

Abstract
Background
Miller syndrome is a rare type of postaxial acrofacial dysostosis caused by biallelic mutations in the DHODH gene, which is characterized mainly by craniofacial malformations of micrognathia, orofacial clefts, cup‐shaped ears, and malar hypoplasia, combined with postaxial limb deformities like the absence of fifth digits.
Methods
In this study, a prenatal case with multiple orofacial‐limb abnormities was enrolled, and a thorough clinical and imaging examination was performed. Subsequently, genetic detection with karyotyping, chromosomal microarray analysis (CMA) and whole‐exome sequencing (WES) was carried out. In vitro splicing analysis was also conducted to clarify the impact of one novel variant.
Results
The affected fetus displayed typical manifestations of Miller syndrome, and WES identified a diagnostic compound heterozygous variation in DHODH, consisting of two variants: exon(1‐3)del and c.819 + 5G > A. We conducted a further in vitro validation with minigene system, and the result indicated that the c.819 + 5G > A variant would lead to an exon skipping in mRNA splicing.
Conclusions
These findings provided with the first exonic deletion and first splice site variant in DHODH, which expanded the mutation spectrum of Miller syndrome and offered reliable evidence for genetic counseling to the affected family.
Keywords: acrofacial dysostosis, DHODH gene, Miller syndrome, whole exome sequencing
This article provided with the first exonic deletion and first splice site variant in DHODH, which expanded the mutation spectrum of Miller syndrome and offered reliable evidence for genetic counseling to the affected family.
1. INTRODUCTION
Human acrofacial dysostosis (AFD) is an umbrella term of a group of at least 18 congenital conditions exhibiting a combination of craniofacial and limb anomalies (Wieczorek, 2013). Among AFDs, the Miller syndrome (also known as Genee‐Wiedemann or Wildervanck‐Smith syndrome, OMIM #263750) is a postaxial type characterized by micrognathia, orofacial clefts, malar hypoplasia, aplasia of the medial lower lid eyelashes, coloboma of the lower eyelid and cup‐shaped ears combined with postaxial limb deformities, including the apparent absence of either the fifth or both the fourth and fifth rays of the hands and feet, with or without ulnar and fibular hypoplasia (Miller et al., 1979). This disorder has been described for more than 50 years, but there were only ~50 associated reports, making it extremely rare (Genée, 1969; Wiedemann, 1973).
This autosomal recessive condition, Miller syndrome, was also famous for being the first entity resolved by exome sequencing (Ng et al., 2010), then responded by another whole genome study (Roach et al., 2010). The DHODH gene (OMIM*126064) was identified as the causative gene of this disease. DHODH is located at chromosome 16q22.2 and contains nine exons encoding dihydroorotate dehydrogenase, which catalyzes the fourth step in de novo pyrimidine biosynthesis (Minet et al., 1992). To date, only 21 DHODH variants in nine studies have been reported to cause Miller syndrome (https://www.hgmd.cf.ac.uk/; pro V2021.10; details see Table 1), demonstrating a low incidence of this condition and a strong conservatism of DHODH gene (Bukowska‐Olech et al., 2020; Duley et al., 2016; Fujikura, 2016; Hou et al., 2020; Kinoshita et al., 2011; Ng et al., 2010; Orozco Rodriguez et al., 2022; Rainger et al., 2012; Roach et al., 2010); in them, missense variants accounted for the most part (~69.6%, Table 1), and the others were base‐level deletion, duplication or frameshift variants. The subjects of previous studies were limited to Caucasian and Japanese populations, and there are no reports on other ethnic groups. Additionally, due to the low number of identified variations, it is still unable to fully establish the genotype–phenotype association of Miller syndrome.
TABLE 1.
Miller syndrome‐related variants in the DHODH gene (cited from HGMD database, in order of genomic position).
| No. | Genomic coordinates | Reference base(s) | Variant base (s) | HGVS description (NM_001361.5) | Protein alteration | Reference (PMID) |
|---|---|---|---|---|---|---|
| 1 | chr16:72045983‐72045983 | G | A | c.56G > A | p.G19E | 19915526 |
| 2 | chr16:72046010‐72046010 | T | C | c.83 T > C | p.L28P | 21851494 |
| 3 | chr16:72046055‐72046055 | C | T | c.128C > T | p.P43L | 33262786 |
| 4 | chr16:72046082‐72046082 | A | G | c.155A > G | p.E52G | 22692683 |
| 5 | chr16:72046089‐72046091 | CCA | C | c.165_166del | p.H55Efs*21 | 27370710 |
| 6 | chr16:72048381‐72048395 | GTTCTGGGCCATAAA | G | c.248_261del | p.L83Pfs*11 | 31980526 |
| 7 | chr16:72048540‐72048540 | C | T | c.403C > T | p.R135C | 19915526 |
| 8 | chr16:72050942‐72050942 | G | A | c.454G > A | p.G152R | 19915526 |
| 9 | chr16:72055100‐72055100 | C | T | c.595C > T | p.R199C | 19915526 |
| 10 | chr16:72055110‐72055110 | G | GC | c.610dupC | p.L204Pfs*55 | 27219052 |
| 11 | chr16:72055110‐72055110 | G | A | c.605G > A | p.G202D | 19915526 |
| 12 | chr16:72055110‐72055110 | G | C | c.605G > C | p.G202A | 19915526 |
| 13 | chr16:72055115‐72055116 | CT | C | c.611delT | p.L204Rfs*7 | 19915526 |
| 14 | chr16:72056285‐72056285 | C | T | c.730C > T | p.R244W | 19915526 |
| 15 | chr16:72057095‐72057095 | C | T | c.851C > T | p.T284I | 19915526 |
| 16 | chr16:72057169‐72057169 | C | T | c.925C > T | p.R309W | 27370710 |
| 17 | chr16:72057375‐72057375 | C | T | c.976C > T | p.R326* | 22692683 |
| 18 | chr16:72057435‐72057435 | C | T | c.1036C > T | p.R346W | 19915526 |
| 19 | chr16:72057438‐72057438 | G | A | c.1039G > A | p.A347T | 21851494 |
| 20 | chr16:72057468‐72057468 | G | A | c.1069G > A | p.A357T | 22692683 |
| 21 | chr16:72058085‐72058085 | A | G | c.1175A > G | p.D392G | 19915526 |
| 22 | chr16:?‐? | Uncertain | del | exon(1‐3)del | Null | This study |
| 23 | chr16:72056379‐72056379 | G | A | c.819 + 5G > A | p.236_264del? | This study |
Note: DHODH gene: NM_001361.5.
Abbreviations: HGMD, The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php); HGVS, Human Genome Variation Society (http://www.hgvs.org/); PMID, PubMed reference ID (https://pubmed.ncbi.nlm.nih.gov/).
In the present study, we identified a fetus with craniofacial and limb abnormalities through prenatal ultrasonography. Subsequently, amniocentesis and prenatal genetic diagnosis were conducted. A heterozygous compound variation in DHODH gene was identified and verified. In vitro study was performed to investigate the specific impact of one potential splicing variant.
2. MATERIALS AND METHODS
This study was approved by the Ethics Committee of Shijiazhuang Obstetrics and Gynecology Hospital. Informed consent was provided by the participants. All procedures performed in the present study were in accordance with the Declaration of Helsinki 1964 and its later amendments or comparable ethical standards.
2.1. Subjects and clinical evaluation
At June 2021, a couple with fetus displaying in utero multiple anomalies was enrolled in our center. Clinical evaluation was made via the combination of ultrasound monitoring and magnetic resonance imaging (MRI) at mid trimester and a comprehensive survey on family history.
2.2. Karyotyping and copy number variation (CNV) analysis
To conduct a prenatal genetic detection, amniocentesis was performed to obtain the amniotic fluid (AF) sample according to routine process. Conventional chromosomal karyotyping by G‐binding was performed to detect overall chromosomal anomalies (Arsham et al., 2017). Genomic DNA was extracted from AF and the parents' peripheral blood using samples QIAamp DNA Midi Kit (Qiagen, Dusseldorf, Germany). Chromosomal microarray analysis (CMA) and testing with CytoScan 750 K SNP‐array (Affymetrix Inc., Santa Clara, USA) were carried out according to the manufacturer's manual workflow on all fetal specimens in order to investigate genomic CNVs with clinical significance.
2.3. Whole‐exome sequencing
Whole‐exome sequencing (WES) was employed to detect the sequence variants in the probands' fetal samples, as described in our previous study (Yang et al., 2022). Briefly, the target‐region sequence enrichment was performed using the Agilent Sure Select Human Exon Sequence Capture Kit (Agilent, Palo Alto, CA, USA).DNA libraries were tested by quantitative PCR, where the size, distribution and concentration were determined using Agilent Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). Along with ~150 bp pair‐end reads, the NovaSeq‐6000 platform (Illumina, Inc., San Diego, CA, USA) was used for sequencing of DNA with ~300 pM per sample with NovaSeq Reagent kit. Sequencing raw reads (quality level Q30% > 90%; and the quality criteria was listed at https://www.illumina.com/science/technology/next‐generation‐sequencing/plan‐experiments/quality‐scores.html; accessed on 25 September 2022) were aligned to the human reference genome (accession No. hg19/GRCh37) using the Burrows Wheeler Aligner tool, and PCR duplicates were removed using Picardv1.57. Variant calling was performed with the Verita Trekker® Variants Detection system (v2.0; Berry Genomics, Beijing, China) and Genome Analysis Tool Kit (https://software.broadinstitute.org/gatk/; accessed on 25 September 2022). Then, variants were annotated and interpreted using ANNOVAR (v2.0) (Wang & Hakonarson, 2010) and Enliven® Variants Annotation Interpretation systems (Berry Genomics), based on the common guidelines by ACMG (American College of Medical Genetics and Genomics) (Richards et al., 2015). To assist with the interpretation of variant pathogenicity, we referred to three frequency databases (ExAC_EAS, http://exac.broadinstitute.org, accessed on 25 September 2022; gnomAD_exome_EAS, http://gnomad.broadinstitute.org, accessed on 25 September 2022; 1000G_2015aug_eas, https://www.internationalgenome.org, accessed on 25 September 2022) and HGMD (Human Gene Mutation Database, pro v2021.10); Revel score (a combined method of pathogenicity prediction) (Ioannidis et al., 2016) and pLI score (representing the tolerance for truncating variants) were also employed.
Sanger sequencing was performed as a validation method with the 3500DX Genetic Analyzer (Applied Biosystems, Thermofisher, LA, CA,USA).
2.4. In vitro study on the novel splicing site variant
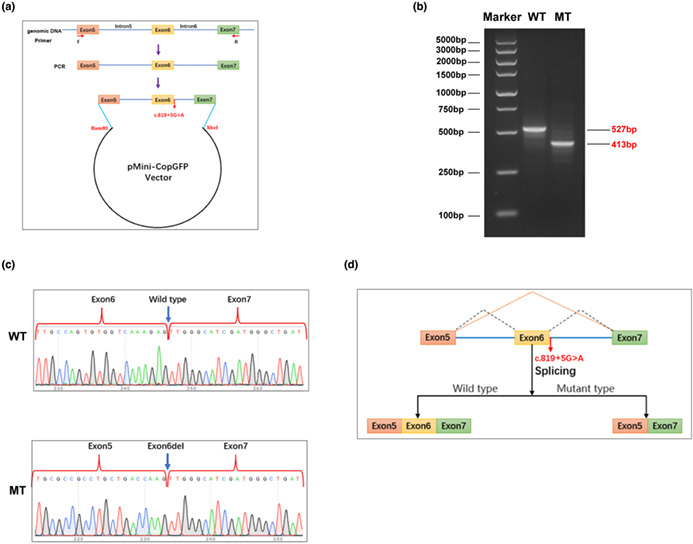
Scince a novel variant (DHODH: c.819 + 5G > A) was adjacent to the splice site of Exon 6, we inferred that it might affect the normal mRNA splicing. Splice site prediction algorithms of DANN and dbscSNV also indicated it to be deleterious (data not shown). Therefore, an in vitro validation experiment was conducted.
In short, the minigene expression plasmids containing the Exon5‐Exon7 segment of either the DHODH wild‐type (WT) or the c.819 + 5G > A mutant (MT) were constructed with a commercially designed expression backbone, pMini‐CopGF. Subsequently, HEK (human embryonic kidney) 293 T tool cells were cultured and transfected by either of these plasmids, respectively. 48 h later, the cells were collected and the RNA sample was extracted and reversely transcripted into cDNA. Subsequently, particular impact on mRNA splicing was analyzed via PCR fragment amplification, AGE (agarose gel electrophoresis), and Sanger sequencing; the primer sequences used for PCR and sequencing were β‐globin intron‐F, 5′‐GATATACACTGTTTGAGATGAGGA‐3′; DHODH‐R, 5′‐TACCAAGGAGAGGAAATCAC‐3′.
3. RESULTS
3.1. Clinical manifestations
The pregnant women, aged 30 with her first pregnancy, came to our center for regular prenatal examination. At the 12th gestational week, ultrasonography revealed that the fetus manifested micrognathia, shortened forearm, wrist angulation (Figure 1a), cleft palate (Figure 1b), enhanced echogenicity in both kidneys (Figure 1c), significantly larger bladder (Figure 1d) and umbilical cord cysts (Figure 1e). At 20th week, the fetus was found to present with low‐set ears, bilateral ulnas absence, adductive and flexed elbow joints, and the fifth digits missing (Figure 1f,g). The subsequent MRI test additionally indicated that the fetus had shortening in distal extremities, ulna and tibia hypoplasia (Figure 1h).
FIGURE 1.
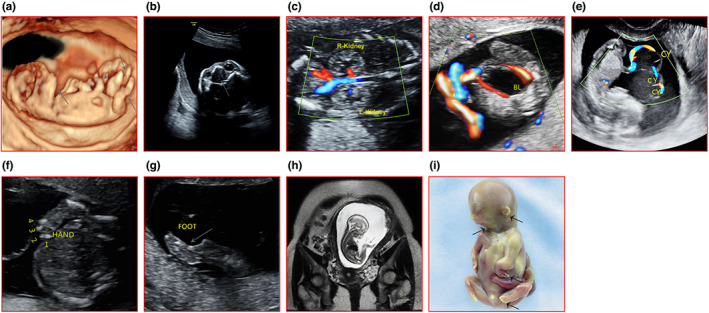
Ultrasonography, MRI, and appearance images of the affected fetus. (a) Micrognathia, forearm shortening, wrist angulation, abnormal angle of wrist and ankle (arrows). (b) Cleft palate (arrow). (c) Slightly stronger echogenicity of both renal parenchyma, mild separation of renal pelvis. (d) Bladder (BL) enlargement. (e) Umbilical cord cysts (CY). (f) Fifth finger absence. (g) Abnormal ankle angle (arrow). (h) MRI: retracted mandible, abnormal ankle angle, and absence of the tibia (arrows). (i) Appearance: micrognathia, zygomatic dysplasia, short palpebral fissures, short wide neck, cup‐shaped ear, hook‐like wrist, fifth digits missing.
The couple chose to induce labor with informed consent at 21th+3d week. The general appearance of the aborted fetus confirmed the results of prenatal imaging, including cleft palate. It was a boy with severe micrognathia, mild zygomatic dysplasia, short palpebral fissures, short wide neck, and lower “cup‐shaped” ears. The forearms were extremely short, the elbows were abducent, and the hands were “hook‐like”. The fifth phalanxes of both hands and feet were absent, and the third and fourth fingers and toes exhibited syndactyly (Figure 1i).
3.2. Genetic finding
The fetal karyotype and CMA results were normal. WES identified a compound heterozygous variation in the DHODH gene, consisting of two variants, namely exon(1‐3)del and NM_001361: c.819 + 5G > A. According to familial validation, it was demonstrated by Sanger sequencing that the former variant was inherited from the pregnant woman, while the latter one was from her husband (Figure 2a,b). We summarized the currently identified variants in the DHODH gene associated with Miller syndrome, exhibited in the form of gene and protein diagrams (Figure 2c, corresponding to Table 1).
FIGURE 2.
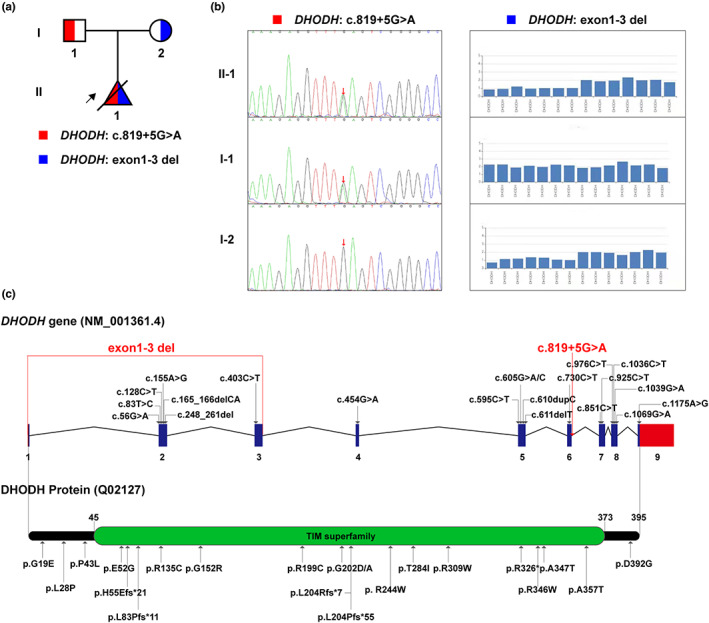
Genetic variations of DHODH in this study and literature. (a) The pedigree of this family. The object of the arrow is the proband. (b) Two variants and their carriage among family members. (c) All the Miller‐syndrome‐associated variants detected in DHODH so far, shown in the form of gene and protein pattern diagrams. The red characters show the variants detected in this study.
3.3. The affect of DHODH : c.819 + 5G > a variant on splicing
In order to elucidate the specific impact of the DHODH: c.819 + 5G > A variant on mRNA splicing, we carried out a minigene expression experiment. And the AGE result indicate that by the wild‐type (WT) transfection, the target band could be amplified with a 527 bp size, as predicted; while by the mutant (MT) transfection, the amplicon size was 413 bp (Figure 3b). This result was consistent with those predicted by the in silico methods. Then, we recycled each gel band and collected the corresponding DNA sample from it for Sanger sequencing validation. Results demonstrated that in the WT the normal transcript was produced, while in the MT there was a exon‐skip at Exon 6 of the expression segment (Figure 3c). A pattern diagram corresponding to this mechanism was shown in Figure 3d.
FIGURE 3.
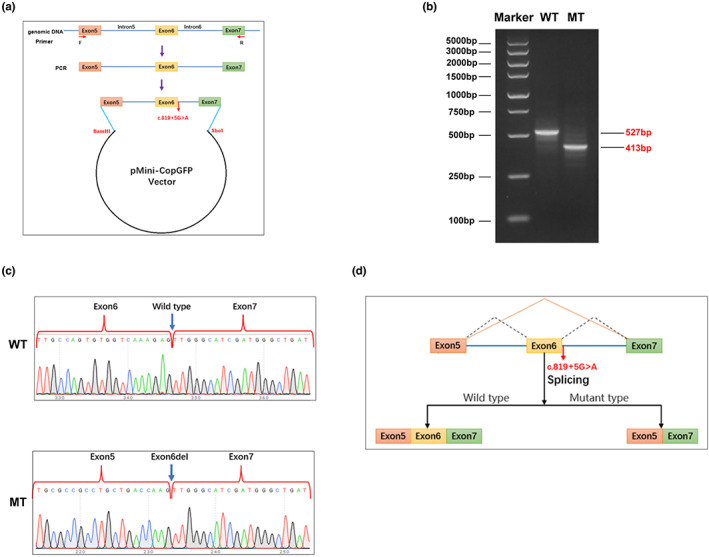
Results of the in vitro minigene experiment. (a) A diagram of the vector construction process, including the restriction endonuclease sites, variant site (red fonts). (b) AGE result showing the bands of the amplified product in either wild‐type (WT) or mutant (MT) transfected cells. (c) Sanger sequencing results of the WT and MT indicating the c.819 + 5G > A variant caused the Exon6 skipping. (d) Model diagram further explaining the results in (b) and (c).
4. DISCUSSION
Human cranial‐maxillofacial development is a complex and elaborate process and is related to hundreds of Mendelian dysmorphologies (Bhat, 2020). The extremely low incidence and mutation amount of DHODH‐caused Miller syndrome, one recessive type of AFD, adds a layer of mystery to its pathogenesis (Duley et al., 2016). There has been even less in‐depth research into the effects of specific mutations of DHODH gene (Orozco Rodriguez et al., 2022).
In this study, we conducted a systematic clinical and radiographic evaluation of a child with prenatal cranial‐maxillofacial and limb abnormalities. The gross examination of the fetus after labor induction also confirmed that the abnormalities in appearance and multiple systems of the fetus were consistent with previous reports (Donnai et al., 1987). Further genetic analysis revealed a novel compound heterozygous DHODH variation involving two variants, namely exon(1‐3)del and c.819 + 5G > A. The interpretation of the former variant was based on the differences in exon capture efficiency presented in our WES data. This variant is most likely to cause the mRNA failed to be properly transcribed, thus to cause a null variation at protein level. To further detect the specific breakpoints which have not been certain by now, other sequence analysis methods such as WGS (whole genome sequencing) are needed. In terms of the c.819 + 5G > A variant, it is clear that the base substitution did not occur at classical splicing site. So, to further clarify its effect on splicing, we designed and performed in vitro validation experiments, and the result showed an Exon6 deletion, which was in line with the in silico predictions. Based on this result, we can predict that in vivo, the variant is most likely to cause a peptide deletion, namely p.236_264del. However, whether this presumption is true still needs to be tested by sequencing in situ specimens. Up to our knowledge, these findings are the first identification of the exonic deletion and splice‐site variant in DHODH, which is of great value for the expansion of its mutation type and the development of future functional research.
In regards of the function of DHODH, its biochemical function is almost clear, which is responsible for the orotate production and significant in the pyrimidine de novo synthesis (Loffler et al., 2015). Yet, its cell biological function is still a hot topic of research in recent years, especially in the field of oncology. Sykes et al. (2016) revealed that DHODH is an important regulator of cell differentiation and metabolism, and its inhibition has a alleviating effect on the differentiation block in the course of acute myeloid leukemia. Similarly, Bajzikova et al. (2019) demonstrated that reactivation of DHODH‐driven pyrimidine synthesis can overcome cell cycle arrest and restore tumor cell growth in a cancer cell model with respiratory defects. These results have been confirmed by more studies in cancer, for example, DHODH can be used as a therapeutic target for small‐cell lung cancer (Li et al., 2019); targeting DHODH‐driven pyrimidine synthesis enhances the molecular therapeutic response of glioblastoma stem cells (Wang et al., 2019); and high DHODH expression in esophageal squamous cell carcinoma promotes cell proliferation by stabilizing the β‐catenin pathway (Qian et al., 2020). Mao et al. (2021) recently clarified that DHODH mediated ferroptosis could inhibit tumor growth and might be a potential therapeutic strategy.
Balasubramaniam et al. (2014) indicated that Genetic aberrations of pyrimidine pathways lead to diverse clinical manifestations including neurological, immunological, hematological, renal impairments, adverse reactions to analogue therapy, and association with malignancies. Yet in developmental biology, the function of DHODH has been relatively poorly studied. By studying zebrafish embryos, White et al. found that DHODH could regulate the transcription elongation of neural crest and melanoma, and the inactivation of DHODH would have a serious impact on the development of neural crest (White et al., 2011). Rainger et al. (2012) showed that DHODH, Cad, and Umps were strongly expressed in the embryonic pharyngeal arch and limb bud, supporting a site‐ and stage‐specific requirement for de novo pyrimidine synthesis. Kondo et al. recently reported that buquinna, an inhibitor of DHODH, induced cell cycle arrest, stemness loss, and cell death in mouse pluripotent stem cells (MPSCS) (Kondo, 2021). Fang et al. found that although the missense mutation of DHODH did not affect the normal mitochondrial localization of the protein, it might lead to the destruction of protein stability or the reduction of enzyme activity (Fang et al., 2012), and speculated that DHODH might induce the phenotype of Miller syndrome through mitochondrial dysfunction (Fang et al., 2013); they also found that the loss of DHODH interfered with mitochondrial function and osteogenic differentiation of osteoblasts (Fang et al., 2016). In animal experiments, Dickinson et al. (2016) found that knockout of the mouse homolog of DHODH is homozygous‐lethal, indicating that this gene is very important for mammalian embryonic development. But even with these efforts, the role of DHODH in developmental biology, especially how it regulates the development of cranial‐maxillofacial tissues, remains somewhat thin.
In conclusion, for the first time in Chinese population, we identified a novel compound heterozygous DHODH variation consisting of one exonic deletion and one splicing variant in a fetus with multiple prenatal abnormalities. This work has constructive implications for further establishing genotype–phenotype association for the DHODH‐caused Miller syndrome and for further understanding of the function of this gene.
AUTHOR CONTRIBUTIONS
Q.G., D.Z., and C.Y. designed and supervised this study. L.F., X.C., and J.Z. recruited the case and conducted the clinical and imaging evaluation. K.Y., Y.Y., and Y.W. performed the genetic analysis. K.Y., X.C.,W.C., and L.F. carried out the in silico analysis. K.Y., X.C. and J.Z. performed the in vitro study. K.Y., X.C., and L.F. wrote the manuscript, and all co‐authors reviewed it and approved its submission.
FUNDING INFORMATION
This work was supported by the Project Funded by China Postdoctoral Science Foundation (No. 2022T150445) and the Beijing Hospitals Authority Youth Programme (No. QML20211401).
CONFLICT OF INTEREST STATEMENT
Kai Yang, Xiao‐yang Chu, Jing Zhang, Ze‐lu Li, Yu‐lan Xiang, Wen‐qi Chen, You‐sheng Yan, Yi‐peng Wang, Dong‐liang Zhang, Cheng‐hong Yin, and Qing Guo declare that they have no conflict of interest.
ETHICAL APPROVAL
The samples used in this study were collected with appropriate informed consent and approval of the ethics committee of Beijing Obstetrics and Gynecology Hospital (No. 2018‐KY‐003‐01). The methods used in this study were carried out in accordance with the approved guidelines.
ACKNOWLEDGMENTS
We acknowledge all the participants in this study.
Yang, K. , Fu, L.‐M. , Chu, X.‐Y. , Zhang, J. , Chen, W.‐Q. , Yan, Y.‐S. , Wang, Y.‐P. , Zhang, D.‐L. , Yin, C.‐H. , & Guo, Q. (2023). Assessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population. Molecular Genetics & Genomic Medicine, 11, e2186. 10.1002/mgg3.2186
Kai Yang, Li‐Man Fu and Xiao‐Yang Chu contributed equally to this study.
Contributor Information
Dong‐Liang Zhang, Email: zhangdongliang@mail.ccmu.edu.cn.
Cheng‐Hong Yin, Email: yinchh@ccmu.edu.cn.
Qing Guo, Email: yfguoqing@163.com.
DATA AVAILABILITY STATEMENT
All the data involved in this study were provided in the manuscript, table, and figures.
REFERENCES
- Arsham, M. S. , Barch, M. J. , & Lawce, H. J. (2017). The AGT cytogenetics laboratory manual. John Wiley & Sons Inc. [Google Scholar]
- Bajzikova, M. , Kovarova, J. , Coelho, A. R. , Boukalova, S. , Oh, S. , Rohlenova, K. , Svec, D. , Hubackova, S. , Endaya, B. , Judasova, K. , Bezawork‐Geleta, A. , Kluckova, K. , Chatre, L. , Zobalova, R. , Novakova, A. , Vanova, K. , Ezrova, Z. , Maghzal, G. J. , Magalhaes Novais, S. , … Neuzil, J. (2019). Reactivation of dihydroorotate dehydrogenase‐driven pyrimidine biosynthesis restores tumor growth of respiration‐deficient cancer Cells. Cell Metabolism, 29(2), 399–416 e310. 10.1016/j.cmet.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam, S. , Duley, J. A. , & Christodoulou, J. (2014). Inborn errors of pyrimidine metabolism: Clinical update and therapy. Journal of Inherited Metabolic Disease, 37(5), 687–698. 10.1007/s10545-014-9742-3 [DOI] [PubMed] [Google Scholar]
- Bhat, M. (2020). The human face: genes, embryological development and dysmorphology. The International Journal of Developmental Biology, 64(4–6), 383–391. 10.1387/ijdb.190312mb [DOI] [PubMed] [Google Scholar]
- Bukowska‐Olech, E. , Materna‐Kiryluk, A. , Walczak‐Sztulpa, J. , Popiel, D. , Badura‐Stronka, M. , Koczyk, G. , Dawidziuk, A. , & Jamsheer, A. (2020). Targeted next‐generation sequencing in the diagnosis of facial dysostoses. Frontiers in Genetics, 11, 580477. 10.3389/fgene.2020.580477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, M. E. , Flenniken, A. M. , Ji, X. , Teboul, L. , Wong, M. D. , White, J. K. , Meehan, T. F. , Weninger, W. J. , Westerberg, H. , Adissu, H. , Baker, C. N. , Bower, L. , Brown, J. M. , Caddle, L. B. , Chiani, F. , Clary, D. , Cleak, J. , Daly, M. J. , Denegre, J. M. , … Murray, S. A. (2016). High‐throughput discovery of novel developmental phenotypes. Nature, 537(7621), 508–514. 10.1038/nature19356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnai, D. , Hughes, H. E. , & Winter, R. M. (1987). Postaxial acrofacial dysostosis (Miller) syndrome. Journal of Medical Genetics, 24(7), 422–425. 10.1136/jmg.24.7.422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duley, J. A. , Henman, M. G. , Carpenter, K. H. , Bamshad, M. J. , Marshall, G. A. , Ooi, C. Y. , Wilcken, B. , & Pinner, J. R. (2016). Elevated plasma dihydroorotate in Miller syndrome: Biochemical, diagnostic and clinical implications, and treatment with uridine. Molecular Genetics and Metabolism, 119(1‐2), 83–90. 10.1016/j.ymgme.2016.06.008 [DOI] [PubMed] [Google Scholar]
- Fang, J. , Uchiumi, T. , Yagi, M. , Matsumoto, S. , Amamoto, R. , Saito, T. , Takazaki, S. , Kanki, T. , Yamaza, H. , Nonaka, K. , & Kang, D. (2012). Protein instability and functional defects caused by mutations of dihydro‐orotate dehydrogenase in Miller syndrome patients. Bioscience Reports, 32(6), 631–639. 10.1042/BSR20120046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, J. , Uchiumi, T. , Yagi, M. , Matsumoto, S. , Amamoto, R. , Takazaki, S. , Yamaza, H. , Nonaka, K. , & Kang, D. (2013). Dihydro‐orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Bioscience Reports, 33(2), e00021. 10.1042/BSR20120097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, J. , Yamaza, H. , Uchiumi, T. , Hoshino, Y. , Masuda, K. , Hirofuji, Y. , Wagener, F. A. , Kang, D. , & Nonaka, K. (2016). Dihydroorotate dehydrogenase depletion hampers mitochondrial function and osteogenic differentiation in osteoblasts. European Journal of Oral Sciences, 124(3), 241–245. 10.1111/eos.12270 [DOI] [PubMed] [Google Scholar]
- Fujikura, K. (2016). Global Carrier Rates of Rare Inherited Disorders Using Population Exome Sequences. PLoS One, 11(5), e0155552. 10.1371/journal.pone.0155552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genée, E. (1969). An extensive form of mandibulo‐facial dysostosis. Journal de Génétique Humaine, 17(1), 45–52. [PubMed] [Google Scholar]
- Hou, Y. C. , Yu, H. C. , Martin, R. , Cirulli, E. T. , Schenker‐Ahmed, N. M. , Hicks, M. , Cohen, I. V. , Jönsson, T. J. , Heister, R. , Napier, L. , Swisher, C. L. , Dominguez, S. , Tang, H. , Li, W. , Perkins, B. A. , Barea, J. , Rybak, C. , Smith, E. , Duchicela, K. , … Caskey, C. T. (2020). Precision medicine integrating whole‐genome sequencing, comprehensive metabolomics, and advanced imaging. Proceedings of the National Academy of Sciences of the United States of America, 117(6), 3053–3062. 10.1073/pnas.1909378117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis, N. M. , Rothstein, J. H. , Pejaver, V. , Middha, S. , McDonnell, S. K. , Baheti, S. , Musolf, A. , Li, Q. , Holzinger, E. , Karyadi, D. , Cannon‐Albright, L. A. , Teerlink, C. C. , Stanford, J. L. , Isaacs, W. B. , Xu, J. , Cooney, K. A. , Lange, E. M. , Schleutker, J. , & Carpten, J. D. (2016). REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics, 99(4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, F. , Kondoh, T. , Komori, K. , Matsui, T. , Harada, N. , Yanai, A. , Fukuda, M. , Morifuji, K. , & Matsumoto, T. (2011). Miller syndrome with novel dihydroorotate dehydrogenase gene mutations. Pediatrics International, 53(4), 587–591. 10.1111/j.1442-200X.2010.03303.x [DOI] [PubMed] [Google Scholar]
- Kondo, T. (2021). Selective eradication of pluripotent stem cells by inhibiting DHODH activity. Stem Cells, 39(1), 33–42. 10.1002/stem.3290 [DOI] [PubMed] [Google Scholar]
- Li, L. , Ng, S. R. , Colon, C. I. , Drapkin, B. J. , Hsu, P. P. , Li, Z. , Nabel, C. S. , Lewis, C. A. , Romero, R. , Mercer, K. L. , Bhutkar, A. , Phat, S. , Myers, D. T. , Muzumdar, M. D. , Westcott, P. M. K. , Beytagh, M. C. , Farago, A. F. , Vander Heiden, M. G. , & Dyson, N. J. (2019). Identification of DHODH as a therapeutic target in small cell lung cancer. Science Translational Medicine, 11(517), eaaw7852. 10.1126/scitranslmed.aaw7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler, M. , Carrey, E. A. , & Zameitat, E. (2015). Orotic acid, more than just an intermediate of pyrimidine de novo synthesis. Journal of Genetics and Genomics, 42(5), 207–219. 10.1016/j.jgg.2015.04.001 [DOI] [PubMed] [Google Scholar]
- Mao, C. , Liu, X. , Zhang, Y. , Lei, G. , Yan, Y. , Lee, H. , Koppula, P. , Wu, S. , Zhuang, L. , Fang, B. , Poyurovsky, M. V. , Olszewski, K. , & Gan, B. (2021). DHODH‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature, 593(7860), 586–590. 10.1038/s41586-021-03539-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. , Fineman, R. , & Smith, D. W. (1979). Postaxial acrofacial dysostosis syndrome. The Journal of Pediatrics, 95(6), 970–975. 10.1016/s0022-3476(79)80285-1 [DOI] [PubMed] [Google Scholar]
- Minet, M. , Dufour, M. E. , & Lacroute, F. (1992). Cloning and sequencing of a human cDNA coding for dihydroorotate dehydrogenase by complementation of the corresponding yeast mutant. Gene, 121(2), 393–396. 10.1016/0378-1119(92)90150-n [DOI] [PubMed] [Google Scholar]
- Ng, S. B. , Buckingham, K. J. , Lee, C. , Bigham, A. W. , Tabor, H. K. , Dent, K. M. , Huff, C. D. , Shannon, P. T. , Jabs, E. W. , Nickerson, D. A. , Shendure, J. , & Bamshad, M. J. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nature Genetics, 42(1), 30–35. 10.1038/ng.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco Rodriguez, J. M. , Wacklin‐Knecht, H. , & Knecht, W. (2022). Protein‐lipid interactions of human dihydroorotate dehydrogenase and three mutants associated with Miller syndrome. Nucleosides, Nucleotides & Nucleic Acids, 1–22. 10.1080/15257770.2022.2039393 [DOI] [PubMed] [Google Scholar]
- Qian, Y. , Liang, X. , Kong, P. , Cheng, Y. , Cui, H. , Yan, T. , Wang, J. , Zhang, L. , Liu, Y. , Guo, S. , Cheng, X. , & Cui, Y. (2020). Elevated DHODH expression promotes cell proliferation via stabilizing beta‐catenin in esophageal squamous cell carcinoma. Cell Death & Disease, 11(10), 862. 10.1038/s41419-020-03044-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainger, J. , Bengani, H. , Campbell, L. , Anderson, E. , Sokhi, K. , Lam, W. , Riess, A. , Ansari, M. , Smithson, S. , Lees, M. , Mercer, C. , McKenzie, K. , Lengfeld, T. , Gener Querol, B. , Branney, P. , McKay, S. , Morrison, H. , Medina, B. , Robertson, M. , … Fitzpatrick, D. R. (2012). Miller (Genee‐Wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Human Molecular Genetics, 21(18), 3969–3983. 10.1093/hmg/dds218 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, J. C. , Glusman, G. , Smit, A. F. , Huff, C. D. , Hubley, R. , Shannon, P. T. , Rowen, L. , Pant, K. P. , Goodman, N. , Bamshad, M. , Shendure, J. , Drmanac, R. , Jorde, L. B. , Hood, L. , & Galas, D. J. (2010). Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science, 328(5978), 636–639. 10.1126/science.1186802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes, D. B. , Kfoury, Y. S. , Mercier, F. E. , Wawer, M. J. , Law, J. M. , Haynes, M. K. , Lewis, T. A. , Schajnovitz, A. , Jain, E. , Lee, D. , Meyer, H. , Pierce, K. A. , Tolliday, N. J. , Waller, A. , Ferrara, S. J. , Eheim, A. L. , Stoeckigt, D. , Maxcy, K. L. , Cobert, J. M. , … Scadden, D. T. (2016). Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell, 167(1), 171–186 e115. 10.1016/j.cell.2016.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from next‐generation sequencing data. Nucleic Acids Research, 38, e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Yang, K. , Wu, Q. , Kim, L. J. Y. , Morton, A. R. , Gimple, R. C. , Prager, B. C. , Shi, Y. , Zhou, W. , Bhargava, S. , Zhu, Z. , Jiang, L. , Tao, W. , Qiu, Z. , Zhao, L. , Zhang, G. , Li, X. , Agnihotri, S. , Mischel, P. S. , … Rich, J. N. (2019). Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Science Translational Medicine, 11(504), eaau4972. 10.1126/scitranslmed.aau4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, R. M. , Cech, J. , Ratanasirintrawoot, S. , Lin, C. Y. , Rahl, P. B. , Burke, C. J. , Langdon, E. , Tomlinson, M. L. , Mosher, J. , Kaufman, C. , Chen, F. , Long, H. K. , Kramer, M. , Datta, S. , Neuberg, D. , Granter, S. , Young, R. A. , Morrison, S. , Wheeler, G. N. , & Zon, L. I. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature, 471(7339), 518–522. 10.1038/nature09882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek, D. (2013). Human facial dysostoses. Clinical Genetics, 83(6), 499–510. 10.1111/cge.12123 [DOI] [PubMed] [Google Scholar]
- Wiedemann, H. R. (1973). Malformation‐retardation syndrome with bilateral absence of the 5th rays in both hands and feets, cleft palate, malformed ears and eyelids, radioulnar synostosis (author's transl). Klinische Pädiatrie, 185(3), 181–186. [PubMed] [Google Scholar]
- Yang, K. , Liu, Y. , Wu, J. , Zhang, J. , Hu, H. Y. , Yan, Y. S. , Chen, W. Q. , Yang, S. F. , Sun, L. J. , Sun, Y. Q. , Wu, Q. Q. , & Yin, C. H. (2022). Prenatal cases reflect the complexity of the COL1A1/2 associated osteogenesis imperfecta. Genes (Basel), 13(9), 1578. 10.3390/genes13091578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data involved in this study were provided in the manuscript, table, and figures.