

Abstract
A monogenic etiology can be identified in up to 40% of people with severe epilepsy. To address earlier and more appropriate treatment strategies, clinicians are required to know the implications that specific genetic causes might have on pathophysiology, natural history, comorbidities, and treatment choices. In this narrative review, we summarize concepts on the genetic epilepsies based on the underlying pathophysiologic mechanisms and present the current knowledge on treatment options based on evidence provided by controlled trials or studies with lower classification of evidence. Overall, evidence robust enough to guide antiseizure medication (ASM) choices in genetic epilepsies remains limited to the more frequent conditions for which controlled trials and observational studies have been possible. Most monogenic disorders are very rare and ASM choices for them are still based on inferences drawn from observational studies and early, often anecdotal, experiences with precision therapies. Precision medicine remains applicable to only a narrow number of patients with monogenic epilepsies and may target only part of the actual functional defects. Phenotypic heterogeneity is remarkable, and some genetic mutations activate epileptogenesis through their developmental effects, which may not be reversed postnatally. Other genes seem to have pure functional consequences on excitability, acting through either loss- or gain-of-function effects, and these may have opposite treatment implications. In addition, the functional consequences of missense mutations may be difficult to predict, making precision treatment approaches considerably more complex than estimated by deterministic interpretations. Knowledge of genetic etiologies can influence the approach to surgical treatment of focal epilepsies. Identification of germline mutations in specific genes contraindicates surgery while mutations in other genes do not. Identification, quantification, and functional characterization of specific somatic mutations before surgery using CSF liquid biopsy or after surgery in brain specimens will likely be integrated in planning surgical strategies and reintervention after a first unsuccessful surgery as initial evidence suggests that mutational load may correlate with the epileptogenic zone. Promising future directions include gene manipulation by DNA or mRNA targeting; although most are still far from clinical use, some are in early phase clinical development.
An increasing number of genetic mutations and genomic rearrangements are being causally associated with a burgeoning spectrum of clinical conditions in which epilepsy is a major feature. A prospective national epidemiologic cohort study conducted in the United Kingdom estimated an overall annual minimum incidence of monogenic epilepsies in children of about 1 per 2,000 live births, with 8 genes accounting for the majority of cases.1 These epidemiologic figures are reflected in the detection rate of potentially pathogenic variants in up to 40% of people with different types of epilepsy, now permitted by next-generation sequencing techniques.2,3
The choice of antiseizure medication (ASM) in clinical practice has benefited from accumulated experience and drug trials in specific genetic conditions and from increased knowledge of the underlying disease mechanisms. Precision medicine, according to the definition promulgated by the NIH, refers to a treatment and prevention approach based on the understanding of individual variability in genetic architecture, environment, and lifestyle. One of the basic assumptions of precision medicine is that the genetic abnormality causes the phenotype, as determined through established frameworks including gene validity and variant pathogenicity. When applied to epilepsy, precision medicine may therefore include treatments addressing seizures (ASM), epileptogenesis (disease-modifying treatments), and comorbidities. However, precision medicine is currently applicable to a narrow number of patients with epilepsy and may target only part of the actual functional defects without reversing their consequences on brain development.4 Most monogenic disorders are rare and high-quality evidence robust enough to inform management strategies remains limited to the more frequent conditions for which controlled trials and observational studies are possible. Model systems and novel bioinformatics approaches have also been used to guide mechanistic and functional understanding of disease mechanisms, and high-throughput functional assays have been implemented for compound screening and profiling of targeted therapies. However, for the majority of rare disorders, treatment choices rely on the hypothesized functional defect or remain confined to symptom relief and general principles of epilepsy management.
In this review, we illustrate the main epilepsy phenotypes associated with monogenic disorders and the currently available treatment strategies in the most prevalent childhood-onset genetic epilepsies based on their epidemiologic framework.1 We also analyze the role of genetic findings in epilepsy surgery and the principles and pitfalls of gene therapy based on the established pathophysiologic mechanisms. Other important related areas such as deep phenotypic characterization, diagnostic pathways, genetic counselling, natural history, and treatment of comorbidities are beyond the scope of this review.
Methods
We searched on PubMed for articles published from inception to January 15, 2021. We conducted our article search and selection in 2 steps. Firstly, we searched for the most prevalent epilepsy genes based on the epidemiologic framework provided in a recent study,1 and any related treatment options. Secondly, we conducted a broader search for less prevalent genetic epilepsies screening for all the genes listed in Tables 1–3 and any related treatment options. The following search terms were used: “seizure + specific gene name + treatment,” “epilepsy + specific gene name + treatment,” “specific gene name + treatment,” “specific gene name + precision medicine,” “gene name + antiseizure,” “gene name + epilepsy + trial.” A similar search with the specific gene names was conducted in the clinicaltrials.gov website for active treatment trials.5 Original research articles published in the last 5 years, that is, between 1 January 2016 and 6 January 2021, were prioritized, and older articles were selected only if including nonredundant information and treatment options still relevant in the field. Reviews were excluded. Articles were also identified through searches of the authors' own files. The search was restricted to articles published in English. The final reference list was generated based on relevance to the scope of this review.
Table 1.
Genes Related to Ion Channels, Receptors, Transporters, Synapse-Related, and Other Mechanisms Associated With Epilepsy Phenotypes With Current and Emerging Therapies When Available at the Time of Writing This Review (July 2021)

Table 2.
Genes Related to Cell Growth, Division, and Proliferation Mechanisms Associated With Epilepsy Phenotypes With Current and Emerging Therapies When Available at the Time of Writing This Review (July 2021)

Table 3.
Genes Related to Cell Metabolism, Protein Biosynthesis and Degradation, and Mitochondrial Function Associated With Epilepsy Phenotypes With Current and Emerging Therapies When Available at the Time of This Review (July 2021)

The Main Categories of Monogenic Epilepsies: From Genotype to Phenotype
Generalized and Focal Epilepsy Syndromes
Like other diseases with complex inheritance, a proportion of idiopathic generalized epilepsy phenotypes are caused by variants in single genes with strong effect size. For example, mutations in the SLC2A1, GABRA1, and GABRAG2 genes account for rare sporadic or familial generalized epilepsies.
Focal epilepsies account for 50%–60% of all epilepsies and are divided into those with a structural etiology and nonacquired focal epilepsies (NAFEs). Examples of focal epilepsies with a structural genetic etiology include COL4A1-related porencephalic cysts and other nonspecific brain abnormalities causing focal epilepsies, GNAQ mosaic mutations causing Sturge-Weber syndrome, and cerebral cavernous malformations caused by mutations in KRIT1, CCM2, or PCDC10. Inherited or de novo germline mutations in the mammalian target of rapamycin (mTOR) pathway genes (i.e., AKT3, DEPDC5, MTOR, NPRL2, NPRL3, PIK3CA, PIK3R2, TSC1, TSC2) cause focal epilepsies, with or without visible brain malformations, often with neurodevelopmental disorders. Somatic mutations in mTOR pathway genes can also cause epileptogenic brain malformations such as type II focal cortical dysplasia (FCD), hemimegalencephaly, and tuberous sclerosis complex (TSC). In some individuals with an already identified germline mutation, a somatic second hit mutation in the same or different genes of the mTOR pathway has been identified in surgically removed brain tissue.6,7 The diagnostic yield of genetic testing in NAFE tends to be low8 and in many the etiology remains unknown. However, a genetic diagnosis should still be pursued as it may prompt specific management and treatment strategies. Examples of monogenic NAFEs include autosomal dominant sleep-related hypermotor epilepsy, caused by mutations in CHRNA4, CHRNA2, CHRNB2, DEPDC5, KCNT1, NPRL2, or NPRL3, familial focal epilepsy occasionally reported with DEPDC5 mutations, autosomal dominant lateral temporal lobe epilepsy caused by LGI1 mutations, and focal epilepsy with rolandic spikes caused by GRIN2A mutations.
Developmental and Epileptic Encephalopathies
Developmental or epileptic encephalopathies (DEE/EEs) are characterized by early onset of seizures, typically pharmacoresistant, often accompanied by severe epileptiform EEG discharges, both of which may affect neurodevelopment. DEE/EEs include syndromes such as early infantile epileptic encephalopathy, West syndrome, epilepsy of infancy with migrating focal seizures, Dravet syndrome (DS), and Lennox-Gastaut syndrome. “Monogenic” deleterious mutations are identified in up to ∼40% of the DEE/EEs.2 Identifying the underlying genetic abnormality of EE/DEEs is critical as some are potentially treatable and pharmacotherapy can be rationalized in a number of conditions. A large number of monogenic epilepsies include channelopathies, where pathogenic mutations result in a gain or loss of function of voltage-gated or ligand-gated ion channels and phenotype severity usually correlates with the degree of functional impairment of the channel involved.9 In addition, gain vs loss of function of the same channel can both, on occasion, result in epilepsy but usually with different phenotypes, as in GRIN2A-, SCN2A-, SCN1A-, and SCN8A-related epilepsies. Monogenic DEE/EE also include metabolic conditions (e.g., GLUT1 deficiency syndrome caused by SLC2A1 mutations), synaptopathies (e.g., VAMP2-related neurodevelopmental disorders), alteration in lysosomal homeostasis (e.g., ATP6V1A-related DEE), cell adhesion molecules (e.g., PCDH19-related EE), transporters (e.g., SLC13A5-related DEE), secreted proteins (e.g., SERPINI1-related progressive myoclonic epilepsy), and alteration in neuronal proliferation, migration, and differentiation (e.g., HECW2-related DEE) (Tables 1–3).
Antiseizure Treatment in the Most Prevalent Monogenic Epilepsies
The identification of a genetic etiology can lead to specific choices of ASM treatment in a small but growing number of genetic epilepsies. Treatments may include conventional ASMs or repurposed therapies (i.e., with specific actions that may have been used in entirely unrelated conditions). Treatment alteration may be guided by empirical clinical observation or targeted to the underlying specific pathophysiologic abnormality determined by the genetic etiology (Figure). We illustrate the most common monogenic childhood-onset epilepsies1 with their most appropriate treatment approaches.
Figure. Schematic Representation of the Disease Mechanisms Associated With Genetic Epilepsies.
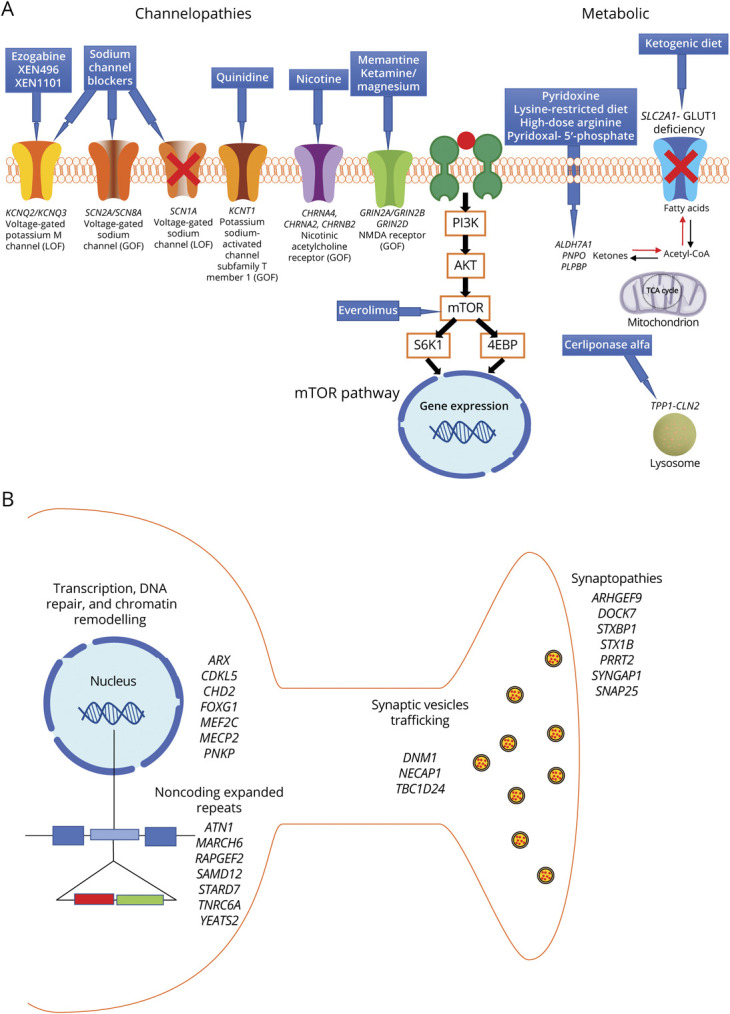
(A) Channelopathies, mTORopathies, and metabolic conditions where there is available targeted treatment based on the established underlying pathophysiology and (B) other genetic mechanisms including alteration of transcription, DNA repair, chromatin remodeling, synaptic pathways, and noncoding expanded repeats, where there are no available precision treatments.
PRRT2-Related Epilepsy
Mutations in PRRT2 cause self-limited (familial) infantile seizures and represent the most common monogenic epilepsy with an incidence of 1 per 9,970 live births.1 PRRT2-related disorders also include paroxysmal kinesigenic dyskinesia, where sodium channel blockers are the first-choice drugs. Seizures are usually responsive to a single ASM, including carbamazepine, oxcarbazepine, valproate, or phenobarbital.10 PRRT2 is thought to be involved in the modulation of synaptic neurotransmitter release, and most mutations lead to haploinsufficiency. Lack of PRRT2 leads to hyperactivity of voltage-dependent sodium channels, therefore causing alteration of neuronal excitability, and this may explain the efficacy of sodium channel blockers.11
Dravet Syndrome due to SCN1A Mutations
In DS, a DEE caused by loss-of-function SCN1A gene mutations, controlled trials have shown the efficacy of stiripentol, cannabidiol, and fenfluramine.12-14 There is no evidence that these drugs target specific pathophysiologic mechanisms in DS. Fenfluramine and cannabidiol are being tested in a variety of other severe epilepsies,15,16 while stiripentol may also be effective in other epilepsies.17 In a randomized, placebo-controlled trial of children with DS on stable doses of clobazam and valproic acid, 71% of those receiving add-on stiripentol vs only 5% receiving placebo achieved a >50% reduction in seizures.12 A 12-year prospective observational open-label study in DS showed that stiripentol improves long-term seizure frequency in approximately 50% of patients and significantly reduces the frequency of status epilepticus.18
Cannabidiol demonstrated efficacy in DS, with median percent reduction in target seizures of 38.9%, significantly higher than placebo treatment.13
After initial observations that that 70% of 12 persons with DS achieved seizure freedom with fenfluramine,19 2 randomized, placebo-controlled trials were conducted. In the first, fenfluramine vs placebo was added to ASM regimens that did not contain stiripentol.14 Higher dose fenfluramine was associated with a significantly greater likelihood of achieving a >50% and a >75% reduction in convulsive seizure frequency. In the second trial, fenfluramine vs placebo was added to children on concurrent stiripentol and those on fenfluramine were significantly more likely to achieve both a >50% and a >75% reduction in convulsive seizures.20 Further active trials are assessing the efficacy, safety, and tolerability of fenfluramine and cannabidiol in children and adults with DS.5 Fenfluramine acts as a serotonin releasing agent but also affects the sigma receptors. There are other serotonin modulators that were shown to be effective in suppressing seizures in DS experimental models, including clemizole and lorcaserin, for both of which there are ongoing multicenter, double-blind, randomized, placebo-controlled trials in DS.5
Recently released but unpublished data on soticlestat, a 24-hydroxylase cholesterol enzyme inhibitor, showed that it significantly reduced convulsive seizures in DS compared with placebo. Currently there is an active phase 2, prospective, multicenter extension study to assess the safety and tolerability of soticlestat in rare epilepsy syndromes, including DS.5
Given the underlying SCN1A loss of function, sodium channel blockers should be avoided in DS.21
KCNQ2-Related Epilepsy
Early-onset DEE due to loss-of-function mutations in KCNQ2, encoding for the Kv7.2 voltage-dependent neuronal potassium channel subunit, responds well to sodium channel blockers and it is thought that early effective treatment may reduce cognitive disability. Voltage-gated sodium channels and KCNQ potassium channels colocalize and are bound at critical neuronal locations (such as the axon initial segment where action potentials are initiated and nodes of Ranvier regulate action potential propagation); blocking sodium channels can therefore redress the functional effect of loss of function of KCNQ2.22 Additional targeted treatments include ezogabine,23 which directly increases the opening of KCNQ2 channels, for which there is an ongoing randomized, double-blind, placebo-controlled, multicenter study in KCNQ2-DEE (EPIK),5 and XEN1101, a small molecule that also selectively modulates the opening of KCNQ2/3 (KV7.2/7.3) potassium channels, for which there is an on ongoing randomized, double-blind, placebo-controlled, multicenter study in adults with focal epilepsy.5
SLC2A1-Related Epilepsy
In epilepsies caused by genetic mutations altering metabolic pathways, correction or replacement of the metabolic deficit can reverse or attenuate the pathophysiologic dysfunction. In GLUT1 deficiency syndrome, caused by heterozygous mutations in SLC2A1, ketogenic diet therapies are effective as they provide an alternative source, namely ketone bodies, for brain energy metabolism, thereby treating the symptoms of neuroglycopenia.24 Early diagnosis and initiation of the ketogenic diet are crucial to improve brain metabolism and seizure control, although the benefit on neurodevelopment remains controversial.25 A novel precision therapeutic option has been proposed through red blood cell exchange transfusion based on the hypothesis that red cells may have impaired glucose uptake; an active single-site proof of concept trial is in progress (early phase 1).5
CDKL5-deficiency disorder
CDKL5 encodes for the cyclin-dependent kinase-like 5 protein, which regulates neuronal morphogenesis and synaptic function. CDKL5 mutations cause a severe early onset DEE for which no targeted treatment exists. A randomized, placebo-controlled trial of ganaxolone, a positive allosteric modulator of the GABAA receptor, has been conducted; the results have not yet been published.5 There is now an expanded access program for compassionate use. A phase 2 extension study to assess the long-term safety and tolerability of soticlestat is including patients with cyclin-dependent kinase-like 5 deficiency disorder.5 Fenfluramine is also being trialed.5 Short-term efficacy with ketogenic diet has been reported in a retrospective uncontrolled study.26
PCDH19-Related Epilepsy
PCDH19 encodes for one of the cadherin superfamily of cell–cell adhesion molecules with diverse roles in neuronal migration, neuronal cell specification, or synaptic function, and its mutation causes PCDH19 girls-clustering epilepsy (PCDH19-GCE). There is evidence that PCDH19 can influence GABA A receptor expression and inhibition of postsynaptic currents in the rat brain. A double-blind, placebo-controlled, phase 3 clinical study is ongoing to evaluate the efficacy and safety of adjunctive ganaxolone, a positive allosteric modulator of the GABA A receptor, in PCDH19-related epilepsy.5 A retrospective study showed that levetiracetam can be effective for seizure control in PCDH19-GCE, including achievement of seizure freedom in a large proportion of patients.27 However, the behavioral side effects of levetiracetam may limit its use in this condition, in which cognitive impairment, autistic features, obsessive-compulsive, and attention-deficit disorders are frequent comorbidities.28
SLC6A1-Related Epilepsy
SLC6A1 encodes the GABA transporter protein type 1 (GAT1), which is one of the major GABA transporters of the human CNS. Mutations in SLC6A1 cause neurodevelopmental disorders most likely through haploinsufficiency. There are no treatment approaches targeting the underlying pathophysiologic mechanisms. A previous observational study showed that valproate was the most effective drug, probably due to its modulation of GABA neuronal concentrations, and most patients had drug-responsive epilepsy.29
TSC1/TSC2- and DEPDC5-Related Epilepsy
The TSC1 and TSC2 genes causing TSC, and the DEPDC5 gene, one of the genes most commonly associated with genetic focal epilepsy, encode for crucial inhibitory regulators of mTOR complex 1 (mTORC1), and their loss results in increased mTOR activity.30
Although TSC was a less frequent cause of epilepsy than DEPDC5 mutations in the population-based study that we used as epidemiologic framework for this review, its incidence was likely underestimated as inclusion criteria envisaged epilepsy to be the clinical presentation.1 Adjunctive treatment with everolimus, an mTOR inhibitor, successfully reduced seizures in a randomized placebo-controlled study including patients with TSC and treatment-resistant seizures31 and an open-label extension phase of the study showed sustained seizure reduction over time.32 Hypothetical mechanisms for the antiseizure effect of mTORC1 inhibition include the inhibition of formation and growth of cortical tubers that are presumed to be epileptogenic or the reduction of inflammation. Vigabatrin is the first-line treatment for children with TSC who present with infantile spasms. Putative mechanisms to explain its efficacy include an enhanced GABA inhibitory neurotransmission and inhibition of mTOR activation.33 A recent multicenter clinical trial showed that vigabatrin may also be used as epilepsy-preventing treatment in TSC as it reduced the risk of clinical seizures, drug-resistant epilepsy, and infantile spasms, compared with conventional ASM initiated after the first electrographic or clinical seizure.34
Chronic mTORC1 inhibition with rapamycin rescues the neurologic phenotype including seizures and premature death in DEPDC5 knockout mice35 and in mice with focal cortical expression of mutant mTOR.36 There is also experimental and clinical evidence of efficacy of the ketogenic diet in mTORopathies.37,38 Although the mechanisms underlying these antiseizure effects are unclear, mTOR pathway inhibition seems to contribute.37 There have not been dedicated medical or dietary treatment trials in patients with epilepsy caused by DEPDC5 mutations.
Epilepsy Surgery for Genetic Epilepsies
Surgical treatment of epilepsy can be undertaken if clinical, EEG, and imaging findings suggest focal localization of the epileptogenic zone and if any resulting neurologic deficit is not more severe than epilepsy itself. The classic principles of epileptogenic zone, ictal onset zone, and epileptogenic lesion based on surgical planning are more recently facing the challenging evidence that medically intractable focal epilepsies may be related to either germline or somatic gene mutations that may confer epileptogenicity to brain areas outside the targeted epileptogenic zone. This risk is particularly high in patients with more than one cortical lesion. In spite of these difficulties, it is recommended that epilepsy surgery be considered promptly in medically refractory TSC after failure of 2 ASMs, even in patients with multiple cortical lesions, multifocal interictal foci, and different types of seizures.39 Cumulative evidence indicates that patients with TSC have a 50%–60% chance of long-term seizure freedom after surgery for epilepsy, including those with infantile spasms.40
Germline or low-allele frequency somatic mutations in mTOR pathway genes can be demonstrated in up to 63% of patients with FCD type II,41 which bears close histopathologic similarities with the tubers of TSC. Somatic mTOR mutations and double mutational hits combining a germline and a somatic mutation of the DEPDC5 gene are most frequently found in FCD II36,41-43 but the overall number of patients described is too limited to draw any conclusion on the prognostic value specific mutations in each of these genes may have. In spite of these limitations, it appears that germline mutations of DEPDC5, PTEN, PIK3CA, AKT3, RHEB, and NPRL2 do not in themselves contraindicate resective surgery if a focal dysplastic lesion is present.41,44,45 Evidence begins to emerge that both mutational load and dysmorphic neuron density correlate with the epileptogenic zone42 and that mTOR mutations with strong hyperactivating properties may carry a higher risk of relapse of seizures after surgery.46 Although identification, quantification, and functional characterization of specific mosaic mutations could until recently be gathered only after surgery, access to cell-free DNA derived from the CSF47 can now demonstrate mosaic mutations before surgery. This type of information will likely become instrumental for better planning of surgical strategies and reintervention after a first unsuccessful surgery.
Experiences of resective epilepsy surgery in patients carrying germline mutations of other epilepsy genes, including sodium channel genes,48-50 or rare copy number variants45 have been rather limited but mostly disappointing, even when an MRI-visible lesion could be targeted.48
Other Targets of Management
Avoiding Seizure Precipitants
Environmental factors may exacerbate seizures in many epilepsy types. In DS and PCDH19-associated epilepsy, fever or hyperthermia are important triggers and in DS, photic stimulation, music, or diaper changing, as well as fatigue or excitement, may trigger seizures. Systematic screening for specific triggers known to cause reflex seizures in genetic epilepsies should be considered to inform management strategies.
Certain ASMs may lead to seizure exacerbation. In DS, sodium channel agents such as lamotrigine, oxcarbazepine, carbamazepine, as well as vigabatrin and tiagabine should be avoided (Table 1).
Treatment of Acute Seizures and Status Epilepticus
In some genetic epilepsies, there is an increased risk of prolonged seizures or status epilepticus, which represent a recurrent or even initial clinical manifestation of the syndrome (Tables 1–3).1,51 In these conditions, early and appropriate treatment of the epilepsy may reduce the incidence of status epilepticus.51 In addition, home rescue therapy should be considered and the adoption of a seizure rescue protocol is recommended, as this can improve outcome by reducing mortality and morbidity. Rescue therapy should be individualized to each patient. For example, young children with DS with a history of recurrent status are often provided rescue at the onset of a convulsive seizure. Although prehospital administration of benzodiazepines may be effective to control seizure clusters or status epilepticus and avoid hospital admission, excessive doses should be avoided due to higher risk of respiratory depression and longer hospitalization.52 The spectrum of action of drugs used in status epilepticus may differ from the epilepsy. For example, although phenytoin is contraindicated in DS, it has been used successfully against status epilepticus in some patients.53
Gene Therapies
There have been considerable recent advances in the development of therapies to target genetic disease. The introduction of new genetic material, modification of the genome, and modification of DNA transcription all fall under the rubric of “gene therapy.” The growth in gene therapies in medicine has partly been realized through the development of safe and effective means of gene delivery using viral vectors. Viral vectors are viruses that have had the genetic instructions for replication removed and replaced with a desired cargo, which consists of a promoter (determining the cell type in which the gene will be expressed) and the target gene. This approach has certain drawbacks. Many of our present viral vectors can only carry a limited amount of DNA, restricting the size of the genes that can be expressed, and most vectors have to be injected into a restricted area of brain. However, improvements in vector design have permitted widespread expression throughout the brain with intraventricular injections and also peripheral injection of vectors that can cross the blood–brain barrier, enabling use in genetic epilepsies.54
Gene therapy treatment approaches can have the advantage of specifically targeting the mutated gene or the consequent protein expression. However, simply introducing unexpressed or underexpressed genes can be problematic. Some genes, such as SCN1A for DS, are too large for our present vectors. Also, there is little way to control “dosing” and there is therefore the risk of overdosing a gene, which could have a detrimental effect on cell viability or excitability.
An alternative approach to introduce a gene is gene editing. One of the simplest and most widely used gene editing tools is the CRISPR-Cas9 system. This makes use of a guide RNA that directs the enzyme Cas9 to a specific part of the genome where it cuts the DNA. This ever-increasingly sophisticated approach can repair or knockout genes, but its clinical translation has been hampered by varying efficiency, off-target effects, and, on occasion, insufficient vector size for the necessary genetic material. From a translational perspective, the dCas9 system may have greater traction. Here Cas9 is mutated to “dead” Cas9, which no longer cuts DNA and is instead fused with gene transcription regulators either activating or repressing genes neighboring the guide RNA binding site. The advantage of this system is that the genetic material can be easily contained in an adeno-associated viral vector. This approach has been used in a mouse model of DS to upregulate Scn1a expression in interneurons using a Scn1a-dCas9 activation system in an adeno-associated viral vector.55,56 This rescued interneuronal excitability, behavior, and attenuated hyperthermic seizures. The disadvantage of the CRISPR-Cas9 system is that Cas9 is a foreign protein and so potentially immunogenic.
Although targeting the abnormal gene or gene products would seem the most obvious strategy, it may not always be the best. This can be because gene mutations often have a developmental effect and so reversing the genetic cause postnatally may not reverse the effect of that mutation. Moreover, it may be possible to successfully treat the seizures but without an effect on the comorbidities. This may apply not only to developmental genes, but also to receptors and channels, some of which can modify brain development. Moreover, a mutation in one gene may have an effect on the expression of multiple other genes, which can then contribute to the development of epilepsy. An example is the mTOR-dependent expression of Kv1.1.57 The mTORopathies have decreased levels of Kv1.1 channel expression; because Kv1.1 is a powerful regulator of neuronal excitability and has potent antiseizure effects,58 upregulating Kv1.1 provides an alternative and a potentially more attractive and more easily translatable strategy to treat the mTORopathies.
An alternative to targeting DNA is to target transcription. mRNA transcription can be regulated using an antisense oligonucleotide (ASO), a single-stranded deoxyribonucleotide, which is complementary to the mRNA target, or double-stranded RNA-mediated interference. The latter is an endogenous system used by cells to regulate gene expression through noncoding small sequences of RNA (small interfering RNA [siRNA]) binding to mRNA, resulting in mRNA degradation. siRNA can be injected directly into the brain/CSF or can be coupled with cell penetrating peptides to enable peripheral administration and penetration across the blood–brain barrier. ASOs are usually administered intrathecally. ASO binding to mRNA can have several different possible effects: it can inhibit mRNA transcription, alter mRNA splicing, or increase mRNA degradation. Intraventricular administration of an ASO directed against the Scn8a transcript in a mouse model of an SCN8A gain-of-function mutation–associated encephalopathy delayed seizure onset and increased survival.59 Following transcription of DNA to produce precursor mRNA, the precursor mRNA is spliced to remove introns and join together exons. This splicing can occur multiple ways, so that most mammalian genes generate multiple mRNA versions (splice variants). Many of these splice variants are not translated and degrade. Because ASOs can regulate splicing, it is possible to use ASOs to increase the production of translated mRNA. This has been termed targeted augmentation of nuclear gene output (TANGO). TANGO has recently been used in an animal model of DS, increasing Scn1a transcript and the production of the sodium channels, reducing seizures and sudden death in epilepsy.60 This approach is undergoing a phase 1 and 2 clinical trial in DS.5
Lastly, a less precise but simpler approach to redress point mutations that prematurely terminate mRNA, and consequently prevent full-length protein expression, has been the emergence of small molecules that induce translational read-through, suppressing stop codons and consequently resulting in the synthesis of full-length proteins. Ataluren is one such drug that is undergoing a trial in a small number of people with nonsense mutation DS or CDKL5 deficiency.5
Clinicians who manage patients with genetic epilepsies should be aware of the implications that specific genetic etiologies may have on pathophysiology, natural history, and the associated comorbidities. The choice of ASMs for these patients is informed by a growing body of knowledge derived from completed and ongoing trials on patients with specific disorders and inferences drawn from observational studies and initial experiences with precision therapies. Despite the lack of robust evidence for most genetic epilepsies, examples of successful precision medicine application are increasing and new methodologies for treatment trials are emerging, such as N-of-1 trials. Ongoing and completed clinical trials for genetic epilepsies can be found online on the clinicaltrials.gov website.5
Although correlations between highly penetrant mutations are easier to grasp and may be seen as elective targets for precision treatment approaches, caution is required in inferring that pathophysiologic mechanisms can be reversed or antagonized after identifying a mutation in a given gene. Phenotypic heterogeneity is remarkable, and some genetic mutations activate epileptogenesis through their developmental effects, which may not be reversed postnatally, while other genes having seemingly pure functional consequences on excitability may act through either loss- or gain-of-function effects, and these may have opposite treatment implications. In addition, the functional consequences of missense mutations may be difficult to predict, making precision treatment approaches considerably more complex than estimated by deterministic interpretations.
Glossary
- ASM
antiseizure medication
- ASO
antisense oligonucleotide
- DEE
developmental epileptic encephalopathy
- DS
Dravet syndrome
- EE
epileptic encephalopathy
- FCD
focal cortical dysplasia
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- NAFE
nonacquired focal epilepsy
- PCDH19-GCE
PCDH19 girls-clustering epilepsy
- siRNA
small interfering RNA
- TANGO
targeted augmentation of nuclear gene output
- TSC
tuberous sclerosis complex
Appendix. Authors
Study Funding
R.G. was supported in part by the Tuscany Region Call for Health 2018 (grant DECODE-EE). S.B. was supported by the Muir Maxwell Trust and the Epilepsy Society.
Disclosure
R. Guerrini has acted as an investigator for studies with Zoogenic, Biocodex, UCB, Angelini, and Eisai Inc.; and has been a speaker and on advisory boards for Zoogenic, Biocodex, Novartis, Biomarin, and GW Pharma. S. Balestrini reports personal fees from Biocodex, Eisai, and UCB Pharma, outside the submitted work. E. Wirrell has served as a consultant for Biocodex and Biomarin and is a member of the Independent Adjudication Committee for the Envision study, supported by Encoded Therapeutics. M. Walker reports grants from Vitaflo, personal fees from UCB Pharma, personal fees from Eisai, personal fees from GSK, personal fees from GW Pharmaceuticals, and personal fees from Marinus Pharmaceuticals, outside the submitted work; and has patents “Composition for the use in the treatment of epilepsy” WO2018189113A1 licensed to Vitaflo, “Combination comprising decanoic acid for the treatment of epilepsy” WO2019002435A1 pending, “Therapeutic use of compounds” EP2642990B1 issued, “Combined use of a vector encoding a modified receptor and its exogenous agonist in the treatment of seizures” US10525103B2 issued, and “Expression vectors comprising engineered genes WO2018229254A1” pending. Go to Neurology.org/N for full disclosures.
References
- 1.Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142(8):2303–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stefanski A, Calle-López Y, Leu C, Pérez-Palma E, Pestana-Knight E, Lal D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta-analysis. Epilepsia. 2021;62(1):143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo MH, Bardakjian TM, Brzozowski MR, et al. Temporal trends and yield of clinical diagnostic genetic testing in adult neurology. Am J Med Genet. Epub 2021 Jun 2. [DOI] [PubMed] [Google Scholar]
- 4.Balestrini S, Chiarello D, Gogou M, et al. Real-life survey of pitfalls and successes of precision medicine in genetic epilepsies. J Neurol Neurosurg Psychiatry. Epub 2021 Apr 26. [DOI] [PMC free article] [PubMed]
- 5.US National Library of Medicine. Accessed July 27, 2021. clinicaltrials.gov/
- 6.Ribierre T, Deleuze C, Bacq A, et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest. 2018;128(6):2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelorosso C, Watrin F, Conti V, et al. Somatic double-hit in MTOR and RPS6 in hemimegalencephaly with intractable epilepsy. Hum Mol Genet. 2019;28(22):3755–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perucca P, Scheffer IE, Harvey AS, et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017;131:1–8. [DOI] [PubMed] [Google Scholar]
- 9.Lauxmann S, Verbeek NE, Liu Y, et al. Relationship of electrophysiological dysfunction and clinical severity in SCN2A-related epilepsies. Hum Mutat. 2018;39(12):1942–1956. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Q, Liu Z, Hu Y, et al. Different experiences of two PRRT2-associated self-limited familial infantile epilepsy. Acta Neurol Belg. 2020;120(4):1025–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fruscione F, Valente P, Sterlini B, et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. 2018;141(4):1000–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial: STICLO study group. Lancet. 2000;356(9242):1638–1642. [DOI] [PubMed] [Google Scholar]
- 13.Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–2020. [DOI] [PubMed] [Google Scholar]
- 14.Lagae L, Sullivan J, Knupp K, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394(10216):2243–2254. [DOI] [PubMed] [Google Scholar]
- 15.Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. 2018;378(20):1888–1897. [DOI] [PubMed] [Google Scholar]
- 16.Lagae L, Schoonjans A-S, Gammaitoni AR, Galer BS, Ceulemans B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia. 2018;59(10):1881–1888. [DOI] [PubMed] [Google Scholar]
- 17.Rosati A, Boncristiano A, Doccini V, et al. Long-term efficacy of add-on stiripentol treatment in children, adolescents, and young adults with refractory epilepsies: a single center prospective observational study. Epilepsia. 2019;60(11):2255–2262. [DOI] [PubMed] [Google Scholar]
- 18.Myers KA, Lightfoot P, Patil SG, Cross JH, Scheffer IE. Stiripentol efficacy and safety in Dravet syndrome: a 12-year observational study. Dev Med Child Neurol. 2018;60(6):574–578. [DOI] [PubMed] [Google Scholar]
- 19.Ceulemans B, Boel M, Leyssens K, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. 2012;53(7):1131–1139. [DOI] [PubMed] [Google Scholar]
- 20.Nabbout R, Mistry A, Zuberi S, et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol. 2020;77(3):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39(5):508–512. [DOI] [PubMed] [Google Scholar]
- 22.Pisano T, Numis AL, Heavin SB, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 2015;56(5):685–691. [DOI] [PubMed] [Google Scholar]
- 23.Millichap JJ, Park KL, Tsuchida T, et al. KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2016;2(5):e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seidner G, Alvarez MG, Yeh JI, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18(2):188–191. [DOI] [PubMed] [Google Scholar]
- 25.Alter AS, Engelstad K, Hinton VJ, et al. Long-term clinical course of Glut1 deficiency syndrome. J Child Neurol. 2015;30(2):160–169. [DOI] [PubMed] [Google Scholar]
- 26.Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: experience of >100 patients. Epilepsia. 2017;58(8):1415–1422. [DOI] [PubMed] [Google Scholar]
- 27.Sadleir LG, Kolc KL, King C, et al. Levetiracetam efficacy in PCDH19 girls clustering epilepsy. Eur J Paediatr Neurol. 2020;24:142–147. [DOI] [PubMed] [Google Scholar]
- 28.Kolc KL, Sadleir LG, Depienne C, et al. A standardized patient-centered characterization of the phenotypic spectrum of PCDH19 girls clustering epilepsy. Transl Psychiatry. 2020;10(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johannesen KM, Gardella E, Linnankivi T, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. 2018;59(2):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crino PB. mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harbor Perspect Med. 2015;5(4):a022442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10056):2153–2163. [DOI] [PubMed] [Google Scholar]
- 32.Franz DN, Lawson JA, Yapici Z, et al. Everolimus for treatment-refractory seizures in TSC: extension of a randomized controlled trial. Neurol Clin Pract. 2018;8(5):412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genom Hum Genet. 2019;20:217–240. [DOI] [PubMed] [Google Scholar]
- 34.Kotulska K, Kwiatkowski DJ, Curatolo P, et al. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann Neurol. 2021;89(2):304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klofas LK, Short BP, Zhou C, Carson RP. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum Mol Genet. 2020;29(8):1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim JS, Kim W, Kang H-C, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21(4):395–400. [DOI] [PubMed] [Google Scholar]
- 37.McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52(3):e7–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S, Lee EJ, Eom S, Kang H-C, Lee JS, Kim HD. Ketogenic diet for the management of epilepsy associated with tuberous sclerosis complex in children. J Epilepsy Res. 2017;7(3):45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curatolo P, Nabbout R, Lagae L, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatric Neurol. 2018;22(5):738–748. [DOI] [PubMed] [Google Scholar]
- 40.Ostrowsky-Coste K, Neal A, Guenot M, et al. Resective surgery in tuberous sclerosis complex, from Penfield to 2018: a critical review. Rev Neurol. 2019;175(3):163–182. [DOI] [PubMed] [Google Scholar]
- 41.Baldassari S, Ribierre T, Marsan E, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138(6):885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee WS, Stephenson SEM, Howell KB, et al. Second-hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol. 2019;6(7):1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirzaa GM, Campbell CD, Solovieff N, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 2016;73(7):836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldassari S, Picard F, Verbeek NE, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21(2):398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevelink R, Sanders MW, Tuinman MP, et al. Epilepsy surgery for patients with genetic refractory epilepsy: a systematic review. Epileptic Disord. 2018;20(2):99–115. [DOI] [PubMed] [Google Scholar]
- 46.Guerrini R, Cavallin M, Pippucci T, et al. Is focal cortical dysplasia/epilepsy caused by somatic MTOR mutations always a unilateral disorder? Neurol Genet. 2021;7(1):e540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye Z, Chatterton Z, Pflueger J, et al. Cerebrospinal fluid liquid biopsy for detecting somatic mosaicism in brain. Brain Commun. 2021;3(1):fcaa235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barba C, Parrini E, Coras R, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014;55(7):1009–1019. [DOI] [PubMed] [Google Scholar]
- 49.Skjei KL, Church EW, Harding BN, et al. Clinical and histopathological outcomes in patients with SCN1A mutations undergoing surgery for epilepsy. J Neurosurg Pediatr. 2015;16(6):668–674. [DOI] [PubMed] [Google Scholar]
- 50.Scheffer IE, Harkin LA, Grinton BE, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. 2007;130(pt 1):100–109. [DOI] [PubMed] [Google Scholar]
- 51.Guerrini R, Parrini E, Marini C, Mei D. What is the role of next generation sequencing in status epilepticus?. Epilepsy Behav. 2019;101(pt B):106373. [DOI] [PubMed] [Google Scholar]
- 52.Spatola M, Alvarez V, Rossetti AO. Benzodiazepine overtreatment in status epilepticus is related to higher need of intubation and longer hospitalization. Epilepsia. 2013;54(8):e99–e102. [DOI] [PubMed] [Google Scholar]
- 53.Tanabe T, Awaya Y, Matsuishi T, et al. Management of and prophylaxis against status epilepticus in children with severe myoclonic epilepsy in infancy (SMEI; Dravet syndrome): a nationwide questionnaire survey in Japan. Brain Dev. 2008;30(10):629–635. [DOI] [PubMed] [Google Scholar]
- 54.Deverman BE, Pravdo PL, Simpson BP, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34(2):204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Colasante G, Lignani G, Brusco S, et al. dCas9-Based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther. 2020;28(1):235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamagata T, Raveau M, Kobayashi K, et al. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis. 2020;141:104954. [DOI] [PubMed] [Google Scholar]
- 57.Raab-Graham KF, Haddick PCG, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314(5796):144–148. [DOI] [PubMed] [Google Scholar]
- 58.Snowball A, Chabrol E, Wykes RC, et al. Epilepsy gene therapy using an engineered potassium channel. J Neurosci. 2019;39(16):3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lenk GM, Jafar‐Nejad P, Hill SF, et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol. 2020;87(3):339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han Z, Chen C, Christiansen A, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12(558):eaaz6100. [DOI] [PubMed] [Google Scholar]