

Abstract
Misalignment of the circadian clock compared to environmental cues causes circadian desynchrony, which is pervasive in humans. Clock misalignment can lead to various pathologies including obesity and diabetes, both of which are associated with pancreatic ductal adenocarcinoma - a devastating cancer with an 80% five-year mortality rate. Although circadian desynchrony is associated with an increased risk of several solid-organ cancers, the correlation between clock misalignment and pancreas cancer is unclear. Using a chronic jetlag model, we investigated the impact of clock misalignment on pancreas cancer initiation in mice harboring a pancreas-specific activated Kras mutation. We found that chronic jetlag accelerated the development of pancreatic cancer precursor lesions, with a concomitant increase in precursor lesion grade. Cell-autonomous knock-out of the clock in pancreatic epithelial cells of Kras-mutant mice demonstrated no acceleration of precursor lesion formation, indicating non-cell-autonomous clock dysfunction was responsible for the expedited tumor development. Therefore, we applied single-cell RNA sequencing over time and identified fibroblasts as the cell population manifesting the greatest clock-dependent changes, with enrichment of specific cancer-associated fibroblast pathways due to circadian misalignment.
Keywords: Circadian, Pancreas, Jetlag, Pancreatic Ductal Adenocarcinoma, Misalignment
Introduction:
Circadian clock proteins coordinate daily biological rhythms with an oscillation of 24 hours (McIntosh et al. 2010). Control from the central clock in the hypothalamus assimilates environmental cues (e.g. day/night cycles) to modulate peripheral organ rhythmic expression of core circadian genes and behavioral patterns (e.g. sleep/wake cycle) (McIntosh et al. 2010). In addition to central control, nearly all cells have an autonomous circadian clock based on transcriptional and translational feedback mechanisms of core clock genes (CCGs) (Lee et al. 2001; Preitner et al. 2002; Sato et al. 2004; Takahashi 2017). Collectively, the CCGs of the central and peripheral clocks orchestrate the transcription of circadian output genes, also known as clock-controlled genes (Takahashi 2017; Cox and Takahashi 2019). The central and peripheral clocks in mammals (and other organisms) have evolved to anticipate environmental changes that occur over the 24-hr day/night cycle, and in doing so, appropriately sequence complex internal processes to align with external cues (Takahashi 2017). For instance, in humans, energy intake, physical activity, and cognitive activities are anticipated to occur during the day while rest and sleep are expected to occur at night (Pittendrigh 1993; Wright et al. 2013). Consequently, this network of circadian control coordinates the optimal timing of critical cell and organ processes such as metabolism, cell division, apoptosis, and immune function (Matsuo et al. 2003; Bass and Takahashi 2010; Lee and Sancar 2011; Scheiermann et al. 2013).
Notably, the internal clock can be adjusted, or re-entrained, based on changes to environmental zeitgebers or ‘time-givers’, such as through alteration of the day/night cycle (Wright et al. 2013). However, persistent misalignment between environmental cues compared to the internal clocks can result in circadian desynchrony with associated aberrant gene signaling (Kettner et al. 2016; West et al. 2017). Unfortunately, this type of clock dysfunction (misalignment or circadian desynchrony) is pervasive in humans and this condition affects a substantial proportion of the human population, particularly shift workers, whose clock timing can be altered dramatically compared to environmental zeitgebers by work schedule requirements (Bass and Lazar 2016). Further, this environmental or behavior-induced desynchrony can have profound implications for human health including the promotion of various pathologies such as the metabolic syndromes of obesity and diabetes (Arble et al. 2009; Roenneberg et al. 2012; Perelis et al. 2016). There is also a strong correlation between circadian clock dysfunction and the risk of developing several different solid organ cancers, including lung cancer, liver cancer, and colon cancer (Schernhammer et al. 2003; Papagiannakopoulos et al. 2016; Kettner et al. 2016). Meanwhile, a history of shiftwork has been shown to increase the risk of pancreatic ductal adenocarcinoma (PDAC) in men over 2-fold (Parent et al. 2012); concordantly, obesity and diabetes are both risk factors for the development of PDAC (Schernhammer et al. 2003; Marcheva et al. 2010; Kettner et al. 2016). Despite this converging evidence and the substantive data linking the circadian clock to pancreas-specific disease processes (Marcheva et al. 2010; Perelis et al. 2015), the correlation between circadian desynchrony and the risk of developing PDAC remains unclear.
We sought to establish the connection between clock misalignment – a process that is highly prevalent in humans – and PDAC because this would have important implications for understanding cancer pathogenesis and risk mitigation in this deadly disease. Risk factor reduction leading to cancer prevention is particularly relevant in this highly lethal malignancy because few people survive after cancer diagnosis: of the 60,430 people estimated to be diagnosed in 2021, approximately 48,220 (~80%) will die of their disease (Siegel et al. 2021). As a foundation for uncovering the link between circadian clock dysfunction and the development of PDAC, we investigated whether circadian misalignment impacted tumor initiation within the pancreas. We used LSL-KrasG12D/+; Pdx1-Cre mice (KC) that harbor a Kras mutation in pancreatic acinar and ductal cells (Hingorani et al. 2003). KC mice develop the full spectrum of neoplastic changes including acinar-to-ductal metaplasia (earliest lesion), pancreatic intra-epithelial neoplasia 1 (PanIN-1), higher grade PanIN lesions (PanIN-2, -3), and ultimately PDAC (Hingorani et al. 2003). We implemented a well-characterized model of chronic jetlag that is known to cause environmental-induced misalignment and phase-shift of the clock (Filipski et al. 2004; Papagiannakopoulos et al. 2016; Schwartz et al. 2021) and is meant to mimic the environmental and behavioral misalignment experienced by humans (Vetter 2020). The abrupt change in timing of the light/dark cycle with chronic jetlag causes a period of desynchrony, with differences in the rate of re-entrainment by internal clocks or behavior (Yamazaki et al. 2000; West and Bechtold 2015). Previously, we have found that behavioral re-entrainment occurs much more rapidly than re-entrainment of the pancreatic clock (Schwartz et al. 2021). Moreover, this type of protocol (chronic jetlag) has been shown to promote the growth of several other cancers, including osteosarcoma, and hepatocellular carcinoma in mice (Filipski et al. 2004; Filipski et al. 2009; Kettner et al. 2016).
In the current study, we hypothesized that circadian clock desynchrony through chronic jetlag would accelerate the initiation of PanIN-1 and progression to higher-grade PanINs or PDAC. We found that the pancreata of KC mice subjected to chronic jetlag developed increased fibroinflammatory infiltrate and neoplastic lesion formation (i.e. PanINs) compared to KC mice under standard conditions. To understand whether cell-autonomous clock dysfunction was responsible for the accelerated PanIN development, we generated KC mice with an abolished clock (Bmal1 knock-out) in the pancreatic ductal and acinar cells (cellular origin of PDAC). These mice did not replicate the phenotype observed in the chronic jetlag group of KC mice, indicating a non-cell-autonomous role of clock dysfunction that is facilitating the PanIN progression. To investigate further, we used single-cell RNA sequencing (scRNA-seq) and identified cancer-associated fibroblasts (CAFs) as the cell population manifesting the greatest clock-dependent changes. This work serves as a foundation for understanding the link between circadian misalignment through chronic jetlag and the acceleration of PDAC development.
Materials and Methods:
Mouse Care and Husbandry
All animal studies were conducted according to an approved protocol (M005959) by the University of Wisconsin School of Medicine and Public Health (UW SMPH) Institutional Animal Care and Use Committee (IACUC). Male and female mice were housed in an Assessment and Accreditation of Laboratory Animal Care (AALAC) accredited selective pathogen-free facility (UW Medical Sciences Center) on corncob bedding with chow diet (Mouse diet 9F 5020; PMI Nutrition International, St. Louis, MO) and water ad libitum.
Conditional Lox-Stop-Lox-KrasG12D/+ (LSL-KrasG12D/+; K) mice (B6.129S4-Kras tm4Tyj/J (#008179)) harboring an oncogenic point mutation in Kras were bred with mice expressing Cre-Recombinase driven by the Pdx1 promoter (C) (B6.FVB-Tg(Pdx1-cre)6Tuv/J) (#014647)) to generate LSL-KrasG12D/+, Pdx1-Cre (KC) mice (Hingorani et al. 2003). The KC mouse has been well characterized as a pancreas pre-cancer model (Hingorani et al. 2003). To evaluate the effect of loss of clock function on KC mouse pathology the conditional Bmal1fx/fx mouse, kindly gifted by Professor Christopher Bradfield (University of Wisconsin-Madison, Madison, WI), was crossed with the KC mouse to generate the Bmal1fx/fx, LSL-KrasG12D/+; Pdx1-Cre (BKC) mouse model (Liu et al. 2014; Johnson et al. 2014). To identify cell types targeted by Pdx1-Cre recombinase in the KC mouse model, the KC mouse was additionally crossed with the LSL-tdTomato reporter (Ai14) mouse (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (#007914)) to produce the KCT mouse (LSL-R26CAG-tdTomato; LSL-KrasG12D/+; Pdx1-Cre). Wild-type C57BL/6J mice (#000664) were used as controls where indicated. The presence of each allele, including activated Kras-mutation, was confirmed with genotyping PCR according to Jackson Laboratory’s (Bar Harbor, ME) protocols as previously described (Liu et al. 2014; Walcheck et al. 2021). The C57BL/6J, K, C, and T mice were purchased from Jackson Laboratory.
Chronic Jetlag Protocol
Starting at 4–6 weeks of age, KC mice were subjected to either a normal circadian (standard lighting) 12-hour/12-hour light: dark (LD) cycle condition, or a chronic jetlag protocol known to induce circadian desynchrony and mimic altered environmental exposure experienced by humans (Filipski et al. 2004; Papagiannakopoulos et al. 2016; Vetter 2020; Schwartz et al. 2021). Chronic jetlag consisted of a 12-hour/12-hour LD cycle, phase-shifted forward 4 hours every 2–3 days. Mice were kept under their respective conditions until the study endpoint at 5 and 9 months of age.
Histologic Analysis
Mouse pancreatic tissue was fixed in 10% neutral buffered formalin for 48 hours and then stored in 70% ethanol until further processing. Tissue was processed, paraffin embedded, sectioned onto slides, and stained with hematoxylin and eosin (H&E) by the University of Wisconsin Carbone Cancer Center (UWCCC) Experimental Animal Pathology Laboratory (EAPL) Core. Each pancreas was sectioned at 200 μm steps for at least 6 slides. All slides processed were assessed for the presence of pancreas cancer precursor lesions (pancreatic intraepithelial neoplasia or PanIN) along with grading on a scale of 1–3 (1 = lowest grade, 2 = intermediate grade, and 3 = high grade) and/or pancreatic ductal adenocarcinoma (PDAC). This was performed by a blinded, board-certified, gastrointestinal pathologist (KAM) at 5 months (n = 36 normal circadian [24 male and 12 female] vs n = 26 chronic jetlag [14 male and 12 female]) and 9 months age (n = 33 normal circadian [19 male and 14 female] vs n = 39 chronic jetlag [23 male and 16 female]). Additional BKC mice (n = 24 [14 male and 10 female]) were assessed at 5 months. Categorical comparisons between groups were made with the Chi-squared or Fisher’s Exact test as appropriate. KC mice develop fibroinflammatory infiltrate associated with adjacent PanINs and are collectively part of the pancreatic changes with neoplasm formation (Hingorani et al. 2003). Quantification of the total percent pancreas involvement by the fibroinflammatory infiltrate and PanINs (referred to collectively as fibroinflammatory infiltrate/PanIN) was made by 3 separate reviewers, and differences were tested using Student’s t-test between groups. For evaluation, two slides were selected from each pancreas comprising the largest area of available tissue. Values are presented as the mean with associated standard deviation (SD). Inter-rater reliability statistics were computed and the intraclass correlation coefficient is reported with associated 95% confidence intervals.
Immunohistochemistry
KCT mouse pancreas (possessing the tdTomato allele) and KC (wild-type control) mouse pancreas samples were formalin-fixed, paraffin embedded, and sectioned by the EAPL Core facility. Immunohistochemistry (IHC) for red fluorescence protein (tdTomato) was performed by EAPL. Sections were deparaffinized in xylenes and hydrated through graded alcohols to distilled water. Antigens were retrieved using citrate buffer pH 6.0 (10 mM Citric Acid and 0.05% tween 20). Endogenous peroxidase was blocked with 0.3% H2O2 and blocking of non-specific binding was performed using 10% goat serum. Sections were incubated with rabbit anti-RFP antibody (600-401-379, Rockland Inc, Pottstown, PA) (1:1600) followed by ImmPRESS goat anti-rabbit IgG (MP-7451, Vector Laboratories, Burlingame, CA). Detection was performed using the DAB substrate kit (8059S, Cell Signaling Technology, Danvers, MA). Samples were counterstained using Mayer’s hematoxylin (MHS32, Millipore-Sigma, St. Louis, MO).
Bmalfx/fx, Bmalfx/fx; Pdx1-Cre, KC, and BKC mouse pancreas samples were formalin-fixed, paraffin embedded, and sectioned by the EAPL Core facility. After sectioning, detection of BMAL1 was performed by the Translational Research Initiatives in Pathology (TRIP) Core facility. Automated immunohistochemistry was performed on the Ventana Discovery Ultra BioMarker Platform (Ventana Medical Systems, Oro Valley, AZ). Deparaffinization was carried out on the instrument, as was heat-induced epitope retrieval with cell conditioner 1 buffer (#950–224, Ventana), an EDTA-based buffer pH 8.4, for 64 minutes at 95 °C. The primary antibody was the BMAL1 polyclonal antibody (NB100-2288, Novus Biologicals, Centennial, CO) diluted 1:100 in Da Vinci Green Diluent (#PD900M, BioCare Medical) and incubated for 60 minutes at 37 °C. Slices were rinsed with reaction buffer (#950–300, Ventana), incubated with Discovery OmniMap anti-rabbit horseradish peroxidase (#760–4311, Ventana) for 20 minutes, and then rinsed with reaction buffer. Discovery ChromoMap DAB detection kit (#760–159, Ventana) was used for visualization. Slides were removed from the instrument, counterstained with Harris hematoxylin (1:5) for 45 seconds, rinsed with dH2O, dehydrated by oven drying and dipping in xylene. Slides were coverslipped by adding 2–3 drops of Mounting Medium (#4112, Thermo Fisher Scientific, Waltham, MA).
scRNAseq Sample Preparation, Library Construction, and Sequencing
Five- and 9-month-old normal circadian and chronic jetlag KC mice were sacrificed at ZT4 with cervical dislocation and the pancreas was rapidly dissected (< 60 seconds) and placed in cold PBS (Thermo Fisher Scientific). One male and female pancreas was pooled from each condition and time point after demonstration of no discernable sex-specific differences (Takele Assefa et al. 2020). Pancreas samples were minced until the tissue was < 1 mm in diameter, and then centrifuged at 900 RPM for 5 minutes to remove fat contamination. Samples were then incubated with 5 mL of Collagenase P (Sigma Aldrich, St. Louis, MO) in HBSS (Thermo Fisher Scientific) at 37° C for 15 minutes while being shaken at 100 RPM. Additionally, samples were manually agitated every 2–3 minutes. The digestion reaction was quenched with 10 mL R10 media. Samples were then centrifuged at 900 RPM and washed twice with 10 mL of R10. Samples were then strained in a gradient fashion using 500 μm, 100 μm, and 40 μm strainers. Samples were then live/dead sorted into single-cell populations and were transferred to the University of Wisconsin Gene Expression Center (GEC) for Library Preparation.
In brief, libraries were constructed according to the Chromium NextGEM Single Cell 3’ Reagent Kit v3.1 User Guide, Rev.D (10x Genomics, Pleasanton, CA). Single-cell suspension cell concentration and viability were quantified on the Countess II (Thermo Fisher Scientific) using 0.4% Trypan Blue (Invitrogen, Carlsbad, CA). Single-cell droplet encapsulation was performed using the Single Cell G Chip and the 10x Genomics Chromium Controller. Following the completion of the Chromium run, the Gel Beads in Emulsion (GEMs) were transferred to emulsion-safe strip tubes for GEM-RT using an Eppendorf MasterCycler Pro thermocycler (Eppendorf, Hamburg, Germany). Following reverse transcription (RT), GEMs were broken, and the pooled single-cell cDNA was collected in Recovery Agent, purified using DynaBeads MyOne Silane beads (Thermo Fisher Scientific), and amplified with the appropriate number of PCR cycles based on the number of cells targeted for recovery. Post-cDNA amplified product was purified using SPRIselect (Beckman Coulter, Brea, CA) and quantified on a Bioanalyzer 2100 (Agilent, Santa Clara, CA) using the High Sensitivity DNA kit. Adapters were then added to the libraries after fragmentation, end repair, A-tailing, and double-sided size selection using SPRIselect. Following adapter ligation, libraries were cleaned up using SPRIselect and sample-specific indexes (Chromium i7 Multiplex Kit, 10x Genomics) were added by sample index PCR using the appropriate number of PCR cycles based on the cDNA yield calculated by the Bioanalyzer quantification. After sample index PCR, samples were double-size selected using SPRIselect and quantified by the Qubit High Sensitivity DNA Kit (Thermo Fisher Scientific). Libraries were sequenced on a NovaSeq6000 (Illumina, San Diego, CA).
scRNAseq Data Analysis
Single-cell RNAseq data were analyzed by the UW Bioinformatics Resource Center. Data were demultiplexed using the Cell Ranger Single Cell Software Suite, mkfastq command wrapped around Illumina’s bcl2fastq. The MiSeq balancing run was quality-controlled using calculations based on UMI-tools (Smith et al. 2017). Sample libraries were balanced for the number of estimated reads per cell and run on an Illumina NovaSeq system. Cell Ranger software was then used to perform demultiplexing, alignment, filtering, barcode counting, UMI counting, and gene expression estimation for each sample according to the 10x Genomics documentation (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). The gene expression estimates from each sample were then aggregated using Cellranger (cellranger aggr) to compare experimental groups with normalized sequencing-depth and expression data.
Gene expression data was then processed using the Seurat version 4 pipeline for data integration (Hao et al. 2021). Data was loaded into Seurat. Low quality (<200 features or >20% mitochondrial gene content) and doublets were removed. Cells expressing more than 1% hemoglobin gene content were filtered out. The normal circadian and chronic jetlag KC data were merged and integrated using the SCTransform v2 wrapper with 3000 unique features (Choudhary and Satija 2022). SCTransform allows for normalization and better batch correction based on a regularized negative binomial regression. The regression additionally allowed for correction of the cell cycle effect and mitochondrial gene content (Nestorowa et al. 2016). A principal component analysis (PCA) was performed with the number of dimensions (principal components) selected based on an elbow plot. Data were visualized using the Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) (McInnes et al. 2020). Cell identities were called based on the expression of known markers for each cell type and scored using the ScType package to cross-validate identities following the default settings for SCT-transformed data in the provided vignette (Ianevski et al. 2022). Differential expression between conditions and time points was performed between clusters or conditions in Seurat using the MAST test based on the default log fold change threshold of 0.25 and a false discovery rate (FDR) corrected p value of p < 0.05 (Hao et al. 2021). The MAST test within Seurat is based on the MAST package, which is specifically designed to handle differential expression in scRNA-seq data and utilizes a hurdle model (Finak et al. 2015). Gene Set Enrichment Analysis (GSEA) was conducted with the fgsea package leveraged against the oncogenic signature gene set (c6.all.v7.5.1.symbols) and biologic process subset of gene ontology (GO BP) (c5.go.v7.5.1.symbols) (Sergushichev 2016). Pseudotime trajectory analysis was performed with the Monocle 3 package (Trapnell et al. 2014). Genes correlated with pseudotime progression were computed using the Moran’s I test and those genes with an FDR p < 0.05 were considered significant. All scRNAseq data is publicly available through gene expression omnibus (GEO) (Accession number: GSE209629). All analyses in this manuscript were conducted in R version 4.2.0 (Vienna, Austria) or GraphPad Prism version 8.3.1 (Dotmatics, Boston, MA).
Results:
Chronic Jetlag Promotes Pancreatic Tumor Initiation
To test the hypothesis that circadian misalignment by chronic jetlag accelerates tumor initiation (i.e. PanIN or pre-cancerous lesion formation) and development of PDAC, we elected to utilize the LSL-KrasG12D/+; Pdx1-Cre (KC) mouse model – a well-characterized PDAC development model – and assessed the burden of neoplastic lesions (i.e. PanIN) and PDAC (Hingorani et al. 2003). We have previously shown that the chronic jetlag protocol results in circadian misalignment with phase-shift of core clock gene (CCG) expression and alteration of diurnally expressed genes in the pancreas compared to mice under normal circadian (standard lighting) conditions (Schwartz et al. 2021). Furthermore, we have demonstrated that after normalizing the time of lights on/lights off (i.e. light/dark cycle) in chronic jetlag mice, complete re-entrainment of the pancreatic clock does not occur out to day 10 post-normalization; this is in contrast to behavioral re-entrainment which occurred by 3 days (Schwartz et al. 2021). Therefore, the chronic jetlag protocol is a robust protocol to evaluate the impact of circadian misalignment on the tumor initiation and progression of pancreatic pre-cancerous lesions.
We first evaluated KC mice at age 5 months, because existing literature demonstrates that 5-month KC mice develop neoplastic changes consisting of early PanINs (i.e. PanIN-1) but most of the pancreas remains normal (Hingorani et al. 2003). Therefore, evaluating the extent of pancreas involvement by PanIN at this time point (not incidence) should allow for the detection of any acceleration in tumor initiation due to circadian misalignment (chronic jetlag). All sections were scored for their degree of pancreatic involvement by neoplastic lesions (e.g. PanINs) and the associated fibroinflammatory infiltrate, hereafter referred to as fibroinflammatory infiltrate/PanIN. This was calculated by determining the percent of the pancreas that was involved with fibroinflammatory infiltrate/PanIN by 3 independent reviewers, and there was an excellent intraclass correlation coefficient of 0.896 (95% CI 0.868–0.919; p < 0.01). We found that KC mice that were exposed to chronic jetlag (n = 26) displayed an approximate 2-fold mean increase (± standard deviation [SD]) in fibroinflammatory infiltrate/PanIN involvement of the pancreas at 5 months compared to KC mice under standard lighting conditions (n = 36) (22.47% ± 19.08 vs 12.73% ± 12.08; p = 0.027, Figure 1). There were no sex-specific differences at 5 months for normal circadian (males 13.14% ± 12.68 vs. females 11.93% ± 11.27; p = 0.77) or chronic jetlag KC mice (males 17.27% ± 12.94 vs. females 28.53% ± 23.55; p = 0.16).
Figure 1: Pancreatic fibroinflammation/PanIN development is increased by chronic jetlag at 5 months.
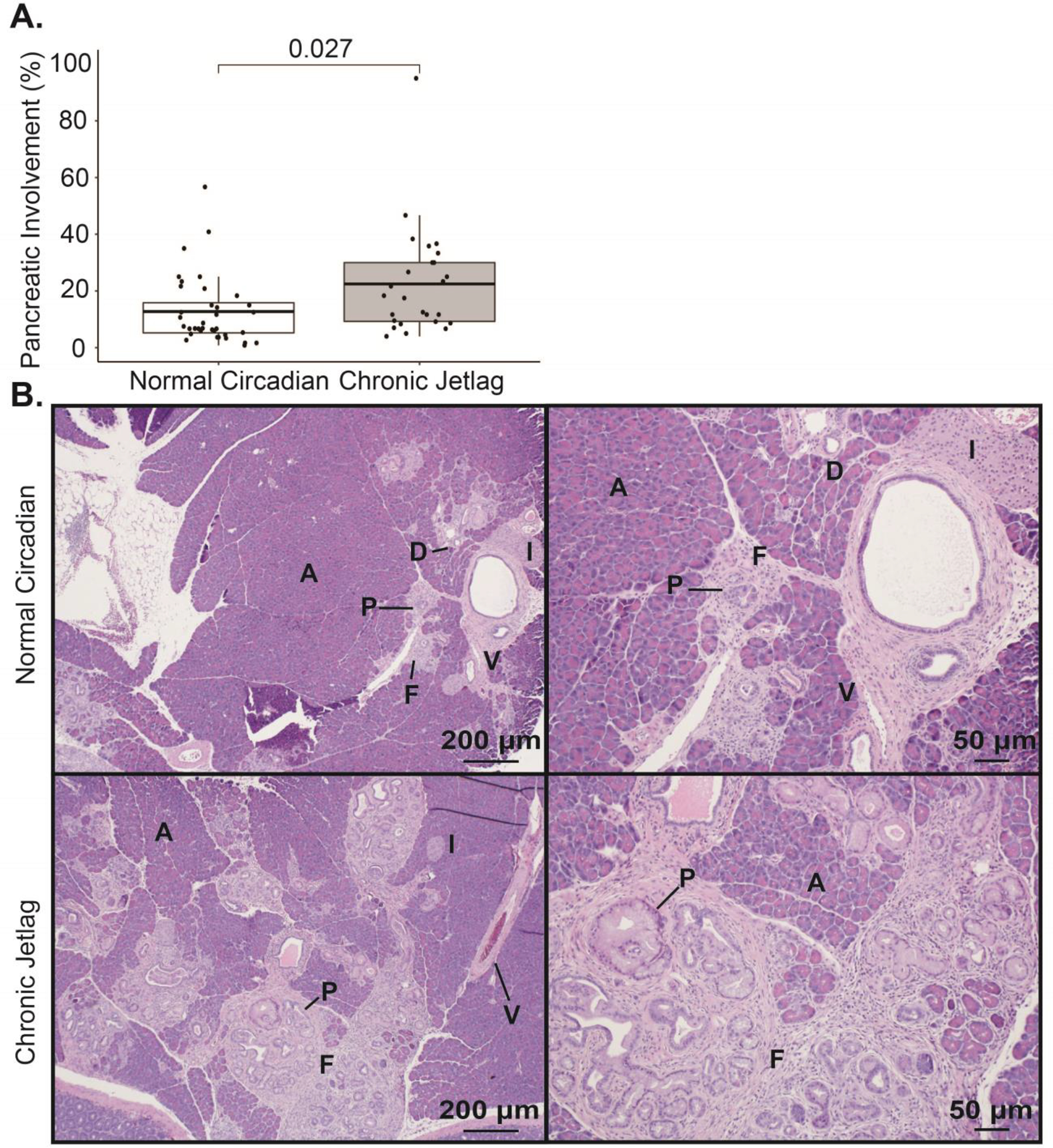
A. Boxplot comparing the mean (with associated 25th and 75th quantiles) percent pancreatic involvement of fibroinflammatory infiltrate/PanIN at 5 months for normal circadian KC (n= 36) and chronic jetlag KC mice (n = 26). B. Representative H&E images of normal circadian (upper) and chronic jetlag (lower) mice at 40x (left) and 100x (right). Scale bars are shown for reference. [Annotation: PanIN (P), acinar (A) cells, islet (I) cells, ductal (D) cells, vascular (V) cells (i.e. endothelial cells), and fibroinflammatory (F) infiltrate]
Due to the expected progression of fibroinflammatory infiltrate/PanIN at 9 months, the extent becomes less relevant (Hingorani et al. 2003). Instead, by 9 months, the incidence of higher grade PanINs (i.e. PanIN-2 and PanIN-3) and development of PDAC better reflects the progression of pre-cancerous lesions into cancer. This is because i) not all PanIN-1s progress to higher-grade lesions, ii) PanIN-2, PanIN-3, and PDAC represent tumor progression, and iii) KC mice are expected to develop a significant incidence of PanIN-2, -3, and PDAC with increasing age (Hingorani et al. 2003). In contrast, higher-grade lesions are rarely observed by approximately 5 months of age (Hingorani et al. 2003). Concordantly, we found that KC mice under normal circadian conditions exhibited almost exclusively PanIN-1, with only 5.6% (n = 2/36) demonstrating a PanIN-2 lesion at age 5 months; no 5-month KC mice under normal conditions developed PanIN-3 or PDAC (Figure 2A/C). Interestingly, although not significant, 7.7% (n = 2/26) of KC mice subjected to chronic jetlag developed PanIN-2, and one mouse developed dedifferentiated PDAC which had replaced the entirety of the pancreatic parenchyma (Figure 2A/C).
Figure 2: Chronic jetlag accelerates the grade of PanIN lesions.
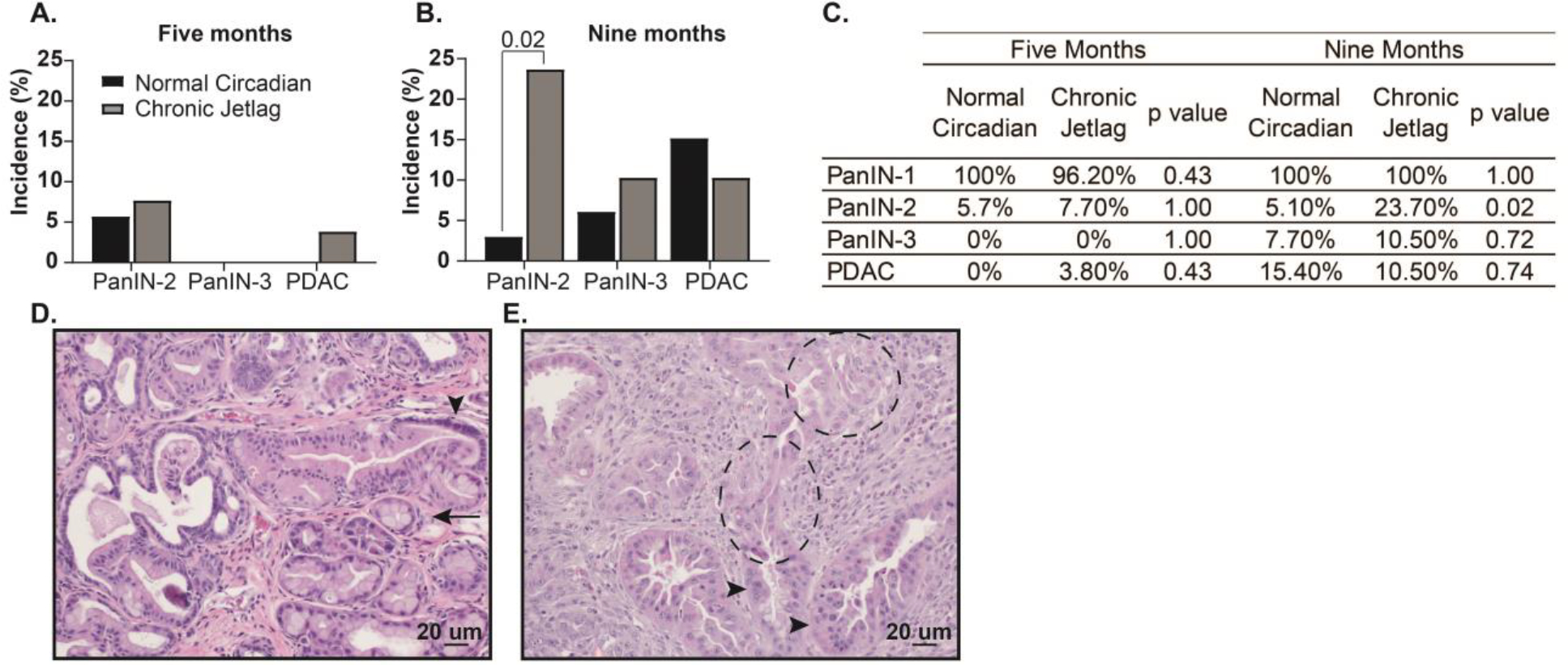
Bar charts indicating the incidence of PanIN-1-3 and PDAC in normal circadian (black) and chronic jetlag (grey) conditions at A. 5 months (normal circadian n = 36 vs. chronic jetlag n = 26) and B. 9 months (normal circadian n = 33 vs. chronic jetlag n = 39). C. Table showing the Fischer’s Exact Test comparisons between 5- and 9-month normal circadian and chronic jetlag mice. Representative H&E images are shown to demonstrate the various pancreatic pathologies being quantified. D. Representative 200x H&E image from a chronic jetlag mouse demonstrating PanIN-1 (arrow) and PanIN-2 lesion (arrowhead). E. Representative 200x H&E image from a chronic jetlag KC mouse demonstrating a PanIN-3 lesion (arrowhead) and PDAC off the same gland (dashed circle). Scale bars are shown for reference.
As expected, the incidence of advanced lesions increased by 9 months of age in KC mice under normal circadian conditions to 3% (n = 1/33) harboring PanIN-2, 6.1% (n = 2/33) developing PanIN-3, and 15.2% (n = 5/33) of mice progressing to PDAC (Figure 2B/C). Notably, although the incidence of PanIN-3 (10.3% [n = 4/39]) and PDAC (10.3% [n = 4/39]) was similar between chronic jetlag and normal circadian KC mice at 9 months, there was a significant increase in PanIN-2 formation in KC mice due to chronic jetlag (23.1% [n = 9/39] vs 3% [n = 1/33], p = 0.02) (Figure 2B–E). There were no sex-specific differences in PanIN-2 incidence for normal circadian (male 0/19 (0%) vs. female 1/14 (7.1%); p = 0.42) and chronic jetlag mice (male 4/23 (17.4%) vs. female 5/16 (31.2%); p = 0.44). Furthermore, there were no differences in the survival between normal circadian (62.2%% [n = 14/37]) and chronic jetlag mice (69.8% [n = 13/43]) at 9 months of age (p = 0.31), and survival was consistent with previous reports in KC mice (Supplemental Data 1) (Hingorani et al. 2003; Walcheck et al. 2022). Collectively, this data demonstrates that circadian desynchrony promoted by chronic jetlag in KC mice caused accelerated development of fibroinflammatory infiltrate/PanIN at 5 months leading to an increased incidence of higher grade PanINs (i.e. PanIN-2) by 9 months, and these findings were independent of sex. This strongly supports the assertion that circadian misalignment (desynchrony) facilitates tumor initiation and progression of the neoplastic PanIN lesions.
Pancreatic Clock Gene Deletion Abrogated Tumor Initiation
Different cell populations within the pancreas can lead to tumor development. For instance, islet cells can transform into neuroendocrine tumors (Zhang et al. 2013; Li and Xie 2022), whereas acinar-to-ductal metaplasia is the initiating process in PanIN formation (and consequently PDAC) (Chuvin et al. 2017). Therefore, we hypothesized that misalignment of the clock within acinar cells was responsible for the accelerated PanIN phenotype seen in chronic jetlag KC mice. In turn, we employed a cell-autonomous Bmal1 knock-out model to test whether circadian disruption in acinar cells facilitates tumor initiation and progression of the pancreas cancer precursor lesions. In support of this hypothesis, previous studies in hepatocellular carcinoma (HCC) have revealed a concordant phenotype between chronic jetlag and cell-autonomous clock disruption (Bmal1 knock-out) in mice predisposed to develop HCC (Kettner et al. 2016). We crossed the KC mouse with a mouse harboring a conditional deletion of the core clock gene Bmal1 to produce Bmal1fx/fx; LSL-KrasG12D/+; Pdx1-Cre (BKC) mice, which possess Bmal1 deletion in cells concomitantly expressing Pdx1 and the KrasG12D mutation (Johnson et al. 2014). BMAL1 is a central transcriptional regulator of the clock and is therefore essential for clock functionality (Bunger et al. 2000). We evaluated the degree of fibroinflammatory infiltrate/PanIN in BKC mice under standard lighting conditions at 5 months and, contrary to our hypothesis, we found that cell-autonomous clock disruption caused resistance to fibroinflammatory infiltrate/PanIN development (Figure 3A–D). Compared to normal circadian KC mice, BKC mice had scant development of fibroinflammatory infiltrate/PanIN (12.73% ± 12.08 (KC) vs 1.59% ± 0.65 (BKC); p < 0.01), with essentially histologically normal pancreas, and no mice exhibited a higher-grade lesion than PanIN-1 (Figure 3A–D). Additionally, this suppressed PanIN development in BKC mice was not sex-dependent (male 1.64% ± 0.74 vs. female 1.52% ± 0.53; p = 0.65).
Figure 3: Pancreatic cell-autonomous deletion of Bmal1 does not accelerate pancreatic fibroinflammatory infiltrate/PanIN development at five months.
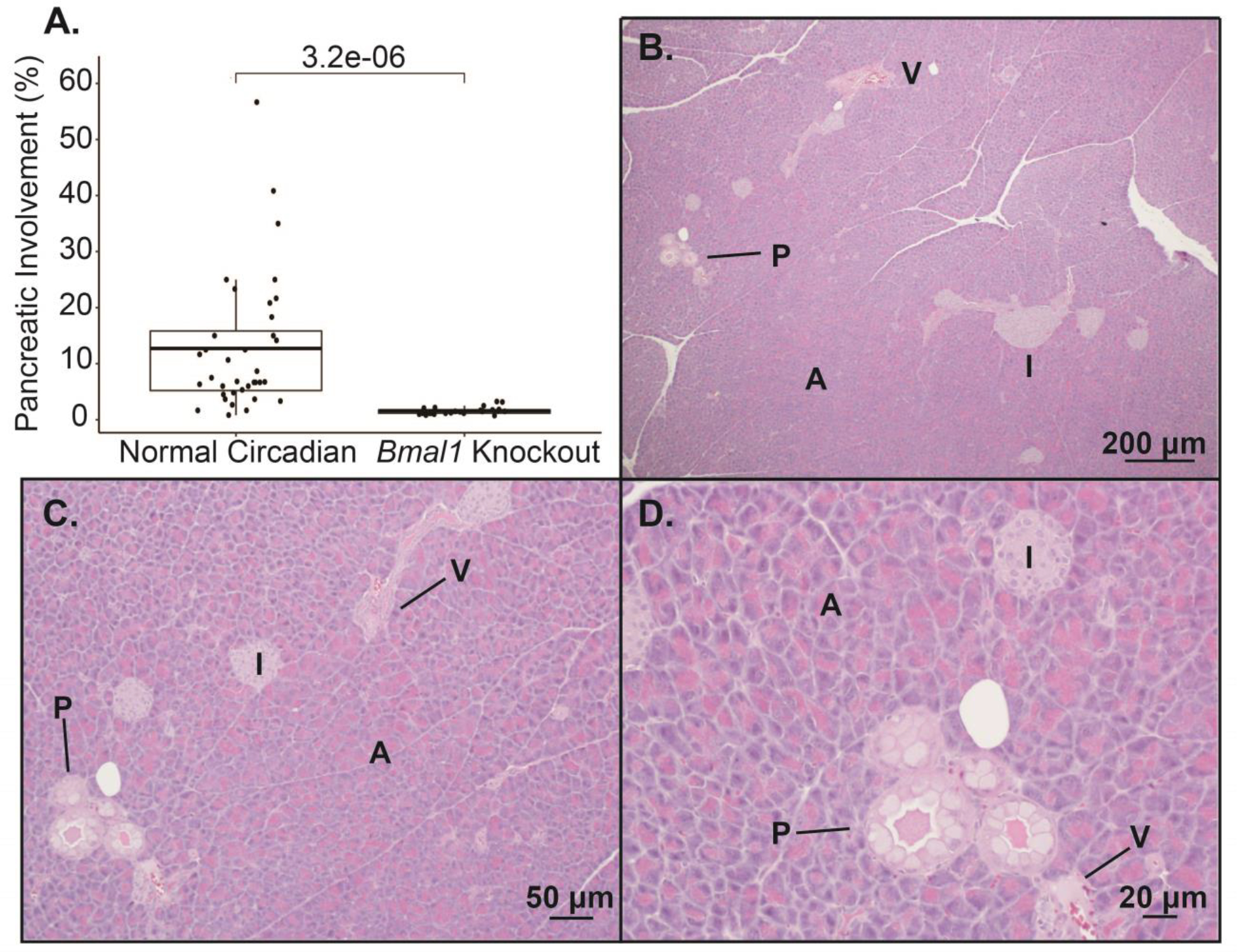
A. Boxplot comparing the mean (with associated 25th and 75th quantiles) percent pancreatic involvement of fibroinflammatory infiltrate/PanIN at 5 months for normal circadian KC (n= 36) and BKC mice (n = 24). Representative H&E images demonstrating the fibroinflammatory infiltrate/PanIN from a BKC mouse at 40x (B), 100x (C), and 200x (D) Scale bars are shown for reference. [annotation: PanIN (P), acinar (A) cells, islet (I) cells, ductal (D) cells, vascular (V) cells (i.e. endothelial cells), and fibroinflammatory (F) infiltrate]
The observed results with cell-autonomous clock disruption were discordant with our expectations and demonstrated that Bmal1 knock-out in the targeted cells did not recapitulate the accelerated PanIN phenotype seen with KC mice under (global) chronic jetlag conditions. Pdx1 is expressed early during pancreatic development and later to commit pancreatic progenitor cells to islets (Offield et al. 1996; Hingorani et al. 2003). Pdx1-Cre expression in pancreatic cells that normally express Pdx1 drives expression from the conditional alleles (i.e., KrasG12D and Bmal1 deletion). To understand which cell populations in BKC mice exhibited loss of Bmal1 (location of Pdx1 expression), we crossed the KC mouse with the Ai14 ‘marker’ mouse (CAG-LSL-tdTomato) that conditionally expresses tdTomato in the presence of Pdx1-driven Cre-recombinase (Figure 4). This revealed tdTomato expression (Kras mutation and Bmal1 deletion) in acinar cells, ductal cells, PanINs, and islet cells. Concomitantly, expression was absent in the fibroinflammatory infiltrate (fibroblasts, collagen, and immune cells) and blood vessels surrounding the PanINs. This result is highly consistent with prior expression patterns of Pdx1 in the pancreas (Ahlgren et al. 1996). To further substantiate BMAL1 loss in the identified cell populations, we performed IHC in Bmal1fx/fx, Bmal1fx/fx; Pdx1-Cre, KC, and BKC mice (Figure 5). We expected to see robust expression of BMAL1 in the control mice (Bmal1fx/fx and KC mice) with loss of BMAL1 protein expression in the comparator groups (Bmal1fx/fx; Pdx1-Cre and BKC mice). Loss of BMAL1 protein detection by IHC in tissue has been previously shown to represent tissue-specific disruption of the clock (Marcheva et al. 2010; Liu et al. 2014; Johnson et al. 2014; Papagiannakopoulos et al. 2016). Indeed, in the Bmal1fx/fx mice (Figure 5, A–D), there was diffuse positivity for BMAL1 as expected, including acinar cells, ductal cells, and islet cell populations. Similarly, the KC mice demonstrated widespread expression of BMAL1 (Figure 5, K–M) including acinar cells, ductal cells, islets, and PanINs. Meanwhile, Bmal1fx/fx; Pdx1-Cre mice (Figure 5, E–J) and BKC mice (Figure 5, N–P) showed loss of BMAL1 protein in each of the pancreatic cell populations except the infiltrating immune cells and fibroblasts (Figure 5, I–J and P) – precisely matching the expected results from the tdTomato analysis. Thus, in mice predisposed to pancreas neoplasm formation, circadian disruption in acinar cells produced a different phenotype compared to chronic jetlag, suggesting a different cell population may be responsible for the observed accelerated PanIN formation and progression in chronic jetlag KC mice.
Figure 4: tdTomato expression in wild-type and reporter strain mice.
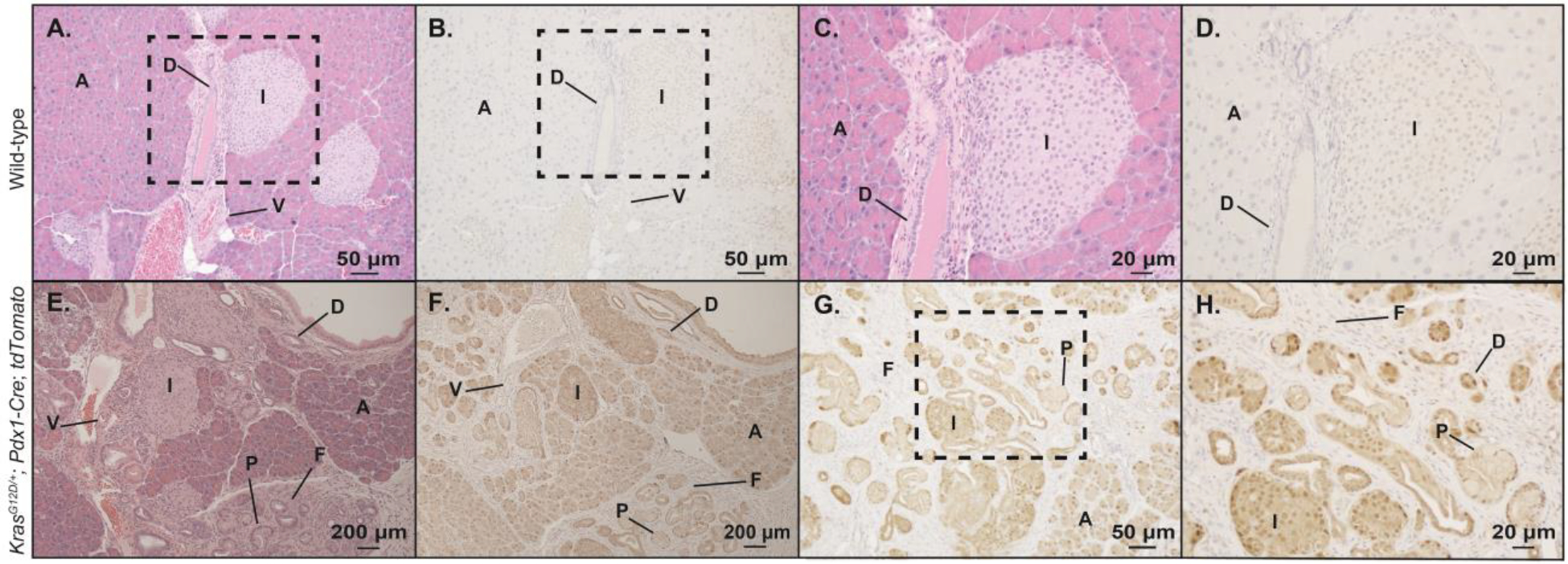
Pancreas from wild-type C57BL/6J mice and from reporter mice (KrasG12D/+; Pdx1-Cre; Ai14) were removed, fixed and embedded, and sectioned for histologic (H&E stain) analysis and immunohistochemistry (IHC) for tdTomato protein expression. Shown are 100x power magnification of H&E and paired IHC for tdTomato in wild-type mice (A-B). Higher power magnification (200x) of the areas from the dotted box are also shown (C-D). H&E stain and IHC for tdTomato are shown (40x magnification) for reporter mice also harboring the mutant Kras allele (E-F). Higher magnification images are shown including 100x (G) with the dotted box pertaining to additional magnification (H, 200x). Scale bars are shown for reference. [annotation: PanIN (P), acinar (A) cells, islet (I) cells, ductal (D) cells, vascular (V) cells (i.e. endothelial cells), and fibroinflammatory (F) infiltrate]
Figure 5: Bmal1 protein expression in pancreas.
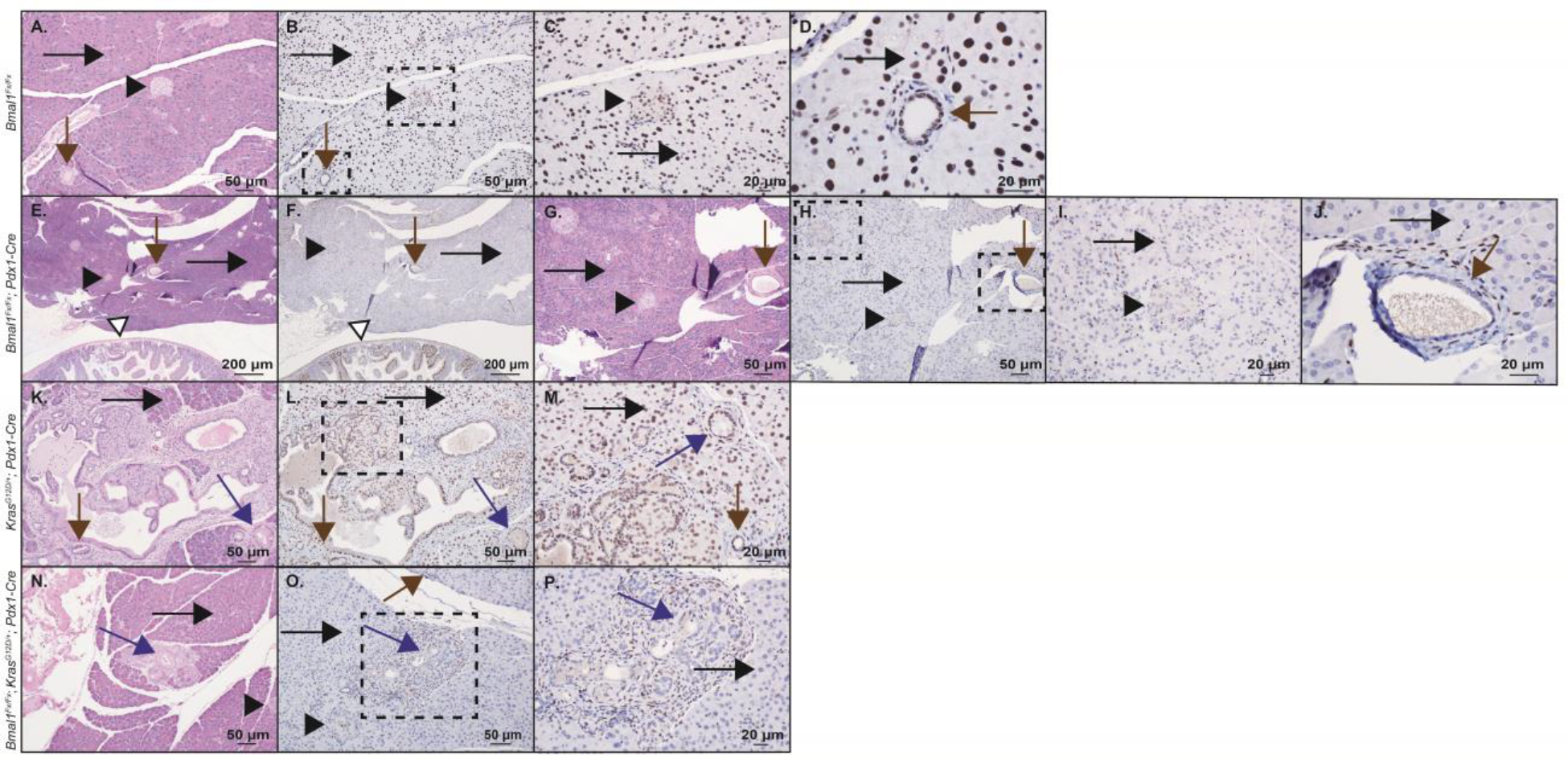
Shown are H&E staining and immunohistochemistry (IHC) staining, including high power fields for BMAL1 protein in Bmal1fx/fx mouse pancreas (wild-type control) (A-B, 100x; C, 200x; D, 400x), Bmal1fx/fx; Pdx1-Cre pancreas (E-F, 40x; G-H, 100x; I, 200x; J, 400x), KrasG12D/+; Pdx1-Cre pancreas (K-L, 100x; M, 200x), and Bmal1fx/fx; KrasG12D/+; Pdx1-Cre pancreas (N-O, 100x; P, 200x). In each of the panels, islet cells are marked with a black arrowhead, acinar cells with a long black arrow, ductal cells with a brown arrow, and PanINs with a blue arrow. In panels (E-F), the duodenal mucosa is also shown as an internal control depicting duodenal mucosal cells that retain BMAL1 expression (white arrow). Dotted boxes indicate the area of subsequent magnification. Scale bars are shown for reference.
Single-cell RNA Sequencing of KC Mice Recapitulates the Highly Heterogeneous Tumor Microenvironment
Prior work in the liver revealed that cell-autonomous knock-out of the clock in liver hepatocytes (Albumin-Cre; Bmal1fx/fx) drove a nearly identical phenotype compared to chronic jetlag mice (non-alcoholic fatty liver disease progressing to HCC) (Kettner et al. 2016). However, we found that in the pancreas of KC mice, this was not evident. We expected that this was a consequence of non-cell autonomous changes to the circadian clock (i.e. not acinar cells or PanINs) – whether by loss of clock function or by misalignment – are responsible for accelerating the neoplastic progression. To examine further, we tested the hypothesis that misalignment of the clock in an alternative cell population caused the accelerated PanIN formation and progression; this was done by evaluating the expression of individual cells in the tumor microenvironment over time. We performed single-cell RNA sequencing (scRNAseq) from pooled pancreatic samples (1 male/female) from both normal circadian and chronic jetlag KC mice at 5 and 9 months (Figure 6A–B). Notably, male and female mice were pooled because we consistently demonstrated no sex-specific effect within either the normal circadian or the chronic jetlag group of KC mice at 5 months or 9 months. After filtering and dimensionality reduction, a total of 14 different heterogeneous cell types were annotated using the ScType package comprising 42,211 cells (Figure 6A–B & Supplemental Data 2–3) (Ianevski et al. 2022). These included acinar cells, several classes of immune cells, endothelial cells, and fibroblast populations.
Figure 6: The single-cell landscape of KC mice recapitulates the developing pancreatic cancer tumor microenvironment.
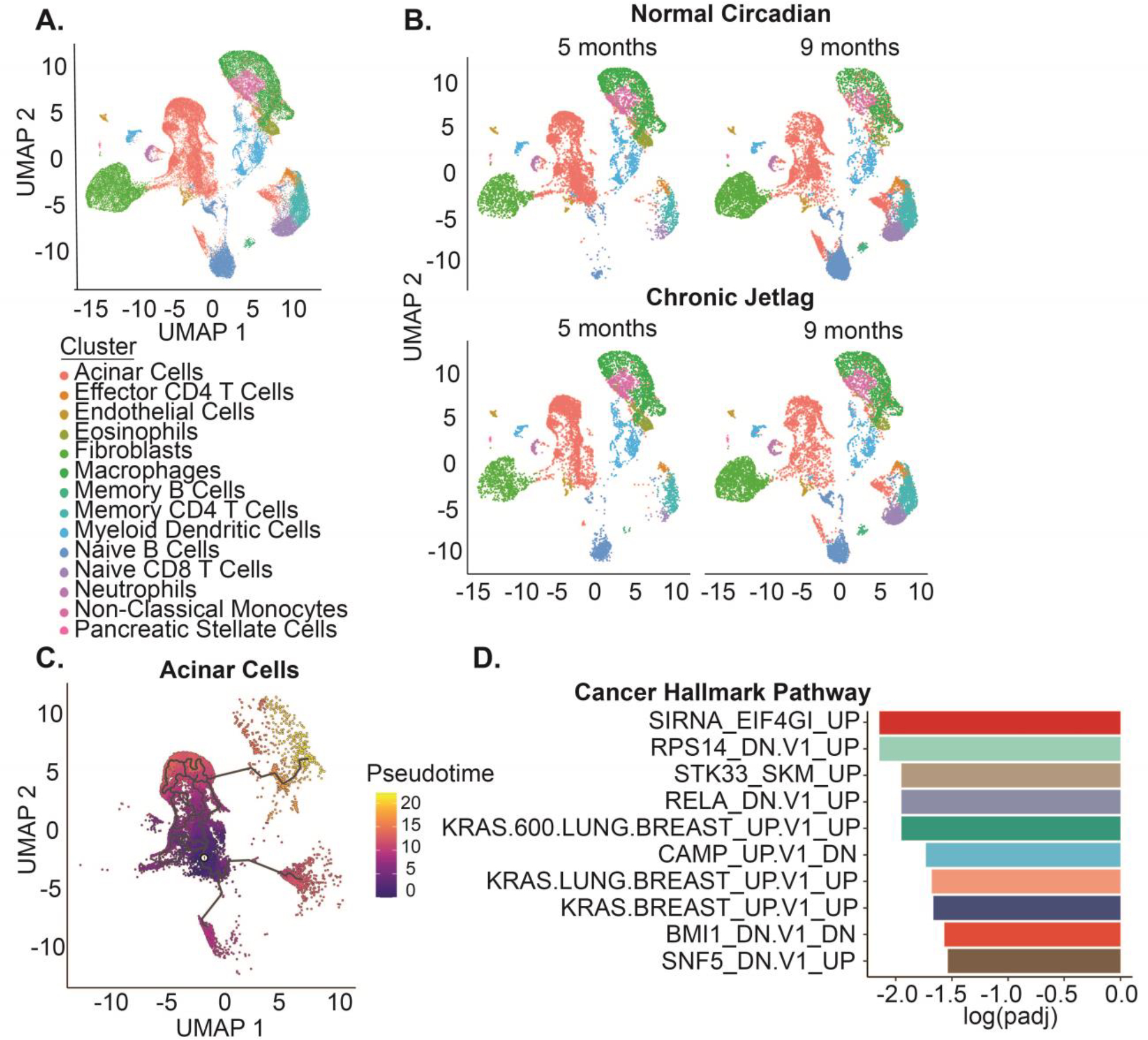
A. UMAP demonstrating the 14 diverse cell types and 42,211 cells in the scRNAseq dataset. B. UMAPs split by time (five and nine months) and condition (normal circadian and chronic jetlag) C. Pseudotime trajectory analysis was performed for the subset of pancreatic acinar cells with zero pseudotime set to the point with the highest fraction of cells at five months. D. Gene Set Enrichment Analysis (GSEA) was performed for the oncogenic signature gene set associated with pseudotime progression and ordered by the log adjusted p value.
We first began by subsetting the pancreatic epithelial cells, which should encompass both normal acinar and developing PanINs, and used pseudotime trajectory analysis to track cell-state changes that occurred from 5 to 9 months. The expectation was that this population would gain signatures of neoplastic transformation over time, given the loss of normal acinar cell density and increase in PanINs from 5 to 9 months. Pseudotime was set to zero at the point with the highest fraction of cells from the 5-month mice and allowed to progress (Figure 6C). Moran’s I autocorrelation analysis was then performed to obtain genes that varied across the trajectory (Supplemental Data 4). Gene set enrichment analysis (GSEA) of the genes that correlated with pseudotime progression revealed an association with multiple Kras-driven cancer cell line signatures – corresponding precisely to the development of Kras-driven pancreatic neoplasia over time (Figure 6D). Therefore, we identified a collection of pancreatic epithelial cells that appeared to represent the transition from acinar cells to PanIN, coinciding with the increased PanIN formation seen in histopathology from 5 to 9 months. These findings established a foundation for the subsequent analysis of the clock to understand the effects of circadian misalignment (chronic jetlag) in the acinar cell and PanIN population, which would then help to support or refute our new hypothesis of a non-cell-autonomous cell population driving the accelerated PanIN phenotype.
Chronic Jetlag Promotes Differential Clock Gene Expression in Fibroblasts
We performed differential gene expression comparing the collective 5- and 9-month normal circadian and chronic jetlag mouse cells, and assessed for differences in the CCG and clock-associated genes (including Tef, Hlf, Bhlhe40, Bhlhe41, Nfil3, Rorc, Dbp, and Ciart) (Supplemental Data 5). We previously showed that chronic jetlag-induced clock misalignment in the pancreas caused differential expression of the core clock genes (Schwartz et al. 2021), and thus we expected populations of cells manifesting misalignment of the clock between normal circadian and chronic jetlag conditions would similarly demonstrate CCG differential expression. In support of our new hypothesis, the acinar cell/PanIN clusters that we identified and confirmed from the pseudotime analysis did not exhibit differential expression of any of the CCGs or the clock-associated genes between clusters derived from normal circadian or chronic jetlag KC mice. We then evaluated the other clusters, and we found that endothelial cells, macrophages, memory CD4 T cells, and monocytes each had a single clock-associated gene differentially expressed (Figure 7A). Interestingly, fibroblasts were the only cell type that exhibited differential expression of CCGs and clock-associated genes as a result of circadian misalignment, including Per2, Per3, Nr1d2, Hlf, Tef, and Dbp (Figure 7A–B). Fibroblasts are an integral part of the PDAC microenvironment and are known to exhibit circadian rhythmicity when compared to naïve fibroblasts (Parascandolo et al. 2020). The lack of other cell types exhibiting changes in clock-specific gene expression suggests fibroblasts may be the target of the chronic jetlag-induced circadian misalignment and accelerated neoplastic phenotype. Concordantly, fibroblasts were among the population of cells that were negative for PDX1 expression (in the KCT ‘marker’ mouse) and retained BMAL1 expression on IHC in the BKC mice; these mice thus had an intact clock function in the fibroblasts and did not manifest the accelerated PanIN formation (Figure 3E–F).
Figure 7: Fibroblasts were preferentially affected by chronic jetlag.
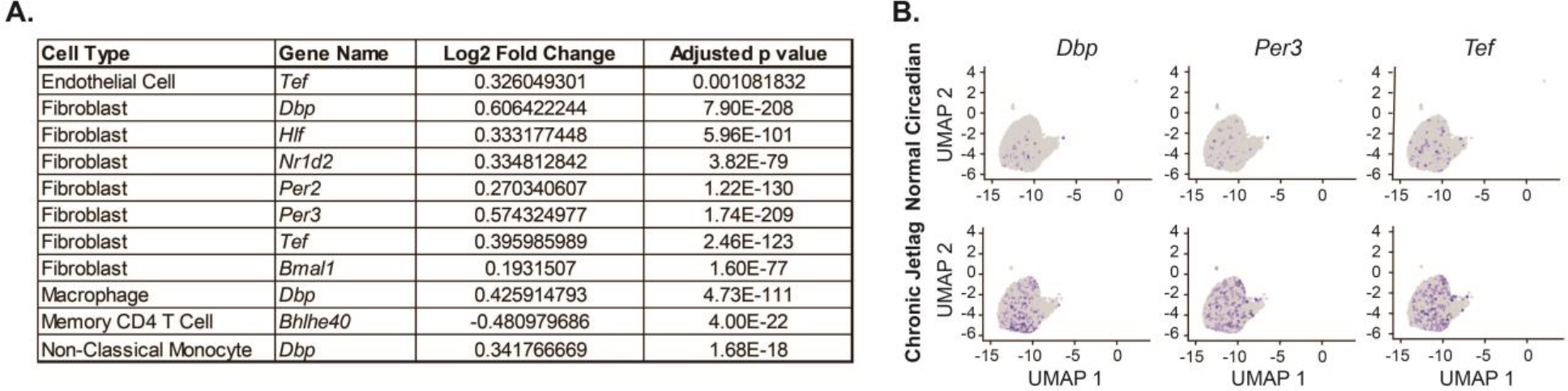
A. Table of core clock genes and clock-associated genes differentially expressed on single-cell RNA sequencing analysis when comparing normal circadian and chronic jetlag conditions of each cell type (all samples collected at ZT4). Although it does not meet our predefined cutoff for differential expression (logFC > 0.25 and false discovery rate p < 0.05), Bmal1 is also shown for reference. B. Representative UMAP dimensionality plots demonstrating cellular expression (blue color) of select core clock genes and clock-associated genes in the subset of fibroblasts.
Fibroblasts Exhibit Time-Dependent Effects Secondary to Chronic Jetlag
To further evaluate the fibroblasts within our dataset, we isolated three different subclusters (Figure 8A). Using existing markers generated for cancer-associated fibroblasts (CAFs), including inflammatory CAFs (iCAFs), myofibroblast CAFs (mCAFs), and antigen-presenting CAFs (apCAFs), we assigned identities with ScType (Supplemental Data 6–7) (Elyada et al. 2019). Of the three clusters, we identified all three as iCAFs – with the expression of known markers, such as Cle3b, Il6, Has1, and Col14a1 (Figure 8B). Fibroblast cluster 3 exhibited some characteristics consistent with apCAFs, including expression of multiple major histocompatibility complex (MHC) II components H2-Aa, H2-Eb1, and H2-ab1 and Cd74, the invariant chain of MHC II (Figure 8B). We then performed differential gene expression and GSEA comparing the iCAFs between normal circadian and chronic jetlag groups at 5 and 9 months (Supplemental Data 8 & Supplemental Data 9). We found that multiple metabolic pathways in fibroblasts were enriched in the chronic jetlag group at 5 months (Supplemental Data 9), and our 9-month comparison revealed enrichment of several apoptosis-related pathways, including ‘NEGATIVE_REGULATION_OF_CELL_DEATH’ and ‘REGULATION_OF_APOPTOTIC_SIGNALING_PATHWAY’. Furthermore, we found that chronic jetlag augmented the expression of multiple cancer-related pathways at five months, including TP53 signaling and STK33 signaling (Supplemental Data 9). To evaluate if chronic jetlag was simply increasing fibrosis (i.e. collagen deposition) to facilitate these changes, we compared the collagen content from 5- and 9-month normal circadian and chronic jetlag KC mice (Supplemental Data 10). We found no differences in the collagen content at either time point. We, therefore, identified that the iCAFs in the developing tumor microenvironment of KC mice had early enrichment of metabolic and cancer pathways in response to chronic jetlag, with late enrichment of apoptosis-related pathways. Enrichment of these pathways in fibroblasts as a consequence of chronic jetlag corresponds with the finding that fibroblasts manifest differential expression of core clock genes (i.e. affected by misalignment of the clock). When combined with the observation that acinar cells/PanINs demonstrated no differential gene expression of CCGs, and abolishing the clock in acinar cells did not replicate the chronic jetlag-induced phenotype, these results suggest chronic jetlag may cause clock desynchrony in the fibroblast population to alter signaling and accelerate the formation and progression of PanINs.
Figure 8: Fibroblasts subclusters in the developing pancreatic tumor microenvironment were identified as inflammatory cancer-associated fibroblasts on single-cell RNA sequencing analysis.
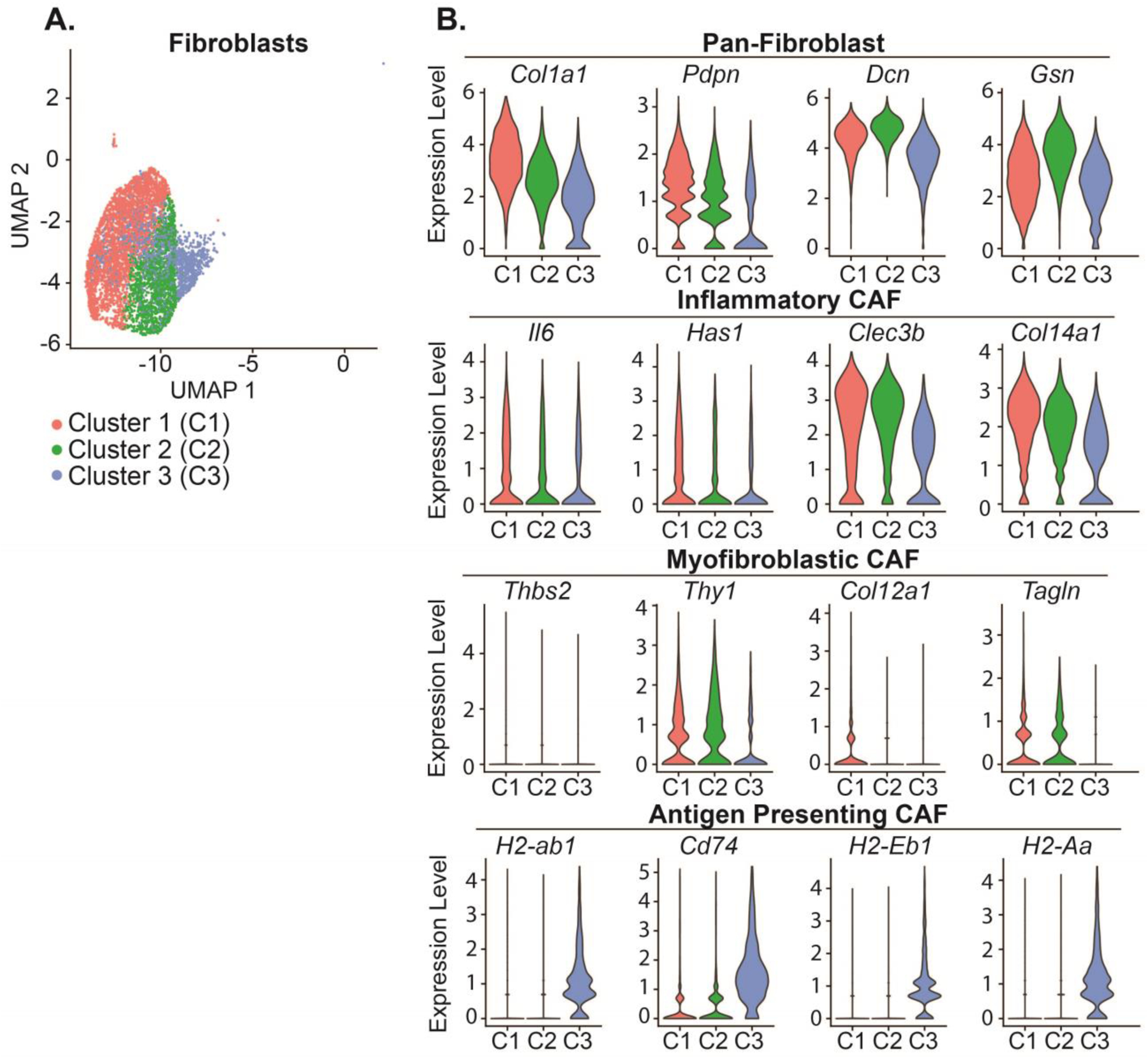
A. A UMAP demonstrating the three fibroblast subclusters identified on dimensionality reduction. B. Violin plots with select markers for Pan-, Inflammatory-, Myofibroblastic-, and Antigen Presenting-Cancer Associated Fibroblasts (CAFs).
Discussion:
There has been a substantial volume of literature examining circadian desynchrony, which encompasses the misalignment between environmental or behavioral cycles and the endogenous clock (Vetter 2020). Persistent misalignment between environmental cues (light/dark cycle) and behavioral cues (feeding) compared to the internal clocks can result in aberrant gene signaling (Kettner et al. 2016; Sulli et al. 2018) with consequent disease pathology (Scheer et al. 2009; Lee et al. 2013; Morris et al. 2015). For instance, in healthy human adults subjected to a forced circadian desynchrony protocol that replicated shift work (12-hour reversal of light/dark cycle for 3 days), there was a misalignment of the circadian clock relative to the timing of the light/dark cycle causing glucose intolerance as well as reduced insulin sensitivity – findings indicative of impaired glucose metabolism that occurs with the onset of type II diabetes (Morris et al. 2015). This coincides with epidemiological findings of an increased risk of type II diabetes with shift work (Pan et al. 2011) and is congruent with the increased risk of obesity and metabolic syndrome in individuals performing rotating shift work schedules (Lin et al. 2009; Antunes et al. 2010). Unfortunately, the metabolic reprogramming and chronic inflammation that ensues following these diagnoses can drive the formation of several cancers such as hepatocellular carcinoma (Campbell et al. 2016), which commences a path of ‘neoplastic change’ consisting of non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, and liver cancer (Simon et al. 2021). The same process of neoplastic transformation in the liver can occur due to circadian misalignment: C57BL/6J mice subjected to chronic jetlag conditions experienced an acceleration of non-alcoholic fatty liver disease and an increased incidence of hepatocellular carcinoma compared to mice under standard lighting conditions (Kettner et al. 2016). Notably, the evidence correlating circadian desynchrony and liver cancer is mirrored in a similar metabolic organ – the pancreas. There is a strong correlation between obesity and diabetes, and risk of developing pancreatic ductal adenocarcinoma (PDAC). A potential link between circadian misalignment (associated with obesity and diabetes) and PDAC can be inferred from this correlation, and this putative link is strengthened by epidemiologic data reporting an increased risk of PDAC in shift workers (Parent et al. 2012). Therefore, based on this cumulative evidence, we sought to test the hypothesis that circadian clock desynchrony through chronic jetlag would accelerate the initiation and progression of PanINs in mice predisposed to develop pre-cancerous lesions (PanIN-1, PanIN-2, PanIN-3) and PDAC.
Our work revealed that circadian misalignment through chronic jetlag increased tumor initiation and progression of PanINs, which are pancreas cancer pre-cursor lesions that can ultimately develop into PDAC. Our findings of accelerated neoplasm progression in the current study are alarming considering the highly lethal nature of PDAC (Siegel et al. 2021), and the prevalence of circadian desynchrony in society (Antunes et al. 2010). This type of clock dysfunction affects a significant percentage of the population, such as shift workers under imposed non-daytime schedules, who experience chronic misalignment between their endogenous clock and environmental cues (Bass and Lazar 2016). A less severe but highly prevalent form of misalignment may also be present through extended exposure to light at night, extended duration of dietary intake, or significant alteration in sleep patterns (social jetlag) (Roenneberg et al. 2012; Chang et al. 2015; Bass and Lazar 2016; Fishbein et al. 2021). To study the potential implications of these types of clock desynchrony in the development of PDAC, we utilized a mouse model in which Kras-mutation in pancreatic acinar cells promotes acinar-to-ductal metaplasia, followed by PanIN formation, and ultimately PDAC. Consequently, a pertinent question for the present work is the applicability of the model to humans. Interestingly, in humans, PanINs are frequently found in the pancreas and their prevalence increases with age (Matsuda et al. 2017). They are a result of acinar metaplastic transformation to a ductal cell phenotype, typically in response to injury or inflammation (De La O et al. 2008; Reichert and Rustgi 2011; Chuvin et al. 2017). In an autopsy study of 173 cases with no evidence of PDAC (mean age 80.5 years), PanIN-1 was present in 77% of pancreas specimens, PanIN-2 was present in 28%, and 4% of the pancreas specimens harbored PanIN-3 (Matsuda et al. 2017). Consistent with our KC model, PanIN-1s were always present in the pancreas when PanIN-2 or PanIN-3 were identified. These autopsy findings were also confirmed in a separate study (Longnecker and Suriawinata 2022). Thus, our model is concordant with the neoplastic process that occurs in humans and relevant for studying PanIN progression. Our findings of chronic jetlag-induced acceleration of advanced PanIN formation could be applicable to many individuals who unknowingly harbor PanINs and experience consistent circadian misalignment.
PanINs are microscopic and early PanINs are unable to be detected with the current imaging modalities (Kanda et al. 2012). Therefore, human studies of circadian misalignment driving PanIN progression have not been feasible. Yet, the in vivo findings in our study warrant consideration of future investigations designed to evaluate risk in humans and possibly risk mitigation. Given the widespread presence of circadian misalignment, and the prevalence of PanINs, a suitable population of individuals to target for risk mitigation of circadian desynchrony may prove tenable. For example, there is convincing evidence that patients who have a family history of PDAC demonstrate a greater propensity for PanIN formation, are more likely to harbor advanced PanINs, and, therefore, are at higher risk for developing PDAC. In a study by Shi and colleagues evaluating patients who underwent pancreatic resection for PDAC, those with a family history of PDAC (without a germline mutation) demonstrated a rate of 1.51 PanIN lesions/cm2 in the pancreas (i.e. non-cancer pancreas) compared to 0.55 PanIN lesion/cm2 in patients with sporadic PDAC (Shi et al. 2009). The majority of the lesions were PanIN-1, with fewer PanIN-2 and PanIN-3 lesions identified (Shi et al. 2009); again mirroring the findings in our KC model where there is multifocal lesion formation, and the majority of the lesions at 9 months were PanIN-1 with fewer PanIN-2 and PanIN-3. Moreover, in the study by Shi et al (Shi et al. 2009), PanIN lesions were found in the pancreas remote from invasive cancer, which supports the findings from autopsy studies (Matsuda et al. 2017; Longnecker and Suriawinata 2022) that not all PanINs progress to cancer. Similar to the etiology of PanINs in our model, greater than 95% of human PanINs harbor a Kras mutation (including 92% of PanIN-1) (Kanda et al. 2012), and it is generally accepted that the fraction of Kras-mutant cells in PanINs increases with PanIN grade, indicating clonal expansion of the Kras-mutant cells that have been ‘transformed’ (Kanda et al. 2012). However, the drivers of this transformation from PanIN-1 to higher-grade PanINs or PDAC are poorly understood. Consequently, our study forms a strong basis for further investigation to understand how circadian misalignment promotes the progression of PanIN lesions. This should lead to prevention strategies (e.g. chemoprevention or behavioral modification) that can then be applied to high-risk patients to prevent the progression of PanINs to cancer (Miller et al. 2016). Incidentally, these high-risk patients are already being captured in high-risk clinics, where individuals with a strong family history or known pre-cursor lesions in the pancreas are being followed to identify cancer transformation at an early stage (Shin et al. 2015; Miller et al. 2016; Dbouk et al. 2022). This would be an ideal group to target for risk mitigation or chemoprevention.
To better understand the basis for our observations of accelerated neoplasm formation with circadian misalignment (chronic jetlag), we implemented a conditional mouse model to achieve cell-autonomous core clock gene knock-out. We chose a conditional knock-out as opposed to a global knock-out because of the known difficulty in disentangling the effects in a global knock-out model (e.g. Bmal1−/−). For example, the opposing effects in glucose metabolism and insulin sensitivity with knock-out of the clock in the pancreas and liver can mask glucose intolerance phenotypes that are otherwise readily apparent with islet-cell-specific clock disruption (Lamia et al. 2008; Marcheva et al. 2010; Perelis et al. 2016). Furthermore, chronic jetlag exposure is a ‘global’ effect and we aimed to home in on the cell population responsible for the observed phenotype. In this way, the use of core clock gene knock-out models has been indispensable in ascertaining clock-dependent mechanisms for various pathologies. Oftentimes, the phenotypes among circadian disrupted groups are concordant, whether by circadian desynchrony or through genetic alteration. For instance, Lee and colleagues found that the cell-autonomous deletion of Bmal1 in β-cells of the pancreas resulted in the loss of glucose-stimulated insulin secretion, and the blunted insulin secretion with peripheral insulin resistance was replicated by environmentally-induced circadian misalignment (i.e. 6-hr light phase-advancement protocol) (Lee et al. 2013). In a separate study, mice predisposed to colorectal cancer development were subjected to circadian misalignment via constant lighting conditions and showed a 2-fold increase in tumor initiation compared to those housed under diurnal conditions (Stokes et al. 2021). In this study, Stokes et al. showed that global and epithelial loss of Bmal1 similarly induced tumor initiation, suggesting that Bmal1 expression in the colon epithelial cells (cell of origin for colorectal cancer) is similarly ‘targeted’ by perturbations in the clock (altered lighting) and by clock disruption (Bmal1 deletion). Comparable results were seen by Kettner et al. when evaluating rates of non-alcoholic fatty liver disease and hepatocellular carcinoma between cell-autonomous Bmal1 knock-out mice and mice subjected to circadian desynchrony (Kettner et al. 2016). Thus, we expected that cell-autonomous clock disruption in KC mice (pancreatic endocrine and exocrine Bmal1 knock-out) would demonstrate a PanIN phenotype concordant with the circadian misaligned KC mice. However, when we crossed our KC mice with the conditional Bmal1fx/fx mouse, we found a discordant phenotype in pancreatic tumor initiation compared to circadian misalignment. Although the results were unanticipated, they did provide insight and enabled us to conclude that disruption of the clock in acinar cells does not facilitate PanIN formation or progression.
To better understand individual cell population involvement, and to build a foundation for discerning how pancreatic neoplasia more rapidly progressed in chronic jetlag conditions, we performed scRNAseq at both 5 and 9 months. We found that the pancreatic epithelial cells, including the PanINs and acinar cells, showed a time-dependent cancer signature on pseudotime analysis consistent with the observed histopathologic changes. Confirming the time-dependent cancer signature enabled us to examine these cells primarily for changes to the clock, to determine if the scRNAseq data from circadian misaligned mice complemented the Bmal1 cell-autonomous knock-out data. Subsequently, we assessed 13 other cell types for evidence of differential gene expression between normal circadian and chronic jetlag, and we found that fibroblasts – not the pancreatic epithelial cells – were the only cell type with multiple CCG and clock-associated gene changes. This finding supports the notion that chronic jetlag augments fibroblasts in the developing tumor microenvironment and not necessarily the epithelial/PanINs themselves. Notably, this also reinforces the differences identified in the cell-autonomous Bmal1 knock-out model versus the circadian desynchrony model – misalignment may be targeting the fibroblast population to accelerate neoplasm formation and progression. Meanwhile, in the Bmal1fx/fx; Pdx1-Cre; LSL-KrasG12D/+ mice, Bmal1 (and thus the clock) is intact in the mesenchymal cells: IHC in KC mice conditionally expressing tdTomato demonstrated that there was Pdx1-Cre-directed expression (indicating BMAL1 loss) in the acinar, ductal, islet, and PanINs, but not the mesenchymal lineage cells, including the fibroblasts and endothelial cells. We similarly found concordant results when directly measuring loss of BMAL1 expression via IHC. We then used additional expression markers to subset the fibroblasts and we identified all 3 clusters as inflammatory CAFs (iCAFs) based on established markers (Elyada et al. 2019). iCAFs secrete IL-6, CXCL9, and TGFβ and can be both immunosuppressive as well as immune-promoting (Sahai et al. 2020). Interestingly, we did not observe any differences in the amount of pancreatic collagen content at 5 or 9 months between normal circadian and chronic jetlag KC mice, suggesting a role for paracrine signaling in mediating the effects of chronic jetlag by fibroblasts rather than extracellular matrix production (i.e. desmoplasia). Furthermore, we identified early enrichment of several cancer pathways in fibroblasts. This includes TP53 signaling, which is a known altered pathway in pancreatic cancer. Others have suggested TP53 signaling may play a clock-dependent role in PDAC progression (Jiang et al. 2016). We also identified strong signatures of STK33 pathway enrichment, which is a serine/threonine kinase involved in Kras-mutant cancers (Luo et al. 2012) and has been shown to correlate with metastatic progression of PDAC as well as poor patient prognosis (Kong et al. 2017). Interestingly, Stk33 upregulation in PDAC has been shown to be HIF1α dependent, and several lines of evidence demonstrate a strong correlation between HIF1α and the circadian clock, including increased levels of HIF1α under hypoxic conditions as a result of circadian disruption (Peek et al. 2017). While the correlation with these cancer signature pathways in our work requires additional consequential investigation and will be the focus of future investigation, it is important to note that other investigators have uncovered the substantive involvement of the fibroblast clock in cancer. In a pan-cancer analysis of The Cancer Genome Atlas tumor samples, Wu and colleagues employed a new computational approach (LTM) to explore clock-controlled pathways in cancer (Wu et al. 2022). The authors found that the variation in clock strength across 9 separate cancers was directly related to the proportion of fibroblasts in the samples and concluded that this was consequent to fibroblasts possessing a more robust circadian clock than the adjacent cancer cells within the tumor microenvironment. Moreover, fibroblasts are known to have an essential role in fostering cancer progression in the pancreas cancer microenvironment (Hanahan and Coussens 2012; Helms et al. 2020). As fibroblast density increases with the advancement of PanINs (i.e. PanIN-1 to PanIN-2/3) towards invasive cancer, the cancer-associated fibroblasts (CAFs) display a ‘wound-healing’ type response that supports neoplastic proliferation through direct and indirect metabolic contributions (Helms et al. 2020). CAFs display differences in rhythmic gene expression compared to naïve fibroblasts (Parascandolo et al. 2020), and the wound healing type response in fibroblasts has been previously shown to be strongly clock dependent (Hoyle et al. 2017), with disruption of the clock profoundly affecting this pathway. Concordantly, after identifying and characterizing the CAFs in our mouse pancreas samples, we performed GSEA at 5 and 9 months. This demonstrated that at 5 months chronic jetlag promoted metabolic gene enrichment in the CAFs, and by 9 months, there was enrichment of multiple cell death and apoptosis-related pathways. It is therefore conceivable that the fibroblasts – which harbor a strong clock and coevolve with PanINs – underlie the circadian desynchrony-induced acceleration of PanIN formation and progression. However, our data is currently limited to differential gene expression in single-cell RNA sequencing analysis and requires much more supportive data to substantiate this possibility. The data does raise an intriguing possibility of fibroblast involvement, and therefore, to investigate further whether circadian disruption in the fibroblasts may recapitulate the circadian misalignment (chronic jetlag) phenotype, in future work we aim to generate Col1a1-Cre; Bmal1fx/fx in FLP recombinase Kras-mutant mice (Pdx1-FlpO; FSF-KrasG12D/+) (Wu et al. 2017). This would be effective in abolishing the fibroblast clock in the developing tumor microenvironment because all the identified fibroblast populations expressed high levels of Col1a1 in the present scRNAseq analysis.
Although we selected a chronic jetlag protocol with a 4-hour phase advancement (corresponding to eastward flight) to be consistent with our prior work in the pancreas (Schwartz et al. 2021; Schwartz et al. 2022), additional experiments will be needed to evaluate phase delay (corresponding to westward flight) and changes in the magnitude of the phase-shift, both of which are known to cause differential reentrainment and could potentially change the observed results (Jud et al. 2005; Leloup and Goldbeter 2013). For example, in an experiment where mice were subjected to a single 4-hour phase advance, aged mice took an average of 7 days longer than young adult mice to reentrain based on wheel running behavior (Valentinuzzi et al. 1997). Conversely, both groups reentrained the following day to a 4-hour phase delay. The implications of this effect are better seen in the work of Davidson et al., who observed that aged mice undergoing a weekly 6-hour advance had increased mortality compared to mice undergoing phase delay – an effect which was accelerated with more frequent shifts (4 vs. 7 days) (Davidson et al. 2006). Thus, the phenotype seen with our 4-hour phase advancement may be more severe compared with a 4-hour phase delay in KC mice (phase shifted backward), but additional studies would need to be performed to evaluate any potential differences.
Another limitation to consider from the scRNAseq data was that we did not identify islet cell clusters and were therefore unable to discern whether differential expression of CCGs was present in this population of cells. However, Pdx1 is expressed in endocrine cells in the pancreas, and therefore the BKC mice (which did not display an accelerated PanIN phenotype) would have a disrupted clock in this cell population (Offield et al. 1996; Hingorani et al. 2003). While clock disruption via Bmal1 knock-out in islet cells produces metabolic changes that could ostensibly alter the formation or growth trajectory of tumors (PanINs), the prevailing phenotype with Bmal1 deficiency in islet cells – and also with circadian misalignment – is glucose intolerance, hypoinsulinemia, and hyperglycemia (i.e. characteristics of diabetes) (Marcheva et al. 2010; Lee et al. 2013; Perelis et al. 2016). Given that diabetes is a risk factor for PDAC development (Abbruzzese et al. 2018), and the lack of phenotype (expedited PanIN formation) in the BKC mice, it seems unlikely that circadian misalignment in the islets would have contributed to accelerated PanIN formation and progression. Regardless, we were not able to evaluate this cell population in the scRNAseq analysis, and so this remains a limitation. To investigate further, one could consider beta-cell specific Bmal1 knock-out (Rip-Cre; Bmal1fx/fx) in FLP recombinase Kras-mutant mice (Pdx1-FlpO; FSF-KrasG12D/+) to determine if the accelerated PanIN development emerges (Wu et al. 2017). Another consideration is the intriguing observation that loss of pancreatic Bmal1 (acinar cells, ductal cells, islets, PanINs) resulted in near abolishment of PanIN formation, and not just an absence of accelerated PanIN formation. This finding speaks to the complex interplay of the circadian clock amongst cell populations in the tumor microenvironment. Adding additional complexity – and highlighting another limitation of our work – these cell populations in the tumor microenvironment can be influenced by neurohumoral factors which may have contributed to the accelerated PanIN phenotype in chronic jetlag versus normal circadian KC mice. For example, circadian misalignment from imposing alterations in the light-dark schedule (8-hr phase delay) was shown to cause alteration in levels of norepinephrine and serotonin in the amygdala of mice (Moriya et al. 2015). Altered neurohumoral stimulation can result in autonomic nervous system dysregulation which has been linked to diabetes (pancreatic pathology associated with PDAC development) and inflammation (Pliquett et al. 2004). Moreover, chronic jetlag causing circadian misalignment results in altered feeding patterns, as demonstrated in our prior work (Schwartz et al. 2022). Given the link between serotonin signaling, timing of food intake and metabolic disorders (risk factor for PDAC) (Versteeg et al. 2015), the dysregulated diurnal feeding pattern can alter the serotonergic-circadian reciprocity to potentially influence PanIN development. Therefore, in future work, we aim to employ various genetically engineered mouse models under time-restricted feeding paradigms, and determine circulating levels of monoamines to disentangle these findings, which should have clinical relevance for the multitude of individuals that harbor PanINs while concomitantly experiencing chronic circadian misalignment.
Although previous evidence suggests a role for the circadian clock in PDAC growth and spread, this is the first study to show perturbations of the clock increases tumor initiation in PDAC (Jiang et al. 2016; Jiang et al. 2018). Our study provides a foundation for future work to expand our understanding of how a dyssynchronous clock orchestrates cancer development in the pancreas.
Supplementary Material
Supplemental Data 1: Survival of KC mice under normal circadian and chronic jetlag conditions
Supplemental Data 2: Marker database for single-cell RNA sequencing cell-type identification
Supplemental Data 3: ScType Scores for overall single-cell RNA sequencing dataset
Supplemental Data 4: Moran’s I pseudotime analysis for the pancreatic acinar cells
Supplemental Data 5: Differential gene expression between normal circadian and chronic jetlag conditions for all cell types
Supplemental Data 6: Marker database for fibroblast subtype identification
Supplemental Data 7: ScType Scores for fibroblast subtype identification
Supplemental Data 8: Differential gene expression between normal circadian and chronic jetlag conditions for fibroblasts at five and nine months
Supplemental Data 9: Transcriptomic effects of chronic jetlag on fibroblasts
Supplemental Data 10: Quantification of collagen content in five-month normal circadian and chronic jetlag KC mice
Acknowledgments:
We would like to take the opportunity to acknowledge the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520, which provides funding for the Experimental Animal Pathology Lab Core.
The author(s) thank the University of Wisconsin Translational Research Initiatives in Pathology laboratory (TRIP), supported by the UW Department of Pathology and Laboratory Medicine, UWCCC (P30 CA014520) and the Office of The Director- NIH (S10 OD023526) for use of its facilities and services.
We would also to acknowledge the Gene Expression Center and the Bioinformatics Resource Center (BRC) at the University of Wisconsin, Madison for their contributions to this work.
Finally, we would like to thank the Michael W. Oglesby Foundation for their funding support of our work in circadian disruption and pancreas pathology.
Grants:
The research reported in this publication was supported by the Department of Defense Peer Reviewed Cancer Research Program Award CA190176 (Grants.gov ID GRANT12935023) and the University of Wisconsin Institute for Clinical and Translational Research KL2 Award (UL1TR002373 and KL2TR002374) [SRK]. Funding support for this work was also provided by the National Institutes of Health under Award Number T32 ES007015 [PBS]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosure: The authors declare no conflict of interest.
Data Statement:
Single-cell RNA sequencing data is made publicly available through gene expression omnibus (GEO) (Accession number: GSE209629). All other data that support the findings of this study are available from the corresponding author, SRK, upon reasonable request.
References:
- Abbruzzese JL, Andersen DK, Borrebaeck CAK, Chari ST, Costello E, Cruz-Monserrate Z, Eibl G, Engleman EG, Fisher WE, Habtezion A, et al. 2018. The Interface of Pancreatic Cancer With Diabetes, Obesity, and Inflammation: Research Gaps and Opportunities: Summary of a National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Pancreas. 47(5):516–525. 10.1097/MPA.0000000000001037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren U, Jonsson J, Edlund H. 1996. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 122(5):1409–1416. 10.1242/dev.122.5.1409 [DOI] [PubMed] [Google Scholar]
- Antunes LC, Levandovski R, Dantas G, Caumo W, Hidalgo MP. 2010. Obesity and shift work: chronobiological aspects. Nutr Res Rev. 23(1):155–168. 10.1017/S0954422410000016 [DOI] [PubMed] [Google Scholar]
- Arble DM, Bass J, Laposky AD, Vitaterna MH, Turek FW. 2009. Circadian timing of food intake contributes to weight gain. Obesity (Silver Spring). 17(11):2100–2102. 10.1038/oby.2009.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J, Lazar MA. 2016. Circadian time signatures of fitness and disease. Science. 354(6315):994–999. 10.1126/science.aah4965 [DOI] [PubMed] [Google Scholar]
- Bass J, Takahashi JS. 2010. Circadian integration of metabolism and energetics. Science. 330(6009):1349–1354. 10.1126/science.1195027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. 2000. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 103(7):1009–1017. 10.1016/s0092-8674(00)00205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PT, Newton CC, Freedman ND, Koshiol J, Alavanja MC, Beane Freeman LE, Buring JE, Chan AT, Chong DQ, Datta M, et al. 2016. Body Mass Index, Waist Circumference, Diabetes, and Risk of Liver Cancer for U.S. Adults. Cancer Res. 76(20):6076–6083. 10.1158/0008-5472.CAN-16-0787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A-M, Aeschbach D, Duffy JF, Czeisler CA. 2015. Evening use of light-emitting eReaders negatively affects sleep, circadian timing, and next-morning alertness. Proc Natl Acad Sci U S A. 112(4):1232–1237. 10.1073/pnas.1418490112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S, Satija R. 2022. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biology. 23(1):27. 10.1186/s13059-021-02584-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuvin N, Vincent DF, Pommier RM, Alcaraz LB, Gout J, Caligaris C, Yacoub K, Cardot V, Roger E, Kaniewski B, et al. 2017. Acinar-to-Ductal Metaplasia Induced by Transforming Growth Factor Beta Facilitates KRASG12D-driven Pancreatic Tumorigenesis. Cell Mol Gastroenterol Hepatol. 4(2):263–282. 10.1016/j.jcmgh.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KH, Takahashi JS. 2019. Circadian clock genes and the transcriptional architecture of the clock mechanism. J Mol Endocrinol. 63(4):R93–R102. 10.1530/JME-19-0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ, Sellix MT, Daniel J, Yamazaki S, Menaker M, Block GD. 2006. Chronic jet-lag increases mortality in aged mice. Curr Biol. 16(21):R914–916. 10.1016/j.cub.2006.09.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dbouk M, Katona BW, Brand RE, Chak A, Syngal S, Farrell JJ, Kastrinos F, Stoffel EM, Blackford AL, Rustgi AK, et al. 2022. The Multicenter Cancer of Pancreas Screening Study: Impact on Stage and Survival. J Clin Oncol. 40(28):3257–3266. 10.1200/JCO.22.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La OJ-P, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. 2008. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 105(48):18907–18912. 10.1073/pnas.0810111105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. 2019. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 9(8):1102–1123. 10.1158/2159-8290.CD-19-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipski E, Delaunay F, King VM, Wu M-W, Claustrat B, Gréchez-Cassiau A, Guettier C, Hastings MH, Francis L. 2004. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 64(21):7879–7885. 10.1158/0008-5472.CAN-04-0674 [DOI] [PubMed] [Google Scholar]
- Filipski E, Subramanian P, Carrière J, Guettier C, Barbason H, Lévi F. 2009. Circadian disruption accelerates liver carcinogenesis in mice. Mutat Res. 680(1–2):95–105. 10.1016/j.mrgentox.2009.10.002 [DOI] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. 2015. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol [Internet]. [accessed 2021 Feb 4] 16. 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein AB, Knutson KL, Zee PC. 2021. Circadian disruption and human health. J Clin Invest. 131(19):e148286. 10.1172/JCI148286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. 2012. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 21(3):309–322. 10.1016/j.ccr.2012.02.022 [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. 2021. Integrated analysis of multimodal single-cell data. Cell. 184(13):3573–3587.e29. 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms E, Onate MK, Sherman MH. 2020. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 10(5):648–656. 10.1158/2159-8290.CD-19-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 4(6):437–450. 10.1016/s1535-6108(03)00309-x [DOI] [PubMed] [Google Scholar]
- Hoyle NP, Seinkmane E, Putker M, Feeney KA, Krogager TP, Chesham JE, Bray LK, Thomas JM, Dunn K, Blaikley J, O’Neill JS. 2017. Circadian actin dynamics drive rhythmic fibroblast mobilization during wound healing. Sci Transl Med. 9(415):eaal2774. 10.1126/scitranslmed.aal2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A, Giri AK, Aittokallio T. 2022. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat Commun. 13(1):1246. 10.1038/s41467-022-28803-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Zhao S, Jiang X, Zhang E, Hu G, Hu B, Zheng P, Xiao J, Lu Z, Lu Y, et al. 2016. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 371(2):314–325. 10.1016/j.canlet.2015.12.002 [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhao S, Shen J, Guo L, Sun Y, Zhu Y, Ma Z, Zhang X, Hu Y, Xiao W, et al. 2018. The MiR-135b-BMAL1-YY1 loop disturbs pancreatic clockwork to promote tumourigenesis and chemoresistance. Cell Death Dis. 9(2):149. 10.1038/s41419-017-0233-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BP, Walisser JA, Liu Y, Shen AL, McDearmon EL, Moran SM, McIntosh BE, Vollrath AL, Schook AC, Takahashi JS, Bradfield CA. 2014. Hepatocyte circadian clock controls acetaminophen bioactivation through NADPH-cytochrome P450 oxidoreductase. Proc Natl Acad Sci U S A. 111(52):18757–18762. 10.1073/pnas.1421708111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda M, Matthaei H, Wu J, Hong S-M, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, Goggins M. 2012. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 142(4):730–733.e9. 10.1053/j.gastro.2011.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, Fu L. 2016. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell. 30(6):909–924. 10.1016/j.ccell.2016.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Kong X, Du Y, Chen Y, Deng X, Zhu J, Du J, Li L, Jia Z, Xie D, et al. 2017. STK33 Promotes Growth and Progression of Pancreatic Cancer as a Critical Downstream Mediator of HIF1α. Cancer Res. 77(24):6851–6862. 10.1158/0008-5472.CAN-17-0067 [DOI] [PubMed] [Google Scholar]
- Lamia KA, Storch K-F, Weitz CJ. 2008. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 105(39):15172–15177. 10.1073/pnas.0806717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 107(7):855–867. 10.1016/s0092-8674(01)00610-9 [DOI] [PubMed] [Google Scholar]
- Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, Nelson DL, Ma K, Moore DD, Yechoor VK. 2013. Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol Cell Biol. 33(11):2327–2338. 10.1128/MCB.01421-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Sancar A. 2011. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc Natl Acad Sci U S A. 108(29):12036–12041. 10.1073/pnas.1108125108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xie K. 2022. Ductal metaplasia in pancreas. Biochim Biophys Acta Rev Cancer. 1877(2):188698. 10.1016/j.bbcan.2022.188698 [DOI] [PubMed] [Google Scholar]
- Lin Y-C, Hsiao T-J, Chen P-C. 2009. Persistent rotating shift-work exposure accelerates development of metabolic syndrome among middle-aged female employees: a five-year follow-up. Chronobiol Int. 26(4):740–755. 10.1080/07420520902929029 [DOI] [PubMed] [Google Scholar]
- Liu Y, Johnson BP, Shen AL, Wallisser JA, Krentz KJ, Moran SM, Sullivan R, Glover E, Parlow AF, Drinkwater NR, et al. 2014. Loss of BMAL1 in ovarian steroidogenic cells results in implantation failure in female mice. PNAS. 111(39):14295–14300. 10.1073/pnas.1209249111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longnecker DS, Suriawinata AA. 2022. Incidence of Pancreatic Intraepithelial Neoplasia in an Autopsy Series. Pancreas. 51(4):305–309. 10.1097/MPA.0000000000002027 [DOI] [PubMed] [Google Scholar]
- Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, Scholl C, Fröhling S, Carr SA, Stern AM, et al. 2012. STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci U S A. 109(8):2860–2865. 10.1073/pnas.1120589109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. 2010. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 466(7306):627–631. 10.1038/nature09253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Furukawa T, Yachida S, Nishimura M, Seki A, Nonaka K, Aida J, Takubo K, Ishiwata T, Kimura W, et al. 2017. The Prevalence and Clinicopathological Characteristics of High-Grade Pancreatic Intraepithelial Neoplasia: Autopsy Study Evaluating the Entire Pancreatic Parenchyma. Pancreas. 46(5):658–664. 10.1097/MPA.0000000000000786 [DOI] [PubMed] [Google Scholar]
- Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. 2003. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 302(5643):255–259. 10.1126/science.1086271 [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J. 2020. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv:180203426 [cs, stat] [Internet]. [accessed 2021 Feb 4]. http://arxiv.org/abs/1802.03426 [Google Scholar]
- McIntosh BE, Hogenesch JB, Bradfield CA. 2010. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol. 72:625–645. 10.1146/annurev-physiol-021909-135922 [DOI] [PubMed] [Google Scholar]
- Miller MS, Allen P, Brentnall TA, Goggins M, Hruban RH, Petersen GM, Rao CV, Whitcomb DC, Brand RE, Chari ST, et al. 2016. Pancreatic Cancer Chemoprevention Translational Workshop: Meeting Report. Pancreas. 45(8):1080–1091. 10.1097/MPA.0000000000000705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya S, Tahara Y, Sasaki H, Ishigooka J, Shibata S. 2015. Phase-delay in the light-dark cycle impairs clock gene expression and levels of serotonin, norepinephrine, and their metabolites in the mouse hippocampus and amygdala. Sleep Med. 16(11):1352–1359. 10.1016/j.sleep.2015.06.020 [DOI] [PubMed] [Google Scholar]
- Morris CJ, Yang JN, Garcia JI, Myers S, Bozzi I, Wang W, Buxton OM, Shea SA, Scheer FAJL. 2015. Endogenous circadian system and circadian misalignment impact glucose tolerance via separate mechanisms in humans. Proc Natl Acad Sci U S A. 112(17):E2225–2234. 10.1073/pnas.1418955112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, Göttgens B. 2016. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood. 128(8):e20–e31. 10.1182/blood-2016-05-716480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. 1996. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 122(3):983–995. 10.1242/dev.122.3.983 [DOI] [PubMed] [Google Scholar]
- Pan A, Schernhammer ES, Sun Q, Hu FB. 2011. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med. 8(12):e1001141. 10.1371/journal.pmed.1001141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T. 2016. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 24(2):324–331. 10.1016/j.cmet.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parascandolo A, Bonavita R, Astaburuaga R, Sciuto A, Reggio S, Barra E, Corcione F, Salvatore M, Mazzoccoli G, Relógio A, Laukkanen MO. 2020. Effect of naive and cancer-educated fibroblasts on colon cancer cell circadian growth rhythm. Cell Death Dis. 11(4):289. 10.1038/s41419-020-2468-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent M-É, El-Zein M, Rousseau M-C, Pintos J, Siemiatycki J. 2012. Night work and the risk of cancer among men. Am J Epidemiol. 176(9):751–759. 10.1093/aje/kws318 [DOI] [PubMed] [Google Scholar]
- Peek CB, Levine DC, Cedernaes J, Taguchi A, Kobayashi Y, Tsai SJ, Bonar NA, McNulty MR, Ramsey KM, Bass J. 2017. Circadian Clock Interaction with HIF1α Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell Metab. 25(1):86–92. 10.1016/j.cmet.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, et al. 2015. Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science. 350(6261):aac4250. 10.1126/science.aac4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelis M, Ramsey KM, Marcheva B, Bass J. 2016. Circadian Transcription from Beta Cell Function to Diabetes Pathophysiology. J Biol Rhythms. 31(4):323–336. 10.1177/0748730416656949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh CS. 1993. Temporal organization: reflections of a Darwinian clock-watcher. Annu Rev Physiol. 55:16–54. 10.1146/annurev.ph.55.030193.000313 [DOI] [PubMed] [Google Scholar]
- Pliquett R, Fasshauer M, Blüher M, Paschke R. 2004. Neurohumoral stimulation in type-2-diabetes as an emerging disease concept. Cardiovasc Diabetol. 3:4. 10.1186/1475-2840-3-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. 2002. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 110(2):251–260. 10.1016/s0092-8674(02)00825-5 [DOI] [PubMed] [Google Scholar]
- Reichert M, Rustgi AK. 2011. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest. 121(12):4572–4578. 10.1172/JCI57131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roenneberg T, Allebrandt KV, Merrow M, Vetter C. 2012. Social jetlag and obesity. Curr Biol. 22(10):939–943. 10.1016/j.cub.2012.03.038 [DOI] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 20(3):174–186. 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA, Hogenesch JB. 2004. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 43(4):527–537. 10.1016/j.neuron.2004.07.018 [DOI] [PubMed] [Google Scholar]
- Scheer FAJL Hilton MF, Mantzoros CS Shea SA. 2009. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 106(11):4453–4458. 10.1073/pnas.0808180106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiermann C, Kunisaki Y, Frenette PS. 2013. Circadian control of the immune system. Nat Rev Immunol. 13(3):190–198. 10.1038/nri3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Fuchs CS, Colditz GA. 2003. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 95(11):825–828. 10.1093/jnci/95.11.825 [DOI] [PubMed] [Google Scholar]
- Schwartz PB, Barrett-Wilt GA, Ronnekleiv-Kelly SM. 2022. Chronic Jetlag Alters the Landscape of the Pancreatic Lipidome. Pancreas. 51(1):80–89. 10.1097/MPA.0000000000001962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PB, Walcheck MT, Berres ME, Nakaya M, Wu G, Carrillo ND, Matkowskyj KA, Ronnekleiv-Kelly SM. 2021. Chronic Jetlag-Induced Alterations in Pancreatic Diurnal Gene Expression. Physiol Genomics. 10.1152/physiolgenomics.00022.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev AA. 2016. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv:060012. 10.1101/060012 [DOI] [Google Scholar]
- Shi C, Klein AP, Goggins M, Maitra A, Canto M, Ali S, Schulick R, Palmisano E, Hruban RH. 2009. Increased Prevalence of Precursor Lesions in Familial Pancreatic Cancer Patients. Clin Cancer Res. 15(24):7737–7743. 10.1158/1078-0432.CCR-09-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EJ, Topazian M, Goggins MG, Syngal S, Saltzman JR, Lee JH, Farrell JJ, Canto MI. 2015. Linear-array EUS improves detection of pancreatic lesions in high-risk individuals: a randomized tandem study. Gastrointest Endosc. 82(5):812–818. 10.1016/j.gie.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2021. Cancer Statistics, 2021. CA Cancer J Clin. 71(1):7–33. 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- Simon TG, Roelstraete B, Sharma R, Khalili H, Hagström H, Ludvigsson JF. 2021. Cancer Risk in Patients With Biopsy-Confirmed Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study. Hepatology. 74(5):2410–2423. 10.1002/hep.31845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, Sudbery I. 2017. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27(3):491–499. 10.1101/gr.209601.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes K, Nunes M, Trombley C, Flôres DEFL, Wu G, Taleb Z, Alkhateeb A, Banskota S, Harris C, Love OP, et al. 2021. The Circadian Clock Gene, Bmal1, Regulates Intestinal Stem Cell Signaling and Represses Tumor Initiation. Cell Mol Gastroenterol Hepatol. 12(5):1847–1872.e0. 10.1016/j.jcmgh.2021.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulli G, Manoogian ENC, Taub PR, Panda S. 2018. Training the Circadian Clock, Clocking the Drugs, and Drugging the Clock to Prevent, Manage, and Treat Chronic Diseases. Trends Pharmacol Sci. 39(9):812–827. 10.1016/j.tips.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS. 2017. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 18(3):164–179. 10.1038/nrg.2016.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takele Assefa A, Vandesompele J, Thas O. 2020. On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genomics. 21(1):312. 10.1186/s12864-020-6721-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 32(4):381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentinuzzi VS, Scarbrough K, Takahashi JS, Turek FW. 1997. Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 273(6):R1957–R1964. 10.1152/ajpregu.1997.273.6.R1957 [DOI] [PubMed] [Google Scholar]
- Versteeg RI, Serlie MJ, Kalsbeek A, la Fleur SE. 2015. Serotonin, a possible intermediate between disturbed circadian rhythms and metabolic disease. Neuroscience. 301:155–167. 10.1016/j.neuroscience.2015.05.067 [DOI] [PubMed] [Google Scholar]
- Vetter C 2020. Circadian disruption: What do we actually mean? Eur J Neurosci. 51(1):531–550. 10.1111/ejn.14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcheck MT, Matkowskyj KA, Turco A, Blaine-Sauer S, Nukaya M, Noel J, Ronnekleiv OK, Ronnekleiv-Kelly SM. 2021. Sex-dependent development of Kras-induced anal squamous cell carcinoma in mice. PLoS One. 16(11):e0259245. 10.1371/journal.pone.0259245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcheck MT, Nukaya M, Ranheim EA, Matkowskyj KA, Ronnekleiv-Kelly S. 2022. Pdx1 expression in hematopoietic cells activates Kras-mutation to drive leukemia in KC (Pdx1-Cre; LSL-KrasG12D/+) mice [Internet]. [accessed 2022 Dec 5]:2022.05.17.492353. 10.1101/2022.05.17.492353 [DOI] [PMC free article] [PubMed]
- West AC, Bechtold DA. 2015. The cost of circadian desynchrony: Evidence, insights and open questions. Bioessays. 37(7):777–788. 10.1002/bies.201400173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AC, Smith L, Ray DW, Loudon ASI, Brown TM, Bechtold DA. 2017. Misalignment with the external light environment drives metabolic and cardiac dysfunction. Nat Commun. 8(1):417. 10.1038/s41467-017-00462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KP, McHill AW, Birks BR, Griffin BR, Rusterholz T, Chinoy ED. 2013. Entrainment of the human circadian clock to the natural light-dark cycle. Curr Biol. 23(16):1554–1558. 10.1016/j.cub.2013.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Ruben MD, Francey LJ, Lee YY, Anafi RC, Hogenesch JB. 2022. An in silico genome-wide screen for circadian clock strength in human samples [Internet]. [accessed 2022 Sep 8]:2022.05.10.491250. 10.1101/2022.05.10.491250 [DOI] [PMC free article] [PubMed]
- Wu J, Liu X, Nayak SG, Pitarresi JR, Cuitiño MC, Yu L, Hildreth BE, Thies KA, Schilling DJ, Fernandez SA, et al. 2017. Generation of a pancreatic cancer model using a Pdx1-Flp recombinase knock-in allele. PLoS One. 12(9):e0184984. 10.1371/journal.pone.0184984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H. 2000. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 288(5466):682–685. 10.1126/science.288.5466.682 [DOI] [PubMed] [Google Scholar]
- Zhang J, Francois R, Iyer R, Seshadri M, Zajac-Kaye M, Hochwald SN. 2013. Current understanding of the molecular biology of pancreatic neuroendocrine tumors. J Natl Cancer Inst. 105(14):1005–1017. 10.1093/jnci/djt135 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data 1: Survival of KC mice under normal circadian and chronic jetlag conditions
Supplemental Data 2: Marker database for single-cell RNA sequencing cell-type identification
Supplemental Data 3: ScType Scores for overall single-cell RNA sequencing dataset
Supplemental Data 4: Moran’s I pseudotime analysis for the pancreatic acinar cells
Supplemental Data 5: Differential gene expression between normal circadian and chronic jetlag conditions for all cell types
Supplemental Data 6: Marker database for fibroblast subtype identification
Supplemental Data 7: ScType Scores for fibroblast subtype identification
Supplemental Data 8: Differential gene expression between normal circadian and chronic jetlag conditions for fibroblasts at five and nine months
Supplemental Data 9: Transcriptomic effects of chronic jetlag on fibroblasts
Supplemental Data 10: Quantification of collagen content in five-month normal circadian and chronic jetlag KC mice
Data Availability Statement
Single-cell RNA sequencing data is made publicly available through gene expression omnibus (GEO) (Accession number: GSE209629). All other data that support the findings of this study are available from the corresponding author, SRK, upon reasonable request.