

Abstract
Purpose
Ulcer is a serious disease that is caused due to different bacteria and over usage of various NSAIDs which caused to reduce the defensive system of stomach. Therefore, some novel series are needed to overcome these issues.
Methods
Oxazole-based imidazopyridine scaffolds (4a-p) were designed and synthesized by two step reaction protocol and then subjected to urease inhibition profile (in vitro). All the newly afforded analogs (4a-p) were found potent and demonstrated moderate to significant inhibition profile.
Results
Particularly, the analogs 4i (IC50 = 5.68 ± 1.66 μM), 4o (IC50 = 7.11 ± 1.24 μM), 4 g (IC50 = 9.41 ± 1.19 μM) and 4 h (IC50 = 10.45 ± 2.57 μM) were identified to be more potent than standard thiourea drug (IC50 = 21.37 ± 1.76 μM). Additionally, the variety of spectroscopic tools such as 1H NMR, 13C NMR and HREI-MS analysis were employed to confirm the precise structures of all the newly afforded analogs.
Discussion
The structure–activity relationship (SAR) studies showed that analogs possess the substitution either capable of furnishing strong HB like –OH or had strong EW nature such as -CF3 & –NO2 groups displayed superior inhibitory potentials than the standard thiourea drug. A good PLI (protein–ligand interaction) profile was shown by most active analogs when subjected to molecular study against corresponding target with key significant interactions such as pi-pi stacking, pi-pi T shaped and hydrogen bonding.
Keywords: Synthesis, Imidazopyridine, Oxazole, Urease and Molecular docking
1. Introduction
Urea is the primary nitrogenous waste product of biological system and is easily metabolized by bacteria (Qin, 1994, Pervez et al., 2008, Smith et al., 2023). The urease enzyme (EC 3.5.1.5) is found in plants, bacteria and fungi. It is crucial to the nitrogen cycle because it catalyzes the hydrolysis of urea into carbon dioxide and ammonia at a rate that is around 1014 times faster than a non-catalyzed reaction (Ara et al., 2007, Arshad et al., 2017, Naz et al., 2020) and enhanced the microorganism growth by providing nitrogen. In the hydrolysis of urea, the urease plays an important role but excessive amounts of it can have negative effects on living things and harm the environment and the economy (Bremner 1996). Urease hyperactivity causes a variety of clinical issues in both humans and other animals, including infection-induced reactive arthritis, peptic ulcers, pyelonephritis and kidney stones (Ashraf et al., 2013). It is also necessary for Helicobacter pylori to colonize human stomach mucosa. The ammonia developed by the urea hydrolysis is hazardous for several stomach cell lines and serves as a favorable environment for the growth of H. pylori, which can cause ulcers and gastric cancer. Additionally, it has been noted that urease promotes the development of infectious kidney stones brought on by Proteus mirabilis and Yersinia enterocolitica (Bayerdörffer and Ottenjann, 1988, Mobley et al., 1995). In agricultural industry, high soil urease concentrations can quickly break down the fertilizer urea and cause phytopathic consequences. Furthermore, crop yields suffer due to the loss of volatilized ammonia (Gioacchini et al., 2002, Lodhi et al., 2013, Taha et al., 2016). Due to the role of urease in a variety of clinical disorders and for agricultural applications, the identification of effective and secure urease inhibitors is a crucial field in pharmaceutical and plant research (Taha et al., 2015, Al-Rooqi et al., 2023). The N-Containing heterocyclic compounds are also crucial for the research of biological activity and for use in pharmaceuticals (Orlemans et al., 1989). Particularly, imidazopyridine with both and imidazole pyridine moieties, which constitutes a typical, favored scaffold, exhibits antifungal activities and serves as an antagonist to the H1 receptor as well as kinase an and antilipases inhibitor (Fig. 1) (Belohlavek and Malfertheiner 1979).
Fig. 1.

Bioactive drugs bearing imidazopyridine skeleton.
Heterocyclic small molecules are essential for understanding cell biology and treating wide range of diseases because they control the function of protein–protein interactions, receptors and enzymes (Zhao et al., 2015, Wang et al., 2023). In this perspective, oxazoles are heterocyclic five-membered molecules with heteroatoms of nitrogen and oxygen that have established a significant class of therapeutic prospects in organic chemistry. Typically, substituted oxazoles containing heterocycles might bind many receptors and enzymes via non-covalent bond and in the biological system to demonstrate a wide range of biological activities (Delost et al., 2018, Warner et al., 2018, Zhang et al., 2018, Ke et al., 2019, Sun et al., 2019, Melfi et al., 2023). There are numerous oxazole-containing drugs have been reported and are widely used in market for the treatment of various kind of diseases, including such as ditazole (platelets aggregation inhibitor), oxaprozin, JTE-5229 COX-2 inhibitors (anti-inflammatory drug), (-)-muscoride A (peptide alkaloid), aleglitazar (antidiabetic), aristoxazole (anti-inflammatory drugs), and AD-5061 (antidiabetic agent) (Fig. 2) (Mukku et al., 2020).
Fig. 2.

Biologically potent drugs containing oxazole moiety in their core skeleton.
Our research groups had reported various N-containing heterocyclic compounds such as benzimidazole, benzoxazole, triazole and thiazole scaffolds for their potentials cholinesterase, urease, alpha-amylase and alpha-glucosidase inhibitors (Hussain et al., 2022, Khan et al., 2022a, Khan et al., 2022b, Khan et al., 2022c). It is noteworthy that oxazole moiety is structurally resemblance to thiazole ring. Further, imidazopyridine-based oxazole analogs have not been reported as urease inhibitors therefore, it is the need of time to incorporate both imidazopyridine and oxazole rings in same molecules to explore their urease activity. Considering the biological importance of oxazole (Georgiades and Rizeq, 2015, Lechel et al., 2019, Rahim et al., 2019) and imidazopyridine (Park et al., 2017, Thakur et al., 2020, Mishra et al., 2021) rings, herein we have designed and synthesized hybrid scaffolds incorporating both imidazopyridine and oxazole rings in the same molecules to further explore urease inhibition profile (in vitro) and further correlated by in silico molecular docking studies (Fig. 3).
Fig. 3.

Rational of the current study.
2. Methods
2.1. Materials and setting
All chemicals were bought from Sigma Aldrich and Merck (Germany) (USA). A Bruker AM spectrometer was used to record the NMR spectra (600 MHz). Mass spectra (EI-MS, HR-EI-MS, and FAB) were recorded on the mass spectrometers JEOL JMS-600H, MAT 312, and MAT 113D. TLC was carried out using precoated silica gel-254 from Merck, Germany. At 366 and 254 nm, dots were visible under UV light. Chemical shift values were measured in ppm, and coupling constants are displayed in Hz, with DMSO‑d6 acting as the reference.
2.2. General procedure for the synthesis of imidazopyridine containing oxazole analogs (4a-l)
In two steps, the hybrid imidazopyridine-based oxazole scaffolds (4a-l) were afforded. Initially, flouro-substituted imidazopyridine-3-carbaldehyde (1 equivalent) in methanol (10 mL) was reacted with flouro-substituted semicarbazide (1) (1 equivalent) and glacial acetic acid (few drops). The reaction mixture was stirred under reflux at refluxing temperature to obtain semicarbazone substrate (2). After completion of reaction, the solvent was removed using a rotary evaporator to obtained the solid intermediate (2). This intermediate then underwent cyclization when it was refluxed and stirred for 16hrs over pre-heated sand bath with different substituted 2-bromoacetophenone (3) (1 equivalent) in ethanol (10 mL) along with few drops of triethylamine as a catalyst. The solid residue so obtained was washed with n-hexane to give targeted imidazopyridine-based oxazole analogs (4a-l) in appropriate yield.
2.3. Spectral analysis (provided in supplementary information)
Scheme 1.

Synthesis of imidazole-fused-pyridine containing oxazole derivatives.
Table 1.
Different substituent (s) and in vitro urease inhibitory profile of synthesized imidazole-fused-pyridine containing oxazole analogs (4a-p).
| S.NO | R | IC50 ± SEMa [μM] | S.NO | R | IC50 ± SEMa [μM] |
|---|---|---|---|---|---|
| 4a | 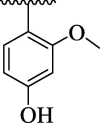 |
18.23 ± 2.05 | 4i | 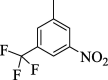 |
5.68 ± 1.66 |
| 4b |  |
15.32 ± 2.46 | 4j | 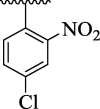 |
11.33 ± 2.01 |
| 4c | 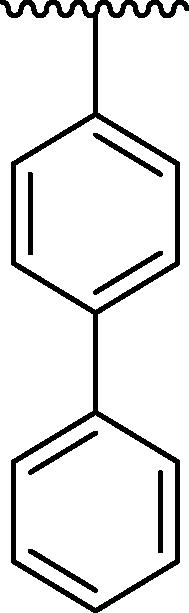 |
81.49 ± 7.18 | 4 k | 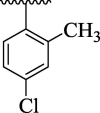 |
21.56 ± 2.24 |
| 4d | 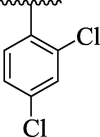 |
16.13 ± 1.65 | 4 l | 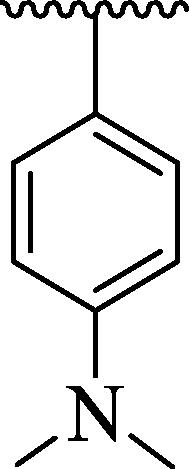 |
74.55 ± 7.18 |
| 4e | 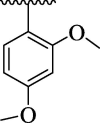 |
43.49 ± 4.20 | 4 m | 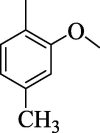 |
48.54 ± 4.18 |
| 4f | 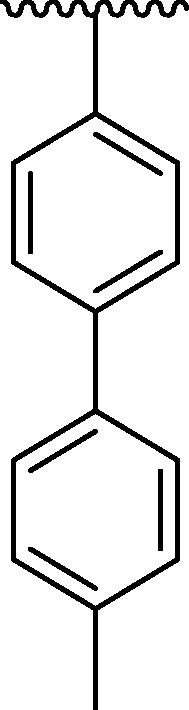 |
71.69 ± 6.13 | 4n | 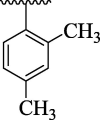 |
58.05 ± 6.69 |
| 4 g | 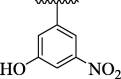 |
9.41 ± 1.19 | 4o | 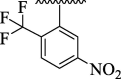 |
7.11 ± 1.24 |
| 4 h | 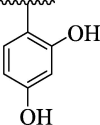 |
10.45 ± 2.57 | 4p | 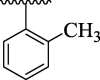 |
63.57 ± 7.25 |
| Standard Thiourea drug | 21.37 ± 1.76 μM | ||||
2.4. Assay protocol for urease inhibition
The protocol was carried by following the previously published study (Mumtaz et al., 2022).
2.5. Assay protocol for molecular docking study
It has also been carried out according to our previously published work (Khan et al., 2022a, Khan et al., 2022b, Khan et al., 2022c, Mumtaz et al., 2022).
3. Results
3.1. Chemistry
The hybrid scaffolds (4a-p) based on imidazopyridine containing oxazole moiety were afforded in two steps. In the first step, semicarbazide (1) was treated with flouro-substituted imidazopyridine-3-carbaldehyde in methanol along with few drops of acetic acid (glacial) and resulting residue was refluxed and stirred at refluxing temperature to afford imidazopyridine-based semicarbazone substrate (2). As the reaction was completed, the solvent was evaporated via rotary evaporator to deliver the solid intermediate (2) which further undergoes cyclization on putting reflux with various substituted 2-bromoacetophenone (3) in ethanol followed by addition of few drops of triethylamine as catalyst. The reaction mixture was refluxed and stirred until the formation of targeted imidazopyridine-based oxazole analogs were complete (conversion were monitored by TLC, refluxed 16 hrs). After being cooled to room temperature, the solvent was removed by applying reduced pressure to give solid residue which was further washed and then recrystallized from ethyl acetate to afford purified targeted imidazopyridine-based oxazole analogs (4a-p). All the newly synthesized scaffolds (4a-p) were characterized structurally by employing variety of spectroscopic techniques including HREI-MS, 1H NMR and 13C NMR.
3.2. Biological analysis (4a-p)
Imidazopyridine-based oxazole scaffolds (4a-p) were subjected to possible urease inhibitory potentials for the first time. It is interesting to mention that the majority of synthesized analogs were found to be active against target urease enzyme with IC50 values ranging from 5.68 ± 1.66 μM to 81.48 ± 7.18 μM. The results obtained were compared to reference thiourea drug with IC50 value 21.37 ± 1.76 μM. Interestingly, the scaffolds 4i (IC50 = 5.68 ± 1.66 μM), 4o (IC50 = 7.11 ± 1.24 μM), 4 g (IC50 = 9.41 ± 1.19 μM) and 4 h (IC50 = 10.45 ± 2.57 μM) were found to be significantly active against urease enzyme as compared to standard thiourea drug. However, the rest of the analogs showed moderate to good urease inhibitory activity.
3.3. Molecular docking study
To establish the correlation between in vitro and in silico studies of all the active synthesized 4i, 4o, 4 h and 4 g analogs, molecular docking study has been performed. Subsequently, all these active analogs bind well in the active cavity of target with different binding affinities and also correlate well with the in vitro studies in (Fig. 4, Fig. 5, Fig. 6, Fig. 7).
Fig. 4.

The PLI (protein–ligand interaction) profile of analog-4i against urease target and its 3D (left) and 2D (right) diagram.
Fig. 5.

The protein–ligand interaction (PLI) profile of analog-4o against urease target and its 3D (left) and 2D (right) diagram.
Fig. 6.

The PLI (protein–ligand interaction) profile of analog-4 h against urease target and its 3D (left) and 2D (right) diagram.
Fig. 7.

The PLI (protein–ligand interaction) profile of analog-4 g against urease target and its 3D (left) and 2D (right) diagram.
4. Discussion
4.1. Urease inhibitory activity
4.1.1. Structure-activity relationship (SAR)
For the sake of simplification of the SAR studies, it was explained based on variation in position, nature and number/s of attached substituent (s) on aryl ring. Electron withdrawing -CF3 and –NO2 groups bearing analogs 4i (holds 3-CF3 & 5-NO2 groups at aryl ring) and 4o (bearing 2-CF3 & 5-NO2 moieties at aryl ring) were identified as excellent inhibitors of urease enzyme. Analog 4i was found to be the most active inhibitor of urease, even more potent than standard thiourea drug. Shifting the –CF3 moiety from meta-position to ortho-position as in the case of analog 4o demonstrates slightly decreased inhibitory potentials compared to scaffold 4i. However, the activity was further declined by replacement of meta-CF3 group with –OH group as in case of compound 4 g (bearing meta-hydroxy and meta-nitro groups at aryl part). Further, the comparison of analog 4 g with analog 4 h (having di-hydroxy at 2,4-position of aryl ring) revealed that changing the nature (–NO2 group was replaced with –OH group) of attached substituent (s) decreased the inhibitory potentials of analog 4 h than its counterpart 4 g. The analog 4 h bearing di-hydroxy groups at 2,4-position of aryl part exhibited less inhibitory potentials than compound 4 g but still found to be more potent than standard urease drug (Fig. 8).
Fig. 8.

SAR studies of 4 g, 4 h, 4o and 4i analogs.
The substituent(s) of electron donating nature such as –OCH3 and –CH3 groups were found to be encouraging for significant urease inhibitory potentials. The analog 4e (bearing di-OCH3 substitution at 2,4-position of aryl part) was identified as an outstanding urease inhibitors, but found less potent when compared to standard thiourea drug. However, the urease inhibitory potential of analog 4e was decreased sharply by replacing 4-OCH3 group with –CH3 group as in case of compound 4 m. This shows that –OCH3 group plays an effective role in inhibition of urease enzyme than –CH3 group. The urease inhibitory potential of scaffold 4e was further reduced by replacing both 2,4-dimethoxy substituents with 2,4-diCH3 moieties as in case of scaffold 4n indicating that –OCH3 groups enhanced the urease inhibitory activity. The compound 4 k (holds ortho-CH3 and para-Cl moieties at aryl part) was interestingly found to be active than analog 4n. This activity comparison confirmed that -Cl substitution at the para-position uplift the inhibitory strength to a good extent (Fig. 9).
Fig. 9.

SAR studies of 4e, 4 k, 4 m and 4n analogs.
Further, comparison of analog 4d (that holds di-Cl moieties at 2,4-position of phenyl ring) with analog 4j (bearing 2-NO2 & 4-Cl group on phenyl ring) shows that analog 4j was emerged as excellent urease inhibitor, even more active than standard thiourea drug. This activity comparison confirmed that –NO2 substitution relative to –Cl substitution uplift the urease inhibitory potential. The scaffold 4b (that holds 2-NO2 & 4-CH3 substitution at phenyl ring) showed slightly decreased urease inhibitory potential than analog 4j. This activity comparison indicates that changing the nature of substituent (para-Cl group is replaced with para-CH3 group) at para-position slightly decreased the urease inhibitory potential. The compound 4a (bearing ortho-methoxy & para-hydroxy substitution at aryl ring) displayed better urease inhibitory potential even more potent than standard thiourea drug. This uplift inhibitory potential of analog 4a might be due to associating nature of –OH group which bind well with urease target active sites and hence enhanced the urease inhibitory potential (Fig. 10).
Fig. 10.

SAR studies of 4a, 4b, 4d and 4j derivatives.
However, the analogues 4c bearing phenyl group at para-position (IC50 = 81.49 ± 7.18 μM) and 4f bearing toluene moiety on para-position (IC50 = 71.69 ± 6.13 μM) were found to be three-fold least potent of urease inhibitor as compared to the standard drug thiourea (IC50 = 21.37 ± 1.76 μM). Because the phenyl, toluene, -N(CH3)2 have bulky nature present at para-position of the aryl part of the oxazole (Gattu et al., 2023, Khan et al., 2023, Vanjare et al., 2023) resulted to reduced inhibitory potentials against urease inhibitor. The analogue 4c bearing para-phenyl moiety at aryl part and analogue 4f having para-toluene moiety at the same para-position were showing least potent competitor against urease inhibitor. Moreover, compound 4 l has -N(CH3)2 at para-position and 4p has methyl group on ortho-position of the aryl ring showing three-fold least potency as compared to the standard drug thiourea. Hence, the bulky groups are responsible for the least potency of molecules (4c, 4f and 4 l) as compared to the standard drug (Fig. 11).
Fig. 11.

SAR studies of 4c, 4f, 4 l and 4p analogs.
On the basis of aforementioned observation, it was summarized form SAR studies that either EW groups such as -CF3, -Cl and –NO2 or group with strong associating nature (–OH) play an important role in significant inhibitory potentials of synthesized analogs against targeted enzyme. However, other groups when placed at certain position and number/s also demonstrated better inhibitory activities.
4.2. Molecular docking study
Subsequently, all these active analogs bind well in the active cavity of target with different binding affinities and also correlate well with the in vitro studies. All these active analogs almost have similar chemistry with slight modification at varied position. These varied functional moieties at varied position of active analogs resulted to different interactions with target urease active sites. The compound 4i was identified as the most active likewise in the vitro analysis. This active analog furnished maximum interactions with target active sites. The detailed PLI of most potent analog 4i shows that this scaffold furnished several significant interactions target urease active sites such as Thr308 (CHB), Val558 (halogen (fluorine)), Lys559 (halogen (fluorine)), Glu560 (halogen (fluorine)), Leu561 (pi-alkyl & alkyl), Gln379 (CHB & carbon hydrogen bond), Ala564 (halogen (fluorine) & pi-alkyl), Phe570 (pi-anion), Phe568 (halogen (fluorine)) and Glu372 (carbon hydrogen bond) interactions (Fig. 4).
The PLI of second most potent scaffold 4o demonstrated that although this analog is quite like most active analog 4i in structure, the only difference is that analog 4o holds –CF3 moiety at meta-position of aryl ring; however, the analog 4i have –CF3 moiety attached at ortho-position of the aryl part of oxazole ring. The discrepancy in the enzymatic potential may perhaps owing to varied position of functional moiety –CF3 around aryl part, which therefore causes both these active 4i & 4o analogs to interacts in different way. This second most potent analog 4o showed less interaction when compared to its structurally similar analog 4i. The detail PLI of second most active scaffold 4o demonstrated that this analog adopted number/s of significant interactions with target urease active sites such as Gln379 (CHB), Leu561 (pi-sigma), Thr308 (carbon hydrogen bond), Glu384 (halogen (fluorine)), Ala564 (pi-alkyl & alkyl), Phe568 (alkyl), Leu375 (alkyl), Met563 (halogen (fluorine)), Phe570 (pi-sigma) and Arg376 (pi-sigma & CHB) interactions (Fig. 5).
From docking analysis, it was also observed that not only the EW groups enhanced the enzymatic inhibition, but also the substituent that forms strong hydrogen bond with the active sites of target also elevate the inhibition profile; therefore, analog 4 h (bearing di-hydroxy at 2,4-position of aryl part) established numerous key interactions target urease active sites including Leu253 (pi-alkyl), His323 ((halogen (fluorine), pi-pi stacked & amide-pi stacked), Ala366 (CHB), Ala170 (pi-alkyl), Cys322 (pi-alkyl), Lys169 (pi-pi T shaped) and Asp224 (pi-anion & carbon hydrogen bond) interactions (Fig. 6).
The analog 4 g bearing 2-hydroxy and 5-NO2 moieties at aryl part of oxazole ring was also emerged as the active inhibitor of urease enzyme and hence displayed several significant important interactions including Ala334 (pi-pi T shaped & amide-pi stacked), Glu331 (carbon hydrogen bond), Leu253 (pi-sigma & carbon hydrogen bond) and Phe335 (pi-pi T shaped & pi-pi stacked) interactions with urease target active sites (Fig. 7).
5. Conclusion
A library of imidazopyridine-based oxazole (4a-l) and synthesized and their chemical structures were elucidated by HREI-MS, 13C NMR and 1H NMR analysis. The anti-urease activity of newly afforded analogs was tested and obtained results were compared with standard thiourea drug. All imidazopyridine-based oxazole analogs, particularly 4i, 4o, 4 g and 4 h with IC50 values of 5.68 ± 1.66, 7.11 ± 1.24, 9.41 ± 1.19 and 10.45 ± 2.57 μM respectively, were identified to be significantly potent and amazing anti-urease agent with strong binding to enzyme that correlated well with the experimental data. While other analogs demonstrated relatively moderate activity. Additionally, the molecular docking studies were performed on the most promising compounds 4i, 4o, 4 g and 4 h in the active sites of urease and results obtained showed that these active scaffolds established several significant interactions with the active sites of targeted urease. There was a good association between in vitro and in silico studies. Moreover, a structure–activity relationship (SAR) study was performed out for all synthesized analogs based on varying substitution (s) around the aryl ring while keeping imidazopyridine and oxazole rings constant and docking results of the active analogs shown excellent urease inhibition profile and these analogs are found to be suitable for the treatment of Ulcer.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia for supporting this research.
Footnotes
Peer review under responsibility of King Saud University. Production and hosting by Elsevier.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsps.2023.05.026.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Al-Rooqi M.M., Mughal E.U., Raja Q.A., et al. Flavonoids and related privileged scaffolds as potential urease inhibitors: a review. RSC advances. 2023;13:3210–3233. doi: 10.1039/d2ra08284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara, R., U. Ashiq, M. Mahroof‐Tahir, et al., 2007. Chemistry, urease inhibition, and phytotoxic studies of binuclear vanadium (IV) complexes. 4, 58-71. [DOI] [PubMed]
- Arshad, T., K. M. Khan, N. Rasool, et al., 2017. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of α-glucosidase and urease enzymes. 72, 21-31. [DOI] [PubMed]
- Ashraf, M., R. Hassan, I. Ahmadand, et al., 2013. Synthesis of N-Substituted Derivatives of N-(4-(N-(5-Chloro-2 methoxyphenyl) sulfamoyl) phenyl) acetamide with Potential Antiurease Activity. 35, 1516.
- Bayerdörffer, E. and R. J. S. J. o. G. Ottenjann, 1988. The role of antibiotics in Campylobacter pylori associated peptic ulcer disease. 23, 93-100. [PubMed]
- Belohlavek, D. and Malfertheiner, P. J. S. J. o. G. S. 1979. The effect of zolimidine, imidazopyridine-derivate, on the duodenal ulcer healing. 54, 44-44. [PubMed]
- Bremner, J., 1996. Recent research on problems in the use of urea as a nitrogen fertilizer. Nitrogen Economy in Tropical Soils: Proceedings of the International Symposium on Nitrogen Economy in Tropical Soils, held in Trinidad, WI, January 9–14, 1994, Springer.
- Delost, M. D., D. T. Smith, B. J. Anderson, et al., 2018. From oxiranes to oligomers: Architectures of US FDA approved pharmaceuticals containing oxygen heterocycles. 61, 10996-11020. [DOI] [PubMed]
- Gattu R., Ramesh S.S., Nadigar S., et al. Conjugation as a Tool in Therapeutics: Role of Amino Acids/Peptides-Bioactive (Including Heterocycles) Hybrid Molecules in Treating Infectious Diseases. Antibiotics. 2023;12:532. doi: 10.3390/antibiotics12030532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades, S. N. and N. J. S. Rizeq, 2015. Synthesis of a ‘propeller-like’oligoheteroaryl with alternating pyridine and oxazole motifs. 26, 489-493.
- Gioacchini, P., A. Nastri, C. Marzadori, et al., 2002. Influence of urease and nitrification inhibitors on N losses from soils fertilized with urea. 36, 129-135.
- Hussain R., Iqbal S., Shah M., et al. Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase. In Vitro Evaluation along with Molecular Docking Study. 2022;27:6457. doi: 10.3390/molecules27196457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, S., Z. Zhang, L. Shi, et al., 2019. An efficient synthesis and bioactivity evaluation of oxazole-containing natural hinduchelins A–D and their derivatives. 17, 3635-3639. [DOI] [PubMed]
- Khan, S., F. Rahim, W. Rehman, et al., 2022. New benzoxazole-based sulphonamide hybrids analogs as potent inhibitors of α-amylase and α-glucosidase: Synthesis and in vitro evaluation along with in silico study. 15, 104341.
- Khan, Y., W. Rehman, R. Hussain, et al., 2022. New biologically potent benzimidazole‐based‐triazole derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors along with molecular docking study.
- Khan S., Rahim F., Rehman W., et al. New benzoxazole-based sulphonamide hybrids analogs as potent inhibitors of α-amylase and α-glucosidase: Synthesis and in vitro evaluation along with in silico study. Arabian Journal of Chemistry. 2022;15 [Google Scholar]
- Khan S., Rahim F., Rehman W., et al. Novel benzoxazole-based thiosemicarbazide derivatives as new inhibitors of urease and β-Glucuronidase: Synthesis, in vivo anti-nematodal activity and ADMET prediction along with in silico study. Journal of Saudi Chemical Society. 2023;27 [Google Scholar]
- Lechel, T., R. Kumar, M. K. Bera, et al., 2019. The LANCA three-component reaction to highly substituted β-ketoenamides–versatile intermediates for the synthesis of functionalized pyridine, pyrimidine, oxazole and quinoxaline derivatives. 15, 655-678. [DOI] [PMC free article] [PubMed]
- Lodhi, M. A., A. Wadood, S. Iqbal, et al., 2013. Three-dimensional quantitative structure–activity relationship (CoMSIA) analysis of bis-coumerine analogues as urease inhibitors. 1, 498-504.
- Melfi F., Carradori S., Campestre C., et al. Emerging compounds and therapeutic strategies to treat infections from Trypanosoma brucei: an overhaul of the last 5-years patents. Expert Opinion on Therapeutic Patents. 2023:1–17. doi: 10.1080/13543776.2023.2193328. [DOI] [PubMed] [Google Scholar]
- Mishra, N. P., S. Mohapatra, C. R. Sahoo, et al., 2021. Design, one-pot synthesis, molecular docking study, and antibacterial evaluation of novel 2H-chromene based imidazo [1, 2-a] pyridine derivatives as potent peptide deformylase inhibitors. 1246, 131183.
- Mobley, H., M. D. Island and R. P. J. M. r. Hausinger, 1995. Molecular biology of microbial ureases. 59, 451-480. [DOI] [PMC free article] [PubMed]
- Mukku N., Madivalappa Davanagere P., Chanda K., et al. A Facile Microwave-Assisted Synthesis of Oxazoles and Diastereoselective Oxazolines Using Aryl-Aldehydes, p-Toluenesulfonylmethyl Isocyanide under. Controlled Basic Conditions. 2020;5:28239–28248. doi: 10.1021/acsomega.0c04130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumtaz S., Iqbal S., Shah M., et al. New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies. Molecules. 2022;27:6580. doi: 10.3390/molecules27196580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz, F., M. Latif, U. Salar, et al., 2020. 4-Oxycoumarinyl linked acetohydrazide Schiff bases as potent urease inhibitors. 105, 104365. [DOI] [PubMed]
- Orlemans, E., W. Verboom, M. Scheltinga, et al., 1989. Synthesis, mechanism of action, and biological evaluation of mitosenes. 32, 1612-1620. [DOI] [PubMed]
- Park, S., H. Kim, J.-Y. Son, et al., 2017. Synthesis of Imidazopyridines via Copper-Catalyzed, Formal Aza-[3+ 2] Cycloaddition Reaction of Pyridine Derivatives with α-Diazo Oxime Ethers. 82, 10209-10218. [DOI] [PubMed]
- Pervez, H., M. S. Iqbal, M. Y. Tahir, et al., 2008. In vitro cytotoxic, antibacterial, antifungal and urease inhibitory activities of some N 4-substituted isatin-3-thiosemicarbazones. 23, 848-854. [DOI] [PubMed]
- Qin, Y., J. M. J. A. b. Cabral and biotechnology, 1994. Kinetic studies of the urease-catalyzed hydrolysis of urea in a buffer-free system. 49, 217-240. [DOI] [PubMed]
- Rahim, F., S. Tariq, M. Taha, et al., 2019. New triazinoindole bearing thiazole/oxazole analogues: Synthesis, α-amylase inhibitory potential and molecular docking study. 92, 103284. [DOI] [PubMed]
- Smith J., Nayak D., Yeluripati J. The potential use of biochar to reduce nitrogen waste from farming systems in India. Current Research in Environmental Sustainability. 2023;5 [Google Scholar]
- Sun, L., Y. Liu, Y. Wang, et al., 2019. An efficient synthesis of oxazolines via a cascade reaction between azaoxyallyl cations and 1, 2-benzisoxazoles. 17, 7526-7530. [DOI] [PubMed]
- Taha, M., N. H. Ismail, M. S. Baharudin, et al., 2015. Synthesis crystal structure of 2-methoxybenzoylhydrazones and evaluation of their α-glucosidase and urease inhibition potential. 24, 1310-1324.
- Taha, M., N. H. Ismail, S. Imran, et al., 2016. Hybrid benzothiazole analogs as antiurease agent: Synthesis and molecular docking studies. 66, 80-87. [DOI] [PubMed]
- Thakur, A., G. Pereira, C. Patel, et al., 2020. Design, one-pot green synthesis and antimicrobial evaluation of novel imidazopyridine bearing pyran bis-heterocycles. 1206, 127686.
- Vanjare B.D., Choi N.G., Eom Y.S., et al. Synthesis, carbonic anhydrase inhibition, anticancer activity, and molecular docking studies of 1, 3, 4-oxadiazole derivatives. Molecular Diversity. 2023;27:193–208. doi: 10.1007/s11030-022-10416-6. [DOI] [PubMed] [Google Scholar]
- Wang J., Zhang J., Wang J., et al. Small-Molecule Modulators Targeting Toll-like Receptors for Potential Anticancer Therapeutics. Journal of Medicinal Chemistry. 2023 doi: 10.1021/acs.jmedchem.2c01655. [DOI] [PubMed] [Google Scholar]
- Warner, K. D., C. E. Hajdin and K. M. J. N. r. D. d. Weeks, 2018. Principles for targeting RNA with drug-like small molecules. 17, 547-558. [DOI] [PMC free article] [PubMed]
- Zhang, H.-Z., Z.-L. Zhao and C.-H. J. E. j. o. m. c. Zhou, 2018. Recent advance in oxazole-based medicinal chemistry. 144, 444-492. [DOI] [PubMed]
- Zhao, Y., A. Aguilar, D. Bernard, et al., 2015. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment: miniperspective. 58, 1038-1052. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.