

Abstract
Cryo-electron microscopy (cryo-EM) has become a major technique for protein structure determination. Many atomic structures have been derived from cryo-EM density maps of about 3Å resolution. Side-chain conformations are well determined in density maps with super-resolutions such as 1–2Å. It is desirable to have a statistical method to detect anomalous side-chains without a super-resolution density map. In this study, we analyzed structures derived from X-ray density maps with higher than 1.5Å resolution and those from cryo-EM density maps with 2–4 Å and 4–6 Å resolutions respectively. We introduce a histogram-based outlier score (HBOS) for anomaly detection in protein models built from cryo-EM density maps. This method uses the statistics derived from X-ray dataset (<1.5Å) as the reference and combines five features involving the distal block distance, side-chain length, phi, psi, and first chi angle of the residue. Higher percentages of anomalies were observed in the cryo-EM models than in the super-resolution X-ray models. Lower percentages of anomalies were observed in cryo-EM models derived after January 2017 than those derived before 2017.
Keywords: protein structure, cryo-electron microscopy, validation, statistics, X-ray, pipe-line, side-chain, anomaly
1. INTRODUCTION
Validation of protein structures is an important problem in the structural biology as more and more atomic models are being resolved from cryo-electron microscopy technique (cryo-EM) [1]. Since early 1990s, many of validation tools [2–10] have been developed to address various aspects such as validation of experimental data, validation of protein models, and validation of the fit between experimental data and protein models. In order to create comprehensive assessment criteria, the Protein Data Bank community convened a Validation Task Force (VTF) to develop standards, formats, and specifications. The wwPDB VTF includes X-ray VTF, NMR VTF, and 3DEM VTF for X-ray, NMR, and 3DEM data respectively. In 2011, X-ray VTF published its recommendation report on how to validate X-ray reconstructed protein structures [15]. After that, 3DEM VTF and NMR VTF released their recommendation reports based on the X-ray VTF report in 2012 [16] and 2013 [17]. wwPDB accepts and curates depositions using the OneDep system that implements the recommendations from wwPDB VTF [18, 33]. wwPDB generates validation reports for new depositions that contain the results from rigorous tests of structure model quality. The validation reports for X-ray structures have been updated by the D&A system in March 2017 with 2016 statistics. The validation reports for NMR and 3DEM structures in PDB were available since May 2016.
The wwPDB OneDep system is an integral validation pipe-line for X-Ray, NMR and EM models. The recommended validation of X-Ray VTF includes bonding geometry, conformation, quality of data set, and fitness to experimental data [18, 33]. Geometric criteria include covalent bond lengths, bond angles, chirality, and planarity. Target values are compared with expected value in wwPDB compilation 2012 to generate Z-scores. Residues with a Z-score greater than 5 are considered over-fitted residues and labeled as outliers. All-atom contacts are evaluated for the non-covalent fit of atomic interactions. The unfavorable overlaps of van der Waals shells are identified as clashes. Conformational criteria include Ramachandran [19] and rotamer [20] analysis. The combinations of phi, psi and side-chain torsion angles are compared with statistics. The residues with unfavorable combinations are labeled as outliers. Both geometry and conformation analysis are carried out by MolProbity [5–7] and the results are listed in model quality section of validation reports. For the quality of data set, X-ray VTF checks include twinning, translational non-crystallographic symmetry (NCS), anisotropic diffraction, and data incompleteness. The data set validation is implemented in phenix.xtriage [8] program of Phenix software [21]. The current criteria for fit of structures to experimental data in validation reports are the z-score of real-space R-value (RSRZ) and Rfree that quantify the fit of an atomic model to electron density [2, 31]. The calculation of RSRZ and Rfree are performed by the Electron-Density Server (EDS) [10] and DCC program [32] respectively. A residue with RSRZ greater than 2 is considered as an outlier. An Rfree much higher than R-value is often a sign of overfitting a model to the experimental density map [33]. EM VTF recommended to validate both EM density maps and models. Validation of EM density maps involves checking absolute hand determination [24, 25], data coverage, the agreement between images and class averages [26], and statistical assessment of maps [27]. Validation of cryo-EM models may either involve tools for X-ray data or fitting methods, such as EMFIT [28], to measure correlation coefficient between a cryo-EM density map and the density map calculated from the model. EM VTF recommended the development of EM model assessment criteria and the corresponding software [16].
In a previous study, we have observed slight distribution difference between the cryo-EM structures and those X-ray structures derived from density maps with higher than 1.5Å resolutions [22]. Here, we describe a statistical method for detecting anomalous conformations of residues in cryo-EM structures.
2. METHOD
The reference dataset X-ray-1.5 contains 9131 atomic structures determined from super-resolution (higher than 1.5Å) density maps using X-ray crystallography. The protein structures in X-ray-1.5 were extracted from PDB website (https://www.rcsb.org/) with the sequence similarity less than 90%. At such resolutions, both backbone and side-chain positions can be located precisely, and they represent most accurately determined conformations of residues. To remove potential bias of non-crystallographic-symmetry (NCS) [23], only the first chain of each protein in X-ray-1.5 was used.
Four EM datasets were collected. The high-resolution EM dataset downloaded in December 2016, referred as EM-2-4-b2017, contains 215 protein structures solved from cryo-EM density maps with resolutions between 2 and 4 Å. The second dataset, referred as EM-4-6-b2017, contains 163 protein structures derived from density maps with resolutions between 4 and 6 Å. The third EM dataset, referred as EM-2-4-a2017, contains 288 proteins solved from January 2017 to March 2018 from cryo-EM density maps with resolutions between 2 and 4 Å. The forth EM dataset, referred as EM-4-6-a2017, has 160 atomic structures solved from January 2017 to March 2018 from cryo-EM density maps with resolutions between 4 and 6 Å. To avoid bias of NCS, chains having over 95% similarity in each protein have been removed from the EM datasets.
Five selected features were used - block length dBlock, side-chain length dSC, backbone torsion angle Phi ϕ and Psi φ, and first side-chain torsion angle χ1, were used to represent residue conformations. The side-chain of each residue is divided into blocks. The blocks in each residue has been defined in our previous study [24]. dBlock is the distance between CA atom on the backbone and the mass center of the distal block in a specific residue. dSC is the distance between CA atom on backbone and the mass center of side-chain. dBlock and dSC are used to represent the side-chain conformation. Note that the range of ϕ and φ in this study is 0° - 360° instead of −180° - +180° in the Ramachandran plot [25]. 18 of 20 residues were used since glycine (GLY) and alanine (ALA) have no χ1 due to their small sizes of side-chains.
For each of the five features, a probability density function (pdf) was generated for each of the 18 residues using X-ray-1.5. The bin size of 5° was used for ϕ, φ, and χ1 respectively, and the bin size of 0.05 Å was used for dBlock and dSC respectively in calculation of the pdf. Each pdf then was normalized by dividing its highest peak value. A total of 90 (5*18) normalized pdf (npdf) plots were generated for each of the five features of 18 residue types in reference dataset X-ray-1.5.
| (1) |
Let npdfi,j(vi) be the normalized density function value for feature i and residue j when i = vi. For example, (3.0) is the function value when dBlock = 3.0 Å for Lys. For each residue, the Histogram-Based Outlier Score (HBOS) of a feature is the log value of its inverted npdf value. If the value of npdfi,j(vi) is less than 0.001, its HBOSj(vi) was assigned a value of 5 to avoid infinite HBOS value. As shown in eq. (1), the HBOS score of a residue is the summation of the five HBOS values from the five features. A residue with a high HBOS has low probability of occurrence and its conformation is unfavorable. An anomaly is labeled if the HBOS of a residue is above a threshold.
The python scripts and MATLAB scripts in the study have been deposited to Github repository at https://github.com/lin-chen-VA/CSBW2018.
3. RESULTS
The HBOS values were calculated for the residues in all five datasets. For each dataset, a probability histogram of HBOS value was generated (Figure 1). The histogram is normalized so that the total area under the curve is 1. The bin size used in generation of the histogram plots is 0.1. Since the protein structures in X-ray-1.5 dataset are determined from super-resolution density maps, they represent most accurate structures currently available. As expected, 99% of the residues in X-ray-1.5 dataset have relative low HBOS value (<6). The probability plot for X-ray-1.5 (red solid line) is a narrow curve that has a HBOS peak at 0.9. In contrast, the plots for four EM datasets have wider distribution with an HBOS peak near 1.1 (Figure 1). The HBOS histograms of four EM datasets have broader distribution with long tails. The five HBOS curves suggest that the four cryo-EM datasets have more conformations with larger HBOS values when they are compared to the configurations derived from super-resolution density maps using X-ray crystallography.
Figure 1. The probability histogram of HBOS values for five datasets.
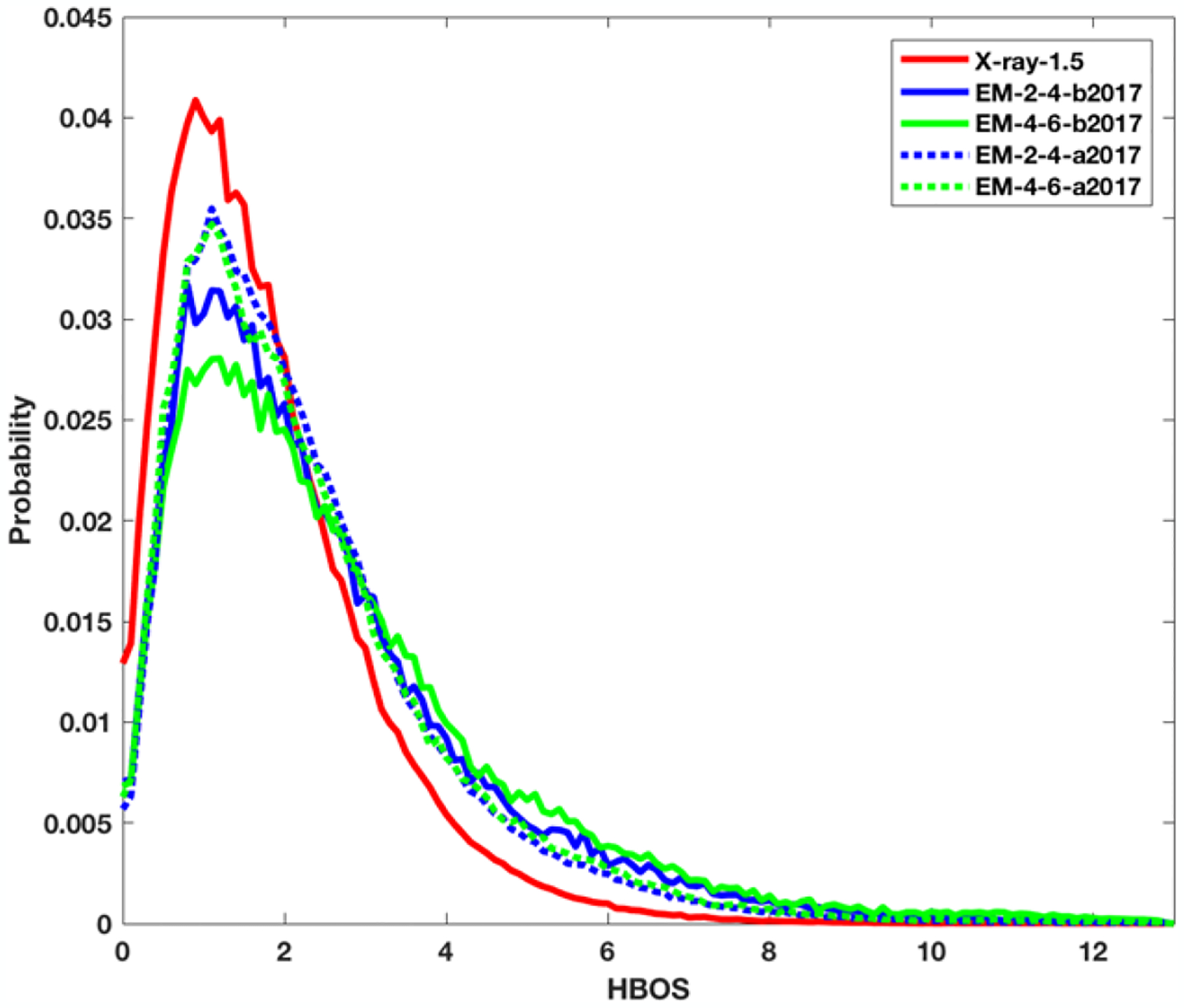
X-ray-1.5 (red solid line), EM-2-4-b2017 (blue solid line), EM-4-6-b2017 (green solid line), EM-2-4-a2017 (blue dashed line), and EM-4-6-a2017 (green dash line).
Although it is challenging to decide an anomaly cutoff value, we studied the effect of HBOS cutoff value on the percentage of anomalies across five datasets (Table 1). For X-ray-1.5 dataset, the percentage of anomalies is 0.23%, 0.13%, and 0.09% of 1,048,576 residues for a HBOS cutoff value at 8, 9, and 10 respectively. Note that there are only 923 of over 1 million residues with a HBOS value over 10. With the same three cutoffs, EM-2-4-b2017 has 2.04%, 1.2%, 0.74% anomalies respectively. The difference in percentage values shows that EM-2-4-b-2017 contains more percentage of unfavorable conformations at the same HBOS cutoff. For EM-4-6-b2017, involving density maps with lower resolutions (4–6Å), the percentages of anomalies are 3.04%, 2.05%, and 1.45%, slightly higher than those models derived from higher resolutions (2–4Å). The higher percentage may indicate that more percentage of residues derived from lower resolution density maps (4–6Å) have unfavorable conformations than those derived from density maps with higher resolutions (2–4Å). Generally, a popular conformation is considered as a favorable conformation and is assigned a low energy. Note that unfavorable conformations still exist in the X-ray-1.5 dataset, but the occurrence rate is much smaller than that in the other four datasets. An anomalous conformation is not necessarily a wrong conformation, rather an unpopular conformation that deserves further investigation. If we use HBOS cutoff of 10, there are 667, 856, 1807, and 1089 anomalous residues in the four datasets respectively (Table 1).
Table 1.
The Number of the Anomalies under Different Cutoffs of HBOS.
| HBOS Cutoff | >8 | >9 | >10 | ||||
|---|---|---|---|---|---|---|---|
| Data Set | #Totala | #b | % c | # d | % e | # f | % g |
| X-ray-1.5 | 1048576 | 2379 | 0.23 | 1354 | 0.13 | 923 | 0.09 |
| EM-2-4-b2017 | 90534 | 1844 | 2.04 | 1089 | 1.2 | 667 | 0.74 |
| EM-4-6-b2017 | 59099 | 1794 | 3.04 | 1210 | 2.05 | 856 | 1.45 |
| EM-2-4-a2017 | 595454 | 5812 | 0.98 | 3115 | 0.52 | 1807 | 0.3 |
| EM-4-6-a2017 | 216142 | 2715 | 1.26 | 1604 | 0.74 | 1089 | 0.5 |
The total number of residues in the dataset;
The number of residues with HBOS greater than 8b, 9d, and 10f is shown and the percentage of residues is shown for HBOS greater than 8c, 9e, and 10g respectively.
Two of the four cryo-EM datasets contain those structures derived after January of 2017, and two have structures before January 2017. We observed that those datasets after 2017 have higher peak probability values at about HBOS of 1.1 (dotted blue and green lines) than those structures obtained before 2017 (solid blue and green lines) when the same resolution range is considered (Figure 1). The percentage of residues with HBOS greater than 10 is also reduced from 0.74% to 0.3% when the structures before 2017 were compared to those structures after 2017 in the resolutions range of 2–4 Å (Table 1 Row 4 and Row 6). Our results may suggest an overall enhanced structure quality after January 2017. Note that the choice of using January 2017 is only due to the convenience of our available dataset previously collected. The enhanced structural quality may be related to the implementation of validation policies in the modeling process sometime before 2017. However, it is noted that the two histograms of EM-2-4-a2017 and EM-4-6-a2017 are almost overlapped. The corresponding percentage of anomalous residues in two datasets are 0.98%, 0.5%, 0.3% and 1.26%, 0.74%, 0.5% respectively in Table 1. The slight difference indicates that the two different resolution ranges does not create distinct difference on the quality of the structures as observed in EM-2-4-b2017 and EM-4-6-b2017. It is not clear if the small difference is related to practices of assigning favorable conformations extracted from known structures in low-resolution dataset EM-4-6-a2017.
Table 2 contains the details of four residues with HBOS over 10. The first three columns identify the labeled residues with PDB ID, chain, index, and residue type, the forth column contains the dataset name, the fifth column is the resolution of the density map, the sixth column is the rank in wwPDB validation report, the seventh column is the HBOS value, the rest five columns are the HBOS values of five validation features for the four residues. The rank of wwPDB validation report ranges from 0 to 3 representing from less likely outliers to most likely outliers. The current validation reports for EM proteins at wwPDB contain metrics considering the clash score, Rmachandran outliers, and side-chain outliers. Rfree and RSRZ values are not included in the validation report of cryo-EM structures, but available for X-ray structures. Figure 2 shows four residues labeled as anomalous residues with the HBOS cutoff 10. Those four residues were selected from EM-2-4-b2017, EM-4-6-b2017, EM-2-4-a2017, EM-4-6a2017 respectively from Fig. A to Fig. D respectively. Fig. 2A contains a residue PHE-126L labeled as anomaly by HBOS score. Its HBOS value of ϕ, φ and χ1 in Table 2 are 0.6797, 0.2357, 0.2136 which are located in the favorable region of those torsion angles. In the validation report of 5a0q [26], PHE-126L is labeled as 0 by geometric quality criteria.
Table 2.
Four Anomalies with HBOS Value Over 10.
| PDB ID | Index/Chain | Residue | Data Set | Resolution (Å) | Validation | HBOS | HBOSj(vi) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dBlock | dSC | Phi | Psi | Chi_1 | |||||||
| 5a0q | 126L | PHE | EM-2-4-b2017 | 2.7 | 0 | 11.129 | 5 | 5 | 0.6797 | 0.2357 | 0.2136 |
| 5lcw | 63K | ARG | EM-4-6-b2017 | 4.2 | 2 | 10.884 | 1.0752 | 1.6206 | 2.1432 | 1.0449 | 5 |
| 5h1s | 133X | LYS | EM-2-4-a2017 | 3.5 | 2 | 10.721 | 5 | 5 | 0.1713 | 0 | 0.5497 |
| 5w9k | 932A | TYR | EM-4-6-a2017 | 4.6 | 1 | 10.358 | 5 | 5 | 0.1412 | 0.2135 | 0.0033 |
Figure 2. Four anomalous examples with HBOS greater than 10.
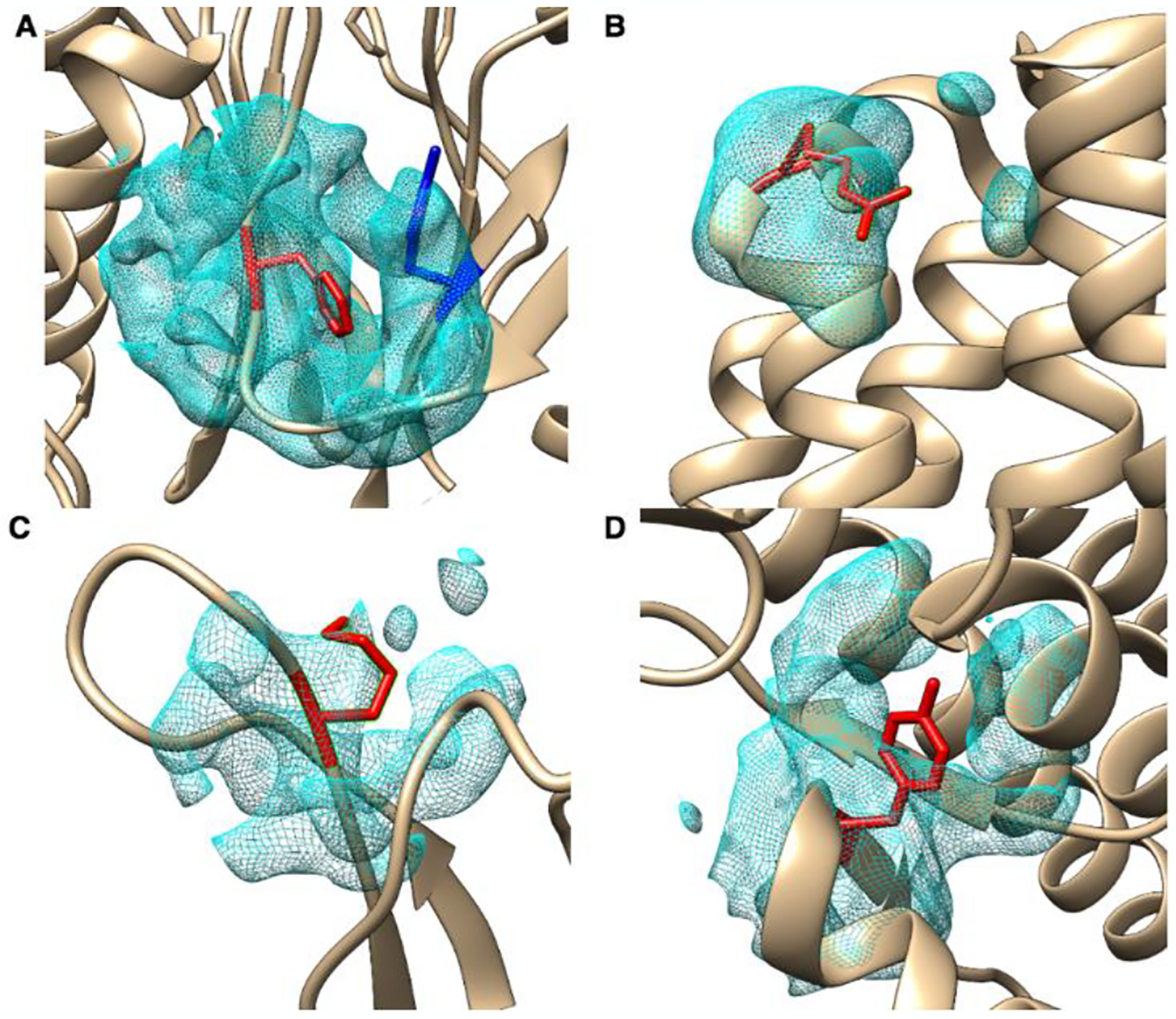
The identified residues (red) are superimposed with the atomic structures (ribbon) and the density at the local region. (A) phenylalanine (PHE) indexed as 126 in chain L of protein 5a0q, referred as PHE-126L; (B) arginine (ARG) indexed as 63 in chain K of protein 5lcw, referred as ARG-63K; (C) lysine (LYS) indexed as 133 in chain X of protein 5h1s, referred as LYS-133X; (D) tyrosine (TYR) indexed as 932 in chain A of protein 5w9k, referred as TYR-932A.
The current validation pipe-line at wwPDB suggests that PHE-126L has low probability of being an outlier. However, PHE-126L has HBOS value of 11.129, a sign of an anomaly if 10 is used as a threshold. This residue has 3.55 Å and 3.18 Å for dBlock and dSC respectively, far away from the favorable length of 3.78 Å and 3.43 Å. In Fig. 2A, PHE-126L (red) is located on an edge strand of a β-sheet and its bending created shorter block length. TYR-113L (blue), on another β-sheet, is closely located to PHE-126L. Being an anomalous residue does not mean the conformation is wrong. However, high HBOS value indicates that the conformation is significantly different from favorable conformations of the residue. Further analysis is needed. Since HBOS uses the length of the distal block and the length of the side-chain that were not used in the pipe-line generating validation report, some differences are expected. ARG-63K in Fig. 2B is in a turn of two consecutive α-helices. In this region, slight unfavorable values of ϕ, φ are can be expected. However, ARG-63K has an extremely high HBOS value 5 for χ1. It would be interesting to see if the unfavorable χ1 is related to fitting of the side-chain of ARG-63K to the density. Avoiding over-fitting of models is challenging in the fitting process. Note that the high HBOS value is mostly contributed from χ1 and slightly high contributions from dBlock and dSC (1.075 and 1.621 respectively) (Table 1).
Based on the HBOS value 10.884, ARG-63K is labeled as an anomaly. In the validation report of 5lcw [29], ARG-63K is ranked 2 and has questionable geometric quality. LYS-133X in Fig. 2C is labeled as anomaly due to its small side-chain size, 5 for both dBlock and dSC. Again, it will be interesting to see if the small side-chain size is related to the need of fitting, since the density cloud at the location of LYS-133X has low density in part of the side-chain region. TYR-932A (Fig. 2D) is at the turn between an α-helix and a β-strand. Its ϕ, φ and χ1 have favorable value. It is ranked 1 in the validation report of 5w9k [30] that represents low risk of being an outlier. However, the combination of those torsion angles causes an extremely small size of side-chain though there appears to have sufficient space to stretch its side-chain. The benzene ring of TYR-932A is exposed outside the density cloud instead of being fitted into the large chunk of density cloud in the opposite direction near TYR-932A.
4. CONCLUSION
Validation of atomic structures is an important but complicated process to maintain the quality of databases. We proposed, in this paper, an approach to monitor HBOS value and we investigated HBOS for five datasets. The distribution of HBOS values shows that the structures derived from super-resolution density maps (higher than 1.5Å) have extremely low occurrences of HBOS values over 10. The highest occurrence percentage were seen from the cryo-EM density maps with 4–6 Å resolutions among the five datasets. Our study suggests that the process of implementing validation criteria may have enhanced the quality of models derived from cryo-EM density maps since 2017. However, it is always desirable to learn from those models derived from super-resolution density maps and to establish practices to enhance the quality even more. We introduce an approach to combine five features to screen for outliers. The combined features may be more sensitive to use than individual features. We observed that the use of two side-chain features dBlock and dSC is simple and sensitive in screening sidechain conformations. The HBOS score function, instead of pipelining validation of one or more features, evaluates the combination of five features including three torsion angles (φ, ψ, χ1) and two distances (dBlock, dSC).
CCS CONCEPTS.
Applied computing → Life and medical sciences → Computational biology → Molecular structural biology
ACKNOWLEDGEMENTS
The work in this paper is supported by NSF DBI-1356621, NIH R01-GM062968, and NIH 4T34GM100831-05.
Contributor Information
Lin Chen, Department of Mathematics & Computer Science, Elizabeth City State University, Elizabeth City, NC 27909.
Jing He, Department of Computer Science, Old Dominion University, Norfolk, VA 23529.
REFERENCES
- [1].Kleywegt GJ and Jones TA (2002). “Homo Crystallographicus—Quo Vadis?” Structure 10(4): 465–472. [DOI] [PubMed] [Google Scholar]
- [2].Jones TA, et al. (1991). “Improved methods for building protein models in electron density maps and the location of errors in these models.” Acta Crystallogr A 47 (Pt 2): 110–119. [DOI] [PubMed] [Google Scholar]
- [3].Laskowski RA, et al. (1993). “PROCHECK: a program to check the stereochemical quality of protein structures.” Journal of Applied Crystallography 26(2): 283–291. [Google Scholar]
- [4].Hooft RW, et al. (1996). “Errors in protein structures.” Nature 381(6580): 272. [DOI] [PubMed] [Google Scholar]
- [5].Lovell SC, et al. (2003). “Structure validation by Calpha geometry: phi,psi and Cbeta deviation.” Proteins 50(3): 437–450. [DOI] [PubMed] [Google Scholar]
- [6].Davis IW, et al. (2004). “MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes.” Nucleic Acids Res 32(Web Server issue): W615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC. (2010). All-atom structure validation for macromolecularcrystallography. Acta Crystallogr. D Biol. Crystallogr 66 Pt 1, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zwart PH, Grosse-Kunstleve RW, & Adams PD (2005). “Xtriage and Fest: automatic assessment of X-ray data and substructure structure factor estimation.” CCP4 newsletter 43. [Google Scholar]
- [9].Bruno IJ, et al. (2004). “Retrieval of crystallographically-derived molecular geometry information.” J Chem Inf Comput Sci 44(6): 2133–2144. [DOI] [PubMed] [Google Scholar]
- [10].Kleywegt GJ, et al. (2004). “The Uppsala Electron-Density Server.” Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1): 2240–2249. [DOI] [PubMed] [Google Scholar]
- [11].Berman H, et al. (2007). “The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data.” Nucleic Acids Res 35(Database issue): D301–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berman HM, et al. (2000). “The Protein Data Bank.” Nucleic Acids Res 28(1): 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Standley DM, et al. (2008). “Protein structure databases with new web services for structural biology and biomedical research.” Brief Bioinform 9(4): 276–285. [DOI] [PubMed] [Google Scholar]
- [14].Ulrich EL, et al. (2008). “BioMagResBank.” Nucleic Acids Res 36(Database issue): D402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Read RJ, et al. (2011). “A new generation of crystallographic validation tools for the protein data bank.” Structure 19(10): 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Henderson R, et al. (2012). “Outcome of the First Electron Microscopy Validation Task Force Meeting.” Structure(London, England:1993) 20–330(2): 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Montelione GT, et al. (2013). “Recommendations of the wwPDB NMR Validation Task Force.” Structure (London, England : 1993) 21(9): 10.1016/j.str.2013.1007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gore S, et al. (2012). “Implementing an X-ray validation pipeline for the Protein Data Bank.” Acta Crystallogr D Biol Crystallogr 68(Pt 4): 478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ramachandran GN, et al. (1963). “Stereochemistry of polypeptide chain configurations.” J Mol Biol 7: 95–99. [DOI] [PubMed] [Google Scholar]
- [20].Shapovalov MV and Dunbrack RL Jr. (2011). “A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions.” Structure 19(6): 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Adams PD, et al. (2010). “PHENIX: a comprehensive Python-based system for macromolecular structure solution.” Acta Crystallogr D Biol Crystallogr 66(Pt 2): 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen L, He J, Sazzed S, and Walker R (2018). An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains. Molecules 23(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].He J, et al. (2001). “Finding and using local symmetry in identifying lower domain movements in hexon subunits of the herpes simplex virus type 1 B capsid.” J Mol Biol 309(4): 903–914. [DOI] [PubMed] [Google Scholar]
- [24].Chen L and He J. (2014). “A distance- and orientation-dependent energy function of amino acid key blocks.” Biopolymers 101(6): 681–692. [DOI] [PubMed] [Google Scholar]
- [25].Ramachandran GN, et al. (1963). “Stereochemistry of polypeptide chain configurations.” J Mol Biol 7: 95–99. [DOI] [PubMed] [Google Scholar]
- [26].daFonseca PCA; Morris EP 2018. Full wwPDB/EMDataBank EM Map/Model Validation Report. Retrieved from https://files.rcsb.org/pub/pdb/validation_reports/a0/5a0q/5a0q_full_validation.
- [27].Penczek PA, Frank J, Spahn CMT.(2006). A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J. Struct. Bio 154, 184–194. [DOI] [PubMed] [Google Scholar]
- [28].Rossmann MG, Bernal R, and Pletnev SV. (2001). Combining Electron Microscopic with X-ray Crystallographic Structures. J. Struct. Biol 136, 190–200. [DOI] [PubMed] [Google Scholar]
- [29].Alfieri C, Chang L, Zhang Z, et al. 2016. Full wwPDB/EMDBBank EM Map/Model Validation Report. Retrieved from https://files.rcsb.org/pub/pdb/validation_reports/lc/5lcw/5lcw_full_validation.pdf.
- [30].Pallesen J, Ward AB 2017. Full wwPDB/EMDBBank EM Map/Model Validation Report. Retrieved from https://files.rcsb.org/pub/pdb/validation_reports/w9/5w9k/5w9k_full_validation.pdf.
- [31].Brünger AT (1992). Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- [32].Yang H, Peisach E, Westbrook JD, Young J, Berman HM, and Burley SK (2016). DCC: a Swiss army knife for structure factor analysis and valida- tion. J. Appl. Cryst 49, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gore S et al. (2017). Validation of Structures in the Protein Data Bank. Structure, 25(12), 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]