

Abstract
Background:
Associations between epigenetic aging with cognitive aging and neuropsychiatric measures are not well-understood.
Objective:
1) To assess cross-sectional correlations between second-generation DNA methylation (DNAm)-based clocks of healthspan and lifespan (i.e., GrimAge, PhenoAge, and DNAm-based estimator of telomere length [DNAmTL]) and cognitive and neuropsychiatric measures; 2) To examine longitudinal associations between change in DNAm markers and change in cognition over 2 years.
Methods:
Participants were members of VITAL-DEP (VITamin D and OmegA-3 TriaL- Depression Endpoint Prevention) study. From previously ascertained cognitive groups (i.e., cognitively normal and mild cognitive impairment), we randomly selected 45 participants, aged ≥ 60 years, who completed in-person neuropsychiatric assessments at baseline and 2 years. The primary outcome was global cognitive score (averaging z-scores of 9 tests). Neuropsychiatric Inventory severity scores were mapped from neuropsychiatric symptoms (NPS) from psychological scales and structured diagnostic interviews. DNAm was assayed using Illumina MethylationEPIC 850K BeadChip at baseline and 2 years. We calculated baseline partial Spearman correlations between DNAm markers and cognitive and NPS measures. We constructed multivariable linear regression models to examine longitudinal relations between DNAm markers and cognition.
Results:
At baseline, we observed a suggestive negative correlation between GrimAge clock markers and global cognition but no signal between DNAm markers and NPS measures. Over 2 years: each 1-year increase in DNAmGrimAge was significantly associated with faster declines in global cognition; each 100-base pair increase in DNAmTL was significantly associated with better global cognition.
Conclusion:
We found preliminary evidence of cross-sectional and longitudinal associations between DNAm markers and global cognition.
Keywords: Alzheimer’s disease, cognition, DNA methylation, epigenetics, neuropsychiatric symptoms
INTRODUCTION
Aging is the strongest known risk factor for dementia [1]. Variation in epigenetic aging has been associated with age-related morbidities including dementia [2, 3]; yet less is known about the interface between epigenetic aging and cognitive aging. Understanding the epigenetic mechanisms through which biological aging processes may affect cognitive function could yield advances in prevention and treatment of dementia.
The second-generation DNA methylation (DNAm)-based epigenetic clocks of healthspan and lifespan, such as PhenoAge [4], GrimAge [5], and DNAm-based estimator of telomere length (DNAmTL) [6], outperform former DNAm clocks (e.g., Horvath’s DNAmAge clock, Hannum’s clock) in predicting numerous age-related morbidities [7]. Few epigenetic studies have examined relations of second-generation DNAm markers to cognitive function (e.g., verbal memory, executive function/attention) in older adults [8–13]; data are limited regarding whether changes in second-generation DNAm markers can be associated with longitudinal changes in cognitive function among community-dwelling older adults. Filling these knowledge gaps could advance understanding of how different paths in biological aging relate to cognitive aging and decline.
Neuropsychiatric symptoms (NPS) are highly prevalent in the ADRD (Alzheimer’s disease and related dementias) spectrum and even appear in the pre-clinical and early stages [14, 15]. Cognitive impairment accompanied by NPS versus without NPS may also relate to higher risk for worse cognitive outcomes [14]. While clinical-level behavioral symptoms or disorders have been significantly related to other aging biomarkers, such as telomeres [16, 17], less is known about the association between epigenetic aging and NPS among persons across the ADRD spectrum. Additionally, data on genome-wide differences in DNAm at specific CpG sites (i.e., leading to differences in gene expression) that may underlie variations in MCI with versus without NPS or in MCI versus CN could yield novel mechanistic insights into the development of cognitive and neuropsychiatric phenotypes.
Thus, we conducted a pilot study of biological aging and cognition by leveraging high-dimensional molecular and phenotypic data. Our study objectives were two-fold: 1) examine cross-sectional correlations between second-generation DNAm-based epigenetic markers and cognitive and neuropsychiatric measures; 2) examine longitudinal associations between changes in DNAm markers and changes in global and domain-specific cognitive functions over 2 years. Exploratorily, we assessed genome-wide differences in DNAm among persons with contrasting cognitive and neuropsychiatric phenotypes.
METHODS
Source of participants and samples
Participants were members of VITAL-DEP (VITamin D and OmegA-3 TriaL-Depression Endpoint Prevention), a late-life depression prevention ancillary study to the VITAL trial; protocol details of VITAL and VITAL-DEP are published elsewhere [18, 19]. VITAL is a completed 2×2 factorial trial of vitamin D and/or marine omega-3 fatty acids (omega-3) supplements for prevention of cardiovascular disease and cancer in 25,871 men and women, aged 50 + and 55 + years, respectively. VITAL established a Clinical Translational Science Center (CTSC) sub-cohort of 1,054 men and women, all of whom were participants in the main trial, who presented for in-person health assessments at baseline and 2-year follow-up. Among these CTSC participants, VITAL-DEP established a subset of 1,046 participants who completed comprehensive neuropsychiatric assessments at baseline and 2 years, as described elsewhere [18]. Participants provided blood samples at baseline and follow-up CTSC visits.
Previously, we ascertained cognitive status in VITAL-DEP CTSC participants at baseline and 2 years. From previously ascertained cognitive groups (i.e., cognitively normal [CN] and mild cognitive impairment [MCI]), we randomly selected 45 VITAL-DEP CTSC participants [aged ≥ 60 years; 20 with CN status and 25 with MCI] who completed cognitive assessments and had at least 1 μg of extracted genomic DNA available for DNAm assay at baseline and 2 years; the sample was balanced by 10-year age groups and sex across cognitive groups. All participants provided written informed consent, and study approvals were obtained from the institutional review board of Mass General Brigham.
Ascertainment of cognitive function status, cognitive outcomes, and neuropsychiatric symptoms
We determined MCI status using the consensus diagnostic method similar to that of the Uniform Dataset protocol [20–22], as implemented by the Alzheimer’s Disease Research Center programs. VITAL enrolled generally healthy and high-functioning men and women; thus, as in prior work [23, 24], we applied the 1.5 standard deviation (SD) below-expected mean cut-point criterion for cognitive test performance to define MCI status in this sample. CN status was determined among those who had no subjective cognitive concerns, had no objective evidence of cognitive impairment, and did not meet the consensus diagnostic criteria for MCI.
The VITAL-DEP CTSC protocol featured in-person detailed neuropsychiatric assessments at baseline and 2 years, as detailed elsewhere [18, 25]. The cognitive battery included nine tests assessing general cognition (Modified Mini-Mental State (3MS; range = 0–100) [26], immediate and delayed verbal memory [27, 28], category fluency [29], executive function and attention) [30]. The primary cognitive outcome was a global cognitive score (averaging z-scores of all tests). Secondary cognitive outcomes were general cognition (3MS score), verbal memory (averaging z-scores of 4 tests: immediate and delayed recalls of both a 10-word list and the East Boston Memory Test), and executive function/attention [averaging z-scores of trail-making tests A and B and 2 category fluency tests (naming animals and vegetables)].
We identified neuropsychiatric symptoms (NPS) at baseline by leveraging both the self-reported psychological scales [e.g., Patient Health Questionnaire (PHQ)-9 [31] for depressive symptoms; Generalized Anxiety Disorder (GAD)-7 [32] for anxiety symptoms] and item-level symptom features from the modules of the Mini International Neuropsychiatric Interview (MINI) [33]. We mapped these symptoms to the Neuropsychiatric Inventory-Questionnaire (NPI-Q) [34] and created an NPI severity score. The presence of NPS was determined using a cut-off of ≥ 2 points on the NPI severity score.
DNA methylation assay and analysis
Genomic DNA was extracted from peripheral blood leukocytes (buffy-coat cryotubes had been stored in the vapor phase of liquid nitrogen freezers at ≤–130°C for later use) using the QIAamp® DNA Blood Mini Kit (Qiagen Inc., Valencia, CA) and PicoGreen DNA quantitation was performed using a Molecular Devices 96-well spectrophotometer. DNA methylation assays on selected samples and quality controls (QCs) were conducted using Illumina Infinium MethylationEPIC BeadArray technology (Methyl850K chip) that allows genome-wide DNA methylation analysis of 866,836 CpG sites; assay details are described elsewhere [35]. To assess QC of the DNA methylation assays, we included four blinded samples randomly placed on the plate, in duplicate for testing of QC replicates, and one in-lab genotyping control. CpG methylation values in the n = 4 pairs of QC replicates were highly correlated (r ≥0.95). All samples passed the QC threshold, and biological sex (X, Y chromosome) was correctly identified. Regarding DNA methylation analysis, we used the minfi Bioconductor package in R for processing functions (performing background correction using negative control probe signal intensities, as well as normalization and correction of dye imbalance) and analyzing the Illumina 850k methylationEPIC data [36, 37]. Probes with a mean detection p-value higher than 0.05, cross-reactive probes, non-CpG probes, and probes bound to SNP (Single Nucleotide Polymorphisms) sites were excluded for downstream analysis. Beta values were normalized using Noob-normalization with the minfi package in R. We computed the second-generation DNAm-based epigenetic markers of lifespan and healthspan—DNAmPhenoAge, DNAmGrimAge, and DNAmTL—using an online age calculator developed by Horvath and colleagues (https://dnamage.genetics.ucla.edu/) [38]. DNAm-PhenoAge is a DNAm-based aging biomarker that utilizes 513 CpGs to predict multifactorial phenotypic age of an individual [4]. DNAmGrimAge, a composite of 12 sub-DNAm-measures that utilizes 1,030 CpGs to predict lifespan and all-cause mortality [5]. DNAmTL uses 140 CpG sites to estimate telomere length in kilobase pairs [6]. Additionally, age-accelerated versions of these 3 clocks (i.e., AgeAccelPheno, AgeAccelGrim, DNAmTLadjAge) can be computed by regressing epigenetic age on chronological age.
Statistical analyses
Descriptive characteristics were shown in the total sample and by cognitive groups. For comparisons, we used Wilcoxon rank sum tests for continuous variables and Fisher exact tests for proportions. We computed Spearman rank correlations between second-generation DNAm markers and chronological age. We computed Spearman partial correlations between DNAm markers and global and domain-specific cognitive scores at baseline; estimates were adjusted by age, sex, estimated blood cell types [i.e., naïve CD8 + T cells, exhausted cytotoxic CD8 + T cells (defined as CD8 positive CD28 negative CD45 R negative), CD4 + T cells, plasma blasts, natural killer cells, monocytes, and granulocytes] [39, 40], body mass index (BMI), cigarette smoking, and Charlson-Deyo comorbidity index [41, 42]. Similarly, we computed partial Spearman correlations between DNAm markers and NPS measures (i.e., PHQ-9, GAD-7, NPI severity score). We performed multivariable linear regression analyses to determine the longitudinal associations between 2-year change in DNAm epigenetic markers and 2-year change in global and domain-specific cognitive scores; we used Δperformance (i.e., cognitive performance at follow-up – cognitive performance at baseline) as the outcome and ΔDNAm as the exposure. Of note, the slope coefficient estimating the magnitude of longitudinal change in primary DNAm clock markers and change in cognitive function (ΔDNAm → Δperformance) is identical to the slope coefficient estimating the magnitude of longitudinal change in age-accelerated versions of DNAm marker and change in cognitive function (ΔAgeAccelDNAm → Δperformance). For simplicity, we present only the regression estimates of longitudinal change in primary DNAm clock markers and cognitive change. Models were adjusted for chronological age, sex, estimated blood cell types, BMI, cigarette smoking, and Charlson-Deyo comorbidity index. Regression coefficients (β) and 95% confidence intervals (CIs) are presented.
Post-hoc analyses
First, we computed Spearman-rank correlations between second-generation DNAm markers and cognitive outcomes according to baseline cognitive status. Second, we used general linear models of response profiles to relate baseline DNAm markers with cognitive change over 2 years. Third, as in prior work [35], we conducted an exploratory analysis of genome-wide differences in DNAm in these contrasting groups: 1) MCI versus CN; 2) MCI without NPS versus MCI with NPS; 3) CN with NPS versus CN without NPS; 4) NPS versus no NPS; ‘DMPFinder’ function in the minfi R package was used. Over-representation analysis was conducted, and annotation was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [43].
Results of secondary cognitive outcomes or post-hoc analyses were not adjusted for multiple hypothesis testing; findings from these analyses are considered exploratory and interpreted with caution. A two-tailed p-value<0.05 was used for statistical significance. All statistical analyses were performed with SAS version 9.4 (SAS, Cary, NC) and R.
RESULTS
Baseline characteristics
Participants’ mean age (standard deviation) was 69.8 (5.5) years and 48.9% were females. Of the n = 45 sample participants, there were 39 non-Hispanic white, 3 Black, and 3 Asian participants. Descriptive characteristics in the total sample and by cognitive groups are shown in Table 1. At baseline, participants with MCI, compared to CN, had significantly lower body mass index (BMI) and lower global and domain-specific cognitive scores. No differences were observed in DNAm-based epigenetic markers by cognitive phenotypes. PHQ-9, GAD-7, and NPI severity scores appeared similar in MCI and CN groups. The prevalence of NPS (≥2 NPI severity score) was 28.9% in this sample. Distributions of estimated blood cell types were similar in both groups except for naïve CD8 + T cells, Plasma blasts, and granulocytes. Figure 1 shows the scatterplot data and Spearman rank correlations between chronological age and second-generation DNAm-based epigenetic markers. DNAmGrimAge had the strongest positive correlation with chronological age [Spearman rho (ρ)=0.84, p < 0.001], followed by DNAmPhenoAge [ρ=0.65, p < 0.001]. There was a significant negative correlation between DNAmTL and chronological age [ρ=−0.57, p < 0.001].
Table 1.
Participant characteristics in the total sample, and by cognitive groups
| Characteristics | Total sample (n = 45) | MCI (n = 25) | CN (n = 20) | pa |
|---|---|---|---|---|
| Age at first cognitive assessment, Mean (SD), y | 69.8 (5.5) | 69.6 (5.3) | 70.0 (5.8) | 0.81 |
| Sex, n (%) | 0.77 | |||
| Male | 23 (51.1) | 12 (48.0) | 11 (55.0) | |
| Female | 22 (48.9) | 13 (52.0) | 9 (45.0) | |
| Self-reported race/ethnicity, n (%) | 0.53 | |||
| Non-Hispanic White | 39 (86.7) | 23 (92.0) | 16 (80.0) | |
| Black | 3 (6.7) | 1 (4.0) | 2 (10.0) | |
| Asian | 3 (6.7) | 1 (4.0) | 2 (10.0) | |
| Education, n (%) | >0.99 | |||
| College or lower | 17 (37.8) | 9 (36.0) | 8 (40.0) | |
| Post-college | 28 (62.2) | 16 (64.0) | 12 (60.0) | |
| BMI at first cognitive assessment, Mean (SD), kg/m2 | 27.3 (5.8) | 24.9 (3.6) | 30.4 (6.5) | 0.001 |
| Total physical activity,b median (IQR), MET-h/week | 21.7 (9.7 − 54.2) | 22.1 (6.9 − 61.1) | 18.5 (12.4 − 36.7) | 0.78 |
| Cigarette smoking, n (%) | >0.99 | |||
| Never | 25 (55.6) | 14 (56.0) | 11 (55.0) | |
| Past or current | 20 (44.4) | 11 (44.0) | 9 (45.0) | |
| Daily alcohol use, n (%) | >0.99 | |||
| Daily | 15 (34.1) | 8 (33.3) | 7 (35.0) | |
| Other than daily | 29 (65.9) | 16 (66.7) | 13 (65.0) | |
| Charlson-Deyo comorbidity index,c n (%) | >0.99 | |||
| 0 point | 40 (88.9) | 22 (88.0) | 18 (90.0) | |
| 1 + points | 5 (11.1) | 3 (12.0) | 2 (10.0) | |
| Estimated blood cell types,d mean (SD) | ||||
| Naїve CD8 + T cells | 175.1 (38.1) | 163.8 (31.5) | 189.3 (41.7) | 0.06 |
| Cytotoxic CD8 + T cellse | 5.9 (3.1) | 6.1 (2.9) | 5.6 (3.5) | 0.26 |
| CD4 + T cells | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.1) | 0.36 |
| Plasma blasts | 1.7 (0.2) | 1.7 (0.2) | 1.6 (0.2) | 0.04 |
| Natural killer cells | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.1) | 0.25 |
| Monocytes | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.0) | 0.26 |
| Granulocytes | 0.5 (0.1) | 0.6 (0.1) | 0.5 (0.1) | 0.04 |
| Second-generation DNAm markers, mean (SD) | ||||
| DNAmPhenoAgef , y | 55.8 (6.2) | 55.7 (6.3) | 55.9 (6.2) | 0.79 |
| AgeAccelPhenof , y | 0.2 (5.0) | 0.2 (5.2) | 0.2 (4.9) | 0.88 |
| DNAmGrimAgeg, y | 69.7 (5.1) | 69.5 (5.2) | 69.8 (5.0) | 0.69 |
| AgeAccelGrimg, y | 0.3 (2.9) | 0.3 (2.9) | 0.3 (2.9) | 0.86 |
| DNAmTLh , per 100-base pairs | 68.0 (2.4) | 68.0 (2.7) | 68.1 (2.0) | 0.92 |
| DNAmTLadjAgeh, per 100-base pairs | 0.0 (2.0) | −0.1 (2.4) | 0.1 (1.4) | 0.81 |
| Cognitive scores, mean (SD), standard units | ||||
| Global cognition scorei | −0.2 (0.8) | −0.7 (0.6) | 0.4 (0.5) | <0.001 |
| General cognition (3MS) score | 94.2 (5.0) | 92.2 (5.6) | 96.7 (2.7) | 0.005 |
| Verbal memory scorej | −0.1 (0.8) | −0.5 (0.6) | 0.5 (0.5) | <0.001 |
| Executive function/attention scorek | −0.2 (0.9) | −0.7 (0.9) | 0.3 (0.7) | <0.001 |
| Neuropsychiatric measures/scores, median (range) | ||||
| PHQ-9 score | 1.0 (0.0−4.0) | 1.0 (0.0 − 4.0) | 0.5 (0.0 − 4.0) | 0.97 |
| GAD-7 score | 0.0 (0.0 − 6.0) | 0.0 (0.0 − 6.0) | 0.5 (0.0 − 3.0) | 0.73 |
| NPI severity score | 1.0 (0.0 − 5.0) | 1.0 (0.0 − 5.0) | 1.0 (0.0 − 4.0) | 0.63 |
| Presence of NPS (≥2 NPI severity score) | 0.52 | |||
| Yes | 13 (28.9) | 6 (24.0) | 7 (35.0) | |
| No | 32 (71.1) | 19 (76.0) | 13 (65.0) |
CN, cognitively normal; MCI, mild cognitive impairment; DNAm, DNA methylation; SD, standard deviation; IQR, interquartile range; BMI, body mass index; MET, metabolic equivalent of task; 3MS, the modified mini-mental state; PHQ, patient health questionnaire; GAD, generalized anxiety disorder; NPS, neuropsychiatric symptoms; NPI, neuropsychiatric inventory.
For comparisons, we used the Wilcoxon-rank sum test for continuous variables and Fisher exact test for categorical variables.
Leisure-time physical activities include walking or hiking; jogging; running; bicycling; aerobic exercise/aerobic dance/exercise machines; lower intensity exercise/yoga/stretching/toning; tennis/squash/racquetball; lap swimming; weightlifting/strength training; other exercise.
The Charlson-Deyo comorbidity index is a weighted comorbidity score derived from the sum of the scores for each of several major medical comorbid conditions; participants were categorized as having 0 and 1 or more points [41, 42].
These are ordinal abundance measures of cell counts estimated in the Epigenetic clock by Horvath and colleagues. The abundance of I CD8 + T cells, exhausted CD8 + T cells, and plasma blasts were derived using the Horvath method [39]. The natural killer cells, monocytes, and granulocytes were derived using the Houseman method [40].
Exhausted CD8 + T cells were defined as CD8 positive CD28 negative CD45 R negative [39].
DNAmPhenoAge is a validated novel epigenetic clock that predicts varying aging outcomes, including all-cause mortality, cancers, healthspan, physical functioning, and Alzheimer’s disease [4]. AgeAccelPheno is an age-accelerated residual measure of DNAmPhenoAge, derived by regressing epigenetic age on chronological age.
DNAmGrimAge is a validated novel DNAm biomarker which predicts lifespan and all-cause mortality [5]. AgeAccelGrim is an age-accelerated residual measure of DNAmGrimAge, derived by regressing epigenetic age on chronological age.
DNAmTL is a DNA methylation measurement of telomere length (unit is per 100-base pairs) [6]. DNAmTLadjAge is an age-accelerated residual measure of DNAmTL, derived by regressing epigenetic age on chronological aIe.
Global cognition score is a composite score for the mean of the z-scores of all cognitive tests.
Verbal memory score is a composite score for the mean of the z-scores of 4 tests: the immediate and delayed recalls of both the 10-word list and the East Boston Memory Test.
Executive function/attention score is a composite score for the mean of the z-scores of 4 tests: trails making tests A and B, and category fluency tests (naming animals and vegetables).
Fig. 1.
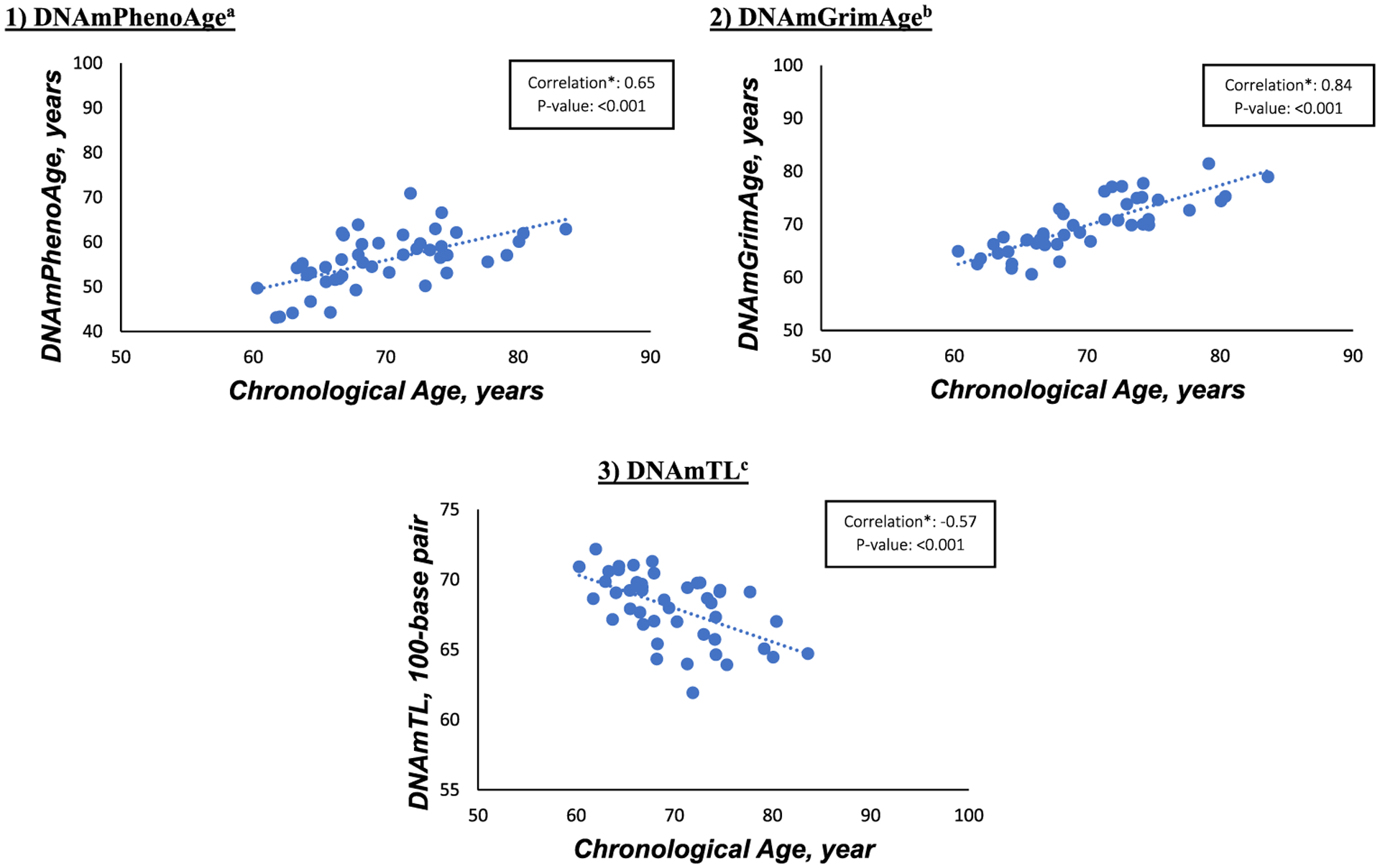
Spearman rank correlations between chronological age and second-generation DNAm markers at baseline. DNAm, DNA methylation. aDNAmPhenoAge is a validated novel epigenetic clock that predicts varying aging outcomes, including all-cause mortality, cancers, healthspan, physical functioning, and Alzheimer’s disease [4]. bDNAmGrimAge is a validated novel DNAm biomarker which predicts lifespan and all-cause mortality [5]. cDNAmTL is a DNA methylation measurement of telomere length (unit is per 100-base pairs) [6].
Cross-sectional relations of DNAm markers with cognitive and neuropsychiatric outcomes
Table 2 shows the partial Spearman rank correlations between second-generation DNAm-based epigenetic markers and cognitive outcomes after adjusting for chronological age, sex, estimated blood cell types, BMI, cigarette smoking, and Charlson-Deyo comorbidity index. We observed a signal of negative partial correlation between DNAmGrimAge and global cognitive score (ρ=−0.36; p = 0.04); partial correlations were similar between AgeAccelGrim and global cognitive score (ρ=−0.40; p = 0.02). There were no signals for partial correlations of DNAmPhenoAge, DNAmTL or their age-accelerated versions with global cognitive score.
Table 2.
Partial Spearman correlations between second-generation DNAm markers and cognitive outcomes at baseline (n = 45)
| Second-generation DNAm marker | Cognitive outcomes | ||||
|---|---|---|---|---|---|
| Primary Global cognitiond | Secondary | ||||
| General cognition (3MS) | Verbal memorye | Executive function/attention scoref | |||
| DNAmPhenoAgea, y | Rho | −0.10 | −0.10 | −0.01 | −0.09 |
| p | 0.59 | 0.57 | 0.95 | 0.62 | |
| AgeAccelPhenoa, y | Rho | −0.01 | −0.01 | 0.06 | −0.03 |
| p | 0.95 | 0.94 | 0.74 | 0.86 | |
| DNAmGrimAgeb, y | Rho | −0.36 | −0.10 | −0.17 | −0.32 |
| p | 0.04 | 0.59 | 0.34 | 0.07 | |
| AgeAccelGrimb, y | Rho | −0.40 | −0.09 | −0.19 | −0.38 |
| p | 0.02 | 0.61 | 0.28 | 0.03 | |
| DNAmTLc, per 100-base pairs | Rho | 0.21 | 0.04 | −0.07 | 0.28 |
| p | 0.24 | 0.83 | 0.71 | 0.12 | |
| DNAmTLadjAgec, per 100-base pairs | Rho | 0.16 | −0.04 | −0.12 | 0.24 |
| p | 0.38 | 0.83 | 0.51 | 0.18 | |
3MS, the Modified-Mini Mental State; DNAm, DNA methylation. Results of secondary cognitive outcomes were not adjusted for multiple hypothesis testing; findings should be interpreted with caution. The Spearman correlations between second-generation DNAm markers and cognitive outcomes were adjusted by chronological age, sex, blood cell proportions, body mass index, cigarette smoking, and Charlson-Deyo comorbidity index.
DNAmPhenoAge is a validated novel epigenetic clock that predicts varying aging outcomes, including all-cause mortality, cancers, healthspan, physical functioning, and Alzheimer’s disease [4]. AgeAccelPheno is an age-accelerated residual measure of DNAmPhenoAge, derived by regressing epigenetic age on chronological age.
DNAmGrimAge is a validated novel DNAm biomarker which predicts lifespan and all-cause mortality [5]. AgeAccelGrim is an age-accelerated residual measure of DNAmGrimAge, derived by regressing epigenetic age on chronological age.
DNAmTL is a DNA methylation measurement of telomere length (unit is per 100-base pair) [6]. DNAmTLadjAge is an age-accelerated residual measure of DNAmTL, derived by regressing epigenetic age on chronological age.
Global cognition score is a composite score for the mean of the z-scores of all cognitive tests.
Verbal memory score is a composite score for the mean of the z-scores of 4 tests: the immediate and delayed recalls of a 10-word list and the East Boston Memory Test.
Executive function/attention score is a composite score for the mean of the z-scores of 4 tests: trails making tests A and B, and category fluency tests (naming animals and vegetables).
Regarding secondary cognitive outcomes, we observed a signal of negative partial correlations between DNAmGrimAge and executive function/attention (ρ=−0.32; p = 0.07); the estimate was similar between AgeAccelGrim and executive function/attention (ρ=−0.38; p = 0.03). There were no signals for cross-sectional associations between PhenoAge and DNAmTL clock measures with verbal memory and executive function/attention. In this sample, second-generation DNAm markers were not correlated with PHQ-9, GAD-7, and NPI severity scores (Supplementary Table 1).
Longitudinal relations of change in DNAm markers and change in cognitive outcomes
We observed significant longitudinal associations between changes in DNAm markers and changes in global cognition (Fig. 2). Each 1-year increase in DNAmGrimAge over 2 years was significantly associated with a faster 2-year decline in global cognitive score [adjusted β (95% CI): −0.09 (−0.15, −0.02); p = 0.02]. Furthermore, each 100-base pair increase in DNAmTL over 2 years was significantly associated with better global cognitive score over 2 years [adjusted β (95% CI): 0.15 (0.03, 0.28); p = 0.02]. There were no significant longitudinal associations between DNAmPhenoAge with global cognitive score. In secondary analyses, we observed variable, weak signals between changes in DNAm markers and changes in verbal memory and executive function/attention (Table 3).
Fig. 2.
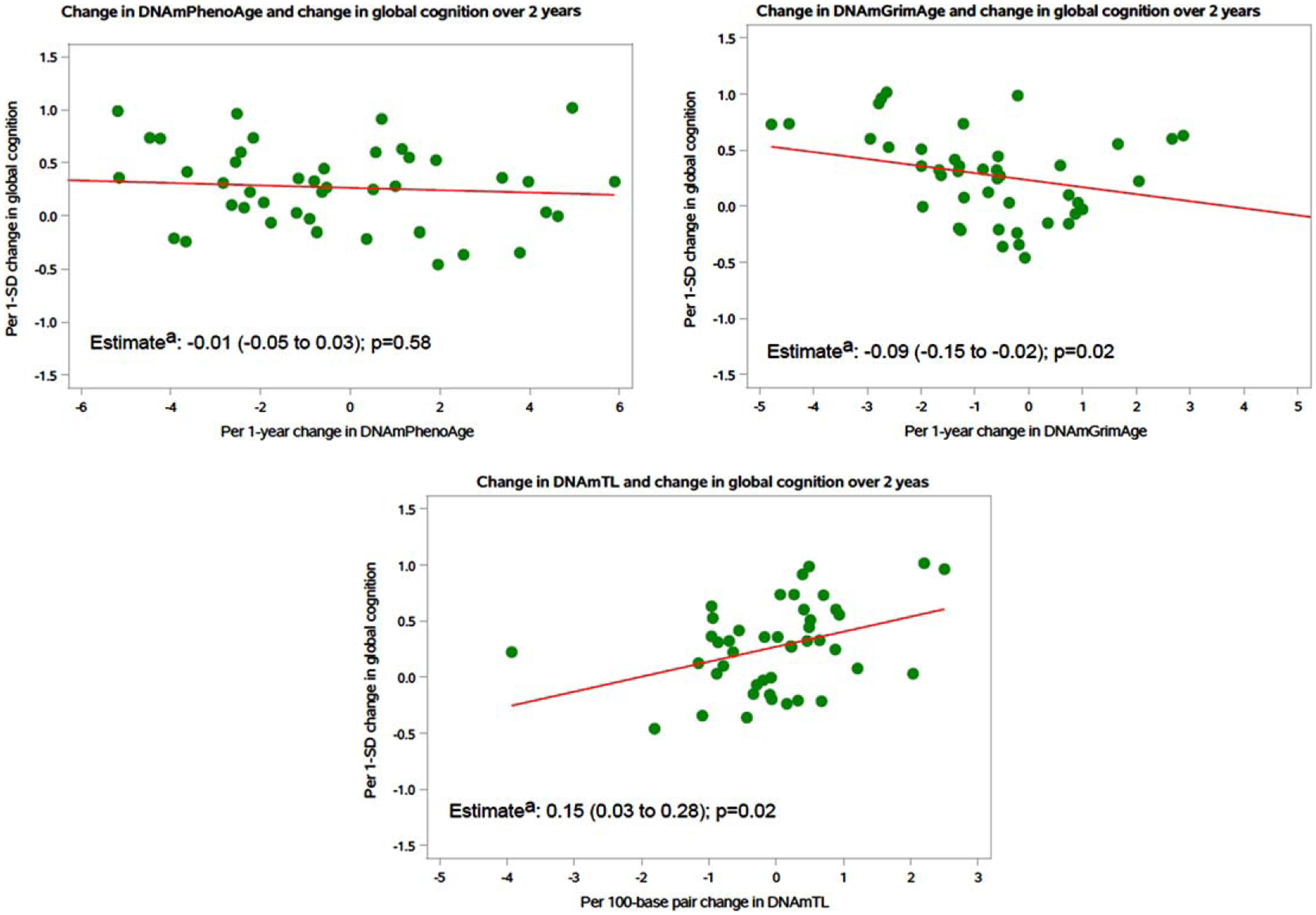
Association between 2-year change in DNAm marker and 2-year change in global cognition (n = 45). DNAm, DNA methylation; CI, confidence interval aRegression estimates and 95% CIs showed in the figures; models were adjusted by chronological age, sex, blood cell proportions, body mass index, cigarette smoking, and Charlson-Deyo comorbidity index.
Table 3.
Associations between 2-year change in second-generation DNAm markers and 2-year change in secondary cognitive outcome scores (n = 45)
| 2-year change in DNAm marker | 2-year change in secondary cognitive outcomes | |||||
|---|---|---|---|---|---|---|
| General cognitive score (3MS) | Verbal memoryd | Executive function/attentione | ||||
| β (95% CI) | p | β (95% CI) | p | β (95% CI) | p | |
| DNAmPhenoAge,a in years | −0.11 (−0.62 to 0.40) | 0.65 | −0.07 (−0.13 to −0.01) | 0.03 | 0.03 (0.003 to 0.06) | 0.03 |
| DNAmGrimAge,b in years | −0.57 (−1.46 to 0.32) | 0.20 | −0.08 (−0.20 to 0.04) | 0.17 | −0.06 (−0.11 to −0.01) | 0.03 |
| DNAmTL,c per 100-base pair | 0.93 (−0.68 to 2.54) | 0.25 | 0.21 (0.01 to 0.41) | 0.04 | 0.07 (−0.03 to 0.17) | 0.15 |
DNAm, DNA methylation; 3MS, the Modified Mini-Mental State; CI, confidence interval. Results of secondary cognitive outcomes were not adjusted for multiple hypothesis testing; findings should be interpreted with caution.
DNAmPhenoAge is a validated novel epigenetic clock that predicts varying aging outcomes, including all-cause mortality, cancers, healthspan, physical functioning, and Alzheimer’s disease [4].
DNAmGrimAge is a validated novel DNAm biomarker which predicts lifespan and all-cause mortality [5].
DNAmTL is a DNA methylation measurement of telomere length (unit is per 100-base pair) [6].
Verbal memory score is a composite score for the mean of the z-scores of 4 tests: the immediate and delayed recalls of both the TICS 10-word list and the East Boston Memory Test.
Executive function/attention score is a composite score for the mean of the z-scores of 4 tests: trails making tests A and B, and category fluency tests (naming animals and vegetables).
Post-hoc analyses
First, results of partial correlations between second-generation DNAm markers and cognitive function scores stratified by baseline cognitive status are shown in Supplementary Table 2; there were modest signals of negative partial correlations of GrimAge clock markers with global cognition and verbal memory in the MCI group but not in the CN group. There were relatively stronger signals of positive correlations of DNAmTL with global cognition and executive function/attention scores in the MCI group but not in the CN group. Second, baseline second-generation DNAm markers were not associated with change in global and domain-specific cognitive function scores over 2 years (Supplementary Table 3). Third, preliminary results from exploratory analyses of genome-wide differences contrasting cognitive and neuropsychiatric phenotypes are included in Supplementary Tables 4 and 5; no CpGs surpassed the threshold for genome-wide significance (p < 5×10 ‒8) in all comparisons. Exploratory results of potential biologic pathways underlying contrasts of MCI versus CN and MCI with versus without NPS are provided in the Supplement.
DISCUSSION
In this pilot study, chronological age was strongly correlated with DNAmGrimAge and moderately correlated with DNAmPhenoAge and DNAmTL. At baseline, we observed negative correlations between GrimAge clock measures with global cognition and executive function/attention. There were no signals for correlations between second-generation DNAm markers and their age-accelerated versions with PHQ-9, GAD-7, or NPI severity scores. Regarding DNAm-global cognition associations over 2-year follow-up: 1) each 1-year increase in DNAmGrimAge was significantly associated with faster decline in global cognition; 2) each 100-base pair increase in DNAmTL was significantly associated with better global cognitive scores. Secondarily, we observed a signal for longitudinal association between increase in DNAmGrimAge and faster declines in executive function/attention over 2 years. Exploratory genome-wide DNAm analysis revealed potential differences comparing MCI versus CN and MCI with versus without NPS.
The second-generation DNAm-based biomarkers of aging were developed recently with the objective of serving as biomarkers for healthspan and lifespan. While second-generation DNAm clocks have been associated with cognitive function and cognitive phenotypes [8–13, 44–46], this study provides new data regarding how changes in these DNAm markers are associated with changes in global and domain-specific cognitive function scores over time among community-dwelling older adults. Recently, a large prospective study found non-significant associations between accelerated second-generation DNAm with incident MCI and dementia [45]. However, differences in sample characteristics (MCI versus no MCI in the baseline sample), and the outcome under study (incident MCI versus cognitive change) limit comparisons between findings from the previous study and our current study.
In this pilot study, we observed accelerated cognitive decline in relation to a per-year increase in DNAmGrimAge and better cognitive change in relation to a per 100-base pair increase in DNAmTL; these results indicate significant correlations between changes in biological indicators of healthspan and lifespan and changes in cognitive function over time. For instance, epigenetic modifications are a prominent mechanism for controlling telomerase activity and regulating the TERT gene [47]. Additionally, experimental evidence suggests that early stages of age-related degenerative phenotypes could be reversed following the reactivation of endogenous telomerase activity [48]. Speculatively, it is possible that an increase in measured telomere length could reactivate telomerase activity through epigenetic modifications, thus, improving longitudinal cognitive function.
We observed no preliminary signals between second-generation DNAm markers and NPS measures in this sample. This pilot study included long-term trial participants who were generally healthy and high-functioning at baseline, and there was a relatively narrow range in PHQ-9, GAD-7, and NPI severity scores. It is also possible that peripheral blood methylation levels may not capture all epigenetic changes that might be related to complex neuropsychiatric manifestations; integration of brain tissue and cerebrospinal fluid could provide additional epigenetic information. Together, these issues may affect the ability to detect significant signals, as there was less variation in the sample than what might be seen in a larger or primarily clinical population. Finally, the exploratory analyses of genome-wide differences in DNAm in MCI versus CN and MCI with versus without NPS suggested some biological pathways (i.e., folate metabolism) reported in previous literature: e.g., altered one-carbon metabolism may impair DNA repair and methylation processes and contribute to microtubule-associated tau protein hyperphosphorylation – mechanisms implicated in the early stages of cognitive dysfunction [49, 50].
Study strengths include a well-characterized sample, cross-sectional and longitudinal study design within an experimental framework, state-of-the-art MethylationEPIC 850K technology, in-person detailed neuropsychiatric assessments, and administration of validated psychiatric and behavioral symptom measures.
Our study results should be interpreted in light of several limitations. First, although we identified signals for possible cross-sectional and longitudinal associations between second-generation DNAm epigenetic markers and global and domain-specific cognition, caution is needed given the exploratory nature of our work and the potential for chance findings. Similarly, results of partial correlations between epigenetic clocks and cognitive outcomes by baseline cognitive status (MCI versus CN) should be interpreted with caution. Second, to avoid statistical overfit in this small pilot sample, we used a limited set of covariates, and ancestry measures (e.g., principal components from the SNP array) were not included in the models. Third, our results regarding telomere length (TL) were obtained using an epigenetic clock, not a direct measurement of relative or absolute TL, so our results cannot be compared with previous studies of actual TL [51]. Fourth, we cannot exclude the possibility of reverse causation bias in observed signals of longitudinal associations between epigenetic aging and cognitive aging, i.e., lower cognitive performance is a cause rather than an effect of accelerated biological aging. Fifth, the sample had limited racial and ethnic diversity; studies with larger numbers of participants from diverse racial and ethnic backgrounds are necessary to improve generalizability of findings.
In conclusion, chronological age was strongly correlated with DNAmGrimAge, followed by DNAmPhenoAge and DNAmTL. Results from this study suggested that, even in the context of small sample size, second-generation DNAm-based clocks of healthspan and lifespan, especially GrimAge, were cross-sectionally and longitudinally related to cognitive aging. Exploration of genome-wide DNAm differences in contrasting cognitive and neuropsychiatric phenotypes raised possible epigenetic mechanisms involved in cognitive aging. Future research is needed to validate our results in larger cohorts and to provide further explorations of mechanistic relationships between newer epigenetic aging markers and cognitive aging. If confirmed, epigenetic aging biomarkers could inform development of targets in prevention studies of dementia.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the invaluable contributions and dedication of the 25,871 participants in VITAL and the entire staff of the VITAL study.
Previous presentation: Portions of the results have been presented at the 2021 Annual Meeting of the American Association for Geriatric Psychiatry and at the 2021 Alzheimer’s Association International Conference.
FUNDING
VITAL-DEP is supported by R01 MH091448 and R56 MH091448 from the National Institute of Mental Health (NIMH). VITAL is supported by grants R01 AT011729, U01 CA138962, and R01 CA138962, which include support from the National Cancer Institute; National Heart, Lung, and Blood Institute (NHLBI); Office of Dietary Supplements; National Institute of Neurological Disorders and Stroke; and the National Center for Complementary and Integrative Health of the National Institutes of Health (NIH). The VITAL ancillary studies and CTSC (Clinical and Translational Science Center) component are supported by grants DK088078 and R01 DK088762 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); R01 HL101932 and R01 HL102122 from NHLBI; R01 AG036755 from the National Institute on Aging (NIA); R01 AR059086 and R01 AR060574 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases; and R01 MH091448 from the NIMH.
This work was conducted with support from the Harvard Catalyst CTSC (UL1TR001102 from the National Center for Advancing Translational Sciences). Dr. Reynolds’ participation also received support from P30 MH090333 from NIMH, and the University of Pittsburgh Medical Center Endowment in Geriatric Psychiatry. Dr. Sadreyev’s participation received support from P30 DK040561 from NIDDK. Pharmavite LLC of Northridge, California (vitamin D) and Pronova BioPharma/BASF of Norway (Omacor® fish oil) donated the study agents, matching placebos, and packaging in the form of calendar packs. VITAL-DEP has been approved by the Institutional Review Board of Partners Healthcare/Brigham and Women’s Hospital, and the VITAL study agents have received Investigational New Drug Approval from the U.S. Food and Drug Administration. Voting members of the Data and Safety Monitoring Board for VITAL and ancillary studies, including VITAL-DEP, included Lawrence S. Cohen, MD; Theodore Colton, ScD; Mark A. Espeland, PhD; Craig Henderson, MD; Alice H. Lichtenstein, ScD; Rebecca A. Silliman, MD, PhD; and Nanette Wenger, MD (chair). Ex-officio members include Josephine Boyington, PhD, MPH; Rebecca Costello, PhD; Cindy Davis, PhD; Peter Greenwald, MD; and Wendy Weber, PhD. VITAL and VITAL-DEP are registered at clinicaltrials.gov (VITAL: NCT01169259; VITAL-DEP: NCT01696435). The VITAL website is http://www.vitalstudy.org.
The NIH, Harvard Catalyst, US FDA, Pharmavite LLC, and Pronova BioPharma/BASF had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
CONFLICT OF INTEREST
Dr. Vyas has received research support from Nestlé-Purina Petcare Company. Dr. Kang has received research support for Pfizer, Inc. Dr. Reynolds receives payment from the American Association of Geriatric Psychiatry as Editor-in-Chief of the American Journal of Geriatric Psychiatry, royalty income for intellectual property as co-inventor of the Pittsburgh Sleep Quality Index and, in the past, a one-time honorarium from Merck for consultation on care pathways for insomnia. Dr. Reynolds also receives royalty income from Oxford University Press and from Up-to-Date.
Dr. Mischoulon has received research support from Nordic Naturals and Heckel Medizintechnik GmbH. He has received honoraria for speaking from the Massachusetts General Hospital Psychiatry Academy, Peerpoint Medical Education Institute, LLC, and Harvard blog. He also works with the MGH Clinical Trials Network and Institute (CTNI), which has received research funding from multiple pharmaceutical companies and NIMH.
Dr. Chang receives royalties from Up-to-Date.
Dr. Manson has received research support from Mars Edge.
Dr. Okereke receives royalties from Springer Publishing for a book on late-life depression prevention.
No other authors have disclosures to report.
Footnotes
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230093.
DATA AVAILABILITY
We recognize and support the principles of data sharing that have been endorsed by the NIH. We maintain a policy that actively promotes new research collaborations that make use of the comprehensive outcomes, covariate and blood-based biomarker data collected during the trial, and will encourage the submission of collaborative studies that include investigators from other departments and institutions. We will also maintain our strong commitment to communicate important study results to participants and the scientific community through the regular VITAL study newsletters, published manuscripts, presentations at national meetings, and interviews for lay publications. The data generated from this research will be made available to affiliated investigators through secure databases. Only investigators with specific IRB approval will have access to any identifiable data. For de-identified datasets, investigators can contact Dr. Olivia I. Okereke (olivia.okereke@mgh.harvard.edu) and Dr. Vyas (cvyas@partners.org). Consent for such data sharing was integral to enrollment in the VITAL study, and our participants have been generous in their willingness to have their data shared to advance health research.
REFERENCES
- [1].Hickman RA, Faustin A, Wisniewski T (2016) Alzheimer disease and its growing epidemic: Risk factors, biomarkers, and the urgent need for therapeutics. Neurol Clin 34, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Salameh Y, Bejaoui Y, El Hajj N (2020) DNA methylation biomarkers in aging and age-related diseases. Front Genet 11, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pagiatakis C, Musolino E, Gornati R, Bernardini G, Papait R (2021) Epigenetics of aging and disease: A brief overview. Aging Clin Exp Res 33, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, Whitsel EA, Wilson JG, Reiner AP, Aviv A, Lohman K, Liu Y, Ferrucci L, Horvath S (2018) An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD, Whitsel EA, Assimes TL, Ferrucci L, Horvath S (2019) DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11, 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lu AT, Seeboth A, Tsai PC, Sun D, Quach A, Reiner AP, Kooperberg C, Ferrucci L, Hou L, Baccarelli AA, Li Y, Harris SE, Corley J, Taylor A, Deary IJ, Stewart JD, Whitsel EA, Assimes TL, Chen W, Li S, Mangino M, Bell JT, Wilson JG, Aviv A, Marioni RE, Raj K, Horvath S (2019) DNA methylation-based estimator of telomere length. Aging (Albany NY) 11, 5895–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bergsma T, Rogaeva E (2020) DNA methylation clocks and their predictive capacity for aging phenotypes and healthspan. Neurosci Insights 15, 2633105520942221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hillary RF, Stevenson AJ, Cox SR, McCartney DL, Harris SE, Seeboth A, Higham J, Sproul D, Taylor AM, Redmond P, Corley J, Pattie A, Hernández M, Muñoz-Maniega S, Bastin ME, Wardlaw JM, Horvath S, Ritchie CW, Spires-Jones TL, McIntosh AM, Evans KL, Deary IJ, Marioni RE (2021) An epigenetic predictor of death captures multi-modal measures of brain health. Mol Psychiatry 26, 3806–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, Corley J, Taylor A, Murphy L, Starr JM, Horvath S, Visscher PM, Wray NR, Deary IJ (2015) The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol 44, 1388–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].O’Shea DM, Alaimo H, Davis JD, Galvin JE, Tremont G (2023) A comparison of cognitive performances based on differing rates of DNA methylation GrimAge acceleration among older men and women. Neurobiol Aging 123, 83–91. [DOI] [PubMed] [Google Scholar]
- [11].Reed RG, Carroll JE, Marsland AL, Manuck SB (2022) DNA methylation-based measures of biological aging and cognitive decline over 16-years: Preliminary longitudinal findings in midlife. Aging (Albany NY) 14, 9423–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sugden K, Caspi A, Elliott ML, Bourassa KJ, Chamarti K, Corcoran DL, Hariri AR, Houts RM, Kothari M, Kritchevsky S, Kuchel GA, Mill JS, Williams BS, Belsky DW, Moffitt TE (2022) Association of pace of aging measured by blood-based DNA methylation with age-related cognitive impairment and dementia. Neurology 99, e1402–e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vaccarino V, Huang M, Wang Z, Hui Q, Shah AJ, Goldberg J, Smith N, Kaseer B, Murrah N, Levantsevych OM, Shallenberger L, Driggers E, Bremner JD, Sun YV (2021) Epigenetic age acceleration and cognitive decline: A twin study. J Gerontol A Biol Sci Med Sci 76, 1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wadsworth LP, Lorius N, Donovan NJ, Locascio JJ, Rentz DM, Johnson KA, Sperling RA, Marshall GA (2012) Neuropsychiatric symptoms and global functional impairment along the Alzheimer’s continuum. Dement Geriatr Cogn Disord 34, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, Cedarbaum J, Brashear R, Miller DS (2011) Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement 7, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Okereke OI, Prescott J, Wong JY, Han J, Rexrode KM, De Vivo I (2012) High phobic anxiety is related to lower leukocyte telomere length in women. PLoS One 7, e40516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Simon NM, Smoller JW, McNamara KL, Maser RS, Zalta AK, Pollack MH, Nierenberg AA, Fava M, Wong KK (2006) Telomere shortening and mood disorders: Preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry 60, 432–435. [DOI] [PubMed] [Google Scholar]
- [18].Okereke OI, Reynolds CF 3rd, Mischoulon D, Chang G, Cook NR, Copeland T, Friedenberg G, Buring JE, Manson JE (2018) The VITamin D and OmegA-3 TriaL-Depression Endpoint Prevention (VITAL-DEP): Rationale and design of a large-scale ancillary study evaluating vitamin D and marine omega-3 fatty acid supplements for prevention of late-life depression. Contemp Clin Trials 68, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Manson JE, Bassuk SS, Lee IM, Cook NR, Albert MA, Gordon D, Zaharris E, Macfadyen JG, Danielson E, Lin J, Zhang SM, Buring JE (2012) The VITamin D and OmegA-3 TriaL (VITAL): Rationale and design of a large randomized controlled trial of vitamin D and marine omega-3 fatty acid supplements for the primary prevention of cancer and cardiovascular disease. Contemp Clin Trials 33, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].The National Alzheimer’s Coordinating Center (NACC). http://www.alz.washington.edu.
- [21].Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, Kukull WA (2004) The National Alzheimer’s Coordinating Center (NACC) Database: An Alzheimer disease database. Alzheimer Dis Assoc Disord 18, 270–277. [PubMed] [Google Scholar]
- [22].Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, Dietrich W, Beekly DL, Kukull WA, Morris JC (2009) The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): The neuropsychologic test battery. Alzheimer Dis Assoc Disord 23, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dickerson BC, Sperling RA, Hyman BT, Albert MS, Blacker D (2007) Clinical prediction of Alzheimer disease dementia across the spectrum of mild cognitive impairment. Arch Gen Psychiatry 64, 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Okereke OI, Copeland M, Hyman BT, Wanggaard T, Albert MS, Blacker D (2011) The Structured Interview & Scoring Tool-Massachusetts Alzheimer’s Disease Research Center (SIST-M): Development, reliability, and cross-sectional validation of a brief structured clinical dementia rating interview. Arch Neurol 68, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kang JH, Vyas CM, Okereke OI, Ogata S, Albert M, Lee IM, D’Agostino D, Buring JE, Cook NR, Grodstein F, Manson JE (2022) Marine n-3 fatty acids and cognitive change among older adults in the VITAL randomized trial. Alzheimers Dement (N Y) 8, e12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Teng EL, Chui HC (1987) The Modified Mini-Mental State (3MS) examination. J Clin Psychiatry 48, 314–318. [PubMed] [Google Scholar]
- [27].Albert M, Smith LA, Scherr PA, Taylor JO, Evans DA, Funkenstein HH (1991) Use of brief cognitive tests to identify individuals in the community with clinically diagnosed Alzheimer’s disease. Int J Neurosci 57, 167–178. [DOI] [PubMed] [Google Scholar]
- [28].Brandt J, Spencer M, Folstein M (1988) The telephone interview for cognitive status. Neuropsychiatry Neuropsychol Behav Neurol 1, 111–117. [Google Scholar]
- [29].Royall DR, Lauterbach EC, Cummings JL, Reeve A, Rummans TA, Kaufer DI, LaFrance WC Jr., Coffey CE(2002) Executive control function: A review of its promise and challenges for clinical research. A report from the Committee on Research of the American Neuropsychiatric Association. J Neuropsychiatry Clin Neurosci 14, 377–405. [DOI] [PubMed] [Google Scholar]
- [30].Reitan R (1992) Trail Making Test. Manual for administration and scoring Reitan Neuropsychological Laboratory, Tucson, AZ. [Google Scholar]
- [31].Kroenke K, Spitzer RL, Williams JB (2001) The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med 16, 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Spitzer RL, Kroenke K, Williams JB, Löwe B (2006) A brief measure for assessing generalized anxiety disorder: The GAD-7. Arch Intern Med 166, 1092–1097. [DOI] [PubMed] [Google Scholar]
- [33].Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, Hergueta T, Baker R, Dunbar GC (1998) The Mini-International Neuropsychiatric Interview (M.I.N.I.): The development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59 Suppl 20, 22–33;quiz 34–57. [PubMed] [Google Scholar]
- [34].Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J (1994) The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 44, 2308–2314. [DOI] [PubMed] [Google Scholar]
- [35].Vyas CM, Hazra A, Chang SC, Qiu W, Reynolds CF 3rd, Mischoulon D, Chang G, Manson JE, De Vivo I, Okereke OI (2019) Pilot study of DNA methylation, molecular aging markers and measures of health and well-being in aging. Transl Psychiatry 9, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fortin JP, Triche TJ Jr., Hansen KD(2017) Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 33, 558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Horvath S, Levine AJ (2015) HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis 212, 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Charlson ME, Pompei P, Ales KL, MacKenzie CR (1987) A new method of classifying prognostic comorbidity in longitudinal studies: Development and validation. J Chronic Dis 40, 373–383. [DOI] [PubMed] [Google Scholar]
- [42].Deyo RA, Cherkin DC, Ciol MA (1992) Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol 45, 613–619. [DOI] [PubMed] [Google Scholar]
- [43].Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fransquet PD, Lacaze P, Saffery R, Shah RC, Vryer R, Murray A, Woods RL, Ryan J (2021) Accelerated epigenetic aging in peripheral blood does not predict dementia risk. Curr Alzheimer Res 18, 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shadyab AH, McEvoy LK, Horvath S, Whitsel EA, Rapp SR, Espeland MA, Resnick SM, Manson JE, Chen JC, Chen BH, Li W, Hayden KM, Bao W, Kusters CDJ, LaCroix AZ (2022) Association of epigenetic age acceleration with incident mild cognitive impairment and dementia among older women. J Gerontol A Biol Sci Med Sci 77, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sibbett RA, Altschul DM, Marioni RE, Deary IJ, Starr JM, Russ TC (2020) DNA methylation-based measures of accelerated biological ageing and the risk of dementia in the oldest-old: A study of the Lothian Birth Cohort 1921. BMC Psychiatry 20, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dogan F, Forsyth NR (2021) Telomerase regulation: A role for epigenetics. Cancers (Basel) 13, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, Horner JW, Maratos-Flier E, Depinho RA (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dayon L, Guiraud SP, Corthésy J, Da Silva L, Migliavacca E, Tautvydaitė D, Oikonomidi A, Moullet B, Henry H, Métairon S, Marquis J, Descombes P, Collino S, Martin FJ, Montoliu I, Kussmann M, Wojcik J, Bowman GL, Popp J (2017) One-carbon metabolism, cognitive impairment and CSF measures of Alzheimer pathology: Homocysteine and beyond. Alzheimers Res Ther 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tapia-Rojas C, Lindsay CB, Montecinos-Oliva C, Arrazola MS, Retamales RM, Bunout D, Hirsch S, Inestrosa NC (2015) Is L-methionine a trigger factor for Alzheimer’s-like neurodegeneration?: Changes in Aβ oligomers, tau phosphorylation, synaptic proteins, Wnt signaling and behavioral impairment in wild-type mice. Mol Neurodegener 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gampawar P, Schmidt R, Schmidt H (2022) Telomere length and brain aging: A systematic review and meta-analysis. Ageing Res Rev 80, 101679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We recognize and support the principles of data sharing that have been endorsed by the NIH. We maintain a policy that actively promotes new research collaborations that make use of the comprehensive outcomes, covariate and blood-based biomarker data collected during the trial, and will encourage the submission of collaborative studies that include investigators from other departments and institutions. We will also maintain our strong commitment to communicate important study results to participants and the scientific community through the regular VITAL study newsletters, published manuscripts, presentations at national meetings, and interviews for lay publications. The data generated from this research will be made available to affiliated investigators through secure databases. Only investigators with specific IRB approval will have access to any identifiable data. For de-identified datasets, investigators can contact Dr. Olivia I. Okereke (olivia.okereke@mgh.harvard.edu) and Dr. Vyas (cvyas@partners.org). Consent for such data sharing was integral to enrollment in the VITAL study, and our participants have been generous in their willingness to have their data shared to advance health research.