N, Frankenberger M, Hummler S, Kahn N, Tello S, Freise J, Welte T, Neuser P, Gunther A, & investigators R (2021). Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med, 9, 476–486. [DOI] [PubMed] [Google Scholar]
- Bhat TA, Kalathil SG, Bogner PN, Lehmann PV, Thatcher TH, Sime PJ, & Thanavala Y (2021). AT-RvD1 Mitigates Secondhand Smoke-Exacerbated Pulmonary Inflammation and Restores Secondhand Smoke-Suppressed Antibacterial Immunity. J Immunol, 206, 1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A, Dobaczewski M, & Frangogiannis NG (2011). TGF-beta signaling in fibrosis. Growth Factors, 29, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerner EB, Cuyas M, Theegarten D, Ohshimo S, Costabel U, & Bonella F (2020). Azathioprine for Connective Tissue Disease-Associated Interstitial Lung Disease. Respiration, 99, 628–636. [DOI] [PubMed] [Google Scholar]
- Borie R, & Crestani B (2019). Familial pulmonary fibrosis: a world without frontiers. J Bras Pneumol, 45, e20190303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveda-Ruiz D, D’Alessandro-Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y, Matsumoto T, Kobayashi T, Gil-Bernabe P, Chelakkot-Govindalayathil AL, Miyake Y, Yasukawa A, Morser J, Taguchi O, & Gabazza EC (2013). Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology, 218, 245–254. [DOI] [PubMed] [Google Scholar]
- Briottet M, Shum M, & Urbach V (2020). The Role of Specialized Pro-Resolving Mediators in Cystic Fibrosis Airways Disease. Front Pharmacol, 11, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan PJ, McNally P, Harvey BJ, & Urbach V (2013). Lipoxin A(4)-mediated KATP potassium channel activation results in cystic fibrosis airway epithelial repair. Am J Physiol Lung Cell Mol Physiol, 305, L193–201. [DOI] [PubMed] [Google Scholar]
- Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, Phipps RP, & Sime PJ (2005). PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol, 288, L1146–1153. [DOI] [PubMed] [Google Scholar]
- Cagnina RE, Duvall MG, Nijmeh J, & Levy BD (2022). Specialized pro-resolving mediators in respiratory diseases. Curr Opin Clin Nutr Metab Care, 25, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra V, Rovati GE, Mangano P, Buccellati C, Murphy RC, & Sala A (2015). Transcellular biosynthesis of eicosanoid lipid mediators. Biochim Biophys Acta, 1851, 377–382. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Komshian S, Sansbury BE, Wu B, Mottola G, Chen M, Spite M, & Conte MS (2017). Biosynthesis of proresolving lipid mediators by vascular cells and tissues. FASEB J, 31, 3393–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, Schnapp A, & Park JE (2007). Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J, 29, 976–985. [DOI] [PubMed] [Google Scholar]
- Chiang N, & Serhan CN (2017). Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med, 58, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, & Serhan CN (2020). Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem, 64, 443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claria J, Nguyen BT, Madenci AL, Ozaki CK, & Serhan CN (2013). Diversity of lipid mediators in human adipose tissue depots. Am J Physiol Cell Physiol, 304, C1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codagnone M, Cianci E, Lamolinara A, Mari VC, Nespoli A, Isopi E, Mattoscio D, Arita M, Bragonzi A, Iezzi M, Romano M, & Recchiuti A (2018). Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol, 11, 35–49. [DOI] [PubMed] [Google Scholar]
- Colas RA, Shinohara M, Dalli J, Chiang N, & Serhan CN (2014). Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol, 307, C39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas DB, Zumwalt RE, Black WC, & Sobonya RE (1994). The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med, 150, 967–972. [DOI] [PubMed] [Google Scholar]
- Croasdell A, Lacy SH, Thatcher TH, Sime PJ, & Phipps RP (2016). Resolvin D1 Dampens Pulmonary Inflammation and Promotes Clearance of Nontypeable Haemophilus influenzae. J Immunol, 196, 2742–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Ramon S, Norris PC, Colas RA, & Serhan CN (2015). Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J, 29, 2120–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Sanger JM, Rodriguez AR, Chiang N, Spur BW, & Serhan CN (2016). Identification and Actions of a Novel Third Maresin Conjugate in Tissue Regeneration: MCTR3. PLoS One, 11, e0149319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, Sanyal S, Brillet PY, Brauner M, Kambouchner M, Huynh S, Naccache JM, Borie R, Piquet J, Mekinian A, Virally J, Uzunhan Y, Cadranel J, Crestani B, Fain O, Lhote F, Dhote R, Saidenberg-Kermanac’h N, Rosental PA, Valeyre D, & Nunes H (2017). Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J, 50. [DOI] [PubMed] [Google Scholar]
- Duggirala VK, Geary K, Hasenmayer D, & Daghigh F (2022). Modulation of Arginase-2 mRNA Levels by omega-3 PUFAs and Aspirin in Asthmatic Human Lung Fibroblasts. J Lipids, 2022, 3062274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvall MG, & Levy BD (2016). DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur J Pharmacol, 785, 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epa AP, Thatcher TH, Pollock SJ, Wahl LA, Lyda E, Kottmann RM, Phipps RP, & Sime PJ (2015). Normal Human Lung Epithelial Cells Inhibit Transforming Growth Factor-beta Induced Myofibroblast Differentiation via Prostaglandin E2. PLoS One, 10, e0135266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas L, Farkas D, Ask K, Moller A, Gauldie J, Margetts P, Inman M, & Kolb M (2009). VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest, 119, 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrand E, Vittinghoff E, Ley B, Butte AJ, & Collard HR (2020). Corticosteroid use is not associated with improved outcomes in acute exacerbation of IPF. Respirology, 25, 629–635. [DOI] [PubMed] [Google Scholar]
- Fernandez BA, Fox G, Bhatia R, Sala E, Noble B, Denic N, Fernandez D, Duguid N, Dohey A, Kamel F, Edwards L, Mahoney K, Stuckless S, Parfrey PS, & Woods MO (2012). A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res, 13, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis N (2020). Transforming growth factor-beta in tissue fibrosis. J Exp Med, 217, e20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdino de Souza D, Santos DS, Simon KS, Morais JAV, Coelho LC, Pacheco TJA, Azevedo RB, Bocca AL, Melo-Silva CA, & Longo JPF (2022). Fish Oil Nanoemulsion Supplementation Attenuates Bleomycin-Induced Pulmonary Fibrosis BALB/c Mice. Nanomaterials (Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, Navarro C, Perez-Padilla R, Vargas MH, Loyd JE, & Selman M (2011). Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med, 105, 1902–1907. [DOI] [PubMed] [Google Scholar]
- Giera M, Ioan-Facsinay A, Toes R, Gao F, Dalli J, Deelder AM, Serhan CN, & Mayboroda OA (2012). Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC-MS/MS. Biochim Biophys Acta, 1821, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XX, Rao DN, Liu YZ, Zhou Y, & Yang HH (2021). Epoxyeicosatrienoic Acids and Fibrosis: Recent Insights for the Novel Therapeutic Strategies. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme RF, Silva J, Waclawiack I, Fraga-Junior VS, Nogueira TO, Pecli C, Araujo-Silva CA, Magalhaes NS, Lemos FS, Bulant CA, Blanco PJ, Serra R, Svensjo E, Scharfstein J, Moraes JA, Canetti C, & Benjamim CF (2023). Pleiotropic antifibrotic actions of aspirin-triggered resolvin D1 in the lungs. Front Immunol, 14, 886601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme RF, Xisto DG, Kunkel SL, Freire-de-Lima CG, Rocco PR, Neves JS, Fierro IM, Canetti C, & Benjamim CF (2013). Pulmonary antifibrotic mechanisms aspirin-triggered lipoxin A(4) synthetic analog. Am J Respir Cell Mol Biol, 49, 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WT, Kelly DR, Zhou Y, Wang D, MacEwen M, Hagood JS, Clancy JP, Ambalavanan N, & Sorscher EJ (2013). Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One, 8, e70196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Cheng N, Gao WW, Zhang M, Zhang YY, Ye RD, & Wang MW (2011). Characterization of Quin-C1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol Sin, 32, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera BS, Kantarci A, Zarrough A, Hasturk H, Leung KP, & Van Dyke TE (2015). LXA4 actions direct fibroblast function and wound closure. Biochem Biophys Res Commun, 464, 1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins G, Fustero Torre C, Tyrrell J, McNally P, Harvey BJ, & Urbach V (2016). Lipoxin A4 prevents tight junction disruption and delays the colonization of cystic fibrosis bronchial epithelial cells by Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol, 310, L1053–1061. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, & Rettig WJ (2008). BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res, 68, 4774–4782. [DOI] [PubMed] [Google Scholar]
- Hiram R, Rizcallah E, Marouan S, Sirois C, Sirois M, Morin C, Fortin S, & Rousseau E (2015). Resolvin E1 normalizes contractility, Ca2+ sensitivity and smooth muscle cell migration rate in TNF-alpha- and IL-6-pretreated human pulmonary arteries. Am J Physiol Lung Cell Mol Physiol, 309, L776–788. [DOI] [PubMed] [Google Scholar]
- Hiram R, Rizcallah E, Sirois C, Sirois M, Morin C, Fortin S, & Rousseau E (2014). Resolvin D1 reverses reactivity and Ca2+ sensitivity induced by ET-1, TNF-alpha, and IL-6 in the human pulmonary artery. Am J Physiol Heart Circ Physiol, 307, H1547–1558. [DOI] [PubMed] [Google Scholar]
- Hiram R, Xiong F, Naud P, Xiao J, Sirois M, Tanguay JF, Tardif JC, & Nattel S (2021). The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc Res, 117, 1776–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R, Creager MA, Serhan CN, & Conte MS (2010). Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol, 177, 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao HM, Thatcher TH, Levy EP, Fulton RA, Owens KM, Phipps RP, & Sime PJ (2014). Resolvin D1 attenuates polyinosinic-polycytidylic acid-induced inflammatory signaling in human airway epithelial cells via TAK1. J Immunol, 193, 4980–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huaux F, Noel S, Dhooghe B, Panin N, Lo Re S, Lison D, Wallemacq P, Marbaix E, Scholte BJ, Lebecque P, & Leal T (2013). Dysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosis. PLoS One, 8, e64341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Kang JY, Rhee CK, Kim YK, & Lee SY (2018). The leukotriene receptor antagonist pranlukast attenuates airway remodeling by suppressing TGF-beta signaling. Pulm Pharmacol Ther, 48, 5–14. [DOI] [PubMed] [Google Scholar]
- Hyldgaard C, Hilberg O, Muller A, & Bendstrup E (2014). A cohort study of interstitial lung diseases in central Denmark. Respir Med, 108, 793–799. [DOI] [PubMed] [Google Scholar]
- Idiopathic Pulmonary Fibrosis Clinical Research N, Raghu G, Anstrom KJ, King TE Jr., Lasky JA, & Martinez FJ (2012). Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med, 366, 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isopi E, Mattoscio D, Codagnone M, Mari VC, Lamolinara A, Patruno S, D’Aurora M, Cianci E, Nespoli A, Franchi S, Gatta V, Dubourdeau M, Moretti P, Di Sabatino M, Iezzi M, Romano M, & Recchiuti A (2020). Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front Immunol, 11, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan N, Smith RA, & Sathananthan M (2021). Mortality Trends of Idiopathic Pulmonary Fibrosis in the United States From 2004 Through 2017. Chest, 159, 228–238. [DOI] [PubMed] [Google Scholar]
- Ji YD, Luo ZL, Chen CX, Li B, Gong J, Wang YX, Chen L, Yao SL, & Shang Y (2018). BML-111 suppresses TGF-beta1-induced lung fi broblast activation in vitro and decreases experimental pulmonary fibrosis in vivo. Int J Mol Med, 42, 3083–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvene CC, Shay AE, Soens MA, Norris PC, Haeggstrom JZ, & Serhan CN (2019). Biosynthetic metabolomes of cysteinyl-containing immunoresolvents. FASEB J, 33, 13794–13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JH, Park SY, Park S, Park HJ, Kim JY, Park GT, Bae SH, Kim JH, & Kim GJ (2021). Formyl Peptide Receptor 2 Alleviates Hepatic Fibrosis in Liver Cirrhosis by Vascular Remodeling. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman NS, Hugo GD, Kahn JM, Zhao SS, Jan N, Mahon RN, & Weiss E (2018). Interobserver reliability in describing radiographic lung changes after stereotactic body radiation therapy. Adv Radiat Oncol, 3, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane MP, Belperio JA, Burdick MD, & Strieter RM (2001). IL-12 attenuates bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol, 281, L92–97. [DOI] [PubMed] [Google Scholar]
- Kendall RT, & Feghali-Bostwick CA (2014). Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol, 5, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JI Jr., Chandler DB, Fulmer JD, Wert MB, & Grizzle WE (1989). Dietary fish oil inhibits bleomycin-induced pulmonary fibrosis in the rat. Exp Lung Res, 15, 315–329. [DOI] [PubMed] [Google Scholar]
- Kim H, Park SH, Han SY, Lee YS, Cho J, & Kim JM (2020). LXA(4)-FPR2 signaling regulates radiation-induced pulmonary fibrosis via crosstalk with TGF-beta/Smad signaling. Cell Death Dis, 11, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Moon SJ, Lee SE, Hwang GW, Yoo HJ, & Song JW (2021). The arachidonic acid metabolite 11,12-epoxyeicosatrienoic acid alleviates pulmonary fibrosis. Exp Mol Med, 53, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolahian S, Fernandez IE, Eickelberg O, & Hartl D (2016). Immune Mechanisms in Pulmonary Fibrosis. Am J Respir Cell Mol Biol, 55, 309–322. [DOI] [PubMed] [Google Scholar]
- Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, & Moore BB (2003). Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol, 29, 537–544. [DOI] [PubMed] [Google Scholar]
- Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, Nathan SD, Grant G, Phipps RP, & Sime PJ (2012). Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med, 186, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal-Bielecka O, Kowal K, Distler O, Rojewska J, Bodzenta-Lukaszyk A, Michel BA, Gay RE, Gay S, & Sierakowski S (2005). Cyclooxygenase- and lipoxygenase-derived eicosanoids in bronchoalveolar lavage fluid from patients with scleroderma lung disease: an imbalance between proinflammatory and antiinflammatory lipid mediators. Arthritis Rheum, 52, 3783–3791. [DOI] [PubMed] [Google Scholar]
- Kronke G, Reich N, Scholtysek C, Akhmetshina A, Uderhardt S, Zerr P, Palumbo K, Lang V, Dees C, Distler O, Schett G, & Distler JH (2012). The 12/15-lipoxygenase pathway counteracts fibroblast activation and experimental fibrosis. Ann Rheum Dis, 71, 1081–1087. [DOI] [PubMed] [Google Scholar]
- Kurahara LH, Hiraishi K, Yamamura A, Zhang Y, Abe K, Yahiro E, Aoki M, Koga K, Yokomise H, Go T, Ishikawa K, Bo Z, Kishi H, Kobayashi S, Aoki-Shoi N, Toru S, Inoue R, & Hirano K (2020). Eicosapentaenoic acid ameliorates pulmonary hypertension via inhibition of tyrosine kinase Fyn. J Mol Cell Cardiol, 148, 50–62. [DOI] [PubMed] [Google Scholar]
- Lacy SH, Woeller CF, Thatcher TH, Maddipati KR, Honn KV, Sime PJ, & Phipps RP (2016). Human lung fibroblasts produce proresolving peroxisome proliferator-activated receptor-gamma ligands in a cyclooxygenase-2-dependent manner. Am J Physiol Lung Cell Mol Physiol, 311, L855–L867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy SH, Woeller CF, Thatcher TH, Pollock SJ, Small EM, Sime PJ, & Phipps RP (2019). Activated Human Lung Fibroblasts Produce Extracellular Vesicles with Antifibrotic Prostaglandins. Am J Respir Cell Mol Biol, 60, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lee EJ, Kim H, Jang A, Koh E, Uh ST, Kim Y, Park SW, & Park CS (2013). Overexpression of apolipoprotein A1 in the lung abrogates fibrosis in experimental silicosis. PLoS One, 8, e55827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Park SJ, Kim SR, Min KH, Lee KY, Choe YH, Hong SH, Lee YR, Kim JS, Hong SJ, & Lee YC (2008). Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur Respir J, 31, 523–531. [DOI] [PubMed] [Google Scholar]
- Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, Israel E, Haley KJ, & Serhan CN (2007). Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol, 178, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hao Y, Zhang H, Ying W, Li D, Ge Y, Ying B, Cheng B, Lian Q, & Jin S (2017). Posttreatment with Protectin DX ameliorates bleomycin-induced pulmonary fibrosis and lung dysfunction in mice. Sci Rep, 7, 46754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li X, Hao Y, Wu C, Fu Y, Su N, Chen H, Ying B, Wang H, Su L, Cai H, He Q, Cai M, Sun J, Lin J, Scott A, Smith F, Huang X, & Jin S (2022). Maresin 1 intervention reverses experimental pulmonary arterial hypertension in mice. Br J Pharmacol, 179, 5132–5147. [DOI] [PubMed] [Google Scholar]
- Li K, Zhao J, Wang M, Niu L, Wang Y, Li Y, & Zheng Y (2021). The Roles of Various Prostaglandins in Fibrosis: A Review. Biomolecules, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Gong Y, Zhang R, Piao L, Li X, Liu Q, Yan S, Shen Y, Guo S, Zhu M, Yin H, Funk CD, Zhang J, & Yu Y (2018). Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J, 32, 5413–5425. [DOI] [PubMed] [Google Scholar]
- Liu T, De Los Santos FG, & Phan SH (2017). The Bleomycin Model of Pulmonary Fibrosis. Methods Mol Biol, 1627, 27–42. [DOI] [PubMed] [Google Scholar]
- Liu T, Gonzalez De Los Santos F, Zhao Y, Wu Z, Rinke AE, Kim KK, & Phan SH (2019). Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J Biol Chem, 294, 8861–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wei L, He C, Chen R, & Meng L (2021). Lipoxin A4 inhibits ovalbumin-induced airway inflammation and airway remodeling in a mouse model of asthma. Chem Biol Interact, 349, 109660. [DOI] [PubMed] [Google Scholar]
- Maeshige N, Torii K, Tabuchi H, Imai M, Koga Y, Uemura M, Aoyama-Ishikawa M, Miyoshi M, Fujino H, Terashi H, & Usami M (2019). Inhibitory Effects of Short-Chain Fatty Acids and omega-3 Polyunsaturated Fatty Acids on Profibrotic Factors in Dermal Fibroblasts. Eplasty, 19, e4. [PMC free article] [PubMed] [Google Scholar]
- Marshall DC, Salciccioli JD, Shea BS, & Akuthota P (2018). Trends in mortality from idiopathic pulmonary fibrosis in the European Union: an observational study of the WHO mortality database from 2001–2013. Eur Respir J, 51. [DOI] [PubMed] [Google Scholar]
- Martins V, Valenca SS, Farias-Filho FA, Molinaro R, Simoes RL, Ferreira TP, e Silva PM, Hogaboam CM, Kunkel SL, Fierro IM, Canetti C, & Benjamim CF (2009). ATLa, an aspirin-triggered lipoxin A4 synthetic analog, prevents the inflammatory and fibrotic effects of bleomycin-induced pulmonary fibrosis. J Immunol, 182, 5374–5381. [DOI] [PubMed] [Google Scholar]
- Mas E, Croft KD, Zahra P, Barden A, & Mori TA (2012). Resolvins D1, D2, and other mediators of self-limited resolution of inflammation in human blood following n-3 fatty acid supplementation. Clin Chem, 58, 1476–1484. [DOI] [PubMed] [Google Scholar]
- Merlin J, Park J, Vandekolk TH, Fabb SA, Allinne J, Summers RJ, Langmead CJ, & Riddy DM (2022). Multipathway In Vitro Pharmacological Characterization of Specialized Proresolving G Protein-Coupled Receptors. Mol Pharmacol, 101, 246–256. [DOI] [PubMed] [Google Scholar]
- Meyer KC (2017). Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med, 11, 343–359. [DOI] [PubMed] [Google Scholar]
- Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, Standiford TJ, Thannickal VJ, & Reddy RC (2008). PPAR-{gamma} agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol, 294, L891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, & Perlman H (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med, 214, 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara T, Runge S, Chatterjee A, Chen M, Mottola G, Fitzgerald JM, Serhan CN, & Conte MS (2013). D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J, 27, 2220–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset J, Johannson KA, Vittinghoff E, Aravena C, Elicker BM, Jones KD, Fell CD, Manganas H, Dube BP, Wolters PJ, Collard HR, Ryerson CJ, & Ley B (2017). Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest, 151, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola G, Chatterjee A, Wu B, Chen M, & Conte MS (2017). Aspirin-triggered resolvin D1 attenuates PDGF-induced vascular smooth muscle cell migration via the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway. PLoS One, 12, e0174936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Sheng W, Michkov A, Sriarm K, Sun R, Dvorkin-Gheva A, Insel PA, & Janssen LJ (2019). Prostaglandin E(2) inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca(2+) signaling. Am J Physiol Lung Cell Mol Physiol, 316, L810–L821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, & Sheppard D (1999). The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell, 96, 319–328. [DOI] [PubMed] [Google Scholar]
- P. TL, Bruggemann TR, R MR, M GM, Cagnina RE, Shay AE, C CG, Nijmeh J, M MT, & Levy BD (2022). Cysteinyl Maresins Reprogram Macrophages to Protect Mice from Streptococcus pneumoniae after Influenza A Virus Infection. mBio, 13, e0126722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Li X, Wang X, Yang L, Chen H, Su N, Wu C, Hao Y, Jin S, & Li H (2021). MCTR1 Intervention Reverses Experimental Lung Fibrosis in Mice. J Inflamm Res, 14, 1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Qi X, Luo Y, Li X, Shu T, Li B, Song M, Liu Y, Wei D, Chen J, Wang J, & Wang C (2021). Multi-omics study of silicosis reveals the potential therapeutic targets PGD(2) and TXA(2). Theranostics, 11, 2381–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D, Gilligan MM, Serhan CN, & Kashfi K (2021). Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther, 227, 107879. [DOI] [PubMed] [Google Scholar]
- Park J, Langmead CJ, & Riddy DM (2020). New Advances in Targeting the Resolution of Inflammation: Implications for Specialized Pro-Resolving Mediator GPCR Drug Discovery. ACS Pharmacol Transl Sci, 3, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perazza LR, Mitchell PL, Lizotte F, Jensen BAH, St-Pierre P, Trottier J, Barbier O, Mathieu P, Geraldes PM, & Marette A (2021). Fish oil replacement prevents, while docosahexaenoic acid-derived protectin DX mitigates end-stage-renal-disease in atherosclerotic diabetic mice. FASEB J, 35, e21559. [DOI] [PubMed] [Google Scholar]
- Podolin PL, Bolognese BJ, Foley JF, Long E 3rd, Peck B, Umbrecht S, Zhang X, Zhu P, Schwartz B, Xie W, Quinn C, Qi H, Sweitzer S, Chen S, Galop M, Ding Y, Belyanskaya SL, Israel DI, Morgan BA, Behm DJ, Marino JP Jr., Kurali E, Barnette MS, Mayer RJ, Booth-Genthe CL, & Callahan JF (2013). In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediat, 104–105, 25–31. [DOI] [PubMed] [Google Scholar]
- Ramon S, Dalli J, Sanger JM, Winkler JW, Aursnes M, Tungen JE, Hansen TV, & Serhan CN (2016). The Protectin PCTR1 Is Produced by Human M2 Macrophages and Enhances Resolution of Infectious Inflammation. Am J Pathol, 186, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchiuti A, Patruno S, Mattoscio D, Isopi E, Pomilio A, Lamolinara A, Iezzi M, Pecce R, & Romano M (2021). Resolvin D1 and D2 reduce SARS-CoV-2-induced inflammatory responses in cystic fibrosis macrophages. FASEB J, 35, e21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach KM, Feghali-Bostwick CA, Amrani Y, & Bradding P (2015). Lipoxin A4 Attenuates Constitutive and TGF-beta1-Dependent Profibrotic Activity in Human Lung Myofibroblasts. J Immunol, 195, 2852–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout-Pitt N, Farrow N, Parsons D, & Donnelley M (2018). Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res, 19, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Sarabia C, Torres M, & Juarez E (2019). Resolvin D1 (RvD1) and maresin 1 (Mar1) contribute to human macrophage control of M. tuberculosis infection while resolving inflammation. Int Immunopharmacol, 74, 105694. [DOI] [PubMed] [Google Scholar]
- Saito A, Okazaki H, Sugawara I, Yamamoto K, & Takizawa H (2003). Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol, 132, 168–176. [DOI] [PubMed] [Google Scholar]
- Santos-Ribeiro D, Lecocq M, de Beukelaer M, Bouzin C, Palmai-Pallag M, Yakoub Y, Huaux F, Horman S, Perros F, Pilette C, & Godinas L (2023). Bleomycin-induced lung injury: Revisiting an old tool to model group III PH associated with pulmonary fibrosis. Pulm Circ, 13, e12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Kitasato H, Murakami Y, Hashimoto A, Endo H, Kondo H, Inoue M, & Hayashi I (2004). Down-regulation of lipoxin A4 receptor by thromboxane A2 signaling in RAW246.7 cells in vitro and bleomycin-induced lung fibrosis in vivo. Biomed Pharmacother, 58, 381–387. [DOI] [PubMed] [Google Scholar]
- Sennello JA, Misharin AV, Flozak AS, Berdnikovs S, Cheresh P, Varga J, Kamp DW, Budinger GR, Gottardi CJ, & Lam AP (2017). Lrp5/beta-Catenin Signaling Controls Lung Macrophage Differentiation and Inhibits Resolution of Fibrosis. Am J Respir Cell Mol Biol, 56, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, & Dalli J (2015). The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin Immunol, 27, 200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, & Gronert K (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med, 192, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, & Petasis NA (2012). Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J, 26, 1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hamberg M, & Samuelsson B (1984). Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci U S A, 81, 5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC, & Hammock BD (2012). Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem, 55, 1789–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin IS, Jeon WY, Shin HK, & Lee MY (2013). Effects of montelukast on subepithelial/peribronchial fibrosis in a murine model of ovalbumin induced chronic asthma. Int Immunopharmacol, 17, 867–873. [DOI] [PubMed] [Google Scholar]
- Shorr AF, Wainright JL, Cors CS, Lettieri CJ, & Nathan SD (2007). Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J, 30, 715–721. [DOI] [PubMed] [Google Scholar]
- Sime PJ, Xing Z, Graham FL, Csaky KG, & Gauldie J (1997). Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest, 100, 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JNP, Witkin MD, Jogasuria AP, Christo KF, Raffay TM, Markowitz SD, & Desai AB (2020). Therapeutic targeting of 15-PGDH in murine pulmonary fibrosis. Sci Rep, 10, 11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, & Weintraub NL (2004). Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res, 43, 55–90. [DOI] [PubMed] [Google Scholar]
- Sun YB, Qu X, Li X, Nikolic-Paterson DJ, & Li J (2013). Endothelial dysfunction exacerbates renal interstitial fibrosis through enhancing fibroblast Smad3 linker phosphorylation in the mouse obstructed kidney. PLoS One, 8, e84063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, & Serhan CN (2007). Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem, 282, 9323–9334. [DOI] [PubMed] [Google Scholar]
- Takezaki A, Tsukumo SI, Setoguchi Y, Ledford JG, Goto H, Hosomichi K, Uehara H, Nishioka Y, & Yasutomo K (2019). A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J Exp Med, 216, 2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Gao C, Long Y, Huang W, Chen J, Fan F, Jiang C, & Xu Y (2017). Maresin 1 Mitigates High Glucose-Induced Mouse Glomerular Mesangial Cell Injury by Inhibiting Inflammation and Fibrosis. Mediators Inflamm, 2017, 2438247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao JH, Liu T, Zhang CY, Zu C, Yang HH, Liu YB, Yang JT, Zhou Y, & Guan CX (2022). Epoxyeicosatrienoic Acids Inhibit the Activation of Murine Fibroblasts by Blocking the TGF-beta1-Smad2/3 Signaling in a PPARgamma-Dependent Manner. Oxid Med Cell Longev, 2022, 7265486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Zhang R, Du R, Yu T, Yang H, Li J, Wang Y, Liu Q, Zuo S, Wang X, Lazarus M, Zhou L, Wang B, Yu Y, & Shen Y (2022). EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher TH, Woeller CF, McCarthy CE, & Sime PJ (2019). Quenching the fires: Pro-resolving mediators, air pollution, and smoking. Pharmacol Ther, 197, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino K, Reed NI, Atakilit A, Ren X, & Sheppard D (2017). Transforming growth factor-beta plays divergent roles in modulating vascular remodeling, inflammation, and pulmonary fibrosis in a murine model of scleroderma. Am J Physiol Lung Cell Mol Physiol, 312, L22–L31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylek K, Trojan E, Regulska M, Lacivita E, Leopoldo M, & Basta-Kaim A (2021). Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: a link to brain pathology. Pharmacol Rep, 73, 1004–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upagupta C, Shimbori C, Alsilmi R, & Kolb M (2018). Matrix abnormalities in pulmonary fibrosis. Eur Respir Rev, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorselaars ADM, Wuyts WA, Vorselaars VMM, Zanen P, Deneer VHM, Veltkamp M, Thomeer M, van Moorsel CHM, & Grutters JC (2013). Methotrexate vs azathioprine in second-line therapy of sarcoidosis. Chest, 144, 805–812. [DOI] [PubMed] [Google Scholar]
- Wagner KM, McReynolds CB, Schmidt WK, & Hammock BD (2017). Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther, 180, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters DM, & Kleeberger SR (2008). Mouse models of bleomycin-induced pulmonary fibrosis. Curr Protoc Pharmacol, Chapter 5, Unit 5 46. [DOI] [PubMed] [Google Scholar]
- Wang F, Xia H, & Yao S (2020). Regulatory T cells are a double-edged sword in pulmonary fibrosis. Int Immunopharmacol, 84, 106443. [DOI] [PubMed] [Google Scholar]
- Wang T, Li Y, Chen J, Xie L, & Xiao T (2018). TGF-beta1/Smad3 signaling promotes collagen synthesis in pulmonary artery smooth muscle by down-regulating miR-29b. Int J Clin Exp Pathol, 11, 5592–5601. [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li R, Chen L, Tan W, Sun Z, Xia H, Li B, Yu Y, Gong J, Tang M, Ji Y, Yuan S, Shanglong Y, & Shang Y (2015). Maresin 1 Inhibits Epithelial-to-Mesenchymal Transition in Vitro and Attenuates Bleomycin Induced Lung Fibrosis in Vivo. Shock, 44, 496–502. [DOI] [PubMed] [Google Scholar]
- Warshamana GS, Pociask DA, Fisher KJ, Liu JY, Sime PJ, & Brody AR (2002). Titration of non-replicating adenovirus as a vector for transducing active TGF-beta1 gene expression causing inflammation and fibrogenesis in the lungs of C57BL/6 mice. Int J Exp Pathol, 83, 183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsenbeek M, Suzuki A, & Maher TM (2022). Interstitial lung diseases. Lancet, 400, 769–786. [DOI] [PubMed] [Google Scholar]
- Willis BC, & Borok Z (2007). TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol, 293, L525–534. [DOI] [PubMed] [Google Scholar]
- Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, & Borok Z (2005). Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol, 166, 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, & Hinz B (2007). Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol, 179, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SH, Wu XH, Lu C, Dong L, & Chen ZQ (2006). Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol, 34, 65–72. [DOI] [PubMed] [Google Scholar]
- Wu SH, Zhang YM, Tao HX, & Dong L (2010). Lipoxin A(4) inhibits transition of epithelial to mesenchymal cells in proximal tubules. Am J Nephrol, 32, 122–136. [DOI] [PubMed] [Google Scholar]
- Xu J, Gonzalez ET, Iyer SS, Mac V, Mora AL, Sutliff RL, Reed A, Brigham KL, Kelly P, & Rojas M (2009). Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A Biol Sci Med Sci, 64, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara T, Tsubouchi K, Gholiof M, Chong SG, Lipson KE, Zhou Q, Scallan C, Upagupta C, Tikkanen J, Keshavjee S, Ask K, & Kolb MRJ (2022). Connective-Tissue Growth Factor Contributes to TGF-beta1-induced Lung Fibrosis. Am J Respir Cell Mol Biol, 66, 260–270. [DOI] [PubMed] [Google Scholar]
- Yang J, Bratt J, Franzi L, Liu JY, Zhang G, Zeki AA, Vogel CF, Williams K, Dong H, Lin Y, Hwang SH, Kenyon NJ, & Hammock BD (2015). Soluble epoxide hydrolase inhibitor attenuates inflammation and airway hyperresponsiveness in mice. Am J Respir Cell Mol Biol, 52, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hu L, Xia H, Chen L, Cui S, Wang Y, Zhou T, Xiong W, Song L, Li S, Pan S, Xu J, Liu M, Xiao H, Qin L, Shang Y, & Yao S (2019). Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol, 316, L1013–L1024. [DOI] [PubMed] [Google Scholar]
- Yao L, Conforti F, Hill C, Bell J, Drawater L, Li J, Liu D, Xiong H, Alzetani A, Chee SJ, Marshall BG, Fletcher SV, Hancock D, Coldwell M, Yuan X, Ottensmeier CH, Downward J, Collins JE, Ewing RM, Richeldi L, Skipp P, Jones MG, Davies DE, & Wang Y (2019). Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ, 26, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, Yang P, Su Y, Zhao J, Zhang S, Yu Q, Ning Q, Xiang X, Xiong W, Wang CY, & Xu Y (2016). Chop Deficiency Protects Mice Against Bleomycin-induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol Ther, 24, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasutomo K (2021). Genetics and animal models of familial pulmonary fibrosis. Int Immunol, 33, 653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatomi M, Hisada T, Ishizuka T, Koga Y, Ono A, Kamide Y, Seki K, Aoki-Saito H, Tsurumaki H, Sunaga N, Kaira K, Dobashi K, Yamada M, & Okajima F (2015). 17(R)-resolvin D1 ameliorates bleomycin-induced pulmonary fibrosis in mice. Physiol Rep, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrough AE, Hasturk H, Stephens DN, Van Dyke TE, & Kantarci A (2022). Resolvin D1 modulates periodontal ligament fibroblast function. J Periodontol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC (2001). Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem, 276, 36059–36062. [DOI] [PubMed] [Google Scholar]
- Zeng C, Liu X, Xiong D, Zou K, & Bai T (2022). Resolvin D1 Prevents Epithelial-to-Mesenchymal Transition and Reduces Collagen Deposition by Stimulating Autophagy in Intestinal Fibrosis. Dig Dis Sci, 67, 4749–4759. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang Y, Wu G, Xiong W, Gu W, & Wang CY (2018). Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res, 19, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Chan-Li Y, Collins SL, Zhang Y, Hallowell RW, Mitzner W, & Horton MR (2014). Pulmonary delivery of docosahexaenoic acid mitigates bleomycin-induced pulmonary fibrosis. BMC Pulm Med, 14, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, D’Souza VK, Bartis D, Dancer RC, Parekh D, Naidu B, Gao-Smith F, Wang Q, Jin S, Lian Q, & Thickett DR (2016). Lipoxin A(4) promotes lung epithelial repair whilst inhibiting fibroblast proliferation. ERJ Open Res, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Wang Q, D’Souza V, Bartis D, Dancer R, Parekh D, Gao F, Lian Q, Jin S, & Thickett DR (2018). ResolvinD(1) stimulates epithelial wound repair and inhibits TGF-beta-induced EMT whilst reducing fibroproliferation and collagen production. Lab Invest, 98, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]