

Abstract
Use of the in vivo alpha-synuclein preformed fibril (α-syn PFF) model of synucleinopathy is gaining popularity among researchers aiming to model Parkinson’s disease synucleinopathy and nigrostriatal degeneration. The standardization of α-syn PFF generation and in vivo application is critical in order to ensure consistent, robust α-syn pathology. Here, we present a detailed protocol for the generation of fibrils from monomeric α-syn, post-fibrilization quality control steps, and suggested parameters for successful neurosurgical injection of α-syn PFFs into rats or mice. Starting with monomeric α-syn, fibrilization occurs over a 7-day incubation period while shaking at optimal buffer conditions, concentration, and temperature. Post-fibrilization quality control is assessed by the presence of pelletable fibrils via sedimentation assay, the formation of amyloid conformation in the fibrils with a thioflavin T assay, and electron microscopic visualization of the fibrils. Whereas successful validation using these assays is necessary for success, they are not sufficient to guarantee PFFs will seed α-syn inclusions in neurons, as such aggregation activity of each PFF batch should be tested in cell culture or in pilot animal cohorts. Prior to use, PFFs must be sonicated under precisely standardized conditions, followed by examination using electron microscopy or dynamic light scattering to confirm fibril lengths are within optimal size range, with an average length of 50 nm. PFFs can then be added to cell culture media or used in animals. Pathology detectable by immunostaining for phosphorylated α-syn (psyn; serine 129) is apparent days or weeks later in cell culture and rodent models, respectively.
The goal of this article is to outline the steps required for the generation of fibrils from monomeric alpha-synuclein, subsequent quality control, and use of the preformed fibrils in vivo.
Keywords: Alpha-Synuclein, Monomers, Preformed Fibrils, Parkinson’s Disease, Quality Control, Stereotaxic Surgery
INTRODUCTION:
Parkinson’s disease (PD) is primarily characterized postmortem by two major pathological features: widespread and progressive alpha-synuclein (α-syn) pathology; and nigrostriatal degeneration. Following injection into wildtype mice or rats, α-syn preformed fibrils (PFFs) induce progressive accumulation of pathological α-syn which can result in protracted degeneration of substantia nigra pars compacta (SNpc) dopamine neurons over the course of many months, as well as sensorimotor deficits 1–7. Neurons are exposed to α-syn fibrils, either via direct intracerebral injection or added to the media of cultured neurons. When the PFFs are taken into the neurons, the PFFs act to “seed” the formation of inclusions through templating, and accumulation of endogenous α-syn into phosphorylated inclusions1, 8–10. Inclusions share similar properties to Lewy bodies: containing α-syn phosphorylated at serine 129 (pSyn), ubiquitin, and p62; possess amyloid quaternary structures as shown with positive thioflavin staining; and are resistant to proteinase K digestion 1, 3, 5, 7, 8–13. PFF exposure leads to α-syn inclusion formation in primary and some immortalized neurons in culture, as well as mice, rats, and non-human primates in vivo 1–10, 14. It is important to note that PFFs will not lead to α-syn inclusion formation in all cell culture models and some cultured neurons will seed better than others.
Another important feature of the in vivo α-syn PFF model is the distinct sequential pathological phases that emerge over several months. In rodents, following intrastriatal injection, α-syn inclusion formation generally peaks within the SNpc and many cortical regions within 1–2 months. This aggregation peak is followed by nigrostriatal degeneration ≈2–4 months later 1, 3, 5, 7. These distinct pathological stages provide researchers the platform with which to study and develop strategies that 1) decrease α-syn aggregation, 2) clear already formed α-syn inclusions, and/or 3) prevent subsequent neurodegeneration. The PFF model offers distinct advantages and disadvantages as compared to neurotoxicant, transgenic, and viral vector mediated α-syn overexpression models as previously reviewed 6. The choice of which model or approach to take should be determined by which model best suits the question the investigators are asking.
Although the PFF model has been successfully utilized by many labs, there are still groups which have experienced inconsistencies with generating fibrils and producing consistent α-syn pathology 15. Examples of inconsistencies range from PFFs that produce little or no α-syn pathology, batch to batch seeding efficiency, and even the failure of fibrils to form. Thus, the standardization of α-syn PFF generation and in vivo application is critical in order to allow for accurate interpretations regarding the impact of novel therapeutic interventions. The following protocol outlines the steps required for the generation of PFFs from α-syn monomers, the in vitro quality control of the PFFs once formed, the sonication and measurement of PFFs prior to use, and suggestions to facilitate successful in vivo injection of PFFs into rats or mice.
PROTOCOL:
All methods involving animals have been approved by the Michigan State University Institutional Animal Care and Use Committee (IACUC).
1. Formation of α-synuclein Preformed Fibrils from Monomers (Figure 1)
Figure 1. Methods for the generation of α-syn fibrils.
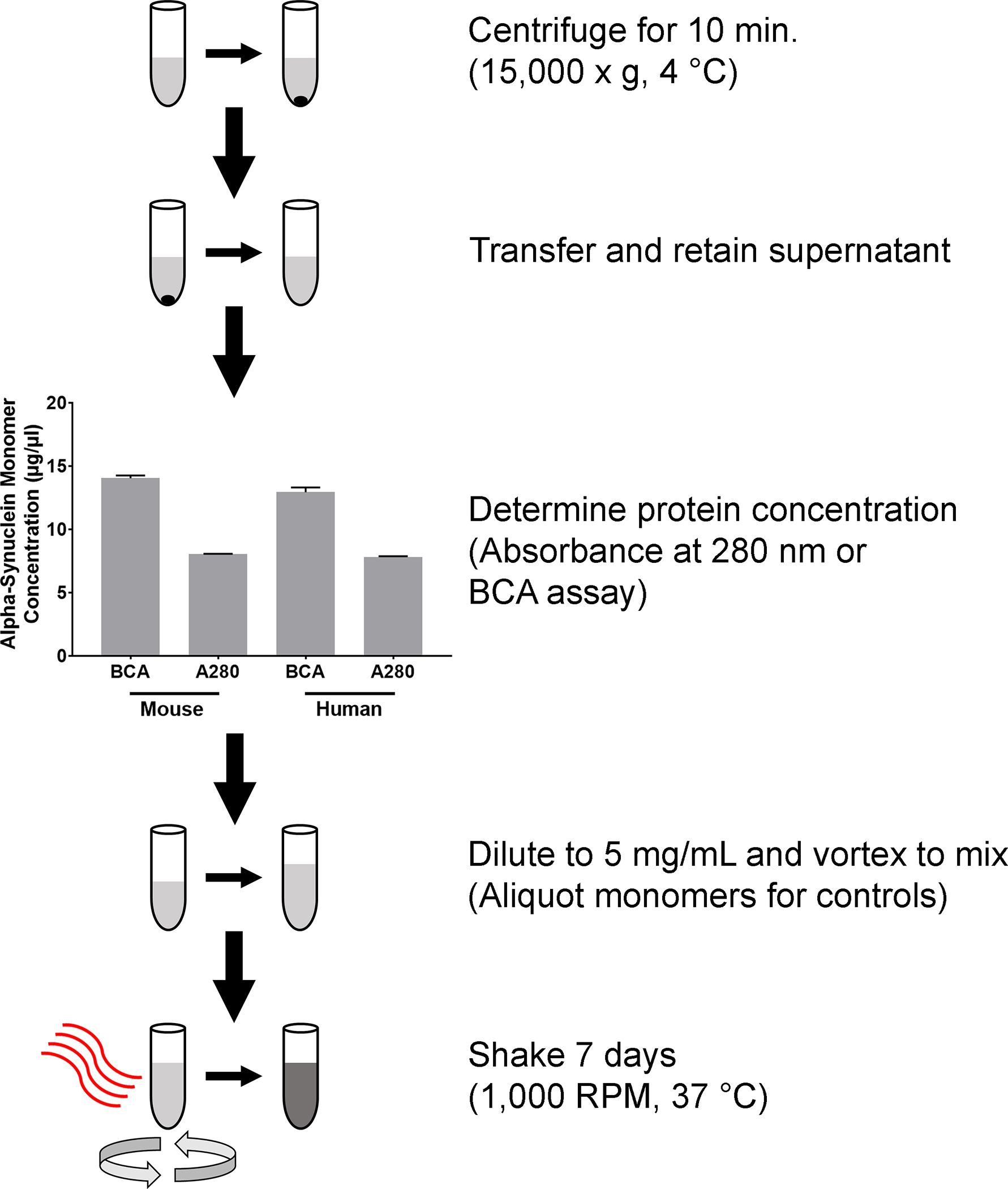
Outline of the steps required to produce fibrils from α-syn monomers. Monomers are centrifuged for 10 min (15,000 × g, at 4 °C). Supernatant is transferred to a clean tube and the protein concentration is determined by either the absorbance at 280 nm, or a BCA assay. Graph shows concentrations from human and mouse α-syn monomers. Columns indicate the group means, error bars represent +1 standard error of the mean. After protein concentration is determined, α-syn monomers are diluted, briefly vortexed and incubated for 37 °C for 7 days, while shaking on an orbital mixer set at 1,000 RPM.
1.1 Thaw α-synuclein monomers on ice, gently resuspend by flicking the tube, and centrifuge at 15,000 × g for 10 min at 4 °C.
Note: α-syn monomer must be specifically formulated for fibrilization. Recombinant monomers can be purchased from commercial sources or generated by protocols on site 4, 10, 15–16. If purchased from commercial sources, the product must state the α-syn monomer is specifically for the generation of fibrils. Regardless of if the monomers are purchased or generated on site, the quality control steps outlined below should be performed with each batch to ensure fibrils have formed and will seed efficiently prior to use in experiments.
1.2 Transfer supernatant to a clean 1.5 mL microcentrifuge tube and record the amount transferred.
Note: Be careful to avoid the pellet, which, if present, will be small.
1.3 Measure the protein concentration of the transferred supernatant by either a standard bicinchoninic acid assay (BCA), or measuring the absorbance at 280 nm with a microvolume spectrophotometer.
Note: The BCA assay is not as accurate for the specific measurement of α-syn and can yield results overestimate protein concentration. As a result, measuring the absorbance at 280 nm is the recommended method for determining protein concentration.
1.3.1.1 To measure with the BCA assay, follow standard BCA protocols and perform with three different dilutions of α-syn monomer.
Note: Suggested dilutions are 1:25, 1:50, and 1:100.
1.3.2.1 To measure with the A280 method, blank the reader with sterile 1x Dulbecco’s phosphate buffered saline (dPBS) without calcium and magnesium, with a salt concentration of approximately 100 mM NaCl, and pH range 7.0–7.3 (Table of Materials).
| Name of Material/Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| 1x Dulbecco’s phosphate buffered saline | Thermo Fisher (Gibco) | 14190144 | |
| Anti-alpha-synuclein (phosphorylated at serine 129) antibody | Abcam | AB184674 | |
| Anti-alpha-synuclein antibody | Abcam | AB15530 | |
| Bicinchonic acid | Thermo Fisher (Pierce) | PI23228 | |
| Clear Medical Shrink Tubing (0.036" inner diameter) | Nordson Medical | 103-0143 | |
| Clear Medical Shrink Tubing (0.044" inner diameter) | Nordson Medical | 103-0296 | |
| Copper sulfate | Thermo Fisher (Pierce) | PI23224 | |
| Eppendorf ThermoMixer C | Eppendorf | 2231000574 | |
| Eppendorf ThermoTop heated lid | Eppendorf | 5308000003 | |
| Formvar/Carbon coated electron microscopy grids | Eletron Microscopy Sciences | FCF300-Cu | |
| Glass capillary tube (0.53 mm outer diameter; 0.09 mm inner diameter; 54 mm length) | Drummond | 22-326223 | |
| Glass needle puller | Narishige | PC-10 | |
| Hamilton syringe | Hamilton | 80000 | |
| Human alpha-synuclein monomer to generate preformed fibrils | Proteos | RP-003 | |
| Mouse alpha-synuclein monomer to generate preformed fibrils | Proteos | RP-009 | |
| Orange Medical Shrink Tubing (0.021" inner diameter) | Nordson Medical | 103-0152 | |
| Parafilm M | Sigma-Aldrich | P7543 | |
| Proteinase K | Invitrogen | 25530015 | |
| Qsonica 3.2 mm tip | Qsonica | 4422 | |
| Qsonica Q125 sonicator | Qsonica | Q125 | |
| Thioflavin S | Sigma-Aldrich | T1892 | |
| Thioflavin T | EMD Millipore | 596200 | |
| ToxinSensor Chromogenic LAL Endotoxin Assay Kit | Genscript | L00350C | |
| Uranyl acetate | Eletron Microscopy Sciences | 22400 |
1.3.2.2 Add 2 μL of sample to the reader and read the absorbance at 280 nm.
1.3.2.3 Use the Beer-Lambert law to determine concentration of the monomers.
Note: Extinction coefficient ε for human α-syn is 5,960 M−1cm−1 and for mouse α-syn is 7,450 M1cm−1. Pathlength is measured in cm. Molecular weight of α-syn (14 kDa) is estimated assuming 1 Da = 1 g/mol.
1.4 Dilute the monomers with 1x dPBS to a final concentration of 5 mg/mL. Use the equation below to calculate the amount of 1x dPBS added to dilute the monomers.
Note: All concentrations are in mg/mL, and volumes in μL. Monomer dilutions should be performed with 1x dPBS. Total volume used to generate fibrils should be between 100 and 500 μL to achieve reproducible fibrilization results.
1.5 Briefly vortex to mix, and centrifuge to collect all liquid at the bottom of the tube. Aliquot monomers for quality control comparison with fibrils as indicated in steps 1.8.1 – 1.8.4.
1.6 Use a tube lock or wax/plastic film (Table of Materials) to secure the microcentrifuge tube lid closed.
1.7 Place the tube in an orbital thermomixer with a lid for 7 days at 37 °C, shaking at 1,000 RPM (Table of Materials).
Note: At the end of the 7 days, the contents of the tube should appear turbid.
Note: The thermomixer must have a lid to prevent condensation formation on the tube lids.
1.8 Gently flick the tube to resuspend the α-syn fibrils. Aliquot fibrils for the following quality control steps as indicated in steps 1.8.1 – 1.8.4.
Note: Fibrils for quality control steps can be stored at RT overnight.
1.8.1 Aliquot 6 μL for the sedimentation assay.
1.8.2 Aliquot 5 μL for the Thioflavin T binding assay.
1.8.3 Aliquot 2 μL for the Transmission electron microscopy for fibril visualization.
1.8.4 Aliquot at least 10 μL for the Endotoxin assay.
Note: For endotoxin assay, a commercial Limulus Amebocyte Lysate (LAL) is used, following manufacturer’s instructions (Table of Materials). Endotoxin levels should be ≤ 0.5 endotoxin units/mL for the fibrils. An endotoxin removal kit can be used if this criterion is not met.
1.9 Aliquot the remaining fibrils and store. For long term storage (12–18 months), rapidly freeze on dry ice, and store at −80 °C.
Note: The amount to aliquot depends on the desired downstream application. In vivo use typically requires more fibrils at higher concentrations than in vitro use, and aliquot volumes should be planned accordingly. Fibrils should be stored towards the back of the freezer to prevent damage from potential freeze/thaw. The protocol can be paused here.
2. Sedimentation Assay
2.1 In a clean microcentrifuge tube, add 6 μL PFF to 54 μL dPBS to dilute fibrils 1:10, and pipet to mix.
2.1.1 In a separate tube, add 6 μL monomer to 54 μL dPBS as an additional control.
2.2 Centrifuge samples at 10,000 × g for 30 min at RT and transfer the supernatant to a clean microcentrifuge tube.
2.3 Add 60 μL dPBS to the remaining pellet and vortex to resuspend.
2.4 Add 30 μL of 3X sample buffer (140 μL 50% Glycerol/0.1% Bromophenol Blue, 40 μL 10% SDS, and 20 μL β-mercaptoethanol) to each tube and vortex to mix.
2.5 Incubate samples at 100 °C for 10 min and allow to cool for 5 min on ice.
2.6 Use a standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) protocol to separate protein by mass. Use a protein ladder with a range that includes 14 kDa (the size band expected for monomeric α-syn).
2.7 Transfer the gel into a staining dish and cover the gel with Coomassie blue stain (0.1% Coomassie brilliant blue, 20% methanol by volume, and 10% acetic acid by volume in ddH2O). Incubate for 3 h at RT while shaking.
2.8 Pour off the Coomassie blue stain and add enough destain (50% methanol by volume, and 10% acetic acid by volume in ddH2O) to cover the gel. Incubate for 30 min. at RT while shaking.
2.9 Pour off destain and repeat the destain step until the gel is clear and bands are visible.
2.10 Wash the gel 3 times for 5 min each by shaking in ddH2O and image the gel.
3. Transmission Electron Microscopy for Fibril Visualization
3.1 Weigh 20 mg uranyl acetate in a microcentrifuge tube and add 1 mL of ddH2O to make a 2% aqueous solution. Vortex until the uranyl acetate is in solution, and allow to sit overnight.
Note: Uranyl acetate should be made the day before preparing samples for transmission electron microscopy.
Note: Exposure to light or agitation can cause the uranyl acetate to precipitate, as such, the tube containing the uranyl acetate solution should be covered in aluminum foil, kept in a dark place, and not shaken after the uranyl acetate is in solution.
3.2 Add 2 μL PFF to 98 μL dPBS to dilute fibrils 1:50, then pipet to mix.
3.3 Prepare a clean wax/plastic film (Table of Materials) covered surface (approximately 50 × 50 mm) on the bench top. On the clean wax/plastic film (Table of Materials), add the following (Figure 2):
Figure 2. Staining methods for transmission electron microscopy.
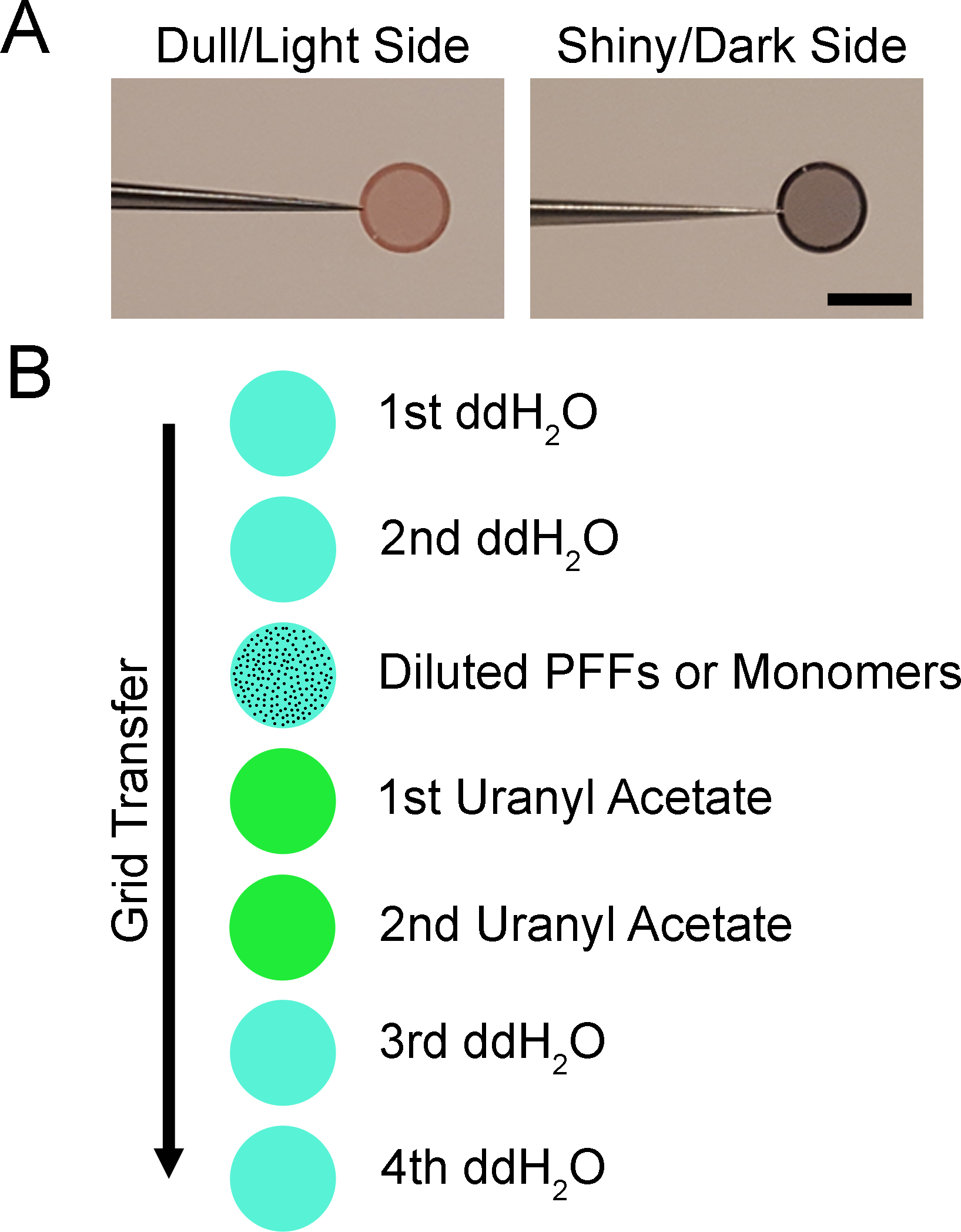
Diagram of negative staining for electron microscopy. A. Images depicting electron microscopy specimen grids. The grid has a dull or light side and a shiny or dark side. The shiny/dark side is coated with a formvar/carbon support film. B. Illustration of staining procedure. The grid is floated shiny/dark side down on the first drop of ddH2O for 1 min and the excess is wicked away with filter paper. The process is repeated with the second drop of ddH2O, diluted PFFs or monomers, two drops of uranyl acetate, and two additional drops of ddH2O. Grids may be stored in a grid box until imaged. Scale bar = 3 mm.
3.3.1 Add 4 × 10 μL drops of ddH2O
3.3.2 Add 1 × 10 μL drop of diluted fibrils or monomer sample
3.3.3 Add 2 × 10 μL drops of 2% uranyl acetate
3.4 Use fine-tipped forceps to pick up a formvar/carbon coated, mesh copper transmission electron microscopy grid (Table of Materials).
Note: Be sure to only pick up the grid by the edge, avoiding the mesh portion in the center (Figure 2A). If the forceps touch the mesh, the formvar/carbon coated film will be damaged, making imaging in that area of the grid impossible.
3.5 Float the grid formvar/carbon coated side (shiny side) down on the first drop of ddH2O. Gently use the forceps to hold the grid and push down to ensure the entire coated surface is in contact with the ddH2O. Let the grid float for 1 min.
3.6 Pick up the grid, and without touching the mesh portion of the grid, wick away the ddH2O with filter paper.
3.7 Float the grid as above on the second drop of ddH2O for 1 min and wick away the ddH2O.
3.8 Float the grid on the drop of diluted fibrils for 1 min and wick away the excess as above.
3.9 Float the grid on the first drop of uranyl acetate for 1 min and wick away the excess.
3.10 Float the grid on the second drop of uranyl acetate for 1 min, wick away the excess.
3.11 Float the grid as above on the third drop of ddH2O briefly and wick away the ddH2O.
3.12 Float the grid as above on the fourth drop of ddH2O briefly and wick away the ddH2O, being sure to remove as much ddH2O as possible.
3.13 Transfer to a grid box for storage until imaging.
Note: Grids should dry for at least 5 min before imaging. Grids can be imaged for at least a year after preparation and should be kept in a dry environment.
4. Thioflavin T Assay
4.1 In a clean microcentrifuge tube, add 5 μL PFF to 245 μL dPBS to dilute fibrils 1:50, and pipet to mix.
4.2 Add 250 μL dPBS to a separate microcentrifuge tube to serve as a negative control.
4.3 Add 5 μL monomer to 245 μL dPBS to serve as the monomer control.
4.4 Add 250 μL thioflavin T in glycine buffer (25 μM thioflavin T, 100 mM glycine, 1% Triton X-100, pH 8.5) to each sample and gently mix.
4.5 Pipet 2 replicates, 200 μL each, into a black 96 well plate. Keep plate in the dark to prevent photobleaching.
4.6 Incubate for 1 h at RT and read the plate using an excitation of 450 nm and emission of 510 nm.
Note: Thioflavin T readings will fluctuate over time. Samples should be read at the same incubation time. If desired, multiple readings can be taken during the hour incubation and plotted over time.
5. Sonication of α-synuclein Preformed Fibrils
Note: CAUTION: The sonicator and all sonication steps are performed in a culture hood to prevent exposure to fibrils that may aerosolize during sonication. The personnel performing the sonication steps should wear personal protective equipment, including gloves, clothing protection in the form of a lab coat, and a face shield while sonicating. Risk of fibril exposure can be reduced by sonicating with a cup horn sonicator, allowing the tube containing fibrils to remain closed during sonication.
Note: Optimal sonication parameters of fibrils are dependent on the model of sonicator used. For this reason, some optimization will need to be performed to ensure fibrils are the correct size. The sonicator used can be found in the Table of Materials and the parameters outlined are based on previous results with this model of sonicator. The parameters below will work for 2–4 μg/μL of PFFs in 200–400 μL of solution. Test sonication with the instrument should be performed and fibrils analyzed to ensure desired results are achieved prior to use of PFFs in experiments.
5.1 Attach a 3.2 mm diameter probe (Table of Materials) to the converter, and set the sonicator parameters as stated below in 5.1.1 – 5.1.3.
5.1.1 Set “amplitude” to 30%.
5.1.2 Set the “Pulse set” to 01 01 (1 s on; 1 s off).
5.1.3 Se the “Time set”: 0:01:00
Note: 1 min of pulsing equates to 60 pulses, and this model of sonicator (Table of Materials) will stop automatically. With other sonicator models, the number of pulses will need to be counted.
5.2 Thaw fibrils at RT and dilute with sterile dPBS in a culture hood.
Note: A clear 0.6 mL microcentrifuge tube works best for sonication and the final fibril concentration depends on intended use. Representative results shown are from a final fibril concentration of 4 μg/μL.
5.3 Wipe the probe of the sonicator with a lab tissue dampened with 70% ethanol to clean the probe. Submerge the tip of the probe in ddH2O and pulse 10 times to further clean the probe, then wipe dry with a lab tissue.
5.4 Place the probe tip into the tube of diluted fibrils, and position the tip at the bottom of the tube.
5.5 Sonicate for 60 pulses (1 s on; 1 s off). Move the probe up and down during each pulse to ensure all of the fibrils in the liquid are sonicated. and clean.
5.6 After 60 pulses, remove the probe from the PFFs, and add 2 μL PFF to 98 μL dPBS in a clean microcentrifuge tube to dilute fibrils 1:50 for sonicated fibril measurement by electron microscopy.
Note: Ideally, a small subset of fibrils should be measured prior to in vivo injection and a more comprehensive measurement of fibrils performed when time permits.
5.7 After sonication, briefly centrifuge PFFs for 1 s at 2,000 × g to collect all liquid off the sides of the tube.
Note: CAUTION: If the tube becomes hot to the touch, stop sonicating after 30 pulses, wait 1 min, and sonicate for the final 30 pulses.
Note: While sonicating, keep the probe tip towards the bottom of the liquid at the start of each pulse, too close to the top of the liquid will cause sample loss.
5.8 Submerge the probe tip in 1% SDS and pulse 10 times to clean the probe. Remove the tip from the SDS, submerge in ddH2O and pulse 10 times.
5.9 Wipe the probe with a lab tissue dampened with 70% ethanol, then wipe the probe with a dry lab tissue. Detach the probe from the converter and store.
5.10 Wipe down all surfaces in the hood with 1% SDS, followed by 70% ethanol.
Note: The 1% SDS solution is used to dissociate fibrils and clean surfaces and equipment 17.
6. Transmission Electron Microscopy for the Measurement of Sonicated Fibrils
Note: If electron microscopy is not feasible, a thioflavin T kinetics assay, and dynamic light scattering can be used as indirect measures of seeding efficiency and fibril size 4, 15.
6.1 Prepare samples using the protocol from the “Electron Microscopy for Fibril Visualization” section above.
6.2 Use a transmission electron microscope to take an image of fibrils from 6 to 10 different grid openings.
Note: Images should be a high enough magnification to measure fibrils, but low enough to visualize multiple fibrils simultaneously. A final magnification of approximately 75,000x should be sufficient.
6.3 Measure the length of a small subset of at least 25 fibrils prior to using fibrils in an experiment.
Note: These measurements can typically be performed using the imaging software associated with the microscope. For the quick validation, the person measuring should select representative fibrils and calculate the average size. Fibril length should average around 50 nm or less, with a more accurate measurement to follow.
6.4 Measure the lengths of 500+ fibrils for more comprehensive results.
Note: The imaging software associated with the microscope can be used for fibril measurements. Alternatively, images can be opened and fibrils measured with image processing software, using the scale bar associated with each image as a size reference with which to compare the fibril lengths.
7. Preparation of Custom Glass Needle Syringes for Stereotactic Injections (Figure 3)
Figure 3. Assembly of custom glass needle syringes.
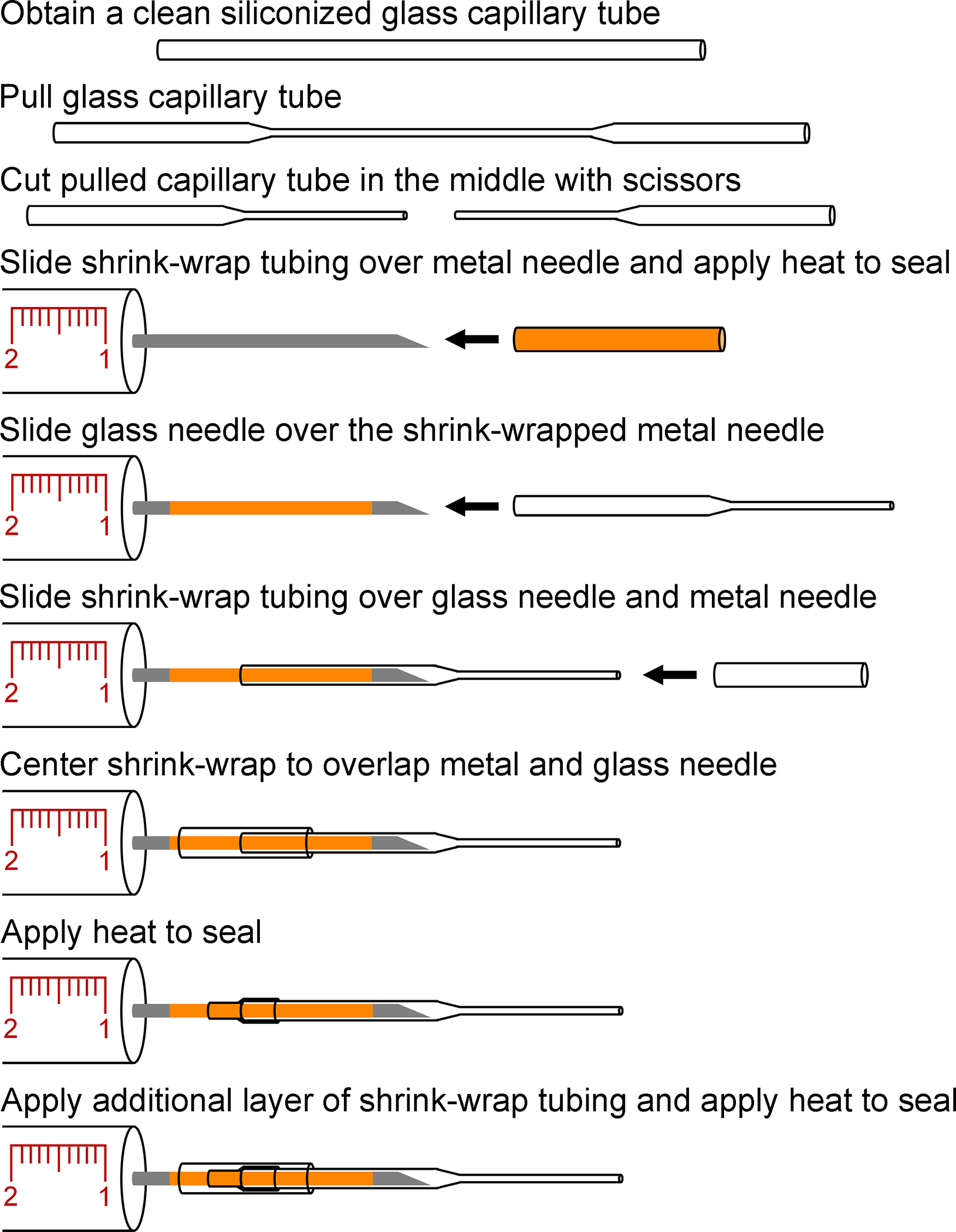
Diagram of the steps required to assemble glass needle attached syringes. Siliconized glass capillary tubes are pulled and cut in the middle to produce glass needles. Shrink wrap tubing is used to prepare the metal needle and form an inner seal when the glass needle is slid onto the metal needle. Two additional layers of shrink-wrap tubing that overlap the base of the glass needle and the metal needle are consecutively added and heat applied to secure the glass needle and form a water-tight seal.
7.1 Add approximately 10 mL of siliconizing reagent (Table of Materials) to a clean 50 mL beaker.
7.2 Place glass capillary tubes (length of 54 mm; outer diameter: 0.86 mm; inner diameter 0.59 mm) vertically in the beaker of siliconizing reagent and allow capillary action to draw the siliconizing reagent up the tube through the lower tube opening submerged in the siliconizing reagent.
7.3 Pipet additional siliconizing reagent into the upper tube opening that is not submerged in siliconizing reagent to completely fill the glass capillary tube.
7.4 Remove the capillary tubes from the siliconizing reagent and blot the open ends of the tubes on a paper towel to remove siliconizing reagent in the tubes. Allow capillary tubes to dry for at least 8 hours.
7.5 Place a siliconized glass capillary tube in a glass needle puller (Table of Materials).
7.6 Turn on the heating element and allow the attached weights to stretch the heated glass capillary tube.
7.7 Cut the pulled glass capillary tube with scissors at the thinnest point in the middle and remove the glass needle from the glass needle puller.
Note: The pulled needle inner and outer diameter should be approximately 80 and 100 μm respectively. Multiple glass needles can be made at one time and stored until ready to attach to a glass syringe with attached metal needle (Table of Materials).
7.8 Cut a length of shrink-wrap tubing (average inner diameter 0.021” and average wall thickness 0.001”) to approximately 40 mm with scissors. Slide the shrink wrap over the metal needle of a 10 μL beveled syringe (Table of Materials).
7.9 Use an open flame to heat and adhere the shrink wrap to the needle while rotating the needle to apply heat evenly.
7.10 Slide the larger end of the pulled glass needle carefully over the metal needle of the syringe.
7.11 Cut a length of shrink-wrap tubing (average inner diameter 0.036” and average wall thickness 0.005”) to approximately 40 mm with scissors and carefully slide over glass needle to overlap the base of the glass needle and the metal needle of the syringe (Table of Materials). Use an open flame to heat the shrink-wrap to secure the glass needle to the metal needle.
7.12 Add an additional layer of shrink-wrap to secure the glass needle. Cut a length of shrink-wrap tubing (average inner diameter 0.044” and average wall thickness 0.005”) to approximately 40 mm with scissors and carefully slide over glass needle to overlap the base of the glass needle and the metal needle of the syringe (Table of Materials). Use an open flame to heat the shrink-wrap to secure the glass needle to the metal needle.
7.13 Trim the glass needle with scissors so that the tip is approximately 8 mm long.
Note: The needle needs to be long enough to target the desired brain regions (required length is dependent on the dorsal/ventral coordinates).
7.14 Use steps 7.14.1 – 7.14.3 to test the needle to insure there are no leaks and there is adequate flow both in withdrawing and dispensing liquid from the glass needle.
7.14.1 Fill a 1 mL syringe with an attached 26 gauge needle with dH20.
7.14.2 Remove the metal plunger from the custom glass needle syringe and insert the needle of the dH20 filled syringe into the base of the syringe. Apply pressure to dispense dH20 from the glass needle. Inspect the interface of the glass needle and the metal needle for leaks and confirm a steady dH20 flow.
Note: If needed, the glass needle can be trimmed to increase the ease of flow and additional layers of shrink-wrap added to patch any leaks from the base of the glass needle.
7.14.3 Fill a microcentrifuge tube with dH20. Use the custom glass needle syringe to draw in the dH20. Inspect the needle to confirm liquid is being taken into the syringe and that there are no bubbles.
Note: If there are bubbles or dH20 is not being drawn into the needle, trimming the needle can help alleviate pressure.
7.15 Carefully store syringe with attached glass needles in the syringe boxes until needed for surgeries.
7.116 Use standard stereotaxic surgery methods for intrastriatal delivery of PFFs at optimized coordinates in mice (one site: AP +0.2 mm and ML +2.0 mm from bregma, DV −2.6 mm from dura) or in rats (two sites: AP +1.6 mm and ML +2.0 mm from bregma, DV −4.0 from dura; AP +0.1 mm and ML +4.2 mm from bregma, DV −5.0 from dura) 1, 7, 15, 18.
Note: These coordinates have been used in C57BL6/C3H mice and Fischer 344 rats. When using other strains, coordinates should be optimized.
REPRESENTATIVE RESULTS:
Generation of fibrils from α-syn monomers begins with determining the concentration of the monomers. Both the BCA assay and measurement of absorbance at 280 nm (A280) can be used to measure protein content, the BCA assay results, however, suggested a higher concentration than the A280 method. PFFs derived from mouse α-syn monomer had a BCA value of 14.05 ± 0.22 and a A280 of 8.05 ± 0.03 μg/μL (Figure 1). Likewise, PFFs derived from human α-syn monomer also appeared to be at a higher concentration, with a BCA value of 12.95 ± 0.38 and a A280 of 7.83 ± 0.05 μg/μL (Figure 1). The A280 measurements are specific to α-syn based on the inclusion of the extinction coefficients and these results were used to dilute the monomers prior to 7-day incubation.
Prior to incubation, the liquid containing the α-syn monomers was clear, but should appear turbid after fibril formation. Examination with transmission electron microscopy confirmed the presence of long fibrils, measuring 10–20 nm wide (Figure 4). In comparison, α-syn monomers were barely visible with no discernible shape apparent (Figure 4). With visual confirmation of fibrillar structures, amyloid conformation of the fibrils is the next feature of PFFs that should confirmed using a thioflavin T assay. Thioflavin T exhibits enhanced fluorescence when binding to amyloid, thus, increased fluorescent signal from the samples indicates presence of amyloid. As an example, Thioflavin in dPBS produced a signal of 3,287 ± 580 relative fluorescent units (RFU), mouse α-syn monomer produced a signal of 4,174 ± 158 RFU, and mouse PFFs produced a signal of 59,754 ± 6,224 RFU (Figure 5). In comparison, human α-syn monomer produced a similar signal of 4,158 ± 105 RFU to mouse monomer, and human PFFs produced a higher signal of 1,235,967 ± 113,747 RFU as compared to mouse PFFs (Figure 5). To assess the presence of pelletable fibrils, a sedimentation assay was performed. Fibrils will pellet with centrifugation. In both the mouse and human PFF samples, the supernatant fraction should have more protein in the pellet than the supernatant (Figure 6). In contrast, the majority of the protein from the mouse and human monomers was present in the supernatant, with little present in the pellet (Figure 6). With the PFFs visibly present by electron microscopy, amyloid structures present, and fibrils pelletable, the PFFs passed all in vitro quality control steps.
Figure 4. Visualization of α-syn monomers and α-syn fibrils via transmission electron microscopy.
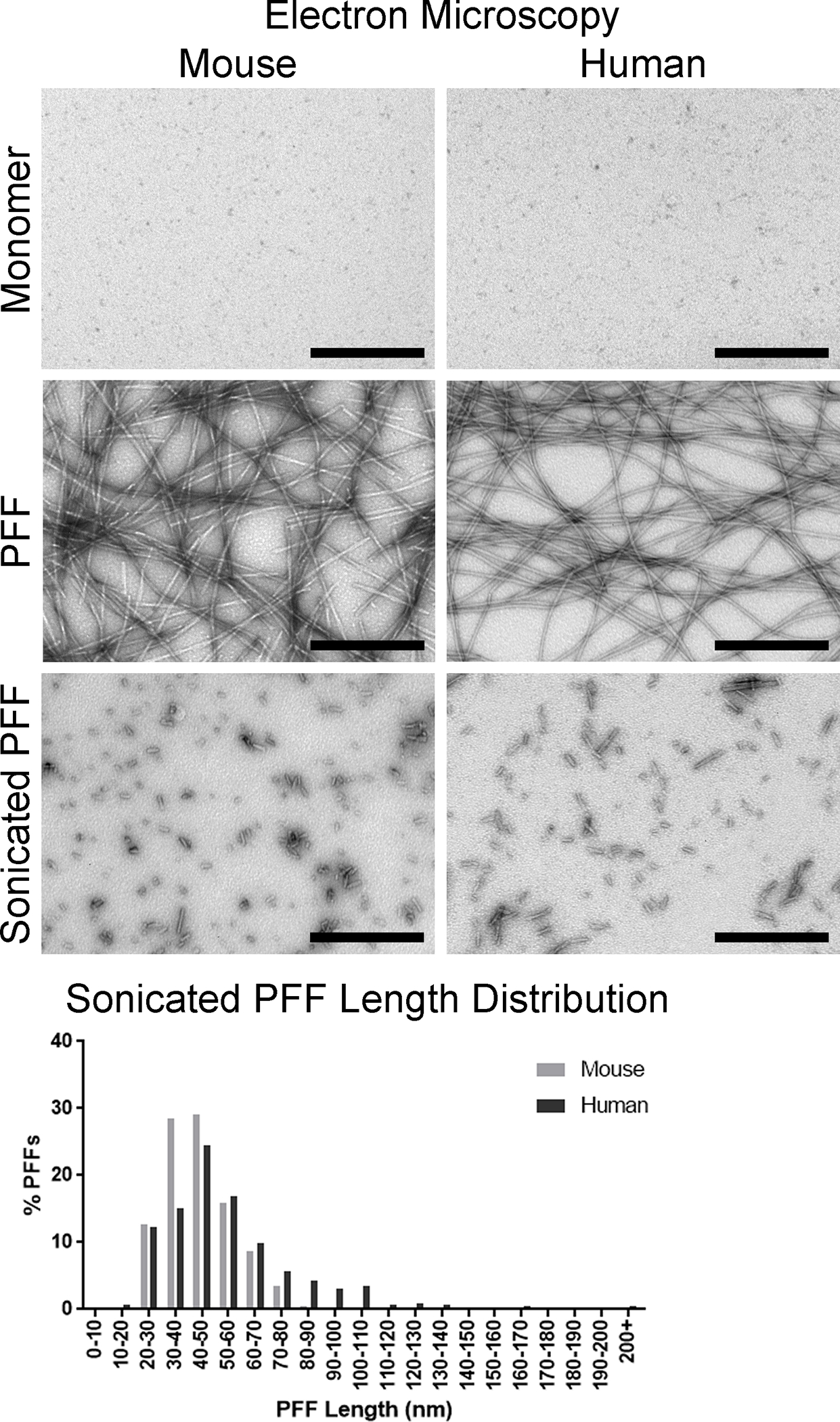
Representative micrographs of α-syn monomers and fibrils. Top panels: Mouse and human α-syn monomer. Middle panels: Full length mouse and human α-syn PFFs. Bottom panels: Mouse and human α-syn PFFs after sonication. Bottom graph: Distribution of sonicated mouse and human α-syn PFF lengths. Scale bar = 500 nm.
Figure 5. Confirmation of amyloid structures by thioflavin T assay.
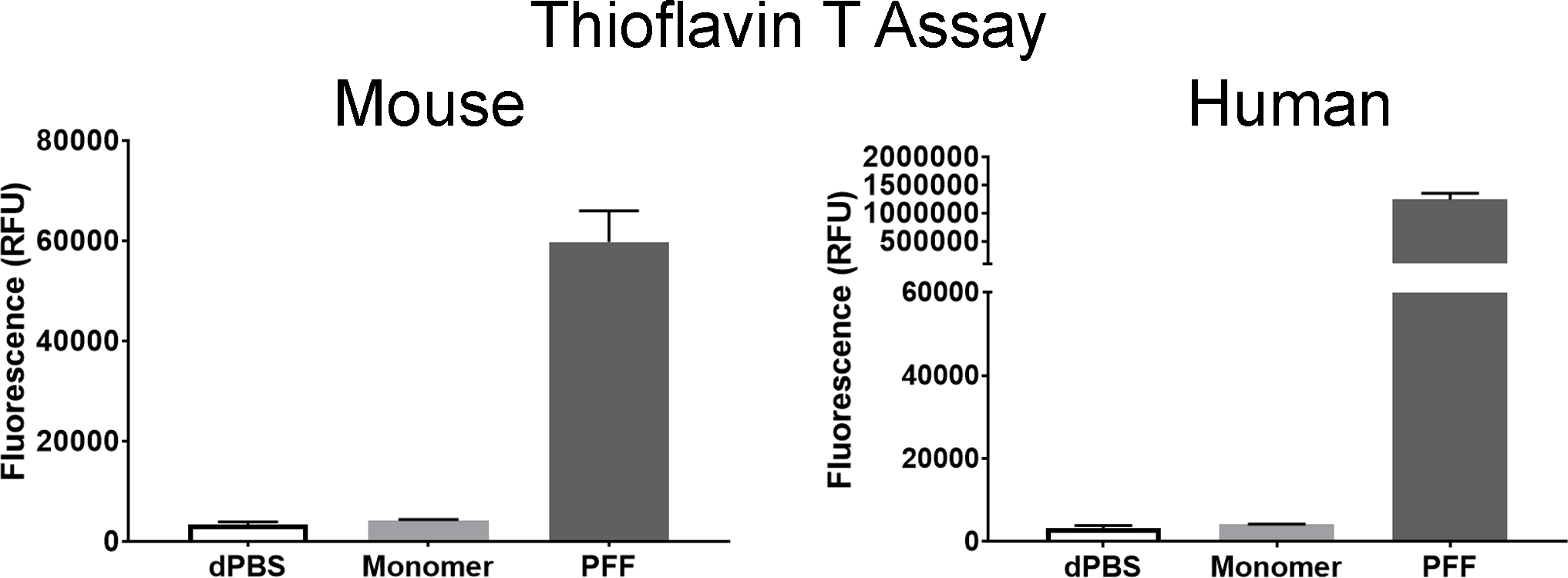
Measurement of fluorescent signal from mouse and human α-syn monomer and PFF samples. Left: Results from mouse α-syn monomers and α-syn PFFs. Right: Results from human α-syn monomers and α-syn PFFs. A dPBS negative control is shown in each graph. All measurements are expressed as relative fluorescent units (RFU). Columns indicate the group means, error bars represent +1 standard error of the mean.
Figure 6. Sedimentation assay for pelletable α-syn.
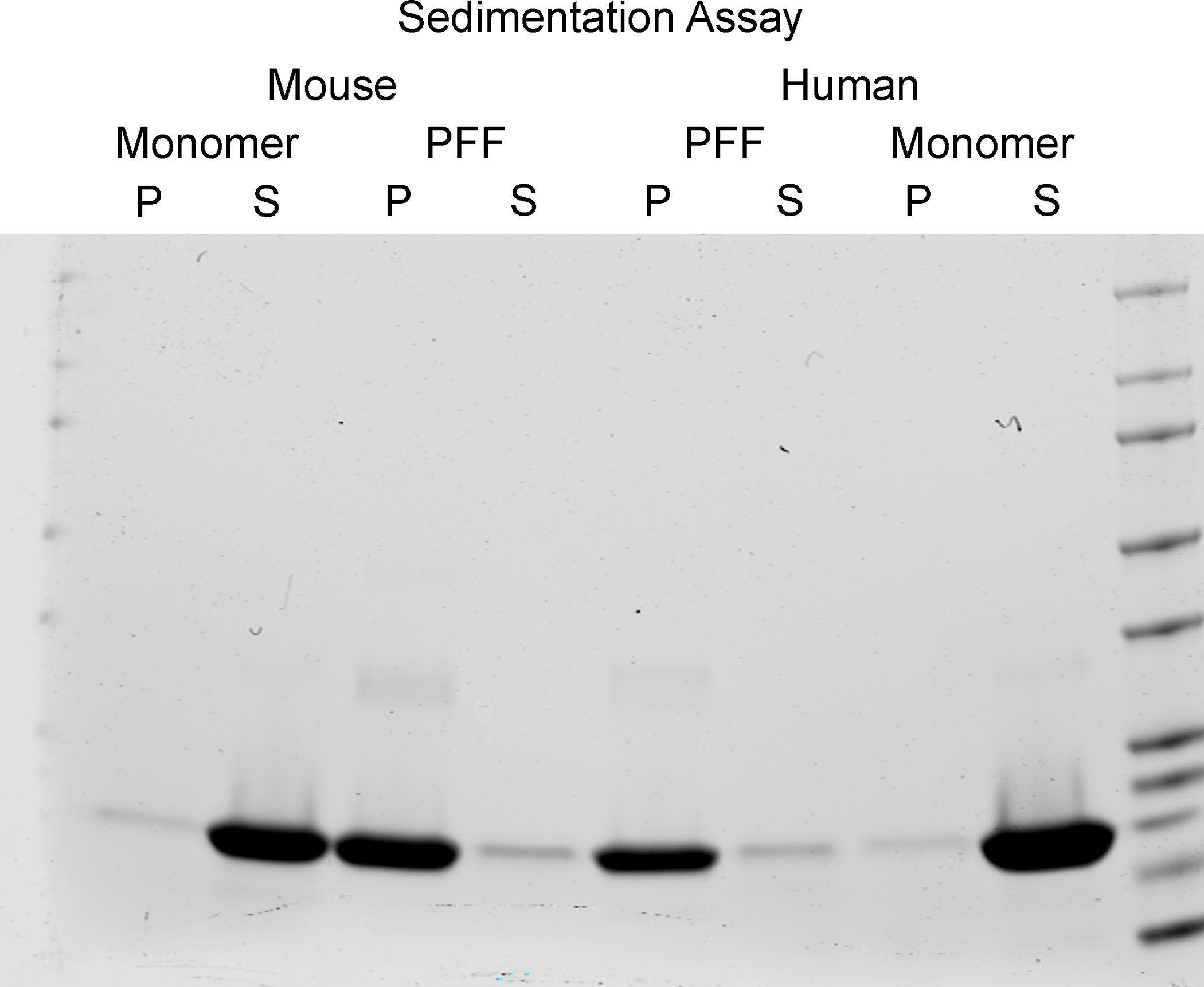
Images from Coomassie stained gels. Bands shown are at approximately 14 kDa based on the protein ladder. Left: Mouse monomer and PFFs. Right: Human monomers and PFFs. For all monomer and PFF samples, the resuspended pellet (P) and supernatant (S) are shown.
Both mouse and human PFFs were sonicated to produce PFFs of appropriate lengths for seeding α-syn inclusions 4, 19. PFFs were diluted to the desired concentration of 4 μg/μL and sonicated. Immediately prior to surgery, 25 representative fibrils were imaged by electron microscopy and measured to spot check the fibril size. The sonicated mouse PFFs measured 48.8 ± 3.1 nm, whereas the human PFFs measured 52.1 ± 4.4 nm in length, PFFs of both species were therefore the appropriate length (50 nm or less) to induce seeding activity. More comprehensive examination of approximately 500 fibrils revealed the average length and length distribution of the sonicated mouse fibrils. The average length was 44.4 ± 0.6 nm, with 86.6% of the PFFs measuring 60 nm or less (Figure 4). In comparison, human PFFs averaged 55.9 ± 1.1 nm with 69.6% of the PFFs measuring 60 nm or less (Figure 4).
Following intrastriatal injection of sonicated mouse PFFs into rats as previously described 3, 5, 7, a series of tissue sections were processed at 2 months post-surgery, when the number of inclusion containing neurons is known to peak in the SNpc, for the confirmation of phosphorylated α-syn inclusions5. Inclusion bearing neurons, as indicated by immunohistochemical staining for pSyn (antibody in Table of Materials) are present within the SNpc (Figure 7), as well as other regions throughout the brain which innervate the striatum (anterior olfactory nucleus, motor, cingulate, piriform, prelimbic, somatosensory, entorhinal, and insular cortices, amygdala, striatum) 1, 3–5, 7, 20. These inclusions share similar properties with Lewy bodies, such as binding thioflavin S, and resistance of total α-syn to proteinase K (Figure 7), as shown by immunohistochemical staining (antibody and proteinase K in Table of Materials). Confirmation of seeding within the brain indicates the in vivo quality control has been passed, and aliquots of PFFs previously frozen and saved from the same batch may be sonicated under identical parameters, with lengths validated, in larger experiments.
Figure 7. Features of inclusions in the rat model confirming α-syn pathology.
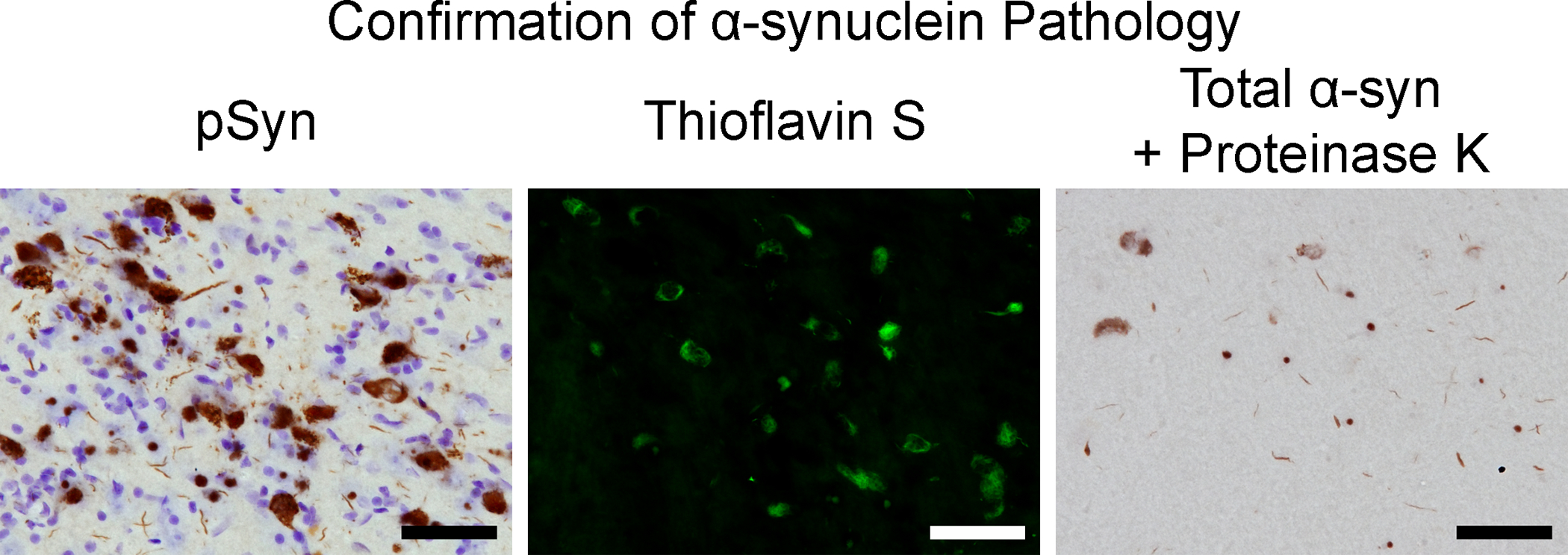
Representative micrographs from the substantia nigra pars compacta at 2 months post-injection. Left: Neurons containing pSyn and counterstained with cresyl violet. Middle: Thioflavin S positive neurons. Right: α-syn-containing inclusions resistant to proteinase K. Scale bar = 50 μm.
DISCUSSION:
Production of α-syn PFFs capable of seeding neurons and leading to Lewy body-like inclusions is dependent on multiple factors and steps. A critical factor, is that the monomers used for generating fibrils need to be specifically formulated for fibrilization4, 10, 15–16. If the monomers are not formulated for fibrilization, fibrils may not form or the fibrils that do form may not produce α-syn pathology. Likewise, the buffer that the monomers are in also influence fibrilization. As such, for the best results, the salt concentration should be approximately 100 mM NaCl and pH between 7.0 and 7.3. An initial step that introduces variability is the method whereby initial protein content is determined, with measurement at A280 likely to produce more accurate results and therefore is the preferred method. The discrepancy in protein concentration can decrease the efficacy of the fibrilization process, as well as alter the assumed PFF concentration used in experiments. Both could lead to a decrease in seeding efficiency and batch variation between experiments.
Initial quality control steps will confirm critical features of the PFFs, specifically that they have a fibrillary conformation (electron microscopy), contain amyloid structures (thioflavin T assay), and are pelletable (sedimentation assay). It is important to note that the results of the thioflavin T assay will fluctuate with time and are not a direct measure of the amount of amyloid structures present, rather, the thioflavin T assay should be used only as an indicator of amyloid structures presence within the sample. Thioflavin T is typically used in in vitro assays, such as the aforementioned assay to show the fibrils contain amyloid structures. Alternatively, Thioflavin S is used in tissue to detect amyloid structures, as shown in Figure 7. In regards to the sedimentation assay, the results show only that the PFFs are found predominantly in the pelletable fraction. As the samples are run in denaturing conditions, a single prominent band of approximately 14 kDa, the size of α-syn monomers, is present on the gels. This is unlike the multiple bands present at higher molecular weights that would be expected with PFFs if a native or non-denaturing gel was used. Lastly, successful passing of all these initial quality control steps does not guarantee α-syn inclusion seeding activity. For this reason, cell culture experiments or a small cohort of surgically-injected animals should be used to test the efficacy of the PFFs before use in larger experiments.
Sonicating is a crucial step in the process and parameters will differ depending on the model of sonicator used. Sonication parameters must be applied and verified to show that short PFF fragments have been produced. Fibril size has bearing on the seeding, with shorter fibrils seeding more efficiently. Though shorter fibrils seed more efficiently, this effect plateaus and the optimal PFF length is approximately 50 nm 4, 19. It is also important not to over-sonicate the samples and expose the PFFs to excessive heat, as this may decrease seeding efficiency. These sonicated PFFs should be tested for efficacy in small cell culture or in vivo experiments prior to use in larger scale experiments. As different sonication sessions have the potential to introduce variability, experimental treatment groups should be planned accordingly.
When delivering PFFs in vivo, the localization of the injection site(s) and the total amount PFFs used can affect the number of neurons that will develop inclusions as well as the extent of neurodegeneration7, 15. The coordinates in the protocol provide a place to start, but should be tested within the lab to ensure the desired target region develops α-syn pathology prior to use in large scale experiments. If desired, tracking dye or fluorescent beads can be used as a way to test regional targeting before using PFFs. The amount of PFFs used in vivo varies between groups, with most groups using a total between 5 to 20 μg of PFFs at one or divided between two injection sites1–8, 15. As the number of injection sites, location of injection sites, and amount of PFFs injected can effect results and progression of the synucleinopathy, the downstream outcomes of the parameters used should be characterized prior to using the model to test potential interventions or examining temporal features of the model.
When selecting a model to use for testing therapeutics or studying disease progression, the model used should be selected to best answer the question asked. Not all models will possess certain disease features of PD, or offer the timeframe needed to test potential interventions. The PFF model recapitulates key features of PD, such as α-syn pathology and neurodegeneration, and can lead to modest motor impairments. The model offers a predictable and protracted time-course, where inclusions form months before neurodegeneration. This allows researchers to examine and exploit the different phases throughout the protracted progression of the synucleinopathy. The current and future use of the model overall is expected to be beneficial in the study of disease progression and development of novel therapies.
ACKNOWLEDGMENTS:
This research was supported by grants from the Michael J. Fox Foundation, the National Institute of Neurological Disorders and Stroke (NS099416) and the Weston Brain Institute.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/59758.
DISCLOSURES:
Joseph Patterson, Kelvin Luk, and Caryl Sortwell are currently involved in contractual arrangements with the Michael J. Fox Foundation to quality control α-syn material generated by Proteos, Inc. Coauthor Nicole Polinski, is an employee of the Michael J. Fox Foundation, which has contracted Proteos, Inc. to produce α-syn monomer. Coauthors Megan Duffy, Laura Volpicelli-Daley, and Nicholas Kanaan have nothing to disclose.
References
- 1.Luk KC et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953, doi: 10.1126/science.1227157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luk KC et al. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Reports 16, 3373–3387, doi: 10.1016/j.celrep.2016.08.053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paumier KL et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiology of Disease 82, 185–199, doi: 10.1016/j.nbd.2015.06.003 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdelmotilib H et al. alpha-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiology of Disease 105, 84–98, doi: 10.1016/j.nbd.2017.05.014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy MF et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. Journal of Neuroinflammation 15, 129, doi: 10.1186/s12974-018-1171-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duffy MF et al. Quality Over Quantity: Advantages of Using Alpha-Synuclein Preformed Fibril Triggered Synucleinopathy to Model Idiopathic Parkinson’s Disease. Frontiers in Neuroscience 12, 621, doi: 10.3389/fnins.2018.00621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patterson et al. Time course and magnitude of alpha-synuclein inclusion formation and nigrostriatal degeneration in the rat model of synucleinopathy triggered by intrastriatal α-synuclein preformed fibrils. Submitted 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luk KC et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences of the United States of America 106, 20051–20056, doi: 10.1073/pnas.0908005106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volpicelli-Daley LA et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71, doi: 10.1016/j.neuron.2011.08.033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volpicelli-Daley LA, Luk KC & Lee VM Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nature Protocols 9, 2135–2146, doi: 10.1038/nprot.2014.143 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuusisto E, Salminen A & Alafuzoff I Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12, 2085–2090 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Neumann M et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. The Journal of clinical investigation 110, 1429–1439, doi: 10.1172/JCI15777 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JY et al. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 25, 1091–1096, doi: 10.1002/mds.23012 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Shimozawa A et al. Propagation of pathological alpha-synuclein in marmoset brain. Acta neuropathologica communications 5, 12, doi: 10.1186/s40478-017-0413-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polinski NK et al. Best Practices for Generating and Using Alpha-Synuclein Pre-Formed Fibrils to Model Parkinson’s Disease in Rodents. Journal of Parkinson’s Disease 8, 303–322, doi: 10.3233/JPD-171248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fares MB et al. Induction of de novo alpha-synuclein fibrillization in a neuronal model for Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America 113, E912–921, doi: 10.1073/pnas.1512876113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenyi A, Coens A, Bellande T, Melki R & Bousset L Assessment of the efficacy of different procedures that remove and disassemble alpha-synuclein, tau and A-beta fibrils from laboratory material and surfaces. Scientific reports 8, 10788, doi: 10.1038/s41598-018-28856-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geiger BM, Frank LE, Caldera-Siu AD & Pothos EN Survivable stereotaxic surgery in rodents. Journal of visualized experiments : JoVE, doi: 10.3791/880 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarutani A et al. The Effect of Fragmented Pathogenic alpha-Synuclein Seeds on Prion-like Propagation. Journal of Biological Chemistry 291, 18675–18688, doi: 10.1074/jbc.M116.734707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wall NR, De La Parra M, Callaway EM & Kreitzer AC Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron 79, 347–360, doi: 10.1016/j.neuron.2013.05.014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]