

Abstract
Background
Congenital adrenal hyperplasia (CAH) due to 21‐hydroxylase (21OH) deficiency is an autosomal recessive inborn error of cortisol biosynthesis, with varying degrees of aldosterone production. There is a continuum of phenotypes which generally correlate with genotype and the expected residual 21OH activity of the less severely impaired allele. CYP21A1P/CYP21A2 chimeric genes caused by recombination between CYP21A2 and its highly homologous CYP21A1P pseudogene are common in CAH and typically associated with salt‐wasting CAH, the most severe form. Nine chimeras have been described (CH‐1 to CH‐9).
Aims
The aim of this study was to genetically evaluate two variant alleles carried by a 22‐year‐old female with the non‐salt‐wasting simple virilizing form of CAH and biallelic 30‐kb deletions.
Methods
The haplotypes of the CYP21A2 heterozygous variants, as well as the chimeric junction sites, were determined by Sanger sequencing TA clones of an allele‐specific PCR product.
Results
Genetic testing revealed two rare CYP21A1P/CYP21A2 chimeras: allele 1 matches the previously described CAH CH‐1 chimera but without the P30L variant, and allele 2, termed here as novel CAH CH‐10, has a junction site between c.293‐37 and c.29314, which is expected to retain partial 21OH activity.
Conclusion
These two variant alleles further document the complex nature of RCCX modules and highlight that not all CYP21A1P/CYP21A2 chimera severely impair 21OH activity.
Keywords: 21‐hydroxylase deficiency, CAH, chimera, congenital adrenal hyperplasia, CYP21A2
CYP21A1P/CYP21A2 chimeric genes caused by recombination between CYP21A2 and its highly homologous CYP21A1P pseudogene are common in congenital adrenal hyperplasia (CAH) due to 21‐hydroxylase (21OH) deficiency and typically are associated with severe salt‐wasting (SW) CAH. We describe a 22‐year‐old female with non‐SW CAH and two rare CYP21A1P/CYP21A2 chimeras, including a novel CAH chimera (named CH‐10) which is expected to retain partial 21OH activity explaining the individual's mild phenotype.
1. INTRODUCTION
Congenital adrenal hyperplasia (CAH) due to 21‐hydroxylase deficiency (21‐OHD, OMIM 201910) caused by CYP21A2 defects (OMIM 613815) is an autosomal recessive inborn error of cortisol biosynthesis, with varying degrees of aldosterone production and corticotropin‐driven adrenal androgen excess (Merke & Auchus, 2020). The majority of individuals are compound heterozygotes. The phenotype is determined by the less severely impaired CYP21A2 allele and the expected residual 21OH activity, with some phenotypic variability described in large cohort studies (Finkielstain et al., 2011; Gurgov et al., 2017; Krone et al., 2000). The phenotypic spectrum is traditionally categorized as classic salt‐wasting (SW), with marked deficiencies in both cortisol and aldosterone and life‐threatening if left untreated; classic simple virilizing (SV), with cortisol deficiency and minimal aldosterone production; and the nonclassic (NC) phenotype with mild manifestations ranging from asymptomatic to late‐onset hyperandrogenism. The SW and SV types are termed classic CAH and result in 46, XX atypical genitalia due to in utero excess androgens. Classic CAH is estimated to affect 1 in 15,000; NC CAH is estimated to affect 1 in 200 (Hannah‐Shmouni et al., 2017).
CYP21A2 encoding 21OH is mapped to chromosome 6p23.1 in a low copy repeat region termed RCCX with homologous genes in tandem: RP1 (STK19) and pseudogene RP2 (STK19P); C4A and C4B; CYP21A2 and pseudogene CYP21A1P; TNXB and pseudogene TNXA. These gene pairs are error‐prone during meiosis with the most common pathogenic outcome CAH. Minor conversions from CYP21A1P to CYP21A2 are responsible for approximately 60% of CAH alleles; chimeric genes, mostly termed “30‐kb deletions,” together with major conversions, account for 30% (Merke & Auchus, 2020). 30‐kb deletions are further categorized based on junction sites into nine CYP21A1P/CYP21A2 chimeras (CAH CH‐1 to CH‐9) (Figure 1) and three CYP21A1P‐TNXA/TNXB chimeras with deletion of the entire CYP21A2 extending to the neighboring TNXB (CAH‐X CH‐1 to CH‐3) (Finkielstain et al., 2011; Merke & Auchus, 2020). Junction site locations can influence gene functionality and the degree of 21OH impairment (Keen‐Kim et al., 2005). Furthermore, the monoallelic presence of a CAH‐X chimera disrupts both CYP21A2 and TNXB which results in hypermobility‐type Ehlers‐Danlos syndrome (Merke et al., 2013). The vast majority of “30‐kb deletions,” including seven of the nine CAH chimeras and all three CAH‐X chimeras, are null variants associated with a SW phenotype. To date, two CAH chimeras (CH‐4 and CH‐9) due to the location of their junction sites correspond to a phenotype which straddles the classic SV–NC boundary and are named attenuated CAH chimeras (Chen et al., 2012).
FIGURE 1.
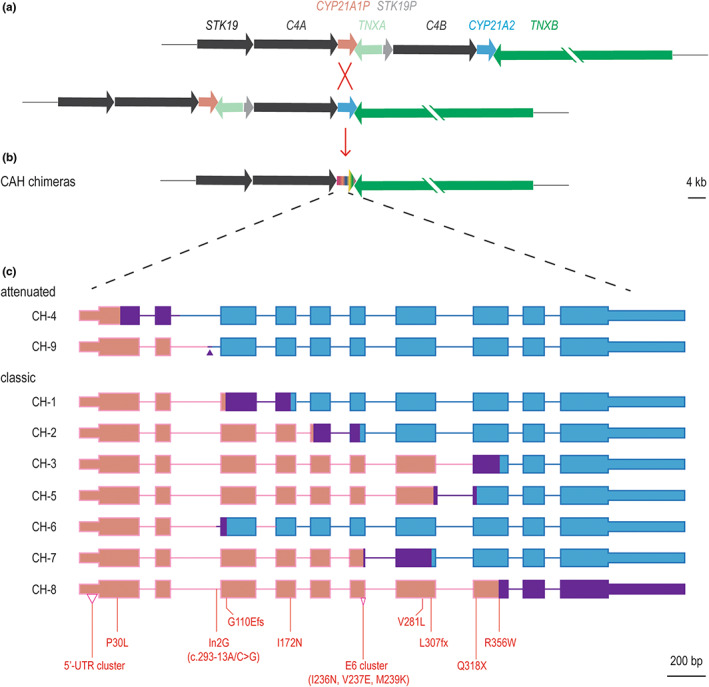
CYP21 chimeras and their junction sites. (a) Unequal crossover between CYP21A1P and CYP21A2 during meiosis, pseudogenes are shown in muted colors. (b) CAH chimeras, shown in chameleon as the unequal crossover outcome. (c) Schematic of all previously known CAH chimeras: gene body of CYP21A1P‐origin and CYP21A2‐origin are in pink and blue, respectively, and junction sites are in purple. The junction site of CAH CH‐9, which is only 7 bp in size, is marked with a purple triangle. The variants of CYP21A1P‐origin commonly used to determine the CAH chimera subtype are marked on CAH CH‐8, which carries all of these variants (Chen et al., 2012). Genes are shown in scale except TNXB, whose size is 68 kb.
Here we report a case of 21‐OHD SV CAH due to biallelic 30‐kb deletions. We identified two rare CAH chimeric genes including a novel attenuated chimera featuring a new junction site. Our findings further document the variability and complexity of CYP21A2 defects causative for 21‐OHD CAH.
2. MATERIALS AND METHODS
2.1. Participants
The subject was enrolled in an ongoing Natural History Study at the NIH Clinical Center in Bethesda, MD, USA (NCT00250159) and gave written informed consent.
2.2. Genetic analyses
CAH genotyping was initially conducted by CLIA‐accredited laboratories with an assay‐based multiplex mini‐sequencing and conversion‐specific PCR (MMCP) (Esoterix Laboratory Services, Inc), (Keen‐Kim et al., 2005) and a comprehensive test based on Sanger methodologies (PreventionGenetics LLC). The haplotypes of the CYP21A2 heterozygous variants, as well as the chimeric junction sites, were then determined by Sanger sequencing TA clones of an allele‐specific PCR product with primers CYP779f/Tena32f (Chen et al., 2012) The allele‐specific PCR, CYP21A2 duplication test, and Sanger sequencing were conducted as previously described (Xu et al., 2013). The TA cloning was conducted with a TOPO XL complete PCR cloning kit (Thermo Fisher Scientific). References for CYP21A2 and CYP21A1P were ENSG00000231852.2 and ENSG00000204338.4, respectively. CAH pathogenic variants on CYP21A2 were presented in common names with Human Genome Variation Society nomenclature shown after their first appearance. A DNASTAR Lasergene software (version 17) was used for sequence analysis.
3. RESULTS
3.1. Clinical findings
A 22‐year‐old Haitian female presented with atypical genitalia at birth. She underwent clitoroplasty at age 3 years, and was treated with prednisone and fludrocortisone until age 10 years, after which she was off medications with limited access to care due to a poor social environment. She started shaving her face around age 13 years and had no breast development and primary amenorrhea. At 21 years, she presented to a community hospital twice with reported possible adrenal crises in the setting of infection and inflammation. Physical examination revealed normal vital signs, hirsutism of the face and trunk, a deep voice, broad shoulders, Tanner 1 breasts, Tanner 5 public hair, malar rash. She was started on dexamethasone 0.5 mg and fludrocortisone 0.1 mg daily. Rheumatological evaluation confirmed a diagnosis of systemic lupus erythematosus (SLE) with constitutional symptoms, serositis (pericarditis), Raynaud's of the hands and feet, positive serology (ANA, anti‐ENA, anti‐SmRNP antibody, anti‐Smith [SM] antibody), low complement, and lymphopenia. Family history reported three unaffected older siblings and a younger brother with CAH who was positive on newborn screening but not on hormone replacement and had never had an adrenal crisis. Due to social reasons, acquisition of genotyping and phenotypic data for family members was not available. At our Institution, early morning laboratory evaluation showed 17‐OHP 32,760 ng/dL, androstenedione 5315 ng/dL [ref 17–175 ng/dL], testosterone 178 ng/dL [ref 8–60 ng/dL], cortisol 3 μg/dL [ref 5–25 μg /dL], and plasma renin activity within the normal range.
3.2. Genetic findings
The subject had two indeterminate 21‐OHD genetic tests from commercial laboratories. Family genotyping was indicated but unavailable. A MMCP‐based test suggested biallelic 30‐kb deletions. Sanger sequencing of CYP779f/Tena32f PCR product detected a cluster of pre‐coding variants of CYP21A1P origin (c.‐210T>C, c.‐199C>T, c.‐190dup, c.‐126C>T, c.‐113G>A, c.‐110T>C and c.‐103A>G), all in homozygous status supporting biallelic 30 kb deletion. Compound heterozygous variants of CYP21A1P origin were also detected, including P30L (c.92C>T, p.Pro31Leu), In2G (c.293‐13A/C>G) and G110Efs (c.332_339del, p.Gly111Valfs*21). The heterozygous status of the compound variants, especially that of P30L, did not support the existence of biallelic 30‐kb deletions.
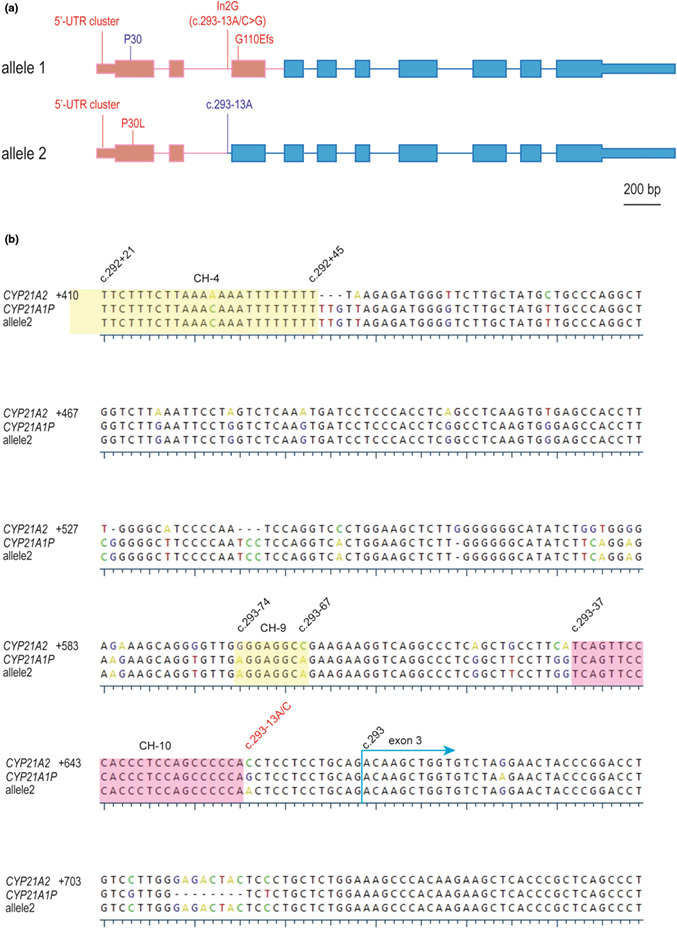
We conducted TA cloning to differentiate the two alleles of the CYP779f/Tena32f PCR product. Plasmids carrying each allele were subject to Sanger sequencing. Eight plasmid clones were analyzed and two CAH chimera alleles (haplotypes) were detected. A CYP21A2 duplication test with negative result indicated these two chimeras were responsible for the subject's CAH phenotype (data not shown). Allele 1 sequence matched chimera CAH CH‐1 except for the missing the P30L variant; allele 2 had all variants of CYP21A1P origin spanning from the pre‐coding region till c.293‐38A of intron 2, only 24 bp upstream of c.293‐13A/C where the In2G (c.293‐13A/C>G) variant occurs (Figure 2a,b).
FIGURE 2.
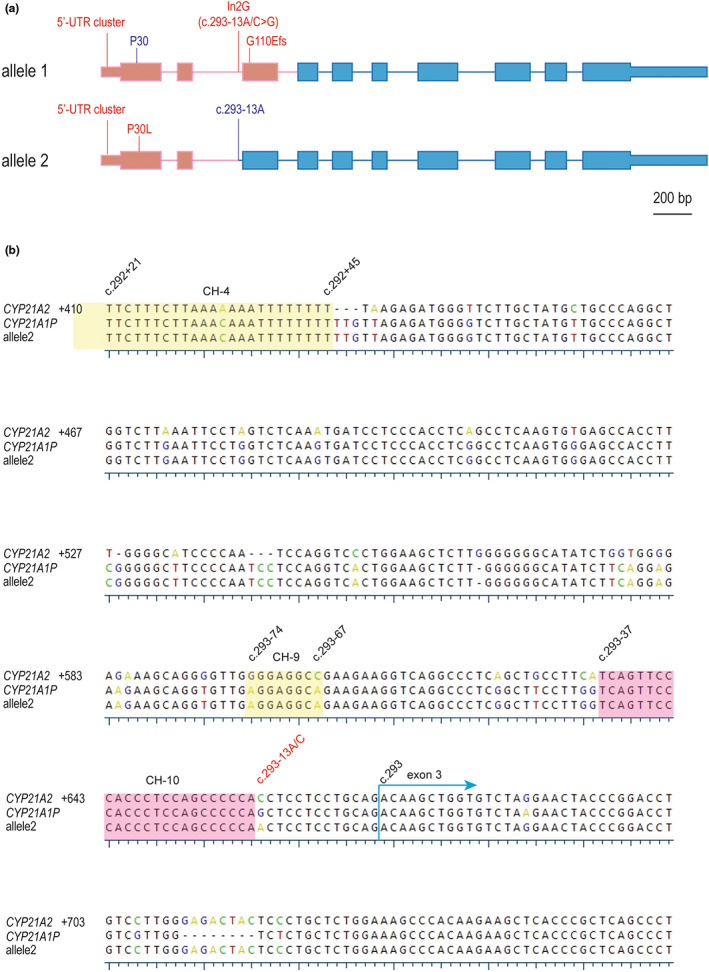
(a) Schematic of two rare CYP21A1P/CYP21A2 chimeras: allele 1 is a classic CAH CH‐1 but without P30L, allele 2 is a novel attenuated CAH chimera termed CH‐10 with junction site between c.293‐37 and c.293‐14. Pink and blue colors represent CYP21A1P and CYP21A2 sequence, respectively, shown in scale. (b) Alignment of CYP21 genes at a locus corresponding to CYP21A2 intron 2 and exon 3, CYP21A2 served as the reference and its numbering is shown at the beginning of each row. Conserved DNA residues are shown in black while non‐conserved are shown in an UGene physiochemical scheme. The junction sites of known attenuated chimeras CH‐4 and CH‐9 are in yellow, and the novel CH‐10 is in pink. Dashed lines denote gaps. Remarkable DNA residues are marked on the top with their respective cDNA nomenclature numberings, including c.293 to start exon 3 and c.293‐13A/C (in red) where In2G (c.293‐13A/C>G) occurs.
4. DISCUSSION
We describe a case of 21‐OHD CAH due to two rare CYP21A1P/CYP21A2 chimeras including a classic CAH CH‐1 chimera without the usual P30L variant, and a novel attenuated chimera termed here CAH CH‐10 with a junction site between c.293‐37 and c.293‐14 upstream of In2G. The subject's unexpected SV phenotype was therefore explained by her genotype. SLE was not entirely unexpected, as 21‐OHD has been associated with an increased prevalence of autoimmune disorders (Falhammar et al., 2019). Our findings further document the complex nature of the RCCX module (Carrozza et al., 2021; Miller, 2020; Pignatelli et al., 2019; Tolba et al., 2022) and highlight that not all CYP21A1P/CYP21A2 chimera severely impair 21OH activity.
Although there are limited studies of the RCCX pseudogenes, CYP21A2‐like variants are commonly found in CYP21A1P, with those corresponding to P30, V281 accounting for 24% and 21% of total CYP21A1P alleles respectively (Tsai & Lee, 2012). Therefore, if CYP21A2‐like CYP21A1P is involved in unequal crossovers, the resulting chimeras could sometimes be missing P30L and V281L (c.844G>T, p.Val282Leu). In fact, a common chimera CAH CH‐5 which has all variants of pseudogene origin spanning from the pre‐coding region to the L307fs (c.923dup, p.Leu308Phefs*6) variant is often missing V281L (Chen et al., 2012). However, CAH chimeras missing P30L have been rarely reported. Our large U.S. cohort of approximately 500 subjects has only one additional family carrying a CAH chimera without P30L (Finkielstain et al., 2011). A report from Brazil described an individual with 21‐OHD carrying a CAH chimera without P30L, but the chimera type was not specified (Coeli et al., 2010).
Unlike classic CAH chimeras that null the corresponding CYP21A2 allele, attenuated chimeras correspond mostly to a SV phenotype (Chen et al., 2012), or sometimes a phenotype with severity between NC and SV, such as a NC form of early onset (l'Allemand et al., 2000). Two attenuated chimeras, CH‐4 and CH‐9, have been reported with junction sites between c.138 and c.292+45 spanning from exon 1 to intron 2, and between c.293‐74 and c.293‐67 inside intron2, respectively. Both chimeras feature a pseudogene 5’‐UTR and a P30L variant as the phenotype determinant. Since the CYP21A1P promoter is known to be weaker compared to that of CYP21A2, the usual phenotype associated with an attenuated chimera is classic SV CAH, more severe than the NC phenotype typically associated with P30L. To date, chimeric CYP21A1P/CYP21A2 genes are termed chronologically after determination of the junction sites (Chen et al., 2012). Here we define and name CH‐10. It is commonly accepted that newly identified junction sites of CYP21A1P/CYP21A2 chimeras based on an array of variants of pseudogene origin define a new chimeric gene, but it is possible that some of these chimeras are formed by minor conversion of a previously described chimera, rather than a deletion or major conversion event. However, the likelihood of this mechanism should be very low since there are no reports of minor conversion events between CYP21A1P/CYP21A2 chimera types.
Our data suggest that the junction site of attenuated chimeras can be anywhere spanning from c.138 (found in CH‐4) to c.293‐14 (found in CH‐10) just upstream of the In2G site. These junction sites might have been historically under‐detected because sequencing analysis of TA clones is often needed to overcome the frequent insert‐deletion interference inside intron 2 in a Sanger assay. Attenuated CAH chimeras, previously considered rare, might be under‐detected by current CYP21A2 testing which commonly excludes the 5’‐UTR and thus can easily confuse an attenuated chimera with a P30L variant (Baumgartner‐Parzer et al., 2020), which is sometimes unexplainably associated with a classic phenotype (Araujo et al., 2005; Finkielstain et al., 2011). A CAH genetic platform that includes the promoter might offer an explanation for these genotype–phenotype discrepancies. Given that all attenuated CYP21A1P/CYP21A2 chimeras identified to date include a CYP21A1P 5’‐UTR, P30L variant, and a junction site between P30L and c.293‐13A/G (absent of In2G), a unified methodological approach should be incorporated into CAH genetic testing. Moreover, our results reinforce that the presence of P30L and the absence of In2G correlates with the attenuated phenotype. Finally, our study agrees with a previous conclusion that Sanger sequencing of CYP779f/Tena32f PCR products offers the most comprehensive genetic test of CYP21A2 in terms of testing and profiling chimeras (Chen et al., 2012).
AUTHOR CONTRIBUTIONS
Qizong Lao and Deborah P. Merke: Conceptualization. Qizong Lao and Deepika D. Burkardt: Methodology. Deborah P. Merke: Resources. Qizong Lao, Sarah Kollender, and Fabio R. Faucz: Data curation. Qizong Lao, Deepika D. Burkardt, and Sarah Kollender: Writing‐original draft. Deborah P. Merke: Writing‐review & editing. Qizong Lao: Visualization. Deborah P. Merke: Supervision. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
This research was supported by the Intramural Research Program at the National Institutes of Health (NIH), Bethesda, Maryland.
CONFLICT OF INTEREST STATEMENT
D.P.M. received unrelated research funds from Diurnal Limited and Neurocrine Biosciences through the NIH Cooperative Research and Development Agreement. The authors declared no conflict of interest.
ETHICS STATEMENT
All procedures were in accordance with good clinical practice and ethical standards. This study was approved by the National Institutes of Health IRB.
ACKNOWLEDGMENTS
Not applicable.
Lao, Q. , Burkardt, D. D. , Kollender, S. , Faucz, F. R. , & Merke, D. P. (2023). Congenital adrenal hyperplasia due to two rare CYP21A2 variant alleles, including a novel attenuated CYP21A1P/CYP21A2 chimera. Molecular Genetics & Genomic Medicine, 11, e2195. 10.1002/mgg3.2195
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available in the article.
REFERENCES
- Araujo, R. S. , Billerbeck, A. E. , Madureira, G. , Mendonca, B. B. , & Bachega, T. A. (2005). Substitutions in the CYP21A2 promoter explain the simple‐virilizing form of 21‐hydroxylase deficiency in patients harbouring a P30L mutation. Clinical Endocrinology, 62(2), 132–136. 10.1111/j.1365-2265.2005.02184.x [DOI] [PubMed] [Google Scholar]
- Baumgartner‐Parzer, S. , Witsch‐Baumgartner, M. , & Hoeppner, W. (2020). EMQN best practice guidelines for molecular genetic testing and reporting of 21‐hydroxylase deficiency. European Journal of Human Genetics, 28(10), 1341–1367. 10.1038/s41431-020-0653-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza, C. , Foca, L. , De Paolis, E. , & Concolino, P. (2021). Genes and pseudogenes: Complexity of the RCCX locus and disease. Frontiers in Endocrinology, 12, 709758. 10.3389/fendo.2021.709758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Xu, Z. , Sullivan, A. , Finkielstain, G. P. , Van Ryzin, C. , Merke, D. P. , & McDonnell, N. B. (2012). Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21‐hydroxylase deficiency. Clinical Chemistry, 58(2), 421–430. 10.1373/clinchem.2011.174037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coeli, F. B. , Soardi, F. C. , Bernardi, R. D. , de Araújo, M. , Paulino, L. C. , Lau, I. F. , Petroli, R. J. , de Lemos‐Marini, S. H. V. , Baptista, M. T. M. , Guerra‐Júnior, G. , & de‐Mello, M. P. (2010). Novel deletion alleles carrying CYP21A1P/A2chimeric genes in Brazilian patients with 21‐hydroxylase deficiency. BMC Medical Genetics, 11(1), 104. 10.1186/1471-2350-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falhammar, H. , Frisén, L. , Hirschberg, A. L. , Nordenskjöld, A. , Almqvist, C. , & Nordenström, A. (2019). Increased risk of autoimmune disorders in 21‐hydroxylase deficiency: A Swedish population‐based National Cohort Study. Journal of the Endocrine Society, 3(5), 1039–1052. 10.1210/js.2019-00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkielstain, G. P. , Chen, W. , Mehta, S. P. , Fujimura, F. K. , Hanna, R. M. , Van Ryzin, C. , McDonnell, N. B. , & Merke, D. P. (2011). Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21‐hydroxylase deficiency. The Journal of Clinical Endocrinology and Metabolism, 96(1), E161–E172. 10.1210/jc.2010-0319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurgov, S. , Bernabé, K. J. , Stites, J. , Cunniff, C. M. , Lin‐Su, K. , Felsen, D. , New, M. I. , & Poppas, D. P. (2017). Linking the degree of virilization in females with congenital adrenal hyperplasia to genotype. Annals of the New York Academy of Sciences, 1402(1), 56–63. 10.1111/nyas.13370 [DOI] [PubMed] [Google Scholar]
- Hannah‐Shmouni, F. , Morissette, R. , Sinaii, N. , Elman, M. , Prezant, T. R. , Chen, W. , Pulver, A. , & Merke, D. P. (2017). Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genetics in Medicine, 19(11), 1276–1279. 10.1038/gim.2017.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen‐Kim, D. , Redman, J. B. , Alanes, R. U. , Eachus, M. M. , Wilson, R. C. , New, M. I. , Nakamoto, J. M. , & Fenwick, R. G. (2005). Validation and clinical application of a locus‐specific polymerase chain reaction‐ and minisequencing‐based assay for congenital adrenal hyperplasia (21‐hydroxylase deficiency). The Journal of Molecular Diagnostics, 7(2), 236–246. 10.1016/s1525-1578(10)60550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krone, N. , Braun, A. , Roscher, A. A. , Knorr, D. , & Schwarz, H. P. (2000). Predicting phenotype in steroid 21‐hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. The Journal of Clinical Endocrinology and Metabolism, 85(3), 1059–1065. 10.1210/jcem.85.3.6441 [DOI] [PubMed] [Google Scholar]
- l'Allemand, D. , Tardy, V. , Grüters, A. , Schnabel, D. , Krude, H. , & Morel, Y. (2000). How a patient homozygous for a 30‐kb deletion of the C4‐CYP 21 genomic region can have a nonclassic form of 21‐hydroxylase deficiency. The Journal of Clinical Endocrinology & Metabolism, 85(12), 4562–4567. 10.1210/jcem.85.12.7018 [DOI] [PubMed] [Google Scholar]
- Merke, D. P. , & Auchus, R. J. (2020). Congenital adrenal hyperplasia due to 21‐hydroxylase deficiency. The New England Journal of Medicine, 383(13), 1248–1261. 10.1056/NEJMra1909786 [DOI] [PubMed] [Google Scholar]
- Merke, D. P. , Chen, W. , Morissette, R. , Xu, Z. , Van Ryzin, C. , Sachdev, V. , Hannoush, H. , Shanbhag, S. M. , Acevedo, A. T. , Nishitani, M. , Arai, A. E. , & McDonnell, N. B. (2013). Tenascin‐X haploinsufficiency associated with Ehlers‐Danlos syndrome in patients with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology and Metabolism, 98(2), E379–E387. 10.1210/jc.2012-3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, W. L. (2020). Tenascin‐X‐discovery and early research. Frontiers in Immunology, 11, 612497. 10.3389/fimmu.2020.612497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli, D. , Carvalho, B. L. , Palmeiro, A. , Barros, A. , Guerreiro, S. G. , & Macut, D. (2019). The complexities in genotyping of congenital adrenal hyperplasia: 21‐hydroxylase deficiency. Frontiers in Endocrinology, 10, 432. 10.3389/fendo.2019.00432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolba, A. , Mandour, I. , Musa, N. , Elmougy, F. , Hafez, M. , Abdelatty, S. , Ibrahim, A. , Soliman, H. , Labib, B. , Elshiwy, Y. , Ramzy, T. , & Elsharkawy, M. (2022). Copy number variations in genetic diagnosis of congenital adrenal hyperplasia children. Frontiers in Genetics, 13, 785570. 10.3389/fgene.2022.785570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, L. P. , & Lee, H. H. (2012). Analysis of CYP21A1P and the duplicated CYP21A2 genes. Gene, 506(1), 261–262. 10.1016/j.gene.2012.06.045 [DOI] [PubMed] [Google Scholar]
- Xu, Z. , Chen, W. , Merke, D. P. , & McDonnell, N. B. (2013). Comprehensive mutation analysis of the CYP21A2 gene: An efficient multistep approach to the molecular diagnosis of congenital adrenal hyperplasia. The Journal of Molecular Diagnostics, 15(6), 745–753. 10.1016/j.jmoldx.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available in the article.