

Abstract
We describe an 11-year old boy with severe global developmental delays, failure to thrive and growth retardation, refractory seizures with recurrent status epilepticus, hypogammaglobulinemia, hypergonadotropic hypogonadism, and duodenal strictures. He had facial and skin findings compatible with trichothiodystrophy, including sparse and brittle hair, thin eyebrows, and dry skin. Exome sequencing showed a hemizygous, truncating variant in RNF113A, c.903_910delGCAGACCA, predicting p.(Gln302fs*12), that was inherited from his mother. Although his clinical features overlap closely with features described in the two previously reported male first cousins with RNF113A loss of function mutations, the duodenal strictures seen in this patient have not been reported. Interestingly, the patient’s mother had short stature and 100% skewed X-inactivation as seen in other obligate female carriers. A second male with developmental delays, microcephaly, seizures, ambiguous genitalia, and facial anomalies that included sparse and brittle hair, thin eyebrows and dry skin was recently reported to have c.897_898delTG, predicting p.(Cys299*) in RNF113A and we provide additional clinical details for this patient. This report further supports deleterious variants in RNF113A as a cause of a novel trichothiodystrophy syndrome.
Keywords: RNF113A, spliceosome, trichothiodystrophy, zinc finger
1 |. CASE REPORTS
In the pregnancy, the patient’s mother was reported to have taken paroxetine during the first month. Decreased fetal movements and pregnancy-induced hypertension were recorded. Labor was induced at 38 weeks and 1 day of gestation due to a nonreassuring fetal stress test with bradycardia and the baby was delivered by C-section with a birthweight of 2,301 g (<3rd centile). A postnatal Ballard assessment was consistent with 35 weeks gestational age. In the neonatal period, continued hospitalization was required for polycythemia (hematocrit 67%) manifesting as respiratory distress and the patient was treated with partial exchange transfusion. He was diagnosed with hypoglycemia due to transient neonatal hyperinsulinism that required treatment with diazoxide until 14 months of age. Cryptorchidism and a small phallus were noted and he underwent orchidopexy. Investigations revealed elevated levels of luteinizing hormone (LH), follicle stimulating hormone (FSH), and prolactin, but testosterone was low, consistent with hypergonadotropic hypogonadism. Assessment of remaining pituitary fuction was normal. A karyotype (46,XY) and subtelomeric probe study were normal and he passed his newborn hearing screen.
In early childhood, his milestones were delayed and he was diagnosed with global developmental delays. At 4 years of age, he was able to crawl and walk with hands held. He had around 10 words and was able to drink and feed himself. He walked independently at 7 years of age. At 11 years of age, he could sign, speak several single words and follow commands, although he was considered largely non-verbal and required special education.
Seizures were first diagnosed at 4 months of age. Episodes have involved cyanosis, facial twitching with eye rolling, drooling, leaning forward and full-body tremor. Electroencephalograms confirmed seizure activity and a magnetic resonance imaging (MRI) scan of the brain revealed thinning of the corpus callosum. He has since developed refractory, focal epilepsy with impairment of consciousness and a history of episodes of status epilepticus. He has required multiple anti-epileptic medications and has had admissions for seizure treatment. A trial of a ketogenic diet was not well tolerated due to weight loss. A vagal nerve stimulator was placed at 11 years of age and resulted in reduced seizure frequency.
He has had slow weight gain and was treated with a gastrostomy tube (G-tube). He developed persistent emesis and inability to tolerate G-tube feeds and total parenteral nutrition was instituted. An upper gastrointestinal tract endoscopy showed multiple strictures in the first, second, and third portions of the duodenum and he underwent laparoscopic gastrojejunostomy and budesonide treatment. Biopsies of the gastrointestinal tract have not shown diagnostic abnormalities. A liver biopsy was performed for transaminitis at 3 months of age and revealed a ductular reaction with possible ductopenia, but electron microscopy studies were normal.
He was diagnosed with hypogammaglobulinemia with a low immunoglobulin (Ig) G level (371 mg/dl; normal range 672–1,760 mg/dl). Testing of IgG subclasses showed a low level of IgG subclass 1 (234 mg/dl; normal range 390–1,235 mg/dl); a repeat sample showed a similarly low level of IgG1 at 252 mg/dl. IgG subclasses 2–4, IgM and IgA levels were in the normal range. In view of frequent infections, he was treated with a trial of monthly intravenous immunoglobulin (IVIG) at 500 mg/kg and his IgG level improved, but did not normalize (636 mg/dl; normal range 672–1,760 mg/dl). He had a protective rubella titer, but low mumps, measles, pneumococcal and tetanus titers. He has developed a mild pancytopenia with low white blood cell (WBC) counts (WBC 2.4 × 10E9/L; normal range 4.5–15.5 × 10E9/L) and a low absolute lymphocyte count of 0.46 × 10E9/L (normal range 1.2–8.0 × 10E9/L). He has also been diagnosed with a macrocytic anemia.
Other medical problems have included acute cholecystitis complicated by a biloma and treated by cholecystectomy. He has also had chronic cholangitis managed by a percutaneous, transhepatic biliary drain insertion. He has been diagnosed with cortical visual impairment, but an ophthalmology examination showed normal ocular structures in the neonatal period and at 4 years of age. There was no history of photosensitivity or ichthyosis.
His mother is of Caucasian ancestry. Few details are known about his father. Consanguinity was denied. The proband has three paternal half-siblings in good health. His mother had short stature with a height of 147 cm (2nd centile) and was otherwise well. The maternal grandmother had short stature with a height of 144.7 cm (1st centile) and was reported to have photosensitivity, but no further details are known.
On examination at 11 years and 10 months of age, growth was severely delayed (Figure 1). Weight was 24.3 kg (0.08th centile; Z score-3.16) and height was 121 cm (<0.01th centile; Z score-3.92). His occipitofrontal circumference was 50 cm (4.62th centile; Z score-1.68) at 7 years and 11 months of age. He has had mild facial anomalies with dolichocephaly, a slightly high anterior hairline and a prominent forehead (Figure 2). His hair was sparse, brittle, thin and “wool-like.” Eyebrows were sparse. He had downslanting palpebral fissures, a well-defined philtrum and a slightly thin vermilion of the upper lip with widely spaced, peg-shaped teeth. Nose, ears and palate were unremarkable. His abdomen was unremarkable except for presence of a G-tube. Genitourinary examination revelaed bilateral undescended testes and a small phallus. He had mild syndactyly of the second to third toes. He had tapered fingers with normal nails and thickened palmar skin. His skin was dry with diffuse cutis marmorata. He had hypotonia and a wide-based gait. He was affectionate with his family.
FIGURE 1.
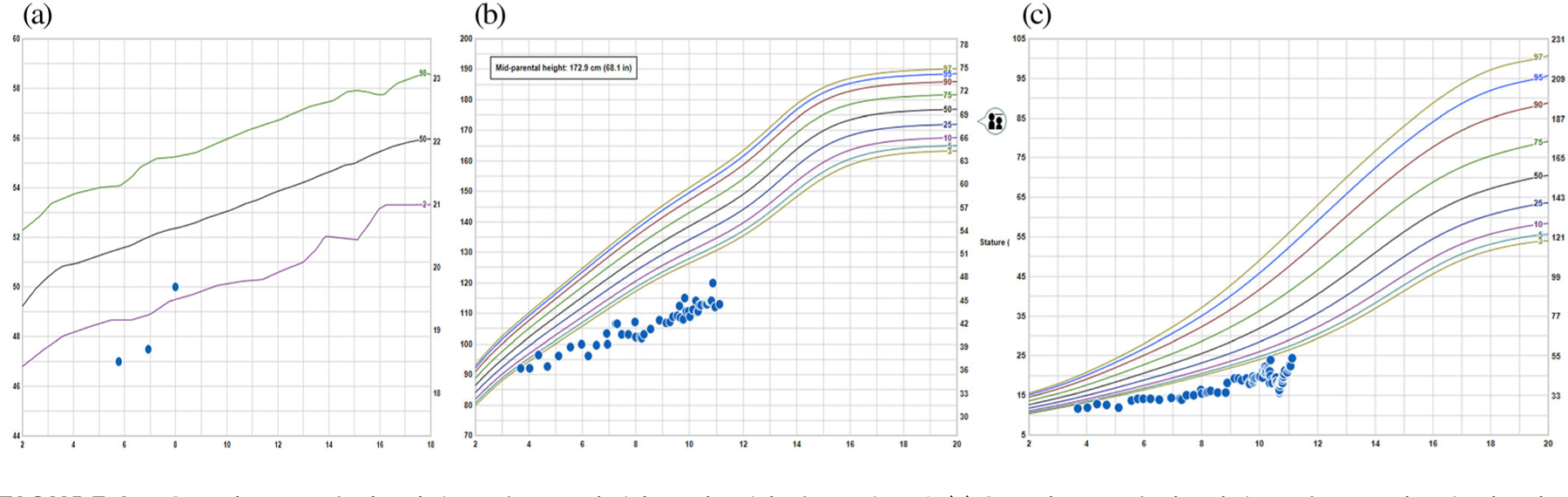
Growth curves for head circumference, height and weight for Patient 1. (a) Growth curve for head circumference showing head circumference measurements below the normal range at 5 years, 9 month and 6 years, 11 months. (b) Growth curve for height, showing height measurements consistently below the normal range from 3 to 4 years of age. Midparental height is estimated to be at the 25th centile. (c) Growth curve for weight, showing weight measurements consistently below the normal range from 3 to 4 years of age
FIGURE 2.
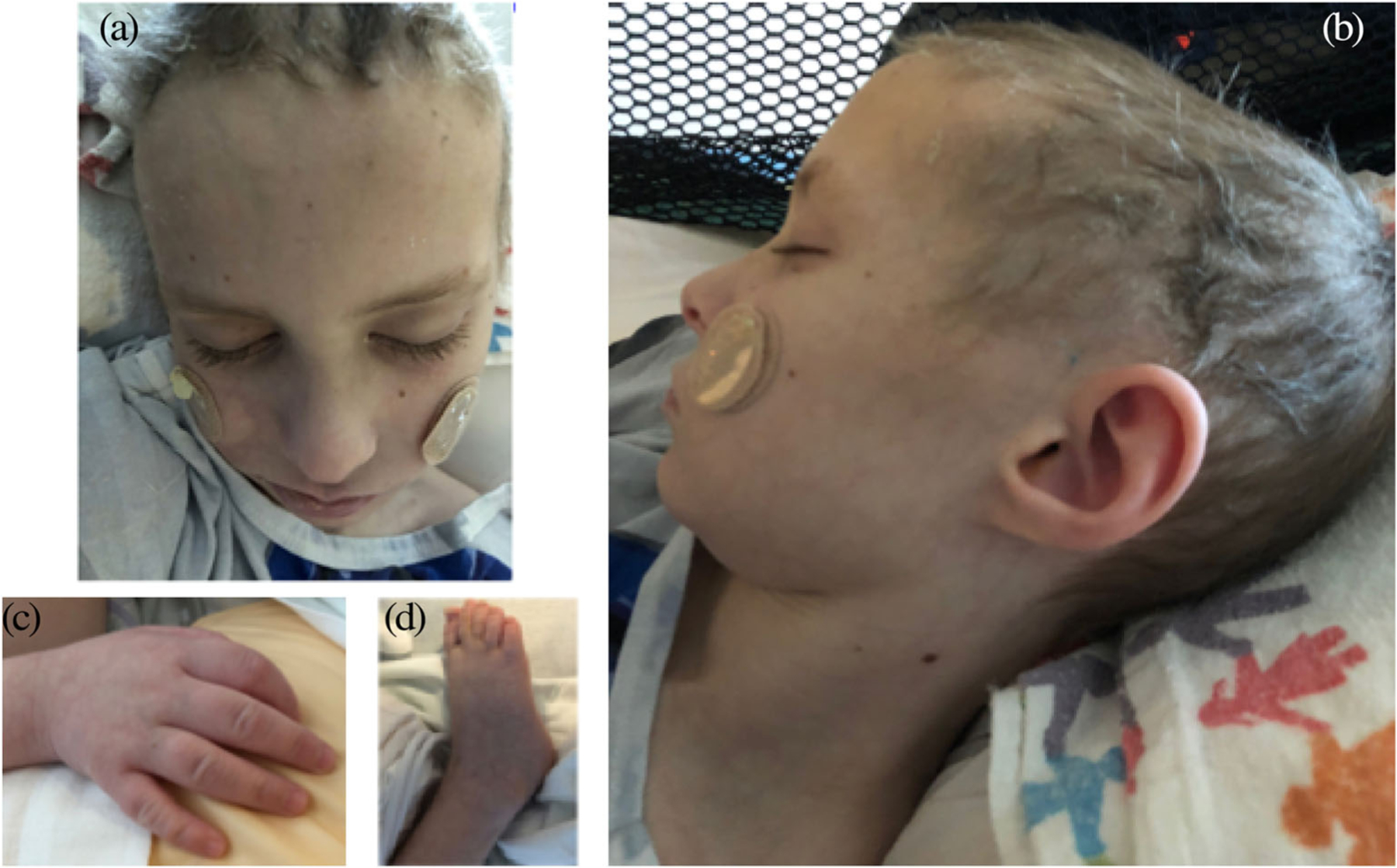
Physical features of Patient 1, an 11-year old male diagnosed with X-linked trichothiodystrophy due to a truncating variant in the RNF113A gene. (a) Facial photograph showing sparse and brittle hair. (b) Profile photograph showing dolichocephaly. He also had downslanting palpebral fissures, low columella, widely spaced, and peg-shaped teeth. (c) Photograph of hand showing tapered fingers. (d) Photograph of foot showing mild 2–3 toe syndactyly
Investigations prior to exome sequencing included a normal microarray (45K oligoarray). Levels of ceruloplasmin (21.7 mg/dl; normal range 19–68 mg/dl) and copper (76 mcg/dl; normal range 27–153 mcg/dl) were within the normal range. Serum amino acids, acylcarnitine profile, ammonia, lactate and pyruvate, very long chain fatty acids, biotinidase, galactose-1-phosphate uridyl transferase, urine organic acids, testing for congenital disorders of glycosylation, Smith–Lemli–Opitz syndrome, and alpha 1 antitrypsin deficiency were nondiagnostic. A sweat test for cystic fibrosis was negative and a TORCH screen was nondiagnostic. Sequencing for Alagille syndrome and for progressive familial intrahepatic cholestasis (PFIC) was negative. A MRI scan of the brain showed hypoplasia of the corpus callosum. Radiographs of the spine showed no vertebral anomalies. Hair analysis with polarized light microscopy showed focal trichorrhexis nodosa and a “vague” pattern of bands, but a tiger-tail banding pattern was not observed. Hair amino acid analysis was not able to be performed.
Clinical whole exome sequencing (ES) was performed as a duo (proband and mother) as part of the Prenatal and Pediatric Genomic Sequencing Program (P3EGs) at the University of California, San Francisco (UCSF). Written, informed consent was obtained for study participation. ES was performed as a clinical test using a bioinformatics pipeline developed by the Institute for Human Genetics (IHG) at UCSF. Exon regions were targeted in extracted genomic DNA from the submitted proband and his mother using the xGen Whole Exome Panel kit (Integrated DNA Technologies). Targeted regions were sequenced using the Illumina HiSeq 2500 sequencing system (v3 chemistry) with 100 bp paired-end reads in rapid run mode. The resulting DNA sequences were mapped to and analyzed in comparison with the published human genome (UCSC hg19 reference sequence). The Ingenuity Variant Analysis (IVA, Qiagen) program was used to filter out likely benign variants and to analyze the proband for candidate de novo, homozygous, compound heterozygous and inherited heterozygous variants that were possibly disease causing. Several filters were applied in a stepwise fashion: confidence filter, common variant filter, predicted deleterious filter, custom filters (elimination of common variants ~3 or more alleles from 80 geographically diverse controls- and pseudo-autosomal regions). The UCSF bioinformatics pipeline utilized five different genotype callers for variant calling. To reduce the high number of false positive calls that originate from variants called by a single variant caller, in performing de novo analysis, only variants called by 2 or more variant callers were analyzed. For inherited heterozygous variants, lower allele frequency cutoff (0.1%) and a patient specific primary gene list were also used for filtering.
Human Gene Mutation Database-Professional (HGMD-Pro), Clin-Var, and Online Mendelian Inheritance in Man (OMIM) databases were evaluated both for gene-specific variants and gene-disease relationships. Pubmed, Pubmed Central, and Google Scholar were also used when no well-defined gene-disease relationship was established in HGMD-Pro and OMIM and if these databases did not include the gene specific variant identified after filtering as described above. Findings were evaluated using the published American College of Medical Genetics and Genomics criteria for variant calling (Richards et al., 2015).
The results of the exome duo showed a maternally inherited, hemizygous variant in RNF113A, c.903_910delGCAGACCA, predicting p.(Gln302fs*12) (NM_006978.3). Although the variant is predicted to cause a premature stop codon, RNF113A comprises a single exon. It is unclear if the variant would induce nonsense mediated decay; however, this variant affects the adjacent Gln residue to p.Gln301*, the only other reported pathogenic variant, and is conserved in all species (Corbett et al., 2015). p.(Gln302fs*12) is predicted to result in disruption of a portion of the really interesting new gene (RING) domain and zinc finger domain of the protein (Corbett et al., 2015). We considered that this variant was likely pathogenic based on location in a mutational hot spot and/or critical and well established functional domain (PM1), absence from control databases (PM2), and the strong phenotypic overlap with other reported patients (PP4; Richards et al., 2015). Maternal X-inactivation studies showed skewed X-inactivation with a ratio of 100:0.
The second patient was a 2-year old male with developmental delays and a truncating variant in RNF113A (Monies et al., 2019). He was delivered at term by C-section for fetal distress to parents of Saudi ethnicity. Birth weight was 1,900 g (<3rd centile). The baby remained in the neonatal intensive care unit for 20 days due to respiratory distress and jaundice. He represented at 16 months of age with global developmental delays, failure to thrive, short stature and microcephaly (Figure 3) and a seizure disorder. Facial anomalies comprised a triangular face with a broad forehead, pointed and cupped, posteriorly rotated and low-set ears and a wide mouth (Figure 4). He had brittle and sparse hair that was light in color, with absent eyelashes and eyebrows and mottled, dry skin. The genitalia were ambiguous, with severe hypospadias and both testes were not palpable. MRI of the brain showed absence of the corpus callosum and abdominal imaging revealed a multicystic, dysplastic kidney. Investigations revealed normal results for karyotype, thyroid function tests, cortisol, adrenocorticotrophic hormone (ACTH) and 17-hydroxyprogesterone. Hair analysis was not performed. Exome sequencing (Monies et al., 2019) showed hemizygosity for c.897_898delTG, predicting p. (Cys299*) in exon 1 of RNF113A (NM_006978.3). This variant was absent from gnomAD, 1000 Genomes, the ExAC database and HGMD and was predicted to result in a frameshift and premature termination codon (MutationTaster; Schwarz, Rodelsperger, Schuelke, & Seelow, 2012).
FIGURE 3.
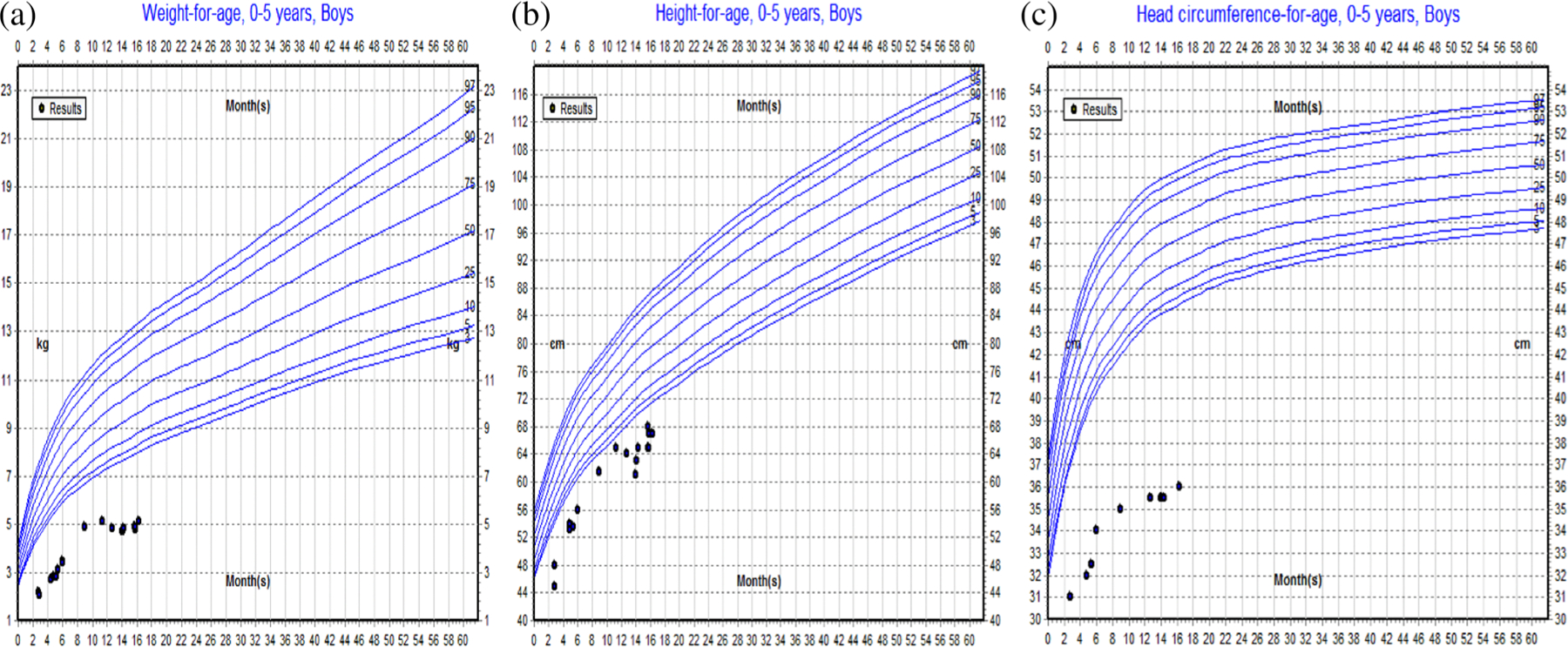
(a) Growth curves for weight, height and head circumference for patient 2. Growth curve for weight, showing weight measurements consistently below the normal range at all ages. (b) Growth curve for height, showing height measurements consistently below the normal range at all ages. (c) Growth curve for head circumference, showing head circumference measurements consistently below the normal range at all ages
FIGURE 4.
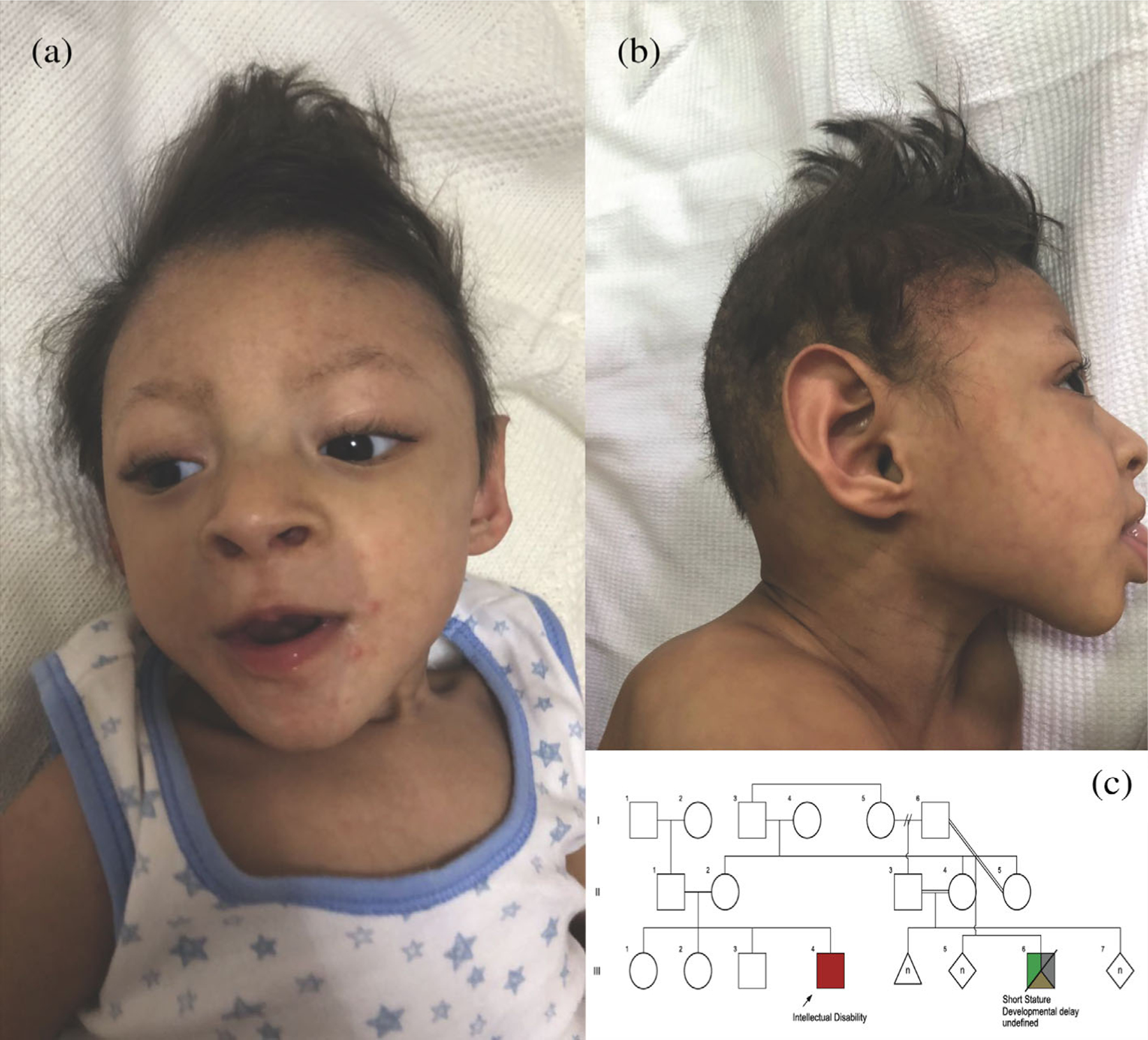
Facial photographs and pedigree of Patient 2, a 2 year old male with a truncating variant in the RNF113A gene. (a) Frontal photograph showing a high anterior hairline abd broad forehead. (b) Profile photograph showing pointed and cupped, posteriorly rotated, and low-set ears. (c) Pedigree of the patient’s family. The patient is indicated by the arrow
The trichothiodystrophies (TTDs) are a heterogeneous group of conditions characterized by sparse, brittle and sulfur-deficient hair that often demonstrates a “tiger-tail” banding pattern visible under polarizing light microscopy (Corbett et al., 2015). The clinical manifestations of TTDs show high phenotypic heterogeneity and affect a wide range of tissues derived from ectoderm or neuroectoderm (Corbett et al., 2015; Faghri, Tamura, Kraemer, & Digiovanna, 2008). We could find only one other description of a truncating, hemizygous variant in RNF113A — c.901C>T, predicting p.(Gln301*) (NM_00 6978.2) that was reported in two male first cousins who were related through the maternal lineage (Corbett et al., 2015). Both cousins manifested intrauterine growth restriction and severe linear growth failure with progressive microcephaly, profound intellectual disability, genital anomalies with a microphallus and absent or rudimentary testes, absence of the posterior portion of the corpus callosum, cerebellar hypoplasia and a Dandy-Walker malformation and ataxic and broad-based gaits (Table 1; Corbett et al., 2015). Other shared findings were sparse, brittle and slow-growing scalp hair and eyebrows with reduced body hair, cutis marmorata, and an aged facial appearance with a high forehead, broad mouth with widely spaced teeth and a prominent chin. One cousin had generalized cerebral atrophy, a short esophagus with a thoracic stomach and upper renal tract obstruction that required surgical intervention. The other cousin had an IgG1 subclass deficiency, recurrent infections and chronic diarrhea. Examination of hair using polarizing light microscopy revealed the characteristic tiger-tail pattern and hair amino acid content analysis showed sulfur deficiency diagnostic of TTD in both cousins (Corbett et al., 2015). Neither cousin had the photosensitivity or ichthyosis that is typical with TTD (Faghri et al., 2008). The clinical overlap between the two cousins and these two patients is significant (Table 1) and implicates the RNF113A variants in the phenotype of these patients. Of interest, the first patient and one of the cousins had an IgG1 subclass deficiency that may require treatment.
TABLE 1.
Clinical features of patients with hemizygous RNF113A variants
| Clinical feature | IV-2 (Corbett et al., 2015) | IV-5 (Corbett et al., 2015) | This paper, patient 1 | This paper, patient 2 |
|---|---|---|---|---|
| Age at last review | 15 years | 9 years | 11 years 10 months | 16 months |
| Birth parameters | 2.18 kg/31 cm @36 weeks (<10th/25th centile) | 2.32 kg/29 cm @38 weeks (<3rd centile) | Not known | 1.9 kg @39 weeks (<1st centile) |
| Growth parameters | ||||
| Height | 112 cm | 117 cm | 121 cm (<1st centile) | 68 cm (<1st centile) |
| Weight | 24 kg | 19.5 kg | 24.3 kg (<1st centile) | 5 kg (<1st centile) |
| OFC | 44.5 cm (<1st centile) | 44 cm (<1st centile) | 50 cm (5th centile) | 36 cm (<1st centile) |
| Development | ||||
| Motor | Delayed | Delayed | Walked at 7 years | Delayed |
| Speech | Delayed | Delayed | Single words | Delayed |
| Gonadal dysgenesis | Microphallus; absent testes | Absent/rudimentary testes | Microphallus/cryptorchidism | Microphallus/cryptorchidism/hypospadias/bifid scrotum |
| Endocrine abnormality | High TSH; normal GH | LowT4/high TSH Low ADH Low cortisol Normal GH High LH, FSH Low testosterone |
High TSH, low/Normal T4 High LH, FSH, prolactin Low testosterone Normal IGF1, IGFBP3 Normal cortisol |
Normal T4/TSH Normal ACTH Normal 17 hydroxyprogesterone Normal cortisol |
| Infections | Frequent hospitalizations | Frequent infections; chronic diarrhea; low IgG1 | Frequent infections; low IgG1 | Frequent infections |
| CNS findings | Cerebellar hypoplasia Cerebral atrophy Absent corpus callosum |
Cerebellar hypoplasia Absent corpus callosum |
Thin corpus callosum | Absent corpus callosum |
| Other structural findings | Short esophagus; ureteric obstruction | Duodenal strictures | Multicystic dysplastic kidney | |
| Hair analysis | Tiger-tail pattern | Tiger-tail pattern | Atypical bands | Not done |
| Photosensitivity | Nil | Nil | Nil | Nil |
| Ichthyosis | Nil | Nil | Nil | Nil |
| Ocular abnormalities | Nil | Nil | Cortical visual impairment | Nil |
| RNF113A variant | p.(Gln301*) | p.(Gln301*) | p.(Gln302fs*12) | p.(Cys299*) |
Abbreviation. CNS, central nervous system.
The reported p.(Gln301*) variant was predicted to remove eight identical residues from the C-terminus of the protein, disrupting a substantial portion of the RING domain (Corbett et al., 2015). A truncated form of RNF113A expressed at a reduced level was demonstrated in both cousins compared to control samples using Western blotting, but immunocytochemistry indicated that the predominantly nuclear localization of the protein was not affected by the p.(Gln301*) variant (Corbett et al., 2015). The frameshift variant described in the first patient in this report is remarkable in that it affects the adjacent Gln residue and can be predicted to have similar effects.
Interestingly, the first patient’s mother had short stature and three obligate female carriers from the family with the two affected cousins also had short stature, with heights recorded at 142 and 151 cm (<3rd centile; Corbett et al., 2015). One female carrier from that family had delayed walking and speech with learning difficulties in school and slow growing hair with some patchiness, but there was no tiger-tail banding on polarized light microscopy (Corbett et al., 2015). All three obligate carriers from the reported family also had 100% skewed X-chromosome inactivation (Corbett et al., 2015), similar to the mother of the first proband reported here, who also demonstrated 100% skewed X-inactivation. This degree of skewing implies that the short stature, delays and slow-growing hair may not be due to the mutant RNF113A allele.
RNF113A was first studied as zinc finger 183 (ZNF183) and mapped to chromosome Xq24–25 (Frattini et al., 1997). Sequence analysis showed an intronless gene with an open reading frame of 343 amino acids and an C3HC4 amino acid motif, or RING finger domain, close to the C-terminus of the predicted protein (Frattini et al., 1997). A Northern blot showed a single transcript of 1.3 kb size that was detected in all tissues tested, including brain, heart, small intestine and colon, testis, liver, muscle and lymphocytes (Frattini et al., 1997). The gene is conserved from yeast to humans (Carney, Struck, & Doe, 2013).
Several animal models have examined the importance of RNF113A function and support the emerging clinical phenotypes noted above. In zebrafish, disruption of rnf113a by transgenic insertional mutagenesis results in a small and slightly necrotic head, small eyes, pericardial oedema, and an underdeveloped liver and gut (Amsterdam et al., 2004). In flies, knockdown of the RNF113A ortholog, midlife crisis (mdlc), leads to reduced proliferation of neuroblasts (Carney et al., 2013). Neuronal differentiation was initiated, but not completed in the mdlc mutants and the expression of Prospero, a pro-differentiation transcription factor, was reduced (Carney et al., 2013). Conservation of function between flies and humans was demonstrated by expression of the full-length human gene that was effective in rescuing the central nervous system defects of the mdlc mutant (Carney et al., 2013). The zinc-finger domain was essential for mdlc function in the nervous system and for viability, but the RING domain was dispensable, despite high conservation of this domain (Carney et al., 2013). In Caenorhabditis elegans, knockdown of RNF-113 sensitizes cells to ultraviolet A (UVA) induced DNA damage and is required for RAD51 focus formation during DNA interstrand crosslink repair (Lee, Alpi, Park, Rose, & Koo, 2013).
RNF113A was found to physically interact with one of the human E2 proteins, UBE2U, and has been thought to function as an E3 ligase (Brickner et al., 2017; van Wijk et al., 2009). There is substantial evidence that RNF113A is involved in spliceosome function (Gatti da Silva, Jurica, Chagas da Cunha, Oliveira, & Coltri, 2018; Wu, Chung, & Cheng, 2017). Cwc24 interacts with the U5-RNA helicase, Brr2, in Saccharomyces cerevisiae and a similar interaction between RNF113A and BRR2 has been noted in human cells (Coltri & Oliveira, 2012; Goldfeder & Oliveira, 2008). The yeast orthologue, Cwc24, transiently associates with the spliceosome after binding of the Prp19-associated complex and is released upon Prp2 action; the spliceosome becomes inactive if remodeling occurs before the addition of Cwc24 (Lardelli, Thompson, Yates III, & Stevens, 2010; Ohrt et al., 2012; Wu et al., 2017). Cwc24 binds directly to pre-mRNA at the 5′ splice site and in the absence of Cwc24, U5, and U6 modes of interaction with the 5′ splice site were altered, causing aberrant cleavage at the 5′ splice site and inefficient splicing (Wu et al., 2017). Studies on yeast showed that the zinc finger domain, but not the RING finger domain, was essential for cellular viability and cellular function (Wu et al., 2017). A model in which RNF113A associates with PRP19, and joins the spliceosome before activation, also interacting with the tri-snRNP complex has been proposed (Gatti da Silva et al., 2018). In the absence of RNF113A, splicing efficiency is severely compromised, possibly because the interactions to assemble the spliceosome are abolished.
In addition to a role in spliceosome function, RNF113A may also be important in the cellular response to alkylating agents. Lymphoblastoid cell lines obtained from the two cousins with the p.(Gln301*) variant were shown to be hypersensitive to the alkylating agent, methyl methanesulphonate (MMS; Brickner et al., 2017). The patient lymphoblastoid cells also had significantly reduced activating signal co-integrator complex 3 (ASCC3) foci formation and this decrease could be rescued with wildtype RNF113A (Brickner et al., 2017). ASCC3 is the largest subunit of ASCC and encodes a 3′−5′ DNA helicase whose activity is crucial for the generation of single-stranded DNA for dealkylation by alpha-ketoglutarate dependent dioxygenase (ALKBH3), an enzyme that is involved in repair of alkylated DNA (Dango et al., 2011). The RING domain of RNF113A promoted BRR2 binding to ASCC2 and recombinant BRR2 was ubiquitinated in vitro by RNF113A in a manner dependent on its RING domain (Brickner et al., 2017). Knockdown of BRR2, or its partner, PRP8, significantly reduced ASCC3 foci formation upon MMS damage and increased sensitivity to the alkylating agent (Brickner et al., 2017). Thus, BRR2 likely represents at least one substrate for RNF113A in the alkylation repair pathway, with RNF113A functioning as the E3 ligase that transduces the alkylation damage signal (Brickner et al., 2017). RNF113A contains a CCCH-type zinc finger and since RNA is also modified by exposure to alkylating agents, it is possible that damaged RNA serves as the initial signal to activate DNA alkylation repair (Brickner et al., 2017).
In summary, we describe a male with significant developmental delays and growth retardation, refractory seizures, hypogonadism, immunodeficiency with low IgG levels, facial anomalies and sparse, brittle hair consistent with TTD. Exome sequencing as a duo revealed a hemizygous variant in RNF113A, c.903_910delGCAGACCA, predicting p.(Gln302fs*12), that was maternally inherited. His clinical features overlap with those described in two male cousins with a variant affecting the adjacent p.Gln residue in RNF113A and we consider that this is the causative variant in this patient. We also describe a second patient with global developmental delays, growth delays and microcephaly, sparse and brittle hair with absent eyebrows, agenesis of the corpus callosum, seizures, ambiguous genitalia and a multicystic, dysplastic kidney (Monies et al., 2019). These findings are also consistent with TTD, and this patient was previously reported to be hemizygous for c.897_898delTG, predicting p.(Cys299*) in exon 1 of RNF113A. RNF113A participates in spliceosome activity and has also been shown to function as the E3 ligase that transduces damage signals in response to alkylating agents. Our report is the third to describe deleterious variants in RNF113A in association with X-linked TTD and thus augments the clinical description of patients with this rare, but distinctive condition.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U01HG009599 (A. Slavotinek; see Amendola et al. (2018)) and by the National Cancer Institute of the National Institutes of Health under Award Number R01CA210561 (D. Beleford). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Amendola et al. (2018). American Journal of Human Genetics, 103 (3):319-327.
Footnotes
CONFLICT OF INTEREST
The authors deny any conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.Thank you.
REFERENCES
- Amendola LM, Berg JS, Horowitz CR, Angelo F, Bensen JT, Biesecker BB, … Jarvik GP (2018). The clinical sequencing evidence-generating research consortium: Integrating genomic sequencing in diverse and medically underserved populations. American Journal of Human Genetics, 103, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, & Hopkins N (2004). Identification of 315 genes essential for early zebrafish development. Proceedings of the National Academy of Sciences of the United States of America, 101, 12792–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner JR, Soll JM, Lombardi PM, Vågbø CB, Mudge MC, Oyeniran C, … Mosammaparast N (2017). A ubiquitin-dependent signaling axis specific for ALKBH-mediated DNA dealkylation repair. Nature, 551, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TD, Struck AJ, & Doe CQ (2013). Midlife crisis encodes a conserved zinc-finger protein required to maintain neuronal differentiation in drosophila. Development, 140, 4155–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltri PP, & Oliveira CC (2012). Cwc24p is a general Saccharomyces cerevisiae splicing factor required for the stable U2 snRNP binding to primary transcripts. PLoS One, 7, e45678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett MA, Dudding-Byth T, Crock PA, Botta E, Christie LM, Nardo T, … Field M (2015). A novel X-linked trichothiodystrophy associated with a nonsense mutation in RNF113A. Journal of Medical Genetics, 52, 269–274. [DOI] [PubMed] [Google Scholar]
- Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, … Shi Y (2011). DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Molecular Cell, 44, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghri S, Tamura D, Kraemer KH, & Digiovanna JJ (2008). Trichothiodystrophy: A systematic review of 112 published cases characterizes a wide spectrum of clinical manifestations. Journal of Medical Genetics, 45, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini A, Faranda S, Bagnasco L, Patrosso C, Nulli P, Zucchi I, & Vezzoni P (1997). Identification of a new member (ZNF183) of the ring finger gene family in Xq24–25. Gene, 192, 291–298. [DOI] [PubMed] [Google Scholar]
- Gatti da Silva GH, Jurica MS, Chagas da Cunha JP, Oliveira CC, & Coltri PP (2018). Human RNF113A participates of pre-mRNA splicing in vitro. Journal of Cellular Biochemistry, 120, 8764–8774. [DOI] [PubMed] [Google Scholar]
- Goldfeder M, & Oliveira CC (2008). Cwc24p, a novel Saccharomyces cerevisiae nuclear ring finger protein, affects pre-snoRNA U3 splicing. The Journal of Biological Chemistry, 283, 2644–2653. [DOI] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR III, & Stevens SW (2010). Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA, 16, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Alpi AF, Park MS, Rose A, & Koo H-S (2013). C. elegans ring finger protein RNF-113 is involved in interstrand DNA crosslink repair and interacts with a RAD51C homolog. PLoS One, 8, e60071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, … Alkuraya FS (2019). Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. American Journal of Human Genetics, 104, 1182–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrt T, Prior M, Dannenberg J, Odenwalder P, Dybkov O, Rasche N, … Lührmann R (2012). Prp2-mediated protein rearrangements at the catalytic core of the spliceosome as revealed by dcFCCS. RNA, 18, 1244–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, & Seelow D (2012). MutationTaster evaluates disease-causing potential of sequence alterations. Nature Method, 7, 575–576. [DOI] [PubMed] [Google Scholar]
- van Wijk SJ, de Vries SJ, Kemmeren P, Huang A, Boelens R, Bonvin AM, & Timmers HT (2009). A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Molecular Systems Biology, 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu NY, Chung CS, Cheng SC. 2017. Role of Cwc24 in the first catalytic step of splicing and fidelity of 5′ splice site selection. Molecular Cell Biology 37:. pii: e00580–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.Thank you.