

Abstract
Data-driven modeling has emerged as a new paradigm for biocatalyst design and discovery. Biocatalytic databases that integrate enzyme structure and function data are in urgent need. Here we describe IntEnzyDB as an integrated structure–kinetics database for facile statistical modeling and machine learning. IntEnzyDB employs a relational database architecture with a flattened data structure, which allows rapid data operation. This architecture also makes it easy for IntEnzyDB to incorporate more types of enzyme function data. IntEnzyDB contains enzyme kinetics and structure data from six enzyme commission classes. Using 1050 enzyme structure–kinetics pairs, we investigated the efficiency-perturbing propensities of mutations that are close or distal to the active site. The statistical results show that efficiency-enhancing mutations are globally encoded and that deleterious mutations are much more likely to occur in close mutations than in distal mutations. Finally, we describe a web interface that allows public users to access enzymology data stored in IntEnzyDB. IntEnzyDB will provide a computational facility for data-driven modeling in biocatalysis and molecular evolution.
Grpahical Abstract:

1. INTRODUCTION
As a holy grail challenge in modern chemical sciences, developing new enzyme catalysts provides solutions to transform chemically challenging reactions,1 expand substrate scope,2 control complex reaction selectivity,3 treat metabolic disorders,4 and degrade inert environmental wastes and pollutants.5 Data-driven modeling methods have been extensively leveraged to innovate the approaches for enzyme catalyst discovery. They help elucidate the mechanisms of enzyme catalysis,6 predict the impact of mutations on enzyme functions,7,8 and even design artificial enzymes.9
Central to data-driven modeling, databases have been established for storing enzyme sequence, structure, and kinetics data (Tables 1 and S1). For example, the Universal Protein Resource Knowledgebase (UniProtKB) contains ~36.7 million unique enzyme sequences.10 The RCSB Protein Data Bank (PDB) contains 108,000 experimentally determined enzyme structures.11 BRENDA12 and Sabio-RK13 store enzyme kinetic parameters, including 80,000 kcat values, 169,000 KM values, and 33,000 kcat/KM values in BRENDA and over 56,000 KM or pseudo-dissociation constant values and more than 52,000 velocity constants (Vmax and kcat) in Sabio-RK. These data cover thousands of Enzyme Commission (EC) classes that span seven enzyme types (i.e., oxidoreductases, transferases, hydrolases, lyases, isomerases, ligases, and translocases). In addition, databases have been established to annotate enzyme functions based on their structural, chemical, and metabolic relevance (e.g., EzCatDB,14 M-CSA,15 KEGG,16 FunCat,17 Reactcome,18 and MetaCyc19); to map enzyme sequence, structure, and function relationships (e.g., PDBSWS,20 SFLD,21 FunTree,22 IntEnz,23 ExploreEnz,24 and ExPASy25); to classify enzymes based structural and functional superfamilies (e.g., CATH26 and SCOP27,28); and to store designed enzymes (e.g., ProtaBank29 and Design2Data4).
Table 1.
A brief summary of enzymology databases.
| Database Type | Databases | Data | UniProtKB | EC Number | PDB ID |
|---|---|---|---|---|---|
| Kinetics | BRENDA | Kinetics | Yes | Yes | Part |
| Sabio-RK | Kinetics | Yes | Yes | No | |
| STRENDA DB | Kinetics with uniform data standard | Yes | Yes | No | |
| Structure | PDB | PDB Structure | Yes | Yes | Yes |
| AlphaFold DB | Predicted Structure | Yes | No | No | |
| UniProt | Sequence with Functional annotation | Yes | Yes | Yes | |
| Kinetics and structure data for designed enzymes | ProtaBank | Kinetics/Structure | Part | Part | Part |
| Design2Data | Kinetics/Structure | Yes | Yes | Yes |
To develop holistic predictive models for enzyme catalysis, an integrated database is needed that merges related enzyme sequence, structure, and function data in one place. However, three challenges are identified. First, collecting data from various sources is difficult because databases involve different designs (e.g., relational, object-oriented, or hybrid), storage hierarchies, query mechanisms, and API protocols. Thus, curating enzyme features consumes significant efforts. Second, data cleaning is tricky due to various data standards adopted by different databases. Although unified data reporting standards have been reported (e.g., STRENDA30 and EnzymeML31), existing enzyme data entries still involve missing or inaccurate mutational spot labels, experimental conditions, or other information. Additionally, manual typos and rounding errors are not uncommon, leading to obstacles for data validation. Third, joining of enzyme structure and kinetics data is challenging because they do not have consistently shared keys. Enzyme kinetics databases store data entries by EC number and do not always have a PDB ID for mapping with the structure database (Table 1). Although UniProtKB is used across databases, one-to-one mapping between structure and kinetics is difficult because one UniProtKB may correspond to tens of PDB IDs.
In the present work, we developed an integrated structure–kinetics enzymology database, IntEnzyDB, for facile data-driven modeling and machine learning. We previously reported the beta version of IntEnzyDB as a hydrolase database.32 In this work, we expanded IntEnzyDB to incorporate data from six EC classes. IntEnzyDB allows fast operation of large amounts of enzyme structure data and enables mapping between enzyme kinetics and structure. Using these data, we analyzed the propensities of catalytic efficiency enhancement, neutrality, and deletion for mutations that are close or distal to the active site. Finally, we developed a web interface for IntEnzyDB that allows public users to freely access and analyze the data.
2. COMPUTATIONAL METHODS
Database Construction.
IntEnzyDB is a relational database with a flattened data structure. IntEnzyDB adopts one data table to store all enzyme records of the same structural hierarchy (i.e., chain, residue, or atom) or property (i.e., kinetics). The current version of IntEnzyDB consists of five data tables: one table storing enzyme kinetic parameters such as Michaelis constants (KM) and apparent turnover numbers (kcat); three tables storing enzyme chain-level, amino acid-level, and atom-level structural information; and one table for one-to-one mapping of enzyme structure, substrate, and kinetics. Notably, the number of data tables can be easily expanded as we further develop IntEnzyDB to incorporate more enzyme properties (e.g., stability, mechanism, etc.). Thus, users can perform various types of joinings under the SQL framework to map enzyme structure and function data based on their need.
Data Collection.
The kinetics data in IntEnzyDB were extracted from BRENDA,12 Sabio-RK,13 ProtaBank,29 and Design2Data,4 the structure data from the PDB,11 and the sequence data from UniProt.10 The enzyme kinetics table contains EC number, UniProtKB entry, organism, substrate, experimental temperature, and mutational information. Using the UniProt Retrieve/ID mapping tool and PDB Data API, we collected 9415 protein structures associated with the PDB IDs under UniProtKB in the kinetics table.
The PDB structure data are stored in three tables. The enzyme chain table stores the general information of a PDB structure, including PDB ID, EC number, enzyme type, enzyme name, mutation, organism, chain ID, resolution, FASTA sequence, active site location, number of residues, and missing residues. The enzyme amino acid table stores the amino acid-level structural information, including PDB ID, chain ID, amino acid name, amino acid index, and center-of-mass coordinate of the amino acid. The enzyme atom table stores the atom-level structural information, including PDB ID, chain ID, atom name, atom index, amino acid name, amino acid index, and atom coordinates. IntEnzyDB will continue incorporating data from other sources, such as STRENDA.30 The database is open to the public and can be accessed through the web interface (https://intenzydb.accre.vanderbilt.edu). Any changes will be posted on the web interface.
Data Curation.
The kinetics data are filtered based on the following criteria: (1) at least one wild-type kinetics parameter (kcat and KM) exists under one UniProtKB; (2) at least one PDB structure exists under one UniProtKB; (3) substrate information exists for each kinetic parameter; (4) experimental temperature and pH values are known for each kinetic parameter; (5) mutation is known for each kinetic parameter; and (6) mutations are single amino acid substitutions. Curation yielded 4243 kcat/KM values derived from 691 enzymes and 2592 enzyme mutants (i.e., single amino acid substitutions) combined with 943 substrates. The experimental temperatures of the kinetic parameters range from 278.1 to 363.1 K (Figure S1). The experimental pH values for the kinetic parameters range from 3 to 11 (Figure S1). These enzymes span six EC classes, including oxidoreductases (EC 1), transferases (EC 2), hydrolases (EC 3), lyases (EC 4), isomerases (EC 5), and ligases (EC 6). Notably, no kinetic data from EC 7 were found in the curated dataset. This is likely associated with the scarcity of translocases in the enzyme population. Although single mutations are the focus of this study, a kinetics data table was curated for mutants with two or more amino acid substitutions (see the Supporting Information).
To conduct one-to-one mapping of enzyme kinetics to structure, we adopted a three-step curation workflow. In step 1, we extracted PDB IDs from the research articles associated with the enzyme kinetics using the text mining method (see the Supporting Information). In step 2, for the kinetic values where the PDB IDs are not available from the research papers, we manually identified the PDB structures by aligning the mutation spot annotations (taken from PDB files). In step 3, under each UniProtKB, we selected the PDB structures with active-site annotation, top resolution, and the least number of missing residues. This three-step approach allowed us to perform one-to-one mapping of the kinetic data with the PDB structure through UniProtKB, yielding 155 PDB structures precisely paired with 1050 kcat/KM values. For the curated dataset, we evaluated the distribution of structure resolution and number of unresolved residues (Figure S2). These data allow in-depth analysis of enzyme structure–function relationship. Notably, the kinetic data table for higher-order mutants will be added to IntEnzyDB in the future after we accomplish the structure–kinetics mapping. These data will enable the analysis of structure–kinetics relationship for enzymes with multiple mutational sites, revealing the structural basis behind epistatic effects. Data collection and curation are performed in Python and R software, and all statistical analyses were performed in R software. The curated kinetic and structural data tables and the data curation codes can be found in the Supporting Information.
3. RESULTS AND DISCUSSION
Design Architecture and Data Processing Efficiency of IntEnzyDB.
Unlike object-oriented databases that store each enzyme record in an individual data table (or file),11 IntEnzyDB adopts a relational database architecture with a flattened data structure (as detailed in Computational Methods). This allows IntEnzyDB to be expandable to incorporate other types of enzyme function data such as stability33 and solubility.34 The database employs five tables to store enzyme kinetics and structure information (Figure 1, top), including three tables for cleaned enzyme structure data derived from the PDB (i.e., ① chain, ② amino acid, and ③ atom), one table for kinetics data derived from BRENDA and Sabio-RK (labeled as ④), and one reference table (labeled as ⑤). The chain, amino acid, and atom tables share PDB ID and Chain ID as foreign keys. The chain table contains general protein structure information, including enzyme name, organism, gene, FASTA sequence, active site, and resolution; the amino acid table stores amino acid attributes, properties, and physiochemical parameters, including residue name, residue sequence number, amino acid weight, center-of-mass coordinates; the atom structure table stores the atom types and coordinates, including atom name, atom sequence number, residue name, residual sequence number, atomic weight, and atom Cartesian coordinates.
Figure 1.
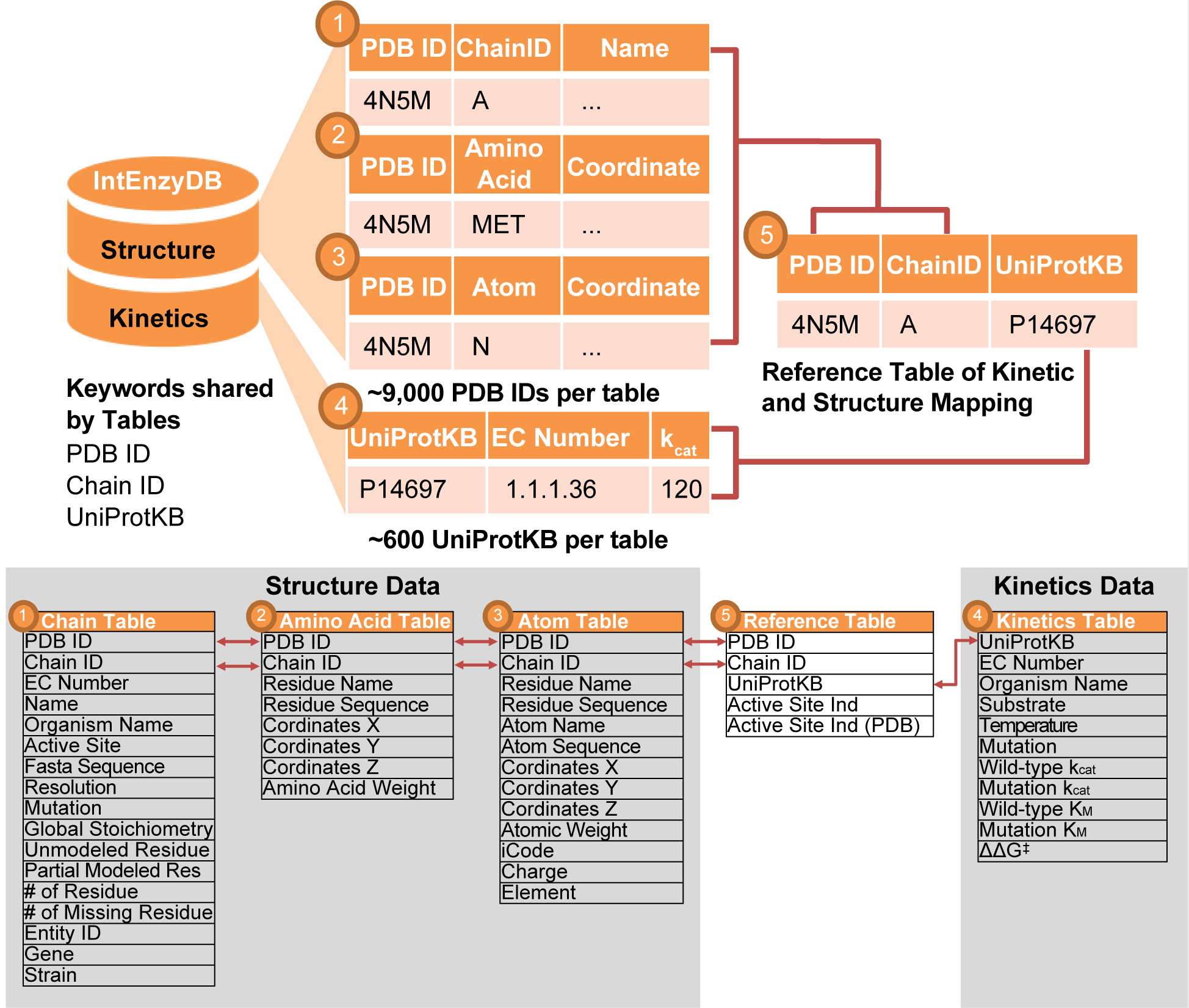
Architecture and relation map for IntEnzyDB. (top) The database architecture involves five tables, including three enzyme structure tables (i.e., chain-level, amino acid-level, and atom-level), one enzyme kinetics table, and one reference table with foreign keys from the structure and kinetics tables. The tables are mapped by the following keys: PDB ID, Chain ID, and UniProtKB. (bottom) Mapping relationship between variables of different tables.
The kinetics table contains kinetic parameters, enzymology assay information, and sequence data, including UniProtKB, EC number, organism, substrate, mutation, experimental temperature, apparent turnover number (kcat), Michaelis constant (KM), enzyme efficiency (kcat/KM), and change of free energy barriers for a mutant compared to the wild-type enzyme (ΔΔG‡, converted from kcat/KM according to eq 1). The kinetics table uses UniProtKB (sequence ID) as the foreign key. The reference table (Table 1 and Figure 1) contains the one-to-one mapping relationship between kinetics data and PDB data based on the foreign keys PDB ID, Chain ID, and UniProtKB (as detailed in Computational Methods). This table can be used to identify the PDB structure for given kinetic data of interest. The data from the table can also be used to investigate the structure–kinetics relationship.
We benchmarked the time of pulling enzyme structure data using IntEnzyDB against a manual curation strategy (Figure 2). Using IntEnzyDB, a user can directly filter and download cleaned and tabulated structural data using SQL language; in contrast, for the manual curation strategy, a user needs to first download data from the PDB, then read and reformat the data by entry, and eventually combine them in one table on the local computer. Figure 2 shows that IntEnzyDB is ~2 times faster than the traditional approach for 200 enzymes (80 vs 173 s) and ~6 times faster for 1000 enzymes (151 vs 905 s). The results indicate that the operating time using IntEnzyDB is nearly independent of data size, which largely outperforms the manual operation strategy when operating on large amounts of structural data (i.e., thousands or more).
Figure 2.
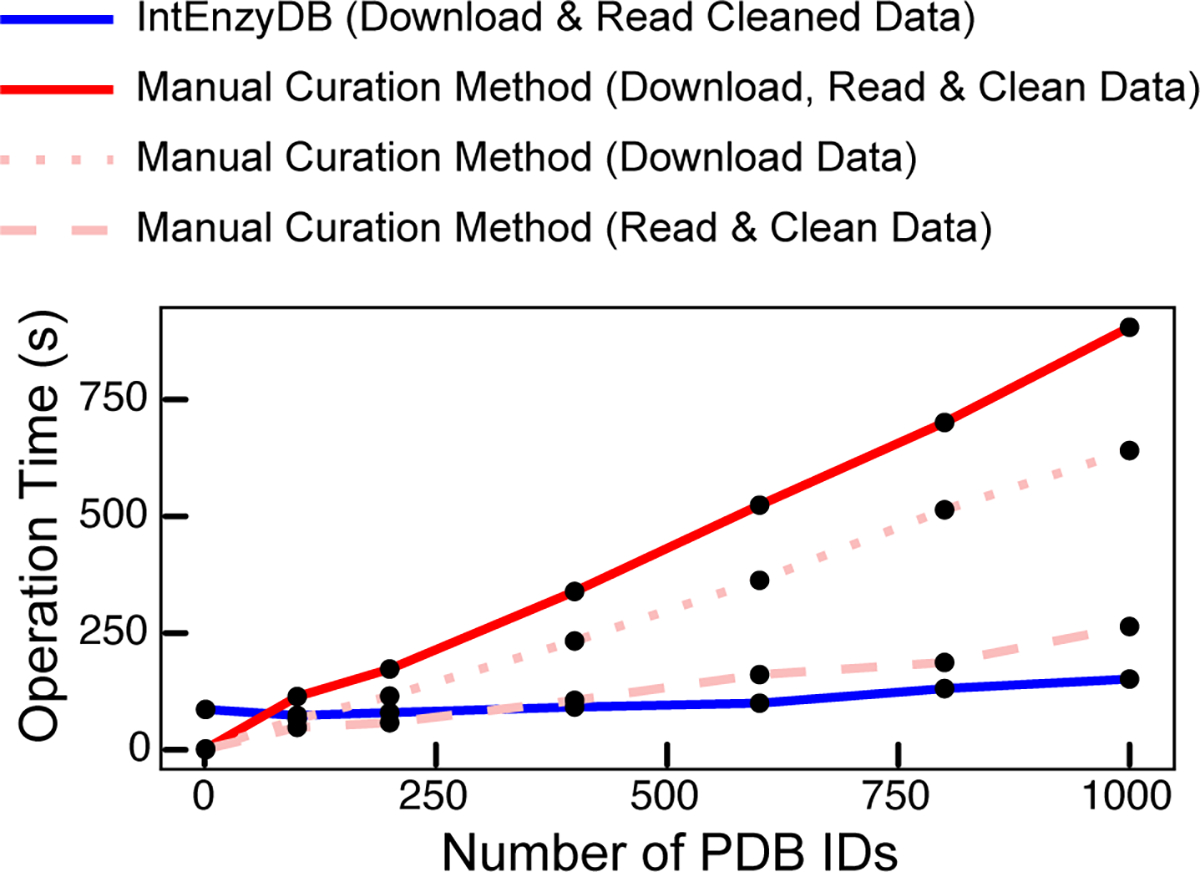
Operation time vs the number of PDB IDs by IntEnzyDB (blue line) and the manual curation method (red line). The operation time for downloading, reading, and cleaning data in a tabulated form is measured for the tasks of processing 1, 100, 200, 400, 600, 800, and 1000 PDB IDs. Data downloading and reading/cleaning are represented by the dotted and dashed lines (in light red), respectively. The total operation time for the manual curation method is shown by the red solid line. All operation times are measured in seconds.
The high data processing efficiency of IntEnzyDB likely results from its flattened data structure. Compared to the traditional approach where data tables and files are accessed serially, IntEnzyDB loads all data entries at one time. This approach makes IntEnzyDB slower when processing smaller amounts of data (e.g., for one enzyme structure, 86 vs 1.9 s) but can save tremendous amounts of time for repeated opening and reading of files when handling large amounts of structure data (e.g., 3.5 min for 5000 structures). Therefore, IntEnzyDB provides an efficient solution for extracting enzyme structural features for statistical analysis or machine learning.
Statistical Analysis of Kinetic Parameters in IntEnzyDB.
From IntEnzyDB, we curated 4243 kcat/KM values for enzymes with single amino acid substitution. The dataset consists of 691 wild-type enzymes, 2592 enzyme mutants, and 943 substrates (as detailed in Computational Methods). The number of kcat/KM values has tripled the size of the hydrolase kinetics data we reported in the prior work (i.e., 1240).32 Among the 4243 kcat/KM values, 29.2% are oxidoreductases (EC 1), 19.4% are transferases (EC 2), 32.6% are hydrolases (EC 3), 9.1% are lyases (EC 4), 4.9% are isomerases (EC 5), and 4.9% are ligases (EC 6) (Figure 3, left). To evaluate the impact of mutation on enzyme catalysis, we investigated the distribution of ΔΔG‡ values derived from 2592 enzyme mutants, where ΔΔG‡ is converted from the ratio of the catalytic efficiency of the mutant to that of the wild-type enzyme (eq 1):
| (1) |
where R, T, kcat, and KM are the gas constant, experimental temperature, turnover number, and Michaelis constant, respectively. Notably, the wild-type enzyme and the mutant have the same substrate, temperature, and pH in the data analysis. The distribution of ΔΔG‡ follows a right-skewed Gaussian that ranges from −5.5 to 11.2 kcal/mol with a mean of 1.3 kcal/mol (Figure 3, right). The breadth of the distribution is wider than that of hydrolases (i.e., −4.2 to 9.4 kcal/mol), but the mean value is similar (i.e., 1.2 kcal/mol).32 We categorized the mutants to be efficiency-enhancing (ΔΔG‡ ≤ −0.5 kcal/mol), -neutral (−0.5 kcal/mol < ΔΔG‡ ≤ 0.5 kcal/mol), and -deleterious (ΔΔG‡ > 0.5 kcal/mol). The efficiency-neutral mutations involve a narrow energy window (i.e., ±0.5 kcal/mol), and their impact on enzyme efficiency falls into the range of experimental error. We observed 11.1% of the mutants to be efficiency-enhancing, 29.7% efficiency-neutral, and 59.2% efficiency-deleterious. As expected, the mutations that decrease the catalytic rate are much more populated than those that are neutral or beneficial to catalysis. The efficiency-enhancing mutations appear to be more abundant in the database than their natural abundance.35,36 This phenomenon might be caused by observational bias. For example, researchers are more likely to perform and report kinetic data when efficiency-enhancing mutants are observed. Another cause is the lack of deleterious mutations, whose kinetic parameters are beyond the detection limit of biochemical assays.
Figure 3.
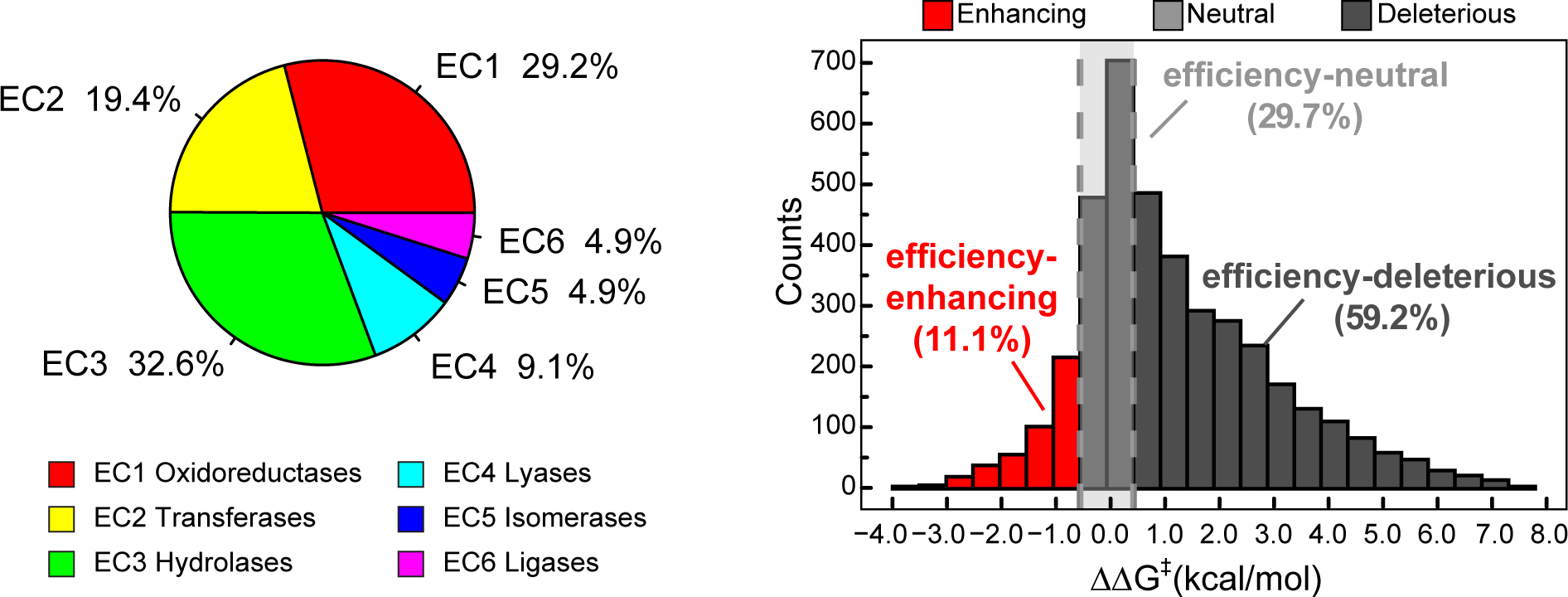
Statistics of kinetics data for enzyme mutants with single amino acid substitution in IntEnzyDB. (left) Distribution of kinetics data for six EC classes. (right) Distribution of ΔΔG‡ values for 2592 enzyme-variant-catalyzed reactions with a bin size of 0.5 kcal/mol. Efficiency-enhancing mutants are defined as those with ΔΔG‡≤ −0.5 kcal/mol (red), efficiency-neutral mutants as those with −0.5 kcal/mol < ΔΔG‡≤ 0.5 kcal/mol (light gray), and efficiency-deleterious mutants as those with ΔΔG‡ > 0.5 kcal/mol (dark gray).
Mutation Effects for Close versus Remote Mutations.
After joining enzyme kinetics with structure data using the reference table (Figure 1), we obtained 1050 reactions with one-to-one-mapped enzyme structure–kinetics pairs, including 385 oxidoreductase reactions, 83 transferase reactions, 355 hydrolase reactions, 114 lyase reactions, 71 isomerase reactions, and 42 ligase reactions. Noticeably, the number of data entries for hydrolases (355) is less than the amount of data curated in our prior work (403).32 This is because in this work we applied a stricter filtration condition that traces every kinetic entry to the corresponding structure in the literature using text mining (as detailed in Computational Methods) rather than simply relying on UniprotKB to map the kinetic entry with the best-resolved structure as done previously, and we removed reaction entries whose wild-type and mutation reaction pH could not be matched. In addition, there are 3193 kcat/KM values from the kinetics table whose corresponding enzyme structures (either wild-type or mutant) or active-site annotation is not known. To address this, we will obtain the missing structures using enzyme structure prediction tools (e.g., AlphaFold237 and RoseTTAFold38) and curate the active-site annotation from the M-CSA database39 or label them manually.
Using 1050 structure–kinetics pairs, we investigated the difference in the efficiency-perturbing propensities for mutations that are spatially close versus distal to the active-site residues (Figure 4). Notably, all of the enzyme structures used in the analysis were assumed to be in the monomeric form, although a dimeric or polymeric form might be the active form in catalysis. This analysis has been conducted for hydrolases in our prior work.32 In contrast, the current dataset involves a greater number of enzymes with a wider converge of enzyme types. As such, the statistical study can potentially inform a more holistic trend for the spatial dependence of efficiency-perturbing mutations. The distance between a mutation spot and the active site was measured between the mutation residue’s Cα coordinate and the geometric center of the active-site residues’ Cα coordinates. Using 15 Å as an empirical cutoff, the efficiency-enhancing propensity of the close mutations (8.0%) is found to resemble that of the distal mutations (8.2%). However, the efficiency-deleterious mutations are much more populated for the close mutations (72.8%) than the distal mutations (47.2%). As a compensation, the efficiency-neutral mutations are about 26% more observed for distal mutations.
Figure 4.
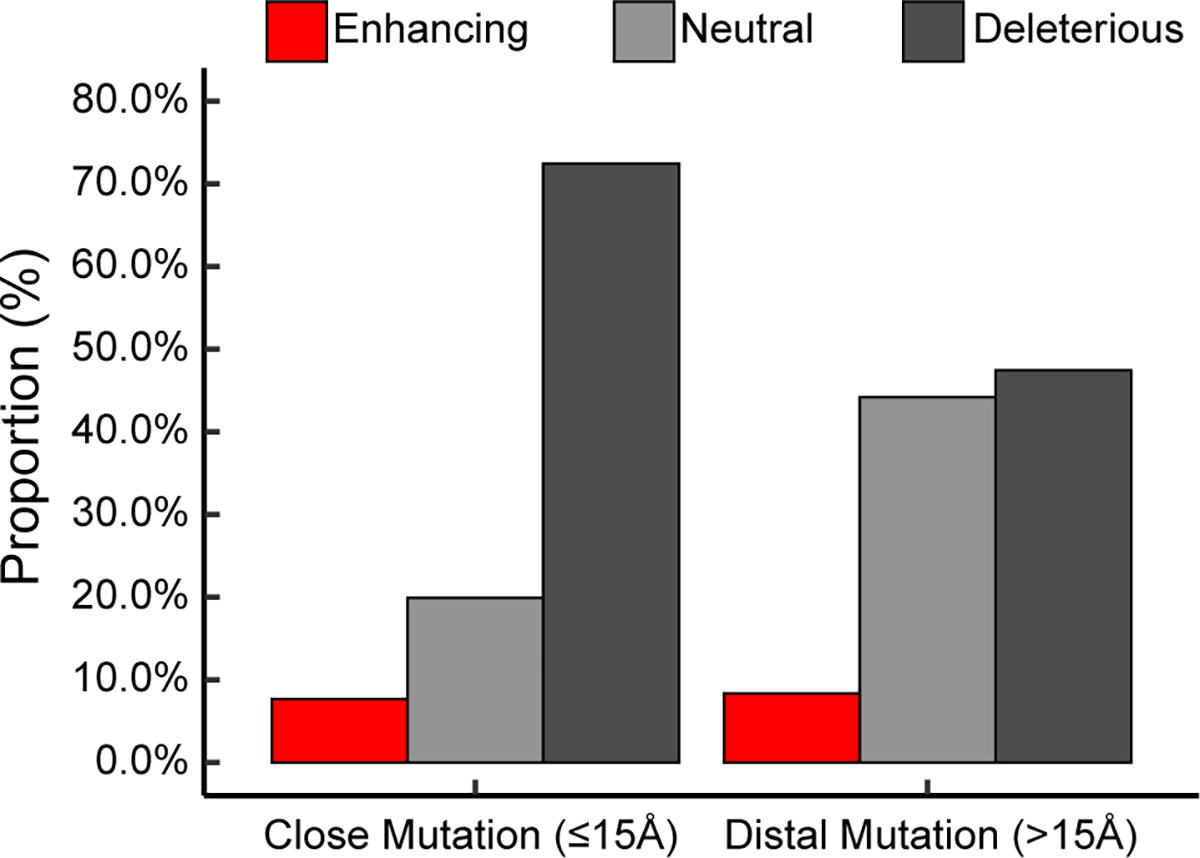
Proportions of efficiency-enhancing (red), -neutral (light gray), and -deleterious (dark gray) mutations for close mutations (≤15 Å) and distal mutations (>15 Å). There are 622 close mutations and 428 distal mutations. The distance is defined as the distance of the mutation residue Cα coordinate to the geometric center of the active-site residues’ Cα coordinates. Efficiency-enhancing mutants are defined as those with ΔΔG‡≤ –0.5 kcal/mol (red), efficiency-neutral mutants as those with –0.5 kcal/mol < ΔΔG‡ ≤ 0.5 kcal/mol (light gray), and efficiency-deleterious mutants as those with ΔΔG‡ > 0.5 kcal/mol (dark gray).
The efficiency-perturbing propensity may be dependent on the choice of the spatial cutoff values. To reduce arbitrariness, we evaluated the proportions for the close versus distal mutations using different spatial cutoffs sampled from 10 to 20 Å with a 1 Å interval (Table S2 and Figure S3). Cutoff values below 10 Å were not tested because of the scarcity of mutations (especially beneficial mutations) falling into the close mutation category. The efficiency-enhancing propensity is estimated to be 7.9% (with lower quartile 6.4% and upper quartile 8.3%) for the close mutations and 8.5% (with lower quartile 7.6% and upper quartile 9.5%) for the distal mutations—they remain highly similar. Despite the fluctuation, the propensity of rate deletion is still much higher for the close mutations (72.8% with lower quartile 69.6% and upper quartile 76.8%) than for the distal mutations (47.2% with lower quartile 42.7% and upper quartile 50.3%). This trend remains to be compensated by the efficiency neutral mutations (19.1% with lower quartile 16.8% and upper quartile 21.9% for close mutations and 44.6% with lower quartile 40.2% and upper quartile 50.2% for distal mutations). Notably, the same trend still exists when the data are separately analyzed for the three major enzyme classes: oxidoreductases, transferases, and hydrolases (Figure S4–S6).
The statistical studies show that close mutations are equally probable in inducing efficiency enhancement as distal mutations, indicating that efficiency-enhancing mutations are globally distributed. This result is consistent with the observation that the Whitehead group reported for Escherichia coli-expressed amidases36 and supports a prior statistical study by the Kazlauskas group (based on 55 rate-enhancing enzyme variants) showing that both close and distal mutations can improve activity.40 For enzyme engineering, given the smaller number of residues in the active site than the distal spots, strategies that emphasize mutagenesis of active-site residues are likely to be more statistically productive, such as the combinatorial active-site saturation test (i.e., CASTing41). In addition, our statistical results show that distal mutations, especially those occurring on the surface residues (Figure S7), are much less likely to induce efficiency deletion than close mutations. This illustrates the important roles of distal mutations in avoiding rate deletion and inducing neutral drift on the fitness landscape, explaining the broadly reported observation of distal mutation in beneficial mutants during directed evolution.42 Finally, we found that mutation of residues on the β-strand is significantly more deleterious than that on the α-helix or coil (Figure S8). This observation might help inspire the development of new design principles for function-enhancing mutations.
IntEnzyDB Web Interface.
To make IntEnzyDB accessible by public users, we developed a web interface that has a back-end link to IntEnzyDB on MongoDB (Figure 5). The web interface allows users to dynamically connect to MongoDB and generate data tables based on search queries. The dynamic connection scheme also makes it easy for users to obtain the most updated data as we continue expanding the database. The website contains general information about the database architecture and scope under the “Home” and “Research” pages. Under the “Database” page, a user can find kinetics data (i.e., the “Kinetics Data” tab), structural data (i.e., the “Structure Data” tab), and mapped structure–kinetics data (i.e., the “Kinetics-Structure Reference” tab). Under the “Kinetics Data” tab, the user can find 4243 curated kinetics data for enzymes with single amino acid substitution where both kcat and KM are available. The data table contains variables including EC number (e.g., 3.1.1.2), UniProtKB (e.g., P27169), organism (e.g., Homo sapiens), substrate (e.g., phenylacetate), mutation (e.g., H115W), experimental temperature (e.g., 298.15 K), and change of free energy barrier ΔΔG‡ (e.g., 1.7 kcal/mol, converted from eq 1). Under the “Structure Data” tab, the user can find general structural information, including the PDB ID (e.g., 1V04), enzyme name (e.g., arylesterase), active-site index (e.g., 115), and resolution (e.g., 2.2 Å). On the “Kinetics-Structure Reference” tab, the mapped kinetics–structure pairs are shown. For each entry in this reference table, the UniProtKB matches an entry in the kinetics table and a PDB ID in the structure table. Under this tab, a user can click on the UniProtKB or PDB ID hyperlink to directly access the UniProt or PDB website for more detailed structure and functional information.
Figure 5.
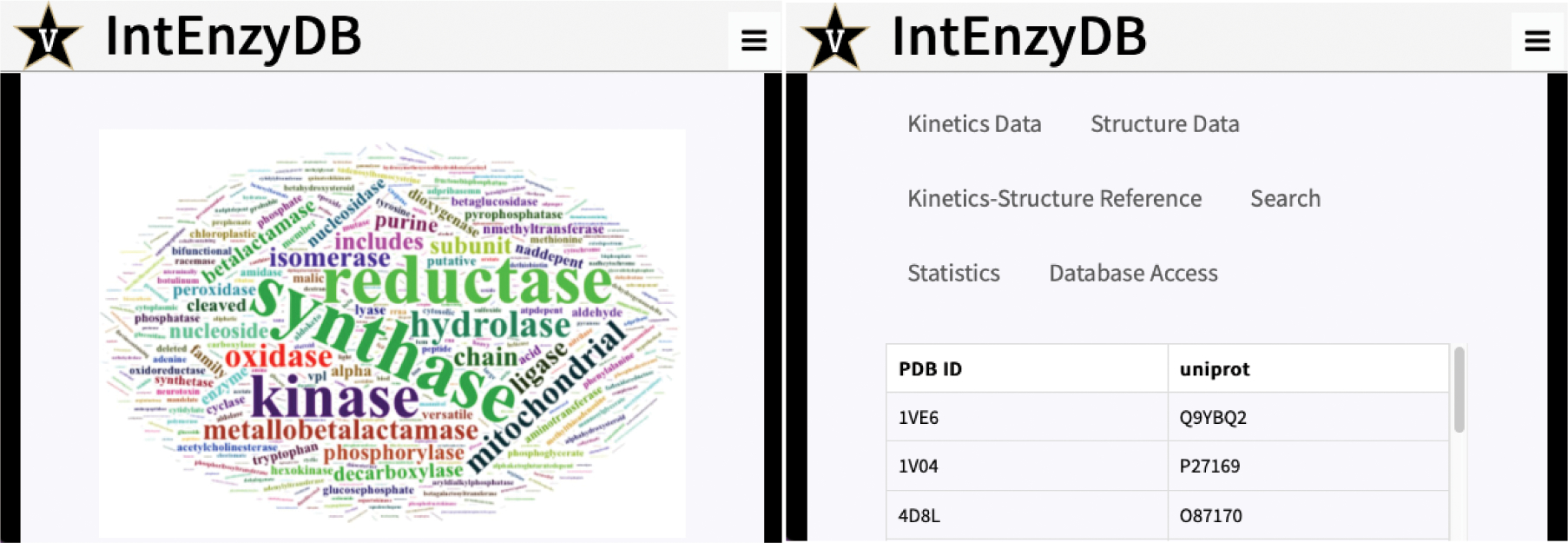
Screenshots of IntEnzyDB web interface: (left) home page for the IntEnzyDB website; (right) database tabs for the website.
Besides the data tables, the user can access the “Search” tab and find specific enzyme data entries in the data tables using UniProtKB, PDB ID, or EC number as search queries. The user can also visualize the statistical analysis of enzyme kinetics data under “Statistics”, including the number of enzymes in each EC class, the distribution of ΔΔG‡, and the frequency of mutations in IntEnzyDB. On the “Database Access” tab, the user can find instructions to directly access IntEnzyDB on MongoDB. This way, the user can access the full database with five tables shown in Figure 1 and query enzymes of interest.
4. CONCLUSION
Here we report IntEnzyDB as an integrated structure–kinetics enzymology database. IntEnzyDB adopts a relational architecture with a flattened data structure. The database consists of five data tables, including one kinetics table, three structure tables, and one structure–kinetics reference table. In the benchmark for processing 1000 protein structures, IntEnzyDB is 6 times faster than the manual curation approach that relies on direct downloading from the PDB website and accessing from a local directory. The high efficiency of IntEnzyDB is due to its flattened data structure: with all of the structure/kinetics data entries read into computer memory in the form of giant data tables, the time for repetitive file input/output operations can be saved.
From IntEnzyDB, we curated 4243 data entries where both kcat and KM are known for enzyme mutants with single amino acid substitution. These data are primarily derived from three enzyme commission classes: oxidoreductases (29.2%), transferases (19.4%), and hydrolases (32.6%). Lyases, isomerases, and ligases are observed to occupy 9.1%, 4.9%, and 4.9% of the population, respectively. Through analysis of mutation effects, we observed 11.1% of the mutants to be efficiency-enhancing, 29.7% efficiency-neutral, and 59.2% efficiency-deleterious.
Using 1050 enzyme structure–kinetics pairs, we investigated the spatial dependence of efficiency-perturbing propensities of mutations. Specifically, we categorized mutations as either close or distal to active-site residues using various spatial cutoff values ranging between 10 and 20 Å with a 1 Å interval; for each cutoff value, we tested the proportions of efficiency-enhancing, -neutral, and -deleterious mutations for both “close” and “distal” mutations. The efficiency-enhancing propensity is estimated to be 7.9% (with lower quartile 6.4% and upper quartile 8.3%) for the close mutations and 8.5% (with lower quartile 7.6% and upper quartile 9.5%) for the distal mutations—they are highly similar. Despite the fluctuation, the propensity of rate deletion is much higher for the close mutations (72.8% with lower quartile 69.6% and upper quartile 76.8%) than for the distal mutations (47.2% with lower quartile 42.7% and upper quartile 50.3%). This trend is compensated by the efficiency-neutral mutations (19.1% with lower quartile 16.8% and upper quartile 21.9% for close mutations and 44.6% with lower quartile 40.2% and upper quartile 50.2% for distal mutations).
Finally, we described the web interface for IntEnzyDB, which employs a back-end link to MongoDB. The web interface allows public users to dynamically access and query data based on their need. Besides the kinetics, structure, and reference data tables, the web interface also contains instructions for users to directly access data tables on IntEnzyDB.
As the next steps for developing IntEnzyDB, we will further expand the mapped structure–kinetics data table by using predicted structures and active-site annotation. Text mining strategies will be implemented to enable more comprehensive data validation and expansion. We will incorporate more types of enzymology data to IntEnzyDB, including stability, solubility, expressibility, and even molecular modeling data derived from high-throughput simulations.43
Supplementary Material
ACKNOWLEDGMENTS
We thank Mr. Qianzhen Shao for annotating the mutation of surface residues in the dataset. We thank Mr. Warren Eckstein for creating and configuring the IntEnzyDB virtual server in the Vanderbilt Data Center ACCRE. This research was supported by a startup grant from Vanderbilt University and a scholarship from the Vanderbilt Institue of Chemical Biology. This work was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award R35GM146982. This work was carried out in part using computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant TG-BIO200057.44
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.2c01139
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c01139.
Combined R and Python code for data collection and cleaning; Python code for text mining workflow; data tables for mapped enzyme structure–kinetics data (ZIP) Summary of enzymology databases; distributions of operating temperatures for enzymatic reactions, numbers of missing residues, and resolution; data table and distributions for the proportion of rate-perturbing mutations under various spatial cutoffs (PDF)
Contributor Information
Bailu Yan, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States; Department of Biostatistics, Vanderbilt University, Nashville, Tennessee 37205, United States.
Xinchun Ran, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States.
Anvita Gollu, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States.
Zihao Cheng, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States.
Xiang Zhou, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States.
Yiwen Chen, Data Science Institute, Vanderbilt University, Nashville, Tennessee 37235, United States.
Zhongyue J. Yang, Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, United States Center for Structural Biology, Vanderbilt Institute of Chemical Biology, and Data Science Institute, Vanderbilt University, Nashville, Tennessee 37235, United States; Department of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, Tennessee 37205.
Data Availability Statement
The data tables used in the analysis can be found in the Supporting Information. The curated data tables can also be accessed through our online IntEnzyDB web interface: https://intenzydb.accre.vanderbilt.edu. The website is maintained by the Yang lab.
REFERENCES
- (1).Yang Y; Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res. 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tang Q; Grathwol CW; Aslan-Üzel AS; Wu S; Link A; Pavlidis IV; Badenhorst CPS; Bornscheuer UT Directed evolution of a halide methyltransferase enables biocatalytic synthesis of diverse SAM analogs. Angew. Chem., Int. Ed. 2021, 60, 1524–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Reetz MT Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew. Chem., Int. Ed. 2011, 50, 138–174. [DOI] [PubMed] [Google Scholar]
- (4).Hou C; Smith P; Huang J; Fell J; Huang P; Vater A; Siegel JB Design to Data for mutants of β-glucosidase B from Paenibacillus polymyxa: Q22T, W123R, F155G, Y169M, W438D, V401A. bioRxiv 2020, DOI: 10.1101/2019.12.23.887380. [DOI] [Google Scholar]
- (5).Yoshida S; Hiraga K; Takehana T; Taniguchi I; Yamaji H; Maeda Y; Toyohara K; Miyamoto K; Kimura Y; Oda K A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 2016, 351, 1196. [DOI] [PubMed] [Google Scholar]
- (6).Bonk BM; Weis JW; Tidor B Machine Learning Identifies Chemical Characteristics That Promote Enzyme Catalysis. J. Am. Chem. Soc. 2019, 141, 4108–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li F; Yuan L; Lu H; Li G; Chen Y; Engqvist MKM; Kerkhoven EJ; Nielsen J Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 2022, 5, 662–672. [Google Scholar]
- (8).Heckmann D; Lloyd CJ; Mih N; Ha Y; Zielinski DC; Haiman ZB; Desouki AA; Lercher MJ; Palsson BO Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nat. Commun. 2018, 9, 5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wu Z; Johnston KE; Arnold FH; Yang KK Protein sequence design with deep generative models. Curr. Opin. Chem. Biol. 2021, 65, 18–27. [DOI] [PubMed] [Google Scholar]
- (10).The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chang A; Jeske L; Ulbrich S; Hofmann J; Koblitz J; Schomburg I; Neumann-Schaal M; Jahn D; Schomburg D BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wittig U; Kania R; Golebiewski M; Rey M; Shi L; Jong L; Algaa E; Weidemann A; Sauer-Danzwith H; Mir S; Krebs O; Bittkowski M; Wetsch E; Rojas I; Müller W SABIO-RK—database for biochemical reaction kinetics. Nucleic Acids Res. 2012, 40, D790–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nagano N EzCatDB: the Enzyme Catalytic-mechanism Database. Nucleic Acids Res. 2004, 33, D407–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Holliday GL; Almonacid DE; Bartlett GJ; O’Boyle NM; Torrance JW; Murray-Rust P; Mitchell JBO; Thornton JM MACiE (Mechanism, Annotation and Classification in Enzymes): novel tools for searching catalytic mechanisms. Nucleic Acids Res. 2007, 35, D515–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kanehisa M; Goto S KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ruepp A; Zollner A; Maier D; Albermann K; Hani J; Mokrejs M; Tetko I; Güldener, U.; Mannhaupt, G.; Münsterkötter, M.; Mewes, H. W. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004, 32, 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gillespie M; Jassal B; Stephan R; Milacic M; Rothfels K; Senff-Ribeiro A; Griss J; Sevilla C; Matthews L; Gong C; Deng C; Varusai T; Ragueneau E; Haider Y; May B; Shamovsky V; Weiser J; Brunson T; Sanati N; Beckman L; Shao X; Fabregat A; Sidiropoulos K; Murillo J; Viteri G; Cook J; Shorser S; Bader G; Demir E; Sander C; Haw R; Wu G; Stein L; Hermjakob H; D’Eustachio P The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Caspi R; Altman T; Billington R; Dreher K; Foerster H; Fulcher CA; Holland TA; Keseler IM; Kothari A; Kubo A; Krummenacker M; Latendresse M; Mueller LA; Ong Q; Paley S; Subhraveti P; Weaver DS; Weerasinghe D; Zhang P; Karp PD The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Martin ACR Mapping PDB chains to UniProtKB entries. Bioinformatics 2005, 21, 4297–4301. [DOI] [PubMed] [Google Scholar]
- (21).Akiva E; Brown S; Almonacid DE; Barber AE 2nd,; Custer AF; Hicks MA; Huang CC; Lauck F; Mashiyama ST; Meng EC; Mischel D; Morris JH; Ojha S; Schnoes AM; Stryke D; Yunes JM; Ferrin TE; Holliday GL; Babbitt PC The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42, D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Furnham N; Sillitoe I; Holliday GL; Cuff AL; Rahman SA; Laskowski RA; Orengo CA; Thornton JM FunTree: a resource for exploring the functional evolution of structurally defined enzyme superfamilies. Nucleic Acids Res. 2012, 40, D776–D782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fleischmann A; Darsow M; Degtyarenko K; Fleischmann W; Boyce S; Axelsen KB; Bairoch A; Schomburg D; Tipton KF; Apweiler R IntEnz, the integrated relational enzyme database. Nucleic Acids Res. 2004, 32, D434–D437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McDonald AG; Boyce S; Tipton KF ExplorEnz: the primary source of the IUBMB enzyme list. Nucleic Acids Res. 2009, 37, D593–D597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Duvaud S; Gabella C; Lisacek F; Stockinger H; Ioannidis V; Durinx C Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Knudsen M; Wiuf C The CATH database. Hum. Genomics 2010, 4, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Andreeva A; Kulesha E; Gough J; Murzin AG The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020, 48, D376–D382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Andreeva A; Howorth D; Chothia C; Kulesha E; Murzin AG SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang CY; Chang PM; Ary ML; Allen BD; Chica RA; Mayo SL; Olafson BD ProtaBank: A repository for protein design and engineering data. Protein Sci. 2018, 27, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Swainston N; Baici A; Bakker BM; Cornish-Bowden A; Fitzpatrick PF; Halling P; Leyh TS; O’Donovan C; Raushel FM; Reschel U; Rohwer JM; Schnell S; Schomburg D; Tipton KF; Tsai M-D; Westerhoff HV; Wittig U; Wohlgemuth R; Kettner C STRENDA DB: enabling the validation and sharing of enzyme kinetics data. FEBS J. 2018, 285, 2193–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Range J; Halupczok C; Lohmann J; Swainston N; Kettner C; Bergmann FT; Weidemann A; Wittig U; Schnell S; Pleiss J EnzymeML—a data exchange format for biocatalysis and enzymology. FEBS J. 2022, 289, 5864–5874. [DOI] [PubMed] [Google Scholar]
- (32).Yan B; Ran X; Jiang Y; Torrence SK; Yuan L; Shao Q; Yang ZJ Rate-Perturbing Single Amino Acid Mutation for Hydrolases: A Statistical Profiling. J. Phys. Chem. B 2021, 125, 10682–10691. [DOI] [PubMed] [Google Scholar]
- (33).Stourac J; Dubrava J; Musil M; Horackova J; Damborsky J; Mazurenko S; Bednar D FireProtDB: database of manually curated protein stability data. Nucleic Acids Res. 2021, 49, D319–D324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Niwa T; Kanamori T; Ueda T; Taguchi H Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8937–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Romero PA; Arnold FH Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wrenbeck EE; Azouz LR; Whitehead TA Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat. Commun. 2017, 8, 15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Baek M; DiMaio F; Anishchenko I; Dauparas J; Ovchinnikov S; Lee GR; Wang J; Cong Q; Kinch LN; Schaeffer RD; Millán C; Park H; Adams C; Glassman CR; DeGiovanni A; Pereira JH; Rodrigues AV; van Dijk AA; Ebrecht AC; Opperman DJ; Sagmeister T; Buhlheller C; Pavkov-Keller T; Rathinaswamy MK; Dalwadi U; Yip CK; Burke JE; Garcia KC; Grishin NV; Adams PD; Read RJ; Baker D Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ribeiro AJM; Holliday GL; Furnham N; Tyzack JD; Ferris K; Thornton J M Mechanism and Catalytic Site Atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2018, 46, D618–D623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Morley K; Kazlauskas R Improving enzyme properties: when are closer mutations better? Trends Biotechnol. 2005, 23, 231–7. [DOI] [PubMed] [Google Scholar]
- (41).Clouthier CM; Kayser MM; Reetz MT Designing New Baeyer-Villiger Monooxygenases Using Restricted CASTing. J. Org. Chem. 2006, 71, 8431–8437. [DOI] [PubMed] [Google Scholar]
- (42).Wilding M; Hong N; Spence M; Buckle AM; Jackson CJ Protein engineering: the potential of remote mutations. Biochem. Soc. Trans. 2019, 47, 701–711. [DOI] [PubMed] [Google Scholar]
- (43).Shao Q; Jiang Y; Yang ZJ EnzyHTP: A High-Throughput Computational Platform for Enzyme Modeling. J. Chem. Inf. Model. 2022, 62, 647–655. [DOI] [PubMed] [Google Scholar]
- (44).Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD; Roskies R; Scott JR; Wilkins-Diehr N XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data tables used in the analysis can be found in the Supporting Information. The curated data tables can also be accessed through our online IntEnzyDB web interface: https://intenzydb.accre.vanderbilt.edu. The website is maintained by the Yang lab.