

Abstract
Much of the human proteome is involved in mRNA homeostasis, but most RNA-binding proteins lack chemical probes. Here, we identifyh electrophilic small molecules that rapidly and stereoselectively decrease the expression of transcripts encoding the androgen receptor and its splice variants in prostate cancer cells. We show by chemical proteomics that the compounds engage C145 of the RNA-binding protein NONO. Broader profiling revealed that covalent NONO ligands suppress an array of cancer-relevant genes and impair cancer cell proliferation. Surprisingly, these effects were not observed in cells genetically disrupted for NONO, which were instead resistant to NONO ligands. Reintroduction of wild-type NONO, but not a C145S mutant, restored ligand sensitivity in NONO-disrupted cells. The ligands promoted NONO accumulation in nuclear foci and stabilized NONO–RNA interactions, supporting a trapping mechanism that may prevent compensatory action of paralog proteins PSPC1 and SFPQ. These findings show that NONO can be co-opted by covalent small molecules to suppress protumorigenic transcriptional networks.
mRNAs are subject to several steps of post-transcriptional regulation, including splicing, transport and modification, which are prerequisites for translation into protein1. Each of these processes is governed by the action of RNA-binding proteins (RBPs), which constitute a large and diverse portion of the human proteome2. RBPs are implicated in many human diseases and oversee the maturation and quality control of mRNAs that encode key oncogenic proteins3,4. In spite of their fundamental roles in human physiology and disease, RBPs are under-explored in terms of chemical probe and drug discovery5,6. Some of the challenges facing the discovery of small-molecule modulators of RBPs include the absence of conventional drug binding pockets and the limited availability of functional assays5,7. Many RBPs also act in the cell as parts of large and dynamic complexes (for example, molecular condensates)8 that are difficult to study in reconstituted biochemical systems. Nonetheless, the discovery of chemical probes and drugs for select RBPs9,10 and RBP–RNA complexes11–13 points to the broader potential of pharmacologically targeting this protein class.
Identifying chemical probes for additional RBPs could provide a way to suppress the expression and activity of diverse disease-relevant proteins, including oncogenic transcription factors, such as the androgen receptor (AR), a principal driver of prostate cancer14. First-line therapies for prostate cancer include AR antagonists, such as enzalutamide15, but resistance to these drugs often emerges16. Approaches to counter resistance include the development of small-molecule degraders (for example, proteolysis targeting chimeras) of AR17, but such candidate therapies may not address cancers with mutations in the androgen ligand-binding site18 or that generate truncated splice variants, such as AR-V7, which lacks the AR ligand-binding domain altogether19. Multiple alternative ways to target AR and AR-V7 have been studied, including small molecules that bind to the N-terminal domain shared by these proteins20,21 and antisense22 and epigenetic23 approaches to suppress AR and AR-V7 expression.
Here, we sought to discover small molecules that rapidly decrease the expression of transcripts encoding AR and its major splice variants in prostate cancer cells in the hopes that such compounds may reveal additional mechanisms for regulating the expression of this oncogenic transcription factor. A focused screen of cysteine-directed electrophilic small molecules identified an α-chloroacetamide compound and analogs thereof that stereoselectively suppressed full-length AR (AR-FL) and AR-V7 splice variant transcript and protein expression. Activity-based protein profiling (ABPP) identified a cysteine (C145) in the Drosophila behavior and human splicing (DBHS) family RBP NONO that stereoselectively and site-specifically reacted with the AR-suppressing compounds. Interestingly, genetic disruption of NONO did not suppress AR transcript or protein expression but instead blocked compound activity. Reintroduction of wild-type (WT) NONO, but not a NONOC145S variant, into NONO-disrupted prostate cancer cells restored the AR-suppressive activity of active compounds. We further show that these compounds also regulate the expression of gene networks enriched in RNA homeostasis and signal transduction and the growth of cancer cells in a NONO-dependent manner. Finally, we provide evidence that the NONO ligands act, at least in part, by stabilizing NONO binding to mRNAs, leading to impaired transcript processing and maturation and frustrating the potential compensatory action of paralog DBHS proteins (PSPC1 and SFPQ) that may overcome the genetic loss of NONO in cancer cells.
Results
Electrophilic compounds that suppress AR expression
We reasoned that identifying new mechanisms for regulating the expression of mRNAs encoding AR-FL and the AR-V7 splice variant in prostate cancer cells may benefit from screening a library of electrophilic compounds, which, due to their covalent mechanism of action, engage diverse structural and functional classes of proteins, including those that are challenging to address with reversibly binding small molecules24–26. We evaluated an in-house collection of ~500 electrophilic compounds bearing cysteine-directed reactive groups (for example, α-chloroacetamides and acrylamides)26 for effects on transcripts encoding AR-FL and AR-V7 in the 22Rv1 human prostate cancer cell line (Fig. 1a). We measured the expression of AR-FL and AR-V7 mRNA at an early time point (6 h) after compound treatment to facilitate identification of mechanisms that directly regulate AR transcript homeostasis, where a hit compound was designated as Q11 reducing the expression of AR-FL and AR-V7 mRNA by >30% (Fig. 1b). We also measured total AR and AR-V7 protein content 24 h after compound treatment by high-content imaging (Fig. 1a) by using 22Rv1 cells stably expressing a small hairpin RNA (shRNA) specific to AR-FL to visualize AR-V7 protein (Extended Data Fig. 1a,b) and required that a hit compound suppress the expression of both AR protein variants by >50% (Fig. 1c). These screens identified a single hit compound (the piperazine chloroacetamide B21 (1; Fig. 1d)) that suppressed AR-FL and AR-V7 transcript and protein expression in 22Rv1 cells without causing substantial cytotoxicity in the 24-h assay window (Fig. 1b,c, Extended Data Fig. 1c, Table 1 and Supplementary Tables 1–3).
Fig. 1 |. Discovery of electrophilic compounds that deplete AR mRNA and protein in prostate cancer cells.

a, Screening workflow to discover compounds that deplete AR-FL and AR-V7 mRNA and protein content in 22Rv1 prostate cancer cells; LBD, ligand-binding domain. The light red bar in AR-V7 marks the alternative exon 3b. b,c, Screening results showing the effects of electrophilic compounds (20 μM) on the expression of AR-FL and AR-V7 mRNA (b; 20 μM compound for 6 h) and protein (c; 10 μM compound for 24 h). Hit compounds that reduced AR-FL and AR-V7 mRNA and protein by ≥30% and ≥50%, respectively, are highlighted in red. Full screens were performed once. See Supplementary Table 3 for structures of other compounds shown in red in b and c. d, Hit compound B21 (1). e, Time course of depletion of AR-FL and AR-V7 mRNA and total AR protein in 22Rv1 cells treated with B21 (25 μM). Data are shown as mean values for two replicates (mRNA) or mean values ± s.e.m. for four replicates (protein) of a single experiment representative of two independent experiments. f, Concentration-dependent effects of B21 and structural analogs (active, 2–5; inactive, 6–9; see Table 1 and Supplementary Table 4 for compound structures) on AR-FL and AR-V7 mRNA expression in 22Rv1 cells. g, Concentration-dependent effects of active (R)-SKBG-1 and inactive (S)-SKBG-1 enantiomers on AR-FL and AR-V7 mRNA expression in 22Rv1 cells. For f and g, compound treatments were for 6 h, and data are shown as mean values for two replicates (f) or mean values ± s.e.m. for four replicates (g) of a single experiment representative of two independent experiments. h, Structures of (R)-SKBG-1 and (S)-SKBG-1. i, Western blots showing AR-FL and AR-V7 protein content in 22Rv1 cells treated with active compounds (5 and (R)-SKBG-1), inactive compounds (9 and (S)-SKBG-1) or DMSO for 24 h. j, Quantification of western blotting data described in i. Data are shown as mean values ± s.e.m. for four independent experiments. P values calculated by one-way ANOVA are indicated above the bars.
Table 1 |.
SAR analysis of analogs of hit compound B21
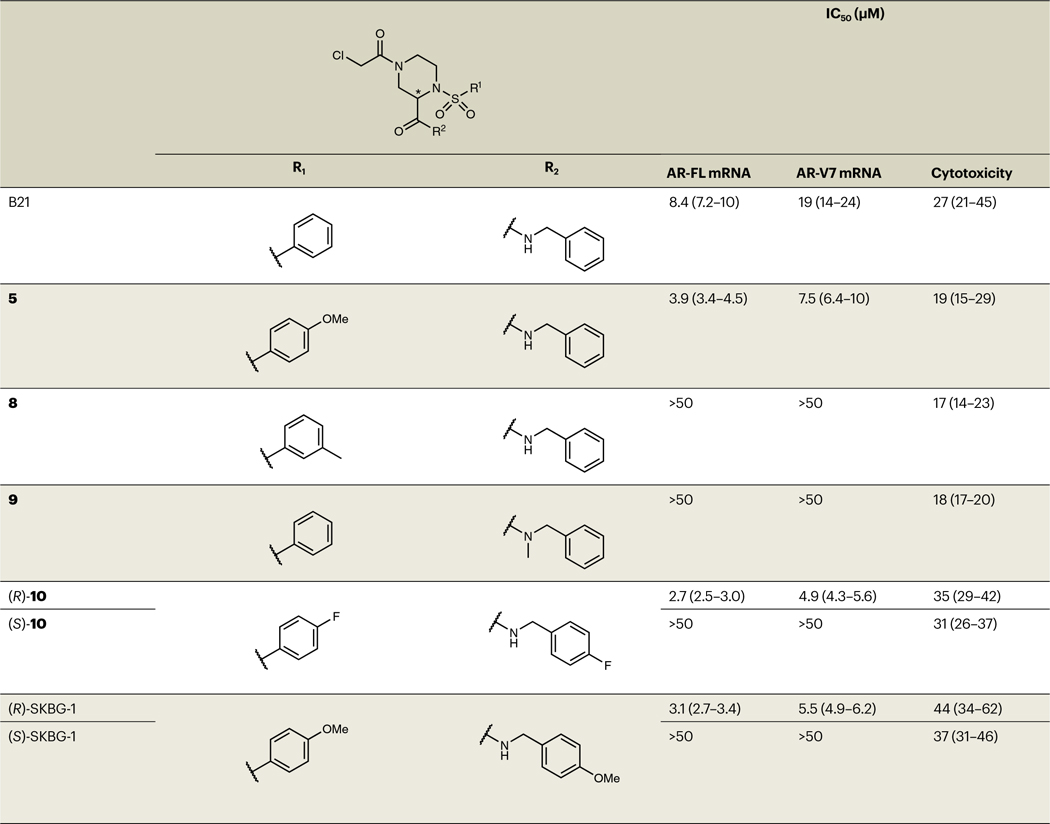
|
Structures of B21 analogs with corresponding IC50 values for depleting AR-FL and AR-V7 mRNA (measured by QuantiGene assays at 6 h after compound treatment) and cytotoxicity estimates (measured by high-content imaging-based cell counting at 24 h after compound treatment). The 95% confidence intervals are shown in parentheses. Data are shown as mean values ± s.e.m. from two experiments, each containing two independent replicates (four replicates total). See Supplementary Table 4 for a complete summary of results for B21 analogs.
Time-course studies revealed that B21 maximally suppressed AR-FL and AR-V7 transcripts by 4–6 h after treatment, followed by a slower reduction in AR protein (Fig. 1e and Extended Data Fig. 1d), suggesting that the direct mode of compound action involved decreasing AR mRNA content. Concentration-dependent profiling of resynthesized B21 and structural analogs confirmed AR transcript suppression for B21 (half-maximum inhibitory concentration (IC50) = 8.4 μM (7.2–10 μM) and 19 μM (14–24 μM) for AR-FL and AR-V7, respectively) and identified compounds with greater potency (compounds 2–5) as well as related inactive compounds (compounds 6–9; Fig. 1f, Table 1 and Supplementary Table 4). The stringent structure–activity relationship (SAR) for AR transcript suppression was exemplified by the differential effects of enantiomeric pairs of compounds, where (R)-10 and (R)-11 decreased AR-FL and AR-V7 transcript expression with IC50 values of 2.7–5.5 μM, while (S)-10 and (S)-11 were inactive (IC50 values of >50 μM; compounds (R)-11 and (S)-11 are referred to hereafter as (R)-SKBG-1 and (S)-SKBG-1, respectively; Fig. 1g,h, Extended Data Fig. 1e, Table 1 and Supplementary Table 4). We furthermore confirmed the importance of the α-chloroacetamide group for compound activity, as acrylamide (12) and propanamide (13) analogs of B21 did not affect the AR transcript content of 22Rv1 cells (Extended Data Fig. 1f,g). Finally, for representative active compounds and structurally related inactive analogs, we confirmed suppression of AR-FL and AR-V7 protein expression in 22Rv1 cells (20 μM compounds, 24 h) by western blotting (Fig. 1i,j).
AR-suppressing compounds engage C145 of NONO
We next sought to identify the protein target(s) of active compounds by chemical proteomics. Specifically, we treated 22Rv1 cells with active (2–5, (R)-10 and (R)-SKBG-1) or inactive control (6–9, (S)-10 and (S)-SKBG-1) compounds (20 μM) for 1 h, after which cells were lysed, and their proteomes were analyzed by cysteine-directed ABPP24,27. Only a modest number (~10) of the >12,500 cysteines (on >5,000 proteins) quantified in the mass spectrometry–ABPP (MS-ABPP) experiments were substantially engaged (>50%) by all of the active compounds, and most of these cysteines were also engaged by one or more inactive control compounds (Fig. 2a, Extended Data Fig. 2a and Supplementary Dataset 1). One clear exception was C145 of NONO, which was substantially engaged by all active compounds but not by the inactive controls (Fig. 2a, Extended Data Fig. 2b and Supplementary Dataset 1). The active and inactive enantiomeric pairs of compounds, such as (R)-SKBG-1 and (S)-SKBG-1, provided particularly useful sets of probes, as across the ~40 proteins possessing cysteines that were substantially (>75%) engaged by the active enantiomer (R)-SKBG-1 after 6 h of treatment, only 4 proteins in addition to NONO were stereoselectively engaged compared to the inactive enantiomer (S)-SKBG-1 (Extended Data Fig. 2c). Among these stereoselective targets of (R)-SKBG-1, only NONO C145 was engaged by each of the other active compounds (2–5 and (R)-10; Extended Data Fig. 2d) and not by the inactive compounds (6–9 and (S)-10; Supplementary Dataset 1).
Fig. 2 |. Active compounds suppress AR expression by targeting the RBP NONO.

a, Heat map showing cysteines that were substantially (>50%) engaged by all active compounds (2–5, (R)-10 and (R)-SKBG-1; 20 μM, 1 h) in 22Rv1 cells as determined by MS-ABPP. Colors in the heat map represent the extent of cysteine engagement in DMSO-treated versus compound-treated cells. Data represent mean values from three independent experiments. See Supplementary Data 1 for full proteomic data. b, Sequence alignment of human and mouse DBHS proteins NONO, PSPC1 and SFPQ for the region around NONO C145 (yellow). The RNA-binding domains RRM1 and RRM2 of human NONO are colored in blue and magenta, respectively. c, Crystal structure of the NONO (cyan)–PSPC1 (green) dimer (Protein Data Bank ID: 3SDE) showing the location of NONO C145 (yellow) in a hinge between the RRM1 (blue) and RRM2 (magenta) domains; red, residues shown to be important for RNA binding30. d, Concentration-dependent effects of active (R)-SKBG-1 and inactive control (S)-SKBG-1 on AR-FL (left) and AR-V7 (right) mRNA content of sgControl and sgNONO cells (6 h of compound treatment). Data are shown as mean values for two replicates of a single experiment representative of two independent experiments. e, Western blots showing AR-FL and AR-V7 protein content in sgControl (NONO+) or sgNONO (NONO–) cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 24 h). Also shown are western blotting signals for NONO and PSPC1. f, Western blots showing signals for endogenous NONO in sgControl (NONO+) 22Rv1 cells and recombinantly expressed FLAG–NONO (WT or C145S) in sgNONO (NONO–) 22Rv1 cells. The FLAG–NONO-expressing sgNONO cells were generated by first stably introducing FLAG–NONO with a silent PAM site mutation and then genetically disrupting endogenous NONO by CRISPR–Cas9. g, Concentration-dependent effects of (R)-SKBG-1 and (S)-SKBG-1 on AR-FL (left) and AR-V7 (right) mRNA content of sgNONO cells expressing NONOWT or NONOC145S. AR mRNAs were quantified by QuantiGene assays at 6 h after compound treatment, and expression was normalized to DMSO-treated NONOWT cells. Data are shown as mean values of two replicates of a single experiment representative of two independent experiments.
NONO (or p54nrb) is a member of the DBHS family of RNA- and DNA-binding proteins that also includes the paralogous proteins PSPC1 and SFPQ (or PSF). The DBHS proteins function as heterodimers and homodimers involved in diverse steps of gene regulation, including DNA unwinding and transcription and mRNA splicing, transport and quality control28,29. NONO shares ~40–50% sequence identity with PSPC1 and SFPQ, but C145 is unique to NONO (Fig. 2b), indicating that the active compounds should selectively engage NONO among the DBHS proteins. A crystal structure of the NONO–PSPC1 heterodimer30 further revealed that C145 is located in a hinge region between the two RNA recognition motifs (RRMs) of NONO and proximal to residues important for RNA binding (Fig. 2b,c).
Previous studies have identified NONO as an AR-interacting protein that supports AR transcriptional activity31,32, and knockdown of NONO or SFPQ by RNA interference decreases AR-FL and AR-V7 mRNA and protein in prostate cancer cells33. To determine whether NONO was a functionally relevant target responsible for mediating AR suppression by active compounds, we generated NONO-disrupted 22Rv1 cells using CRISPR–Cas9 technology. The 22Rv1 cell populations containing NONO-targeting single guide RNAs (sgRNAs; sgNONO cells) displayed substantial loss of NONO compared to sgControl cells (Extended Data Fig. 2e). Surprisingly, AR-FL or AR-V7 mRNA (<20% change; Extended Data Fig. 2f) and protein (Extended Data Fig. 2e) were generally unaltered in sgNONO cells compared to in sgControl cells. Instead, we discovered that sgNONO cells were resistant to the effects of active compounds suppressing AR-FL and AR-V7 mRNA (Fig. 2d and Extended Data Fig. 2g) and protein (Fig. 2e and Extended Data Fig. 2h,i) expression. The ability of active compounds to suppress AR expression was restored in sgNONO cells expressing NONOWT but not in cells expressing a NONOC145S mutant (Fig. 2f,g and Extended Data Fig. 2j–m). We also confirmed direct and site-specific modification of NONO by active compounds using an alkyne analog 14, which maintained AR-suppressive activity and labeled and enriched NONO from NONOWT-, but not NONOC145S-, expressing cells (Extended Data Fig. 3a–d and Supplementary Dataset 1). We further used alkyne 14 in competitive gel ABPP assays to determine an in situ IC50 value of 5.7 μM (4.5–7.2 μM) for (R)-SKBG-1 engagement of NONO C145 (Extended Data Fig. 3e,f). Taken together, these data indicate that the active compounds promote AR loss through a mechanism that depends on covalent modification of C145 of NONO, and this effect is prevented, rather than replicated, by the genetic disruption of NONO.
Transcriptomic and growth effects of NONO ligands
We next sought to determine the scope of transcriptomic and proteomic effects of the NONO ligands in cancer cells. RNA-sequencing (RNA-seq) of sgControl and sgNONO 22Rv1 cells treated with DMSO or active ((R)-SKBG-1) and inactive control ((S)-SKBG-1) compounds (20 μM, 4 h) identified a broad set of mRNAs (~1,000 transcripts) that were substantially (log2 (fold change) < −0.5) and significantly (adjusted P value (Padj) < 0.01) decreased by (R)-SKBG-1 compared to (S)-SKBG-1 or DMSO-treated sgControl cells (Fig. 3a,b, Extended Data Fig. 4a–d and Supplementary Dataset 2). Strikingly, the vast majority of the changes were not observed in (R)-SKBG-1-treated sgNONO cells (Fig. 3a,b and Supplementary Dataset 2) or in untreated sgNONO cells (Fig. 3b, Extended Data Fig. 4b and Supplementary Dataset 2). We interpret the very strong correlation between the global gene expression differences observed in (R)-SKBG-1- and (S)-SKBG-1-treated sgControl cells and (R)-SKBG-1-treated sgControl and (R)-SKBG-1-treated sgNONO cells (Pearson r = 0.82; Fig. 3a) as indicating that most of the transcriptomic changes specifically caused by (R)-SKBG-1 required the presence of NONO in 22Rv1 cells. This conclusion was also supported by a principal-component analysis (PCA), which revealed that (R)-SKBG1-treated sgControl cells were substantially separated from the other compound-treated control groups ((R)-SKBG-1-treated sgNONO cells and (S)-SKBG-1-treated sgControl and sgNONO cells), which all clustered together (Fig. 3c).
Fig. 3 |. Global effects of NONO ligands on the transcriptomes and proteomes of cancer cells.

a, Scatter plot of mRNA log2 (fold change) values for sgControl 22Rv1 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (x axis) versus mRNA log2 (fold change) values for sgControl or sgNONO cells each treated with (R)-SKBG-1 (y axis). Cells were treated with compounds at a concentration of 20 μM for 4 h. Red dotted brackets designate genes decreased (log2 (fold change) ≤ –0.5) or increased (log2 (fold change) ≥ 0.5) in expression by (R)-SKBG-1 in a NONO-dependent manner. Data are shown as mean values for protein-coding genes with >50 counts in each of three independent replicates. b, Heat map showing transcripts that were substantially and significantly changed (log2 (fold change) ≤ −0.5 or log2 (fold change) ≥ 0.5, Padj ≤ 0.01) by (R)-SKBG-1/(S)-SKBG-1 in sgControl cells or DMSO-treated sgNONO/sgControl cells. c, PCA plot of RNA-seq data for the indicated groups, revealing that sgControl 22Rv1 cells treated with (R)-SKBG-1 are substantially separated from the other three compound-treated groups ((R)-SKBG-1-treated sgNONO cells and (S)-SKBG-1-treated sgControl and sgNONO cells). d, Scatter plot showing proteins that were specifically decreased in expression by (R)-SKBG-1 in a NONO-dependent manner. Cells were treated with compounds at a concentration of 20 μM for 24 h. The following are the displayed plotted proteins: (1) ≥30% decrease in (R)-SKBG-1-treated sgControl cells relative to DMSO control, (2) a log2 (fold change) for (R)-SKBG-1/(S)-SKBG-1 in sgControl cells of ≤−0.5 and (3) a log2 (fold change) for (R)-SKBG-1 in sgControl cells/(R)-SKBG-1 in sgNONO cells of ≤−0.5. Data are shown as average values from two independent experiments each containing two replicates. e, GSEA showing the ten most significantly downregulated pathways in (R)-SKBG-1-treated sgControl 22Rv1 cells compared to (R)-SKBG-1-treated sgNONO cells. f, Effects of (R)-SKBG-1 and (S)-SKBG-1 on the growth of sgControl and sgNONO cells. Cells were treated with the indicated concentrations of compounds for 6 d, and cell growth was determined by CellTiterGlo. Data are shown as mean values ± s.e.m. normalized to DMSO-treated control cells for four independent replicates. EC50 values are reported with 95% confidence intervals shown in parentheses.
Proteomic experiments performed at a 24-h time point after compound exposure identified several proteins with decreased expression in (R)-SKBG-1-treated sgControl cells compared to either (S)-SKBG1-treated sgControl cells or (R)-SKBG-1-treated sgNONO cells (Fig. 3d and Supplementary Dataset 3). Consistent (R)-SKBG-1-induced mRNA and protein changes were observed for many gene products (Extended Data Fig. 4e), including AR (Extended Data Fig. 4f), while we noted that instances where (R)-SKBG-1 caused a more dramatic mRNA reduction may correspond to proteins with half-lives much greater than 24 h (for example, SRSF4 with a half-life of >80 h (ref. 34); Extended Data Fig. 4g). Finally, we determined that NONO ligands caused similar global transcriptomic changes in a second human cancer cell line, MCF7 human breast cancer cells (Extended Data Fig. 5a–d and Supplementary Dataset 2). Exceptional transcripts that were regulated by NONO ligands predominantly in one of the two cancer lines generally showed preferential expression in those lines (for example, AR in 22Rv1 cells and RXRA in MCF7 cells; Extended Data Fig. 5e,f).
Pathway analysis of NONO-dependent transcriptional changes caused by (R)-SKBG-1 in 22Rv1 cells revealed an enrichment of hormone signaling, RNA synthesis and GTPase signal transduction pathways (Fig. 3e and Supplementary Dataset 2). Among these affected pathways, we found that some of the earliest changes observed at the proteomic level (24 h after compound treatment; Fig. 3d) were transcriptional regulators, including (in addition to AR) other oncogenic transcription factors, such as the breast cancer dependency protein TRPS1 (ref. 35) and the Wnt–β-catenin pathway component AFF3 (ref. 36) as well as the CGCG-motif-binding transcriptional activator BANP37 and the histone demethylase KDM4B38 (Fig. 3d). These results underscore the potential for NONO ligands to remodel gene regulatory networks in cancer cells, an outcome that is correlated with cell growth inhibition that is both stereoselective and dependent on the presence of NONO (Fig. 3f and Extended Data Fig. 6a). Introduction of NONOWT, but not NONOC145S, into sgNONO 22Rv1 cells restored sensitivity to the stereoselective growth inhibitory effects of (R)-SKBG-1 (Extended Data Fig. 6b). The stereoselective cell growth impairment caused by (R)-SKBG-1 was not restricted to hormone-sensitive breast (MCF7) and prostate (22Rv1) cancer cells and was also observed in additional human cancer lines (HeLa and HT1080 cells; Extended Data Fig. 6c).
In attempts to better understand whether NONO ligands may act in a dominant-negative manner, we ectopically expressed recombinant NONOWT or NONOC145S (or an empty vector control) in 22Rv1 cells expressing endogenous NONOWT (Fig. 4a). These experiments revealed that recombinant NONOWT and NONOC145S bidirectionally modulate AR expression (Fig. 4b) and global transcriptomic (Fig. 4c) and growth (Fig. 4d) effects of (R)-SKBG-1, with NONOWT expression leading to enhanced (R)-SKBG-1 effects and NONOC145S expression suppressing compound activity. This outcome can be visualized in a PCA plot, where the transcriptomic effects of (R)-SKBG-1 in empty vector control cells or cells coexpressing exogenous and endogenous NONOWT segregated together, while the transcriptomic effects of (R)-SKBG-1 in cells coexpressing exogenous NONOC145S and endogenous NONOWT more closely resembled those of the inactive enantiomer (S)-SKBG-1 (Fig. 4e). The partial blockade of (R)-SKBG-1 effects by NONOC145S suggests that, while covalent ligands require NONO for activity, their pharmacological effects are not fully dominant and can be at least partly rescued by coexpression of a ligand-insensitive NONO mutant. We note, however, that, despite the strong overall reduction in (R)-SKBG-1 activity in cells coexpressing exogenous NONOC145S and endogenous NONOWT, there was a subset of transcripts for which stereoselective suppression by (R)-SKBG-1 was preserved in these cells (Fig. 4f,g).
Fig. 4 |. Ectopic NONOC145S attenuates the activity of (R)-SKBG-1 in cancer cells coexpressing endogenous NONOWT.

a, Western blot showing coexpression of endogenous NONO (lower-molecular-weight band) and exogenous FLAG-tagged NONOWT or NONOC145S (higher-molecular-weight band). b, Concentration-dependent effects of (R)-SKBG-1 on AR-FL (left) and AR-V7 (right) mRNA content of 22Rv1 cells coexpressing exogenous NONOWT or NONOC145S. Data for sgControl, sgNONO and empty vector control cells are shown for comparison. AR mRNAs were quantified by QuantiGene assays at 6 h after compound treatment, and expression was normalized relative to DMSO treatment for each experimental group. Data are shown as mean values for two replicates of a single experiment representative of two independent experiments. c, Number of genes that were substantially and significantly decreased by (R)-SKBG-1/(S)-SKBG-1 (log2 (fold change) ≤ −0.75, Padj ≤ 0.01) in 22Rv1 cells coexpressing exogenous NONOWT or NONOC145S compared to empty vector control cells. d, Effects of (R)-SKBG-1 and (S)-SKBG-1 on the growth of 22Rv1 cells coexpressing exogenous NONOWT or NONOC145S compared to empty vector control cells. Cells were treated with the indicated concentrations of compounds or DMSO, and, after 6 d, cell growth was determined by CellTiterGlo. Data are shown as mean values ± s.e.m. normalized to DMSO-treated control cells for two independent experiments with four replicates per experiment. EC50 values are reported with 95% confidence intervals shown in parentheses. e, PCA plot of RNA-seq data for the indicated cell models, revealing that (R)-SKBG-1 effects in 22Rv1 cells coexpressing exogenous NONOC145S shift to resemble the effects of the inactive enantiomer (S)-SKBG-1. f, Pie chart showing the effects of coexpression of exogenous NONOC145S and endogenous NONOWT in 22Rv1 cells on genes stereoselectively regulated by (R)-SKBG-1 in empty vector 22Rv1 cells (genes with (R)-SKBG-1/(S)-SKBG-1 log2 (fold change) ≤ −0.75, Padj ≤ 0.01). Rescue refers to genes for which NONOC145S coexpression substantially nullified the effects of (R)-SKBG-1. g, Bar graphs for representative (R)-SKBG-1-regulated genes that were unaffected by coexpression of NONOC145S. Data are shown as mean values ± s.e.m. for three independent replicates; TPM, transcript per million.
Alterations in DBHS paralog proteins in NONO-disrupted cells
Genetic disruption of NONO did not, on its own, affect the growth of cancer cells (Extended Data Fig. 6d), and sgNONO cells showed fewer transcriptional changes than cancer cells treated with NONO ligands (Extended Data Fig. 4b). Previous studies have reported an increase in the expression of the DBHS paralogs PSPC1 and/or SFPQ in Nono–/– mouse embryonic fibroblasts39 and cancer cells treated with short interfering RNAs targeting Nono40, pointing to a potential mechanism to compensate for NONO loss. Here too, we observed increases in PSPC1 and SFPQ mRNA (Extended Data Fig. 7a and Supplementary Dataset 2) and protein (Fig. 2e, Extended Data Fig. 7b and Supplementary Dataset 3) in sgNONO cells. Interestingly, (R)-SKBG-1-treated sgControl cells also showed strong elevations in PSPC1 and SFPQ compared to DMSO- or (S)-SKBG-1-treated sgControl cells (Fig. 2e and Extended Data Fig. 7a,b). These data thus indicate that genetic or acute chemical perturbation of NONO leads to increased expression of PSPC1 and SFPQ, but the potential for these paralogs to compensate for NONO deficiency may have been subverted in cells treated with NONO ligands.
To our knowledge, a model for compensatory action among DBHS proteins has not been experimentally tested, but we noted that three of the human cancer cell lines in the Cancer Dependency Map that showed the strongest growth effects following genetic deletion of NONO each possessed predicted deleterious mutations in SFPQ or PSPC1 (Extended Data Fig. 7c). This correlation indicated that the genetic loss of NONO may preferentially impair proliferation in cancer cells where compensation by PSPC1 and/or SFPQ is not possible. To explore this concept further, we treated the sgControl or sgNONO 22Rv1 cell lines generated by lentiviral transduction with a control sgRNA or sgRNAs targeting PSPC1 or SFPQ using the Alt-R CRISPR–Cas9 nucleofection system41. We confirmed substantial loss of expression of each targeted DBHS protein by 72 h after nucleofection (Extended Data Fig. 7d), at which time we evaluated transcriptomes by RNA-seq and monitored cell growth. These experiments revealed that dual sgNONO–sgSFPQ cells displayed the largest transcriptomic (Extended Data Fig. 7e and Supplementary Dataset 2) and growth (Extended Data Fig. 7f) defects. Genetic disruption of SFPQ alone (sgControl–sgSFPQ), but not PSPC1 alone (sgControl–sgPSPC1), also caused substantial gene expression (Extended Data Fig. 7e) and growth (Extended Data Fig. 7f) effects, while dual sgNONO–sgPSPC1 cells displayed an intermediate phenotype. Notably, the transcripts decreased in sgNONO–sgSFPQ and sgControl–sgSFPQ cells showed a striking (approximately four- to fivefold) enrichment in transcripts stereoselectively decreased by (R)-SKBG-1 compared to all quantified transcripts (Extended Data Fig. 7g), while other gene-disrupted cell models (sgNONO–sgControl, sgControl–sgPSPC1 and sgNONO–sgPSPC1) did not display this type of enrichment for (R)-SKBG-1-regulated transcripts. Taken together, we interpret these data as supportive of a model where genetic disruption of NONO can be compensated for, at least in part, by elevations in other DBHS proteins and that (R)-SKBG-1 may circumvent this compensation by producing pharmacological effects that resemble the combined disruption of both NONO and SFPQ. We next sought to explore further how (R)-SKBG-1 engagement of NONO might mechanistically cause such gene expression and growth effects.
Covalent ligands stabilize NONO–mRNA interactions in cells
NONO functions in paraspeckles42, which are phase-separated subnuclear bodies organized by the binding of DBHS proteins to long noncoding RNAs43 and are thought to regulate gene expression through nuclear retention of specific mRNAs44. Immunofluorescence studies revealed that NONO ligands ((R)-SKBG-1 and compound 5), but not inactive control compounds ((S)-SKBG-1 and compound 8), induced the formation of large subnuclear foci of NONO that were detectable by 1 h and prevalent by 4 h and continued to intensify up to 24 h after treatment (Fig. 5a,b and Extended Data Fig. 8a). The NONO foci colocalized near the nucleolar marker fibrillarin (Extended Data Fig. 8b–e) and also contained PSPC1 and SFPQ, albeit less frequently and prominently than NONO (Extended Data Fig. 8f–h). Previous studies have shown that DBHS family proteins continuously traffic between paraspeckles and perinucleolar caps43, suggesting that NONO ligands may disrupt this normal, dynamic trafficking, leading to accumulation of NONO in perinucleolar bodies.
Fig. 5 |. Effects of (R)-SKBG-1 on NONO localization and RNA interactions.

a, Time-dependent effects of (R)-SKBG-1 and (S)-SKBG-1 (20 μM) on the localization of NONO in nuclear foci (yellow wedges) in 22Rv1 cells. NONO was imaged with a primary antibody (Bethyl Laboratories) and an Alexa 488 secondary antibody; scale bar, 20 μm. b, Quantification of NONO nuclear foci induced by (R)-SKBG-1 and (S)-SKBG-1. The percentage of cells with NONO foci was determined manually in a blinded manner. Data are shown as mean values ± s.e.m. for five representative images from two independent experiments where 17–124 cells (median: 58 cells) were analyzed per image. c, Volcano plot comparing enrichment of mRNAs stereoselectively decreased by (R)-SKBG-1 in 22Rv1 cells (as described in Fig. 3a) among the RNAs interacting with 104 RBPs in previous eCLIP experiments performed in HepG2 cells45,46. A bound transcript was defined as ≥1 eCLIP IDR peak for a given RBP. d,e, eCLIP–seq significant peak quantity and regional distribution (d) and relative information content (e) determined for transcripts bound to NONO in 22Rv1 cells treated with (R)-SKBG-1 and (S)-SKBG-1 (20 μM, 4 h). Relative information defined as , where and are the fractions of total reads of a given transcript in the NONO immunoprecipitation and SM-Input, respectively, that map to element . Results are from a single experiment representative of two independent experiments; UTR, untranslated region; CDS, coding sequence; miRNA, microRNA; SS, splice site. f, Effects of (R)-SKBG-1 and (S)-SKBG-1 (20 μM, 4 h) on alternative splicing events in sgControl and sgNONO 22Rv1 cells, as determined by RNA-seq. Bars indicate whether the alternative splicing event was included or excluded, respectively, by the indicated first condition compared to the second condition listed on the y axis. Significant alternative splicing events had an FDR of <0.1 and an | InclusionLevelDifference | of >0.05 (n = 3); SE, skipped exons; MXE, mutually exclusive exons; A5SS, alternative 5′ splice site; A3SS, alternative 3′ splice site; RI, retained intron. g, Trapping model for how covalent ligands targeting NONO C145 affect mRNA processing in cancer cells and subvert the compensatory action of paralogous proteins PSPC1 and SFPQ.
We next investigated whether NONO ligands promote or stabilize NONO binding to other RBPs or RNAs. We first determined by coimmunoprecipitation–MS experiments of 22Rv1 cells stably expressing a FLAG epitope-tagged NONO that (R)-SKBG-1 did not alter the extent of association of NONO with PSPC1 or SFPQ compared to cells treated with DMSO or (S)-SKBG-1 (Extended Data Fig. 9a). This result may be consistent with the location of C145 in the NONO structure (Fig. 2c), which appears better positioned to influence NONO interactions with RNAs than associated DBHS partners.
Leveraging publicly available enhanced UV cross-linking and immunoprecipitation followed by sequencing (eCLIP–seq) datasets from HepG2 cells that evaluated 104 total RBPs45,46, we observed that transcripts stereospecifically decreased by (R)-SKBG-1 in 22Rv1 cells were statistically enriched in RNA targets of NONO (odds ratio of 7.8, false discovery rate (FDR) of <10−5), SFPQ (odds ratio of 9.7, FDR of <10−5) and MATR3 (odds ratio of 7.6, FDR of <10−5), which is another NONO-binding protein47, as well as a handful of other RBPs implicated in mRNA maturation and splicing (HNRNPM (odds ratio of 7.5, FDR of <10−5), SUGP2 (odds ratio of 7.2, FDR of <10−5) and QKI (odds ratio of 7.0, FDR of <10−5; note that PSPC1 eCLIP data were not available; Fig. 5c, Extended Data Fig. 9b and Supplementary Dataset 4). By contrast, randomly selected sets of 22Rv1 transcripts of similar number and expression level to (R)-SKBG-1-decreased mRNAs were not enriched for binding NONO or SFPQ (Extended Data Fig. 9c). These results support that (R)-SKBG-1 preferentially suppresses the expression of transcripts that are direct targets of NONO and NONO-related protein complexes.
To evaluate the effects of (R)-SKBG-1 on RNA interactions of NONO, we performed NONO eCLIP–seq experiments in 22Rv1 cells treated with DMSO, (R)-SKBG-1 or (S)-SKBG-1 (20 μM compound, 4 h) with accompanying size-matched input (SM-Input) controls46. These experiments revealed substantially more NONO–RNA interaction peaks in (R)-SKBG1-treated cells than observed in DMSO- or (S)-SKBG-1-treated cells, the majority of which occurred within proximal and distal intronic regions (Fig. 5d and Supplementary Dataset 5). A motif analysis revealed that NONO-binding sites enhanced by (R)-SKBG-1 generally maintained the G/U-rich sequence motifs previously described for NONO and SFPQ28 (Extended Data Fig. 9d). These data indicated that (R)-SKBG-1 strengthens, but does not substantially alter, the sequence preferences of RNA binding by NONO.
(R)-SKBG-1 did not appear to alter the broad categories of transcript types and regions bound to NONO (Extended Data Fig. 9e and Supplementary Dataset 5), but instead, a metagene analysis48 showed that (R)-SKBG-1 impacted the positional preference of NONO within these transcripts by enhancing interactions with the first exons and 5′ end of first introns of RNAs in comparison to (S)-SKBG-1 or DMSO controls (Fig. 5e). Interestingly, this effect was most pronounced for transcripts stereospecifically depleted by (R)-SKBG-1 (Extended Data Fig. 9f), including AR (Extended Data Fig. 9g), compared to transcripts bound to NONO in eCLIP–seq experiments but not decreased in abundance in (R)-SKBG-1-treated cells (Extended Data Fig. 9f–h). Lastly, we observed that a statistically enriched fraction of transcripts specifically decreased by (R)-SKBG-1 were direct RNA targets of NONO in (R)-SKBG-1-treated cells (Extended Data Fig. 9i and Supplementary Dataset 5; chi-squared test, FDR < 10−5). Notably, these (R)-SKBG-1-regulated transcripts were also bound to NONO in control cells (DMSO- or (S)-SKBG-1-treated cells; Extended Data Fig. 9i and Supplementary Dataset 5), supporting that they represent physiological NONO–RNA interactions (versus ‘neo’ interactions induced by (R)-SKBG-1).
Deep RNA-seq and rMATs analysis revealed that NONO ligands also perturbed mRNA splicing. For instance, we quantified substantially more included and excluded exon events in (R)-SKBG-1-treated sgControl cells than in DMSO- or (S)-SKBG-1-treated sgControl cells or (R)-SKBG-1-treated sgNONO cells (Fig. 5f and Supplementary Dataset 6). Further analyses revealed that the (R)-SKBG-1-regulated exon events did not demonstrate significant differences in splice site sequence composition (Extended Data Fig. 10a,b) or splice site strength (Extended Data Fig. 10c) compared to control unchanged exons. We next used the algorithm HOMER to identify motifs in regions surrounding the cassette exons, which revealed that the inclusion events promoted by (R)-SKBG-1 displayed an enrichment of AU-rich sequences in the neighboring intronic regions and within the cassette exon itself compared to unchanged exons (Extended Data Fig. 10d). Finally, analysis of NONO’s positional preferences proximal to (R)-SKBG-1-induced splicing perturbations using the RBP-Maps tool indicated that these events were unlikely to be caused by direct alterations of NONO binding to the perturbed splice sites (Extended Data Fig. 10e). However, the regions surrounding the cassette exons included after (R)-SKBG-1 treatment with ENCODE HepG2 eCLIP irreproducible discovery rate (IDR) peaks revealed an enrichment of binding for BCLAF1 and KHSRP (Extended Data Fig. 10f). BCLAF1 does not have a well-known motif, while KHSRP is documented to bind AU-rich elements49. Interestingly, our RNA-seq analyses revealed that BCLAF1 expression is stereoselectively decreased by (R)-SKBG-1 in a NONO-dependent manner, along with several other splicing and transcriptional regulatory factors (Supplementary Dataset 2). It is therefore possible that some of the splicing changes caused by (R)-SKBG-1 reflect a larger remodeling of RBP/ splicing/transcriptional networks.
Discussion
We report herein the identification of chemical probes that site-specifically and stereoselectively react with the RBP NONO to remodel the transcriptomes and proteomes of cancer cells through an unusual mechanism involving the ligand-induced accumulation of NONO in nuclear foci and at the 5′ ends of immature transcripts. The transcriptomic changes caused by NONO ligands occurred within 4 h after compound treatment, indicating the rapid turnover potential of mRNAs when their normal processing and maturation is disrupted. That these effects correlate with a blockade in the growth of cancer cells from diverse lineages suggests a broad impact of the NONO chemical probes on cell proliferation. While this could limit translational paths to exploit this pharmacological mechanism for cancer therapy, if, for instance, normal cell growth is also affected, we wonder whether deeper exploration of the SAR of NONO ligands might uncover compounds that have more restricted effects on the transcriptome by, for instance, stabilizing a subset of the NONO–mRNA interactions impacted by (R)-SKBG-1. Toward this end, understanding if NONO ligands physically bind to not only NONO but also its RNA interactors is an important future objective, as this type of ‘molecular glue’50 mechanism has been described for the pateamine A and rocaglate natural products that bind at eIF4A1–RNA interfaces to differentially regulate the translation of specific mRNAs11,12 and for the spinal muscular atrophy drug candidate branaplam, which stabilizes the SMN2 premRNA and U1 small nuclear ribonucleic protein complex13. If the current set of chemical probes could also be converted into pure antagonists that prevent rather than stabilize NONO binding to RNA, such compounds might display preferential antiproliferative activity in cancer cells harboring mutations in other DBHS proteins (PSPC1 and SFPQ), as suggested by the Cancer Dependency Map.
Other areas for potential improvement of NONO ligands include, for instance, substituting the α-chloroacetamide with a less reactive electrophile or even converting such ligands to reversibly binding compounds to improve selectivity across the proteome. Our limited initial exploration of these concepts has not yet yielded active compounds (Extended Data Fig. 1f,g), but screening larger small-molecule libraries for binding to the NONO C145 pocket may prove more fruitful. At present, we believe (R)-SKGB-1, when used in combination with additional active and inactive compounds and cells that have been genetically disrupted for NONO (and ideally reconstituted with the NONOC145S mutant), can serve as a versatile chemical probe for studying NONO function in diverse biological contexts.
From a biological perspective, it is interesting to consider whether the mRNAs regulated by NONO ligands represent physiological or novel (neo) interactions with NONO. Given that the mRNAs stereoselectively decreased by (R)-SKBG-1 showed a clear enrichment for binding to NONO compared to other RBPs in eCLIP–seq experiments (Fig. 5c) and that these mRNAs were consistently bound to NONO across different treatment groups ((R)-SKBG-1 and control (DMSO and (S)-SKBG-1) groups; Extended Data Fig. 9i), we hypothesize that a substantial fraction of these transcripts constitute physiological interactors with NONO that are perturbed in their normal processing by ligand-induced trapping in NONO complexes. In this trapping model (Fig. 5g), elevations in PSPC1 and/or SFPQ are unable to compensate for alterations in NONO function because the covalently liganded NONO remains bound to RNAs, ultimately leading to their impaired processing and degradation. Our discovery that the NONOC145S mutant can attenuate the activity of (R)-SKBG-1 in cancer cells coexpressing endogenous NONOWT further suggests that the stabilization of NONO–RNA interactions by (R)-SKBG-1 is likely reversible (for example, representing a ‘kinetic trap’), such that a ligand-insensitive variant of NONO can restore a substantial proportion of the normal transcriptional output of cancer cells. However, the rare (R)-SKBG-1-regulated transcripts that were insensitive to coexpression of the NONOC145S mutant might reflect highly stabilized or even neo interactions with (R)-SKBG-1-bound NONOWT that cannot be rescued by NONOC145S coexpression.
Our studies of cancer cells with various combinations of genetic disruptions of DBHS proteins suggest that the transcriptional and growth effects of NONO ligands most closely resemble those caused by dual loss of NONO and SFPQ. Whether this implies that NONO ligands preferentially stabilize RNA interactions with NONO–SFPQ heterodimers is an interesting question for future investigation. Another intriguing topic for further consideration is the mechanism for elevations in PSPC1 and SFPQ expression in cells with pharmacological or genetic perturbations of NONO. Until the present work, these changes in NONO paralogs had only been observed in constitutive genetic models of NONO loss39,40, but we have found that elevations in PSPC1 and SFPQ occur rapidly, within 24 h of treatment with (R)-SKBG-1. Examination of our eCLIP data did not reveal direct binding of NONO or (R)-SKBG-1-modified NONO to PSPC1 or SFPQ transcripts, suggesting that an alternative mechanism may underlie this cross-talk among DBHS paralogs.
Contemplating the broader implications of our findings, we believe that they highlight the value of selective chemical probes for studying proteins with functions that may be obscured following genetic disruption due to the compensatory actions of paralogs. More generally, our work points to covalent chemistry as a potentially versatile source of chemical probes for RBPs that play important roles in sculpting the transcriptional landscape of cancer cells.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-023-01270-0.
Methods
Antibodies
The following antibodies were used: rabbit anti-NONO (Bethyl Laboratories, A300–587A), mouse anti-NONO (BD Biosciences, 611278), rabbit anti-AR (Cell Signaling Technology, 5153S), anti-actin-horseradish peroxidase (HRP; Santa Cruz Biotechnology, sc-47778 HRP), mouse anti-PSPC1 (Sigma-Aldrich, SAB4200503), mouse anti-SFPQ (Sigma-Aldrich, WH0006421M2), mouse anti-FLAG (Sigma-Aldrich, F1804), rabbit anti-fibrillarin (Cell Signaling Technology, 2639S), anti-mouse HRP (Cell Signaling Technology, 7076S), anti-rabbit HRP (Cell Signaling Technology, 7074S), anti-rabbit Alexa 488 (Invitrogen, A11034), anti-mouse Alexa 488 (Invitrogen, A32723), anti-rabbit Alexa 647 (Invitrogen, A32733), anti-mouse Alexa 647 (Invitrogen, A21236) and anti-AR Alexa 488 (Cell Signaling Technology, 7395).
Cell culture
The 22Rv1, MCF7, HT1080, HEK293T and HeLa cell lines were obtained from ATCC and grown and maintained in RPMI-1640 medium (Thermo Fisher Scientific) containing 10% (vol/vol) fetal bovine serum (FBS; Omega Scientific) and GlutaMAX (22Rv1 cells) or DMEM (Thermo Fisher Scientific) containing 10% (vol/vol) FBS (Omega Scientific), 2 mM l-glutamine and 100 U ml–1 penicillin–streptomycin (GE Life Sciences; MCF7, HT1080, HEK293T and HeLa cells) at 37 °C with 5% CO2. For QuantiGene and high-content imaging assays, 22Rv1 cells were cultured in RPMI-1640 (32404014, Thermo Fisher) medium supplemented with 10% FBS (S181H-500, Biowest), 2 mM glutamine (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), 4.5 g liter–1 glucose (D8769, Sigma-Aldrich), 10 mM HEPES (Sigma-Aldrich) and 50 μg ml–1 gentamycin (Sigma-Aldrich).
Cloning and mutagenesis
Plasmid amplification and purification.
All plasmids were amplified in Stbl3 cells (Thermo Fisher) using the vendor’s procedures and were purified by miniprep (QIAGEN), with the exception of the pRK5 and pLenti6.2-ccdB-3×FLAG-V5 Gateway destination vectors, which were amplified in ccdB Survival T1 cells (Invitrogen).
Mutagenesis.
Site-directed mutagenesis was performed on the pDONR 223-NONO construct from the human ORFome V8.1 library (Dharmacon) using the Quikchange mutagenesis kit (Agilent). All mutations were verified by DNA sequencing. See Supplementary Table 5 for mutagenesis primers.
Plasmids for transient expression.
The desired pDONR 223-NONO construct was cloned into a pRK5-derived plasmid generated using the Gateway vector conversion system (Invitrogen). Final plasmids contained a cytomegalovirus (CMV) promoter followed by a Gateway cloning linker, a start methionine, an N-terminal FLAG tag, the NONO open reading frame and a second Gateway cloning linker. A stop codon was introduced at the NONO C terminus to prevent translation of extra amino acids from the Gateway linker. All gene constructs were verified by DNA sequencing.
Plasmids for stable expression.
The desired pDONR 223-NONO construct was cloned into pLenti6.2-ccdB-3×FLAG-V5 (Addgene, 87071) using the Gateway vector conversion system (Invitrogen). Final plasmids contained a CMV promoter followed by a Gateway cloning linker, a start methionine, the NONO open reading frame without a stop codon, a C-terminal FLAG tag and a second Gateway cloning linker. All gene constructs were verified by DNA sequencing.
Generation of the 22Rv1 shRNA AR-FL cell line
Cultures of 22Rv1 cells were diluted to 125,000 cells per ml in RPMI1640 medium, and 400 μl of cell suspension was added to each well of a 24-well microtiter plate. Lentiviral vectors containing shRNAs targeting AR exon 8 (AR-FL, PLKO.5-puro (vector) CCGGCCAGC-CACACAAACGTTTACTCTCGAGAGTAAACGTTTGTGTGGCTG-GTTTTTG), negative-control shRNA or no shRNA was added in duplicate to each well in a volume of 5 μl per well. Plates were incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. To one of each duplicate well, either 400 μl of RPMI-1640 medium containing 2 μg ml–1 puromycin (A1113803, Thermo Fisher) or 400 μl of medium was added and incubated an extra 3 d. Subsequently the transduction efficiency was determined by adding Alamar Blue (Thermo Fisher) to the wells (10% final concentration), and cells were incubated for 4 h at 37 °C and 5% CO2. One hundred microliters of each supernatant was added to the wells of a 96-well black-walled, clear-bottom plate, and fluorescence was measured in a Spectramax M2 microplate reader (Molecular Devices). Transduction efficiency (%) was calculated as the percentage of Alamar Blue signal measured under the condition with puromycin divided by the no-puromycin condition.
NONO knockout by CRISPR–Cas9
NONO-knockout cells were generated using previously described protocols51. In brief, non-targeting control sgRNAs or sgRNAs targeting NONO (see Supplementary Table 5 for sequences) were designed (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design ) and cloned into Lenti-CRISPR v2 plasmid (Addgene). One microgram of sgRNA-encoding plasmids was cotransfected with ΔVPR envelope (0.9 μg) and CMV VSV-G (0.1 μg) packaging plasmids into 2.5 × 106 HEK293T cells using Fugene 6 transfection reagent (Promega). Virus-containing supernatants were collected 48 h after transfection, passed through a 0.45-μm filter and used to infect target cells in the presence of 10 μg ml–1 polybrene (Santa Cruz). Twenty-four hours after infection, fresh medium was added to the target cells, which were allowed to recover for an additional 24 h. Puromycin (1 μg ml–1) was then added to cells. Following 10 d of puromycin selection, NONO expression in NONO sgRNA cells was determined by Western blotting compared to control sgRNA-transfected cells. For 22Rv1 cells, NONO sgRNA 5 was used for further experiments. For MCF7 cells, NONO sgRNA 1 and sgRNA 3 were used for further experiments.
Alt-R CRISPR knockout of PSPC1 and SFPQ
Control sgRNA and NONO sgRNA 22Rv1 cells described above were treated with control guide RNA or two predesigned guide RNAs (IDT) targeting PSPC1 or SFPQ (see Supplementary Table 5 for sequences) and Alt-R s.p. Hifi Cas (IDT, 1081061). Each gRNA was resuspended to 100 μM in nuclease-free water. The ribonucleoprotein (RNP) complex was formed by adding PBS (0.9 μl), corresponding sgRNAs (2.4 μl each) and Cas9 (2 μl) to a 1.5-ml Eppendorf tube and left to incubate at room temperature for 30 min. During this incubation, 22Rv1 cells were trypsinized from a 15-cm plate and counted. Five million cells were collected for each treatment condition and washed with PBS. The cells were resuspended in 100 μl of nucleofection buffer from the Lonza cell line nucleofector kit V (VCA-1003) and added to the RNP complex in an Eppendorf tube. One hundred microliters was then transferred to the nucleofection cuvette carefully without introducing bubbles, and the ATCC program (X-001) was run on the Lonza electroporator. Cells were then transferred to a 10-cm plate that contained medium with no antibiotics. After 24 h, medium was changed to medium containing antibiotics.
Generation of 22Rv1 NONO C-FLAG WT or C145S cell lines (coexpression with endogenous NONO and knockout of endogenous NONO lines)
One microgram of pLenti6.2-NONO-C-FLAG-encoding plasmid (WT or C145S) with a silent PAM site mutation was cotransfected with ΔVPR envelope (0.9 μg) and CMV VSV-G (0.1 μg) packaging plasmids into 2.5 × 106 HEK293T cells using Fugene 6 transfection reagent (Promega). Virus-containing supernatants were collected 48 h after transfection, passed through a 0.45-μm filter and used to infect target cells in the presence of 10 μg ml–1 polybrene (Santa Cruz). Twenty-four hours after infection, fresh medium was added to the target cells, which were allowed to recover for an additional 24 h. Blasticidin (10 μg ml–1) was then added to cells. Following 10 d of blasticidin selection, NONO– FLAG expression was determined by western blotting and compared to that in empty vector-transfected cells. If indicated, endogenous NONO was then knocked out by CRISPR–Cas9 using NONO sgRNA 5 as described above.
QuantiGene assay for AR mRNA expression screen
Parental 22Rv1 cells or 22Rv1 cells with stable expression of shRNA targeting AR-FL were seeded at 15,000 cells per well in 384-well plates and 24 h later were treated with compounds in 0.2% DMSO or with 0.2% DMSO only. After 6 h of treatment, cells were directly collected by adding 0.5 volumes of the QuantiGene lysis mixture (Thermo Fisher) and processed immediately for hybridization or stored at −80 °C.
The QuantiGene assay was performed according to the manufacturer’s protocol. Briefly, 40 μl of the prewarmed cell lysate was incubated with blocking reagent, protease K, capture beads and the probe set overnight at 54 °C. The next day, the beads were washed, and the signal was amplified using Branch DNA technology by serial incubation with preamplification, amplification and labeling reagent for 1 h at 50 °C each and visualization by incubation with SAPE for 30 min at room temperature, as indicated by the manufacturer’s protocol. Beads were measured using FLEXMAP 3D. Data interpretation followed a three-step normalization/transformation flow in which raw median fluorescent gene signals were transformed to fold change values with the use of housekeeping genes (SUCLA2 and TCEB2) and then transformed to expression (percentage of DMSO) by normalizing to DMSO controls. This in-house-developed method was previously published52, and the R-based analysis tool QGProfiler has been made publicly available at https://qgprofiler.openanalytics.eu/app/QGprofiler.
QuantiGene probes and blocking reagents were designed and provided by Thermo Fisher using the following RefSeq ID numbers: AR-FL, NM_000044, Homo sapiens AR transcript variant 1; AR-V7, HM055487, H. sapiens AR isoform 8 (AR8); SUCLA2, NM_003850, H. sapiens succinate-CoA ligase ADP-forming β-subunit (SUCLA2) and TCEB2, NM_007108, H. sapiens elongin B (ELOB), transcript variant 1.
High-content imaging
Cultures of 22Rv1 cells were seeded in 384 Cell Carrier Ultra plates (6057300, PerkinElmer) at 5,000 cells per well in 30 μl of medium and incubated overnight at 37 °C. On the following day, cells were treated with compounds (final DMSO = 0.2% (vol/vol)) and incubated for 24 h. Cells were fixed by incubation with 4% formaldehyde for 20 min. The medium was completely removed, and the cells were permeabilized by the addition of 20 μl of ice-cold methanol for 10 min at 20 °C, followed by extensive washing using 1× PBS (11666789001, Roche) to remove the methanol. The cells were incubated with AR antibody conjugated with Alexa 488 (Cell Signaling Technology) in 1× PBS supplemented with 5% bovine serum albumin (wt/vol; Sigma) overnight at 20 °C in the dark. The cells were washed three times with 1× PBS and incubated with 0.625 μg ml–1 CellMask Deep Red (Thermo Fisher) and 2 μg ml–1 Hoechst 33258 (Thermo Fisher) in 1× PBS for 1 h at 20 °C in the dark followed by three washes with 1× PBS. The cells were imaged on a Cell Voyager 7000 (Yokogawa) using a ×40/0.95-NA dry objective lens.
PerkinElmer Acapella/Columbus image analysis software was used to identify the boundaries of individual nuclei based on the Hoechst channel and the surrounding cytoplasm based on the CellMask Deep Red channel. Mean intensity of AR expression per cell median aggregated over all cells per well and cell counts per well were then calculated. These measurements together with the original images and segmentation masks of the nuclei and cells were imported to Phaedra53 (www.phaedra.io) for visual and statistical quality control and plate-wise normalization as a percentage of the median DMSO control value.
Western blotting
For western blotting experiments, 2 × 106 cells per treatment were seeded in a 10-cm plate for 24 h. Medium was then aspirated and replaced with 5 ml of medium containing the indicated compounds or 0.04% DMSO for the indicated times. Following this incubation period, the cells were washed in cold DPBS, scraped, collected and pelleted in 1.5-ml tubes. Cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C until further analysis. On the day of the analysis, the cell pellets were thawed on ice, resuspended in cold PBS (300–400 μl) and lysed by sonication with a Branson probe sonicator (two sets of eight pulses, output of 4 and duty cycle of 40%). Protein concentrations for all samples were measured by DC assay (Bio-Rad) and adjusted to 1.5 mg ml–1, 4× loading buffer was added, and the samples were heated at 95 °C for 5 min, followed by centrifugation for 1 min at 20,000g. Proteins were resolved by SDS–PAGE (10% acrylamide gel) and transferred to 0.45-μm nitrocellulose membranes (GE Healthcare). The membrane was blocked with 5% milk in Tris-buffered saline (20 mM Tris-HCl (pH 7.6) and 150 mM NaCl) with 0.1% Tween 20 (TBST) buffer at room temperature for 1 h or at 4 °C overnight, washed with TBST and incubated with primary antibodies in 5% milk in TBST at 4 °C overnight. All primary antibody dilutions were 1:1,000 except for actin-HRP (1:2,500) and mouse anti-FLAG (1:3,000). Following additional TBST washes (three 3-min washes), the membrane was incubated with secondary antibody (1:3,000 in 5% milk in TBST) at room temperature for 1 h. The membrane was washed with TBST (three 3-min washes) and developed with a femto (for AR, SFPQ and PSPC1) or standard (for NONO and actin) ECL western blotting detection reagent kit (Thermo Scientific), and chemiluminescence was imaged on a ChemiDoc MP system (Bio-Rad). Relative band intensities were quantified using Image Lab software (Bio-Rad).
For western blot AR analysis in shRNA AR-FL 22Rv1 cells, cells were lysed in MPER buffer (Thermo Fisher) supplemented with phosphatase/ protease inhibitor cocktail (Thermo Fisher), 0.5 mM EDTA (pH 8.0; Thermo Fisher) and benzonase (Sigma). After clearance by centrifugation at 20,817g for 10 min at 4 °C, the lysate was analyzed in WES (Protein Simple) according to the manufacturer’s protocol. Briefly, the lysate was diluted in 0.1× sample buffer and 1× fluorescence master mix. The samples and biotinylated ladder were denatured by incubating at 70 °C for 10 min and were added to an assay plate. Primary antibodies diluted in antibody diluent 2 and luminol-S and peroxide mix were added to the appropriate wells, and the plate was loaded onto WES after centrifugation at 1,100g for 5 min at 20 °C. Data were analyzed using CompassX software (Protein Simple).
Gel-based activity-based protein profiling
HEK293T cells (5 × 105) were seeded into a six-well plate and allowed to adhere overnight at 37 °C. One microgram of empty pRK5 vector or pRK5-NONO N-FLAG WT or C145S was transfected using PEI (1 mg ml–1, linear 40,000 molecular weight) for 48 h. Transfected cells were then treated with DMSO (0.1%) or the indicated compound for 2 h in situ, followed by treatment with 20 μM B21 alkyne probe in situ for 1 h at 37 °C. Following this incubation period, the cells were washed in cold DPBS, scraped, collected and pelleted in 1.5-ml tubes (2,000g, 5 min, 4 °C). Cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C until further analysis. On the day of the analysis, the cell pellets were thawed on ice, resuspended in cold PBS (with Roche cOmplete, mini, EDTA-free protease inhibitor cocktail, 1 tablet in 10 ml of PBS) and lysed by sonication with a Branson probe sonicator (eight pulses, output of 3 and duty cycle of 30%). Protein concentrations for all the samples were measured by DC assay (Bio-Rad) and adjusted to 2 mg ml–1. TAMRA-PEG3-N3 (1 μl per reaction, 1.25 mM in DMSO), CuSO4 (1 μl per reaction, 50 mM in water), tris(benzyltriazolylmethyl)amine ligand (TBTA; 3 μl per reaction, 1.7 mM in DMSO/tert-butanol (1:4 (vol/vol))) and tris(2-carboxyethyl)phosphine (TCEP; 1 μl per reaction, 50 mM in water, freshly prepared) were premixed. Six microliters of this click reagent mixture was immediately added to 50 μl of each alkyne probe-labeled sample and incubated for 1 h at room temperature. The reactions were quenched by adding 4× SDS–PAGE loading buffer. The quenched samples were loaded on a 10% acrylamide gel for separation by SDS–PAGE. Samples were visualized by in-gel fluorescence scanning using the ChemiDoc MP system (Bio-Rad), and fluorescent band intensity was quantified with Image Lab software (Bio-Rad).
Cysteine-directed mass spectrometry-activity-based protein profiling
For cysteine-directed MS-ABPP, 2 × 106 cells per treatment were seeded in a 10-cm plate and grown for 48 h. Medium was then aspirated and replaced with medium containing the indicated compounds or 0.04% DMSO for the indicated times. Following this incubation period, the cells were collected, lysed and normalized to 2 mg ml–1 protein in 500 μl of PBS, as described above. Remaining sample preparation, tandem mass tag (TMT) labeling, fractionation, liquid chromatograhy–MS (LC–MS) analysis and data analysis and filtering were conducted as previously described26.
Whole proteomics
For the whole proteomics studies, 2 × 106 cells per treatment were seeded in a 10-cm plate and grown for 24 h. Medium was then aspirated and replaced with 5 ml of medium containing the indicated compounds or 0.04% DMSO for the indicated times. Following this incubation period, the cells were collected and lysed as described above. Remaining sample preparation, TMT labeling, fractionation, LC–MS analysis and data analysis and filtering were conducted as previously described26.
Enriched proteomics with alkyne probe 14
A total of 2 × 106 cells per treatment were seeded in a 10-cm plate and grown for 48 h. Medium was then aspirated and replaced with 5 ml of medium containing the indicated compounds or 0.04% DMSO for 2 h. Alkyne probe 14 (20 μM) or 0.04% DMSO was then added for 1 h. Following this incubation period, the cells were collected, lysed and normalized to 2 mg ml–1 protein in 500 μl of PBS as described above.
Modified proteins in 500 μl of lysates were then conjugated to biotin-azide. Biotin-PEG4-azide (ChemPep; 5 μl of 10 mM stock in DMSO), TBTA (30 μl of 1.7 mM stock in DMSO:tert-butanol (1:4)), TCEP (10 μl of fresh 50 mM stock in water) and CuSO4 (10 μl of 50 mM stock in water) were combined in an Eppendorf tube, vortexed and added to the proteomes (55 μl of solution per 500-μl sample). The reaction was allowed to proceed at room temperature for 1 h, and the samples were each added to a separate 15-ml conical Falcon tube on ice containing 4 ml of cold methanol, 1 ml of CHCl3 and 2 ml of water. Eppendorf tubes from the reaction mixtures were washed with additional water (1 ml of each), and the washes were added to the same Falcon tube (final ratio of methanol:CHCl3:water = 4:1:4). Following centrifugation (5,000g, 10 min, 4 °C), a protein disk formed at the interface of CHCl3 and aqueous layers. Both layers were aspirated without perturbing the disk, which was resuspended in cold methanol (2 ml) and CHCl3 (1 ml) by vortexing. The proteins were pelleted (5,000g, 10 min, 4 °C), and 500 μl of PBS containing 6 M urea was added to the resulting pellets. A 50-μl mixture of 100 mM TCEP and 300 mM K2CO3 was then added, followed by 30 μl of 10% SDS in PBS. The pellets were resuspended with sonication and heated at 37 °C in a shaker for 30 min. Then, 70 μl of 400 mM iodoacetamide in PBS was added to each sample, and the mixture was incubated in the dark at room temperature for 30 min. Samples were then rotated with streptavidin beads for 2 h at room temperature. Beads were then washed with 0.2% SDS (1×), PBS (2×) and water (2×) with centrifugation in between each wash. Beads were then transferred to a 1.5-ml Lo-Bind Eppendorf tube in 1 ml of water and pelleted. Twenty micrograms of trypsin was resuspended in 2 ml of 200 mM EPPS containing 2 M urea. Twenty microliters of 100 mM CaCl2 in water was added to the trypsin solution. Two hundred microliters of the trypsin solution was added to the beads in each Eppendorf tube, and proteins were digested by shaking at 37 °C for 14 h. Beads were removed from digests by Bio-Spin filtration columns. Subsequent TMT labeling, desalting with peptide desalting spin columns (Thermo, 89852), LC–MS analysis and data analysis and filtering were conducted as previously described26.
NONO immunoprecipitation–mass spectrometry
Cultures of 22Rv1 cells stably expressing C-Flag NONOWT or empty vector control were treated in situ with DMSO, 20 μM (R)-SKBG-1 or 20 μM (S)-SKBG-1 for 4 h. Cells were collected, washed with ice-cold PBS and lysed in immunoprecipitation lysis buffer (50 mM NaHPO4, 150 mM NaCl, 1.5 mM MgCl2, 1% NP-40 and 1× protease inhibitor). Lysates were normalized to 3 mg ml–1, and 500 μl of each lysate was aliquoted to a clean Eppendorf tube.
Lysates were incubated with 25 μl of Pierce anti-DYKDDDDK magnetic agarose beads overnight at 4 °C. Beads were washed three times with immunoprecipitation washing buffer (50 mM NaHPO4, 150 mM NaCl and 0.2% NP-40) and then twice with PBS, followed by elution with 8 M urea in PBS at 65 °C for 10 min. Eluates were reduced with DTT at 65 °C for 15 min (2.5 μl of 200 mM = 12.5 mM final concentration), alkylated with iodoacetamide at 37 °C for 30 min (2.5 μl of 400 mM = 25 mM final concentration) and diluted to 2 M urea by the addition of 115 μl of EPPS (pH 8.0; total volume = 160 μl). Samples were then trypsinized at 37 °C overnight (2 μg of trypsin per sample).
Seventy microliters of acetonitrile was added to each sample (30% final, 230 μl), TMT labeled with 6 μl of corresponding TMT tag (20 μg μl–1), vortexed and incubated at room temperature for 1 h and 15 min. TMT labeling was quenched with the addition of hydroxylamine (6 μl, 5% solution in water) and incubated for 15 min at room temperature. Samples were then acidified with 6 μl of formic acid, combined and split back into five tubes of approximately 500 μl and dried using a SpeedVac. Samples were resuspended to 600 μl with water and desalted and fractionated using peptide desalting spin columns (Thermo, 89852) to a final of three fractions and analyzed by MS as described previously26.
RNA-seq
Sample preparation.
Cells (2 × 106 cells per treatment) were seeded in 10 ml of medium in a 10-cm plate for 24 h. Medium was then aspirated and replaced with 5 ml of medium containing the indicated compounds or 0.04% DMSO for 4 h or 24 h. Following this incubation period, the cells were washed in cold DPBS, scraped, collected and pelleted in 1.5-ml tubes (2,000g, 3 min, 4 °C). Cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C until further analysis. Total RNA from thawed cells was isolated using an RNeasy Plus kit (QIAGEN) with QIAshredder columns for cell lysis (QIAGEN) according to the manufacturer’s protocol and stored at −80 °C until further analysis. RNA concentration was measured by Nanodrop, and 1–2 μg was used for sequencing.
Library preparation with poly(A) selection and HiSeq sequencing.
Library preparations and sequencing reactions were conducted at GENEWIZ using the following protocol. Extracted RNA samples were quantified using a Qubit 2.0 fluorometer (Life Technologies), and RNA integrity was checked using an Agilent TapeStation 4200 (Agilent Technologies).
RNA-seq libraries were prepared using the NEBNext Ultra RNA library prep kit for Illumina following the manufacturer’s instructions (NEB). Briefly, mRNAs were first enriched with oligo(dT) beads. Enriched mRNAs were fragmented for 15 min at 94 °C. First-strand and second-strand cDNAs were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3′ ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on an Agilent TapeStation (Agilent Technologies) and quantified by using a Qubit 2.0 fluorometer (Invitrogen) and quantitative PCR (KAPA Biosystems).
The sequencing libraries were clustered on two lanes of a flowcell. After clustering, the flowcell was loaded on an Illumina HiSeq instrument (4000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150-base pair (bp) paired-end configuration. Image analysis and base calling were conducted by the HiSeq control software. Raw sequence data (.bcl files) generated from the Illumina HiSeq were converted into fastq files and demultiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Data analysis.
Any samples with >20% rRNA content were excluded from further analysis. Statistical analysis was performed in R (v4.0.4). Transcript abundance was quantified using Salmon (v1.3.0)54 with GENCODE v37 annotation. Gene level quantification was performed using tximeta (v1.8.4)55. Differential gene expression was analyzed by DESeq2 (v1.30.1)56. All log2 (fold change) values were estimated using apeglm (v1.12.0), and P values were adjusted using the Benjamini–Hochberg procedure to control for multiple hypothesis testing. Gene set enrichment analysis (GSEA) was implemented in fgsea (v1.16.0) using gene sets from MsigDB (v7.1)57. Redundant Gene Ontology terms from GSEA were removed with Revigo (http://revigo.irb.hr/).
Immunofluorescence
Approximately 150,000 cells were seeded onto UV-sterilized 6-mm circular glass coverslips in 1 ml of medium in a 24-well plate. Cells were grown for 24 h before treatment with 0.04% DMSO or the indicated compound for the indicated time. Cells were washed two times with PBS and fixed by incubating with 4% formaldehyde (Sigma-Aldrich) in PBS for 10 min at room temperature. Cells were then washed two times with PBS for 5 min at room temperature. Next, cells were permeabilized by incubating with 0.1% Triton X-100 (Thermo Fisher) in PBS for 30 min at room temperature. After permeabilization, the cells were washed two times with PBS and blocked with 10% goat serum (Thermo Fisher) for 1 h at room temperature. The blocking buffer was decanted, and primary antibodies in blocking buffer (1% goat serum in PBS) were added (primary antibody dilutions (vol/vol): AR, 1:300; fibrillarin, 1:400; NONO mouse, 1:300; NONO rabbit, 1:200; PSPC1, 1:300; SFPQ, 1:300). After incubation at 4 °C overnight, the cells were washed two times with PBS for 5 min at room temperature and incubated for 1 h at room temperature with the indicated Alexa Flour 488- or Alexa Fluor 647-conjugated secondary antibodies (Thermo Fisher; diluted 1:500 (vol/vol) in 1% goat serum (Thermo Fisher)) protected from light. The cells were then washed two times with PBS for 5 min at room temperature, dried for 5 min, mounted with Prolong Diamond antifade mountant containing DAPI (Life Technologies) and allowed to cure in the dark for 24 h. The slides were sealed with clear nail polish and visualized with a Zeiss LSM 880 microscope (25-mW multiline Ar laser for Alexa Flour 488, 5-mW HeNe 633 laser for Alexa Fluor 647 and 30-mW diode UV laser for DAPI) containing a ×63/1.4-NA oil α-Plan-Apochromat objective. Images were processed with ZEN 3.0 (blue edition) software (Zeiss).
Cell growth inhibition assays
Cells were seeded at 5,000 cells per well in white, clear-bottom 96-well plates (Greiner) for 16 h. Then, 0.1% DMSO or α-chloroacetamide compound in 0.1% DMSO (threefold dilution starting from 50 μM, eight concentrations) was then added for 4–6 d until DMSO control cells were confluent. At 4–6 d, plates were removed from the incubator and allowed to cool to room temperature before CellTiterGlo reagent (Promega) was added for 30 min. Luminescence was then measured on a Clariostar plate reader (BMG Labtech). Relative cell growth was determined by normalizing the luminescence reading to the DMSO-treated control. Viability curves were plotted in GraphPad Prism and fitted with a four-parameter variable slope curve to determine half-maximum effective concentration (EC50) values. Each plate contained four replicate wells per given compound concentration.
For growth assays with double-knockout cells, cells were seeded at 10,000 cells per well (100 μl of medium) in 96-well flat-bottom white plates 72 h after electroporation of RNP complex. After seeding, a reference plate to determine baseline at time zero was assayed using CellTiterGlo reagent (50 μl added to each well). One replicate plate was read every 24 h for 5 d. Raw values were normalized to time zero and graphed using GraphPad Prism.
Alternative splicing analysis with rMATS
RNA-seq libraries from compound-treated WT NONO and NONO-KO 22Rv1 cells from the protocol above were resequenced to achieve 80–100 million reads. RNA-seq reads were mapped to hg38 using STAR Aligner. Alternative splicing analysis was completed using rMATS (v3.2.5)58. All alternative splicing events were filtered using an FDR of <0.1 and inclusion level difference of >0.05. Numbers of events were then counted per comparison.
Comparison of mRNA affected by (R)-SKBG-1 to RBP binding profiles
Differentially expressed genes were identified as described above. Genes were grouped into three categories: increased (Padj < 0.01, log2 (fold change) ≥ 0.5, n = 82), decreased (Padj < 0.01, log2 (fold change) ≤ −0.5, n = 619) and no change (Padj > 0.5, | log2 (fold change) | < 0.5, n = 9,638). To identify if differentially expressed genes are enriched in NONO-binding sites, we downloaded all HepG2 eCLIP IDR peaks from the ENCODE portal (https://www.encodeproject.org/ )45. The IDR peaks represent the highest-confidence binding sites for each RBP. For each RBP, we counted how many transcripts from each group intersect with IDR peaks using pyBedTools 0.8.1 (bedtools: a powerful toolset for genome arithmetic — bedtools 2.30.0 documentation). A chi-squared test was used to determine if the decreased or increased genes were enriched in RBP binding sites. To control for potential confounders in both eCLIP–seq and RNA-seq experiments that may lead to spurious statistical enrichment, we randomly sampled from the transcripts of no differential expression 30 times, with matched expression level to the downregulated genes (discretize into ten equal-sized bins based on baseMean) and performed the same test.
Enhanced UV cross-linking and immunoprecipitation followed by sequencing
Library preparation.
A total of 2 × 106 cells per plate (two plates for each compound) were seeded in a 10-cm plate for 48 h. Medium was then aspirated and replaced with 5 ml of medium containing the indicated compounds or 0.04% DMSO for 4 h. Following this incubation period, medium was aspirated, and NONO–RNA interactions were stabilized with UV cross-linking (254 nm, 400 mJ cm–2) in cold DPBS, and cells were collected and frozen as described above. Thawed pellets were then lysed in 1 ml of iCLIP lysis buffer and digested with RNase I (Ambion). Immunoprecipitation of NONO–RNA complexes with NONO primary antibody (Bethyl; 10 μg per ml of lysate) was performed using magnetic beads with precoupled secondary antibody (M-280 sheep anti-rabbit IgG Dynabeads, Thermo Fisher Scientific, 11204D), and beads were stringently washed. After dephosphorylation with FastAP (Thermo Fisher) and T4 PNK (NEB), a barcoded RNA adapter was ligated to the 3′ end (T4 RNA Ligase, NEB). On-bead ligations were performed (to allow washing away of unincorporated adapter) in a high concentration of PEG8000. Samples were then run on standard protein gels and transferred to nitrocellulose membranes, and a region 75 kDa (∼220 nucleotides of RNA) above the protein size was isolated and proteinase K (NEB) treated to isolate RNA. RNA was reverse transcribed with Superscript III (Invitrogen) and treated with ExoSAP-IT (Affymetrix) to remove excess oligonucleotides. A second DNA adapter (containing a random-mer of 10 (N10) random bases at the 5′ end) was then ligated to the cDNA fragment 3′ end (T4 RNA Ligase, NEB), performed with high concentration of PEG8000 (to improve ligation efficiency) and DMSO (to decrease inhibition of ligation due to secondary structure). After cleanup (Dynabeads MyOne Silane, Thermo Fisher), an aliquot of each sample was first subjected to quantitative PCR (to identify the proper number of PCR cycles), and then the remainder was PCR amplified (Q5, NEB) and size selected via agarose gel electrophoresis. Samples were sequenced on an Illumina NovaSeq 6000 platform as two single-end 75-bp reads.
Data analysis.
After standard HiSeq demultiplexing, eCLIP libraries with distinct in-line barcodes were demultiplexed using custom scripts, and the random-mer was appended to the read name for later usage. Reads were then adapter trimmed (cutadapt v1.9.dev1), and reads less than 18 bp were discarded. Mapping was then first performed against human elements in RepBase (v18.05) with STAR (v2.4.0i), repeat-mapping reads were segregated for separate analysis, and all others were then mapped against the full human genome (hg19), including a database of splice junctions with STAR (v2.4.0i). Uniquely mapped reads were then run through a custom-built PCR duplicate removal script, removing duplicate reads based on sharing identical read 1 start position, read 2 start position and random-mer sequence to leave ‘Usable’ reads. eCLIP datasets with multiple in-line barcodes were merged at the usable read stage, and cluster identification was performed on usable reads using CLIPper11 (available at https://github.com/YeoLab/clipper/releases/tag/1.0) with options –s GRCh38 –o –bonferroni –superlocal–threshold-method binomial–save-pickle, considering read 2 only (the read that is enriched for termination at the cross-link site). For visualization on the Univeristy of California Santa Cruz Genome Browser, all tracks were reads per million normalized against the total number of usable reads in that dataset.
Finally, the number of post-PCR duplicate removal read pairs mapping to each class was counted in both the immunoprecipitation and paired input samples and normalized for sequencing depth (using the total number of post-PCR duplicate read pairs from both unique genomic mapping and repeat mapping as the denominator to calculate the fraction of reads). Significance of peaks was determined by Fisher’s exact test or Pearson’s chi-squared test if all expected and observed values were five or more. Peaks were filtered using a Padj of <10–3 and a fold change of >8.
Because the number of significant peaks is a function of read depth, the deduplicated reads were further downsampled to roughly equal numbers across the library using samtools 1.9. The peak calling procedure was performed in the same way with the downsampled BAM file.
Peak regional distribution.
Significant peaks were further annotated with the gene name and the transcriptomic regions (coding sequence, untranslated region and so on) using our in-house annotator pipeline (https://github.com/YeoLab/annotator). This pipeline prioritizes overlapping transcript regions. For example, if a feature overlaps both exon and introns, exons will be prioritized over introns.
eCLIP signal distribution analysis.
We realized that peaks called by our current pipeline are sometimes not sensitive enough; therefore, we have an alternative tool to analyze the distribution of eCLIP signals. In each transcript, relative information content of each nucleotide position was calculated as , where and are the fraction of total reads in immunoprecipitation and SM-Input, respectively, that map to element . Relative information content here serves as an approximation of binding distribution in the transcript by considering the non-specific backgrounds captured by the SM-Input library.
To compare transcripts in groups (that is, the (R)-SKBG-1 decreased RNAs and the no-change group), we generated the Metagene plots, where we averaged the relative information over all transcripts in a group. For each transcript feature, such as ‘first intron (the 5′-most intron)’ and ‘first exon’, we sliced a fixed-length window at the start and end of each feature. The Metagene plots were generated using a custom package Metadensity (https://github.com/YeoLab/Metadensity)48.
Motif analysis.
To test if (R)-SKBG-1 alters the sequence preference of NONO, we used the analyze_motif script in our in-house pipeline to find motifs (https://github.com/YeoLab/clip_analysis_legacy). Briefly, this pipeline starts by extracting sequences from the peaks and stratifies peaks by transcriptomic features (as they exhibit different sequence content). Finally, the HOMER motif-finding algorithm59 looks for global motifs and feature-specific motifs.
Repetitive element analysis.
Reads mapping to Repbase segregated from genome-mapped reads were quantified into families of repetitive elements (https://github.com/YeoLab/repetitive-element-mapping), including transcriptomic feature groups, such as proximal introns, exons and various species of small RNAspetitive elem and repeat elements. The fold enrichment values for each family were calculated by first normalizing the total reads in the repeat pipeline and then dividing the immunoprecipitation fraction to the SM-Input fraction.
Correlating alternative splicing events with eCLIP data.
To understand the relationship between NONO binding and (R)-SKBG-1-induced alternative splicing events, we performed RBP-maps analysis60 (https://github.com/YeoLab/rbp-maps), which summarizes the eCLIP read distribution around the significantly alternatively spliced exons/ introns and compares them to a set of control exons/introns. To test if the distribution of skipped exon events was biased toward the 5′ end, the relative position of exons (middle nucleotides normalized to gene length) was calculated for (R)-SKBG-1-specific skipped exon events and (S)-SKBR-1-induced skipped exon events. A two-sided Kolmogorov–Smirnov test was used to determine if the two distributions were different.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism version 9 for Windows (GraphPad Software; https://www.graphpad.com/) unless otherwise noted. Statistical values, including the n and statistical significance, are reported in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD039283 (cysteine MS-ABPP with 6 h of compound treatment and FLAG–NONO coimmunoprecipitation–MS) and PXD032087 (all other experiments). Raw RNA-seq and eCLIP–seq data have been deposited to NCBI under Gene Expression Omnibus accession codes GSE222217 and GSE198212, respectively. Processed proteomic data are provided in Supplementary Datasets 1 and 3, RNA-seq data are provided in Supplementary Datasets 2 and 6, and eCLIP–seq data are provided in Supplementary Datasets 4 and 5. Source data are provided with this paper.
Code availability
All software used for data analysis is publicly available and detailed in the Methods. Custom scripts that were used for analyzing eCLIP data are linked in the Methods and Reporting Summary.
Extended Data
Extended Data Fig. 1 |. Discovery of electrophilic compounds that deplete AR mRNA and protein.

a, AR-FL and AR-V7 mRNA content in parental 22Rv1 cells and 22Rv1 cells stably expressing an shRNA targeting AR-FL (shRNA_ARFL cells) as quantified by QuantiGene assays and normalized relative to a set of housekeeping genes. Data are mean values ± s.e.m. for 96 data points from two independent experiments. p values from unpaired t-test (two-tailed) are indicated above the bars. b, Western blot showing AR-FL and AR-V7 protein content in parental 22Rv1 cells and shRNA_AR-FL 22Rv1 cells. Data are from a single experiment representative of at least two independent experiments. c, AR protein content and cell counts (used as a cytotoxicity estimate counter screen) measured by high content imaging (HCI) after treatment of parental (upper) or shRNA_AR-FL (lower) 22Rv1 cells with electrophilic compounds (10 μM, 24 h). Hit compounds depleting AR protein content by ≥ 50% while maintaining cell counts ≥ 50% relative to DMSO are highlighted in red. See Supplementary Table 3 for structures of other hit compounds in addition to B21. d, Representative HCI images of time-dependent effects of hit compound B21 (25 μM) on total AR protein in 22Rv1 cells. See Fig. 1e for quantification of these data. Scale bar: 50 μM. e, Concentration-dependent effects of active (R)-10 and inactive (S)-10 enantiomers (see Table 1 for compound structures) on AR-FL and AR-V7 mRNA in 22Rv1 cells. Compound treatments were for 6 h, and Data are mean values ± s.e.m. for four replicates of a single experiment representative of two independent experiments. f, Structures of B21 (1) and analogues where the α-chloroacetamide reactive group was replaced with an acrylamide (12) or non-electrophilic propanamide (13). g, Concentration-dependent effects of B21, 12, and 13 on ARFL and AR-V7 mRNA in 22Rv1 cells. Compound treatments were for 6 h, and Data are mean values for two replicates of a single experiment representative of two independent experiments.
Extended Data Fig. 2 |. Active compounds suppress AR expression by targeting NONO.

a, Global cysteine reactivity profiles of 22Rv1 cells treated with DMSO or (R)-SKBG-1 (20 μM, 1 h) as determined by MS-ABPP. Data are mean values from three independent experiments, where cysteines were quantified in at least two experiments. Maximum quantifiable ratio values were set at 10 and 0.1. The ratio for NONO_C145 is marked in red. b, Quantification of NONO_C145 engagement by active (2–5, (R)-10 and (R)-SKBG-1) and inactive (6–9, (S)-10 and (S)-SKBG-1) compounds as determined by MS-ABPP. Data are mean values ± s.e.m for three independent experiments. c Heat map showing cysteines substantially (> 75%) engaged by (R)-SKBG-1 (20 μM) at 6 h in 22Rv1 cells and their corresponding engagement by (S)-SKBG-1 as determined by MS-ABPP. Blue and white respectively designate cysteines engaged ≥ 50% or < 50% by the indicated compounds. d, Engagement of the stereoselective targets of (R)-SKBG-1 from (c) by other active compounds 2–5 (20 μM, 1 h) in MS-ABPP experiments. For c, d, data represent mean values from three independent experiments. For a-d, see Supplementary Data 1 for full MS-ABPP data. e, Western blot showing NONO and AR-FL and V7 protein content of sgControl and sgNONO 22Rv1 cell populations generated by CRISPR-Cas9. sgControl 1 and sgNONO 5 cells were used for subsequent experiments. Data are from a single experiment representative of two independent experiments. f, AR-FL and AR-V7 mRNA content of sgControl and sgNONO cells. Data are mean values ± s.e.m. for 14 data points from two independent experiments. p values from unpaired t-tests. g, Concentrationdependent effects of active compound 5 and inactive control 8 (6 h treatments) on AR-FL (left) and AR-V7 (right) mRNA content of sgControl and sgNONO cells. Data are mean values for two replicates of a single experiment representative of two independent experiments. h, Western blots showing AR-FL and AR-V7 protein content in sgControl (NONO+) or sgNONO (NONO-) cells treated with active compound 5 and inactive control 9 (20 μM, 24 h). Also shown are western blotting signals for NONO and PSPC1. i, Quantification of western blotting data for AR-FL and AR-V7 in h and Fig. 2e. Data are mean values ± s.e.m. for four independent experiments. p values from two-way ANOVA. j, Concentration-dependent effects of compound 5 (6 h treatment) on AR-FL and AR-V7 mRNA content of sgNONO cells expressing WT-NONO or C145S-NONO. Data are mean values for two replicates of a single experiment representative of two independent experiments. For g and j, AR mRNAs were quantified by QuantiGene. k, l, Western blots showing AR-FL and AR-V7 protein content of sgNONO cells expressing WT-NONO or C145SNONO treated with active compounds (R)-SKBG-1 (k) or 5 (l) and inactive controls (S)-SKBG-1 (k) or 9 (l) (20 μM compounds, 24 h). m, Quantification of western blotting data for AR-FL and AR-V7 in k and l. Data are mean values ± s.e.m. for four independent experiments. p values from two-way ANOVA.
Extended Data Fig. 3 |. Confirming direct engagement of NONO_C145 by active compounds using alkyne probe 14.

a, Structure of alkyne probe 14. b, Western blots showing effects of alkyne probe 14 on AR-FL and AR-V7 protein content of 22Rv1 cells. Active compounds 5 and (R)-SKBG-1 and inactive control compounds 8 and (S)-SKBG-1 are shown for comparison. Cells were treated with 20 μM of each compound for 24 h. Also shown are western blotting signals for PSPC1. Data are from a single experiment representative of two independent experiments. c, Waterfall plot showing global protein enrichment profiles from MS-ABPP experiments performed with alkyne 14-treated sgNONO 22Rv1 cells expressing FLAG-WT- or C145S-NONO. Cells were treated with 20 μM of 14 for 1 h prior to lysis and enrichment and quantification of 14-labeled proteins (see Methods section for more details). Data are presented as the ratio of enrichment of proteins from WT-NONO/C145S-NONO cells. Data are average values for all quantified peptides for each protein from two independent replicates. d, Gel-based ABPP experiment showing that alkyne 14 labels WT-NONO, but not C145S-NONO expressed as FLAG epitope-tagged proteins in sgNONO 22Rv1 cells. sgNONO cells (with or without exogenous expression of WT- or C145S-NONO) were pretreated with DMSO or (R)-SKBG-1 (20 μM, 2 h) followed by DMSO or alkyne 14 (20 μM, 1 h), lysed, conjugated to a rhodamine-azide (Rh-N3) by copper-catalyzed azide-alkyne cycloaddition chemistry, and analyzed by SDS-PAGE and in-gel fluorescence. e, Concentration-dependent effects of (R)-SKBG-1 and (S)-SKBG-1 on alkyne 14 labeling of FLAG-tagged WT-NONO expressed in HEK cells. Cells were treated with (R)-SKBG-1 or (S)-SKBG-1 for 2 h, followed by 14 (20 μM, 1 h), and then processed as described in d. Mock (empty vector-transfected) cells and cells expressing FLAG-tagged C145S-NONO are shown as controls. For d and e, red asterisk marks band corresponding to the MW of FLAG-NONO. IB, immunoblot. f, Quantification of blockade of 14 labeling of recombinant FLAG-WT-NONO by (R)-SKBG-1. Data are mean values ± s.e.m. for four independent experiments. 95% confidence interval or calculated IC50 value is shown in brackets.
Extended Data Fig. 4 |. Global effects of NONO ligands on the transcriptomes and proteomes of cancer cells.

a, Volcano plot of mRNA log2 fold changes in (R)-SKBG-1-treated sgControl cells/(S)-SKBG-1-treated sgControl cells. Dotted lines indicate cutoffs for genes with significantly decreased (log2 fold change ≤ −0.5, padj ≤ 0.01) or increased (log2 fold change ≥ 0.5, padj ≤ 0.01) expression. log2 fold change values are mean values from three independent replicates. Plotted are protein-coding genes with >50 counts in each of three independent replicates and a coefficient of variation for gene counts < 0.5. b, Volcano plot of genes significantly decreased by (R)-SKBG-1-treated sgControl cells relative to DMSO-treated sgControl cells (left) and of genes significantly decreased by DMSO-treated sgNONO cells relative to DMSO-treated sgControl cells (right). Significantly decreased was defined as log2 fold change ≤ −0.5, padj ≤ 0.01. log2 fold change values are mean values from three replicates. Plotted are proteincoding genes with at least 50 counts in at least three samples, and a coefficient of variation for gene counts < 0.5 among the three replicates. c, Volcano plot of mRNA log2 fold change in (R)-SKBG-1-treated sgControl cells / DMSO-treated sgControl cells. d, Volcano plot of mRNA log2 fold change in (S)-SKBG-1-treated sgControl cells / DMSO-treated sgControl cells. For a–d, differential expression p values were adjusted with the Benjamini-Hochberg procedure for multiple comparisons. e, Scatter plot comparing mRNA log2 fold changes (x-axis) versus protein log2 fold changes (y-axis) in (R)-SKBG-1-treated sgControl / (R)-SKBG1-treated sgNONO 22Rv1 cells for proteins found by MS-based proteomics to be specifically decreased by (R)-SKBG-1 in a NONO-dependent manner (see Fig. 3d). Gene products inside the red dotted brackets show consistent mRNA and protein changes. f, g, Comparison of mRNA and protein content for AR (f) and SRSF4 (g) in the indicated treatment groups as measured by RNA-seq (4 h post-treatment with compounds) or MS-based proteomics (24 h post-treatment with compounds), respectively. Data are mean values ± s.e.m. for three (RNA-seq) or four (MS-based proteomics) independent replicates. p values from one-way ANOVA analysis are indicated above the bars.
Extended Data Fig. 5 |. (R)-SKBG-1 causes similar transcriptomic changes in human prostate and breast cancer cells.

a, Western blot showing NONO content of sgControl and sgNONO MCF7 cell populations generated by CRISPRCas9. sgControl 1 and sgNONO 1 or 3 cells were used for subsequent experiments. b, Quantification of NONO_C145 engagement by (R)-SKBG-1 versus (S)-SKBG-1 in MCF7 cells determined by MS-ABPP (indicated compound concentrations, 3 h). Data are mean values for two independent cell treatments analyzed in a single MS-ABPP experiment. See Supplementary Data 1 for complete proteomic data. c, Scatter plot of mRNA log2 fold changes for sgControl MCF7 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (x-axis) versus mRNA log2fold changes for sgControl or sgNONO cells each treated with (R)-SKBG-1 (y-axis). Cells were treated with compounds (5 μM) for 4 h prior to RNA-seq analysis. Genes inside the red dotted brackets were designated as decreased (log2 fold change ≤ −0.5) or increased (log2 fold change ≥ 0.5) by (R)-SKBG-1 in a NONO-dependent manner. Data are mean values for protein-coding genes with at least 50 counts in each of two-three independent replicates. d, Scatter plot of mRNA log2fold changes in (R)-SKBG-1-treated sgControl / (R)-SKBG-1-treated sgNONO 22Rv1 cells (x-axis) versus (R)-SKBG-1-treated sgControl / (R)-SKBG-1-treated sgNONO MCF7 cells (y-axis). MCF7 and 22Rv1 cells were treated with 5 and 20 μM of compounds, respectively, for 4 h, to match the respective potency of NONO_C145 engagement by (R)-SKBG-1 in each cell line (see panel b and Extended Data Fig. 2b). Data are mean values for protein-coding genes with >50 counts in each of two-three independent replicates. e, f, Quantification of AR (e) and RXRA (f) mRNA from RNA-seq experiments performed in (R)-SKBG-1 or (S)-SKBG-1-treated sgControl or sgNONO 22Rv1 and MCF7 cells. Data are mean values ± s.e.m. for two-three independent replicates. TPM, transcripts per million reads. p values from oneway ANOVA analysis are indicated above the corresponding bars, with relative percent decreases in mRNA caused by (R)-SKBG-1 in sgControl cells shown in parentheses.
Extended Data Fig. 6 |. NONO ligand (R)-SKBG-1 impairs cancer cell growth in a stereoselective and NONO C145-dependent manner.

a, Effects of (R)-SKBG-1 and (S)-SKBG-1 on the growth of sgControl and sgNONO MCF7 cells. b, Effects of (R)-SKBG-1 and (S)-SKBG-1 on the growth of sgNONO 22Rv1 cells expressing WT-NONO or C145S-NONO. c, Effects of (R)-SKBG-1 and (S)-SKBG-1 on the growth of HeLa and HT1080 cells. Cells were treated with DMSO or (R)-SKBG-1 or (S)-SKBG-1. After 6 days (22Rv1), 5 days (MCF7) or 4 days (HeLa and HT1080), cell growth was determined by CellTiterGlo, normalized to cells treated with only DMSO. Data are mean values ± s.e.m. normalized to DMSO-treated control cells for four independent replicates. EC50 values are reported with 95% confidence intervals shown in brackets. d, sgControl and sgNONO 22Rv1 cells show similar growth rates. 10,000 cells were seeded overnight, and then cell growth was measured by CellTiterGlo, normalized to day 0 signal. Data are mean values ± s.e.m. for four independent replicates.
Extended Data Fig. 7 |. Crosstalk between NONO and DBHS paralogs PSPC1 and SFPQ in cells genetically or chemically disrupted for NONO.

a, Quantification of PSPC1 (left) and SFPQ (right) mRNA from RNA-seq experiments performed in (R)-SKBG-1 or (S)-SKBG-1-treated sgControl or sgNONO 22Rv1 and MCF7 cells. See Fig. 3a for more details on RNA-seq experiments. Data are mean values ± s.e.m. shown as % of the DMSO-treated sgControl groups for two-three independent replicates. p values from one-way ANOVA analysis are indicated above the bars. b, Quantification of PSPC1 (left) and SFPQ (right) protein from MS-based proteomic experiments performed in (R)-SKBG-1 or (S)-SKBG-1 -treated sgControl or sgNONO 22Rv1 and MCF7 cells. MCF7 and 22Rv1 cells were treated with 2.5 and 20 μM of compounds, respectively, for 24 h. Data are mean values ± s.e.m. for four independent replicates. p values from one-way ANOVA analysis are indicated above the bars. c, Dependency Map data showing that the three human cancer cell lines showing greatest dependency on NONO have predicted deleterious mutations in SFPQ or PSPC1. d, Western blot showing efficiency of disruption of PSPC1 and SFPQ expression in sgControl and sgNONO 22Rv1 cells following 72 h treatment with sgRNAs targeting PSPC1 or SFPQ using the Alt-R CRISPR-Cas9 nucleofection system. e, Number of transcripts that were strongly decreased (log2 fold change ≤ −0.75, padj ≤ 0.01) in the indicated sgRNA cell models relative to sgControl_sgControl cells. Red and blue bars correspond to transcripts decreased in sgControl_sgXXX (sgControl_ sgPSPC1, sgControl_sgSFPQ) and sgNONO_sgXXX (sgNONO_sgControl, sgNONO_sgPSPC1, sgNONO_sgSFPQ) cell models, respectively. f, Cell growth of indicated sgRNA cell models measured by seeding 10,000 cells overnight (72 h post-nucleofection) and following growth over a five-day additional time period as determined by CellTiterGlo, normalized to day 0 signal. Data are mean values ± s.e.m. for five independent replicates. g, Bar graph showing the percentage of overall transcripts or transcripts stereoselectively decreased by (R)-SKBG-1 (log2 fold change (R)-SKBG-1 / (S)-SKBG-1 ≤ −0.75, padj ≤ 0.01) that were also decreased in the indicated sgRNA cell models.
Extended Data Fig. 8 |. NONO ligands induce accumulation of NONO and the paralogous proteins PSPC1 and SFPQ into nuclear foci.
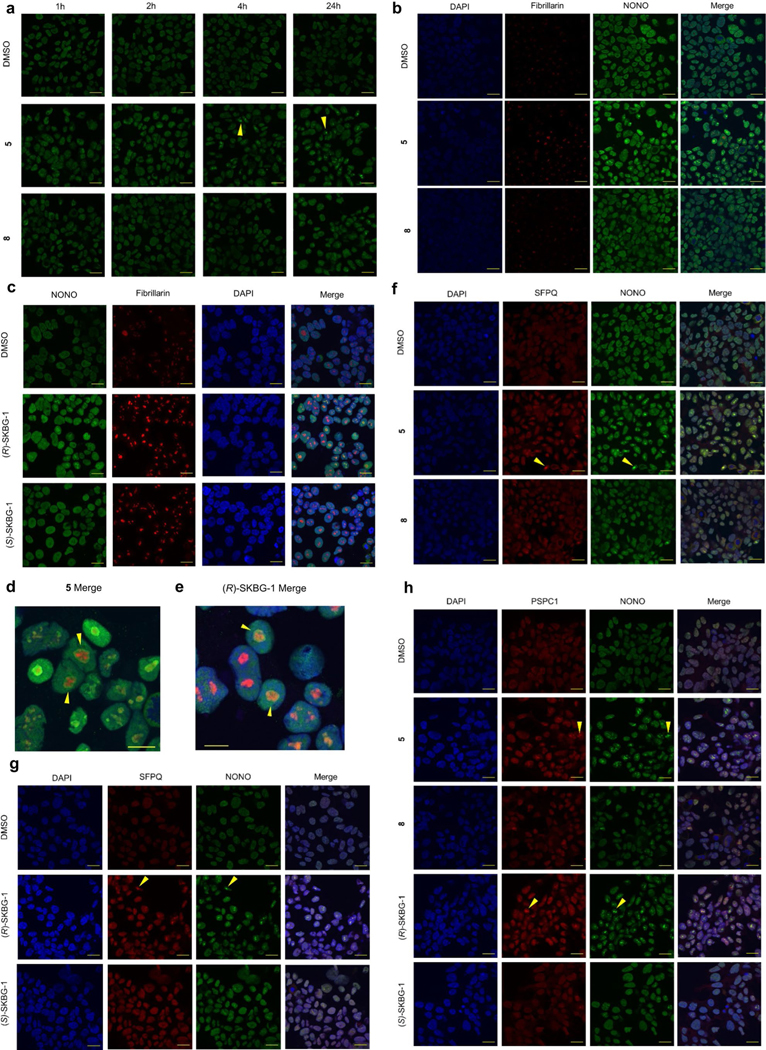
a, Immunofluorescence data showing time-dependent effects of active compound 5 and inactive control 8 (20 μM) on the localization of NONO in nuclear foci (yellow wedge) in 22Rv1 cells. NONO was imaged with primary antibody (Bethyl Laboratories, A300–587A) and an Alexa-488 secondary antibody. Scale bar: 20 μm. b, c, NONO foci induced by compounds 5 (b) and (R)-SKBG-1 (c) localize proximal to the nucleolar marker fibrillarin (20 μM compound treatment, 24 h). NONO was imaged with primary antibody (BD Biosciences, 611278) and an Alexa-488 secondary antibody. Fibrillarin was imaged with primary antibody (Cell Signaling, 2639 S) and an Alexa-647 secondary. Scale bar: 20 μm. d, e, Zoomed-in merged images of NONO (green) and fibrillarin (red) localization from compounds 5 (d) or (R)-SKBG-1 (e) treated 22Rv1 cells (24 h time point). DAPI stain, blue. Scale bar: 10 μm. f, g, Immunofluorescence data showing effects of active compounds 5 (f) and (R)-SKBG-1 (g) and inactive controls 8 and (S)-SKBG-1 on the localization of SFPQ in nuclear foci (yellow wedge) in 22Rv1 cells (20 μM of each compound, 24 h). NONO was imaged with primary antibody (Bethyl Laboratories, A300–587A) and an Alexa-488 secondary antibody. SFPQ was imaged with primary antibody (Sigma-Aldrich, WH0006421M2) and an Alexa-647 secondary. Scale bar: 20 μm. h, Immunofluorescence data showing effects of active compounds 5 and (R)-SKBG-1 and inactive controls 8 and (S)-SKBG-1 on the localization of PSPC1 in nuclear foci (yellow wedge) in 22Rv1 cells (20 μM of each compound, 24 h). NONO was imaged with primary antibody (Bethyl Laboratories, A300–587A) and an Alexa-488 secondary antibody. PSPC1 was imaged with primary antibody (SigmaAldrich, SAB4200503) and an Alexa-647 secondary. Scale bar: 20 μm.
Extended Data Fig. 9 |. Effects of NONO ligands on NONO-protein and NONO-mRNA interactions.

a, Quantification of PSPC1 and SFPQ from anti-FLAG immunoprecipitation-MS experiments of FLAG-NONO-expressing cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h). Data are mean values ± s.e.m.for four independent replicates. b, (R)-SKBG-1 effects on the abundance of transcripts previously shown in eCLIP experiments to bind NONO or NONO-associated RBPs (SFPQ, MATR3) versus transcripts bound to any RBP or not bound to RBPs45,46. Transcript abundances determined by RNA-seq of 22Rv1 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h). Transcripts stereoselectively decreased or increased by (R)-SKBG-1 determined as described in Fig. 3a. A bound transcript was defined as having ≥ 1 eCLIP IDR peak for a given RBP. c, Volcano plot comparing enrichment of mRNAs stereoselectively decreased by (R)-SKBG-1 in 22Rv1 cells among NONO and SFPQ-bound transcripts (in black) to 30 sets of randomly selected transcripts of matched number and expression levels (in red). Enrichment odds ratio and FDR values were determined as in Fig. 5c. d, Motifs enriched in NONO eCLIP-seq peaks from cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h) or DMSO. e, Scatter plot showing repetitive element distribution between (R)-SKBG-1 and (S)-SKBG-1-treated eCLIP-seq. f, eCLIP-seq relative information content determined for transcripts bound to NONO in 22Rv1 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h) that were stereoselectively decreased or not decreased by (R)-SKBG-1. g, h, eCLIP-seq reads for total mRNA for AR (g) or TAF4 (h) in 22Rv1 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h). TAF4 is a representative mRNA that was bound to NONO but not altered in abundance by (R)-SKBG-1. For f–h, relative information content was determined as described in Fig. 5e. i, Percent of transcripts from the indicated categories bound to NONO in 22Rv1 cells treated with (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h) or DMSO. d-i, results are from a single eCLIP-seq experiment representative of two independent replicates.
Extended Data Fig. 10 |. Effects of NONO ligands on mRNA splicing.

a, b, 5’ splice site sequence upstream (a) and 3’ splice site sequence downstream (b) of cassette exons in different contexts. Included, excluded or no change refers to the effect on cassette exons upon (R)-SKBG-1 treatment compared to (S)-SKBG-1 treatment in sgControl or sgNONO cells. c, Splice site strength of the splice sites surrounding the alternatively-spliced exons was determined using MaxEntScore. No difference was observed comparing the included/excluded group to the no change group. d, Motif enrichment comparing inclusion/exclusion events following (R)-SKBG-1 treatment to cassette exons with no change, spread across surrounding regions. Intronic sequences are defined as 500 b.p. surrounding the splice site e, Normalized eCLIP density around alternatively spliced exons for transcripts bound to NONO in 22Rv1 cells under the three indicated treatment conditions (DMSO or (R)-SKBG-1 or (S)-SKBG-1 (20 μM, 4 h treatment)), and control exons with various inclusion levels (ψ). f, Overlapping ENCODE HepG2 eCLIP IDR peaks with regions near cassette exons included after (R)-SKBG-1 treatment. Coloring shows the log2 (odds ratio) comparing included exons to regions with no change.
Supplementary Material
Acknowledgements
This work was supported by the NIH (R35 CA231991), a fellowship to S.G.K. from the National Cancer Institute (CA228436), a fellowship to H.-L.H. from the J. Yang Foundation Fellowship, a fellowship to H.L. from the Damon Runyon Foundation and a fellowship to J.R.R. from the American Cancer Society (PF-18-217-01-CDD). Part of this work was supported by Janssen R&D Integrated Technology Strategy Funding. We thank G. Van Hecke, H. De Wolf, G. Laenen and H. Murray for technical help, R. de Hoogt for providing the engineered cell line and S. Allen, I. Heo, H. Murray, P. Abeywickrema, A. Thompson, S. Sharma, P. J. Connolly and J. P. Edwards for insightful discussions. G.W.Y. is supported by NIH grants U41 HG009889, U01 HG009417, R01 HL137223 and R01 HG004659. G.W.Y. is supported as an Allen Distinguished Investigator, a Paul G. Allen Frontier Group advised grant to the Paul G. Allen Family Foundation.
B.F.C. is a founder and scientific advisor to Vividion Therapeutics.
G.W.Y. is an SAB member of Jumpcode Genomics and a cofounder, a member of the Board of Directors, on the SAB, an equity holder and a paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y. is a visiting professor at the National University of Singapore. G.W.Y.’s interests have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies.
Footnotes
Competing interests
The authors declare no other competing financial interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41589-023-01270-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-023-01270-0.
Peer review information Nature Chemical Biology thanks Benjamin Blencowe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Bentley DL Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet 15, 163–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerstberger S, Hafner M.& Tuschl T.A census of human RNA-binding proteins. Nat. Rev. Genet 15, 829–845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gebauer F, Schwarzl T, Valcárcel J.& Hentze MW RNA-binding proteins in human genetic disease. Nat. Rev. Genet 22, 185–198 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Pereira B, Billaud M.& Almeida R.RNA-binding proteins in cancer: old players and new actors. Trends Cancer 3, 506–528 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Julio AR & Backus KM New approaches to target RNA binding proteins. Curr. Opin. Chem. Biol 62, 13–23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu P.Inhibition of RNA-binding proteins with small molecules. Nat. Rev. Chem 4, 441–458 (2020). [DOI] [PubMed] [Google Scholar]
- 7.D’Agostino VG et al. Screening approaches for targeting ribonucleoprotein complexes: a new dimension for drug discovery. SLAS Discov. 24, 314–331 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Alberti S.& Hyman AA Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol 22, 196–213 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Han T.et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Kotake Y.et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol 3, 570–575 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Naineni SK et al. Functional mimicry revealed by the crystal structure of an eIF4A:RNA complex bound to the interfacial inhibitor, desmethyl pateamine A. Cell Chem. Biol 28, 825–834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwasaki S.et al. The translation inhibitor rocaglamide targets a bimolecular cavity between eIF4A and polypurine RNA. Mol. Cell 73, 738–748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palacino J.et al. SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 11, 511–517 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Lonergan P.& Tindall D.Androgen receptor signaling in prostate cancer development and progression. J. Carcinog 10, 20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanford M.Enzalutamide: a review of its use in metastatic, castration-resistant prostate cancer. Drugs 73, 1723–1732 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Kregel S.et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 22, 111–119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salami J.et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol 1, 100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joseph JD et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 3, 1020–1029 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Antonarakis ES et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen RJ et al. Regression of castrate-recurrent prostate cancer by a amall-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17, 535–546 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Xie J.et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat. Chem. Biol 18, 1341–1350 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Luna Velez MV, Verhaegh GW, Smit F, Sedelaar JPM & Schalken JA Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene 38, 3696–3709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melnyk JE et al. Targeting a splicing-mediated drug resistance mechanism in prostate cancer by inhibiting transcriptional regulation by PKCβ1. Oncogene 41, 1536–1549 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vinogradova EV et al. An activity-guided map of electrophile-cysteine interactions in primary human T Cells. Cell 182, 1009–1026 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weerapana E.et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knott GJ, Bond CS & Fox AH The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 44, 3989–4004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emili A.et al. Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA 8, 1102–1111 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passon DM et al. Structure of the heterodimer of human NONO and paraspeckle protein component 1 and analysis of its role in subnuclear body formation. Proc. Natl Acad. Sci. USA 109, 4846–4850 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishitani K.et al. p54nrb acts as a transcriptional coactivator for activation function 1 of the human androgen receptor. Biochem. Biophys. Res. Commun 306, 660–665 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Dong X, Sweet J, Challis JRG, Brown T.& Lye SJ Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Biol. Cell 27, 4863–4875 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takayama K-I et al. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc. Natl Acad. Sci. USA 114, 10461–10466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieson T.et al. Systematic analysis of protein turnover in primary cells. Nat. Commun 9, 689 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witwicki RM et al. TRPS1 is a lineage-specific transcriptional dependency in breast cancer. Cell Rep. 25, 1255–1267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lefèvre L.et al. Combined transcriptome studies identify AFF3 as a mediator of the oncogenic effects of β-catenin in adrenocortical carcinoma. Oncogenesis 4, e161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grand RS et al. BANP opens chromatin and activates CpG-island-regulated genes. Nature 596, 133–137 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Young LC, McDonald DW & Hendzel MJ Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ irradiation. J. Biol. Chem 288, 21376–21388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S.et al. Double-strand break repair deficiency in NONO knockout murine embryonic fibroblasts and compensation by spontaneous upregulation of the PSPC1 paralog. Nucleic Acids Res. 42, 9771–9780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stagsted LVW, O’Leary ET, Ebbesen KK & Hansen TB The RNA-binding protein SFPQ preserves long-intron splicing and regulates circRNA biogenesis in mammals. eLife 10, e63088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim S, Kim D, Cho SW, Kim J.& Kim J-S Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox AH, Bond CS & Lamond AL P54nrb forms a heterodimer with PSP1 that localizes to paraspeckles in an RNA-dependent manner. Mol. Biol. Cell 16, 5304–5315 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox AH et al. Paraspeckles: a novel nuclear domain. Curr. Biol 12, 13–25 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Prasanth KV et al. Regulating gene expression through RNA nuclear retention. Cell 123, 249–263 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Van Nostrand EL et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salton M, Lerenthal Y, Wang S-Y, Chen DJ & Shiloh Y.Involvement of matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 9, 1568–1576 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Her H, Boyle E.& Yeo GW Metadensity: a background-aware Python pipeline for summarizing CLIP signals on various transcriptomic sites. Bioinform. Adv 2, vbac083 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briata P.et al. Diverse roles of the nucleic acid-binding protein KHSRP in cell differentiation and disease. Wiley Interdiscip. Rev. RNA 7, 227–240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schreiber SL The rise of molecular glues. Cell 184, 3–9 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Shalem O.et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verbist B.et al. Analyzing magnetic bead QuantiGene Plex 2.0 gene expression data in high throughput mode using QGprofiler. BMC Bioinformatics 20, 378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornelissen F, Cik M.& Gustin E.Phaedra, a protocol-driven system for analysis and validation of high-content imaging and flow cytometry. J. Biomol. Screen 17, 496–506 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C.Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love MI et al. Tximeta: reference sequence checksums for provenance identification in RNA-seq. PLoS Comput. Biol 16, e1007664 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love MI, Huber W.& Anders S.Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subramanian A.et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen S.et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl Acad. Sci. USA 111, E5593–E5601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinz S.et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yee BA, Pratt GA, Graveley BR, Van Nostrand EL & Yeo GW RBP-Maps enables robust generation of splicing regulatory maps. RNA 25, 193–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD039283 (cysteine MS-ABPP with 6 h of compound treatment and FLAG–NONO coimmunoprecipitation–MS) and PXD032087 (all other experiments). Raw RNA-seq and eCLIP–seq data have been deposited to NCBI under Gene Expression Omnibus accession codes GSE222217 and GSE198212, respectively. Processed proteomic data are provided in Supplementary Datasets 1 and 3, RNA-seq data are provided in Supplementary Datasets 2 and 6, and eCLIP–seq data are provided in Supplementary Datasets 4 and 5. Source data are provided with this paper.