

SUMMARY
The lateral entorhinal cortex (LEC) provides multisensory information to the hippocampus, directly to the distal dendrites of CA1 pyramidal neurons. LEC neurons perform important functions for episodic memory processing, coding for contextually salient elements of an environment or experience. However, we know little about the functional circuit interactions between the LEC and the hippocampus. We combine functional circuit mapping and computational modeling to examine how long-range glutamatergic LEC projections modulate compartment-specific excitation-inhibition dynamics in hippocampal area CA1. We demonstrate that glutamatergic LEC inputs can drive local dendritic spikes in CA1 pyramidal neurons, aided by the recruitment of a disinhibitory VIP interneuron microcircuit. Our circuit mapping and modeling further reveal that LEC inputs also recruit CCK interneurons that may act as strong suppressors of dendritic spikes. These results highlight a cortically driven GABAergic microcircuit mechanism that gates nonlinear dendritic computations, which may support compartment-specific coding of multisensory contextual features within the hippocampus.
Graphical abstract
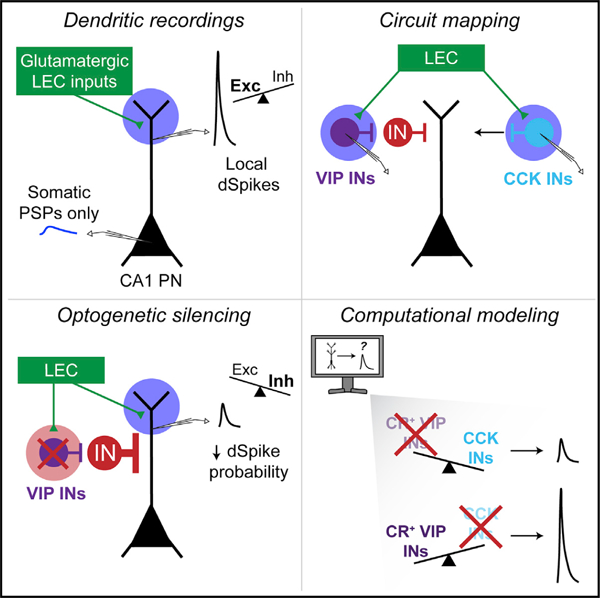
In brief
Bilash et al. found that long-range cortical inputs can drive local dendritic spikes in hippocampal pyramidal neurons. These inputs recruit a local GABAergic microcircuit to disinhibit dendritic spikes. These findings identify circuit mechanisms where the dynamic interaction of compartment-specific excitation, inhibition, and disinhibition supports supralinear neuronal computations.
INTRODUCTION
The formation of multisensory episodic memories relies on functional interactions between the hippocampus and the entorhinal cortex.1–5 The medial entorhinal cortex (MEC) and lateral entorhinal cortex (LEC) provide functionally distinct spatial and nonspatial multisensory inputs, respectively.6–8 Several studies have explored the circuit interactions between the MEC and major hippocampal output area CA1 in the context of place coding and episodic memory formation.4,9–16 Meanwhile, we know little about the circuit organization and function of LEC inputs in shaping CA1 activity. The LEC is a key interaction partner of the hippocampus,8,17,18 poised to participate in contextual memory associations. Recent in vivo studies show that LEC neurons code for odors,3,19–21 object novelty,22,23 object-place associations,24–26 contextual salience,27 temporal structure,28 and cue-reward associations.29 At the circuit level, the LEC sends glutamatergic14,21 and GABAergic27 projections directly to area CA1, which projections play distinct roles in associative, context-dependent learning. However, we still lack a basic understanding of how long-range, excitatory cortical inputs influence single-neuron computations and local microcircuit dynamics to support hippocampal memory processing. We define how direct glutamatergic inputs from the LEC to CA1 sculpt compartment-specific activity by recruiting distinct GABAergic microcircuits.
We hypothesize that glutamatergic LEC-CA1 inputs sculpt nonlinear neuronal computations, such as dendritic spikes, by tuning the compartment-specific excitation-inhibition balance in CA1. Dendritic spikes underlie vital neural processes such as burst activity,30–32 long-term plasticity,33–35 and feature-selective tuning.36–39 We lack a mechanistic understanding of the circuits that gate input-driven dendritic spikes, due to the experimental difficulty in combining dendritic recordings and circuit manipulations. Past studies used computational modeling to infer the ionic, synaptic, and anatomical determinants of dendritic spikes and their contribution to single-neuron computations.37,40–45 However, such models often cannot capture important dendritic, microcircuit, and input pathway characteristics due to a lack of experimental data used to constrain model parameters. Thus, the circuit interactions that shape dendritic non-linearities have remained especially elusive. GABAergic inhibitory neurons (INs) can serve as effective circuit switches to rapidly and precisely shape single-cell computations.27,31,46–48 Given their functional diversity and wiring specificity,49,50 INs can modulate compartment-specific activity to expand the computational coding capacity of single neurons. Various models have predicted important effects of inhibition on local spiking in single neurons.51–55 However, these models were not parameterized to investigate how specific inputs shape compartment-specific activity within neurons. Combining experiments and computational modeling to study LEC-CA1 circuit interactions allows for a powerful investigation into how synaptic inputs recruit diverse GABAergic microcircuits to dynamically shape compartment-specific input-output transformations.
In this study, we used in vitro electrophysiology, optogenetics, and computational modeling to examine the cellular and circuit mechanisms by which long-range glutamatergic LEC projections influence the local activity in the hippocampus. We recorded LEC-driven responses from various neuronal populations in area CA1. LEC inputs could drive local dendritic spikes in CA1 pyramidal neuron (PN) distal dendrites, even with inhibition present, and directly recruit vasoactive intestinal polypeptide (VIP)-expressing and cholecystokinin (CCK)-expressing INs. Optogenetic silencing of VIP INs led to a decreased incidence of LEC-driven dendritic spikes, suggesting a disinhibitory gating of dendritic activity by local INs. To dissect the relative contributions of specific IN subpopulations, we developed a data-driven computational model of an LEC-driven CA1 PN and surrounding microcircuitry. The model replicated our experimental data and further predicted that calretinin-expressing VIP INs drive the disinhibition, while CCK INs drive inhibition onto CA1 PN dendrites. Together, our findings demonstrate that a VIP IN-mediated disinhibitory microcircuit gates cortically driven, local dendritic spikes in area CA1. Our study establishes a circuit mechanism by which the LEC promotes supralinear dendritic computations in the hippocampus.
RESULTS
LEC directly excites CA1 pyramidal neurons, driving depolarization of dendrites and somata
We investigated the direct glutamatergic projections from the LEC to hippocampal area CA1. Using acute hippocampal slices, we performed optogenetic circuit mapping of the LEC inputs to CA1 PNs and INs. We unilaterally injected a recombinant adeno-associated virus (rAAV-CaMKII-ChR2-eYFP) into the superficial layers of the LEC to express ChR2 in glutamatergic LEC neurons and their axons (Figures 1A, S1A, and S1B). Confocal imaging confirmed that ChR2-eYFP expression was restricted to the LEC (Figure 1B). Local photostimulation reliably evoked photocurrents in LEC neurons, confirming ChR2 function (Figures S1B–S1D). In area CA1, LEC axons projected into the distal dendrite layer (stratum lacunosum moleculare, SLM), (Figures 1B and 1C).14,17,18,56,57
Figure 1. LEC inputs provide direct excitation to CA1 pyramidal neuron compartments.
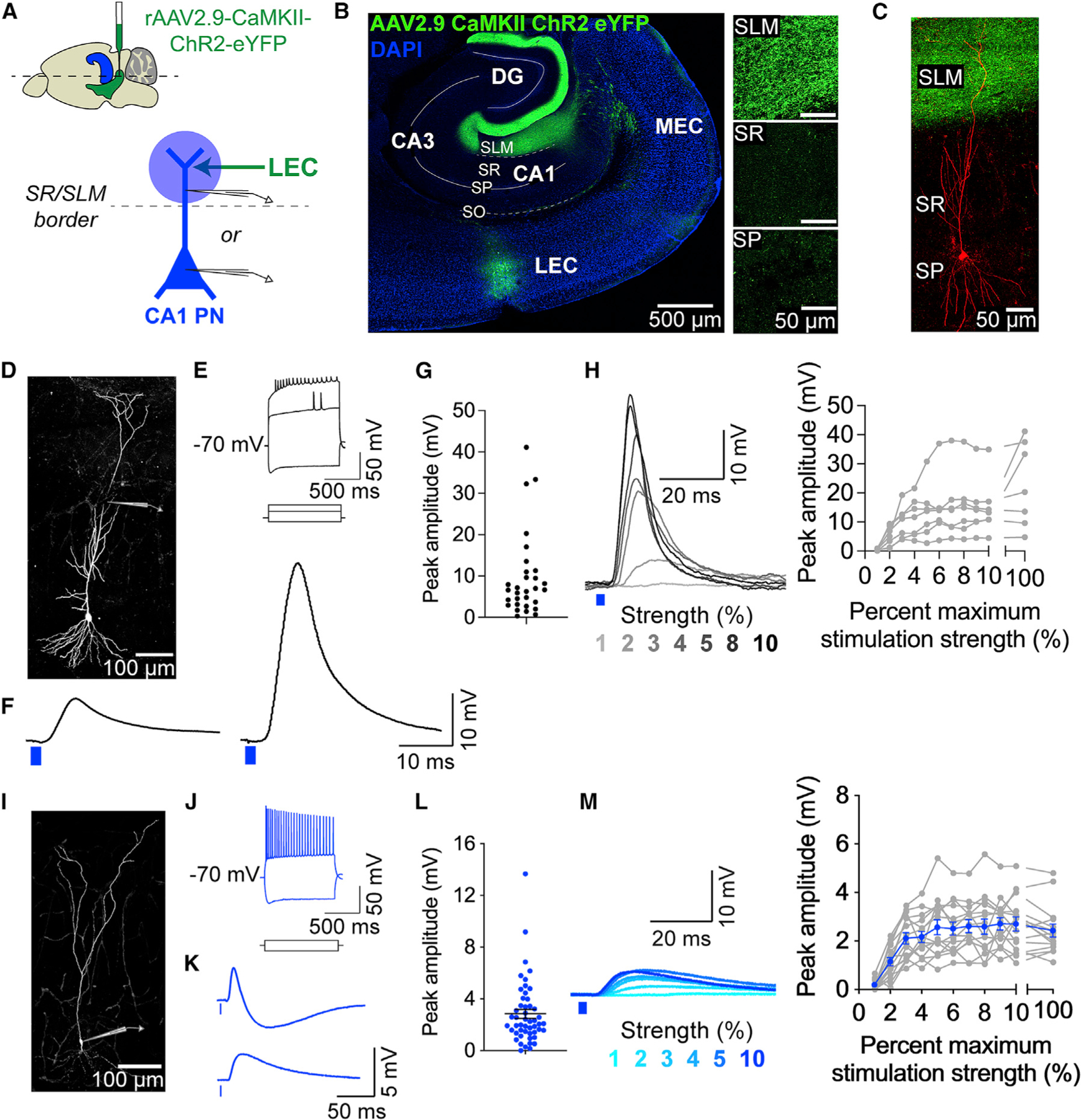
(A) Experimental design. Top: LEC injection site (green) and hippocampus (blue) in a mouse brain (sagittal view). Bottom: Photostimulation (blue circle) of ChR2+ LEC axons (green arrow) during whole-cell recordings from a CA1 PN (blue) dendrite or soma.
(B) Confocal image of a horizontal brain slice demonstrating the LEC injection site and LEC axons in the hippocampus (green) and in various CA1 layers (right).
(C) Confocal image of a patched CA1 PN (red) surrounded by LEC axons. The PN was filled with biocytin and counterstained with streptavidin-AF647.
(D–H) LEC-driven dendritic responses from CA1 PNs. (D) Example CA1 PN, patched at the dendrite (cartoon pipette). Counterstained as in (C). (E) Example electrophysiological properties of a CA1 PN distal dendrite, measured in response to dendritic current injections (700, 400, −325 pA). The membrane potential was maintained at −70 mV. (F) Example LEC-driven dendritic post-synaptic responses, showing putative subthreshold (left) and suprathreshold (right) dendritic responses. (G) Peak amplitudes of LEC-driven dendritic responses. Total n = 17 dendrites, 16 slices, 13 mice. (H) Input-output transformation. Left: example LEC-driven dendritic responses (1%–10% maximum strength photostimulation). Putative suprathreshold response seen at 8% maximum photostimulation strength. Right: response amplitudes, including both sub- and suprathreshold responses. n = 7 dendrites, 7 slices, 7 mice.
(I–M) LEC-driven somatic responses from CA1 PNs. (I) Same as (D) but patched at the soma. (J) Same as (E) but measured in the soma; 400 and −375 pA somatic currents injected. (K) Same as (F) but showing somatic PSPs. Two example responses shown: with (top) or without (bottom) a visible IPSP component. (L) Same as (G), but from the soma. Total n = 50 somata, 48 slices, 33 mice. (M) Same as (H), but from the soma. Data from individual somata are shown in gray. Mean ± SEM data are shown in blue. n = 15 pyramidal neuron somata, 14 slices, 11 mice.
To determine the synaptic connectivity and strength of the glutamatergic LEC inputs, we locally photostimulated the LEC axon terminals in the SLM of area CA1 with 470 nm light (single 2 ms pulse, maximum LED strength 56 mW, 50 μm diameter beam spot) and used intracellular, whole-cell recordings in current clamp to record light-evoked responses from CA1 PNs (Figures 1 and 2). The LEC-driven responses persisted in the presence of tetrodotoxin (TTX; 1 μM) and 4-aminopyridine (4-AP; 100 μM) (Figure S2I), a pharmacological condition that eliminates polysynaptic inputs and feedforward inhibition,58 indicating monosynaptic connectivity between LEC and CA1 PNs. We next characterized the LEC-driven responses in CA1 PNs under more physiological, drug-free conditions, where inhibition and polysynaptic connections remained intact. Glutamatergic LEC axons synapse onto CA1 PN distal dendrites, so to directly access LEC-driven activity in the dendritic compartment, we performed dendritic patch-clamp recordings from CA1 PN distal dendrites (Figures 1D–1H and S2A–S2D). We recorded around the stratum radiatum (SR)/SLM border, 225–400 μm from the soma. Maximum-strength photostimulation resulted in dendritic postsynaptic responses that varied in amplitude and kinetics (Figures 1F and 1G). To measure the input-output transformation of the responses, we photostimulated the LEC axons across a range of lower-strength light intensities to recruit an increasing number of LEC axons (Figure 1H). The amplitudes of LEC-driven dendritic responses saturated after 6% maximum stimulation strength. Some response amplitudes exceeded 30 mV and resembled supra-threshold dendritic spikes.
Figure 2. Optogenetic stimulation of LEC inputs can drive dendritic spikes in CA1 pyramidal neurons.
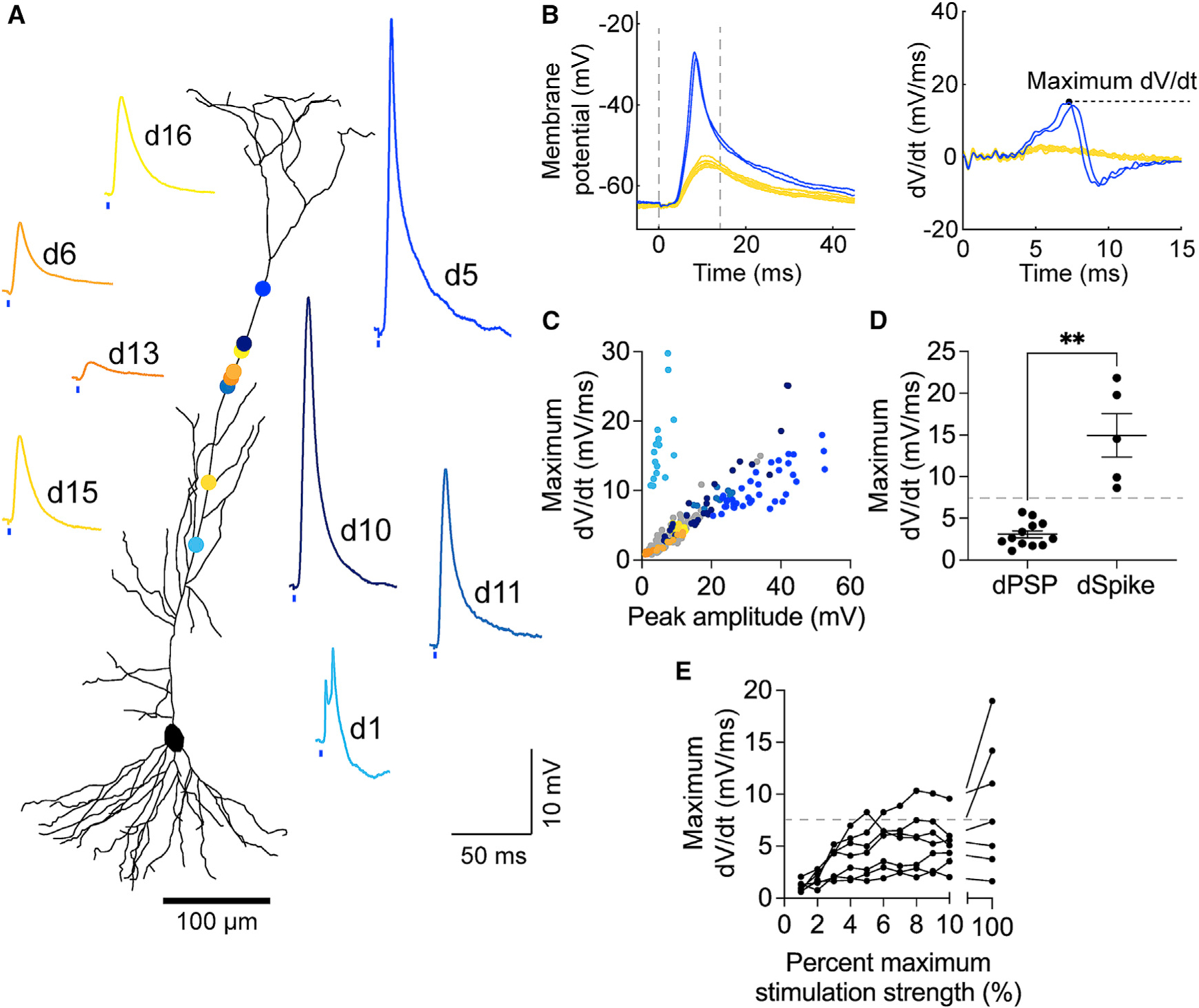
(A) Example LEC-driven dendritic responses, numbered in the order they were recorded (same dataset as in Figures 1D–1H). Putative dPSPs shown in yellow. Putative dSpikes shown in blue. Circle colors correspond to the dendritic recording sites. Neuronal morphology was reconstructed from the neuron in Figure 1D.
(B) Left: example LEC-driven dSpikes (blue) and dPSPs (yellow). Right: corresponding derivative traces and maximum dV/dt value (dashed black line), shown from t = 0–15 ms (left graph, dashed gray line).
(C) Peak amplitude versus maximum dV/dt values of every recorded LEC-driven dendritic response sweep. Data from the example dendrites are color codedasin (A).
(D) Average maximum dV/dt values of LEC-driven dPSPs versus dSpikes (**p = 0.0097). Unpaired t test with Welch’s correction; n = 14 dPSPs and 5 dSpikes. Dashed line denotes the threshold dV/dt value (7.5 mV/ms).
(E) Input-output transformation of the maximum dV/dt values, corresponding to Figure 1H dendrites. Some responses become suprathreshold with increased photostimulation strength.
In a separate set of experiments, we recorded from CA1 PN somata to assess the propagation of LEC-driven responses (Figures 1I–1M and S2E–S2H), using the same photostimulation paradigms as for dendritic recordings. Similarly, the somatic postsynaptic responses exhibited a range of amplitudes and kinetics (Figures 1K and 1L), and the amplitudes saturated after 6% maximum stimulation strength (Figure 1M). Notably, single-pulse photostimulation of LEC inputs never led to enough depolarization to elicit LEC-driven action potentials (APs) in CA1 PN somata. In summary, we demonstrated that glutamatergic LEC inputs drive depolarization in both CA1 PN compartments, but without leading to somatic APs.
LEC drives supralinear output in dendrites but not in somata
As stated, some LEC-driven dendritic responses resembled suprathreshold dendritic spikes previously described in the literature,27,59,60 such as putative sodium spikes (Figure 2A, d5, d10, d11)33,40,61–63 and complex calcium spikes (Figure 2A, d1).64,65 To validate that these were LEC-driven dendritic spikes, rather than large-amplitude dendritic postsynaptic potentials (dPSPs), we analyzed the derivative traces40,60,62,66 and phase plots of all LEC-driven responses (Figure S3). Both analyses consistently revealed obvious visual differences between the dPSPs and the dendritic spikes. The derivative traces of LEC-driven dendritic spikes exhibited a recognizable, high-amplitude peak that reached a maximum dV/dt value before dipping back below zero. In contrast, the derivative traces of LEC-driven dPSPs exhibited a smooth, flattened curve (Figures 2B and 2C). Accordingly, we set a classification criterion for our dataset: we categorized dendritic responses with maximum dV/dt > 7.5 mV/ms as suprathreshold dendritic spikes (dSpikes) and those with maximum dV/dt < 7.5 mV/ms as subthreshold dPSPs. This criterion provided the clearest, quantifiable delineation of subthreshold versus suprathreshold LEC-driven dendritic responses (Figures 2D and S4A–S4H).
Interestingly, LEC-driven dSpike incidence was not strictly dependent on recording location (Figures 2A and S4B), and LEC-driven dSpike generation was possible at lower photostimulation strengths (4%–10% maximum; Figure 2E). LEC-driven dPSPs were larger and faster than LEC-driven somatic PSPs (Figures S4I–S4O), which is expected given the proximity of the dendritic recordings to the LEC axon terminals and biophysical properties of the distal dendrites. Together, our results indicate that glutamatergic LEC inputs can drive suprathreshold dSpikes in CA1 PN distal dendrites. The complete lack of LEC-driven somatic APs suggests that these are local dSpikes.
We briefly investigated whether somatic APs could emerge under specific conditions (Figure S5). We first measured the presynaptic short-term plasticity dynamics of the LEC inputs in CA1 PN somata by activating the inputs using low-strength photostimulation trains delivered across physiologically relevant frequencies (see STAR Methods) (Figures S5A–S5E). Photostimulation at 4–10 Hz was particularly effective at facilitating the inputs (Figures S5D and S5E). Simplistically, the facilitation suggested that the glutamatergic LEC axons may have a low release probability, although we cannot rule out the contribution of shifts in excitation-inhibition balance. Nevertheless, a low release probability in glutamatergic LEC axons may explain why a single light pulse did not drive APs. We next tested whether maximum-strength photostimulation trains could evoke APs (Figures S5F–S5K). Although the photostimulation elicited significant summation of the responses, it did not drive LEC-driven APs (Figures S5I–S5K and S5O). Meanwhile, maximum-strength photostimulation produced significant summation of LEC-driven dendritic responses but had a modest effect on dSpike probability (Figures S5L–S5O). This further confirmed that LEC-driven dSpikes remain local, even during repeated activity in the LEC-CA1 circuit.
The generation of distal dSpikes in CA1 PNs frequently requires coincident inputs,27,42,67 burst activity,33,35,67 inhibition blockade,33,61,67,68 and/or simultaneous depolarizing current injections.12,33,46 Electrical stimulation of EC inputs in area CA1 has been shown to drive dSpikes, while inhibition remains intact,61 but such a stimulation paradigm would have activated long-range glutamatergic and disinhibitory GABAergic inputs from the LEC and MEC,27,69 as well as axons of local interneurons in the SLM. Thus, it was interesting that pathway-specific photostimulation of glutamatergic LEC inputs with a single light pulse could elicit dSpikes with inhibition intact. Nevertheless, the circuit mechanisms gating these cortical input-driven dSpikes remain elusive.
LEC inputs recruit strong feedforward inhibition onto both compartments of CA1 PNs
What are the local circuit mechanisms underlying LEC-driven activity in CA1 PNs? We turned our attention to inhibition. GABAergic inputs synapse onto the entire somatodendritic axis of CA1 PNs and can shape compartment-specific activity.51,70–72 To assess how feedforward inhibition (FFI) regulates LEC-driven depolarization in CA1 PNs, we pharmacologically blocked GABAA and GABAB receptors with bath application of the antagonists SR95531 (2 μM) and CGP55845 (1 μM) (Figures 3A and 3F). In these experiments, we severed the CA3-CA1 collaterals in the slices to prevent the propagation of any hyperexcitability. We directly measured the LEC-driven excitatory PSP (EPSP) at the dendrite or soma in current clamp and derived the underlying inhibitory PSP (inferred IPSP) for subthreshold responses27,72,73 (see STAR Methods, Figures 3B and 3G). This strategy was necessary to investigate LEC-driven FFI onto the dendrites and gives a reliable estimate of somatic and dendritic IPSPs.73 Voltage clamp is not effective in dendrites,74,75 so isolating inhibitory postsynaptic currents (IPSCs) by holding the dendritic membrane potential at +10 mV was not feasible.
Figure 3. LEC inputs recruit strong feedforward inhibition onto both compartments of CA1 PNs.
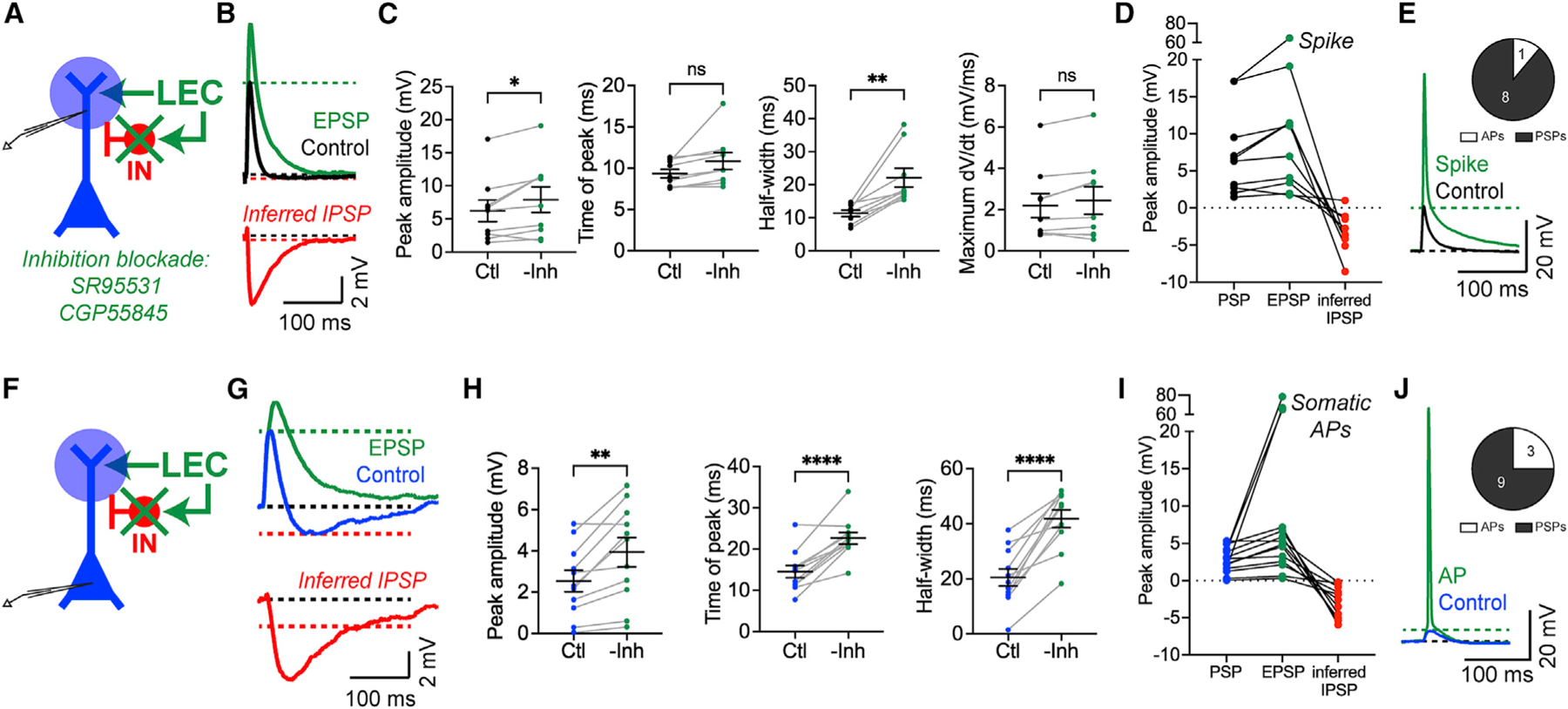
(A) Experimental strategy. Photostimulation of LEC axons during whole-cell recordings from a CA1 PN dendrite as SR95531 and CGP55845 are perfused onto the brain slice to block inhibition (green x).
(B) Example LEC-driven dendritic PSPs before (black, control) and after pharmacological inhibition blockade (green, EPSP). The inferred IPSP (red) was calculated post hoc from the two traces.
(C) LEC-driven dendritic response parameters before and after inhibition blockade. Left to right: peak amplitude (*p = 0.025), time of peak (p = 0.0612 [ns]), half-width (**p = 0.0022), maximum dV/dt (p = 0.0866 [ns]). Paired t test; n = 9 dendrites.
(D) LEC-driven dendritic PSP, EPSP, and inferred IPSP amplitudes. Total n = 9 dendrites, 9 slices, 5 mice. One dendrite spiked after inhibition blockade.
(E) Example LEC-driven spike in the dendrite after inhibition blockade. Inset: response type after inhibition blockade.
(F) Same as (A) but patched at the soma.
(G) Same as (B) but with somatic PSPs.
(H) Same as (C) but from somatic responses. Left to right: peak amplitude (**p = 0.0023), time of peak (****p < 0.0001), half-width (****p < 0.0001). Paired t test; n = 11 somata.
(I) Same as (D) but from somatic responses. Total n = 12 somata, 12 slices, 9 mice. Three somata spiked after inhibition blockade.
(J) Same as (E) but showing a somatic AP after inhibition blockade.
Blocking GABA receptors demonstrated that LEC-driven FFI sculpts the responses in both neuronal compartments (Figures 3C and 3H). After inhibition was removed, LEC-driven PSPs exhibited significantly higher amplitudes and wider half-widths compared with control conditions (dendritic amplitude p = 0.025, dendritic half-width p = 0.0022, somatic amplitude p = 0.0023, somatic half-width p < 0.0001, paired t test, n = 9 dendrites and 11 somata). In addition, LEC-driven somatic PSPs showed significant delays (time to peak p < 0.0001). There were no significant changes to the time of peak or maximum dV/dt of LEC-driven dPSPs (time of peak p = 0.0612, maximum dV/dt p = 0.087). In some cases, inhibition blockade enabled LEC-driven spikes in CA1 PNs (Figures 3D, 3E, 3I, and 3J), hinting at a complex interaction between the excitation-inhibition balance within the circuit and the propagation of suprathreshold activity. Together, our results demonstrate that LEC-driven FFI limits the amount and duration of depolarization in CA1 PN dendrites and somata, sculpting the activity in both neuronal compartments.
LEC inputs directly recruit VIP and CCK interneuron populations in CA1
Next, we asked which GABAergic microcircuit(s) could mediate the FFI to shape LEC-driven responses and whether any of them could specifically modulate dendritic excitability in CA1 PNs. There are over 20 types of INs in area CA1, each with distinct characteristics and functions.49,50,76,77 We investigated three genetically defined78 IN populations that were likely to be targeted by LEC projections and could modulate dendritic activity in CA1 PNs. These were the VIP-expressing INs and CCK-expressing INs, whose somata and dendrites are located in the SLM,27,73,79–84 and the somatostatin (SST)-expressing INs, which classically modulate PN dendritic activity.31,85,86 Although parvalbumin (PV)-expressing INs are known to mediate FFI in area CA1, they were not investigated because they classically modulate somatic and axonal activity81,87–89 and because there is mixed evidence for whether they receive input from EC.21,46,90
To test whether these CA1 INs are recruited by glutamatergic LEC inputs, we photostimulated the LEC axons in the SLM, as previously described, and performed targeted patch-clamp recordings from tdTomato-labeled VIP, CCK, or SST INs in hippocampal slices derived from IN-specific transgenic mice (see STAR Methods) (Figure 4).
Figure 4. LEC inputs recruit VIP INs and CCK INs in CA1.
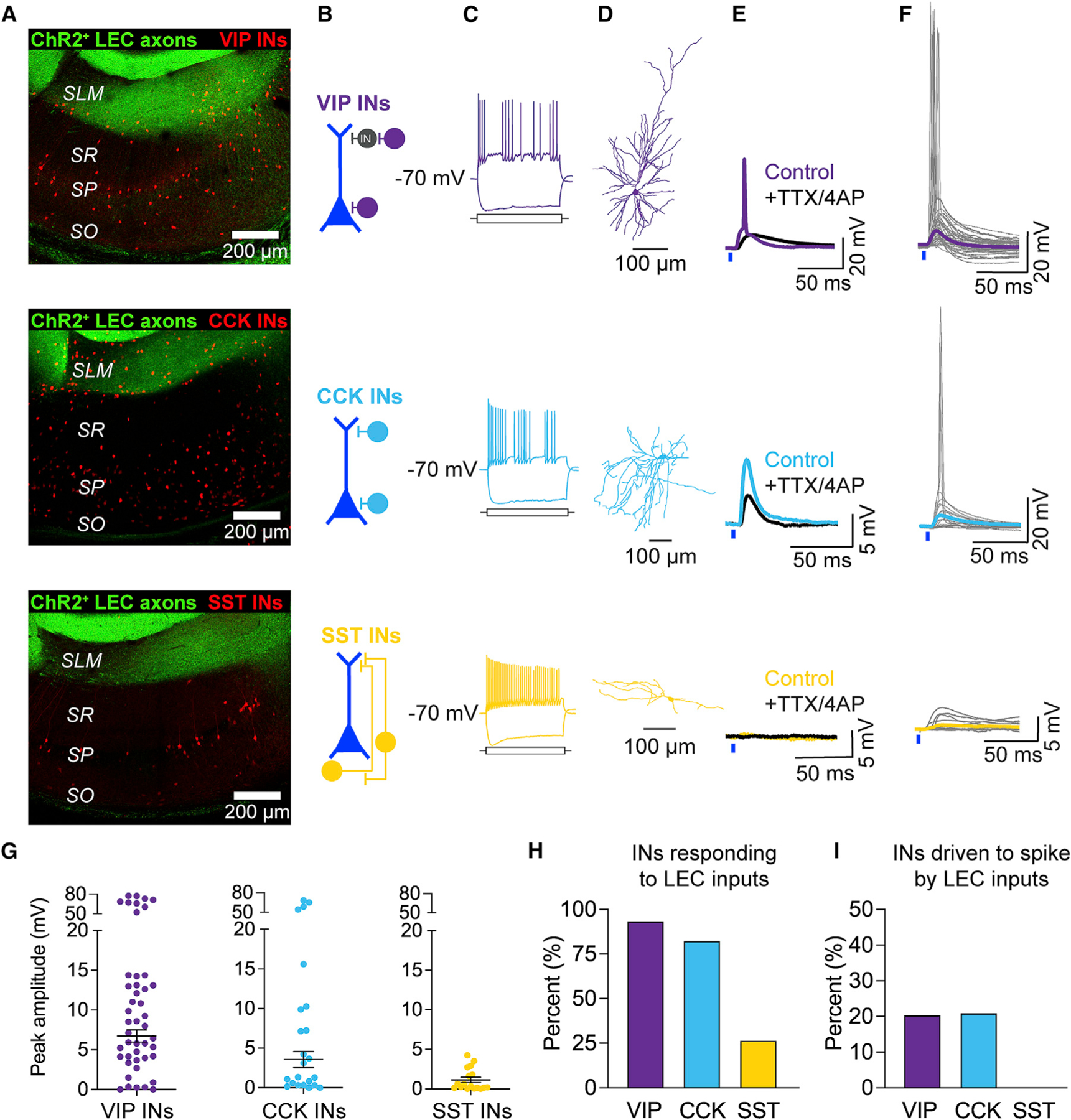
(A–F) Top to bottom: VIP INs (purple), CCK INs (blue), SST INs (yellow). (A) Confocal images of hippocampal area CA1 with LEC axons (green) and tdTomato-labeled INs (red). VIP-Cre/Ai14, CCK-Cre/Dlx-Flp/Ai65, and SST-Cre/Ai14 mice were used. (B) Predominant location of the IN somata in CA1. (C) Example electrophysiological properties, generated similar to Figure 1E. VIP IN, 175 and −150; CCK IN, 100 and −200; SST IN, 175 and −250 pA somatic current injected. (D) Example neuronal morphologies. (E) Example LEC-driven responses before (color) and after (black) application of TTX and 4-AP. (F) All LEC-driven post-synaptic responses recorded in the INs. Traces from individual neurons are shown in gray. Average PSP (color) was calculated from subthreshold PSPs only. Total n = 46 VIP INs, 23 CCK INs, 15 SST INs.
(G) LEC-driven postsynaptic response amplitudes. Mean ± SEM PSP amplitude is shown in black.
(H) Percentage of INs that responded to LEC axon photostimulation. Total n = 46 VIP INs, 23 CCK INs, 15 SST INs.
(I) Percentage of LEC-driven INs that exhibited LEC-driven APs; n = 44 VIP INs, 19 CCK INs, 4 SST INs.
VIP INs in CA1 were mainly located in the SLM and stratum pyramidale (SP) (Figures 4A, 4B, and S6A). LEC-driven responses persisted in the presence of TTX and 4-AP, confirming monosynaptic connectivity (Figure 4E). Recordings under drug-free conditions showed robust LEC-driven excitation in VIP INs, with 20% of the recorded VIP INs exhibiting LEC-driven APs (Figures 4F–4I). The vast majority of VIP INs driven to spike by LEC inputs were located in the SLM layer (Figure S6C).
CCK INs in CA1 were located mainly around the SR/SLM border and in the SP (Figures 4A, 4B, and S6A). Using pharmacology, we confirmed monosynaptic connectivity (Figure 4E). Under drug-free conditions, the majority of CCK INs responded to photostimulation of LEC inputs, with 21% of the recorded CCK INs exhibiting LEC-driven APs (Figures 4F–4I). The CCK INs driven to spike by LEC inputs were located exclusively at the SR/SLM border region (Figure S6C). These overlap with the interneuron subpopulation that has been shown to mediate FFI onto CA1 PN dendrites,80,81,91 and that is targeted by long-range GABAergic projections from LEC.27
In contrast to the other candidate INs, SST INs in CA1 were found almost exclusively in the stratum oriens (SO) and SP (Figures 4A, 4B, and S6A). They were not monosynaptically recruited or driven to spike by LEC inputs (Figures 4E–4I). SST INs in CA1 consist of two main subpopulations: oriens-lacunosum moleculare (OLM) INs, which mediate feedback inhibition,85 and bistratified SST INs, which modulate CA3-driven FFI.31 OLM INs are recruited by spiking CA1 PNs and inhibit CA1 PN distal dendrites. Because glutamatergic LEC inputs do not recruit CA1 SST INs or drive CA1 PNs to spike, our results suggest that SST INs do not modulate the generation of local LEC-driven dSpikes in our experiments. Our results highlight that glutamatergic LEC inputs recruit VIP INs and CCK INs to shape LEC-driven activity in the LEC-CA1 circuit.
LEC-driven VIP INs provide disinhibition to promote dendritic activity and dSpikes
VIP INs classically mediate disinhibition in the hippocampus83,92,93 and cortex,94–96 posing an intriguing possibility that they gate LEC-driven dSpike generation. We tested this experimentally by silencing local VIP INs during LEC-driven activity in CA1 PNs (Figure 5). First, we injected a Cre-dependent Jaws virus into hippocampal area CA1 of VIP-Cre/Ai14 mice to express the far-red-shifted inhibitory opsin Jaws97 in local CA1 VIP INs (Figures 5A and 5B). Jaws effectively silenced VIP INs throughout the injected region (Figures 5C and S7A–S7F).
Figure 5. Local VIP INs disinhibit LEC-driven dendritic spikes in CA1 PNs.
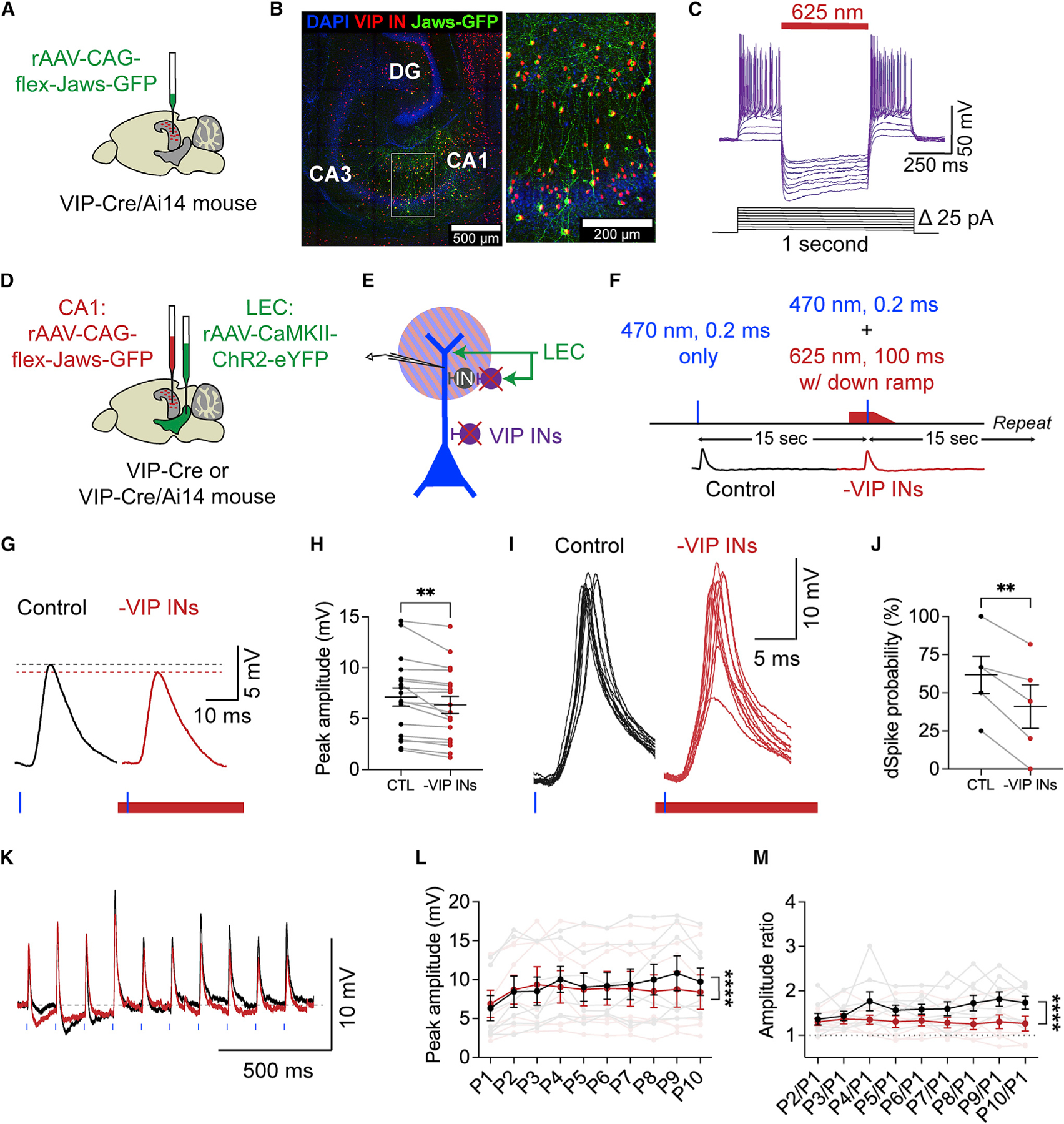
(A) Injection strategy. Cre-dependent Jaws virus (green) is injected into area CA1 of a VIP-Cre/Ai14 mouse.
(B) Confocal image of the hippocampus, demonstrating the CA1 injection site (left) and expression of Jaws-GFP (green) in tdTomato+ VIP INs (red) (right inset).
(C) Example trace illustrating that 625 nm light photostimulation silences Jaws+ VIP INs in CA1.
(D) Injection strategy. Co-injection of the Jaws virus into CA1 (now red) and ChR2 virus into LEC (green) in a VIP-Cre or VIP-Cre/Ai14 mouse.
(E) Dual-color optogenetics strategy. Simultaneous activation of ChR2+ LEC axons (green) and silencing of local Jaws+ VIP INs (purple) while recording from a CA1 PN distal dendrite. Blue and red stripes represent photostimulation with 470 and 625 nm light, respectively.
(F) Experimental strategy. LEC axons were photostimulated alone or with simultaneous silencing of VIP INs: 470 nm light, 0.2 ms, 20%–90% maximum strength; 625 nm light, 100 ms, maximum strength, starting earlier, ending with a down ramp. Control and –VIP INs conditions alternated every 15 s.
(G) Example LEC-driven dPSPs before (black) and during (red) VIP IN silencing. Dotted lines indicate the respective peak amplitudes.
(H) LEC-driven dPSP peak amplitudes before and during VIP IN silencing (**p = 0.0016). Paired t test; n = 18 dendrites, 18 slices, 13 mice.
(I) Example LEC-driven dSpikes before (black) and during (red) VIP IN silencing. Multiple sweeps are shown to illustrate the varying LEC-driven responses in a spiking dendrite.
(J) Dendritic spike probability before and during VIP IN silencing (**p = 0.0047). Paired t test; n = 5 dendrites, 5 slices, 4 mice.
(K) Example LEC-driven dendritic responses after maximum-strength 8 Hz photostimulation before (black) and during (red) VIP IN silencing.
(L and M) Peak amplitudes (L) and amplitude ratios (M) of LEC-driven dendritic responses after repeated photostimulation of LEC inputs before (black) and during (red) VIP IN silencing. Individual data are shown in faded colors. Mean ± SEM is shown in full opacity. Peak amplitudes (****p = 0.0312) and amplitude ratios (****p < 0.0001). Two-way ANOVA; n = 7 dendrites.
Next, we co-injected the Cre-dependent Jaws virus into CA1 and the CaMKII-driven ChR2-eYFP virus into the LEC of VIP-Cre mice (Figure 5D). This allowed us to use dual-color optogenetics to simultaneously activate glutamatergic LEC axons and silence local VIP INs in area CA1 in a circuit-specific, temporally precise manner (see STAR Methods) (Figure 5E). We combined this approach with dendritic recordings to test whether VIP INs disinhibit LEC-driven dendritic activity in CA1 PNs. We alternated between control (470 nm light) and experimental (470 and 625 nm light) conditions every 15 s (Figure 5F). The 625 nm light pulse began before the 470 nm stimulus, to ensure maximum hyperpolarization of Jaws+ VIP INs during the LEC-driven response in the CA1 PN (Figures S7G–S7I) and ended with a down ramp to prevent rebound firing of the VIP INs 97. We photostimulated with both wavelengths over the SLM.
To investigate how VIP INs shape subthreshold versus suprathreshold dendritic activity, we categorized the LEC-driven responses into dPSPs and dSpikes based on their maximum dV/dt values in the control condition (see Figure 2D). Optogenetic silencing of VIP INs led to a significant decrease in dPSP amplitude (p = 0.0016, paired t test, n = 18), but did not change dPSP kinetics, compared with the control conditions (Figures 5G, 5H, S8A, and S8B). Notably, VIP IN silencing had a significant effect on suprathreshold activity (Figures 5I, 5J, S8C, and S8D). In dendrites that exhibited LEC-driven dSpikes under control conditions, VIP IN silencing led to a significant decrease in dSpike probability (p = 0.0047, paired t test, n = 5). There was no significant difference in dSpike amplitudes (p = 0.323, Figure S8D). Finally, we tested how VIP INs affect repetitive LEC-driven activity within the LEC-CA1 circuit. VIP IN silencing led to a significant decrease in the amplitude of repeated LEC-driven responses in CA1 PN distal dendrites compared with control conditions (peak amplitude p = 0.0312, amplitude ratio p < 0.0001, two-way ANOVA, n = 7, Figures 5K–5M). Thus, local VIP INs can dynamically boost repetitive dendritic inputs in CA1 PNs. VIP IN silencing had no effect on LEC-driven somatic activity (Figures S8E and S8F). Moreover, Jaws expression levels were correlated with the VIP IN silencing effect size in the dendrites but not somata (Figures S7J and S7K), further suggesting that the LEC-driven VIP IN microcircuit acts in a compartment-specific manner.
Our circuit mapping also revealed that LEC inputs recruit CCK INs in area CA1. CCK INs have been shown to modulate CA1 PN activity,27,73,98,99 but their role in gating cortical input-driven dSpikes has not been investigated. We tested three strategies to silence CCK INs to investigate their influence over LEC-driven dendritic activity in CA1 PNs. The CCK peptide is also expressed in CA1 PNs,78 so two strategies involved an intersectional approach to specifically target CCK INs (see STAR Methods). Unfortunately, none of these strategies worked to exclusively target or sufficiently hyperpolarize CCK INs (Figure S9). Given the lack of appropriate intersectional tools, CCK IN silencing is not possible at this time. Nevertheless, our VIP IN silencing experiment demonstrated that VIP INs serve a disinhibitory function to promote LEC-driven dSpike generation in CA1 PNs.
LEC inputs recruit disinhibitory and inhibitory VIP IN subpopulations in CA1
Hippocampal VIP INs are extremely heterogeneous: the co-expression of the molecular marker calretinin (CR) or CCK roughly categorizes VIP INs as disinhibitory or inhibitory, respectively.83,92 Because our VIP IN silencing strategy used VIP-Cre transgenic mice, we silenced both VIP IN subpopulations with Jaws. This may explain why the dPSP amplitude effect size was small. Although we demonstrated that LEC inputs recruit the general VIP IN population, it was unclear whether this included one or both functional subpopulations.
To investigate the recruitment of the VIP IN subpopulations, we photostimulated the LEC axons in the SLM, as previously described, and performed targeted patch-clamp recordings from tdTomato-expressing CR+ VIP INs or CCK+ VIP INs in hippocampal slices derived from intersectional triple transgenic mice (see STAR Methods) (Figures 6A–6G). We found that LEC inputs monosynaptically recruit and drive AP firing in both VIP IN subpopulations (Figures 6A–6D). About 14% of the recorded CR+ VIP INs and 33% of the recorded CCK+ VIP INs exhibited LEC-driven APs (Figures 6E and 6F). Only the neurons found around the SR/SLM border were driven to spike by LEC inputs (Figure S6). Interestingly, the LEC-driven AP probability was higher in the CCK+ VIP IN subpopulation than in the CR+ VIP IN subpopulation. CCK+ VIP INs overlap with the general CCK IN population, which we found was recruited by LEC inputs (Figure 4).
Figure 6. LEC inputs recruit both VIP IN subpopulations in CA1.
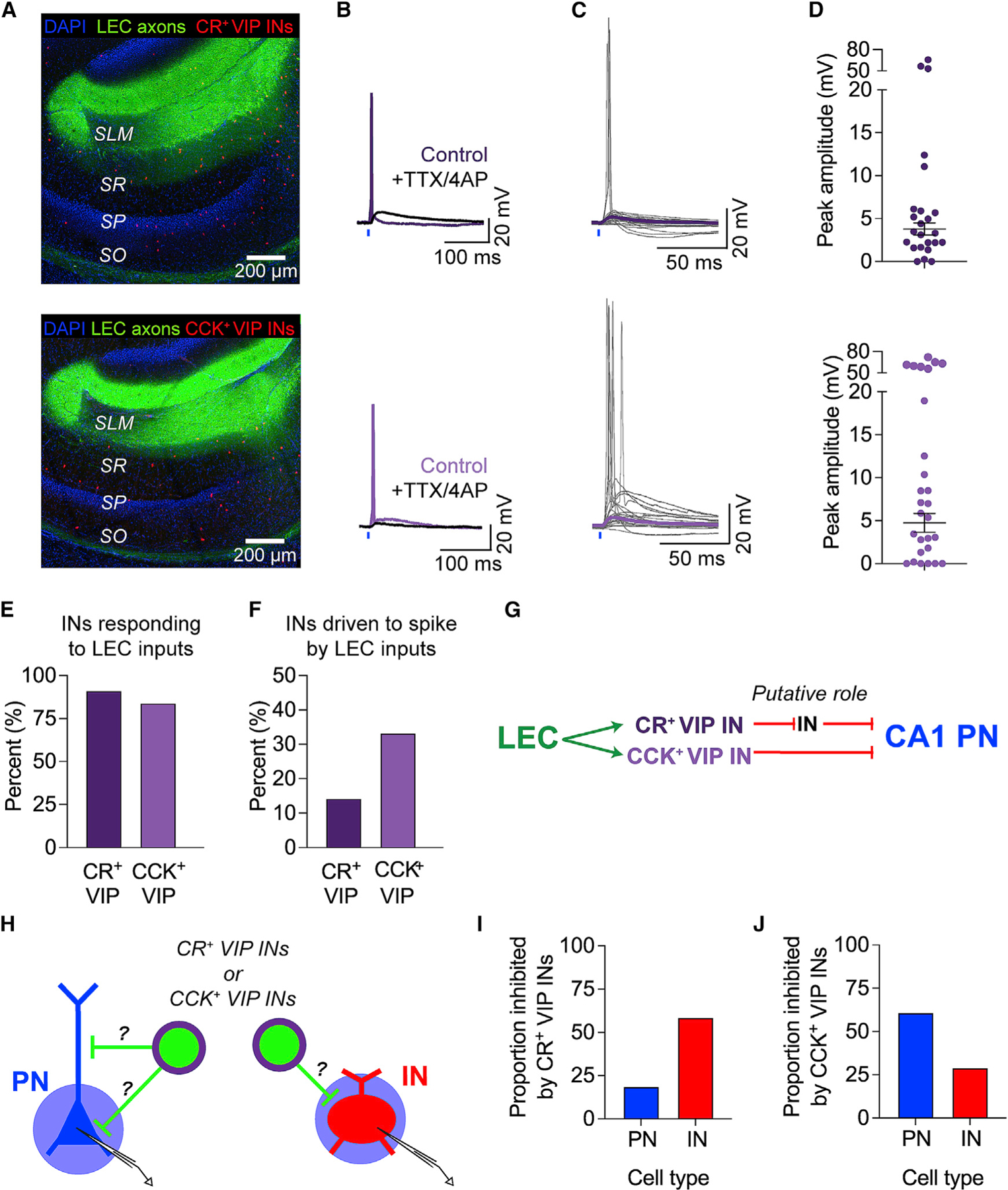
(A–D) Top to bottom: CR+ VIP INs (dark), CCK+ VIP INs (light). (A) Confocal images of hippocampal area CA1 with LEC axons (green) and tdTomato-labeled INs (red). VIP-Flp/CR-Cre/Ai65 and VIP-Flp/CCK-Cre/Ai65 mice were used. (B) Example LEC-driven responses before (purple) and after (black) application of TTX and 4-AP. (C) All LEC-driven postsynaptic responses recorded in the INs. Traces from individual neurons are shown in gray. Average PSP (purple) was calculated from subthreshold PSPs only. Total n = 23 CR+ VIP INs, 25 CCK+ VIP INs. (D) All LEC-driven postsynaptic response amplitudes. Mean ± SEM PSP amplitude is shown in black.
(E) Percentage of INs that responded to LEC axon photostimulation. Total n = 23 CR+ VIP INs, 25 CCK+ VIP INs.
(F) Percentage of LEC-driven INs that exhibited LEC-driven APs. n = 21 CR+ VIP INs, 21 CCK+ VIP INs.
(G) Proposed circuit diagram, based on our findings and the literature.
(H) Experimental strategy. Photostimulation (blue circle) of CatCh+ VIP IN subtypes (green) during somatic voltage-clamp recordings from putative CA1 PNs (blue) and INs (red). Neurons were held at +10 mV to record IPSCs.
(I) Proportion of neurons that were inhibited by CR+ VIP INs. Total n = 16 PNs and 29 INs tested.
(J) Proportion of neurons that were inhibited by CCK+ VIP INs. Total n = 23 PNs and 31 INs tested.
In addition, we probed the local postsynaptic targets of the VIP IN subpopulations to confirm whether they contribute to disinhibitory or inhibitory microcircuits (Figures 6H–6J). We used intersectional triple-transgenic mice to express the blue-light-sensitive excitatory opsin CatCh100 in CR+ VIP INs or CCK+ VIP INs (see STAR Methods). Using optogenetics and somatic voltage-clamp recordings, we photostimulated a specific VIP IN subpopulation and checked for the presence of IPSCs in downstream PNs and INs throughout area CA1. Consistent with previous findings, CR+ VIP INs predominantly targeted INs, whereas the CCK+ VIP INs predominantly targeted PNs (Figures 6H–6J and S10).83,101 Neuronal reconstructions of the downstream neurons further revealed that CR+ VIP INs inhibit dendrite-targeting perforant path-associated (PPA) INs, Schaffer collateral-associated (SCA) INs, and OLM INs (Figure S10D). Morphologically defined PPA and SCA INs are found along the SR/SLM border and can express CCK.80,81,91 These INs likely overlap with the LEC-driven CCK IN population and may inhibit LEC-driven dendritic activity in CA1 PNs. We also found some instances where CR+ VIP INs inhibited putative CCK+ VIP IN basket cells, revealing a complex interconnectivity between the disinhibitory and the inhibitory microcircuits recruited by LEC inputs in area CA1. Although PN-targeting VIP INs are known to inhibit the perisomatic region of CA1 PNs,79,83,92 it is possible that some VIP INs directly inhibit CA1 PN dendrites. However, we cannot verify this possibility because our somatic voltage-clamp recordings likely did not detect dendritic IPSCs. Nevertheless, our data are consistent with previously reported functions of the VIP IN subpopulations.
In summary, we found that glutamatergic LEC inputs recruit various GABAergic interneuron (sub)populations, some with opposing functions. By recruiting VIP INs, LEC inputs drive disinhibition onto CA1 PN distal dendrites, likely through the CR+ VIP IN subpopulation. On the other hand, by targeting CCK INs (including CCK+ VIP INs), LEC inputs likely drive FFI onto CA1 PNs.
Computational modeling dissects the relative contributions of VIP IN subpopulations and CCK INs in gating excitation in CA1 PNs
Given the limitations posed by the lack of available intersectional silencing tools, we used a data-driven computational model to test how various intersectional interneuron types modulate LEC-driven dendritic activity in CA1 PNs (Figures 7 and S14A–S14D). We built a multicompartmental model of an LEC-driven CA1 PN interconnected with various IN types and tested how the simulated IN deletion affects LEC-driven dSpike generation in the PN (see STAR Methods). The simplified neuron morphology and IN connectivity features of the canonical circuit were adapted from previous modeling studies.102,103 Biophysical and connectivity properties of all model neurons were heavily constrained by and updated to match experimental data collected in this study. The model CA1 PN received excitatory LEC inputs onto its distal dendrites and general inhibitory inputs throughout its somatodendritic axis to provide perisomatic and dendritic inhibition (Figure 7A). The model recapitulated the local LEC-driven dSpike incidence under control conditions, as well as the effects of inhibition blockade on LEC-driven responses in CA1 PN dendrites and somata (Figures 7B, S11, and S12). Simulations of compartment-specific inhibition blockade further revealed that distal dendritic inhibition significantly limits the generation of LEC-driven dSpikes, while perisomatic and proximal dendritic inhibition do not have an effect (Figure S12B). Our canonical circuit model included one of each of the following: CR+ VIP IN, CCK+ VIP IN, CCK IN found at the SR/SLM border, and SST-expressing OLM IN (Figures 7C and S13). Experimental data (Figures 4, 6, and S6) were used to inform the recruitment and spiking probability of these INs by LEC inputs, so the OLM IN did not exhibit APs. Experimental and published data were used to inform the downstream connectivity of the candidate interneurons (Figures 6 and S10).27,51,76,83,93,104 Data from the optogenetic VIP IN silencing experiment (Figure 5) were not used to validate and constrain the model.
Figure 7. Computational modeling suggests that CR+ VIP INs disinhibit and CCK INs inhibit dendritic activity.
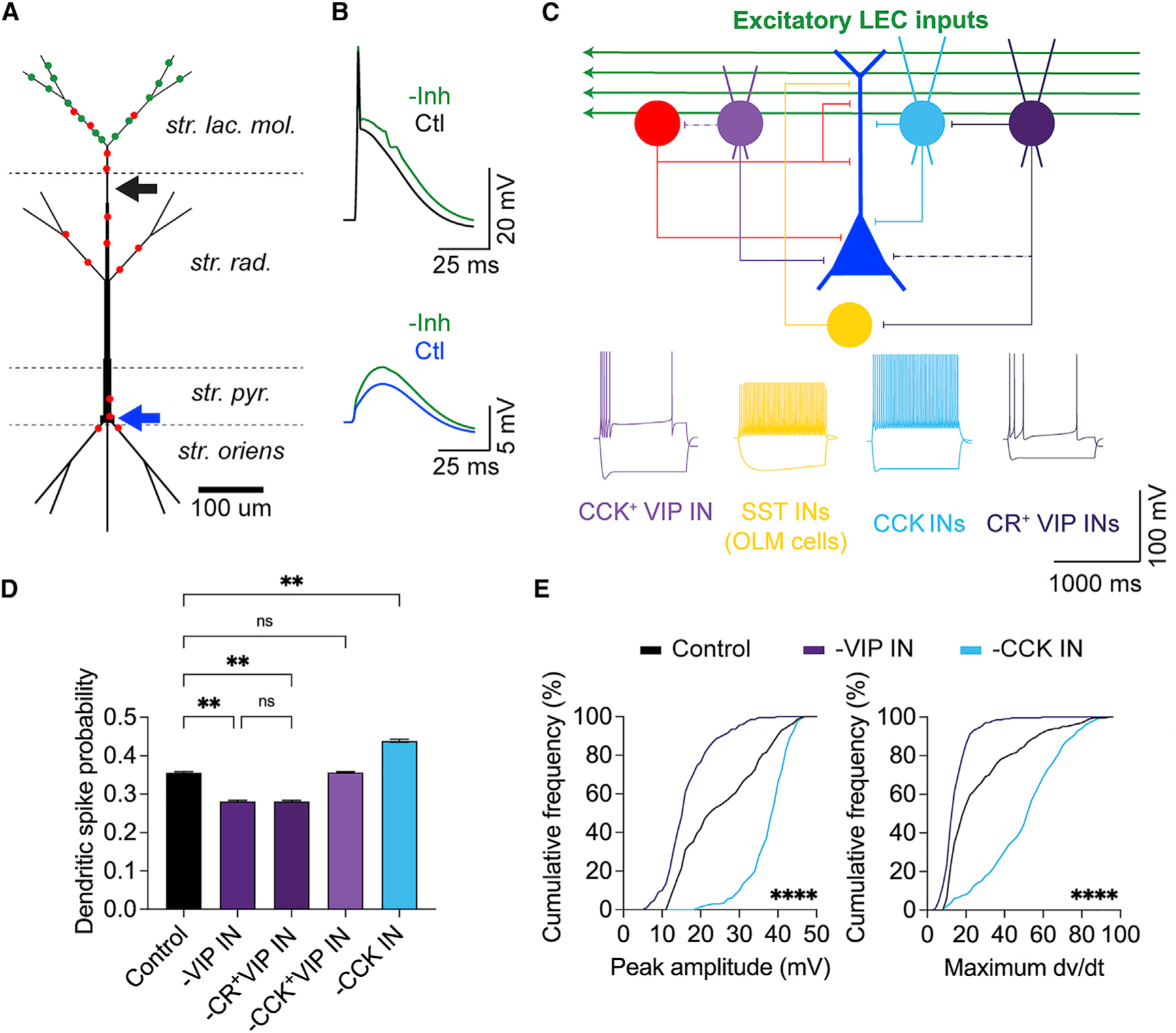
(A) Multicompartment CA1 PN model, receiving excitatory LEC inputs (green) and general inhibitory inputs (red). The dendritic recording site is indicated by a black arrow (300 μm from soma). The somatic recording site is indicated by a blue arrow.
(B) Simulated LEC-driven dendritic (top) and somatic (bottom) responses in the CA1 PN before (black and blue, respectively) and after inhibition removal (green).
(C) Model circuit schematic. LEC-driven CA1 PN (blue) surrounded by local GABAergic microcircuitry: general feedforward inhibition (red), CCK+ VIP IN (light purple), SST OLM IN (yellow), SR/SLM CCK IN (light blue), and CR+ VIP IN (dark purple). Weaker and/or less common connections are shown as dotted lines. Bottom: example electrophysiological properties of the model INs (see Figure S13 for details).
(D) Dendritic spike probability after silencing LEC-driven INs. Dunn’s multiple comparisons test; n = 20 × 1,000 trials.
(E) Cumulative frequency distributions of all simulated LEC-driven dendritic spike amplitudes (left) and maximum dV/dt values (right). Friedman test; n = 200 trials. **p < 0.01, ****p < 0.0001.
First, we deleted the entire VIP IN population (CR+ VIP INs and CCK+ VIP INs) to confirm that the model could independently recreate the effects of our optogenetic silencing experiments. Consistent with our previous results, VIP IN deletion led to a significant decrease in LEC-driven dSpike probability (p = 0.0056, Dunn’s multiple comparisons test, n = 20 trials, 1,000 runs per trial, Figure 7D) and LEC-driven dPSP amplitude (p < 0.0001, Dunn’s multiple comparisons test, n = 200 trials, Figure S14D) compared with control conditions.
Next, we deleted each of the intersectional INs to predict what role the VIP IN subpopulations and CCK INs have on LEC-driven dendritic activity in CA1 PNs. Deleting the CR+ VIP IN or CCK+ VIP IN allowed us to parse the influence of the disinhibitory and inhibitory microcircuits on LEC-driven dSpike generation. CR+ VIP IN deletion led to a significant decrease in dSpike probability (p = 0.0056), whereas CCK+ VIP IN deletion produced no significant change (p = 0.99, Dunn’s multiple comparisons test, n = 20 trials, 1,000 runs per trial, Figure 7D). This result suggests that the effect of LEC-driven disinhibition onto CA1 PN dendrites likely stems from the activity of the CR+ VIP IN subpopulation, in line with previous predictions for EC-driven excitation.102 CCK+ VIP INs generally have a soma-targeting, basket cell morphology,80,91 which may limit the extent of their influence on LEC-driven dendritic activity. CCK+ VIP IN deletion resulted in a significant increase in the LEC-driven somatic PSP amplitude (Figure S14D), suggesting that the downstream effect of each VIP IN subpopulation is localized to a specific neuronal compartment. Meanwhile, deleting the SR/SLM CCK IN led to a significant increase in LEC-driven dSpike probability (p = 0.0039) and LEC-driven dPSP amplitude (p < 0.0001) (Figures 7D and S14C), showing the effect opposite to VIP IN silencing. This is consistent with the idea that CCK INs found in the SR/SLM border region mediate cortically driven FFI onto CA1 PN distal dendrites.27,80,81,91 The dSpike amplitudes and maximum dV/dt values were similarly affected by VIP IN and CCK IN deletions in the model (peak amplitude p < 0.0001, maximum dV/dt p < 0.0001, Friedman test, n = 200 trials, Figures 7E and S14B).
Our data-driven model confirmed that LEC-driven VIP INs act as a disinhibitory gate to promote local dSpikes in CA1 PNs, likely due to the CR+ VIP INs. Our model further predicts that LEC-driven CCK INs mediate dendritic FFI to curb dendritic excitation. Together, our experimental data and computational modeling results reveal a circuit mechanism by which excitatory inputs from the LEC recruit local GABAergic microcircuits to gate supralinear computations in CA1 PNs and shape activity dynamics in hippocampal area CA1.
DISCUSSION
In summary, we combined dendritic recordings, optogenetic circuit manipulations, and computational modeling to demonstrate that a long-range cortical input pathway activates a hippocampal disinhibitory microcircuit to modulate the generation of local dSpikes. With inhibition intact, photostimulation of glutamatergic LEC inputs could elicit spikes in CA1 PN dendrites but not somata. In parallel, LEC inputs recruit strong FFI onto CA1 PN dendrites and somata. Targeted circuit mapping identified CA1 VIP INs and CCK INs as the key GABAergic interneurons recruited by LEC inputs. Optogenetic silencing of VIP INs uncovered their disinhibitory role in gating LEC-driven dSpikes in CA1 PNs. From the various VIP IN subpopulations, LEC inputs recruit both the disinhibitory CR+ VIP INs, which inhibit dendrite-targeting INs, and the inhibitory CCK+ VIP INs, which target PN somata. A detailed computational model predicted that LEC-driven CCK INs mediate FFI to suppress local dSpikes, whereas LEC-driven CR+ VIP INs suppress dendritic inhibition to disinhibit the dSpikes in CA1 PNs. Overall, our study provides mechanistic insight into the circuit interactions underlying the dynamic gating of supralinear dendritic functions and single-neuron computations.
Activation of glutamatergic LEC inputs can drive local dendritic spikes
Photostimulation of glutamatergic LEC inputs with a single light pulse could elicit dSpikes in 20%–30% of dendritic recordings (5/17 in Figure 2, 5/23 in Figure 5), even in the presence of inhibition. A greatly reduced photostimulation intensity (5% maximum) or pulse width (0.2 ms) could still drive dSpikes. Meanwhile, our various photostimulation paradigms never elicited somatic APs, indicating that LEC-driven dSpikes are local events. While the ionic nature of these dSpikes requires further investigation, we speculate that they are sodium spikes,40,61–63 as opposed to long-duration plateau potentials and complex spikes with calcium and NMDA components.12,27,105 One dendrite exhibited multipeaked putative calcium spikes (Figure 2, d1), but its small amplitude suggests it attenuated from a more distal origin than our recording site.
Input-driven disinhibitory microcircuit gates dendritic spikes
Our findings uncover additional possibilities for dSpike generation. LEC-driven dSpikes bypassed the usual requirements for dSpike generation involving strong, repetitive activity, integration with other coincident inputs, and/or inhibition blockade.27,31,33,35,40,42–44,61,63,67,68,73,106 The experimental conditions used in previous studies predominantly served to raise the dendritic membrane potential to suprathreshold levels. Dendritic inhibition limits dSpike generation,31,107–109 so any mechanism that reduces this inhibition is uniquely positioned to influence nonlinear dendritic computations.27,31 We demonstrate that input-driven GABAergic microcircuits can also serve this function. Recording LEC-driven activity with inhibition intact enabled us to capture the VIP IN-mediated disinhibitory microcircuit in action. Thus, input-driven disinhibition can overcome the dendritic FFI to permit dSpikes in CA1 PNs. Such a circuit motif can serve as a powerful gain modulation mechanism to permit dendritic nonlinearities in a fast, dynamic, and pathway-specific manner. Moreover, it may work in concert with long-range disinhibitory inputs from the entorhinal cortex (EC)27,69 to further boost dendritic excitation of PNs.
Recruitment of diverse GABAergic microcircuits can dynamically modulate excitation-inhibition balance
Our study uncovered multiple microcircuit routes by which glutamatergic LEC inputs can recruit excitation, inhibition, and disinhibition in area CA1 to bidirectionally modulate compartment-specific activity in CA1 PNs. LEC inputs drive dendritic and perisomatic inhibition by recruiting SR/SLM CCK INs, which target CA1 PN dendrites,27 and CCK+ VIP INs, which target CA1 PN somata,80,110 respectively. In parallel, LEC inputs also drive dendritic disinhibition by recruiting CR+ VIP INs, which target CA1 INs, including dendrite-targeting PPA and SCA INs and OLM INs (Figure S10).83,92,101 PPA and SCA INs can express CCK80,81,91 and likely overlap with LEC-driven CCK INs in the SR/SLM (Figures 4 and S6). Thus, LEC-driven VIP IN-mediated disinhibition can counteract dendritic inhibition to boost dendritic activity and local dSpike generation. It is unclear whether different LEC neurons recruit disinhibitory VIP INs versus inhibitory CCK INs in CA1. If LEC simultaneously recruits counteracting IN subpopulations, then dSpike generation may depend on the relative number of recruited IN types and/or the synaptic weights of upstream and downstream connections within CA1. Alternatively, if multiple groups of LEC neurons differentially recruit disinhibitory versus inhibitory microcircuits, then dSpike generation may be strongly influenced by sensory stimuli or behavioral state in vivo,20,27,111 which activate specific LEC neuron ensembles.
A data-driven computational model links dendritic nonlinearities with excitatory and inhibitory input pathways in CA1
Our computational model extensively explored the role of specific microcircuit elements in the LEC-CA1 circuit. Existing CA1 PN models cater to MEC-CA1 interactions,44,112–116 whereas our circuit model simulated LEC-mediated effects, readily reproducing experimentally observed LEC-driven dSpike and IN activity. The model allowed us to simulate compartment-specific inhibition blockade (Figure S12) and the deletion of intersectional INs (Figures 7 and S14). This revealed that dSpikes are gated by CR+ VIP INs and CCK INs, which could not be silenced experimentally. Unlike previous highly simplified117–119 or detailed104 CA1 circuit models, we opted for an intermediate level of model complexity. We inserted detailed biophysical profiles into simplified neuronal morphologies that accounted for key dendritic features of CA1 PNs, constrained by experimental data. This way, we maximized tractability and computational efficiency while closely fitting our own data. This minimized nonbiological variability and ensured very high model accuracy. The properties of our deterministic model make it a valuable tool for the community, as it can simulate difficult experiments across subcellular, single-neuron, and circuit levels. Our study demonstrates a powerful, mutually beneficial experiment-modeling loop approach to investigate dendritic nonlinearities and their control by circuit mechanisms.
Local dendritic spikes: Causes and consequences
Our distal dendritic recordings captured LEC-driven dSpikes in the neuronal compartment closest to the location of glutamatergic LEC synapses. The same photostimulation paradigm failed to generate somatic APs, suggesting that LEC-driven dSpikes are local. This phenomenon may result from attenuation along the dendrite68,120 or compartment-specific inhibition that restricts dSpike propagation and somatic spike output.27,31,46,51,53,121 During hippocampal theta oscillations, EC-driven depolarization of CA1 PN distal dendrites coincides with somatic hyperpolarization, thereby failing to generate somatic firing.121
The local nature of LEC-driven dSpikes does not make them functionally inconsequential. Local dSpikes have been reported in vivo in the hippocampus and cortex36,37,121–124 and linked to feature-selective tuning.36–38,48 In CA1, large-amplitude dSpikes, resembling our local dSpikes, occur during coordinated oscillatory activity from CA3 and the EC, without triggering somatic APs.32 Local dSpikes may serve a function distinct from that of somatic APs by enhancing the electrical and biochemical dendrite-soma coupling. For example, dendritic nonlinearities broaden temporal integration windows by activating NMDARs and by triggering Ca2+ influx and downstream intracellular signaling cascades to influence long-term plasticity at the soma.33–35,73,106,125,126 Ultimately, soma-independent, branch-specific dendritic activity can greatly expand the computational capacity of a neuron.44,127–133
Integration with other inputs may enable the LEC to drive somatic output and plasticity
Under what conditions could excitatory LEC inputs drive somatic spike output? High-frequency burst-like activity and/or integration of coincident inputs could elicit APs by sufficiently depolarizing the somatic compartment. We expanded our computational model and demonstrated that high-frequency, in vivo-like patterns of activity from excitatory LEC inputs, together with somatic depolarization, can lead to APs in the model CA1 PN (Figures S14E and S14F). At higher rates of LEC input activity, somatic spike output is differentially modulated by LEC-driven interneurons: disinhibitory VIP INs promote APs, while CCK INs and CCK+ VIP INs limit APs.
Glutamatergic LEC inputs may integrate with other inputs in CA1 PNs, including glutamatergic MEC12 and CA3 inputs, and long-range GABAergic (disinhibitory) inputs from the LEC27 and MEC.69 Integration of co-active inputs may additionally boost dendritic depolarization to aid the propagation of LEC-driven dSpikes to drive somatic APs, burst firing, and timing-dependent plasticity rules.27,33,39,67,73,125,134 Co-active inputs may also differentially recruit local circuit elements to further diversify computations in the hippocampus.
LEC inputs may support hippocampal memory representations in vivo by gating contextually relevant sensory activity
There is mounting evidence to suggest that the LEC provides a multisensory, behaviorally relevant signal to shape context-dependent coding in the hippocampus.3,6,8,19–29,86,135 LEC and CA1 activity synchronizes during odor sampling of context-place association tasks.3 Disinhibitory GABAergic LEC-CA1 projections are activated by rewarding and aversive stimuli and are requiredfor precise contextual discrimination and novelty detection.27 In vivo, LEC-driven dSpikes may assign contextual salience to sensory cues in the environment, especially when further boosted by co-active inputs or neuromodulatory signals.29,86,136,137 Because dSpikes serve as a predictive signal for place cell tuning,12,39,138 our findings also pose an interesting possibility that LEC inputs may influence context-dependent formation and/or remapping of hippocampal place cells.139,140 This may explain why MEC silencing only moderately affects dSpike output,12 place cell properties,11,15 and spatial memory behavior.4,9 Although the LEC has been classically associated with nonspatial sensory processing,6,22 recent studies suggest that LEC neurons display spatially modulated activity.24,25,141 Together, our and others’ findings warrant further investigation to link cellular-level dendritic computations to behaviorally relevant network dynamics and episodic memory processing supported by corticohippocampal interactions.
Limitations of the study
Although our study presents a detailed characterization of the LEC-CA1 circuit, and uncovers a disinhibitory microcircuit that gates local dSpikes, there are limitations to our experiments and interpretation. These primarily stem from the challenges of studying diverse sources of inhibition. We used existing transgenic mice to identify genetically defined IN types that are recruited by glutamatergic LEC inputs in CA1: VIP INs (including CR+ and CCK+ subpopulations) and CCK INs. However, hippocampal area CA1 harbors many IN subpopulations,50 with no clear genetic strategy to label every one of them. Creating a complete LEC-CA1 IN connectivity map is technically challenging and beyond the scope of the present study. Our optogenetic mapping and silencing experiments both used the general VIP-Cre mouse line and therefore included all VIP IN subpopulations: disinhibitory CR+ VIP INs, inhibitory CCK+ VIP INs, and VIP INs that lack CR and CCK expression. The CR−/CCK− VIP INs are considered disinhibitory ‘‘interneuron-specific type II (IS2) cells’’ and are found in the SLM.79,92,93 Although IS2 cells likely contributed to the disinhibition we observed experimentally, their properties are poorly understood. Our model did not include IS2 cells, so it likely underestimated the net disinhibitory effect of LEC-driven VIP INs. On the other hand, optogenetic silencing of multiple, counteracting VIP IN subpopulations in area CA1 may have masked the observed disinhibitory effect upon dendritic and somatic activity. Nevertheless, our experimental and modeling results together suggest that glutamatergic LEC inputs recruit a delicate balance of inhibition and disinhibition onto CA1 PN dendrites. Finally, given the lack of appropriate intersectional viral or transgenic silencing strategies, we could not experimentally test how CCK INs or CR+ VIP INs modulate LEC-driven dSpikes in area CA1. Nevertheless, using our own data to extend and refine our computational model allowed us to predict the roles of these GABAergic neuron subpopulations with the highest possible accuracy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Jayeeta Basu (jayeeta.basu@nyulangone.org).
Materials availability
This study used some custom-designed viral reagents. Please contact the corresponding author for further details.
Data and code availability
Electrophysiology and microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited on ModelDB: http://modeldb.yale.edu/267221 and Zenodo: https://doi.org/10.5281/zenodo.7413215 and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments were conducted in accordance with the National Institutes of Health guidelines and with the approval of the New York University Grossman School of Medicine and NYU Langone Health Institutional Animal Care and Use Committee (IACUC). Mouse lines were obtained from the Jackson Laboratory (JAX) and crossed to reporter lines in-house. Slice electrophysiology experiments were done on acute brain slices from CaMKII-Cre145 or interneuron-specific mouse lines. To fluorescently label VIP- or SST-expressing interneurons, homozygous VIP-Cre and SST-Cre mice78 were crossed with homozygous Cre-dependent Ai14-tdTomato reporter mice,146 respectively. To fluorescently label CCK-expressing interneurons, CCK-Cre drive mice78 were first crossed with the Cre-/Flp-dependent Ai65-tdTomato reporter strain,147 and the resulting progeny were crossed with the Dlx-Flp driver mice (gift from Gordon Fishell, NYU/Harvard,148), producing a triple transgenic CCK-Cre/Dlx-Flp/Ai65 mouse line with tdTomato expression restricted to GABAergic CCK-expressing interneurons.73 To fluorescently label VIP IN subpopulations, double homozygous VIP-Flp/CR-Cre or VIP-Flp/CCK-Cre mice78,149 were crossed with homozygous Cre/Flp-dependent Ai65-tdTomato reporter mice.147 To express excitatory opsin CatCh100 in VIP IN subpopulations, double homozygous VIP-Flp/CR-Cre or VIP-Flp/CCK-Cre mice were crossed with homozygous Cre-/Flp-dependent Ai80-CatCH-expressing reporter mice.150 Litter mates were co-housed up to 5 mice per cage and were provided with food and water ad libitum. Male and female mice were used, ages 7–12 weeks (PN somatic data: ~40% males/60% females; PN dendritic date: ~60% males/40% females). No significant differences were found between data collected from male versus female mice (data not shown).
METHOD DETAILS
Viruses
Commercially-generated recombinant adeno-associated viruses (rAAVs) were injected into the lateral entorhinal cortex (LEC) or hippocampal area CA1 (dorsal and intermediate) region under stereotactic control. Viruses were stored at −80°C in 2–4 μL aliquots and were thawed prior to use. Co-injected viruses were combined <16 h before viral injections. For optogenetic activation experiments, AAV2.9-CaMKIIa-hChR2(H143R)-eYFP142 was injected into LEC. For targeted interneuron recordings, the virus was injected into LEC of interneuron-specific transgenic mice. Slice electrophysiology experiments were conducted 7–10 days after viral injection. For optogenetic silencing experiments, AAV2.5-CAG-flex-Jaws-GFP97 was injected into hippocampal area CA1 of VIP-Cre or VIP-Cre/Ai14 mice, targeting multiple CA1 layers. The Jaws virus incubated for 15–17 days before slice electrophysiology experiments. Meanwhile, the mice were additionally injected into the ipsilateral LEC with AAV2.9-CaMKIIa-hChR2(H143R)-eYFP, as described above, and slice electrophysiology experiments were conducted 7–10 days after injecting the ChR2 virus. For the CCK IN silencing experiments, three viral strategies were tested. For strategy #1, AAV2.8-nEF-Con/Fon-iC++-eYFP143 was injected into CCK-Cre/Dlx-Flp/Ai65 triple transgenic mice into area CA1 and incubated for 19–22 days after injection. For strategy #2, a custom-made AAV2.9-hDlx-Flex-Jaws-TdTomato virus (hDLX-flex-tdTomato vector, generous gift of Gordon Fishell, Harvard Medical School, and Jordane Dimidschstein, MIT,151 and Jaws sequence, both sub-cloned and packaged by Canadian Neurophotonics Platform Viral Vector Core Facility) was injected into area CA1 of CCK-Cre mice and incubated for 2–6 weeks. For strategy #3, AAV2.5-CAG-flex-Jaws-GFP was injected in various dilutions into hippocampal area CA1 of CCK-Cre mice and incubated for 8–9 or 15–17 days before slice electrophysiology experiments.
Viral injections
Viral injection pipettes were prepared using thin glass pipette (Drummond Scientific), pulled by a micropipette puller (Sutter P-1000, Sutter Instrument Company) and fire polished using a microforge to have a long taper and 7–10 μm tip diameter. Pipettes were back-filled with mineral oil, then front-filled with the virus using a Nanoject II injector (Drummond Scientific). Adult mice (6–11 weeks old) were injected using stereotactic surgery and sterile technique. The animal was deeply anesthetized with inhaled isoflurane (5% for induction, 1.5–3% for maintenance during surgery, Matrx VIP 3000 calibrated vaporizer), and was given an IP injection of 0.05–0.1 mg/kg Buprenorphine before surgery. After head-fixation of the animal in a stereotactic frame (Stoelting), the surgical area was prepared with aseptic technique. The hair on the mouse’s head was shaved with a razor, and the exposed skin was locally disinfected with ethanol and betadine. Before incision, Ropivacaine (1–3 drops) was applied to the skin as a local analgesic. An incision was made to expose the skull, and hydrogen peroxide (0.1%) was applied to the surface of the skull to clean it. The level of the skull was adjusted such that Bregma and Lambda were aligned in the z-axis.
A unilateral craniotomy was made by thinning the skull with a dental drill. After gaining access to the surface of the brain, the injection pipette was lowered into either the lateral entorhinal cortex (LEC) or into the dorsal and intermediate portions of hippocampal area CA1. LEC: A/P −3.2 ± 0.2, M/L 4.5 ± 0.2, D/V −2.5 ± 0.2 mm from Bregma (6 sites); 46 nL injected per site, left hemisphere, specifically targeting superficial layers (L2/3). Dorsal and intermediate CA1, targeting local VIP INs in various CA1 strata: A/P −2.3 ± 0.3, M/L 1.5 ± 0.2, D/V −1.3 ± 0.2 mm from Bregma and A/P −3.2 ± 0.2, M/L 3.7, D/V −2.5 ± 0.2 mm from Bregma (11 sites total); 46 nL injected per site. The pipette was lowered into the brain, and the virus was injected into each coordinate (2 x 23 nL for a total of 46 nL injected per site). After the final coordinate in the injection site, the pipette was left in place for 10–15 min to allow for adequate dispersion of the virus. After slowly withdrawing the injection pipette from the brain, the incision was sutured (Henry Schein) and antibiotic ointment (Neosporin) was applied. Animals were injected with 0.5–1.0 mL saline solution to rehydrate and were allowed to recover for 1–3 weeks to allow for adequate viral expression.
Slice preparation and electrophysiology
Slice preparation
Adult mice were deeply anesthetized with 5% isoflurane and transcardially perfused with ice-cold sucrose-enriched dissection artificial cerebrospinal fluid (dACSF). After the dissected brain was cut down the midline, the two hemispheres were tilted ventro-medially at a 10° angle, and a vibratome (Leica VT1200S) was used to cut 400 μm-thick horizontal brain slices. The slices were incubated in a 1:1 mixture of sucrose-enriched dACSF and standard ACSF solution for 15 min at 34°C and 45 min at room temperature before being transferred to the recording chamber containing standard ACSF.
Solutions
Sucrose-enriched dissection ACSF (dACSF) consisted of (in mM): Sucrose 195.0, NaCl 10.0, Glucose 10.0, NaHCO3 25.0, KCl 2.5, NaH2PO4 1.25, Sodium pyruvate 2.0, CaCl2 0.5, MgCl2 7.0, saturated with 95% O2 and 5% CO2. Standard artificial cerebrospinal fluid (ACSF) consisted of (in mM): NaCl 125.0, Glucose 22.5, NaHCO3 25.0, KCl 2.5, NaH2PO4 1.25, Sodium pyruvate 3.0, Ascorbic Acid 1.0, CaCl2 2.0, MgCl2 1.0, saturated with 95% O2 and 5% CO2. The intracellular solution contained the following (in mM): KMeSO4 135 (for current clamp recordings) or CsMeSO4 135 (for voltage clamp recordings), KCl 5, EGTA 0.1, HEPES 10, NaCl 2, Mg-ATP 5, Na2-GTP 0.4, Na2Phosphocreatine 10, and Neurobiotin or Biocytin (0.2%). The pH of the intracellular solution was adjusted to 7.2 with KOH as needed. Osmolarity ranged from 270 to 290, and was adjusted with sterifiltered MilliQ water. In a subset of experiments, the following pharmacological agents were used via bath application in the ACSF: TTX (1 μM), 4-AP (100 μM), SR95531 (2 μM), and CGP55845 (μ mM).
Electrophysiology setup
Slices were visualized with an LNscope microscope from Luigs & Neumann, equipped with Infrared Dodt Gradient Contrast optics (Luigs & Neumann), 5x air and 60x/0.8 nA water immersion objective (Olympus), 1–4x zoom optics (Luigs & Neumann), and a Hamamatsu ORCA-R2 CCD camera using ImageJ Micromanager imaging acquisition software. A MultiClamp 700B amplifier (Axon Instruments), Digidata 9 Digitizer (Axon Instruments), and two constant current stimulators (Digitimer Ltd.) using pClamp 9 software (Molecular Devices) were used for data acquisition. Optogenetic stimulation and fluorescence-guided targeted patch-clamp recordings were performed using a Thorlabs LED system (405/470/565/635 nm wavelengths). Patch-clamp recordings were performed using junior micromanipulators (Luigs & Neumann) on movable motorized shifting tables (Luigs & Neumann). Recordings were performed at 34°C in standard ACSF, maintained by an inline heater (Warner Instruments) and saturated with 95% O2 and 5% CO2. Fire-polished borosilicate glass pipettes (Sutter) were used with tip resistances of 3.0–5.5 MΩ for somatic recordings (up to 6.5 MΩ for VIP IN somata) and 13–17 MΩ for dendritic recordings, pulled with a micropipette puller (Sutter P-1000). Current clamp recordings were obtained with access resistances up to 20 MΩ for somata and up to 40 MΩ for dendrites, compensated in bridge balance mode. The series resistance was not compensated for voltage-clamp recordings.
Electrophysiology recordings
All whole-cell patch-clamp recordings (dendritic, somatic) were performed in current clamp mode, with the exception of the recordings in Figures 6H–6J and S10.
To confirm adequate expression and function of ChR2 in infected LEC neurons, slices containing LEC were photostimulated over LEC using two protocols. First, a train of 470 nm light was given in cell-attached mode (10 Hz, 2 ms, 100% LED power) to test for time-locked action potentials. Next, a 500 ms-long light pulse was delivered in whole-cell configuration to check for a large, excitatory photocurrent. These protocols were also used at the beginning of every recording to confirm the lack of off-target viral expression in area CA1. If local CA1 photostimulation produced a large photocurrent in the recorded CA1 neuron, then all data from that animal were discarded.
CA1 neurons were targeted for recording throughout the proximal-distal of area CA1 with a focus on the middle and distal (close to subiculum) thirds, where glutamatergic LEC inputs are most dense (Figures S1E and S6A), as previously described.14,17 Deep and superficial CA1 pyramidal neurons were patched throughout the radial axis of layer SP. Candidate interneuron (sub)populations were targeted for recording in the CA1 layers that were most densely populated by that IN type (Figure S6A).
To measure the electrophysiological properties (firing and sag) of CA1 neurons (Figures 1E, 1J and 4C), current was first injected into the neuron to maintain a membrane potential of −70 mV for the following protocols. Depolarizing or hyperpolarizing current steps (1 s) were then injected into the neuron to measure the firing and sag properties, respectively. The firing was measured as the number of spikes during the 1 s depolarizing current step. The sag ratio was calculated as ((VPeak–VSteadyState)/VPeak)*100%, where VPeak and VSteadyState represent the change in membrane potential from baseline (−70 mV) to the peak sag and steady state response, respectively.
To measure light-evoked post-synaptic responses, CA1 neurons were kept at their resting membrane potential (RMP). Light-evoked responses were recorded every 15 s. Unless otherwise stated, LEC inputs were activated using a single 2 ms light pulse (470 nm) delivered at 100% maximum photostimulation strength (56 mW, 50 μm diameter beam spot). To measure the input-output transformation of light-evoked responses, the stimulation strength was incrementally adjusted, keeping pulse duration constant. To measure the short-term plasticity dynamics of LEC-CA1 inputs, five low intensity light pulses (2–3% maximum strength, 2 ms each) were delivered at physiologically-relevant frequencies (1–10 Hz, ≤ theta frequency).152,153 To measure the summation of LEC-driven post-synaptic responses, ten maximum strength light pulses were delivered at 2–10 Hz. For experiments involving pharmacology, light-evoked responses were recorded in control conditions (at least 5 min), during drug infusion (at least 10 min, while monitoring cell health), and in the experimental condition (at least 5 min). The amount of current injected was continuously monitored and adjusted to keep the neuron’s membrane potential close to the resting membrane potential in control conditions. Before pharmacological inhibition blockade experiments, the Schaffer collaterals (CA3-CA1 connection) were manually cut with a scalpel before the start of recording to prevent runaway epileptiform activity during the experiment.
For the optogenetic silencing experiments, LEC-driven post-synaptic responses were recorded every 15 s, alternating between control and experimental conditions. In the control condition, 470 nm light was used to photostimulate glutamatergic LEC axons only. In the experimental condition, 470 nm and 625 nm light were used simultaneously to optogenetically activate glutamatergic LEC axons and silence local CA1 VIP INs, respectively. Although the excitation spectra of ChR2 and Jaws show minimal overlap,97,154 we reduced the strength and duration of the 470 nm light (0.2 ms, 20–90% maximum strength) to decrease the chance of cross-talk with the Jaws expressed in VIP INs. To ensure maximum silencing of VIP INs during photostimulation of LEC inputs, the 625 nm light pulse (100 ms, maximum photostimulation) began 50–60 ms before the 470 nm light pulse. The 625 nm light pulses ended with a down ramp (100 ms) to decrease the probability of rebound firing in the VIP INs. When photostimulating LEC axons with an 8 Hz train of light pulses (0.2 ms each, 40–60% maximum photostimulation strength to produce subthreshold responses only), VIP INs were silenced with continuous 625 nm light for 1 s, starting 50–60 ms before the first 470 nm light pulse. Data from slices with insufficient Jaws expression (Figure S14J) were discarded.
For the somatic voltage clamp recordings in Figures 6 and S10, CA1 PNs or INs were held at +10 mV to measure inhibitory post-synaptic currents (IPSCs).
Immunohistochemistry
Animals were deeply anesthetized with 5% isoflurane for 5 min, followed by an injection of a mixture of ketamine (150 mg/kg) and xylazine (10 mg/kg). Animals were then perfused with 1X PBS, followed by 4% paraformaldehyde in PBS. The brain was removed and fixed overnight in 4% paraformaldehyde in 4°C and then sectioned into 100 μm or 400 μm slices using a microtome (Leica VT1000S) after washing. Alternatively, after slice electrophysiology experiments, brain slices were drop-fixed in 4% paraformaldehyde in PBS and left overnight at 4°C. The slices were washed once with 0.3 M PBS-Glycine for 15–20 min and three times in PBS. Slices were left in PBS at 4°C until immunostaining.
Slices that underwent CUBIC tissue clearing155 were thoroughly washed with 0.1M PB and then moved to CUBIC#1 tissue clearing solution for two days. Next, slices were thoroughly washed in 0.1M PB before undergoing the standard immunostaining protocol. Slices were permeabilized in 0.5% PBS-Triton X (PBST), blocked in 3% Normal Goat Serum (2–4 h), incubated with primary antibodies (overnight, 4°C), washed, and incubated with secondary antibodies (1–2 days, 4°C). The antibodies were diluted in blocking solution (1X PBS, 0.5% Triton, 3% NGS). After the final antibody incubation step, the slices were thoroughly washed in 0.1M PB and moved to CUBIC#2 tissue clearing solution for 1 h, after which the slices were mounted on glass slides in CUBIC#2 solution. Slices that did not undergo tissue clearing immediately started with the standard immunostaining protocol, beginning with the permeabilization in 0.5% PBST and ending with the secondary antibody incubation step. These slices were then thoroughly washed in 1X PBS and mounted in Vectashield Hard Set Mounting Medium with DAPI (Vector Laboratories).
Slices containing ChR2-eYFP+ LEC axons and/or Jaws-GFP+ VIP INs were stained using a rabbit anti-GFP primary antibody (1:1000; Invitrogen) and a goat anti-rabbit Alexa Fluor 488 secondary antibody (1:1000; Invitrogen). Alexa Fluor 593 or 647 streptavidin (1:500; Thermo Fischer Scientific) was added with the secondary antibody to counterstain the cells filled with biocytin during intracellular recordings. For slices that underwent the combined tissue clearing and immunostaining steps, Neurotrace 435/455 was added with the secondary antibody to label somata in the brain slice (1:200; Thermo Fischer Scientific). Data from animals with inadequate, mistargeted, or retrograde viral infection (of ChR2 and/or Jaws) were discarded.
Confocal imaging
Slices were imaged using an upright Zeiss 520 Meta Confocal Microscope, using (magnification/NA) 10x/0.3 air, 20x/0.8 air, and/or 40x/1.30 oil immersion objectives (Zeiss) and 405, 488, 594, and 647 nm lasers for excitation. 512 x 512 pixel images were acquired every 5–10 μm throughout the depth of the tissue (or every 2 μm for neuronal reconstructions). Tiled images were stitched using Zen Microscopy Software (Zeiss), and the tiled z-stacks were subsequently processed using Fiji (ImageJ).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis
In vitro electrophysiology data were analyzed using Axograph (Version 1.7.0). After measuring the baseline membrane potential, all traces were zeroed at the start of photostimulation (x = 0) and baseline membrane potential (y = 0). Subthreshold light-evoked responses were averaged in Axograph before measuring the amplitude and kinetics of the average response. Suprathreshold light-evoked responses were measured first, and then the values were averaged.
For all light-evoked post-synaptic responses, peak amplitude was measured as the maximum depolarization from the baseline. The time of peak was measured as the duration from the start of photostimulation (x = 0) to peak amplitude. The rise time was measured as the duration from 10% to 90% of peak amplitude. The half-width was measured as the duration between the 50% peak amplitude during the rise and decay of depolarization. The IPSP amplitude was measured as the maximum negative amplitude of the hyperpolarization component of the post-synaptic response from the baseline.
For dendritic recordings, derivative voltage traces and phase plots were generated from dendritic post-synaptic response traces using MATLAB R2019b (MathWorks). Maximum dV/dt values were calculated as the peak amplitude of the derivative traces and were averaged per dendrite to categorize the dendritic response. Within our experimental dataset, light-evoked dendritic responses with dV/dt < 7.5 mV/ms were categorized as dendritic post-synaptic potentials (dPSPs), whereas those with dV/dt > 7.5 mV/ms were categorized as dendritic spikes (dSpikes). Dendritic recording distance was measured in ImageJ using a 5x image taken after completing the dendritic recording.
Paired pulse ratio (PPR) was calculated as the Pulse n (Pn) amplitude divided by the Pulse 1 (P1) amplitude, with all amplitudes measured from the membrane potential immediately preceding each pulse. PPR >1 indicates short-term facilitation, whereas PPR <1 indicates short-term depression of inputs. Amplitude ratios were calculated to measure the post-synaptic summation of inputs. The amplitude ratios were calculated as the ratio of Pulse n (Pn) amplitude divided by the Pulse 1 (P1) amplitude, with all amplitudes measured from the membrane potential immediately preceding P1. Photostimulation protocols were run three times, and the resulting PPR or amplitude ratio values were averaged per cell.
For pharmacological inhibition blockade experiments, data that suggested that the recorded cell was experiencing the effects of runaway epileptiform activity were discarded. Inferred IPSP traces were generated post hoc in Axograph as the post-synaptic potential recorded in control conditions (PSP) minus the post-synaptic potential recorded after inhibition blockade (EPSP).
During targeted recordings from CA1 interneurons, the location of the patched soma was noted as: SLM, SR/SLM border, SR, SP, or SO (Figure S6). Interneurons were counted as responding to LEC inputs if they exhibited a reliable light-evoked post-synaptic response and were counted as driven to spike by LEC inputs if they responded with a light-evoked action potential.
During the voltage clamp experiments investigating the downstream targets of VIP IN subpopulations, neurons were counted as targeted by a VIP IN subpopulation if they exhibited a reliable light-evoked inhibitory post-synaptic current (IPSC). Because IPSC amplitudes were often less than 20 pA, connectivity was determined while blinded to the identity of the recorded neuron.
During the VIP IN silencing experiments, individual dendritic responses were categorized as subthreshold versus suprathreshold based on the maximum dV/dt value. Data were analyzed by comparing average amplitude and kinetics values, calculated under control versus experimental conditions, per cell. In dendrites that exhibited LEC-driven dSpikes in control conditions, dendritic spike probability was calculated as the number of dendritic spikes per number of recorded sweeps.
Statistical analysis
No statistical methods were used to predetermine the sample size. Dataset normality was determined using the Shapiro-Wilk test. Data were analyzed using appropriate parametric or non-parametric tests, as stated in the text and figure legends. When necessary, analyses were corrected for multiple comparisons using post hoc tests, as indicated. Data are shown as mean ± SEM, unless otherwise stated. Box and whisker plots are shown as median ± interquartile range. Significance level was set at p < 0.05. Prism (GraphPad) was used for plotting data and statistical analyses. Figures were generated using Adobe Illustrator.
Computational modeling
The neuronal models were developed based on prior computational models of the hippocampus.102,103 Specifically, we implemented the simplified morphology and ionic mechanisms for the individual neuronal models (i.e., pyramidal cells, OLM INs, non-basket cell CCK INs in the SR/SLM region, CCK+ VIP INs, and CR+ VIP INs) based on Cutsuridis and Poirazi, 2015.55 Individual cells were revalidated to match experimental data (experimental data from Figures 1,2, 3, 4, 6, S2, S4, S6, S10; validation in S11, S12 and S13), specifically the active and passive membrane properties, such as input resistance, resting potential, and rheobase current. LEC provided input to the network as a single spike or a Poisson-distributed spike train (see sections below). Data from the optogenetic VIP IN silencing experiment were not used to validate and constrain the model. For all simulations, we used the NEURON (v8.0.0) simulation environment,144 while analysis was performed in python (v3.9.7) using custom-based software. The simulations were performed on a server with Intel(R) Xeon(R) Silver 4210R CPU at 2.40GHz and 126 GB of shared RAM under CentOS (v 7.9) operating system. The code is available on ModelDB (http://modeldb.yale.edu/267221) and Zenodo (https://doi.org/10.5281/zenodo.7413215).
Model neurons
Individual neuron types were modeled as simplified biophysical neurons with various active dendrites using the Hodgkin-Huxley formalism. Specifically, the pyramidal cells incorporated 27 apical and basal dendrites to capture their complex morphology, while most interneurons were simulated with a simplistic morphology consisting of apical and basal dendrites in an ‘x’ shape. The OLM IN consisted of a soma, an axon, and two sister dendrites.156 The morphological features of all neuronal types are listed in Tables S1 and S2 for interneurons and pyramidal cells, respectively. The model SR/SLM CCK IN was generated de novo, as described below. For all simulations, the temperature was set to 34°C, and we used a variable time step integration method (i.e., CVode). The time step was initially set to 0.025 ms. The model did not include noise or spontaneous activity.
Random distributions
To account for the variability observed in experiments, all synaptic numbers were drawn from probability distributions. We used the Poisson (Equation 1), the Exponential (Equation 2), and the Gaussian (Equation 3) distributions. The synaptic location on the post-synaptic neuron was chosen based on a uniform distribution, i.e., all sites were equally probable to contain a synapse.
| (Equation 1) |
where x denotes the number of events, and λ denotes the mean of the distribution. •! is the factorial of a number.
| (Equation 2) |
where β is the scale parameter which represents the mean of the distribution.
| (Equation 3) |
where μ denotes the mean and σ the standard deviation.
We returned the rounded to the closer integer value for all distributions, as the number of synapses cannot be a float number.
Model CA1 pyramidal neuron (PN)
The model CA1 PN was adapted from Turi et al.,102 and modified to increase its complexity. Specifically, we increased the number of compartments representing the apical trunk and adjusted all compartments’ lengths (Table S2). The diameters of all compartments were updated to follow the d3/2 rule. The model cell consists of five apical dendritic compartments simulating the apical trunk (radProx, radMed, radDisti, and six dendritic compartments simulating the basal tree, each containing a Ca2+ pump and buffering mechanism, Ca2+ activated slow and medium AHP potassium (K+) currents, an HVA L-type Ca2+ current, an HVA R-type Ca2+ current, an LVA T-type Ca2+ current, an h current, a fast Na+ and a delayed rectifier K+ current, a slowly inactivating K+ M-type current and a fast inactivating K+ A-type current.43,44 The proximal and distal apical dendrites consisted of eight compartments, respectively, each containing a fast Na+, a delayed rectifier K+ and a fast-inactivating K+ A-type current. The model PN was topologically oriented: its soma was located in the simulated SP layer, its basal dendrites in the SO layer, and its proximal and distal apical dendrites in the SR and SLM layers, respectively. The type and distribution of ionic mechanisms in the model PN are described below, while each channel’s maximum conductance is given in Tables S3 and S4.
Model SR/SLM CCK IN
The non-basket, SR/SLM CCK IN had 17 compartments, containing a leak conductance, a sodium current, a fast-delayed rectifier K+ current, an A-type K+ current, L- and N-type Ca2+ currents, a calcium-dependent K+ current, and calcium- and voltage-dependent K+ current (Table S5, Figure S13). The CCK IN received excitatory connections from LEC to their distal SLM dendrites and from the CR+ VIP IN to their soma. To reproduce the LEC-induced activity on CCK INs (see Figures 4G–4I, S13B and S13C), we drew the number of excitatory synapses from a Poisson distribution with mean equals 10. At approximately 17% of the trials (n = 10 x 200 trials), the CCK IN receives no LEC connections (Figure 4H). The CR+ VIP IN also targeted the SR/SLM CCK IN, with the synaptic number drawn from an exponential distribution with mean equals 60. Using this value, the CCK IN was silenced when the number of inhibitory synapses was chosen to be high enough, but in other cases remained intact.
Model OLM IN
The OLM IN had four compartments, which included a Na+ current, a delayed rectifier K+ current, an A-type K+ current, and an h-current (Table S5, Figure S13). The OLM IN received excitatory connections from CA1 PN in their basal dendrites and inhibitory connections from the CR+ VIP IN in their soma. The number of excitatory synapses from LEC was drawn from a Poisson distribution, with lambda equals 0.3, to explain the low activation and no induction of spike shown in experiments (Figures 4G–4I, S13B and S13C). The number of excitatory connections from CA1 PN was drawn from an exponential distribution with mean equals 10. Using this distribution, we generated activity on the OLM IN when using the Poisson protocol (see sections below). The OLM IN also received an inhibitory connection from the CR+ VIP IN, the number drawn from an exponential distribution with mean equals 60, a value sufficient to induce a hyperpolarization to the OLM IN soma.
Model CCK+ VIP IN
The CCK+ VIP IN had 17 compartments, containing a leak conductance, a sodium current, a fast-delayed rectifier K+ current, an A-type K+ current, L- and N-type Ca2+ currents, a Ca2+- dependent K+ current, and a Ca2+- and voltage-dependent K+ current (Table S5, Figure S13). To replicate the irregular firing, which was experimentally observed, we tuned the calcium influx of this model, making the decay of Ca2+ current slower (Figure S13A). The CCK+ VIP IN received excitatory connections from LEC to the distal SLM dendrites and made inhibitory synapses onto the CA1 PN soma. The number of excitatory connections was drawn from a Poisson distribution with mean equals 21, while in 16% of the trials (n = 10 x 200 trials), the CCK+ VIP IN received no connections from LEC (Figures 6D–6F, S13B and S13C).
Model CR+ VIP IN
The CR+ VIP IN consisted of 17 compartments, including mechanisms for slow K+ current, fast Ca2+-activated K+ current, and N-type Ca2+ current (Table S5, Figure S13). Each CR+ VIP IN received excitatory input from LEC. The number of excitatory connections from LEC was drawn from a Poisson distribution with a mean equal to 5, while in 9% of the trials (n = 10 x 200 trials), the CR+ VIP IN did not receive any connection from LEC (Figures 6D–6F, S13B and S13C). The spread of the dendritic tree of the CR+ VIP IN model cell was larger than that of the CCK+ VIP IN model cell, as per Tyan et al.,83 (Table S1).
Validation
Passive (intrinsic) and active (spiking) properties of each neuronal type were validated against experimental data. For the validation, we used the frequency vs. injected current (f-I) curve and the sag-ratio as a function of injected currents (hyperpolarization) (Figure S2).
Modeling synapses
AMPA, NMDA and GABAA, and GABAB synapses were included in the model. NMDA synapses were used only in CA1 PNs. The interneurons received excitatory afferents via AMPA receptors. AMPA, NMDA, GABAA, and GABAB synapses were simulated as a sum of two exponentials. All synaptic properties (i.e., conductance g, rise time τr, decay time τd, synaptic delay) were validated with experimental evidence (see Figures 4, S4).
AMPA and GABA synapses
The AMPA, GABAA, and GABAB synapses were simulated as a two-state kinetic scheme, described by the rise and decay phases. The following equations give the synaptic current through these synapses:
assuming that at t = 0 we have a presynaptic spike. f is a normalization factor to bound gsyn at 1, and it is calculated by the following set of equations:
NMDA synapse
The NMDA synapse was simulated as a two-state kinetic scheme involving a factor representing the voltage dependency. The corresponding equations were:
where gNMDA was calculated as above, and the voltage dependence is given by:
where Vm denotes the membrane voltage.
In order to validate the synaptic weights, we used the experimental data shown in Figures 4, S4. All synaptic properties are summarized in Table S6.
Single-cell simulations
Various synapses, excitatory and inhibitory, were randomly placed in the model CA1 PN. The excitatory synapses representing the synapses from LEC were placed in the apical tuft at the SLM layer. In contrast, the inhibitory synapses were placed either in SLM, SR, or SP based on random distribution. The number of excitatory synapses was drawn from an exponential distribution with a mean of 12. Each excitatory synapse consisted of one AMPA and one NMDA component. The 60% of inhibitory synapses were located at SLM (distal dendritic inhibition), 20% at SR (proximal dendritic inhibition), and 20% were located at SP (somatic inhibition). The synapses located at SR and SLM were GABAA and GABAB based on uniform distribution. The total number of inhibitory synapses was selected from a random Gaussian distribution with a mean of 40 and std 4. The simulation duration was set at t = 2sec, and the activation of the pre-synaptic axons was set at t = 700ms. For the various deletion protocols, we set the respective weights to zero. All other parameters remained the same so that to compare the conditions pairwise.
Canonical microcircuit
The CA1 canonical circuitry consisted of the following cell types: 1 CA1 PN, 1 SR/SLM CCK IN, 1 OLM IN, 1 CCK+ VIP IN, and 1 CR+ VIP IN. The LEC input was given to all neuronal cells with several synapses drawn from distributions, as explained above. All neuronal models were held at their resting membrane potential. At t = 700ms, we stimulated the circuitry with a single spike. The simulation duration was set to 2sec. For the various deletion protocols, we set the weights of the afferents of the deleted neurons to zero. The rest parameters remained the same across the deletion so that the results were paired. We repeated the simulations using a Poisson-like, theta-filtered spike train. The theta filter equation is:
where is the theta cycle frequency set to 8Hz, the phase of the cycle set to zero, and tspike denotes the spike-time, which came from the Poisson distribution. If the probability is greater than 0.7, a spike is generated for this specific train. Also, 20% of out-of-theta-cycle spikes were included in the final spike train to have noise in the spike trains.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Neurotrace 435/455 Blue Fluorescent Nissl Stain | Fisher Scientific | Cat# N21479 |
| Rabbit anti-GFP (polyclonal) | Invitrogen | Cat# A6455; RRID: AB_221570 |
| Goat anti-rabbit 488 | Invitrogen | Cat# A11008; RRID: AB_143165 |
| Biocytin | Invitrogen | Cat# B1592 |
| Alexa Fluor 647-conjugated Streptavidin | Thermo Fisher Scientific | Cat# S32357 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pAAV-CaMKIIa-hChR2(H143R)-eYFP | Lee et al.142 | Addgene Cat# 26969-AAV9; RRID: Addgene_26969 |
| rAAV-CAG-FLEX-rc [Jaws-KGC-GFP-ER2] | Chuong et al.97 | Addgene Cat# 84445-AAV5; RRID: Addgene_84445 |
| AAV2.8-nEF-Con/Fon-iC++-eYFP | Fenno et al.143 | Addgene Cat# 137155; RRID: Addgene_137155 |
| AAV2.9-hDlx-Flex-Jaws-TdTomato | Custom-made by Neurophotonics Facility, using plasmid reagents from Gordon Fishell (NYU/Harvard) and Addgene | RRID: SCR_016477 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tetrodotoxin (TTX) | Hello Bio | Cat# HB1035 |
| 4-aminopyridine (4-AP) | Sigma | Cat# A0152 |
| SR 95531 hydrobromide | Hello Bio | Cat# HB0901 Cat# 1248 |
| CGP 55845 hydrochloride | Tocris | Cat# 1248 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| CaMKII-Cre | Jackson Laboratory | JAX: 005359; RRID: IMSR_JAX: 005359 |
| VIP-IRES-Cre | Jackson Laboratory | JAX: 010908 and 031628; RRID: IMSR_JAX:010908 and IMSR_JAX:031628 |
| SST-IRES-Cre | Jackson Laboratory | JAX: 013044; RRID: IMSR_JAX:013044 |
| CCK-IRES-Cre | Jackson Laboratory | JAX: 012706; RRID: IMSR_JAX: 012706 |
| CR-IRES-Cre | Jackson Laboratory | JAX: 010774; RRID: IMSR_JAX: 010774 |
| VIP-IRES-Flp | Jackson Laboratory | JAX: 028578; RRID: IMSR_JAX: 028578 |
| Dlx-Flp | Jackson Laboratory | JAX: 010815; RRID: IMSR_JAX:010815 |
| Ai14(RCL_tdT)-D | Jackson Laboratory | JAX: 007914; RRID: IMSR_JAX: 007914 |
| Ai65D(RCFL-tdT)-D | Jackson Laboratory | JAX: 021875; RRID: IMSR_JAX: 021875 |
| Ai80D(RCFL-CatCh)-D | Jackson Laboratory | JAX: 025109; RRID: IMSR_JAX: 025109 |
|
| ||
| Software and algorithms | ||
|
| ||
| pClamp 9 | Molecular Devices | N/A |
| AxoGraph (v1.7.0) | Axograph | N/A |
| MATLAB R2019b | Mathworks | N/A |
| GraphPad Prism 9 | GraphPad | N/A |
| NEURON (v8.0.0) | Carnevale and Hines144 | N/A |
| Python (v3.8.5) | https://www.python.org/ | N/A |
| ImageJ (FIJI) | NIH | N/A |
| Illustrator | Adobe | N/A |
| Custom data-driven LEC-CA1 circuit model | This paper | https://doi.org/10.5281/zenodo.7413215 |
Highlights.
Lateral entorhinal cortex can drive local dendritic spikes in CA1 pyramidal neurons
LEC inputs recruit VIP INs and CCK INs to gate compartment-specific inhibition
A disinhibitory VIP IN microcircuit boosts LEC-driven dendritic spike generation
A computational model further predicts which CA1 IN subtypes gate dendritic spikes
ACKNOWLEDGMENTS
This work was supported by grants from the NIH NINDS BRAIN Initiative-1R01NS109994, NINDS 1R01NS109362-01, McKnight Foundation, Mathers Foundation, Klingenstein Fund-Simons Foundation, Alfred P. Sloan Foundation, Whitehall Foundation, American Epilepsy Society, Blas Frangione Foundation, NYU Whitehead Fellowship, and Leon Levy Foundation to J.B.; NIH 5T32NS86750 and P30 AG066512 to O.B.; and the European Commission FET OPEN NEUREKA (GA 863254), NIH 1R01MH124867-01, and Einstein Foundation Berlin-EVF-2019-508 to P.P. We thank the Rudy lab (NYULH) for generously providing VIP/CR and VIP/CCK IN-specific transgenic mice and Drs. Tanvi Butola, Adam Carter, Dmitri Chklovskii, Melissa Hernández Frausto, Robert Machold, Jason Moore, John Rinzel, Vincent Robert, and Richard Tsien for feedback on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111962.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of nonhuman subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We avoided ‘‘helicopter science’’ practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
REFERENCES
- 1.Basu J, and Siegelbaum SA (2015). The corticohippocampal circuit, synaptic plasticity, and memory. Cold Spring Harb. Perspect. Biol 7, a021733. 10.1101/cshperspect.a021733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brun VH, Otnass MK, Molden S, Steffenach HA, Witter MP, Moser MB, and Moser EI (2002). Place cells and place recognition maintained by direct entorhinal-hippocampal circuitry. Science 296, 2243–2246. 10.1126/science.1071089. [DOI] [PubMed] [Google Scholar]
- 3.Igarashi KM, Lu L, Colgin LL, Moser MB, and Moser EI (2014). Co-ordination of entorhinal-hippocampal ensemble activity during associative learning. Nature 510, 143–147. 10.1038/nature13162. [DOI] [PubMed] [Google Scholar]
- 4.Kitamura T, Pignatelli M, Suh J, Kohara K, Yoshiki A, Abe K, and Tonegawa S (2014). Island cells control temporal association memory. Science 343, 896–901. 10.1126/science.1244634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Remondes M, and Schuman EM (2004). Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature 431, 699–703. 10.1038/nature02965. [DOI] [PubMed] [Google Scholar]
- 6.Hargreaves EL, Rao G, Lee I, and Knierim JJ (2005). Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science 308, 1792–1794. 10.1126/science.1110449. [DOI] [PubMed] [Google Scholar]
- 7.Kerr KM, Agster KL, Furtak SC, and Burwell RD (2007). Functional neuroanatomy of the parahippocampal region: the lateral and medial entorhinal areas. Hippocampus 17, 697–708. 10.1002/hipo.20315. [DOI] [PubMed] [Google Scholar]
- 8.Witter MP, Doan TP, Jacobsen B, Nilssen ES, and Ohara S (2017). Architecture of the entorhinal cortex A review of entorhinal anatomy in rodents with some comparative notes. Front. Syst. Neurosci 11, 46. 10.3389/fnsys.2017.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suh J, Rivest AJ, Nakashiba T, Tominaga T, and Tonegawa S (2011). Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science 334, 1415–1420. 10.1126/science.1210125. [DOI] [PubMed] [Google Scholar]
- 10.Zhang SJ, Ye J, Miao C, Tsao A, Cerniauskas I, Ledergerber D, Moser MB, and Moser EI (2013). Optogenetic dissection of entorhinal-hippocampal functional connectivity. Science 340, 1232627. 10.1126/science.1232627. [DOI] [PubMed] [Google Scholar]
- 11.Hales JB, Schlesiger MI, Leutgeb JK, Squire LR, Leutgeb S, and Clark RE (2014). Medial entorhinal cortex lesions only partially disrupt hippocampal place cells and hippocampus-dependent place memory. Cell Rep 9, 893–901. 10.1016/j.celrep.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S, and Magee JC (2015). Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat. Neurosci 18, 1133–1142. 10.1038/nn.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitamura T, Sun C, Martin J, Kitch LJ, Schnitzer MJ, and Tonegawa S (2015). Entorhinal cortical ocean cells encode specific contexts and drive context-specific fear memory. Neuron 87, 1317–1331. 10.1016/j.neuron.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masurkar AV, Srinivas KV, Brann DH, Warren R, Lowes DC, and Siegelbaum SA (2017). Medial and lateral entorhinal cortex differentially excite deep versus superficial CA1 pyramidal neurons. Cell Rep 18, 148–160. 10.1016/j.celrep.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlesiger MI, Boublil BL, Hales JB, Leutgeb JK, and Leutgeb S (2018). Hippocampal global remapping can occur without input from the medial entorhinal cortex. Cell Rep 22, 3152–3159. 10.1016/j.celrep.2018.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jun H, Bramian A, Soma S, Saito T, Saido TC, and Igarashi KM (2020). Disrupted place cell remapping and impaired grid cells in a knockin model of Alzheimer’s disease. Neuron 107, 1095–1112.e6. 10.1016/j.neuron.2020.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witter MP, Groenewegen HJ, Lopes da Silva FH, and Lohman AH (1989). Functional organization of the extrinsic and intrinsic circuitry of the parahippocampal region. Prog. Neurobiol 33, 161–253. [DOI] [PubMed] [Google Scholar]
- 18.van Groen T, Miettinen P, and Kadish I (2003). The entorhinal cortex of the mouse: organization of the projection to the hippocampal formation. Hippocampus 13, 133–149. 10.1002/hipo.10037. [DOI] [PubMed] [Google Scholar]
- 19.Xu W, and Wilson DA (2012). Odor-evoked activity in the mouse lateral entorhinal cortex. Neuroscience 223, 12–20. 10.1016/j.neuroscience.2012.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leitner FC, Melzer S, Lütcke H, Pinna R, Seeburg PH, Helmchen F, and Monyer H (2016). Spatially segregated feedforward and feedback neurons support differential odor processing in the lateral entorhinal cortex. Nat. Neurosci 19, 935–944. 10.1038/nn.4303. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Xu J, Liu Y, Zhu J, Liu N, Zeng W, Huang N, Rasch MJ, Jiang H, Gu X, et al. (2017). A distinct entorhinal cortex to hippocampal CA1 direct circuit for olfactory associative learning. Nat. Neurosci 20, 559–570. 10.1038/nn.4517. [DOI] [PubMed] [Google Scholar]
- 22.Deshmukh SS, and Knierim JJ (2011). Representation of non-spatial and spatial information in the lateral entorhinal cortex. Front. Behav. Neurosci 5, 69. 10.3389/fnbeh.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson DIG, Langston RF, Schlesiger MI, Wagner M, Watanabe S, and Ainge JA (2013). Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus 23, 352–366. 10.1002/hipo.22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsao A, Moser MB, and Moser EI (2013). Traces of experience in the lateral entorhinal cortex. Curr. Biol 23, 399–405. 10.1016/j.cub.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Chen X, Lee H, Deshmukh SS, Yoganarasimha D, Savelli F, and Knierim JJ (2018). Egocentric coding of external items in the lateral entorhinal cortex. Science 362, 945–949. 10.1126/science.aau4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuruvilla MV, Wilson DIG, and Ainge JA (2020). Lateral entorhinal cortex lesions impair both egocentric and allocentric object-place associations. Brain Neurosci. Adv 4, 2398212820939463. 10.1177/2398212820939463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basu J, Zaremba JD, Cheung SK, Hitti FL, Zemelman BV, Losonczy A, and Siegelbaum SA (2016). Gating of hippocampal activity, plasticity, and memory by entorhinal cortex long-range inhibition. Science 351, aaa5694. 10.1126/science.aaa5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsao A, Sugar J, Lu L, Wang C, Knierim JJ, Moser MB, and Moser EI (2018). Integrating time from experience in the lateral entorhinal cortex. Nature 561, 57–62. 10.1038/s41586-018-0459-6. [DOI] [PubMed] [Google Scholar]
- 29.Lee JY, Jun H, Soma S, Nakazono T, Shiraiwa K, Dasgupta A, Nakagawa T, Xie JL, Chavez J, Romo R, et al. (2021). Dopamine facilitates associative memory encoding in the entorhinal cortex. Nature 598, 321–326. 10.1038/s41586-021-03948-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grienberger C, Chen X, and Konnerth A (2014). NMDA receptor-dependent multidendrite Ca(2+) spikes required for hippocampal burst firing in vivo. Neuron 81, 1274–1281. 10.1016/j.neuron.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, and Losonczy A (2012). Regulation of neuronal input transformations by tunable dendritic inhibition. Nat. Neurosci 15, 423–430. 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- 32.Kamondi A, Acsády L, and Buzsáki G (1998). Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J. Neurosci 18, 3919–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golding NL, Staff NP, and Spruston N (2002). Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418, 326–331. 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- 34.Gambino F, Pagès S, Kehayas V, Baptista D, Tatti R, Carleton A, and Holtmaat A (2014). Sensory-evoked LTP driven by dendritic plateau potentials in vivo. Nature 515, 116–119. 10.1038/nature13664. [DOI] [PubMed] [Google Scholar]
- 35.Kim Y, Hsu CL, Cembrowski MS, Mensh BD, and Spruston N (2015). Dendritic sodium spikes are required for long-term potentiation at distal synapses on hippocampal pyramidal neurons. Elife 4, e06414. 10.7554/eLife.06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia H, Rochefort NL, Chen X, and Konnerth A (2010). Dendritic organization of sensory input to cortical neurons in vivo. Nature 464, 1307–1312. 10.1038/nature08947. [DOI] [PubMed] [Google Scholar]
- 37.Smith SL, Smith IT, Branco T, and Häusser M (2013). Dendritic spikes enhance stimulus selectivity in cortical neurons in vivo. Nature 503, 115–120. 10.1038/nature12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cichon J, and Gan WB (2015). Branch-specific dendritic Ca(2+) spikes cause persistent synaptic plasticity. Nature 520, 180–185. 10.1038/nature14251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bittner KC, Milstein AD, Grienberger C, Romani S, and Magee JC (2017). Behavioral time scale synaptic plasticity underlies CA1 place fields. Science 357, 1033–1036. 10.1126/science.aan3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasparini S, Migliore M, and Magee JC (2004). On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J. Neurosci 24, 11046–11056. 10.1523/JNEUROSCI.2520-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J, Papoutsi A, Ash RT, Marin MA, Poirazi P, and Smirnakis SM (2019). Contribution of apical and basal dendrites to orientation encoding in mouse V1 L2/3 pyramidal neurons. Nat. Commun 10, 5372. 10.1038/s41467-019-13029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pissadaki EK, Sidiropoulou K, Reczko M, and Poirazi P (2010). Encoding of spatio-temporal input characteristics by a CA1 pyramidal neuron model. PLoS Comput. Biol 6, e1001038. 10.1371/journal.pcbi.1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poirazi P, Brannon T, and Mel BW (2003). Pyramidal neuron as two-layer neural network. Neuron 37, 989–999. 10.1016/s0896-6273(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 44.Poirazi P, Brannon T, and Mel BW (2003). Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron 37, 977–987. 10.1016/s0896-6273(03)00148-x. [DOI] [PubMed] [Google Scholar]
- 45.Poirazi P, and Papoutsi A (2020). Illuminating dendritic function with computational models. Nat. Rev. Neurosci 21, 303–321. 10.1038/s41583-020-0301-7. [DOI] [PubMed] [Google Scholar]
- 46.Milstein AD, Bloss EB, Apostolides PF, Vaidya SP, Dilly GA, Zemelman BV, and Magee JC (2015). Inhibitory gating of input comparison in the CA1 microcircuit. Neuron 87, 1274–1289. 10.1016/j.neuron.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 47.Grienberger C, Milstein AD, Bittner KC, Romani S, and Magee JC (2017). Inhibitory suppression of heterogeneously tuned excitation enhances spatial coding in CA1 place cells. Nat. Neurosci 20, 417–426. 10.1038/nn.4486. [DOI] [PubMed] [Google Scholar]
- 48.Moore JJ, Robert V, Rashid SK, and Basu J (2021). Assessing local and branch-specific activity in dendrites. Neuroscience 489, 143–164. 10.1016/j.neuroscience.2021.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klausberger T, and Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic inhibitory interneurons. Physiol. Rev 97, 1619–1747. 10.1152/physrev.00007.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bloss EB, Cembrowski MS, Karsh B, Colonell J, Fetter RD, and Spruston N (2016). Structured dendritic inhibition supports branch-selective integration in CA1 pyramidal cells. Neuron 89, 1016–1030. 10.1016/j.neuron.2016.01.029. [DOI] [PubMed] [Google Scholar]
- 52.Gidon A, and Segev I (2012). Principles governing the operation of synaptic inhibition in dendrites. Neuron 75, 330–341. 10.1016/j.neuron.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 53.Jadi M, Polsky A, Schiller J, and Mel BW (2012). Location-dependent effects of inhibition on local spiking in pyramidal neuron dendrites. PLoS Comput. Biol 8, e1002550. 10.1371/journal.pcbi.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilmes KA, Sprekeler H, and Schreiber S (2016). Inhibition as a binary switch for excitatory plasticity in pyramidal neurons. PLoS Comput. Biol 12, e1004768. 10.1371/journal.pcbi.1004768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cutsuridis V, and Poirazi P (2015). A computational study on how theta modulated inhibition can account for the long temporal windows in the entorhinal-hippocampal loop. Neurobiol. Learn. Mem 120, 69–83. 10.1016/j.nlm.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Desmond NL, Scott CA, Jane JA Jr., and Levy WB (1994). Ultra-structural identification of entorhinal cortical synapses in CA1 stratum lacunosum-moleculare of the rat. Hippocampus 4, 594–600. 10.1002/hipo.450040509. [DOI] [PubMed] [Google Scholar]
- 57.Kajiwara R, Wouterlood FG, Sah A, Boekel AJ, Baks-te Bulte LTG, and Witter MP (2008). Convergence of entorhinal and CA3 inputs onto pyramidal neurons and interneurons in hippocampal area CA1–an anatomical study in the rat. Hippocampus 18, 266–280. 10.1002/hipo.20385. [DOI] [PubMed] [Google Scholar]
- 58.Petreanu L, Mao T, Sternson SM, and Svoboda K (2009). The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145. 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Häusser M, Spruston N, and Stuart GJ (2000). Diversity and dynamics of dendritic signaling. Science 290, 739–744. 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- 60.Remy S, Csicsvari J, and Beck H (2009). Activity-dependent control of neuronal output by local and global dendritic spike attenuation. Neuron 61, 906–916. 10.1016/j.neuron.2009.01.032. [DOI] [PubMed] [Google Scholar]
- 61.Golding NL, and Spruston N (1998). Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron 21, 1189–1200. 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- 62.Losonczy A, and Magee JC (2006). Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron 50, 291–307. 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 63.Remy S, and Spruston N (2007). Dendritic spikes induce single-burst long-term potentiation. Proc. Natl. Acad. Sci. USA 104, 17192–17197. 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stuart G, Schiller J, and Sakmann B (1997). Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol 505, 617–632. 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Golding NL, Jung HY, Mickus T, and Spruston N (1999). Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J. Neurosci 19, 8789–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Müller C, Beck H, Coulter D, and Remy S (2012). Inhibitory control of linear and supralinear dendritic excitation in CA1 pyramidal neurons. Neuron 75, 851–864. 10.1016/j.neuron.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 67.Takahashi H, and Magee JC (2009). Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62, 102–111. 10.1016/j.neuron.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 68.Golding NL, Mickus TJ, Katz Y, Kath WL, and Spruston N (2005). Factors mediating powerful voltage attenuation along CA1 pyramidal neuron dendrites. J. Physiol 568, 69–82. 10.1113/jphysiol.2005.086793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melzer S, Michael M, Caputi A, Eliava M, Fuchs EC, Whittington MA, and Monyer H (2012). Long-range-projecting GABAergic neurons modulate inhibition in hippocampus and entorhinal cortex. Science 335, 1506–1510. 10.1126/science.1217139. [DOI] [PubMed] [Google Scholar]
- 70.Glickfeld LL, Roberts JD, Somogyi P, and Scanziani M (2009). Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat. Neurosci 12, 21–23. 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Megías M, Emri Z, Freund TF, and Gulyás AI (2001). Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102, 527–540. 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- 72.Pouille F, and Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163. 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 73.Basu J, Srinivas KV, Cheung SK, Taniguchi H, Huang ZJ, and Siegelbaum SA (2013). A cortico-hippocampal learningrule shapes inhibitory microcircuit activity to enhance hippocampal information flow. Neuron 79, 1208–1221. 10.1016/j.neuron.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beaulieu-Laroche L, and Harnett MT (2018). Dendritic spines prevent synaptic voltage clamp. Neuron 97, 75–82.e3. 10.1016/j.neuron.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 75.To MS, Honnuraiah S, and Stuart GJ (2021). Voltage clamp errors during estimation of concurrent excitatory and inhibitory synaptic input to neurons with dendrites. Neuroscience 489, 98–110. 10.1016/j.neuroscience.2021.08.024. [DOI] [PubMed] [Google Scholar]
- 76.Bezaire MJ, and Soltesz I (2013). Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity. Hippocampus 23, 751–785. 10.1002/hipo.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geiller T, Vancura B, Terada S, Troullinou E, Chavlis S, Tsagkatakis G, Tsakalides P, Ócsai K, Poirazi P, Rózsa BJ, and Losonczy A, (2020). Large-scale 3D two-photon imaging of molecularly identified CA1 interneuron dynamics in behaving mice. Neuron 108, 968–983.e9. 10.1016/j.neuron.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Acsády L, Arabadzisz D, and Freund TF (1996). Correlated morphological and neurochemical features identify different subsets of vasoactive intestinal polypeptide-immunoreactive interneurons in rat hippocampus. Neuroscience 73, 299–315. [DOI] [PubMed] [Google Scholar]
- 80.Cope DW, Maccaferri G, Márton LF, Roberts JDB, Cobden PM, and Somogyi P (2002). Cholecystokinin-immunopositive basket and Schaffer collateral-associated interneurones target different domains of pyramidal cells in the CA1 area of the rat hippocampus. Neuroscience 109, 63–80. 10.1016/s0306-4522(01)00440-7. [DOI] [PubMed] [Google Scholar]
- 81.Pawelzik H, Hughes DI, and Thomson AM (2002). Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippo campus. J. Comp. Neurol 443, 346–367. 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- 82.Klausberger T (2009). GABAergic interneurons targeting dendrites of pyramidal cells in the CA1 area of the hippocampus. Eur. J. Neurosci 30, 947–957. 10.1111/j.1460-9568.2009.06913.x. [DOI] [PubMed] [Google Scholar]
- 83.Tyan L, Chamberland S, Magnin E, Camiré O, Francavilla R, David LS, Deisseroth K, and Topolnik L (2014). Dendritic inhibition provided by interneuron-specific cells controls the firing rate and timing of the hippocampal feedback inhibitory circuitry. J. Neurosci 34, 4534–4547. 10.1523/JNEUROSCI.3813-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Francavilla R, Luo X, Magnin E, Tyan L, and Topolnik L (2015). Co-ordination of dendritic inhibition through local disinhibitory circuits. Front. Synaptic Neurosci 7, 5. 10.3389/fnsyn.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leão RN, Mikulovic S, Leão KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort ABL, and Kullander K (2012). OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat. Neurosci 15, 1524–1530. 10.1038/nn.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, and Losonczy A, (2014). Dendritic inhibition in the hippocampus supports fear learning. Science 343, 857–863. 10.1126/science.1247485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sik A, Penttonen M, Ylinen A, and Buzsáki G (1995). Hippocampal CA1 interneurons: an in vivo intracellular labeling study. J. Neurosci 15, 6651–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takács VT, Szőnyi A, Freund TF, Nyiri G, and Gulyás AI (2015). Quantitative ultrastructural analysis of basket and axo-axonic cell terminals in the mouse hippocampus. Brain Struct. Funct 220, 919–940. 10.1007/s00429-013-0692-6. [DOI] [PubMed] [Google Scholar]
- 89.Dudok B, Klein PM, Hwaun E, Lee BR, Yao Z, Fong O, Bowler JC, Terada S, Sparks FT, Szabo GG, et al. (2021). Alternating sources of perisomatic inhibition during behavior. Neuron 109, 997–1012.e9. 10.1016/j.neuron.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dudok B, Szoboszlay M, Paul A, Klein PM, Liao Z, Hwaun E, Szabo GG, Geiller T, Vancura B, Wang BS, et al. (2021). Recruitment and inhibitory action of hippocampal axo-axonic cells during behavior. Neuron 109, 3838–3850.e8. 10.1016/j.neuron.2021.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Klausberger T, Marton LF, O’Neill J, Huck JHJ, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, et al. (2005). Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J. Neurosci 25, 9782–9793. 10.1523/JNEUROSCI.3269-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Acsády L, Görcs TJ, and Freund TF (1996). Different populations of vasoactive intestinal polypeptide-immunoreactive interneurons are specialized to control pyramidal cells or interneurons in the hippocampus. Neuroscience 73, 317–334. [DOI] [PubMed] [Google Scholar]
- 93.Chamberland S, and Topolnik L (2012). Inhibitory control of hippocampal inhibitory neurons. Front. Neurosci 6, 165. 10.3389/fnins.2012.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee S, Kruglikov I, Huang ZJ, Fishell G, and Rudy B (2013). A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci 16, 1662–1670. 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci 16, 1068–1076. 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, and Kepecs A, (2013). Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524. 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chuong AS, Miri ML, Busskamp V, Matthews GAC, Acker LC, Sørensen AT, Young A, Klapoetke NC, Henninger MA, Kodandaramaiah SB, et al. (2014). Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat. Neurosci 17, 1123–1129. 10.1038/nn.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Del Pino I, Brotons-Mas JR, Marques-Smith A, Marighetto A, Frick A, Marín O, and Rico B (2017). Abnormal wiring of CCK(+) basket cells disrupts spatial information coding. Nat. Neurosci 20, 784–792. 10.1038/nn.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Whissell PD, Bang JY, Khan I, Xie YF, Parfitt GM, Grenon M, Plummer NW, Jensen P, Bonin RP, and Kim JC (2019). Selective activation of cholecystokinin-expressing GABA (CCK-GABA) neurons enhances memory and cognition. eNeuro 6, ENEURO.0360, 18.2019. 10.1523/ENEURO.0360-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C, and Bamberg E (2011). Ultra light-sensitive and fast neuronal activation with the Ca(2)+-permeable channelrhodopsin CatCh. Nat. Neurosci 14, 513–518. 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- 101.Guet-McCreight A, Skinner FK, and Topolnik L (2020). Common principles in functional organization of VIP/calretinin cell-driven disinhibitory circuits across cortical areas. Front. Neural Circuits 14, 32. 10.3389/fncir.2020.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Turi GF, Li WK, Chavlis S, Pandi I, O’Hare J, Priestley JB, Grosmark AD, Liao Z, Ladow M, Zhang JF, et al. (2019). Vasoactive intestinal polypeptide-expressing interneurons in the Hippocampus support goal-oriented spatial learning. Neuron 101, 1150–1165.e8. 10.1016/j.neuron.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shuman T, Aharoni D, Cai DJ, Lee CR, Chavlis S, Page-Harley L, Vetere LM, Feng Y, Yang CY, Mollinedo-Gajate I, et al. (2020). Break-down of spatial coding and interneuron synchronization in epileptic mice. Nat. Neurosci 23, 229–238. 10.1038/s41593-019-0559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bezaire MJ, Raikov I, Burk K, Vyas D, and Soltesz I (2016). Interneuronal mechanisms of hippocampal theta oscillations in a full-scale model of the rodent CA1 circuit. Elife 5, e18566. 10.7554/eLife.18566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Larkum ME, Nevian T, Sandler M, Polsky A, and Schiller J (2009). Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science 325, 756–760. 10.1126/science.1171958. [DOI] [PubMed] [Google Scholar]
- 106.Gómez González JF, Mel BW, and Poirazi P (2011). Distinguishing linear vs. Non-linear integration in CA1 radial oblique dendrites: it’s about time. Front. Comput. Neurosci 5, 44. 10.3389/fncom.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miles R, Tóth K, Gulyás AI, Hájos N, and Freund TF (1996). Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16, 815–823. 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- 108.Larkum ME, Zhu JJ, and Sakmann B (1999). A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398, 338–341. 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- 109.Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, and Larkum ME (2009). Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature 457, 1137–1141. 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- 110.Glickfeld LL, and Scanziani M (2006). Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat. Neurosci 9, 807–815. 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bitzenhofer SH, Westeinde EA, Zhang HXB, and Isaacson JS (2022). Rapid odor processing by layer 2 subcircuits in lateral entorhinal cortex. Elife 11, e75065. 10.7554/eLife.75065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hsu CL, Zhao X, Milstein AD, and Spruston N (2018). Persistent sodium current mediates the steep voltage dependence of spatial coding in hippocampal pyramidal neurons. Neuron 99, 147–162.e8. 10.1016/j.neuron.2018.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tomko M, Benuskova L, and Jedlicka P (2021). A new reduced-morphology model for CA1 pyramidal cells and its validation and comparison with other models using HippoUnit. Sci. Rep 11, 7615. 10.1038/s41598-021-87002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schulz JM, Knoflach F, Hernandez MC, and Bischofberger J (2018). Dendrite-targeting interneurons control synaptic NMDA-receptor activation via nonlinear alpha5-GABAA receptors. Nat. Commun 9, 3576. 10.1038/s41467-018-06004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Migliore M, De Simone G, and Migliore R (2015). Effect of the initial synaptic state on the probability to induce long-term potentiation and depression. Biophys. J 108, 1038–1046. 10.1016/j.bpj.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mergenthal A, Bouteiller JMC, Yu GJ, and Berger TW (2020). A computational model of the cholinergic modulation of CA1 pyramidal cell activity. Front. Comput. Neurosci 14, 75. 10.3389/fncom.2020.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cutsuridis V, and Hasselmo M (2012). GABAergic contributions to gating, timing, and phase precession of hippocampal neuronal activity during theta oscillations. Hippocampus 22, 1597–1621. 10.1002/hipo.21002. [DOI] [PubMed] [Google Scholar]
- 118.Ferguson KA, Chatzikalymniou AP, and Skinner FK (2017). Combining theory, model, and experiment to explain how intrinsic theta rhythms are generated in an in vitro whole Hippocampus preparation without oscillatory inputs. eNeuro 4, ENEURO.0131, 17.2017. 10.1523/ENEURO.0131-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Udakis M, Pedrosa V, Chamberlain SEL, Clopath C, and Mellor JR (2020). Interneuron-specific plasticity at parvalbumin and somatostatin inhibitory synapses onto CA1 pyramidal neurons shapes hippocampal output. Nat. Commun 11, 4395. 10.1038/s41467-020-18074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spruston N, Jaffe DB, and Johnston D (1994). Dendritic attenuation of synaptic potentials and currents: the role of passive membrane properties. Trends Neurosci 17, 161–166. 10.1016/01662236(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 121.Kamondi A, Acsady L, Wang XJ, and Buzsaki G (1998). Theta oscillations in somata and dendrites of hippocampal pyramidal cells in vivo: activity-dependent phase-precession of action potentials. Hippocampus 8, 244–261. . [DOI] [PubMed] [Google Scholar]
- 122.Lavzin M, Rapoport S, Polsky A, Garion L, and Schiller J (2012). Nonlinear dendritic processing determines angular tuning of barrel cortex neurons in vivo. Nature 490, 397–401. 10.1038/nature11451. [DOI] [PubMed] [Google Scholar]
- 123.Moore JJ, Ravassard PM, Ho D, Acharya L, Kees AL, Vuong C, and Mehta MR (2017). Dynamics of cortical dendritic membrane potential and spikes in freely behaving rats. Science 355, eaaj1497. 10.1126/science.aaj1497. [DOI] [PubMed] [Google Scholar]
- 124.Kerlin A, Mohar B, Flickinger D, MacLennan BJ, Dean MB, Davis C, Spruston N, and Svoboda K (2019). Functional clustering of dendritic activity during decision-making. Elife 8, e46966. 10.7554/eLife.46966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dudman JT, Tsay D, and Siegelbaum SA (2007). A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron 56, 866–879. 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li B, Suutari BS, Sun SD, Luo Z, Wei C, Chenouard N, Mandelberg NJ, Zhang G, Wamsley B, Tian G, et al. (2020). Neuronal inactivity Co-opts LTP machinery to drive potassium channel splicing and homeostatic spike widening. Cell 181, 1547–1565.e15. 10.1016/j.cell.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Augusto E, and Gambino F (2019). Can NMDA spikes dictate computations of local networks and behavior? Front. Mol. Neurosci 12, 238. 10.3389/fnmol.2019.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Branco T, and Häusser M (2010). The single dendritic branch as a fundamental functional unit in the nervous system. Curr. Opin. Neurobiol 20, 494–502. 10.1016/j.conb.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 129.Chavlis S, and Poirazi P (2021). Drawing inspiration from biological dendrites to empower artificial neural networks. Curr. Opin. Neurobiol 70, 1–10. 10.1016/j.conb.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 130.Häusser M, and Mel B (2003). Dendrites: bug or feature? Curr. Opin. Neurobiol 13, 372–383. 10.1016/s0959-4388(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 131.Losonczy A, Makara JK, and Magee JC (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441. 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- 132.Moore JJ, Robert V, Rashid SK, and Basu J (2022). Assessing local and branch-specific activity in dendrites. Neuroscience 489, 143–164. 10.1016/j.neuroscience.2021.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Poirazi P, and Mel BW (2001). Impact of active dendrites and structural plasticity on the memory capacity of neural tissue. Neuron 29, 779–796. 10.1016/s0896-6273(01)00252-5. [DOI] [PubMed] [Google Scholar]
- 134.Jarsky T, Roxin A, Kath WL, and Spruston N (2005). Conditional dendritic spike propagation following distal synaptic activation of hippocampal CA1 pyramidal neurons. Nat. Neurosci 8, 1667–1676. 10.1038/nn1599. [DOI] [PubMed] [Google Scholar]
- 135.Doan TP, Lagartos-Donate MJ, Nilssen ES, Ohara S, and Witter MP (2019). Convergent projections from perirhinal and postrhinal cortices suggest a multisensory nature of lateral, but not medial, entorhinal cortex. Cell Rep 29, 617–627.e7. 10.1016/j.celrep.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 136.Palacios-Filardo J, Udakis M, Brown GA, Tehan BG, Congreve MS, Nathan PJ, Brown AJH, and Mellor JR (2021). Acetylcholine prioritises direct synaptic inputs from entorhinal cortex to CA1 by differential modulation of feedforward inhibitory circuits. Nat. Commun 12, 5475. 10.1038/s41467-021-25280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ito HT, and Schuman EM (2007). Frequency-dependent gating of synaptic transmission and plasticity by dopamine. Front. Neural Circuits 1, 1. 10.3389/neuro.04.001.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sheffield MEJ, and Dombeck DA (2015). Calcium transient prevalence across the dendritic arbour predicts place field properties. Nature 517, 200–204. 10.1038/nature13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Buzsáki G, and Moser EI (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat. Neurosci 16, 130–138. 10.1038/nn.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Robinson NTM, Descamps LAL, Russell LE, Buchholz MO, Bicknell BA, Antonov GK, Lau JYN, Nutbrown R, Schmidt-Hieber C, and Häusser M (2020). Targeted activation of hippocampal place cells drives memory-guided spatial behavior. Cell 183, 1586–1599.e10. 10.1016/j.cell.2020.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Save E, and Sargolini F (2017). Disentangling the role of the MEC and LEC in the processing of spatial and non-spatial information: contribution of lesion studies. Front. Syst. Neurosci 11, 81. 10.3389/fnsys.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, Fenno LE, Ramakrishnan C, and Deisseroth K (2010). Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature 465, 788–792. 10.1038/nature09108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fenno LE, Ramakrishnan C, Kim YS, Evans KE, Lo M, Vesuna S, Inoue M, Cheung KYM, Yuen E, Pichamoorthy N, et al. (2020). Comprehensive dual- and triple-feature intersectional single-vector delivery of diverse functional payloads to cells of behaving mammals. Neuron 107, 836–853.e11. 10.1016/j.neuron.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Carnevale NT, and Hines ML (2006). The NEURON Book (Cambridge University Press; ). [Google Scholar]
- 145.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, and Tonegawa S (1996). Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87, 1317–1326. 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 146.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJB, Battiste J, Johnson JE, Machold RP, and Fishell G (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci 30, 1582–1594. 10.1523/JNEUROSCI.4515-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and tools for combinatorial targeting of GABAergic neurons in mouse cerebral cortex. Neuron 91, 1228–1243. 10.1016/j.neuron.2016.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell 174, 465–480.e22. 10.1016/j.cell.2018.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi GA, Guo L, Xu Q, Liu R, Lu C, Chu J, et al. (2016). A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci 19, 1743–1749. 10.1038/nn.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Frank LM, Brown EN, and Wilson MA (2001). A comparison of the firing properties of putative excitatory and inhibitory neurons from CA1 and the entorhinal cortex. J. Neurophysiol 86, 2029–2040. 10.1152/jn.2001.86.4.2029. [DOI] [PubMed] [Google Scholar]
- 153.Pilkiw M, Insel N, Cui Y, Finney C, Morrissey MD, and Takehara-Nishiuchi K (2017). Phasic and tonic neuron ensemble codes for stimulus-environment conjunctions in the lateral entorhinal cortex. Elife 6, e28611. 10.7554/eLife.28611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lin JY (2011). A user’s guide to channelrhodopsin variants: features, limitations and future developments. Exp. Physiol 96, 19–25. 10.1113/expphysiol.2009.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Susaki EA, Tainaka K, Perrin D, Yukinaga H, Kuno A, and Ueda HR (2015). Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat. Protoc 10, 1709–1727. 10.1038/nprot.2015.085. [DOI] [PubMed] [Google Scholar]
- 156.Cutsuridis V, Cobb S, and Graham BP (2010). Encoding and retrieval in a model of the hippocampal CA1 microcircuit. Hippocampus 20, 423–446. 10.1002/hipo.20661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Electrophysiology and microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited on ModelDB: http://modeldb.yale.edu/267221 and Zenodo: https://doi.org/10.5281/zenodo.7413215 and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.