

Summary
Background
Lenalidomide is a cornerstone of maintenance therapy in patients with newly diagnosed multiple myeloma after autologous stem-cell transplantation. We aimed to compare the efficacy and safety of maintenance therapy with carfilzomib, lenalidomide, and dexamethasone versus lenalidomide alone in this patient population.
Methods
This study is an interim analysis of ATLAS, which is an investigator-initiated, multicentre, open-label, randomised, phase 3 trial in 12 academic and clinical centres in the USA and Poland. Participants were aged 18 years or older with newly diagnosed multiple myeloma, completed any type of induction and had stable disease or better, autologous stem-cell transplantation within 100 days, initiated induction 12 months before enrolment, and an Eastern Cooperative Oncology Group performance status of 0 or 1. Patients were randomly assigned (1:1) using permuted blocks of sizes 4 and 6 and a web-based system to receive up to 36 cycles of carfilzomib, lenalidomide, and dexamethasone (28-day cycles of carfilzomib 20 mg/m2 administered intravenously in cycle one on days 1 and 2 then 36 mg/m2 on days 1, 2, 8, 9, 15, and 16 in cycles one to four and 36 mg/m2 on days 1, 2, 15, and 16 from cycle five up to 36 [per protocol]; lenalidomide 25 mg administered orally on days 1–21; and dexamethasone 20 mg administered orally on days 1, 8, 15, and 22) or lenalidomide alone (10 mg administered orally for the first three cycles and then at the best tolerated dose [≤15 mg for 28 days in 28-day cycles]) until disease progression or unacceptable toxicity as maintenance therapy. After 36 cycles, patients in both treatment groups received lenalidomide maintenance. Randomisation was stratified by response to previous treatment, cytogenetic risk factors, and country. Investigators and patients were not masked to treatment allocation. Patients in the carfilzomib, lenalidomide, and dexamethasone group with no detectable minimal residual disease after cycle six (as per International Myeloma Working Group criteria) and standard-risk cytogenetics were switched to lenalidomide maintenance as of cycle nine. The primary endpoint was progression-free survival in the intention-to-treat population (defined as all randomly assigned patients). Safety was analysed in all randomly assigned patients who received at least one dose of study treatment. This unplanned interim analysis was triggered by the occurrence of 59 (61%) of the expected 96 events for the primary analysis and the results are considered preliminary. This trial is registered with ClinicalTrials.gov, NCT02659293 (active, not recruiting) and EudraCT, 2015–002380–42.
Findings
Between June 10, 2016, and Oct 21, 2020, 180 patients were randomly assigned to receive either carfilzomib, lenalidomide, and dexamethasone (n=93) or lenalidomide alone (n=87; intention-to-treat population). The median age of patients was 59∙0 years (IQR 49∙0–63∙0); 84 (47%) patients were female and 96 (53%) were male. With a median follow-up of 33·8 months (IQR 20·9–42·9), median progression-free survival was 59·1 months (95% CI 54·8–not estimable) in the carfilzomib, lenalidomide, and dexamethasone group versus 41·4 months (33·2–65·4) in the lenalidomide group (hazard ratio 0·51 [95% CI 0·31–0·86]; p=0·012). The most common grade 3 and 4 adverse events were neutropenia (44 [48%] in the carfilzomib, lenalidomide, and dexamethasone group vs 52 [60%] in the lenalidomide group), thrombocytopenia (12 [13%] vs six [7%]), and lower respiratory tract infections (seven [8%] vs one [1%]). Serious adverse events were reported in 28 (30%) patients in the carfilzomib, lenalidomide, and dexamethasone group and 19 (22%) in the lenalidomide group. One treatment-related adverse event led to death (respiratory failure due to severe pneumonia) in the carfilzomib, lenalidomide, and dexamethasone group.
Interpretation
This interim analysis provides support for considering carfilzomib, lenalidomide, and dexamethasone therapy in patients with newly diagnosed multiple myeloma who completed any induction regimen followed by autologous stem-cell transplantation, which requires confirmation after longer follow-up of this ongoing phase 3 trial.
Funding
Amgen and Celgene (Bristol Myers Squibb).
Introduction
The role of extended treatment is well established in patients with newly diagnosed multiple myeloma. For patients eligible for transplant, there is clear evidence that maintenance with a single agent after autologous stem-cell transplantation prolongs progression-free survival and, in some studies, overall survival.1–4 Lenalidomide maintenance is the most well established among single agents and considered globally as the standard of care based on several randomised studies and meta-analyses showing an overall survival benefit for lenalidomide compared with observation.5 However, access to lenalidomide as maintenance therapy and reimbursement might be limited in many low-income and middle-income countries. Bortezomib might be an alternative for maintenance after autologous stem-cell transplantation but based on less direct evidence (ie, a landmark analysis of a randomised trial and no clear overall survival benefit).6,7 Ixazomib showed a small progression-free survival benefit, but no overall survival compared with observation only. Use of ixazomib in maintenance is scarce but has increased during the COVID-19 pandemic as a convenient oral alternative to bortezomib.8
Attempts to improve progression-free survival and overall survival after autologous stem-cell transplantation using short consolidation therapy or tandem transplantation (or both), with or without subsequent maintenance with a single agent, have generated mixed results.9,10 The EMN02/HOVON95 study11 showed a progression-free survival benefit with short-course bortezomib, lenalidomide, and dexamethasone consolidation after autologous stem-cell transplantation, whereas the STaMINA trial10 did not; differences in study populations and induction regimens might be partly responsible for these disparate findings. The EMN02/HOVON95 study11 also showed a benefit with tandem versus single autologous stem0cell transplantation, favouring patients at high risk. The intention-to-treat population in STaMINA showed no benefit of tandem transplantation, although a substantial proportion of patients did not receive a second transplant. An updated, post-hoc analysis of STaMINA, which included patients who received planned tandem transplantation, corroborated with the observations from EMN02/HOVON95 and showed that tandem autologous stem-cell transplantation benefits patients at high risk.12
In the GEM2014MAIN trial,13 the addition of ixazomib to lenalidomide and dexamethasone for 2 years followed by minimal residual disease-guided discontinuation of maintenance therapy did not result in improved progression-free survival, whereas the CASSIOPEIA trial14 showed a progression-free survival benefit with daratumumab maintenance every 8 weeks compared with observation in patients who did not receive daratumumab before autologous stem-cell transplantation. Several ongoing randomised trials (including phase 3 studies from the German Multicentre Myeloma Group [NCT02495922 and NCT03617731], the Southwest Oncology Group [NCT04071457], the Canadian Myeloma Research Group [NCT04786028], and an industrysponsored trial [NCT03901963]) are evaluating the role of elotuzumab, daratumumab, or isatuximab plus lenalidomide maintenance—results from these trials are eagerly awaited.
With these trials still in progress, there is a scarcity of definitive findings from phase 3 trials to show that any alternative maintenance therapy is superior to lenalidomide alone. In a phase 2 trial,15 extended treatment with 14 cycles of carfilzomib, lenalidomide, and dexamethasone after autologous stem-cell transplantation led to deep responses with median progression-free survival exceeding 5 years since diagnosis, which suggests that this regimen has potential to surpass the current standard of care. Results from the phase 2 FORTE trial16 further support this hypothesis as maintenance with carfilzomib plus lenalidomide after a second randomisation, following carfilzomib-based induction and consolidation with or without autologous stem-cell transplantation, improved progression-free survival compared with lenalidomide alone.
In this unplanned interim analysis of an ongoing trial, we compare the efficacy and safety of maintenance therapy with carfilzomib, lenalidomide, and dexamethasone versus lenalidomide alone in patients with newly diagnosed multiple myeloma who received autologous stem-cell transplantation after any induction regimen.
Methods
Study design
This study is an unplanned interim analysis of ATLAS, which is an investigator-initiated, multicentre, open-label, randomised, phase 3 trial coordinated by the University of Chicago in collaboration with the Polish Myeloma Consortium and conducted in 12 academic and clinical centres in the USA and Poland (appendix p 8). The study, protocol, and amendments were approved by an institutional review board or ethics committee at each participating centre and the Office for Registration of Medicinal Products, Medical Devices, and Biocidal Products in Poland. This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation of Good Clinical Practice guidelines. The study protocol is shown in the appendix.
Participants
Eligible participants were aged 18 years or older with newly diagnosed multiple myeloma who completed any type of induction (up to two regimens allowed if the first induction generated a suboptimal response or the regimen was not well tolerated) followed by single autologous stem-cell transplantation. Patients were included if they had stable disease or better (according to the International Myeloma Working Group [IMWG] response criteria),17 received autologous stem-cell transplantation within 100 days, initiated induction 12 months before enrolment, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, an absolute neutrophil count of at least 1 × 109 cells per L, a platelet count of at least 70 × 109 platelets per L, adequate liver function (bilirubin ≤1·5 times the upper limit of normal and aspartate and alanine aminotransferases ≤3∙0 times the upper limit of normal), and creatinine clearance of at least 50 mL per min or serum creatinine lower than 2 mg/dL. Women of childbearing potential had to have two negative pregnancy tests within 14 days before treatment initiation and agree to use two reliable forms of contraception simultaneously or practice abstinence. Men had to agree to use latex condoms during sexual intercourse. All participants in the USA had to provide consent to be registered into the Lenalidomide Risk Evaluation and Mitigation Strategy programme. Key exclusion criteria were progressive disease (as per IMWG response criteria17) at any time after the initiation of induction, treatment with consolidation or maintenance therapy after transplantation, grade 2 or worse peripheral neuropathy (as defined by the National Cancer Institute Common Terminology Criteria for Adverse Events [NCI-CTCAE]; version 4.0), uncontrolled hypertension or diabetes, myocardial infarction within 6 months before randomisation, uncontrolled angina, heart failure (New York Heart Association Functional Classification 3 or 4), severe coronary artery disease, severe uncontrolled ventricular arrhythmias, electrocardiographic evidence of acute ischaemia or active conduction system abnormalities, other malignancy within the past 3 years (except adequately treated basal cell carcinoma, squamous cell carcinoma, thyroid cancer, cervical carcinoma in situ, prostate cancer [Gleason score 6 with stable prostate-specific antigen concentrations], or cancer considered cured using surgical resection alone), or any substantial medical condition that could interfere with protocol adherence. Complete inclusion and exclusion criteria are listed in the protocol (appendix). All patients provided written informed consent.
Randomisation and masking
Patients were randomly assigned (1:1) to receive carfilzomib, lenalidomide, and dexamethasone or lenalidomide alone as maintenance therapy, using permuted blocks (random block sizes of 4 and 6). Randomisation was performed between day 70 and day 120 after autologous stem-cell transplantation using a web-based system (REDCap, Vanderbilt University, Nashville, TN, USA). Enrolment was done independently by the lead principal investigators (AJJ and DD). Randomisation was stratified by response to previous treatment at study entry (less than very good partial response vs very good partial response or better), cytogenetic risk factors (presence vs absence of del(13) (q14), t(4;14)(p16;q32), t(14;16)(q32;q23), del(17)(p13.1), or hypodiploidy), and site location (USA vs Poland). Investigators and patients were not masked to treatment allocation.
Procedures
Treatment was to be initiated between day 80 and day 130 after autologous stem-cell transplantation (appendix p 1). Patients in the carfilzomib, lenalidomide, and dexamethasone group received 8–36 cycles (as guided by the adaptive study design) of carfilzomib (Amgen, Thousand Oaks, CA, USA) 20 mg/m2 administered intravenously in cycle one on days 1 and 2 then 36 mg/m2 on days 1, 2, 8, 9, 15, and 16 in cycles one to four and 36 mg/m2 on days 1, 2, 15, and 16 from cycle five up to 36 cycles; lenalidomide (Bristol-Myers Squibb [Celgene], New York, NY, USA) 25 mg administered orally on days 1–21; and dexamethasone (Adamed, Pienkow, Poland, and Pfizer, New York, NY, USA) 20 mg administered orally on days 1, 8, 15, and 22 in 28-day cycles. To limit the risk of toxic effects and financial burden of the treatment, patients in the carfilzomib, lenalidomide, and dexamethasone group with no detectable minimal residual disease after cycle six (defined by the IMWG as at least 10−5 sensitivity and at least a complete response)18 and protocol-defined, standard-risk cytogenetics (ie, no high-risk cytogenetic features) were switched to lenalidomide maintenance (best tolerated dose up to 15 mg) as of cycle nine, as part of the risk-adapted and minimal residual disease-directed study design. Patients in the lenalidomide group received lenalidomide 10 mg administered orally for the first three cycles and then at the best tolerated dose (≤15 mg for 28 days in 28-day cycles until disease progression or unacceptable toxicity). After 36 protocol-defined cycles, patients in both treatment groups received or continued to receive lenalidomide maintenance. Patients were removed from treatment in either group if they had disease progression, unacceptable toxic effects, were non-compliant with the study procedures, withdrew consent, or required alternative therapy. All patients received herpes zoster and venous thromboembolism prophylaxis for the duration of treatment, and other concomitant medications were permitted at the investigator’s discretion.
Dose reductions, delays, or interruptions were permitted for any of the study drugs as per protocol guidelines after the occurrence of haematological or non-haematological adverse events (appendix). In cases when dexamethasone was discontinued due to toxic effects, carfilzomib and lenalidomide were continued without dexamethasone if tolerated.
Radiographic assessment (x-ray skeletal survey, whole-body CT, or whole-body MRI) was performed within 30 days before treatment initiation. Imaging was repeated throughout the trial, if clinically indicated. Basic clinical chemistry and haematology tests were performed at the beginning of each cycle and when clinically indicated. To characterise disease response, serum and 24 h urine M-protein, quantitative immunoglobulins, and serum free light chain concentrations were reported at baseline before induction and assessed at screening and day 1 of each cycle (starting from cycle two). Bone marrow samples for plasma cell quantification, fluorescence in situ hybridisation, and minimal residual disease analyses were collected at screening, at the end of cycles 6, 12, 18, 24, and 36, at assessment of response for suspected complete response, at the end of treatment, and then once per year for up to 5 years from random assignment. Minimal residual disease was assessed by centralised next-generation sequencing (NGS) using clonoSEQ (Adaptive Biotechnologies, Seattle, WA, USA; limit of detection 6·8 × 10−7 with input of 20 μg DNA) as the primary method. If NGS was not available, centralised and standardised multiparametric flow cytometry was performed (minimum sensitivity of 10−⁵ based on the method of the EuroFlow Consortium19) either at the University of Chicago (Chicago, IL, USA) or Poznan University of Medical Sciences (Poznan, Poland). If both NGS and flow cytometry results were available, we used the most sensitive method. Adverse events were monitored at every study visit and graded according to the NCI-CTCAE from the first dose until 30 days after the last dose.
Outcomes
The primary endpoint was progression-free survival measured from randomisation to disease progression (as assessed by investigators) or death from any cause, whichever occurred first. Secondary endpoints were the rate of minimal residual disease negativity at 6, 12, 18, 24, and 36 months, correlation of minimal residual disease negativity with progression-free survival, duration of minimal residual disease negativity, depth of response (according to the IMWG criteria), improvement in the depth of response at 6 and 12 months, overall survival (defined as time from randomisation to death from any cause), and safety.
The results of minimal residual disease at 12 months and onwards are not available at the time of this analysis and will be provided, along with the improvement in the depth of response at 12 months and the duration of minimal residual disease at the final analysis.
Statistical analysis
The ATLAS study aimed to enrol 180 patients (90 in each treatment group) based on previous studies3,15 showing a progression-free survival of 65% at 4 years with carfilzomib, lenalidomide, and dexamethasone versus 43% with lenalidomide maintenance therapy. Sample size was estimated based on detecting a hazard ratio (HR) of 0·51 (ie, 1/1∙96) for the carfilzomib, lenalidomide, and dexamethasone group relative to the lenalidomide group, which corresponds to the hypothesised difference in 4-year progression-free survival under exponential assumptions, with 88% power at a two-sided significance level of 0·05 and an estimate of 96 events assuming 1·5 years of accrual and 4 years of subsequent follow-up. An independent data and safety monitoring board evaluated the study conduct and interim safety and efficacy data every 6 months and made recommendations to the sponsor regarding further conduct of the study. This unplanned interim analysis was triggered by the occurrence of 59 (61%) of the expected 96 events for the primary analysis and was motivated partly by results from the FORTE study.16 The p values are not adjusted for the interim nature of the comparison. Patients will continue to be followed up until the primary analysis (time at which the last enrolled patient will reach 4 years of follow-up), without any additional analyses of interim data.
Efficacy was analysed in the intention-to-treat population (ie, all randomly assigned patients) and safety was analysed in the safety population (defined as all randomly assigned patients who received at least one dose of study treatment).
The proportional hazard assumption was assessed with the Schoenfeld residuals method. For progression-free survival and overall survival analyses, the Kaplan-Meier method was used to calculate survival rates and the log rank test was performed to compare treatment groups. The differences in minimal residual disease negativity rates, depth of response per IMWG criteria, and improvement in depth of response between groups were analysed using the χ2 or Mann-Whitney U tests. To evaluate the consistency of treatment effects, post-hoc subgroup analyses of progression-free survival and minimal residual disease negativity rates were conducted based on age (younger than 60 years vs 60 years or older), sex (female vs male), ECOG performance status (0 vs 1), International Staging System (ISS; I–II vs III), response after autologous stem-cell transplantation (less than very good partial response vs very good partial response or better), cytogenetics (standard risk vs high risk), and the number of induction regimens (one vs two). Another post-hoc analysis compared progression-free survival between treatment groups in patients with standard cytogenetic risk. We also conducted a post-hoc landmark analysis after cycle six of progression-free survival (excluding patients who progressed or died before cycle six) in patients with standard-risk cytogenetics and no detectable minimal residual disease that were switched from carfilzomib, lenalidomide, and dexamethasone to lenalidomide as of cycle nine versus those in the lenalidomide group with standard-risk cytogenetics and negative minimal residual disease after cycle six. All the survival analyses, excluding landmark analysis, were performed in the intention-to-treat population. Minimal residual disease negativity for rate calculations required that patients had at least a complete response as defined in the IMWG criteria and assessments were reported according to international harmonisation criteria.20 Patients without trackable clonotype by NGS and unsuccessful assessment by flow cytometry were excluded from the evaluable population, which included all other enrolled patients. Depth of minimal residual disease assessed by NGS was analysed post hoc. GraphPad Prism (version 9.1) was used for all statistical analyses.
This trial is registered with ClinicalTrials.gov, NCT02659293 and EudraCT, 2015–002380–42.
Role of the funding source
The funders of the study had an opportunity to review the protocol and manuscript, but had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Results
Between June 10, 2016, and Oct 21, 2020, 193 patients with newly diagnosed multiple myeloma were assessed for eligibility, of which 13 patients were ineligible. 180 were randomly assigned to receive either carfilzomib, lenalidomide, and dexamethasone (n=93) or lenalidomide alone (n=87; intention-to-treat population; figure 1). 178 (99%) received at least one treatment dose (92 in the carfilzomib, lenalidomide, and dexamethasone group vs 86 in the lenalidomide group; safety population). Baseline characteristics were balanced between the groups (table 1). The median age of patients was 59∙0 years (IQR 49∙0–63∙0). 84 (47%) patients were female and 96 (53%) were male.
Figure 1: Trial profile.
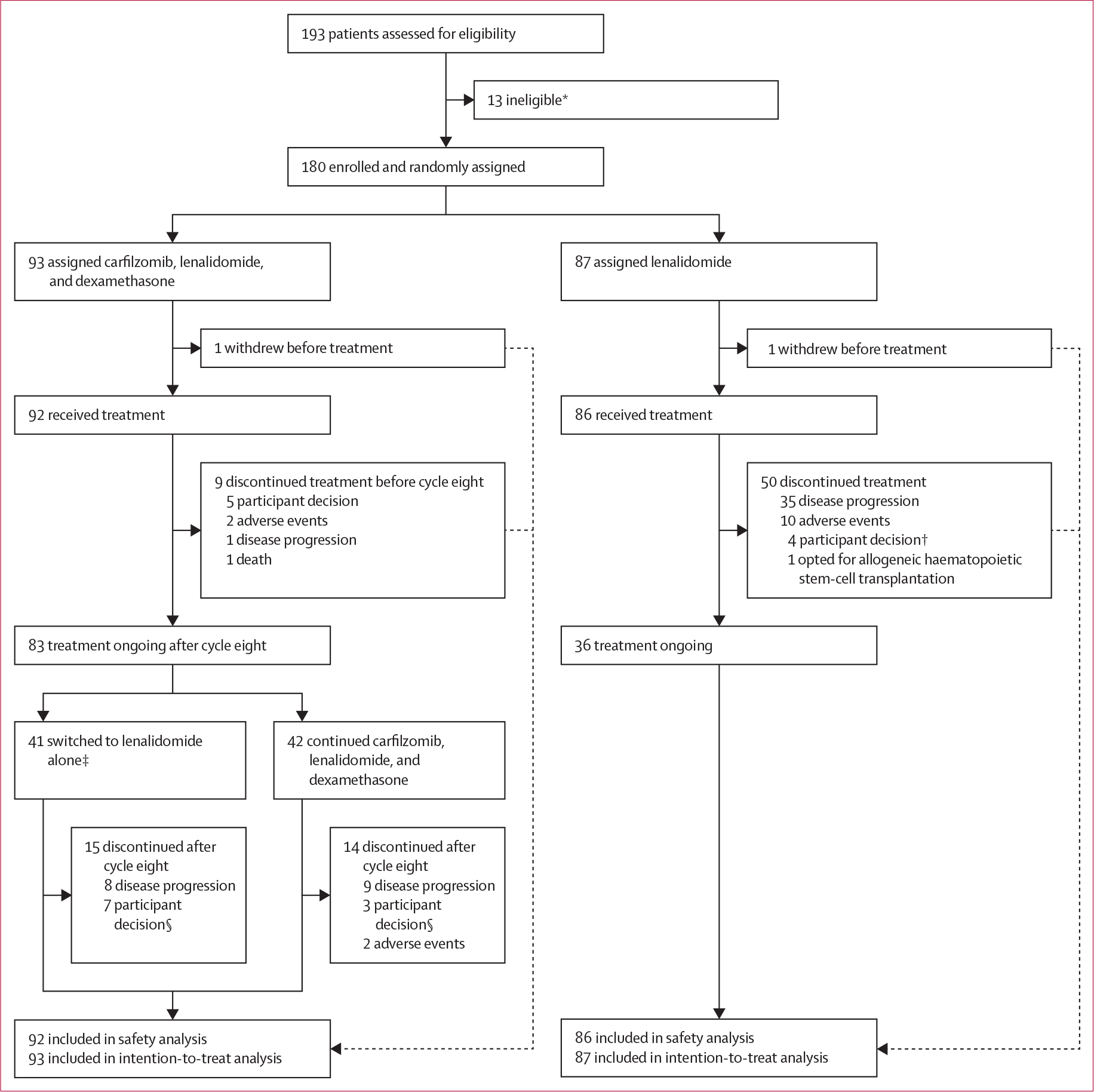
*Two patients had progressive disease, one withdrew consent, one did not have cytogenetics results, one had suspected other malignancy, one had cardiac contraindications, and seven did not meet eligibility criteria. †Two of four patients completed 36 cycles per protocol and refused further treatment with lenalidomide. ‡35 met criteria for treatment deescalation (standard-risk cytogenetics and minimal residual disease negativity after cycle six). Of the remaining six patients (included in analyses of all patients in the carfilzomib, lenalidomide, and dexamethasone group), two had high-risk cytogenetics (analysed together with other high-risk patients in subset analyses) and four had standard-risk cytogenetics (analysed with standard-risk patients in subset analyses) and minimal residual disease negativity (but not a complete response or better). §Four of seven and two of three patients in the carfilzomib, lenalidomide, and dexamethasone group completed 36 cycles per protocol and refused further treatment with lenalidomide.
Table 1:
Baseline characteristics
| Carfilzomib, lenalidomide, and dexamethasone (n=93) | Lenalidomide (n=87) | |
|---|---|---|
|
| ||
| Age, years | 57.0 (49.0-63.5) | 59.0 (49.0-63.0) |
| Sex | ||
| Female | 49 (53%) | 35 (40%) |
| Male | 44 (47%) | 52 (60%) |
| Race | ||
| White | 86 (92%) | 81 (93%) |
| Black | 5 (5%) | 4 (5%) |
| Not reported | 2 (2%) | 2 (2%) |
| Ethnicity | ||
| Non-Hispanic | 91 (98%) | 84 (97%) |
| Not reported | 2 (2%) | 3 (3%) |
| Site location | ||
| Poland | 82 (88%) | 77 (89%) |
| USA | 11 (12%) | 10 (11%) |
| Baseline ECOG performance status | ||
| 0 | 46 (49%) | 33 (38%) |
| 1 | 47 (51%) | 54 (62%) |
| ISS stage | ||
| I | 39 (42%) | 28 (32%) |
| II | 39 (42%) | 42 (48%) |
| III | 15 (16%) | 17 (20%) |
| VGPR or better at enrolment | 82 (88%) | 80 (92%) |
| Cytogenetic profile | ||
| Standard risk | 72 (77%) | 69 (79%) |
| High risk* | 21 (23%) | 18 (21%) |
| Time from autologous stem cell transplantation, days | 92.0 (74.5-114.5) | 98.0 (80.0-124.0) |
| Type of induction | ||
| Bortezomib, thalidomide, and dexamethasone | 64 (69%) | 53(61%) |
| Bortezomib, cyclophosphamide, and dexamethasone | 14 (15%) | 17 (20%) |
| Other† | 15 (16%) | 17 (20%) |
| Previous treatment | ||
| Lenalidomide-containing regimen | 10 (11%) | 11 (13%) |
| Carfilzomib-containing regimen | 4 (4%) | 5 (6%) |
| Number of induction regimens | ||
| One | 86 (92%) | 82 (94%) |
| Two | 7 (8%) | 5 (6%) |
Data are median (IQR) or n (%). ECOG=Eastern Cooperative Oncology Group. ISS=International Staging System. VGPR=very good partial response.
Presence of del(13)(q14), t(4;14)(p16;q32), t(14;16)(q32;q23), del(17)(p13.1), or hypodiploidy.
Other induction regimens included carfilzomib, lenalidomide, and dexamethasone; bortezomib and dexamethasone; bortezomib, thalidomide, dexamethasone, cisplatin, doxorubicin, cyclophosphamide, and etoposide; bortezomib, doxorubicin, and dexamethasone; and bortezomib, lenalidomide, and dexamethasone.
After cycle eight, 41 (44%) of 93 patients in the intention-to-treat population were switched from carfilzomib, lenalidomide, and dexamethasone to lenalidomide alone. Two (5%) of these 41 patients had high-risk cytogenetics and four (10%) did not meet minimal residual disease negativity criteria per IMWG (had less than a complete response). Hence, 35 (85%) patients had standard-risk cytogenetics and minimal residual disease negativity. The median duration of follow-up from randomisation was 33·8 months (IQR 20·9–42·9). At the data cutoff on Dec 31, 2021, in addition to one patient in each group who withdrew before treatment, 38 (41%) of 93 patients in the carfilzomib, lenalidomide, and dexamethasone group and 50 (57%) of 87 in the lenalidomide group discontinued treatment mainly due to disease progression and adverse events (figure 1). Dose reductions of any drug were observed in 32 (34%) patients in the carfilzomib, lenalidomide, and dexamethasone group and 35 (40%) patients in the lenalidomide group (appendix p 9).
For the primary endpoint analysis, the proportional hazard assumption was met (data not shown). In the intention-to-treat population, median progression-free survival was 59·1 months (95% CI 54·8–not estimable [NE]; events reported for 21 [23%] of 93 patients) in the carfilzomib, lenalidomide, and dexamethasone group versus 41·4 months (33·2–65·4; events reported for 38 [44%] of 87 patients) in the lenalidomide group (hazard ratio [HR] 0·51 [95% CI 0·31–0·86]; p=0·012; figure 2). In a post-hoc subgroup analysis, progression-free survival in patients with standard-risk disease was longer with carfilzomib, lenalidomide, and dexamethasone than with lenalidomide alone (median not reached [54·8–NE] vs 65·4 months [33·2–65·4]; HR 0·37 [0·20–0·70]; appendix p 3). In a post-hoc landmark analysis of patients with standard-risk cytogenetics and no detectable minimal residual disease after cycle six (35 [38%] patients meeting these criteria were switched from carfilzomib, lenalidomide, and dexamethasone to lenalidomide alone as of cycle nine and 20 [23%] in lenalidomide group continued lenalidomide), a progression-free survival benefit was observed in the carfilzomib, lenalidomide, and dexamethasone group (median not reached [NE–NE] vs not reached [15·7–NE]; HR 0·25 [0·08–0·84]; appendix p 4). A post-hoc forest plot of progression-free survival stratified by baseline characteristics is shown in figure 3.
Figure 2: Kaplan-Meier estimates of progression-free survival (intention-to-treat population).
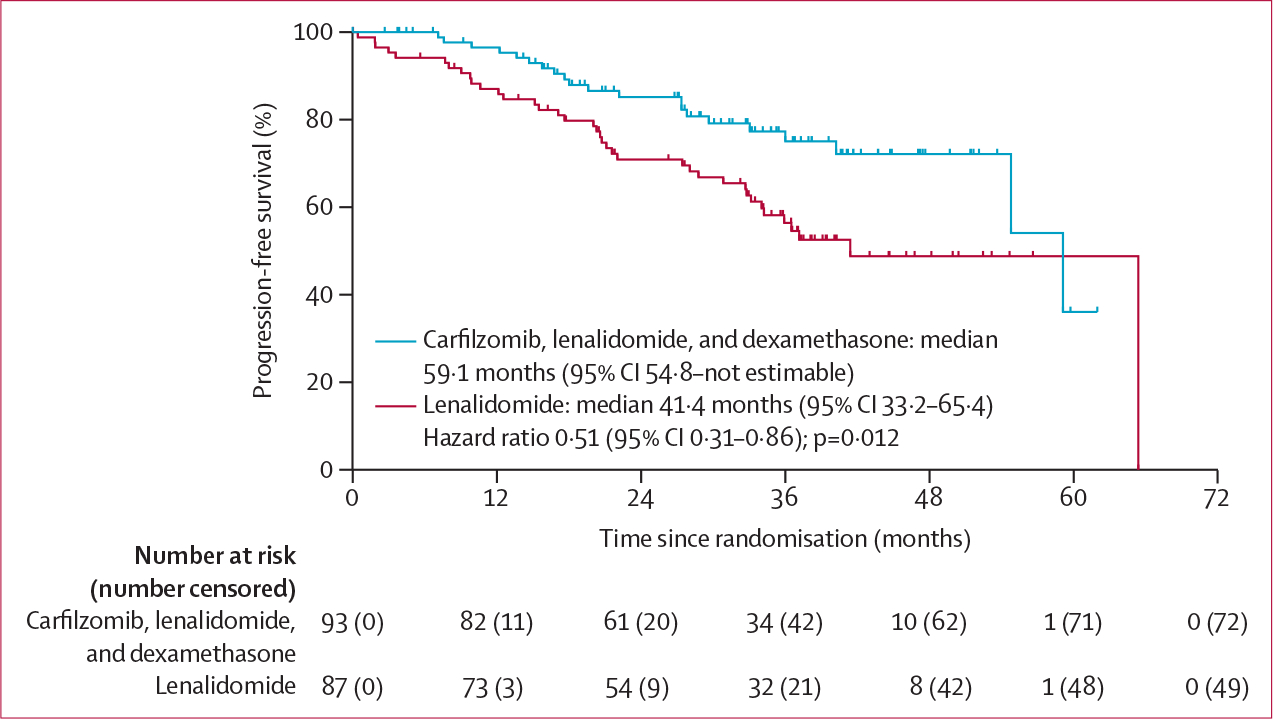
Figure 3: Post-hoc subgroup analysis of progression-free survival.
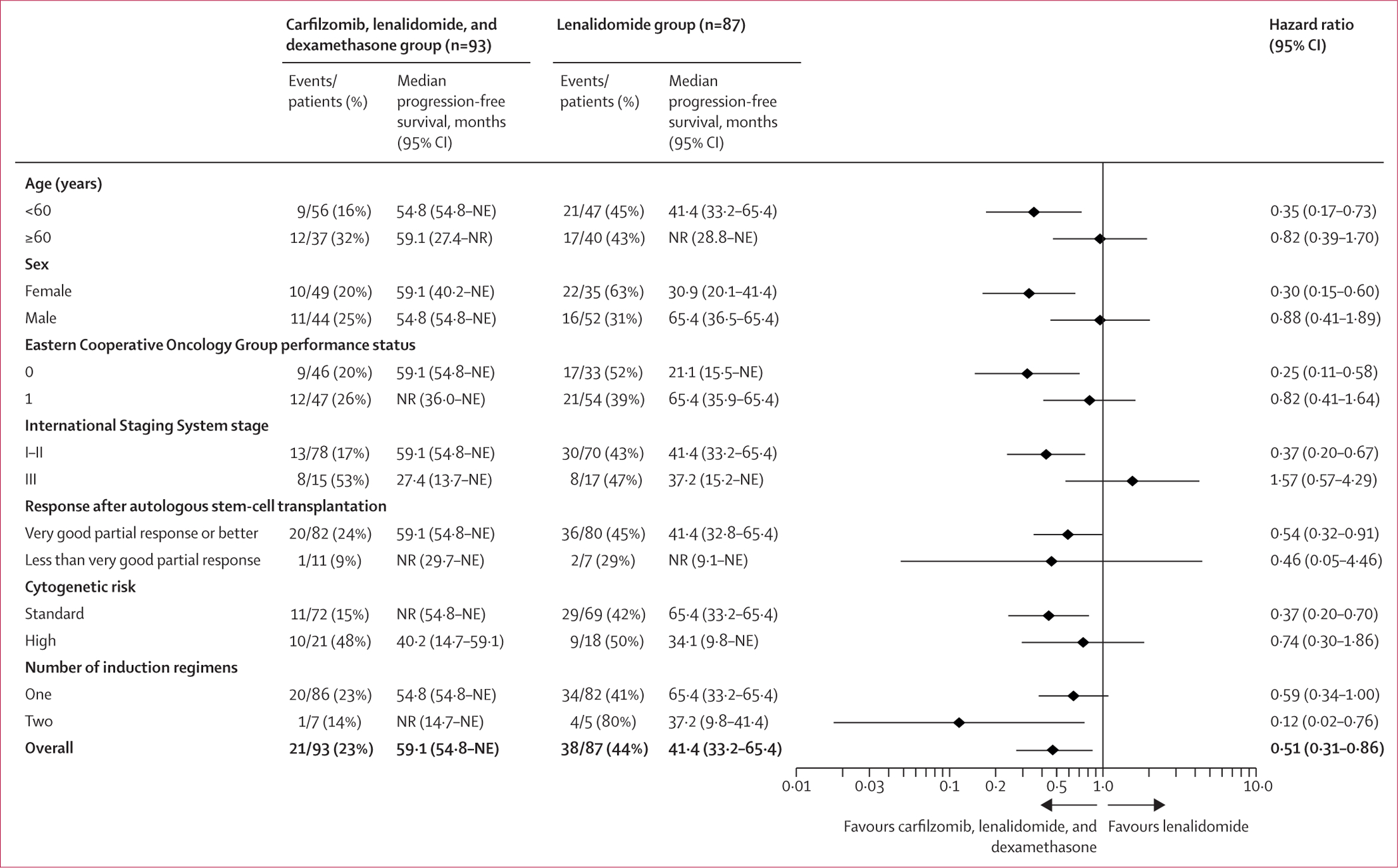
NE=not estimable. NR=not reached.
Overall survival was not significantly different between treatment groups (HR 0·83 [95% CI 0·35–2·00]; p=0·68; median not reached [NE–NE], nine patients [10%] died in the carfilzomib, lenalidomide, and dexamethasone group vs 61·8 months [61·8–NE], 11 [13%] died in the lenalidomide group; appendix p 2).
At the data cutoff, 71 (76% [95% CI 66–85]) patients in the carfilzomib, lenalidomide, and dexamethasone group versus 62 (71% [61–80]) in the lenalidomide group reached complete response or better, but no significant difference was observed in terms of depth of response (p=0·71; data not shown). Improvement in depth of response after 6 months compared with baseline assessment after autologous stem-cell transplantation, was observed in 50 (54%) versus 38 (44%) patients (p=0·18; appendix p 7). Patients without trackable clonotype by NGS and unsuccessful assessment by flow cytometry were excluded from the minimal residual disease analysis (two [2%] patients in the carfilzomib, lenalidomide, and dexamethasone group and three [3%] in the lenalidomide group). The rate of minimal residual disease negativity evaluated after cycle six, according to the IMWG criteria,18 was higher in the carfilzomib, lenalidomide, and dexamethasone group (48 [53%] of 91) than in the lenalidomide group in all evaluable patients (26 [31%] of 84; p=0·0035; appendix p 5). Post-hoc analysis of minimal residual disease after cycle six in standard-risk patients and rates assessed by NGS are shown in the appendix (pp 5–6). Minimal residual disease rates stratified by baseline characteristics are also shown in the appendix (p 10).
Adverse events in the safety population are summarised in table 2. Grade 1–2 adverse events were slightly more frequent in the carfilzomib, lenalidomide, and dexamethasone group (86 [93%] of 92 vs 71 [83%] of 86 in the lenalidomide group), including higher proportions of lymphopenia (11 [12%] vs six [7%]) and anaemia (11 [12%] vs one [1%]) for haematological toxic effects and upper respiratory tract infections (51 [55%] vs 36 [42%]), fever (16 [17%] vs two [2%]), and diarrhoea (27 [29%] vs 17 [20%]) for non-haematological toxic effects. Grade 3 and 4 adverse events occurred in 70 (76%) patients in the carfilzomib, lenalidomide, and dexamethasone group versus 63 (73%) in the lenalidomide group and were generally balanced between groups. The most common grade 3 and 4 adverse events were neutropenia (44 [48%] vs 52 [60%]), thrombocytopenia (12 [13%] vs six [7%]), and lower respiratory tract infections (seven [8%] vs one [1%]; appendix pp 11–12). Adverse events that led to treatment discontinuation occurred in four (4%) patients in the carfilzomib, lenalidomide, and dexamethasone group and ten (12%) in the lenalidomide group (figure 1). Serious adverse events were reported in 28 (30%) patients in the carfilzomib, lenalidomide, and dexamethasone group and 19 (22%) in the lenalidomide group, with lower respiratory tract infections as the most common serious adverse event in both groups (11 [12%] vs three [3%]; appendix p 13). One treatment-related adverse event led to death (respiratory failure due to severe pneumonia) in the carfilzomib, lenalidomide, and dexamethasone group. There were three second primary malignancies reported (one in the carfilzomib, lenalidomide, and dexamethasone group vs two in the lenalidomide group).
Table 2:
Summary of adverse events in all patients who received treatment (safety population)
| Carfilzomib, lenalidomide, and dexamethasone (n=92) |
Lenalidomide (n=86) |
|||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
|
| ||||||
| Overall | 86 (93%) | 57 (62%) | 13 (14%) | 71 (83%) | 60 (70%) | 3 (3%) |
| Haematological | 28 (30%) | 46 (50%) | 11 (12%) | 16 (19%) | 51 (59%) | 2 (2%) |
| Anaemia | 11 (12%) | 3 (3%) | 1 (1%) | 1 (1%) | 0 | 0 |
| Neutropenia | 4 (4%) | 36 (39%) | 8 (9%) | 3 (3%) | 50 (58%) | 2 (2%) |
| Thrombocytopenia | 11 (12%) | 8 (9%) | 4 (4%) | 8 (9%) | 5 (6%) | 1 (1%) |
| Lymphopenia | 11 (12%) | 5 (5%) | 0 | 6 (7%) | 2 (2%) | 0 |
| Non-haematological | 85 (92%) | 27 (29%) | 2 (2%) | 71 (83%) | 26 (30%) | 1 (1%) |
| Diarrhoea | 27 (29%) | 1 (1%) | 0 | 17 (20%) | 2 (2%) | 0 |
| Nausea or vomiting | 12 (13%) | 0 | 0 | 3 (3%) | 0 | 0 |
| Fatigue | 10 (11%) | 0 | 0 | 4 (5%) | 0 | 0 |
| Fever | 16 (17%) | 0 | 0 | 2 (2%) | 0 | 0 |
| Pain | 13 (14%) | 0 | 0 | 11 (13%) | 0 | 0 |
| Influenza-like symptoms | 11 (12%) | 1 (1%) | 0 | 5 (6%) | 1 (1%) | 0 |
| Febrile neutropenia | 0 | 3 (3%) | 1 (1%) | 0 | 5 (6%) | 0 |
| Upper respiratory tract infection | 51 (55%) | 5 (5%) | 0 | 36 (42%) | 3 (3%) | 0 |
| Lower respiratory tract infection | 8 (9%) | 6 (7%) | 1 (1%)* | 4 (5%) | 1 (1%) | 0 |
| Aminotransferase increase | 20 (22%) | 5 (5%) | 0 | 9 (10%) | 0 | 0 |
| Electrolyte abnormalities | 11 (12%) | 2 (2%) | 0 | 6 (7%) | 2 (2%) | 0 |
| Hyperglycaemia | 5 (5%) | 2 (2%) | 0 | 3 (3%) | 0 | 0 |
| Skin infection | 5 (5%) | 1 (1%) | 0 | 9 (10%) | 1 (1%) | 0 |
| Neuropathy | 8 (9%) | 0 | 0 | 10 (12%) | 1 (1%) | 0 |
| Thromboembolic events | 1 (1%) | 1 (1%) | 0 | 1 (1%) | 2 (2%) | 0 |
| Second primary malignancies† | 1 (1%) | 0 | 0 | 2 (2%) | 0 | 0 |
Data are n (%). Grade 1–2 adverse events in 10% or more patients and grade 3–4 in two or more patients in either treatment group are shown.
There was a treatment-related death associated with this adverse event (grade 5). No other grade 5 adverse events were recorded.
Second primary malignancies are not graded.
Discussion
In this interim analysis of the ATLAS study, we showed that minimal residual disease-directed and risk-adapted treatment with carfilzomib, lenalidomide, and dexamethasone for 8–36 cycles might offer a progression-free survival benefit compared with lenalidomide alone as maintenance therapy after autologous stem-cell transplantation in patients with newly diagnosed multiple myeloma. To our knowledge, this study is the first randomised phase 3 trial suggesting superiority of an alternative maintenance therapy to lenalidomide alone.
Our results are in line with the randomised phase 2 FORTE trial, in which the addition of carfilzomib to lenalidomide as maintenance therapy improved progression-free survival compared with lenalidomide alone in patients who completed four cycles of carfilzomib-based consolidation after autologous stem-cell transplantation or 12 cycles of carfilzomib-based induction in those who deferred transplantation.16 Despite differences, both studies showed that extended treatment with carfilzomib and lenalidomide with or without dexamethasone after initial therapy can improve progression-free survival in transplantation candidates with newly diagnosed multiple myeloma. Indirectly, our results are also in line with results from the phase 3 CASSIOPEIA21 and phase 2 GRIFFIN trials.22 Both of these studies indicate a progression-free survival benefit with extended daratumumab-based multidrug treatment after autologous stem-cell transplantation, although the comparator group in CASSIOPEIA was observation only and no second randomisation was performed in GRIFFIN. Although these two studies evaluated maintenance strategies with defined induction courses, this ATLAS study enrolled patients after any induction regimen, making our results possibly more generalisable to current clinical practice. Notably, most patients in our study received triplet induction with a proteasome inhibitor, an immunomodulatory drug, and dexamethasone, which reflected the standard of care for induction at the time of enrolment.
In this interim analysis, a reduction in the risk of disease progression or death by adding carfilzomib and dexamethasone to lenalidomide was observed for all enrolled patients and in subsets of patients younger than 60 years, females, and those with ISS stage I–II disease, ECOG performance status 0, very good partial response or better after transplantation, two induction regimens, and standard cytogenetic risk. The effects on other subsets, including subsets of patients with high-risk cytogenetics and ISS stage III disease, were not statistically significant. These findings might appear counterintuitive, especially considering the results of the phase 2 trial of carfilzomib, lenalidomide, and dexamethasone with autologous stem-cell transplantation15 and the FORTE trial,16 which indicated a progression-free survival benefit with extended carfilzomib and lenalidomide with or without dexamethasone for a subset of high-risk patients after autologous stem-cell transplantation. Different induction regimens between studies, an insufficiently small number of events in this subset analysis to draw definite conclusions, and a study design not powered to answer these questions might explain our results. Including del(13)(q14) or hypodiploidy in the high-risk subset (both no longer considered high-risk features in the mSMART guidelines by the Mayo Clinic) should not affect our results because none of the enrolled patients had either of these two as a sole lesion. Longer follow-up, which would include additional evaluations of minimal residual disease at later timepoints, might provide a more definitive assessment for patients with these characteristics.
The ATLAS study design included minimal residual disease-directed and risk-adapted duration of carfilzomib, lenalidomide, and dexamethasone treatment, which was based on observations from previous studies.15,23 The post-hoc landmark analysis showed that patients without high-risk disease and no detectable minimal residual disease have a reduced risk of disease progression or death when given carfilzomib, lenalidomide, and dexamethasone for eight cycles and then lenalidomide versus those who received lenalidomide alone. Planned assessment of minimal residual disease at later timepoints, especially sustained minimal residual disease negativity in both groups, might provide additional information to improve the evaluation of outcomes. To fully validate this approach, a randomised comparison of patients who receive carfilzomib, lenalidomide, and dexamethasone deescalation with those who have the same characteristics but are treated for 36 cycles with carfilzomib, lenalidomide, and dexamethasone would be needed. To date, results seem to support an adaptive approach to prevent overtreatment of a considerable number of patients. Minimal residual disease-guided treatment deescalation or cessation in patients with standard-risk disease or with only one risk factor showed progression-free survival rates of 91% for standard risk (97% for one risk factor) at 2 years in the MASTER trial24 and 86% for standard risk at 3 years in a phase 2 study by Derman and colleagues.25
An understandable concern might arise that extended treatment with carfilzomib, lenalidomide, and dexamethasone after transplantation might be too toxic or difficult to sustain compared with the generally well tolerated and more convenient treatment with lenalidomide. Analysis of adverse events indicates that the incidence of grade 1 and 2 events, including the rates of cardiac and vascular events, was slightly higher in the carfilzomib, lenalidomide, and dexamethasone group. There was also slightly higher incidence of grade 3 and 4 haematological events, infections, and serious adverse events in the carfilzomib, lenalidomide, and dexamethasone group compared with the lenalidomide group. However, we observed a similar rate of discontinuation due to adverse events and low but similar rates of dose modifications in both groups. Assessing to what extent the shortening of carfilzomib, lenalidomide, and dexamethasone treatment in patients with standard-risk and minimal residual disease negativity at cycle six contributed to limiting toxicities was not possible with this study design. Overall, the toxicity and efficacy results indicate a favourable therapeutic index for carfilzomib, lenalidomide, and dexamethasone, which might be considered when making informed decisions about treatment after autologous stem-cell transplantation.
The main limitation of our results is the interim nature in the absence of a prespecified stopping rule for the efficacy evaluation. This interim analysis was prompted by results from the FORTE trial16 and investigators’ impression that carfilzomib, lenalidomide, and dexamethasone treatment generates superior results to lenalidomide at the time when the study reached 59 (61%) of the expected 96 events, which was deemed justifiable for this analysis. Other limitations include the low number of patients in both groups, which was sufficient for the evaluation of primary progression-free survival but not powered for statistical comparisons among subgroups, and the short follow-up. Using two different methods for minimal residual disease assessment can also be perceived as a limitation, with the flow method used when NGS was not available. As per the study design, minimal residual disease results were required for de-escalation of treatment after cycle eight, provided that the IMWG criteria were met, regardless of the method used. Therefore, using two methods of minimal residual disease assessment at cycle six should have minimal effect on the results. Not including a cohort of patients with standard-risk disease and minimal residual disease negativity at cycle six who continued carfilzomib, lenalidomide, and dexamethasone treatment for 36 cycles also limits the evaluation of this approach. Therefore, these findings need to be interpretated with caution while awaiting the final analysis planned at 4 years of follow-up from the time of enrolment of the last patient.
In conclusion, this interim analysis provides support for considering carfilzomib, lenalidomide, and dexamethasone therapy in patients with newly diagnosed multiple myeloma who completed any induction regimen followed by autologous stem-cell transplantation. The treatment regimen and outcomes require further confirmation with a longer follow-up of this ongoing trial (NCT02659293).
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed for articles published between database inception, and Aug 1, 2022, using the search terms “myeloma”, “maintenance”, “transplant”, or “lenalidomide”. Several studies investigated maintenance strategies for patients with multiple myeloma after autologous stem-cell transplantation. Continuous lenalidomide maintenance has been established as the standard of care in this setting. Bortezomib, ixazomib, and daratumumab showed an improvement of progression-free survival after transplantation compared with placebo or no treatment, but none of these therapies have been directly compared with lenalidomide maintenance. The value of an alternative to maintenance therapy with lenalidomide alone or as a combination, initiated after autologous stem-cell transplantation or as part of an extended treatment schedule (including induction or consolidation, or both, after autologous stem-cell transplantation followed by maintenance), is being investigated in several ongoing studies (NCT02495922, NCT03617731, NCT04071457, NCT04786028, and NCT03901963). Carfilzomib, lenalidomide, and dexamethasone is indicated for the treatment of patients with relapsed or refractory multiple myeloma and recommended in some guidelines for patients with newly diagnosed myeloma. The use of carfilzomib, lenalidomide, and dexamethasone for patients with newly diagnosed myeloma as part of extended treatment before and after autologous stem-cell transplantation (NCT01816971), provided rationale for using this regimen as maintenance therapy in our study. A short course of bortezomib, lenalidomide, and dexamethasone as consolidation therapy after autologous stem-cell transplantation improved progression-free survival compared with lenalidomide alone in the phase 3 EMN02/HOVON95 trial but not in the STaMINA trial. The phase 2 FORTE trial, published in 2021, showed an improvement of progression-free survival after carfilzomib was added to lenalidomide compared with lenalidomide alone in patients who completed carfilzomib, lenalidomide, and dexamethasone or carfilzomib, cyclophosphamide, and dexamethasone induction and consolidation before and after autologous stem-cell transplantation or extended induction and consolidation without autologous stem-cell transplantation.
Added value of this study
We report preliminary results of the ATLAS study, which to our knowledge is the first randomised phase 3 study to compare the efficacy and safety of maintenance therapy with carfilzomib, lenalidomide, and dexamethasone versus lenalidomide alone (standard of care) after autologous stem-cell transplantation. These early results suggest that ATLAS will meet the primary endpoint of improved progression-free survival. As expected, we observed a slightly greater number of toxic effects (but an acceptable level of safety and tolerability) in the carfilzomib, lenalidomide, and dexamethasone group than in the lenalidomide alone group, which indicates an overall favourable therapeutic index. Additionally, using a minimal residual disease-directed and risk-adapted design, we show that to reduce the number of toxic effects with extended carfilzomib, lenalidomide, and dexamethasone treatment, patients with standard-risk disease who reach minimal residual disease negativity after six cycles of treatment can deescalate to maintenance with lenalidomide alone while maintaining the benefit conferred by carfilzomib, lenalidomide, and dexamethasone.
Implications of all the available evidence
When interpreted together with the FORTE trial, our results further support a role of carfilzomib and lenalidomide with or without dexamethasone as the possible new standard of care in maintenance therapy after autologous stem-cell transplantation. Additionally, we show that cytogenetic risk or minimal residual disease-guided therapy (or both) might contribute to limiting toxicity while maintaining efficacy, contributing to further development of treatment strategies with some results already reported (MASTER trial and the NCT02969837 trial) and several ongoing phase 3 trials (with risk-guided and minimal residual disease-guided study designs). Similar to the key lenalidomide maintenance studies, this study evaluated maintenance in patients who completed any induction regimen, making the results more generalisable for the selection of maintenance therapy after autologous stem-cell transplantation, which could be implemented in a clinical setting. Longer follow-up of ATLAS will allow confirmation of these results and assessment of sustained minimal residual disease negativity, efficacy in patients with high-risk disease, and overall survival.
Acknowledgments
This study was funded by Amgen and Celgene (Bristol Myers Squibb). The University of Chicago (Chicago, IL, USA) sponsored the ATLAS trial and delegated this role to the Polish Myeloma Consortium to conduct the study in Poland. Medical writing assistance was provided by Marcin Balcerzak of Medink (Mysiadlo, Poland) and funded by the Polish Myeloma Consortium. We thank the patients and their families, study coordinators, and support staff.
Footnotes
Declaration of interests
DD reports speaker honoraria and participation in advisory boards for Amgen and Celgene (Bristol Myers Squibb) and had conference fees paid by Amgen. TW reports honoraria from AbbVie, Amgen, BeiGene, Celgene (Bristol Myers Squibb), Gilead, GlaxoSmithKline, Janssen-Cilag, Novartis, Pfizer, Roche, and Takeda; conference fees or travel support (or both) from AbbVie, Amgen, Celgene (Bristol Myers Squibb), Gilead, GlaxoSmithKline, Janssen-Cilag, Pfizer, Roche, and Takeda; and advisory board participation for AbbVie, Amgen, BeiGene, Celgene (Bristol Myers Squibb), Gilead, GlaxoSmithKline, Janssen-Cilag, Pfizer, Roche, Novartis, and Takeda. KJ reports grants paid to his institution and payment for expert testimony from Janssen; honoraria from Amgen, Celgene (Bristol Myers Squibb), GlaxoSmithKline, Janssen, Sandoz, and Takeda; financial support for attending meetings or travel support (or both) from Amgen, Celgene, and Janssen; and consulting and advisory board participation for Amgen, Celgene (Bristol Myers Squibb), Janssen, Sandoz, and Takeda. TK reports honoraria from AbbVie and Celgene (Bristol Myers Squibb) and financial support for attending meetings or travel support (or both) from Sanofi. AD-S reports honoraria for presentations and lectures from Amgen and Celgene (Bristol Myers Squibb); and financial support for attending meetings or travel support (or both) and advisory board participation from Celgene (Bristol Myers Squibb). KrG reports honoraria from Amgen and Celgene (Bristol Myers Squibb). AN reports fees for lectures and presentations from Amgen, Celgene (Bristol Myers Squibb), and Janssen; and financial support for attending meetings or travel support (or both) from Amgen, Celgene (Bristol Myers Squibb), Janssen, and Teva. BP reports grants or contracts from AbbVie, Amgen, Celgene (Bristol Myers Squibb), and Janssen; consultation fees from AbbVie, Roche, and Sandoz; honoraria for lectures and presentations from AbbVie, Amgen, Celgene (Bristol Myers Squibb), and Janssen; and financial support for attending meetings or travel support (or both) from AbbVie and Celgene (Bristol Myers Squibb). JR reports speaker honoraria and advisory board participation from Amgen, Celgene (Bristol Myers Squibb), and Janssen. LU-Z reports financial support for attending meetings or travel support (or both) from Janssen. AK reports payment for clinical trial participation from Janssen. JMZ reports grants from Celgene (Bristol Myers Squibb); financial support for attending meetings or travel support (or both) from AbbVie and Takeda; and participation on a data safety monitoring board or advisory board for Amgen, Celgene (Bristol Myers Squibb), Roche, and Takeda. JW reports personal and institutional grants from GlaxoSmithKline, Novartis, and Roche; and personal consulting, lecture and presentation fees, speaker bureaus, and educational events fees from AbbVie, Gilead, Novartis, Roche, and Takeda. DM reports lecture and presentation fees from Amgen and Janssen. TR reports research grants from Amgen and Celgene (Bristol Myers Squibb). OBL reports participation on an advisory board for MorphoSys. JAZ reports an institutional grant from Celgene (Bristol Myers Squibb); personal consulting fees from Alnylam, Celgene (Bristol Myers Squibb), Janssen, Prothena, and Regeneron; and participation on an advisory board for Takeda. KeG reports personal payment for the statistical design of this study and consulting fees related to other studies from the University of Chicago. BAD reports consulting fees from COTA Healthcare, Janssen, and Sanofi; payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from MJH Life Sciences and Plexus Communications. AJJ reports consulting fees and honoraria for lectures and presentations from AbbVie, Amgen, Celgene (Bristol Myers Squibb), Gracell, GlaxoSmithKline, Janssen, and Sanofi-Aventis. All other authors declare no competing interests.
Contributor Information
Dominik Dytfeld, Poznan University of Medical, Sciences, Poznan, Poland.
Tomasz Wróbel, Wroclaw, Medical University, Wroclaw, Poland.
Krzysztof Jamroziak, Institute of Hematology, and Blood Transfusion, Warsaw, Poland.
Tadeusz Kubicki, Poznan University of Medical, Sciences, Poznan, Poland.
Paweł Robak, Medical University of Lodz, Lodz, Poland.
Adam Walter-Croneck, Medical University of Lublin, Lublin, Poland.
Jarosław Czyż, Department of Hematology, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University, Torun, Poland.
Agata Tyczyńska, Department of Hematology and Transplantology, Medical University of Gdansk, Gdansk, Poland.
Agnieszka Druzd-Sitek, Maria Sklodowska-Curie National, Research Institute of Oncology, Warsaw, Poland.
Krzysztof Giannopoulos, Department of Experimental Hematooncology, Medical University of Lublin, Lublin, Poland.
Adam Nowicki, Poznan University of Medical, Sciences, Poznan, Poland.
Tomasz Szczepaniak, Poznan University of Medical, Sciences, Poznan, Poland.
Anna Łojko-Dankowska, Poznan University of Medical, Sciences, Poznan, Poland.
Magdalena Matuszak, Poznan University of Medical, Sciences, Poznan, Poland.
Lidia Gil, Poznan University of Medical, Sciences, Poznan, Poland.
Bartosz Puła, Institute of Hematology, and Blood Transfusion, Warsaw, Poland.
Justyna Rybka, Wroclaw, Medical University, Wroclaw, Poland.
Maciej Majcherek, Wroclaw, Medical University, Wroclaw, Poland.
Lidia Usnarska-Zubkiewicz, Wroclaw, Medical University, Wroclaw, Poland.
Łukasz Szukalski, Department of Hematology, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University, Torun, Poland.
Agnieszka Końska, Institute of Hematology, and Blood Transfusion, Warsaw, Poland.
Jan Maciej Zaucha, Department of Hematology and Transplantology, Medical University of Gdansk, Gdansk, Poland.
Jan Walewski, Maria Sklodowska-Curie National, Research Institute of Oncology, Warsaw, Poland.
Damian Mikulski, Medical University of Lodz, Lodz, Poland.
Olga Czabak, Medical University of Lublin, Lublin, Poland.
Tadeusz Robak, Medical University of Lodz, Lodz, Poland.
Oscar B Lahoud, Memorial Sloan-Kettering, Cancer Institute, New York, NY, USA.
Jeffrey A Zonder, Karmanos Cancer Institute, Detroit, MI, USA.
Kent Griffith, University of Michigan, Ann Arbor, MI, USA.
Andrew Stefka, University of Chicago Medical Center, Chicago, IL, USA.
Ajay Major, University of Chicago Medical Center, Chicago, IL, USA; University of Colorado School of Medicine, Aurora, CO, USA.
Benjamin A Derman, University of Chicago Medical Center, Chicago, IL, USA.
Andrzej J Jakubowiak, University of Chicago Medical Center, Chicago, IL, USA.
Data sharing
Data sharing requests will be considered on a case-by-case basis. Requests by academic study groups for deidentified patient data with the intent to achieve aims of the original proposal can be forwarded to the corresponding author.
References
- 1.Attal M, Harousseau J-L, Leyvraz S, et al. Maintenance therapy with thalidomide improves survival in patients with multiple myeloma. Blood 2006; 108: 3289–94. [DOI] [PubMed] [Google Scholar]
- 2.Jackson GH, Davies FE, Pawlyn C, et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2019; 20: 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366: 1770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med 2012; 366: 1782–91. [DOI] [PubMed] [Google Scholar]
- 5.McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol 2017; 35: 3279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivaraj D, Green MM, Li Z, et al. Outcomes of maintenance therapy with bortezomib after autologous stem cell transplantation for patients with multiple myeloma. Biol Blood Marrow Transplant 2017; 23: 262–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosiñol L, Oriol A, Teruel AI, et al. Bortezomib and thalidomide maintenance after stem cell transplantation for multiple myeloma: a PETHEMA/GEM trial. Leukemia 2017; 31: 1922–27. [DOI] [PubMed] [Google Scholar]
- 8.Dimopoulos MA, Gay F, Schjesvold F, et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2019; 393: 253–64. [DOI] [PubMed] [Google Scholar]
- 9.Sonneveld P, Dimopoulos MA, Beksac M, et al. Consolidation and maintenance in newly diagnosed multiple myeloma. J Clin Oncol 2021; 39: 3613–22. [DOI] [PubMed] [Google Scholar]
- 10.Stadtmauer EA, Pasquini MC, Blackwell B, et al. Autologous transplantation, consolidation, and maintenance therapy in multiple myeloma: results of the BMT CTN 0702 trial. J Clin Oncol 2019; 37: 589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol 2020; 7: e456–68. [DOI] [PubMed] [Google Scholar]
- 12.Hari P, Pasquini MC, Stadtmauer EA, et al. Long-term follow-up of BMT CTN 0702 (STaMINA) of postautologous hematopoietic cell transplantation (autoHCT) strategies in the upfront treatment of multiple myeloma (MM). Proc Am Soc Clin Oncol 2020; 38: 8506 (abstr). [Google Scholar]
- 13.Rosinol L, Oriol A, Ríos Tamayo R, et al. Ixazomib plus lenalidomide/dexamethasone (IRd) versus lenalidomide/dexamethasone (Rd) maintenance after autologous stem cell transplant in patients with newly diagnosed multiple myeloma: results of the Spanish GEM2014MAIN trial. Blood 2021; 138 (suppl 1): 466. [Google Scholar]
- 14.Moreau P, Hulin C, Perrot A, et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): an open-label, randomised, phase 3 trial. Lancet Oncol 2021; 22: 1378–90. [DOI] [PubMed] [Google Scholar]
- 15.Jasielec JK, Kubicki T, Raje N, et al. Carfilzomib, lenalidomide, and dexamethasone plus transplant in newly diagnosed multiple myeloma. Blood 2020; 136: 2513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gay F, Musto P, Rota-Scalabrini D, et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. Lancet Oncol 2021; 22: 1705–20. [DOI] [PubMed] [Google Scholar]
- 17.Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20: 1467–73. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016; 17: e328–46. [DOI] [PubMed] [Google Scholar]
- 19.Kalina T, Flores-Montero J, van der Velden VHJ, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012; 26: 1986–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa LJ, Derman BA, Bal S, et al. International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials. Leukemia 2021; 35: 18–30. [DOI] [PubMed] [Google Scholar]
- 21.Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet 2019; 394: 29–38. [DOI] [PubMed] [Google Scholar]
- 22.Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood 2020; 136: 936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paiva B, Gutiérrez NC, Rosiñol L, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood 2012; 119: 687–91. [DOI] [PubMed] [Google Scholar]
- 24.Costa LJ, Chhabra S, Medvedova E, et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. J Clin Oncol 2022; 40: 2901–12. [DOI] [PubMed] [Google Scholar]
- 25.Derman BA, Kansagra A, Zonder J, et al. Elotuzumab and weekly carfilzomib, lenalidomide, and dexamethasone in patients with newly diagnosed multiple myeloma without transplant intent: a phase 2 measurable residual disease-adapted study. JAMA Oncol 2022; 8: 1278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing requests will be considered on a case-by-case basis. Requests by academic study groups for deidentified patient data with the intent to achieve aims of the original proposal can be forwarded to the corresponding author.