

Abstract
Muscle-invasive urothelial cancer (MUC), characterized by high aggressiveness and significant heterogeneity, is currently lacking highly precise individualized treatment options. We used a computational pipeline to synthesize multiomics data from MUC patients using 10 clustering algorithms, which were then combined with 10 machine learning algorithms to identify molecular subgroups of high resolution and develop a robust consensus machine learning-driven signature (CMLS). Through multiomics clustering, we identified three cancer subtypes (CSs) that are related to prognosis, with CS2 exhibiting the most favorable prognostic outcome. Subsequent screening enabled identification of 12 hub genes that constitute a CMLS with robust predictive power for prognosis. The low-CMLS group exhibited a more favorable prognosis and greater responsiveness to immunotherapy and was more likely to exhibit the “hot tumor” phenotype. The high-CMLS group had a poor prognosis and lower likelihood of benefitting from immunotherapy, but dasatinib and romidepsin may serve as promising treatments for them. Comprehensive analysis of multiomics data can offer important insights and further refine the molecular classification of MUC. Identification of CMLS represents a valuable tool for early prediction of patient prognosis and for screening potential candidates likely to benefit from immunotherapy, with broad implications for clinical practice.
Keywords: MT: Bioinformatics, muscle-invasive urothelial cancer, multiomics, machine learning, immunotherapy, prognosis
Graphical abstract
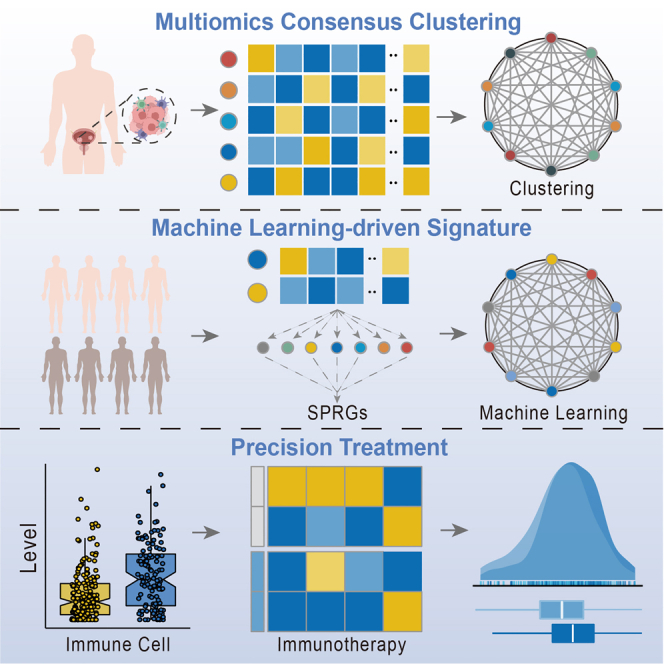
Niu and colleagues first combined 5 types of multiomics data, 10 clustering algorithms, and 99 machine learning algorithm combinations to refine the molecular subtype of MUC and build a robust prognostic signature, which will improve precise treatment and treatment options for MUC.
Introduction
Urothelial carcinoma is highly malignant and often leads to a poor prognosis for patients.1 The most common urothelial carcinoma is bladder cancer, which is the second most common urological malignancy in the world, with over 430,000 people diagnosed and 170,000 deaths worldwide each year.1,2 Bladder cancers mainly progress along two distinct pathways. Nonmuscle-invasive bladder cancer (NMIBC) accounts for 70% of the incidence of bladder cancer. It progress slowly but has a high probability of recurrence and requires lifelong monitoring. Muscle-invasive bladder cancer (MIBC) tends to progress rapidly and metastasize and has a higher mortality rate.2,3,4
With the improvement of current treatment technology, people are constantly trying to use advanced treatment methods for innovative treatment of urinary tumors.5 Nonetheless, strategies aimed at treating advanced muscle-invasive urothelial cancer (MUC) have so far made limited progress. For instance, immunotherapy has been the focus of substantial research efforts and has demonstrated favorable outcomes in certain patients. However, a significant proportion of patients still fails to derive clinical benefits from this approach. This may be due to the marked heterogeneity of urothelial cancer, and molecular subtypes may help address this difficulty.6,7 Given the substantial costs associated with immunotherapy and the potential for severe adverse reactions, there is a compelling need to leverage large-scale multiomics data with advanced machine learning algorithms to identify biomarkers capable of facilitating effective outcome prediction and immunotherapy management in patients with MUC.
In this study, we combined mRNA, long non-coding RNA (lncRNA), and microRNA (miRNA) expression profiles, genomic mutations, and epigenomic DNA methylation data to develop an integrated consensus subtype of MUC using 10 multiomics integration strategies. Subsequently, we identified 32 stable prognosis-related genes (SPRGs) based on differential expression across subtypes and employed 10 machine learning algorithms to construct the consensus machine learning-driven signature (CMLS). In training and validation cohorts, the CMLS demonstrated significant prognostic value while exhibiting robust performance in predicting response to immunotherapy and drug-based therapies. Our findings represent a critical reference point for refining the molecular subtypes of MUC and enhancing precision stratification and individualized treatment approaches for this malignancy.
Results
Multiomics consensus prognosis-related molecular subtypes of MUC
The workflow of this research is shown in Figure 1. Following effective preprocessing of all data, we further verified our results by employing a principal-component analysis (PCA) to analyze the data before and after processing the batch effect (Figures S1A and S1B). We independently identified three subtypes from 10 multiomics ensemble clustering algorithms, and the number of subtypes was determined by comprehensively referring to the cluster prediction index, gap statistical analysis, silhouette score, and previous research experience (Figures S2 and S3). Then, the clustering results were further combined through the consensus ensemble approach with distinctive molecular patterns of expression across transcriptomes (mRNA, lncRNA, and miRNA), epigenetic methylation, and somatic mutations (Figures 2A–2C). Our classification system was closely related to overall survival (OS) (p < 0.001; Figure 2D). Notably, cancer subtype 2 (CS2) exhibited the most favorable survival outcomes among all subtypes evaluated.
Figure 1.

The computational framework for establishing the CMLS
The top 1,500 genes with the greatest degree of variation were screened from the omics data of each dimension, and then the genes associated with prognosis were selected as clustering candidate genes based on Cox regression analysis. For mutation data, we screened candidate genes based on mutation frequency. Stable MUC prognostic subtypes were obtained by consensus clustering using 10 multiomics clustering algorithms: iClusterBayes, moCluster, Cancer Integration via MultIkernel LeaRning (CIMLR), Integrative Clustering of Multiple Genomic Dataset (IntNMF), ConsensusClustering, Cluster-Of-Cluster-Assignments (COCA), NEighborhood based Multi-Omics clustering (NEMO), PINSPlus, Similarity network fusion (SNF), and Landscape Reconstruction Algorithm (LRA). To further identify stable prognostic genes according to subtypes, all combinations of machine learning algorithms (stepwise Cox, CoxBoost, ridge regression, RSF, GBM, Survival-SVM, LASSO, SuperPC, plsRcox, and Enet) were used to screen out the hub signature with the highest C-index. Finally, CMLS was constructed based on the combination of CoxBoost and stepwise Cox. The relationship between CMLS and prognosis, tumor immune microenvironment, immunotherapy response, and potential therapeutic agents was comprehensively determined.
Figure 2.

The multiomics integrative consensus subtypes of MUC
(A) Comprehensive heatmap of consensus ensemble subtypes, including mRNA, lncRNA, miRNA, DNA CpG methylation site, and mutant gene. (B) Clustering of MUC patients through 10 cutting-edge multiomics clustering methods. (C) Consensus clustering matrix for three novel prognostic subtypes based on the 10 algorithms. (D) Different survival outcomes among the three subtypes.
Partitioning of MUC integrative consensus molecular subtypes
At present, most of the molecular subtypes of MUC are classified based on molecular expression levels and may be related to specific biological functions. Therefore, we also tried to explore the different molecular features of these CSs. The single-sample gene set enrichment analysis (ssGSEA) algorithm measured the enrichment of different molecular signatures in the sample. Interestingly, we found that pathways such as luminal differentiation and urothelial differentiation were significantly enriched in CS2, while pathways such as basal differentiation and immune differentiation were significantly enriched in CS3, suggesting that CS2 may be more prone to the currently recognized luminal-like subtypes and that CS3 is more inclined to the basal-like subtypes. Moreover, there were also large differences in response to specific treatments among different subtypes, and pathways such as immune-inhibited oncogenic pathways were significantly enriched in CS2, while CS3 may be more likely to benefit from treatments such as radiotherapy or targeted therapy (Figure 3A).
Figure 3.

Molecular landscape and validation of MUC CSs
(A) The enrichment of three subtypes for different treatment-related signatures and bladder cancer-related signatures. (B) Regulon activity profiles for 23 TFs (top) and potential regulators associated with chromatin remodeling (bottom) of three subtypes. (C) Immune profiles in the TCGA-MUC cohort. The top annotation of the heatmap shows the immune enrichment score, stromal enrichment score, and DNA methylation of tumor-infiltrating lymphocytes. The top panel shows the expression of canonical immune checkpoint genes, and the bottom panel shows the enrichment levels of 24 TME-related immune cells. (D) Validation of MUC CSs in the nearest template of the META-MUC cohort. (E) Survival analysis of MUC CSs in the META-MUC cohort. (F) The consistency of CSs with NTP in the TCGA-MUC cohort. (G) The consistency of CSs with PAM in the TCGA-MUC cohort. (H) The consistency of NTP with PAM in the META-MUC cohort. (I) The consistency of NTP with PAM in the IMvigor-MUC cohort.
To further research transcriptome differences, we analyzed potential regulators associated with cancer chromatin remodeling and 23 transcription factors (TFs) of MUC (Figure 3B). The close correlation of regulator activity with CSs confirmed the biological relevance of CSs. Androgen Receptor (AR), ERBB2, Fibroblast Growth Factor Receptor 3 (FGFR3), and FOXA1 regulators were significantly activated in CS1 and CS2, while EGFR, TP63, HIF1A, and STAT3 were specifically enriched in CS3. The regulon activity profiles associated with cancerous chromatin remodeling further highlight potential patterns of differential regulation between CSs, suggesting that epigenetically driven transcriptional networks may be important differentiating factors for these molecular subtypes. Given the pivotal role of tumor immunity in tumorigenesis and progression, we quantified the infiltration levels of microenvironmental cells and showed that immune cell infiltration was significantly increased in CS1 and CS3 but relatively lower in CS2 (Figure 3C). Based on the results of differential expression analysis between subtypes (Table S1), we selected 20 genes specifically upregulated for each subtype as classifiers and validated them in multiple external cohorts to further validate the stability of subtypes. The nearest template prediction (NTP) classifies each sample in the external cohort as one of the identified CSs. Consistent with this, CS2 in the meta-muscle invasive urothelial cancer cohort (META-MUC) cohort (combined with 8 cohorts) had the best prognosis of all subtypes (p < 0.005), and similar results were obtained in other external cohorts (Figures 3D and 3E). The consistency of CSs with NTP and partition around medoids (PAM) algorithms was also evaluated (p < 0.005; Figures 3F–3J).
Development of the CMLS
We performed univariate Cox regression analysis to screen 32 SPRGs whose expression was significantly related to OS from the consensus genes IMvigor 210 cohort (IMvigor)-MUC, The Cancer Genome Atlas (TCGA)-MUC, and META-MUC. Subsequently, SPRGs were included in the ensemble framework to perform CMLS. In the IMvigor-MUC training cohort, we built consistent models based on 99 algorithmic combinations and calculated the average C-index for each model in all cohorts to assess the predictive power of all models (Figure 4A). As shown in the 99 models, the algorithm consisting of CoxBoost and stepwise Cox (direction = forward) maintained the highest mean C-index to build the final model. The CoxBoost algorithm identified the most valuable SPRGs, and the stepwise Cox algorithm filtered the most valuable model, which was constructed by 12 hub genes (Figures 4B and 4C; Table S2). Then, we calculated the CMLS score per sample for all cohorts. High-CMLS patients had poor clinical outcomes in the TCGA, META, IMvigor, and combined cohorts (Figures 4D–4G).
Figure 4.

The generation and prognostic value of CMLS
(A) Through a comprehensive computational framework, a combination of 99 machine learning algorithms was generated. The C-index of each model was calculated through the TCGA-MUC, IMvigor-MUC, and META-MUC cohorts and sorted by the average C-index of the validation set. (B) The hub gene selected through the CoxBoost algorithm. (C) The univariate Cox regression analysis results of hub genes in training and validation cohorts. (D–G) Survival analysis of MUC patients with high CMLS and low CMLS in the IMvigor-MUC, META-MUC, TCGA-MUC, and combined-MUC cohorts.
For the hub genes of CMLS, we further verified the prognostic value of these hub genes in bladder cancer through the Biomarker Exploration for Solid Tumors (BEST) database (https://rookieutopia.com/app_direct/BEST/) using a Kaplan-Meier analysis, and the obtained results were basically consistent with the results we calculated by the Cox algorithm (Figure S4). We also observed that these genes were significantly associated with progression-free survival (PFS) and disease-specific survival (DSS) of bladder cancer, underscoring their close prognostic relevance for patients (Figures S5 and S6). Subsequently, we utilized the GSCALite public server (http://bioinfo.life.hust.edu.cn/web/GSCALite/) to systematically examine the multiomics phenotypes of CMLS across 33 distinct cancer types in TCGA. The results showed that, in cancer types with more than 10 tumor and normal samples, FJX1, FSCN1, EREG, BNC1, SIRPG, and PTHLH genes were found to be highly expressed in several cancer tissues (Figure S7A). We also found a positive correlation between mRNA expression levels and copy number variations (CNVs) of CMLS genes in most cancer types, especially for FJX1 (Figure S7B). Through analysis of CNV frequency changes, the CNVs of CMLS genes showed significant differences in various cancer types, among which SIRPG and FSCN1 had the highest CNV frequencies, mainly copy number heterozygous amplification (Figures S7C and S7D). In addition, we found that the methylation levels of CMLS genes in most cancer specimens were significantly different between tumor and normal samples (Figure S8A). Methylation levels of CMLS genes were negatively correlated with mRNA expression levels of these genes in most cancers (Figure S8B). All of these indicated CMLS genes may have an influence on the prognosis of patients through epigenetic changes. CMLS genes could activate the pan-cancer epithelial-mesenchymal transition (EMT) pathway and had a significant inhibitory effect on the hormone AR inhibition pathway (Figures S8C and S8D).
Comparison of prognostic signatures in MUC
With the advent of next-generation sequencing technologies, numerous gene expression-based prognostic signatures have been reported extensively in recent years. To enable a comprehensive comparison between CMLS and other signatures, we conducted a systematic search for relevant literature published within the past 5 years and ultimately incorporated 22 distinct signatures into our study (Table S3). These features are associated with different biological processes, such as immunotherapy response, immune infiltration, and glycolysis. Notably, CMLS showed better C-index performance than almost all models in the TCGA-MUC, IMvigor-MUC, and META-MUC datasets (Figures 5A–5C). Considering the clinical application prospects of CMLS, we screened potential independent prognostic factors for MUC by independent prognostic analysis (Figure S9) and integrated them to construct a comprehensive nomogram in the form of a web calculator (https://the-nomogram.shinyapps.io/CMLS-DynNomapp/; Figure 5D). The calibration curve proved that the nomogram has an accuracy that is consistent with the actual situation (Figure 5E). Decision Curve Analysis (DCA) indicated that the clinical benefit of the nomogram for patients was significantly higher than that of CMLS alone (Figures 5F and 5G), and the time-dependent C-index further proved that the nomogram had better prediction performance (Figure 5H).
Figure 5.

Clinical practice value of CMLS
(A–C) Comparison between the CMLS and the other 22 published models in the TCGA-MUC, META-MUC, and IMvigor-MUC cohorts. (D) A comprehensive nomogram constructed based on CMLS and presented in the form of a web calculator for enhanced usability. (E) Calibration curve for the comprehensive nomogram. (F and G) Net decision curve analyses demonstrating the benefit for the comprehensive nomogram in clinical practice for MUC patients. (H) Comparison of the time-dependent C-index between the comprehensive nomogram and CMLS.
Immune characteristics related to CMLS
Employing the Immuno-Oncology Biological Research (IOBR) R package, we conducted a comprehensive analysis of the tumor microenvironment (TME) of MUC and observed that immune cell infiltration levels, including T cells, B cells, and macrophages, were significantly higher in low-CMLS patients than in high-CMLS patients, indicative of an immune activation state (Figure 6A). These findings suggest that MUCs characterized by low CMLS levels are more likely to be classified as “hot tumors.” Fibroblasts and neutrophils were mainly enriched in high-CMLS patients, and molecular markers related to immunosuppression and exclusion, such as the EMT pathway, were also mainly enriched in high groups, showing an immunosuppressive state (Figures 6B and 6C). This means that high-CMLS MUC was more inclined to be a “cold tumor.” As we expected, previously reported signatures associated with better immunotherapy were also significantly enriched in the low-CMLS group (Figure 6D). Tumor mutational burden (TMB) and tumor neoantigen burden (TNB) are currently recognized biomarkers for evaluating the response of patients to immunotherapy, and Zeng et al.8 also proposed a special role of M1 macrophages in bladder cancer immunotherapy. Therefore, we analyzed the differences in the content of these biomarkers between the two groups. The low-CMLS group had higher enrichment of TMB, TNB, and M1 macrophages, which means that the low group may have higher immunogenicity (Figure 6E–6H). Survival analysis also indicated that the CMLS could serve as an effective complementary factor for TMB, TNB, and M1 macrophages to differentiate patient outcomes (Figures 6I–6K). Lower CMLS with higher TMB or TNB or M1 macrophage infiltration tended to have a better survival prognosis for MUC patients.
Figure 6.

The TME-related molecular characteristics of high- and low-CMLS patients
(A) The distribution of TME immune cell type signatures between high- and low-CMLS patients. (B) The distribution of immune suppression signatures between high- and low-CMLS patients. (C) The distribution of immune exclusion signatures between high- and low-CMLS patients. (D) The distribution of immunotherapy biomarkers between high- and low-CMLS patients. (E) The distribution of TMB between high- and low-CMLS patients. (F) The distribution of TNB between high- and low-CMLS patients. (G) The distribution of M1 macrophages between high- and low-CMLS patients. (H) The relationship between CMLS and M1 macrophages. (I–K) Survival analysis combined CMLS with TMB, TNB, and M1 macrophages.
The CMLS has excellent predictive power for immunotherapy response
To comprehensively assess the role of CMLS in MUC immunotherapy, we conducted a systematic analysis. Initially, we conducted a detailed analysis of the IMvigor-MUC cohort in view of the comprehensive prognostic and treatment-related information available for this patient population. Unlike many prior studies, we accounted for the delayed clinical effects of immunotherapy by comparing the restricted mean survival (RMS) between the two groups at 6 and 12 months while evaluating long-term survival differences among patients after 3 months of treatment (p < 0.05; Figures 7A and 7B). The lower group showed better prognostic outcomes, which indicates that the benefit of immunotherapy is greater. The distribution of CMLS among patients with different response degrees also showed that the CMLS score of the responder group (complete response [CR]/partial response [PR]) was significantly lower than that of the nonresponder group (progressive disease [PD]/stable disease [SD]) (p < 0.05; Figure 7C). Then, we calculated the tracking tumor immunophenotype (TIP) to explore the potential biological mechanisms associated with CMLS, and as we expected, the low-CMLS group showed significant differences mainly at step 4 (tumor immune-infiltrating cell recruitment), step 5 (immune cell infiltration), and step 7 (cancer cell killing), consistent with the results of our above analysis (Figure 7D). In addition, the tumor immune dysfunction and exclusion (TIDE) algorithm was used to assess patient response to immunotherapy and showed better responsiveness in the low CMLS group (P [Fisher's exact test] = 5.38e−06; Figure 7E). The subclass mapping algorithm was performed with another group of melanoma patients receiving immunotherapy, and the results also showed that low CMLS indicated a better response to PD-1 therapy (Bonferroni-corrected p = 0.008’ Figure 7F). Finally, we revalidated our conclusions in multiple immunotherapy validation cohorts with prognostic information. Low CMLS tended to have better prognostic outcomes in the post-immunotherapy population (GSE78220, p = 0.015 [Figure 7G]; GSE135222, p = 0.026 [Figure 7H]), and low CMLS tended to be associated with better immunotherapy outcomes (GSE91061, p = 0.032; Figure 7I).
Figure 7.

The value of CMLS in predicting immunotherapy response in MUC patients
(A) The restricted mean survival (RMS) time difference by 6 months and 1 year after treatment between high- and low-CMLS groups. (B) The long-term survival (LTS) difference after 3 months of treatment between high- and low-CMLS groups. (C) The distribution of CMLS in different immunotherapy response groups. (D) Differences in the degree of activation between high- and low-CMLS groups at each step of TIP. (E) The TIDE algorithm predicts response to immunotherapy between high- and low-CMLS groups. (F) The subclass mapping algorithm predicts response to immunotherapy between high- and low-CMLS groups. (G) Survival analysis of high- and low-CMLS groups in GSE78220. (H) Survival analysis of high- and low-CMLS group in GSE135222. (I) Distribution of CMLS in different immunotherapy response groups of GSE91061.
Screening of potential therapeutic drugs
There were significant differences in prognosis between high- and low-CMLS populations, and GSEA also showed that angiogenesis, EMT, hypoxia, and other pathways were significantly activated in high-CMLS patients (Figure 8A). In view of the poor response to immunotherapy in patients with high CMLS, we used Cancer Therapeutics Response Portal (CTRP) and Profiling Relative Inhibition Simultaneously in Mixtures (PRISM) to screen potential therapeutic drugs for patients with high CMLS. To ensure the robustness of our methodology, we employed cisplatin, a widely utilized treatment for bladder cancer, as a means of validating whether algorithm-derived sensitivities were consistent with established clinical practices. A previous report indicated that ERCC1 is a prognostic biomarker in patients with advanced bladder cancer receiving cisplatin-based chemotherapy.9 Our algorithm yielded similar findings, demonstrating that patients characterized by low ERCC1 expression levels exhibited a more robust response to cisplatin therapy, thereby conferring potential benefits for patient chemotherapy (Figure 8B). Then, we systematically explored potential drugs for high-CMLS patients according to the previous studies (Figure 8C). Finally, we screened one CTRP-derived agent (dasatinib; Figure 8D) and two PRISM-derived agents (romidepsin and ispinesib; Figure 8E). We then assessed the differences in the expression levels of target genes for drug candidate action in tumor tissues and normal tissues (including paired and unpaired analyses) (Figures 8F and 8G). A higher fold change indicates greater potential for drug candidate therapy (dasatinib: ABL1, FYN Proto-Oncogene, Src Family Tyrosine Kinase [FYN], KIT Proto-Oncogene, Receptor Tyrosine Kinase [KIT], STAT5B; romidepsin: HDAC1). Finally, we searched for evidence of candidate compounds in PubMed (https://www.ncbi.nlm.nih.gov/PubMed/). Overall, dasatinib and romidepsin are considered promising potential drugs for treatment of high-CMLS patients.
Figure 8.

Potential agents for patients with high CMLS
(A) Discovery of pathways significantly activated in the high-CMLS group through the GSEA algorithm. (B) Pre-predicting the sensitivity of cisplatin to validate the feasibility of the computational algorithm. (C) The comprehensive computational pipeline for the screen of potential agents. (D and E) The correlation and differential analysis of drug sensitivity for potential drugs screened from the CTRP and PRISM datasets. (F and G) The unpaired and paired differential expression analyses for potential target gene of screened drugs in normal and tumor tissue. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Gene expression is finely regulated by various genetic/epigenetic processes, such as methylation, mutation, and histone modifications.10 Therefore, comprehensive analysis of multiomics data from patients will help us gain insights into disease-specific regulatory mechanisms.10,11 However, most studies to date have focused primarily on individual -omics research.12 The choice of clustering methods for omics is also largely based on personal preference, which further expands the limitations of a certain method with expansion of the scope of use. Our research tries to make up for this. We integrated the latest 10 clustering algorithms to identify three types of prognostic subtypes with different characteristics, which may have certain value for accurate stratified treatment of MUC patients. The novel subtypes were proven to be stable in multiple cohorts. In addition, we found that CS2 may have a certain similarity to the currently known luminal molecular subtypes, and our classification may further refine the traditional luminal/basal classification method.
Machine learning algorithms are currently an effective method for analyzing multiomics data.13 To gain an understanding of the differences in molecular characteristics among different prognostic subtypes and improve clinical practical value, we included 10 MUC multicenter cohorts in this study and finally combined them into 1 training set and 2 validation sets and then selected the best CMLS through 99 algorithm combinations to overcome the limitations caused by algorithm selection. At present, when algorithms such as artificial intelligence are deeply integrated with a large amount of biological big data, overfitting is an important problem that cannot be ignored in the process of model construction.14 It is not uncommon for models that demonstrate favorable performance in the training set to prove challenging to generalize effectively to other validation sets. To avoid the trouble caused by overfitting of the training cohort, we used the average of the C-index of multiple validation cohorts as the sorting criterion. As found in the study, when trained with random survival forest (RSF), it has excellent performance in the training set, but it is difficult to generalize in validation sets. Along similar lines, we observed that CMLS, which had been meticulously screened, exhibited robust prognostic value in each cohort compared with other published signatures. Conversely, while four other signatures demonstrated marginally superior performance than CMLS in the training set, their predictive performance markedly decreased across all validation sets, which was potentially related to overfitting of the model.
Through the GSEA algorithm and the IOBR R package, we analyzed the enrichment of dozens of immune-related signatures between the two groups. We found that various oncogenic pathways were significantly activated in the high-CMLS group, which was more prone to the cold tumor phenotype.15 The low-CMLS group has higher TMB and TNB, is rich in immune cell types, and may have stronger antitumor immunity.16 Survival analysis also shows better prognostic outcomes, and this is also validated in multiple immunotherapy cohorts in conclusion. Two widely used predictive tools, TIDE and subclass mapping, also demonstrated a better response to immunotherapy in the low-CMLS group, which is consistent with our analysis and indicates that CMLS may be useful for early identification of immunotherapy-sensitive populations.
In view of the current situation of poor prognosis response to immunotherapy in the high-CMLS group, we systematically screened its potential therapeutic drugs using a comprehensive screening framework that has been proven effective in previous studies,17,18 and finally screened dasatinib and romidepsin as possible candidates for the high-CMLS group. Previous reports have found that the combination therapy of anti-PD-1 and dasatinib can lead to a reduction in tumor burden in tumor-bearing mice.19 Dasatinib combined with a selective FGFR inhibitor could be an option to overcome the intrinsic drug resistance of urothelial cancer patients with FGFR3 alterations.20 In addition, romidepsin has also been shown to effectively and safely control urothelial carcinoma by compensatory combination of gemcitabine and cisplatin,21 and their combination with 2,3-dimethoxycinnamoyl azide can also enhance the resistance of romidepsin to bladder cancer in vitro and in vivo.22
Compared with earlier published research, our study features several notable distinctions. First, recognizing the marked heterogeneity of urothelial cancer, we conducted our analyses specifically in MUC, affording more precise patient stratification and treatment. Second, we incorporated omics information from all five dimensions of MUC and utilized a comprehensive set of 10 clustering algorithms to fully leverage the informative content of each omics dimension while mitigating the impact of clustering method selection preferences on our analyses. Third, our modeling factors were selected based on SPRGs derived from multiple cohorts, enhancing the stability and prognostic value of our model genes. Fourth, by systematically incorporating data from 10 MUC multicenter cohorts and leveraging 10 widely used machine learning algorithms, we identified the model with the best average C-index performance to establish CMLS, aiming to minimize the potential impact of overfitting on our findings. Nonetheless, we acknowledge that our study still has some limitations. For instance, the cohorts we included differed in size and sequencing platform despite utilizing correction algorithms to mitigate these differences. Additionally, the specific mechanisms underlying the tumorigenesis-associated activity of CMLS genes warrant further exploration. Furthermore, the clinical value of CMLS should be validated more extensively in larger, prospective multicenter cohorts.
Conclusions
This study identified three molecular subtypes of MUC via multiomics consensus clustering, revealing significant differences in prognosis among them and potentially refining the molecular typing of MUC. Leveraging a machine learning algorithm framework, we defined CMLS, which exhibited superior performance across multiple cohorts for robustly predicting patient prognosis while demonstrating close associations with immunotherapy response. Given the observed poor prognoses and low immunotherapy responses among high-CMLS groups, we further explored the potential therapeutic benefits of dasatinib and romidepsin for this population. Through integrating multiomics data and cutting-edge computational algorithms, this study provides a foundation for early diagnosis and precise treatment of MUC patients.
Materials and methods
Multiomics data of MUC and data preprocessing of multicenter cohorts
We first collected multiomics data of MUC from the TCGA (https://portal.gdc.cancer.gov) Bladder Cancer (BLCA) cohort, including patients with complete transcriptome expression, DNA methylation, somatic mutations, and clinical information available. The transcriptome profiles of mRNA and lncRNA were obtained by the TCGAbiolinks package.23 The ID of the mature miRNA of TCGA was noted via the miRBaseVersions.db package. The somatic mutations were also obtained by TCGAbiolinks and were processed through the maftools package. The DNA methylation profile and clinical information were downloaded from UCSC xena (https://xenabrowser.net/). In addition, we collected full information on MUC from nine other cohorts, including six from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo; GEO: GSE13507,24 GSE31684,25 GSE32548,26 GSE32894,27 GSE48075,28 and GSE482763), one from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/; E-MTAB-180329), one from the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra, Su et al.30), and one clinical trial from http://research-pub.gene.com/IMvigor210CoreBiologies, which is available under the Creative Commons 3.0 license.31 The high-throughput sequencing of the transcriptome was converted to transcripts per kilobase million (TPM), and all expression profiling by array was deduplicated and standardized. In this study, we defined samples with Ta- or T1-stage tumors as NMIBC that were initially removed. Patients with an OS time of less than 1 month were also excluded to enhance the robustness of downstream analyses.32
For the data from microarrays, the expression matrix and clinical information were downloaded from the official website. After downloading, the data were processed using the robust limma package for background correction, log 2 transformation, and quantile normalization and visualized using the boxplot function to assess the uniformity of sample expression abundance value distribution.33 Similar methods have been applied in previous studies on glioma,34 esophageal cancer,35 liver cancer,36 thyroid cancer,37 and lung adenocarcinoma.38 When multiple probes were mapped to a single gene symbol, the probe with the highest expression was annotated as gene expression. For RNA sequencing (RNA-seq) data from high-throughput sequencing, we used TPM values because they are more similar to gene expression from microarrays and enhance the comparability between samples.39 For the cohort of Su et al.,30 we downloaded the raw data from the BioProject (PRJNA678814), used fastqc to quality control the raw data, and used trim-galore for filtering. Then the data were compared through hisat2 with the gencode27 version of the genome. Next, we used featureCounts to quantify the bam files and filter the low-expressed genes according to the criteria of Su et al.30 The clinical information was obtained from the supplementary files reported by Su et al.30 For the merging of different datasets, the “ComBat” function in the sva package was used to adjust for batch effects from non-biological technical biases of each dataset using an empirical Bayes framework.40,41 Similar algorithms have been reported in many high-quality studies, ensuring the scientific validity of our methods.13,42,43,44,45,46 To further validate the effectiveness of data merging, similar to the study conducted by Liu et al.,47 we also performed PCA on the merged samples before and after merging.
Multiomics consensus ensemble analysis
To effectively perform a comprehensive analysis, we first matched the omics information of the five dimensions through the sample ID (n = 386). The TPM expression data were transformed by log2. For DNA methylation data, we selected probes of promoter CpG islands. For the gene mutation matrix, we identified the gene mutated when it contained any of the following nonsynonymous variations: frameshift insertion or deletion, in-frame insertion or deletion, nonsense or missense or nonstop mutation, or splice site or translation start site mutation.
In this study, we utilized the “getElites” function of the Multi-Omics integration and VIsualization in Cancer Subtyping (MOVICS) package to screen gene features. For continuous variables (mRNA, lncRNA, miRNA, and methylation), we set the “method” parameter of the “getElites” function to “mad” to screen for the top 1,500 genes with the greatest degree of variation. We then set the “method” parameter to “cox” and combined clinical data to identify prognostic genes with a significance level of p < 0.05 in each data dimension. For binary variable gene mutation data, we first used the “oncoPrint” function of the maftools package to screen for the top 5,000 genes with the highest degree of mutation. We then further screened based on mutation frequency by setting the “method” parameter to “freq” to identify the top 5% most frequently mutated genes. The resulting data from these five dimensions were included for further analysis in our study.
After preliminary feature selection, we further determined the optimal number of clusters for our study. As is well known, the optimal number of clusters for data should be small enough to reduce noise but large enough to retain important information. Therefore, we employed the “getClustNum” function from the MOVICS package, which integrates Clustering prediction index (CPI), Gaps-statistics, and Silhouette score to estimate the number of subgroups.48,49 We also incorporated our prior knowledge of urothelial carcinoma from previous studies and ultimately decided to classify it into three subtypes. Subsequently, we applied the “getMOIC” function for clustering analysis. We used 10 clustering algorithms (CIMLR, ConsensusClustering, SNF, iClusterBayes, PINSPlus, moCluster, NEMO, IntNMF, COCA, and LRA) as input for the “methodslist” parameter and utilized the default parameters provided by the MOVICS package. As a result, we obtained the clustering results for each method. After computing the clustering results for the 10 methods, we integrated the results from different algorithms using the “getConsensusMOIC” function based on the concept of consensus clustering to improve the robustness of the clustering.; he “distance” parameter was set to “euclidean,” and the '”inkage” parameter was set to “average.”50,51 The final clustering result was obtained through this integration process.
Specific molecular characteristics and stability of consensus subtypes
Through gene set variation analysis (GSVA),52 we calculated the enrichment scores of multitherapy-related signatures, including signatures positively associated with the response to atezolizumab in BLCA from Mariathasan et al.,53 BLCA molecular subtype-related signatures from the Bladder Cancer Molecular Taxonomy Group, and signatures associated with targeted therapy and radiotherapy. These gene signatures were collected from the research of Hu et al.54 The transcriptional regulatory networks (regulons) were constructed through the Reconstruction of Transcriptional regulatory Networks and analysis of regulons (RTN) R package, including 23 induced/repressed target-associated TFs and 71 candidate regulators related to cancerous chromatin remodeling collected from Lu et al.45 Then, the distribution of immune checkpoints among these subgroups was compared, and the ESTIMATE R package was used to estimate the immune/stromal score of tumor tissue. The score of DNA methylation of tumor-infiltrating lymphocytes (MeTIL) was calculated according to published protocols. The enrichment of 24 kinds of tumor immune microenvironment cells was also evaluated through GSVA. For the stability of subtypes, we first validated the clustering results using subtype-specific biomarkers in the validation cohort and then compared the consistency of consensus clustering with the NTP and PAM classifier.55
Establishment of a consensus machine learning-driven prognostic signature
To enhance the comparability via different cohorts, we performed Z score processing on all data in advance. Then, to evaluate the relationship among CMLS, immunotherapy, and prognosis, we selected the IMvigor-MUC cohort with relatively complete treatment information as the training set and the other cohorts were used as validation sets. Considering that some cohorts had fewer sample accounts, we combined 8 cohorts into the META-MUC cohort and removed batch effects using the sva package. To construct CMLS with high accuracy and generalizability, we integrated 10 machine learning algorithms, including CoxBoost, stepwise Cox, Lasso, Ridge, elastic net (Enet), survival support vector machines (survival-SVMs), generalized boosted regression models (GBMs), supervised principal components (SuperPC), partial least Cox (plsRcox) and RSF. Notably, algorithms such as stepwise Cox, RSF, CoxBoost, and Lasso possess feature selection capabilities. During the model-building phase, we used the IMvigor210 cohort as the training set for preliminary model construction.
For the CoxBoost model, we first called the “optimCoxBoostPenalty” function to determine the optimal penalty (shrinkage) value. We then combined this with cross-validation to perform 10-fold cross-validation on the CoxBoost model to search for the best number of boosting steps. Finally, the “CoxBoost” function was used to fit the model. The stepwise cox analysis was performed using the survival package, and the complexity of the statistical model was evaluated based on the Akaike information criterion (AIC). We calculated all possible combinations for the direction parameter, including “both,” “backward,” and “forward.” Lasso, Ridge, and Enet models were implemented using the glmnet package and the “cv.glmnet” function. The regularization parameter lambda was determined through 10-fold cross-validation, while the tradeoff parameter alpha was set between 0 and 1 (interval = 0.1). When alpha is equal to 1, Lasso is executed, while Ridge is executed when alpha is equal to 0. For other values of alpha, Enet is executed. The survival-SVM model was implemented using the “survivalsvm” function from the survivalsvm package, which employs support vector analysis on datasets with survival outcomes. The GBM model was implemented using the gbm package. The “gbm” function was used with 10-fold cross-validation to fit a GBM. The SuperPC model was implemented using the superpc package, which is an extension of PCA. The “superpc.cv” function was also used with 10-fold cross-validation. For the plsRcox model, the “cv.plsRcox” function of the plsRcox package was used. For the RSF model, we utilized the randomForestSRC package and used the “rfsrc” function with two important parameters: “ntree” and “nodesize.” The parameter “ntree” represents the number of trees in the random forest, while “nodesize” represents the minimum size of the terminal nodes. In this study, we set “ntree” to 1,000 and “nodesize” to 5.
The CMLS development pipeline is as follows.
-
•
We conducted univariate Cox analysis in the IMvigor-MUC, TCGA-MUC, and META-MUC cohorts. Genes with p < 0.05 and the same hazard ratio (HR) orientation in all cohorts were considered SPRGs.
-
•
Ten machine learning algorithms were used. Ninety-nine combinations of these algorithms were used to construct the most predictive CMLS with the best C-index performance.
-
•
After model establishment on the training set, we further tested all validation cohorts. We calculated the average C-index for each model, where the model with the highest value was considered optimal.
Prognostic value of CMLS and potential clinical application
We scored each sample in the training and validation sets according to the resulting model and divided the samples into high- and low-CMLS groups based on the score. The prognostic significance of CMLS was evaluated through the Kaplan-Meier survival curve. In addition, we systematically retrieved 22 prognostic features associated with urothelial cancer and calculated the score for each sample based on the published coefficients. The ability of all signatures to predict prognosis was assessed in each cohort by the C-index. To enhance the clinical utility value of CMLS, we constructed a nomogram using factors obtained after multivariate Cox regression. The time-dependent C-index curve and calibration curve were drawn to describe the accuracy, and the decision curve was used to calculate the clinical benefit to patients.
Comprehensive analysis of immune-omics molecular characterization and immunotherapy response based on CMLS
Based on the IOBR package,56 we collected dozens of previously published signatures related to TME cell types, immunotherapy responses, immune suppression, and immune exclusion and used a unified method to calculate the enrichment score for each sample, which comprehensively analyzed the immunological differences between high- and low-CMLS patients. Differences in the distribution of TMB, TNB, and M1 macrophages between these two groups were compared, and patients were regrouped in combination with CMLS. For the immunotherapy response, we first evaluated the patients’ delayed response survival to immunotherapy and combined the TIP algorithm, the subclass mapping, and TIDE algorithm to estimate the immunotherapy response.57,58,59 This was further verified in GSE78220,60 GSE135222,61 and GSE91061.61
In silico analysis to screen potential therapy agents for patients with high CMLS
The activation status of oncogenic pathways between high- and low-CMLS patients was analyzed through the GSEA algorithm.62 Expression data for human cancer cell lines (CCLs) were obtained from the Broad Institute Cancer Cell Line Encyclopedia (CCLE). The CTRP v.2.0 (https://portals.broadinstitute.org/ctrp) and PRISM Repurposing datasets (19Q4; https://depmap.org/portal/prism/) were used to obtain drug sensitivity data for CCLs. The area under the dose-response curve (AUC) values acted as a measure of drug sensitivity.
Statistical analysis
For comparisons of two groups, the normally distributed variables were tested by unpaired Student’s t tests, and the non-normally distributed variables were tested by the Wilcoxon rank-sum test. For comparisons of more than two groups, the parametric and nonparametric variables were tested by one-way ANOVA and Kruskal-Wallis tests, respectively. A two-sided Fisher’s exact test was performed for the contingency tables. The cutoff value of the CMLS score was determined through the “surv-cutpoint” function of the survminer package. We repeatedly tested all possible cutoff points to identify the maximum rank statistics and then used the two-classification method to classify the CMLS score. According to the maximum selected logarithmic rank statistics, patients could be distinguished into high- and low-score groups in each cohort to reduce the computational batch effect, which is similar to previous research.63,64,65 The differential expression analysis was analyzed by the limma package, and multiomics clustering was completed through the MOVICS package.55 All statistical analyses were performed in R v.4.1.0.
Acknowledgments
This work was supported by the National Natural Science Foundation of China under grant 82071750 and the Taishan Scholar Program of Shandong Province under grant tstp20221165. We sincerely acknowledge all authors who greatly contributed to this study. Figure 1 was created with the help of BioRender. We thank Elsevier Language Editing Services for linguistic editing and proofreading of the manuscript.
Author contributions
G.C. and X.J. designed the whole study, analyzed the data, and drafted the manuscript. H.N. and Y.W. revised the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.06.001.
Contributor Information
Yonghua Wang, Email: wangyonghua@qdu.edu.cn.
Haitao Niu, Email: niuht0532@126.com.
Supplemental information
Data availability
All data used in this work can be acquired from TCGA (https://portal.gdc.cancer.gov), GEO (http://www.ncbi.nlm.nih.gov/geo), ArrayExpress (https://www.ebi.ac.uk/arrayexpress/), and SRA (https://www.ncbi.nlm.nih.gov/sra). The key code and parameters of this study were uploaded to GitHub (https://github.com/BioUro/MTNA-MUC).
References
- 1.Patel V.G., Oh W.K., Galsky M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA A Cancer J. Clin. 2020;70:404–423. doi: 10.3322/caac.21631. [DOI] [PubMed] [Google Scholar]
- 2.Compérat E., Amin M.B., Cathomas R., Choudhury A., De Santis M., Kamat A., Stenzl A., Thoeny H.C., Witjes J.A. Current best practice for bladder cancer: a narrative review of diagnostics and treatments. Lancet. 2022 doi: 10.1016/S0140-6736(22)01188-6. [DOI] [PubMed] [Google Scholar]
- 3.Choi W., Porten S., Kim S., Willis D., Plimack E.R., Hoffman-Censits J., Roth B., Cheng T., Tran M., Lee I.L., et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran L., Xiao J.-F., Agarwal N., Duex J.E., Theodorescu D. Advances in bladder cancer biology and therapy. Nat. Rev. Cancer. 2021;21:104–121. doi: 10.1038/s41568-020-00313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J., Yang X., Chu G., Feng W., Ding X., Yin X., Zhang L., Lv W., Ma L., Sun L., et al. European Urology; 2022. Application of Improved Robot-Assisted Laparoscopic Telesurgery with 5G Technology in Urology. [DOI] [PubMed] [Google Scholar]
- 6.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sjödahl G., Abrahamsson J., Holmsten K., Bernardo C., Chebil G., Eriksson P., Johansson I., Kollberg P., Lindh C., Lövgren K., et al. Different responses to neoadjuvant chemotherapy in urothelial carcinoma molecular subtypes. Eur. Urol. 2022;81:523–532. doi: 10.1016/j.eururo.2021.10.035. [DOI] [PubMed] [Google Scholar]
- 8.Zeng D., Ye Z., Wu J., Zhou R., Fan X., Wang G., Huang Y., Wu J., Sun H., Wang M., et al. Macrophage correlates with immunophenotype and predicts anti-PD-L1 response of urothelial cancer. Theranostics. 2020;10:7002–7014. doi: 10.7150/thno.46176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellmunt J., Paz-Ares L., Cuello M., Cecere F.L., Albiol S., Guillem V., Gallardo E., Carles J., Mendez P., de la Cruz J.J., et al. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann. Oncol. 2007;18:522–528. doi: 10.1093/annonc/mdl435. [DOI] [PubMed] [Google Scholar]
- 10.Oh M., Park S., Kim S., Chae H. Machine learning-based analysis of multi-omics data on the cloud for investigating gene regulations. Briefings Bioinf. 2021;22:66–76. doi: 10.1093/bib/bbaa032. [DOI] [PubMed] [Google Scholar]
- 11.Reel P.S., Reel S., Pearson E., Trucco E., Jefferson E. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol. Adv. 2021;49:107739. doi: 10.1016/j.biotechadv.2021.107739. [DOI] [PubMed] [Google Scholar]
- 12.Ma C., Wu M., Ma S. Analysis of cancer omics data: a selective review of statistical techniques. Briefings Bioinf. 2022;23:bbab585. doi: 10.1093/bib/bbab585. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z., Liu L., Weng S., Guo C., Dang Q., Xu H., Wang L., Lu T., Zhang Y., Sun Z., Han X. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 2022;13:816. doi: 10.1038/s41467-022-28421-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L., Liu Z., Liang R., Wang W., Zhu R., Li J., Xing Z., Weng S., Han X., Sun Y.-L. Comprehensive machine-learning survival framework develops a consensus model in large-scale multicenter cohorts for pancreatic cancer. Elife. 2022;11:e80150. doi: 10.7554/eLife.80150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 16.Borst J., Ahrends T., Bąbała N., Melief C.J.M., Kastenmüller W. CD4 T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 17.Chu G., Shan W., Ji X., Wang Y., Niu H. Multi-omics analysis of novel signature for immunotherapy response and tumor microenvironment regulation patterns in urothelial cancer. Front. Cell Dev. Biol. 2021;9:764125. doi: 10.3389/fcell.2021.764125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang C., Chen J., Li Y., Huang X., Liu Z., Wang J., Jiang H., Qin W., Lv Y., Wang H., Wang C. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: an in silico strategy towards precision oncology. Briefings Bioinf. 2021;22:bbaa295. doi: 10.1093/bib/bbaa164. [DOI] [PubMed] [Google Scholar]
- 19.Tu M.M., Lee F.Y.F., Jones R.T., Kimball A.K., Saravia E., Graziano R.F., Coleman B., Menard K., Yan J., Michaud E., et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci. Adv. 2019;5:eaav2437. doi: 10.1126/sciadv.aav2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lima N.C., Atkinson E., Bunney T.D., Katan M., Huang P.H. Targeting the src pathway enhances the efficacy of selective FGFR inhibitors in urothelial cancers with FGFR3 alterations. Int. J. Mol. Sci. 2020;21:3214. doi: 10.3390/ijms21093214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattarawat P., Hong T., Wallace S., Hu Y., Donnell R., Wang T.-H., Tsai C.-L., Wang J., Wang H.-C.R. Compensatory combination of romidepsin with gemcitabine and cisplatin to effectively and safely control urothelial carcinoma. Br. J. Cancer. 2020;123:226–239. doi: 10.1038/s41416-020-0877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan J., Stanfield J., Guo Y., Karam J.A., Frenkel E., Sun X., Hsieh J.-T. Effect of trans-2,3-dimethoxycinnamoyl azide on enhancing antitumor activity of romidepsin on human bladder cancer. Clin. Cancer Res. 2008;14:1200–1207. doi: 10.1158/1078-0432.CCR-07-1656. [DOI] [PubMed] [Google Scholar]
- 23.Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T.S., Malta T.M., Pagnotta S.M., Castiglioni I., et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim W.-J., Kim E.-J., Kim S.-K., Kim Y.-J., Ha Y.-S., Jeong P., Kim M.-J., Yun S.-J., Lee K.M., Moon S.-K., et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol. Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riester M., Taylor J.M., Feifer A., Koppie T., Rosenberg J.E., Downey R.J., Bochner B.H., Michor F. Combination of a novel gene expression signature with a clinical nomogram improves the prediction of survival in high-risk bladder cancer. Clin. Cancer Res. 2012;18:1323–1333. doi: 10.1158/1078-0432.CCR-11-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindgren D., Sjödahl G., Lauss M., Staaf J., Chebil G., Lövgren K., Gudjonsson S., Liedberg F., Patschan O., Månsson W., et al. Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS One. 2012;7:e38863. doi: 10.1371/journal.pone.0038863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sjödahl G., Lauss M., Lövgren K., Chebil G., Gudjonsson S., Veerla S., Patschan O., Aine M., Fernö M., Ringnér M., et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012;18:3377–3386. doi: 10.1158/1078-0432.CCR-12-0077-T. [DOI] [PubMed] [Google Scholar]
- 28.Guo C.C., Bondaruk J., Yao H., Wang Z., Zhang L., Lee S., Lee J.-G., Cogdell D., Zhang M., Yang G., et al. Assessment of luminal and basal phenotypes in bladder cancer. Sci. Rep. 2020;10:9743. doi: 10.1038/s41598-020-66747-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rebouissou S., Bernard-Pierrot I., de Reyniès A., Lepage M.-L., Krucker C., Chapeaublanc E., Hérault A., Kamoun A., Caillault A., Letouzé E., et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci. Transl. Med. 2014;6:244ra291. doi: 10.1126/scitranslmed.3008970. [DOI] [PubMed] [Google Scholar]
- 30.Su X., Lu X., Bazai S.K., Compérat E., Mouawad R., Yao H., Rouprêt M., Spano J.-P., Khayat D., Davidson I., et al. Comprehensive integrative profiling of upper tract urothelial carcinomas. Genome Biol. 2021;22:7. doi: 10.1186/s13059-020-02230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balar A.V., Galsky M.D., Rosenberg J.E., Powles T., Petrylak D.P., Bellmunt J., Loriot Y., Necchi A., Hoffman-Censits J., Perez-Gracia J.L., et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389:67–76. doi: 10.1016/S0140-6736(16)32455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu X., Meng J., Zhu J., Zhou Y., Jiang L., Wang Y., Wen W., Liang C., Yan F. Prognosis stratification and personalized treatment in bladder cancer through a robust immune gene pair-based signature. Clin. Transl. Med. 2021;11:e453. doi: 10.1002/ctm2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cong P., Wu T., Huang X., Liang H., Gao X., Tian L., Li W., Chen A., Wan H., He M., et al. Identification of the role and clinical prognostic value of target genes of m6A RNA methylation regulators in glioma. Front. Cell Dev. Biol. 2021;9:709022. doi: 10.3389/fcell.2021.709022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H., Shi X., Ren L., Zhuo H., Zeng L., Qin Q., Wan Y., Sangdan W., Zhou L. Identification of the miRNA-mRNA regulatory network associated with radiosensitivity in esophageal cancer based on integrative analysis of the TCGA and GEO data. BMC Med. Genom. 2022;15:249. doi: 10.1186/s12920-022-01392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z., Pan L., Guo D., Luo X., Tang J., Yang W., Zhang Y., Luo A., Gu Y., Pan Y. A novel five-gene signature predicts overall survival of patients with hepatocellular carcinoma. Cancer Med. 2021;10:3808–3821. doi: 10.1002/cam4.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X., Peng W., Li C., Qin R., Zhong Z., Sun C. Identification of an immune-related signature indicating the dedifferentiation of thyroid cells. Cancer Cell Int. 2021;21:231. doi: 10.1186/s12935-021-01939-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C., Li X., Shao H., Li D. Identification and validation of two lung adenocarcinoma-development characteristic gene sets for diagnosing lung adenocarcinoma and predicting prognosis. Front. Genet. 2020;11:565206. doi: 10.3389/fgene.2020.565206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner G.P., Kin K., Lynch V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor. Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 40.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X., Shi M., Chen T., Zhang B. Characterization of the immune cell infiltration landscape in head and neck squamous cell carcinoma to aid immunotherapy. Mol. Ther. Nucleic Acids. 2020;22:298–309. doi: 10.1016/j.omtn.2020.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao R., Ma B., Wang G., Xiong Y., Tian Y., Yuan L. Characterization of hypoxia response patterns identified prognosis and immunotherapy response in bladder cancer. Mol. Ther. Oncolytics. 2021;22:277–293. doi: 10.1016/j.omto.2021.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang M., Liu L., Zhu J., Jin T., Chen Y., Xu L., Cheng W., Ruan X., Su L., Meng J., et al. Identification of immune-related subtypes and characterization of tumor microenvironment infiltration in bladder cancer. Front. Cell Dev. Biol. 2021;9:723817. doi: 10.3389/fcell.2021.723817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu X., Meng J., Su L., Jiang L., Wang H., Zhu J., Huang M., Cheng W., Xu L., Ruan X., et al. Multi-omics consensus ensemble refines the classification of muscle-invasive bladder cancer with stratified prognosis, tumour microenvironment and distinct sensitivity to frontline therapies. Clin. Transl. Med. 2021;11:e601. doi: 10.1002/ctm2.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Zhang Y., Zhao L., Wang J., Li J., Wang X., Zhang M., Hu X. Exploitation of tumor antigens and construction of immune subtype classifier for mRNA vaccine development in bladder cancer. Front. Immunol. 2022;13:1014638. doi: 10.3389/fimmu.2022.1014638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z., Guo C., Dang Q., Wang L., Liu L., Weng S., Xu H., Lu T., Sun Z., Han X. Integrative analysis from multi-center studies identities a consensus machine learning-derived lncRNA signature for stage II/III colorectal cancer. EBioMedicine. 2022;75:103750. doi: 10.1016/j.ebiom.2021.103750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chalise P., Fridley B.L. Integrative clustering of multi-level 'omic data based on non-negative matrix factorization algorithm. PLoS One. 2017;12:e0176278. doi: 10.1371/journal.pone.0176278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tibshirani R., Walther G., Hastie T. Estimating the number of clusters in a data set via the gap statistic. J. Roy. Stat. Soc. B. 2001;63:411–423. [Google Scholar]
- 50.Strehl A., Ghosh J. Cluster ensembles - a knowledge reuse framework for combining multiple partitions. J. Mach. Learn. Res. 2002;3:583–617. [Google Scholar]
- 51.Lu X., Meng J., Zhou Y., Jiang L., Yan F. MOVICS: an R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics. 2021;36:5539–5541. doi: 10.1093/bioinformatics/btaa1018. [DOI] [PubMed] [Google Scholar]
- 52.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., Koeppen H., Astarita J.L., Cubas R., et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu J., Yu A., Othmane B., Qiu D., Li H., Li C., Liu P., Ren W., Chen M., Gong G., et al. Siglec15 shapes a non-inflamed tumor microenvironment and predicts the molecular subtype in bladder cancer. Theranostics. 2021;11:3089–3108. doi: 10.7150/thno.53649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu X., Meng J., Zhou Y., Jiang L., Yan F. MOVICS: an R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics. 2020;36:5539–5541. doi: 10.1093/bioinformatics/btaa1018. [DOI] [PubMed] [Google Scholar]
- 56.Zeng D., Ye Z., Shen R., Yu G., Wu J., Xiong Y., Zhou R., Qiu W., Huang N., Sun L., et al. IOBR: multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 2021;12:687975. doi: 10.3389/fimmu.2021.687975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu L., Deng C., Pang B., Zhang X., Liu W., Liao G., Yuan H., Cheng P., Li F., Long Z., et al. TIP: a web server for resolving tumor immunophenotype profiling. Cancer Res. 2018;78:6575–6580. doi: 10.1158/0008-5472.CAN-18-0689. [DOI] [PubMed] [Google Scholar]
- 58.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoshida Y., Brunet J.-P., Tamayo P., Golub T.R., Mesirov J.P. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One. 2007;2:e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung H., Kim H.S., Kim J.Y., Sun J.-M., Ahn J.S., Ahn M.-J., Park K., Esteller M., Lee S.-H., Choi J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019;10:4278. doi: 10.1038/s41467-019-12159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng D., Li M., Zhou R., Zhang J., Sun H., Shi M., Bin J., Liao Y., Rao J., Liao W. Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures. Cancer Immunol. Res. 2019;7:737–750. doi: 10.1158/2326-6066.CIR-18-0436. [DOI] [PubMed] [Google Scholar]
- 64.Zhang B., Wu Q., Li B., Wang D., Wang L., Zhou Y.L. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer. 2020;19:53. doi: 10.1186/s12943-020-01170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chong W., Shang L., Liu J., Fang Z., Du F., Wu H., Liu Y., Wang Z., Chen Y., Jia S., et al. m6A regulator-based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics. 2021;11:2201–2217. doi: 10.7150/thno.52717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this work can be acquired from TCGA (https://portal.gdc.cancer.gov), GEO (http://www.ncbi.nlm.nih.gov/geo), ArrayExpress (https://www.ebi.ac.uk/arrayexpress/), and SRA (https://www.ncbi.nlm.nih.gov/sra). The key code and parameters of this study were uploaded to GitHub (https://github.com/BioUro/MTNA-MUC).