

Adipose tissues function in metabolic, endocrine and immune roles to regulate energy balance and maintain health. Adipocytes reside within adipose tissues (so-called depots) together with fibroblasts, adipose progenitors, endothelial cells and immune cells. Collaboration among these multiple cell types is necessary for the tissue to remodel and maintain homeostasis in the face of metabolic challenges (1). In non-obese individuals, resident adipose tissue macrophages (ATMs) are mainly M2-like and function to remodel the tissue as needed for growth, maintenance and repair. With chronic overnutrition and obesity, adipocytes expand in size to accommodate the need for energy storage and become dysfunctional. As a result, monocytes are recruited into adipose tissue in a CCL2 (MCP-1) dependent manner. These ATMs localize around dead or dying adipocytes in so-called crown-like structures where they become M1 polarized and increase production of inflammatory cytokines (2). The result is a feed-forward cycle within the adipose tissue, which is thought to lead to adipocyte metabolic and endocrine dysfunction, most notably excessively high release of potentially lipotoxic fatty acids and lower production of the anti-inflammatory hormone adiponectin. The resulting adipocyte and systemic insulin resistance and dyslipidemia mechanistically links obesity to disease risk.
Virtually all studies that led the macrophage recruitment hypothesis used obesity-prone male C57BL/6 mice fed a very high fat diet (HFD, usually 60% fat calories) for a prolonged period, at least 3 months, to induce obesity. However, analysis of the established obese state cannot reflect the dynamics of its development and hence its etiology, as Muir et al (3) have accomplished in this issue of the journal. Their paper provides convincing evidence that the initial response to nutrient overload occurs within 3 days, long before significant obesity develops and involves the local proliferation of resident ATMs.
Muir et al. (3) performed careful time course experiments and show that during the initial stages of overnutrition, within 3d of exposure to an obesifying HFD and prior to any increase in body weight, body fat depots have already expanded (~2x) by adipocyte hypertrophy and there is a 2-fold increase in the number of ATMs as well dendritic cells (ATDC) per depot in both epididymal adipose tissue (eWAT) and inguinal subcutaneous (iWAT). Ki67 labeling showed that the HFD increased proliferation of resident M2-like CD11c-ATMs and endothelial cells, but not CD11c+ ATMs or adipose tissue dendritic cells (ATDCs). In addition, the HFD also decreased the quantity of apoptotic ATM, which also contributes to the increased number of ATMs. Multiple lines of evidence, including elegant studies with inducible genetic labeling of resident ATMs, support the conclusion that the accumulation of resident ATMs results from a CCR2 and GM-CSF independent proliferation of the resident ATMs, and also that this increase does not require B or T cells. The increase in tissue resident ATMs and endothelial cells at this time early point suggest that their function is to remodel the extracellular matrix (ECM) of the tissue in process normally associated with tissue growth and so-called ‘healthy expansion’ (1). The increased number of endothelial cells in both depots after 7 to 14d of HFD is thought to maintain blood flow to expanding adipocytes and prevent local hypoxia, which can lead to oxidative stress and impaired metabolic homeostasis. A similar remodeling process occurs, albeit more slowly, in the subcutaneous depots (2; 4). The study Muir et al. (3) highlights the need for future studies of how nutrition-induced changes in ATM subtypes modulate time-dependent changes in ECM composition, vascularization and adipocyte function.
With continued HFD, adipocyte hypertrophy leveled off at a maximum from 28 – 56 days and CD11c-ATM proliferation remained high. CD11c+ macrophages and ATDCs increased in number, but without local proliferation, i.e. they were recruited to the tissue. After 28 d of diet, lipid-laden CD11c+ ATMs were observed in eWAT, consistent with the hypothesis they function to remove lipid from dead adipocytes and buffer the excessive release of fatty acids from adipocytes. This lipid buffering concept was proposed by the Ferrante lab (5); ATMs take up and esterify excess fatty acids mobilized by enlarged adipocytes so that net FA release from the tissue is decreased. However, the amount of lipid that is phagocytosed from dead adipocytes vs. synthesized from released fatty acids remains to be determined. How the increased availability of fatty acid fuel within the expanding adipose tissue influences the functional phenotype of ATMs will be an important avenue for immunometabolism research.
In addition to their comprehensive evaluation of ATM and ATDC origins, the experimental design employed by the Lumeng lab in this study has a number of strengths. Importantly, they examined more than one fat depot and showed distinct, time-dependent responses. In contrast to eWAT, in iWAT, they found no increase in high lipid CD11c+ ATMs perhaps in part because this depot has smaller adipocytes that were still capable of additional hypertrophy without adipocyte dysfunction (2; 4). Also, unlike most prior studies, they used markers that could distinguish ATMs from ATDCs. They found that the HFD caused a greater relative increase (4-fold) in the number of ATDCs per depot in iWAT compared to eWAT (2-fold), and suggested they produce a unique signal for adipose expansion in this depot that merits further investigation.
It is noteworthy that the authors calculated the number of immune cells in each population as ‘quantity’ per depot. This calculation avoids the problem of expressing data only as a % of stromal cells as this denominator is the sum of multiple cells types and therefore only provides a relative frequency, or per gm of adipose tissue weight, which represents fewer adipocytes as they enlarge.
As pointed out by the authors, the signals that lead to the rapid proliferation of resident ATMs in response to the diet remain unknown. ATMs may detect specific dietary components such as fatty acids, or the concomitant changes in hormonal or paracrine factors such as insulin or cytokines. Consistent with this concept, Muir et al. also found that weight loss reversed changes in ATM proliferation and turnover. It will be important to assess time-dependent functions of recruited versus local ATMs function in tissue homeostasis in response to substantial weight change as evidence suggests a pro-inflammatory state persists (6).
The current results have relevance to the ‘expansion hypothesis’ in the field of obesity (7). Average adipocyte size and tissue weight plateaued from days 28 to 56 in both eWAT and iWAT, so it is unlikely that there was a net increase in adipocyte number over this time, consistent with prior studies (4). A transient increase in preadipocyte numbers and proliferation at day 3–7 was detected by Muir et al. With time, these preadipocytes may have replaced dead/dying adipocytes. ATMs may play a role in the remodeling of the extracellular matrix needed for adipogenesis and the maximal adipocyte size that can be achieved without dysfunction (1; 8), so studies of their roles in the dynamics of adipocyte growth and turnover will be of high interest in future studies.
The paper of Muir et al. (3) has some limitations that point to important directions for future work. The control diet (standard chow) used is different in many respects from the HFD (e.g. fiber content and source, fatty acid composition, micronutrients, protein source) that could impact the microbiome and hence metainflammation independent of obesity per se. A carefully controlled diet design will be needed to dissect these mechanisms that increase the rapid proliferation of resident ATMs. In addition, female mice do not develop HFD-induced inflammation over the same rapid time course or with the same intensity as observed in male eWAT (4). Prior studies from the Lumeng lab (9) document sex differences in leukocyte responses to HFDs that should be extended to the rapid response of macrophage proliferation that they have discovered.
In summary, in response to an acute nutrient overload, the proliferative response of resident ATMs documented by Muir et al. may function to remodel the extracellular matrix and achieve homeostatic adipose expansion or remodeling and thus optimal function. Knowledge of how chronic nutrient excess or deficiency, or other environmental challenges, diverts this adaptive remodeling and repair process into a pathological one are clearly needed. Muir et al (3) provide critical groundwork for needed advancements in our understanding of metabolic inflammation in obesity.
Figure 1: Model of dynamic changes in ATMs in adipose tissue in response to overnutrition.
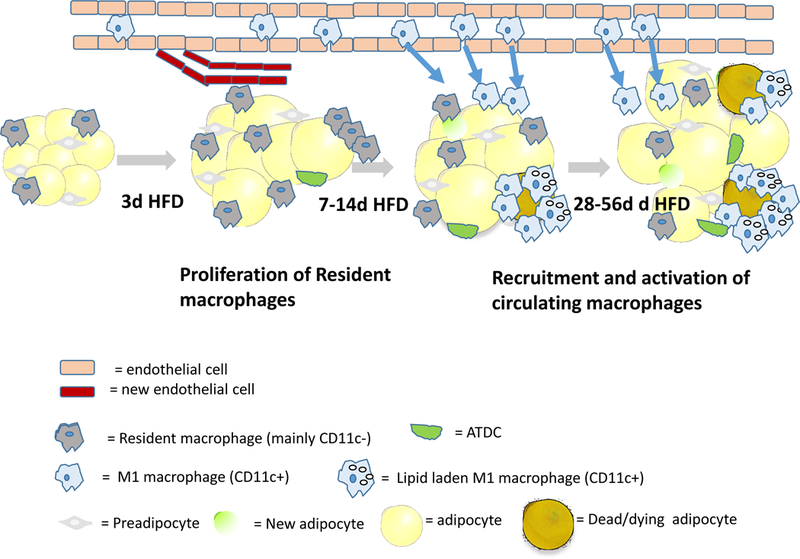
Muir et al found the HFD leads to a rapid (within 3d) expansion of adipose tissues of male mice and an increase in the proliferation of resident macrophages. Only later, as obesity develops, are circulating monocyte precursors recruited from the circulation into the tissue in a CCR2 dependent manner where they become activated (M1-like, CD11c+) and drive inflammation and tissue dysfunction. From 28–56 d of HFD, adipocyte size plateaus and CD11c+ ATMs accumulate lipid droplets. The magnitude and time course of these changes were more dramatic in eWAT than iWAT, but otherwise similar. The adipose expansion was due to increased adipocytes size, with only an early transient increase in preadipocyte proliferation and numbers, but this result does not preclude the possibility that some were differentiated into adipocytes, as illustrated. The results of Muir et al reinforce the importance of analyzing the dynamic changes in the cross-talk among leukocytes, endothelial cells and adipose progenitors in adipose tissue rsemodeling in response to acute and chronic overnutrition.
Footnotes
Conflict of Interest
The author declares no conflict of interest
REFERENCES
- 1.Crewe C, An YA, Scherer PE. 2017. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127:74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, et al. 2007. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56:2910–8 [DOI] [PubMed] [Google Scholar]
- 3.Muir LA, Kiridena S, Griffin C, DelProposto JB, Geletka L, Martinez-Santibañez G, Zamarron BF, Lucas H, Singer K, O’Rourke RW and Lumeng CN. 2018. Rapid adipose tissue expansion triggers unique proliferation and lipid accumulation profiles in adipose tissue macrophages. Journal of Leukocyte biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Lee MJ, Ido Y, Fried SK. 2017. High-fat diet-induced obesity regulates MMP3 to modulate depot-and sex-dependent adipose expansion in C57BL/6J mice. Am J Physiol Endocrinol Metab 312:E58–E71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. 2013. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18:816–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamarron BF, Mergian TA, Cho KW, Martinez-Santibanez G, Luan D, et al. 2017. Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 66:392–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pellegrinelli V, Carobbio S, Vidal-Puig A. 2016. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia 59:1075–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarti P, Kim JY, Singh M, Shin YK, Kim J, et al. 2013. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol Cell Biol 33:3659–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singer K, Maley N, Mergian T, DelProposto J, Cho KW, et al. 2015. Differences in Hematopoietic Stem Cells Contribute to Sexually Dimorphic Inflammatory Responses to High Fat Diet-induced Obesity. J Biol Chem 290:13250–62 [DOI] [PMC free article] [PubMed] [Google Scholar]