

Abstract
Background
There is accumulating evidence that progressive changes in brain structure and function take place as schizophrenia unfolds. Among many possible candidates, oxidative stress may be one of the mediators of neuroprogression, grey matter loss and subsequent cognitive and functional impairment. Antioxidants are exogenous or endogenous molecules that mitigate any form of oxidative stress or its consequences. They may act from directly scavenging free radicals to increasing anti‐oxidative defences. There is evidence that current treatments impact oxidative pathways and may to some extent reverse pro‐oxidative states in schizophrenia. The existing literature, however, indicates that these treatments do not fully restore the deficits in antioxidant levels or restore levels of oxidants in schizophrenia. As such, there has been interest in developing interventions aimed at restoring this oxidative balance beyond the benefits of antipsychotics in this direction. If antioxidants are to have a place in the treatment of this serious condition, the relevant and up‐to‐date information should be available to clinicians and investigators.
Objectives
To evaluate the effect of antioxidants as add‐on treatments to standard antipsychotic medication for improving acute psychotic episodes and core symptoms, and preventing relapse in people with schizophrenia.
Search methods
We searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials which is based on regular searches of CINAHL, BIOSIS, AMED, Embase, PubMed, MEDLINE, PsycINFO, and registries of clinical trials. There are no language, time, document type, or publication status limitations for inclusion of records in the register. We ran this search in November 2010, and again on 8 January 2015. We also inspected references of all identified studies for further trials and contacted authors of trials for additional information.
Selection criteria
We included reports if they were randomised controlled trials (RCTs) involving people with schizophrenia who had been allocated to either a substance with antioxidant potential or to a placebo as an adjunct to standard antipsychotic treatment.
Data collection and analysis
We independently extracted data from these trials and we estimated risk ratios (RR) or mean differences (MD), with 95% confidence intervals (CI). We assessed risk of bias for included studies and created a 'Summary of findings' table using GRADE.
Main results
The review includes 22 RCTs of varying quality and sample size studying Ginkgo biloba, N‐acetyl cysteine (NAC), allopurinol, dehydroepiandrosterone (DHEA), vitamin C, vitamin E or selegiline. Median follow‐up was eight weeks. Only three studies including a minority of the participants reported our a priori selected primary outcome of clinically important response. Short‐term data for this outcome (measured as at least 20% improvement in scores on Positive and Negative Syndrome Scale (PANSS)) were similar (3 RCTs, n = 229, RR 0.77, 95% CI 0.53 to 1.12, low quality evidence). Studies usually reported only endpoint psychopathology rating scale scores. Psychotic symptoms were lower in those using an adjunctive antioxidant according to the PANSS ( 7 RCTS, n = 584, MD ‐6.00, 95% CI ‐10.35 to ‐1.65, very low quality evidence) and the Brief Psychiatric Rating Scale (BPRS) (8 RCTS, n = 843, MD ‐3.20, 95% CI ‐5.63 to ‐0.78, low quality evidence). There was no overall short‐term difference in leaving the study early (16 RCTs, n = 1584, RR 0.73, 95% CI 0.48 to 1.11, moderate quality evidence), or in general functioning (2 RCTs, n = 52, MD ‐1.11, 95% CI ‐8.07 to 5.86, low quality evidence). Adverse events were generally poorly reported. Three studies reported useable data for 'any serious adverse effect', results were equivocal (3 RCTs, n = 234, RR 0.65, 95% CI 0.19 to 2.27, low quality evidence). No evidence was available for relapse, quality of life or service use.
Authors' conclusions
Although 22 trials could be included in this review, the evidence provided is limited and mostly not relevant to clinicians or consumers. Overall, although there was low risk of attrition and selective data reporting bias within the trials, the trials themselves were not adequately powered and need more substantial follow‐up periods. There is a need for larger trials with longer periods of follow‐up to be conducted. Outcomes should be meaningful for those with schizophrenia, and include measures of improvement and relapse (not just rating scale scores), functioning and quality of life and acceptability and, importantly, safety data.
Plain language summary
Antioxidants as add‐on treatment for people with schizophrenia
Antioxidants are substances that protect cells from the damage caused by unstable molecules known as free radicals (causing oxidative stress). It is well known that adding antioxidant‐rich fruits and vegetables to your daily diet will strengthen your ability to fight infection and disease. There is recent evidence that progressive brain changes take place as schizophrenia unfolds. Among many possible explanations, oxidative stress may be one of the factors contributing to the deterioration of the brain and its grey matter, leading to difficulties in people’s thinking and everyday functioning. The aim of this review is to evaluate the effect of antioxidants as an add‐on treatment to standard antipsychotic medication. In particular, by reducing (or lessening) psychotic episodes and core symptoms, and preventing relapse
Searches for randomised trials were run in 2010 and 2012, review authors found 22 relevant trials that randomised a total of 2041 people with schizophrenia. The trials compared the effects of taking a variety of antioxidants (allopurinol, Ginkgo biloba, N‐acetyl cysteine (NAC), selegiline, vitamins C and E) compared with placebo. Most results showed no real differences between the antioxidants and placebo although there was evidence Ginkgo biloba had a positive effect on psychotic symptoms in the short term. The quality of this evidence was moderate.
However, overall, the trials suffered from a lack of real‐world outcomes, such as clinical response, rates of relapse, quality of life, functioning, safety and satisfaction or acceptability of treatment. Adverse effects were also poorly reported with some studies not providing any data for adverse effects.
Ginkgo biloba and NAC emerged from the trials as the most promising, so should have priority in the design of future trials that are larger, longer and better reported than the 22 studies available at the present time.
Ben Gray, Senior Peer Researcher, McPin Foundation: http://mcpin.org/
Summary of findings
Summary of findings for the main comparison. Adjunctive antioxidants for schizophrenia versus placebo.
| Adjunctive antioxidants for schizophrenia | ||||||
| Patient or population: people with schizophrenia Settings: Inpatients and outpatients Intervention: Adjunctive antioxidants | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Adjunctive antioxidants | |||||
| Global state: improvement, short term PANSS Follow‐up: 6 to 8 weeks | Study population | RR 0.77 (0.53 to 1.12) | 229 (3 studies) | ⊕⊕⊝⊝ low1,2 | ‐ | |
| 1000 per 1000 | 690 per 1000 (360 to 1000) | |||||
| Moderate | ||||||
| 1000 per 1000 | 690 per 1000 (360 to 1000) | |||||
| Total PANSS scores, short term PANSS Follow‐up: 6 to 12 weeks | The mean total PANSS scores in the intervention groups was 6.0 lower (10.53 lower to 1.65 lower) | 584 (7 studies) | ⊕⊝⊝⊝ very low1,2,3 | ‐ | ||
| Total BPRS scores, short term BPRS Follow‐up: 6 to 16 weeks | The mean total BPRS scores in the intervention groups was 3.2 lower (5.63 lower to 0.78 lower) | 843 (8 studies) | ⊕⊕⊝⊝ low1,3,4 | ‐ | ||
| General functioning ‐ short term GAS Follow‐up: 6 weeks | The mean general functioning ‐ short term in the intervention groups was 1.11 lower (8.07 lower to 5.86 higher) | 52 (2 studies) |
⊕⊕⊝⊝ low1,5 | ‐ | ||
| General functioning ‐ medium term GAS Follow‐up: 24 weeks | The mean general functioning ‐ medium term in the intervention groups was 2.84 higher (2.09 lower to 7.77 higher) | 135 (1 study) | ⊕⊕⊕⊕ high | ‐ | ||
| Leaving the study early, short term Follow‐up: 6 to 16 weeks | Study population | RR 0.73 (0.48 to 1.11) | 1584 (16 studies) | ⊕⊕⊕⊝ moderate1,4 | ‐ | |
| 96 per 1000 | 72 per 1000 (39 to 132) | |||||
| Moderate | ||||||
| 91 per 1000 | 68 per 1000 (37 to 126) | |||||
| Adverse effects: 1. Serious (any time point) ‐ any serious adverse effect Various methods | Study population | RR 0.65 (0.19 to 2.27) | 234 (3 studies) | ⊕⊕⊝⊝ low1 | ‐ | |
| 60 per 1000 | 39 per 1000 (11 to 137) | |||||
| Moderate | ||||||
| 59 per 1000 | 38 per 1000 (11 to 134) | |||||
1 Unclear process of allocation concealment in included studies 2 High heterogeneity 3 Difference in rating scale scores not necessarily reflects meaningful clinical change 4 Heterogeneity high, but reduced when results are grouped by specific agent tested 5 Very limited number of patients available
Background
Description of the condition
Schizophrenia is a severely debilitating and progressive illness. Although not especially highly prevalent, it carries a disproportionate share of illness‐related disability, partially due to its early age at onset, associated impact in functioning, and chronic course. This effect is even more devastating as the illness tends to be deteriorating, with increased disability and personal and societal burden (Berk 2009; Lieberman 2006). Furthermore, only a minority of this burden is currently averted with standard treatment (Rossler 2005).
There is accumulating evidence that progressive changes in brain structure and function take place as the disease unfolds (DeLisi 2008). Among many possible candidates, oxidative stress may be one of the mediators of neuroprogression, grey matter loss and subsequent cognitive and functional impairment (Dean 2009; Lieberman 2006; Wood 2009). Specifically, oxidative imbalance has been evidenced by the increased levels of 8‐OH deoxyguanosine, (an indicator of DNA damage and potentially of apoptotic (programmed cell death) events), protein carbonylation (leading to cellular dysfunction), and lipid peroxidation (potentially leading to alterations in membrane structure and permeability) shown in individuals with schizophrenia. Oxidative defences have also been shown to be impaired, including decreased glutathione (the primary antioxidant in the brain) levels, polymorphisms in gene pathways associated with oxidative defence and changes in other antioxidants including superoxide, dismutase, catalase and glutathione peroxidase (Wood 2009).
Description of the intervention
Antioxidants are exogenous or endogenous molecules that mitigate any form of oxidative stress or its consequences. They may act from directly scavenging free radicals to increasing anti‐oxidative defences (Uttara 2009). In this fashion, antioxidants with different mechanisms have been studied in different progressive illnesses (Berk 2009).
There is evidence that current treatments impact on oxidative pathways and may to some extent reverse pro‐oxidative states in schizophrenia. Second generation antipsychotics have been shown to have effects in neuroprotection, the existing literature, however, indicates that these treatments do not fully restore the deficits in antioxidant levels or restore levels of oxidants in schizophrenia (Lieberman 2005; Padurariu 2010; Wang 2008).
How the intervention might work
Oxidative stress occurs when there is an overproduction of free radicals or a deficiency in antioxidant defences (Wood 2009). This has had theoretical appeal to neurodegenerative disorders, since the brain is considered particularly vulnerable to oxidative damage. This process has been implicated in many psychiatric disorders, and most robust evidence of its importance comes from studies of schizophrenia (Ng 2008). The underlying mechanisms underpinning the process of disease neuroprogression and subsequent brain changes are incompletely understood. There is, however, some evidence pointing to central and peripheral pro‐oxidative changes, including lower oxidative defences and oxidative damage to proteins, lipids and DNA (Wood 2009).
As such, there has been interest in developing interventions aimed at restoring this oxidative balance beyond the benefits of antipsychotics in this direction (Dean 2009). Some work has been done investigating the modulation of antioxidants as a therapeutic target for the treatment of schizophrenia. Similarly, mechanisms of action are believed to vary between compounds. Omega‐3 for example, is proposed to have some direct antioxidant properties, but works primarily by protecting against oxidative attack by reinforcing lipid membranes and lipid‐associated structures (such as myelin). Alternatively, N‐acetyl cysteine (NAC) is believed to act predominantly on the glutathione pathway and may also modulate glutamate function (Dean 2009).
Why it is important to do this review
The investigation on oxidative stress in schizophrenia has increased exponentially in the past decade (Ng 2008). If antioxidants are to have a place in the treatment of this serious condition, the relevant and up‐to‐date information should be available to clinicians and investigators.
Objectives
To evaluate the effect of antioxidants as add‐on treatments to standard antipsychotic medication for improving acute psychotic episodes and core symptoms, and preventing relapse in people with schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised trials. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week. As the level of blindness has not been definitely linked to bias (Petiti 2000), studies with any level of blinding were eligible. Non‐English language was not an obstacle to inclusion.
Types of participants
Adults (18+ years) with schizophrenia or other types of schizophrenia‐like psychosis (e.g. schizophreniform and schizoaffective disorders), irrespective of diagnostic criteria used. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994).
We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible so if information was provided in the trials, we proposed to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and as to whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Antioxidants
Any pharmacologically active substance explicitly administered with the purpose of antioxidation.
2. Placebo
As antipsychotics are interventions with a large evidence‐base of efficacy, we included only add‐on studies. In these, participants already using a first line agent are randomised to an antioxidant or placebo in addition to their previous treatment. Although we required that participants were on antipsychotics, more 'naturalistic' studies including those on 'poly‐therapy' could be included, provided participants are randomised to placebo or an antioxidant. Alternatively, for maintenance studies they could be randomised to stay on the antioxidant or have it substituted for a placebo.
Types of outcome measures
We grouped outcomes into immediate (four weeks or less), short‐term (4‐12 weeks), medium‐term (13‐26 weeks) and long‐term (over 26 weeks).
Primary outcomes
1. Global state
1.1 No clinically important response as defined by the individual studies
For example, global impression less than much improved or less than 50% reduction on a rating scale.
Secondary outcomes
1. Leaving the studies early
1.1 Any reason, adverse events, or inefficacy of treatment
2. Global state
2.1 No clinically important change in global state (as defined by individual studies)
2.2 Relapse (as defined by the individual studies)
3. Mental state
3.1 No clinically important change in general mental state score
3.2 Average endpoint general mental state score
3.3 Average change in general mental state scores
3.4 No clinically important change in specific symptoms (positive symptoms of schizophrenia, negative symptoms of schizophrenia)
3.5 Average endpoint specific symptom score
3.6 Average change in specific symptom scores
4. General functioning
4.1 No clinically important change in general functioning
4.2 Average endpoint general functioning score
4.3 Average change in general functioning scores
5. Quality of life/satisfaction with treatment
5.1 No clinically important change in general quality of life
5.2 Average endpoint general quality of life score
5.3 Average change in general quality of life scores
6. Cognitive functioning
6.1 No clinically important change in overall cognitive functioning
6.2 Average endpoint of overall cognitive functioning score
6.3 Average change of overall cognitive functioning scores
7. Service use
7.1 Number of participants hospitalised
7.2 Duration of hospitalisation
8. Adverse effects
8.1 Number of participants with at least one adverse effect
8.2 Clinically important specific adverse effects (cardiac effects, death, movement disorders and associated effects, sedation, seizures, weight gain, effects on white blood cell count)
8.3 Average endpoint in specific adverse effects
8.4 Average change in specific adverse effects
9. Laboratory data
9.1 Change in tests of oxidative stress
9.2 Change in tests of antioxidant defences / potential
'Summary of findings table
We anticipated including the following short‐ or medium‐term outcomes in a Table 1.
1. Global state
1.1 Clinically significant response ‐ as defined by each of the studies
1.2 Relapse
2. Mental state
2.1 Clinically significant response in mental state ‐ as defined by each of the studies
3. Service utilisation outcome
3.1 Hospital admission
3.2 Days in hospital
4. Adverse effect
4.1 Any important adverse event
5. Quality of life
5.1 Improved to an important extent
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Trials Register
On January 08, 2015, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials using the following search strategy:
*Antioxidant* in Intervention Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected the reference lists of all retrieved articles, previous reviews and major text books of schizophrenia for additional trials.
2. Personal contact
If necessary, we contacted the authors of significant papers, as well as other experts in the field, and asked for their knowledge of further studies, published or unpublished, relevant to the review. We noted any responses in the Characteristics of included studies
Data collection and analysis
Selection of studies
Review authors PVM and OD independently inspected citations from the searches and identified relevant abstracts. MB independently re‐inspected a random 20% sample of the citations and abstracts to ensure reliability. PVM and OD obtained full reports of the abstracts meeting the review criteria and inspected them. Again, MB re‐inspected a random 20% of these full reports in order to ensure reliable selection. There were no disputes whether any particular study should be included.
Data extraction and management
1. Extraction
Review author PVM extracted data from all included studies. In addition, to ensure reliability, OD independently extracted data from all studies. We made attempts to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
2. Management
2.1 Forms
We extracted data extracted onto standard, pre‐designed, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, in Description of studies we noted if this is the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We planned to combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to relevant data before inclusion.
Change data
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We would have presented and entered change data into statistical analyses.
Endpoint data
Endpoint scores on scales often have a finite start and end point and we applied the following rules:
standard deviations (SDs) and means are reported in the paper or obtainable from the authors;
when a scale starts from the finite number zero, the standard deviation (SD), when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution) (Altman 1996);
if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) (Kay 1986), which can have values from 30 to 210), the calculation described above would be modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
Had we found endpoint data from studies of fewer than 200 participants we planned to present these as other data within the data and analyses section rather than entering such data into statistical analyses.
Skewed endpoint data pose less of a problem when looking at means if the sample size is large and if we found skewed endpoint data from studies of over 200 participants we entered such data from these studies into statistical analyses.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month), although no such occasions appeared.
2.6 Conversion of continuous data to binary data
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
We entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome adjunctive antioxidant. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we reported data where the left of the line indicates an unfavourable outcome.
2.8 Multiple doses
When a study investigating a number of fixed doses of an antioxidant was included, we used the method described in section 16.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to combine data from multiple groups.
Assessment of risk of bias in included studies
Again review authors PM and OD worked independently to assess risk of bias using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. There was no disagreement in quality assessment.
The level of risk of bias was noted in both the text of the review in the Table 1, and in Figure 1 and Figure 2.
1.
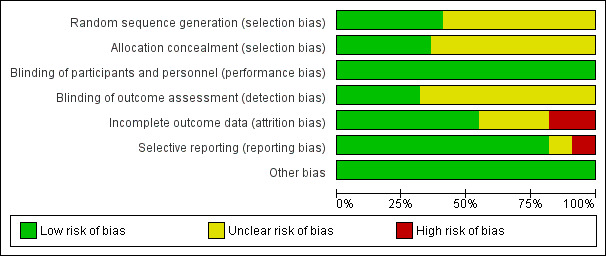
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.
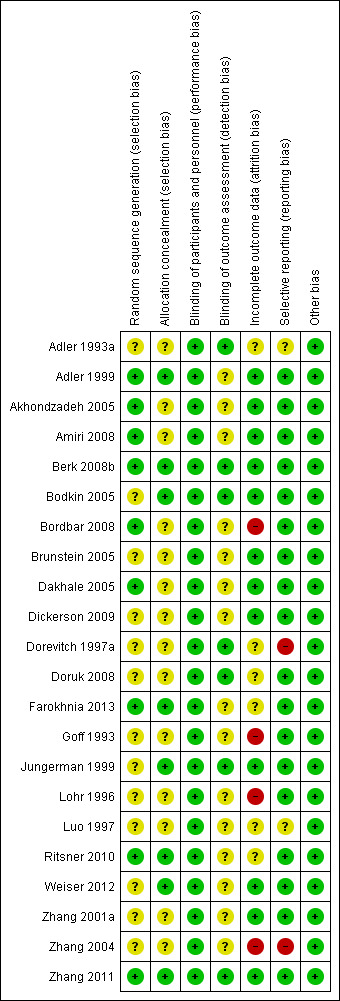
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The Number Needed to Treat/Harm (NNT/H) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic, both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference SMD) unless necessary. However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
There were no cluster‐randomised trials included in the review.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase, the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we included randomised cross‐over trials, but only used data form the first phase.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary, we simply added these and combined within the two‐by‐two table.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 40% of data be unaccounted for, we would not reproduce these data or use them within analyses.
2. Binary
In the case where attrition for a binary outcome is between 0% and 40% and where these data are not clearly described, data would be presented on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). Those leaving the study early are all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. A sensitivity analysis was planned but was not undertaken since very few studies reported on binary improvement and those that reported used ITT analysis.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 40% and completer‐only data were reported, we reproduced these.
3.2 Standard deviations
There were no instances where standard deviations were not reported.
3.3 Last observation carried forward (LOCF)
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within study reports. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data were used in the trial, if less than 50% of the data had been assumed, we reproduced these data and indicated that they are the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups existed, they are discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We inspected all studies for clearly outlying methods which we had not predicted would arise. We discussed such methodological outliers if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. a 'P' value from Chi2 test, or a confidence interval for I2). an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there are 10 or fewer studies, or where all studies were of similar sizes.
Data synthesis
Although the choice of model for meta‐analysis remains controversial (Freemantle 1999), a random‐effects model (DerSimonian 1986), which assumes that studies analysed actually comes from pool of hypothetical studies, was used in this meta‐analysis. The random‐effects methods does put added weight onto the smaller of the studies ‐ those that may be most prone to bias. We nevertheless favoured using a random‐effects model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup
We conducted subgroup analyses regarding the specific antioxidant used.
We were interested in making sure that information is as relevant to the current care of people with schizophrenia and broad clinical groups (see Types of participants). We intended to be able to present these data according to current clinical state, stage and for people with particular problems. This was not possible, however, because none of the studies reported relevant subgroups.
2. Investigation of heterogeneity
2.1 Unanticipated heterogeneity
If inconsistency was high, we reported this.
2.2 Anticipated heterogeneity
We did not anticipate important levels of heterogeneity.
Sensitivity analysis
1. Quality
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation, but there were no such instances.
2. Handling of missing observations
Where assumptions have had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only.
3. Statistical model for the synthesis
For the primary outcome, if there was substantial heterogeneity, we checked if using a fixed‐effect model substantively changed the final result.
Results
Description of studies
Please see Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification.
Results of the search
Electronic searches in 2010, 2012 and 2015 identified 147 references with no additional records identified through personal contact or inspecting references of retrieved articles. After duplicates were removed, we screened 109 records. Forty‐six potentially relevant records were obtained and scrutinised and 24 of these reports either did not meet the inclusion criteria and had to be excluded (seven) or were unclear and are awaiting classification pending further information (17). Twenty‐two trials are therefore included (Figure 3).
3.
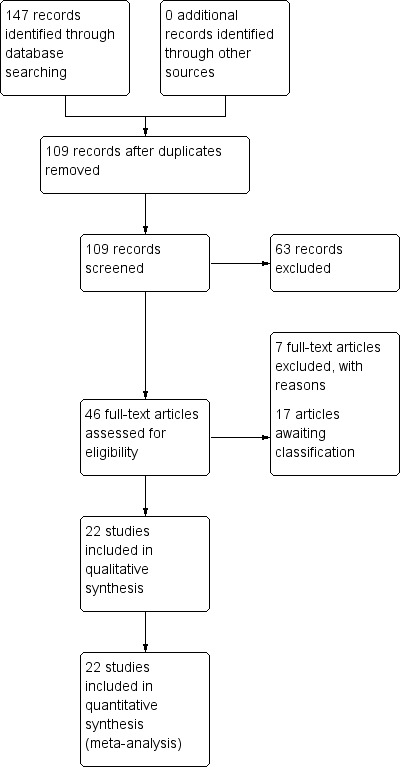
Study flow diagram.
Included studies
1. Allocation
All included studies were stated to be randomised and double‐blinded. All were, as per protocol, add‐on trials.
2. Design
All included studies had a parallel longitudinal design. Only two of 22 studies used a cross‐over design with two periods and had data available from the first period (Brunstein 2005; Dorevitch 1997a).
3. Duration
All but three studies were short‐term studies. Berk 2008b was a medium‐term study (24 weeks) and Adler 1993a and Adler 1999 were long‐term studies. We made the decision to group Luo 1997 with short‐term studies, since it was the only study with a 16‐week duration and it was similar to other ginkgo studies in this review in terms of diagnosis and other methodological aspects. Overall, the median follow‐up was eight weeks.
4. Participants
Participants total 2041 people. The number of participants ranged from 16 to 545 (median 46). Nineteen of the 22 studies reported using Diagnostic and Statistical Manual (DSM‐III, DSM‐IIIR or DSM‐IV) criteria to diagnose schizophrenia. Six studies also required patients to present with tardive dyskinesia for inclusion. Of the studies that reported the patients' sex, men were over‐represented in most; slightly over two men to one woman.
5. Setting
Six trials were conducted in the USA, four in China, four in Iran, three in Israel and one each in Brazil, India, Romania, Australia and Turkey. Nine trials were conducted in inpatient and 11 in outpatient settings, two in mixed settings.
6. Interventions
6.1 Adjunctive antioxidants
Four studies employed adjunctive Ginkgo biloba extract, from 120 to 360 mg/day (Doruk 2008; Luo 1997; Zhang 2001a; Zhang 2011); five selegiline, from 5 to 15 mg/day (Amiri 2008; Bodkin 2005; Bordbar 2008; Goff 1993; Jungerman 1999); four allopurinol, from 300 to 600 mg/day (Akhondzadeh 2005; Brunstein 2005; Dickerson 2009; Weiser 2012); five, vitamin E (Adler 1993a; Adler 1999; Dorevitch 1997a; Lohr 1996; Zhang 2004), from 1200 IU to 1600 IU/day; two N‐acetyl cysteine (NAC) (Berk 2008b; Farokhnia 2013); and one each dehydroepiandrosterone (DHEA) (Ritsner 2010) and vitamin C (Dakhale 2005).
6.2 Comparison group
An adjunctive placebo was used as a comparison group, with no further details given in most studies.
7. Outcomes
7.1 General
Although we intended to report on a host of clinically relevant outcomes, only a few clinical outcomes, generally derived from rating scales and quite limited oxidative stress data were reported in the included studies.
Some outcomes were presented in graphs, P values or a statement of significant or non‐significant difference. This made it impossible to use data for synthesis.
Notably, very few studies (only Berk 2008b, Brunstein 2005 and Dickerson 2009) reported any clinically meaningful measure of clinical response.
7.2 Scales used to measure psychopathology, functioning and adverse effects
The two scales most employed to measure symptoms were the Brief Psychiatric Rating Scale (BPRS) and the Positive and Negative Syndrome Scale (PANSS).
7.2.1 BPRS (Overall 1962 The BPRS is a one‐page, 16‐ or 18‐item rating scale developed more than 50 years ago. It assesses a range of psychotic and affective symptoms. The original purpose was rapid evaluation of clinical change irrespective of origin in the broad range of psychiatric patients.
7.2.2 PANSS (Kay 1986). The PANSS originated from a need to reduce the heterogeneity of schizophrenia, based on a positive–negative dichotomy for explaining and understanding variability in the aetiology of schizophrenia, treatment and prognosis.
7.2.3 SANS (Andreasen 1990) Also often employed to determine positive and negative symptoms were Andreasen's Schedule for the Assessment of Positive Symptoms (SAPS) and Schedule for the Assessment of Negative Symptoms .
7.2.4 GAS (Endicott 1976) and GAF (Jones 1995).
The Global Assessment of Functioning (GAF), and its older version, the Global Assessment Scale (GAS) were used in two studies. These are general functioning scales developed to be clinically useful and easy to administer, but with the caveat that they do not separate functioning from symptoms.
7.2.5 TESS (Guy 1991)
The Treatment Emergent Symptom Scale (TESS) measures side‐effects severity. It can be used as a total score or as six subscores that gather symptoms related to different body systems.
7.3 Laboratory methods used to measure oxidative stress and antioxidant defences
Three studies reported on laboratory methods. Dakhale 2005 reported serum malondialdehyde (MDA) levels, according to methods described by Satoh (Satoh 1978). MDA is an oxidative stress marker, the final product of peroxidation processes and oxidative damage in cells (Valko 2007). Zhang 2001a and Zhang 2004 reported on superoxide dysmutase (SOD, an antioxidant enzyme) serum levels with radioimmunoassay, described in Zhang 2001a.
Excluded studies
We excluded seven studies, either because they did not measure any outcome of interest (Dorevitch 1997; Elkashef 1990; Suresh 2007), were not adjunctive or randomised trials (Bhavani 1962; Kapur 1991), randomised participants to combined interventions (Rees 1951) or participants were not diagnosed with schizophrenia (Eranti 1998).
Awaiting classification
There are 17 trials awaiting classification pending provision of more information.
Ongoing studies
There are currently no ongoing studies we are aware of.
Risk of bias in included studies
Figure 1 and Figure 2 are graphical overviews of the risk of bias in the included studies.
Allocation
All 22 included studies were randomised but not all described how this was achieved, and were not generally clear about allocation concealment. Only nine (Adler 1999; Akhondzadeh 2005; Amiri 2008; Berk 2008b; Bordbar 2008; Dakhale 2005; Farokhnia 2013; Ritsner 2010; Zhang 2011) gave further details about sequence generation. Eight studies (Adler 1999; Berk 2008b; Bodkin 2005; Farokhnia 2013; Jungerman 1999; Ritsner 2010; Weiser 2012; Zhang 2011) reported a satisfactory method of concealment.
Blinding
All studies were reported as double‐blind trials with participants blinded to treatment. Not all studies were clear about rater blinding. One study is in the Studies awaiting classification section because it is unclear whether it was a single‐blind or open‐label study (Xu 2002).
Incomplete outcome data
Several studies reported using some form of intention‐to‐treat analysis to account for incomplete data, usually last observation carried forward (LOCF) (Adler 1999; Akhondzadeh 2005; Amiri 2008; Berk 2008b; Bodkin 2005; Brunstein 2005; Dakhale 2005; Dickerson 2009; Weiser 2012; Zhang 2001a; Zhang 2011). One study explicitly reported all participants completed the trial (Jungerman 1999). Two studies were classified as high risk for stating using a completer analysis (Bordbar 2008; Goff 1993) or failing to report any attrition (Lohr 1996; Zhang 2004).
Selective reporting
Selective reporting was not a major issue for this review. Most studies reported primary outcomes of interest. We classified two studies as high risk primarily because of endpoint rating scale data not reported (Dorevitch 1997a; Zhang 2004).
Other potential sources of bias
We found no other obvious potential sources of bias.
Effects of interventions
See: Table 1
COMPARISON 1: ADJUNCTIVE ANTIOXIDANTS versus PLACEBO
1.1 Global state: 1. No clinically important response (20% improvement PANSS)
Studies tended not to include categorical measures of response (only three out of the 20 included studies). Only two short‐term studies and one medium‐term study reported clinical response as a 20% reduction in the PANSS. These results did not demonstrate an advantage for adjunctive antioxidants 3 RCTS, n = 229, risk ratio (RR) 0.77, 95% confidence interval (CI) 0.53 to 1.12 Analysis 1.1),
1.1. Analysis.
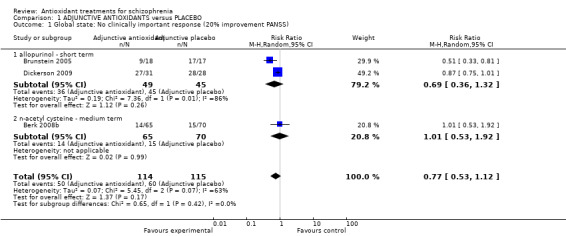
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 1 Global state: No clinically important response (20% improvement PANSS).
1.1.1 Allopurinol: PANSS improvement: short term
In this subgroup we found two relevant trials (n = 94). There was no significant difference between adjunctive allopurinol and placebo (RR 0.69 95% CI 0.36 to 1.32). This subgroup had important levels of heterogeneity (Chi2 = 7.36; df = 1; P = 0.007; I2 = 86%).
1.1.2 N‐acetyl cysteine (NAC): PANSS improvement: medium term
In this subgroup we only found one relevant trial (n = 135). There was no significant difference between adjunctive NAC and placebo (RR 1.01 95% CI 0.53 to 1.92).
1.2 Leaving the study early:
1.2.1 Short term (all antioxidants)
Use of any adjunctive antioxidant did not change the risk of leaving the study early in the short term (16 RCTs, 1584 people, RR 0.73, 95% CI 0.48 to 1.11 Analysis 1.2). There was also no significant difference in retention in the medium term (1 RCT, n = 140, RR 0.99, 95% CI 0.67 to 1.48 Analysis 1.3), or in two long‐term studies (2 RCTS, n = 195, RR 0.90 95% CI 0.59 to 1.35 Analysis 1.4 ).
1.2. Analysis.
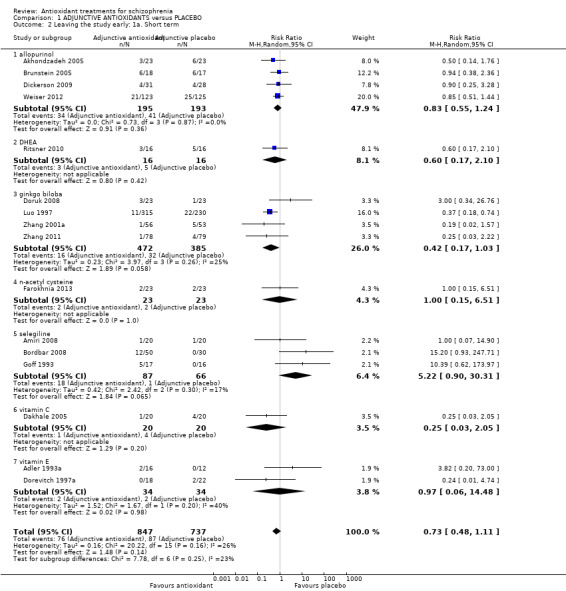
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 2 Leaving the study early: 1a. Short term.
1.3. Analysis.
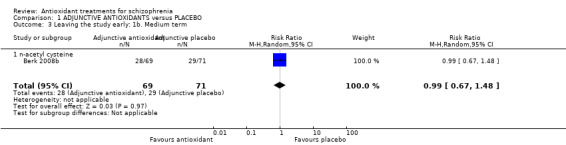
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 3 Leaving the study early: 1b. Medium term.
1.4. Analysis.
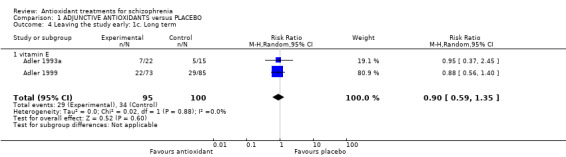
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 4 Leaving the study early: 1c. Long term.
Data could be subgrouped into individual antioxidant treatments (Figure 1).
1.2.1.1 Allopurinol
In this subgroup we found four relevant trials (n = 388). There was no significant difference between adjunctive allopurinol and placebo (RR 0.83 95% CI 0.55 to 1.24).
1.2.1.2 Ginkgo biloba
In this subgroup we found four relevant trials (n = 857). There was no significant difference between adjunctive ginkgo and placebo (RR 0.42 95% CI 0.17 to 1.03).
1.2.1.3 Selegiline
In this subgroup we found three relevant trials (n = 153). There was no significant difference between adjunctive selegiline and placebo (RR 5.22 95% CI 0.90 to 30.31).
1.2.1.4 Vitamin C
In this subgroup we only found one relevant trial (n = 40) (Dakhale 2005). There was no significant difference between adjunctive vitamin C and placebo (RR 0.25 95% CI 0.03 to 2.05).
1.2.1.5 Vitamin E
In this subgroup we found two relevant trials (n = 68). There was no significant difference between adjunctive vitamin E and placebo (RR 0.97 95% CI 0.06 to 14.48) This subgroup had moderate levels of heterogeneity (Chi2 = 1.67; df = 1; P = 0.197; I2 = 40%).
1.2.1.6 NAC
In this subgroup we only found one relevant trial (n = 46) (Farokhnia 2013). There was no significant difference between adjunctive NAC and placebo (RR 1.00 95% CI 0.15 to 6.51).
1.2.1.7 DHEA
In this subgroup we only found one relevant trial (n = 32) (Ritsner 2010). There was no significant difference between adjunctive DHEA and placebo (RR 0.60 95% CI 0.17 to 2.10).
1.2.2 Medium term (NAC)
Medium‐term data for leaving the study early were provided by only one relevant trial (n = 140) (Berk 2008b). There was no significant difference between adjunctive NAC and placebo (RR 0.99 95% CI 0.67 to 1.48, Analysis 1.3).
1.2.3 Long term (Vitamin E)
Two trials (n = 195) provided long‐term data for leaving the study early. There was no significant difference between adjunctive vitamin E and placebo (RR 0.9 95% CI 0.59 to 1.35, Analysis 1.4).
1.3 Mental state
Thirteen studies measured mental state using the BPRS or PANSS. Pooling individual antioxidant short‐term data favoured antioxidants, however there were substantial levels of heterogeneity. Results varied for individual antioxidants. Medium‐term data were presented by one trial only, as were long‐term data; both studies found no significant advantage of antioxidant over placebo at these time points.
1.3.1 Mental state : 1a. General ‐ short‐term BPRS total endpoint
Eight studies provided short‐term BPRS endpoint data; a favourable effect for antioxidants was found (n = 843, mean difference (MD) ‐3.20,95% CI ‐5.63 to ‐0.78) I2 = 74%). There was substantial heterogeneity for this outcome.(Chi² = 27.41, df = 7 (P = 0.0003); I² = 74%). Analysis 1.5
1.5. Analysis.
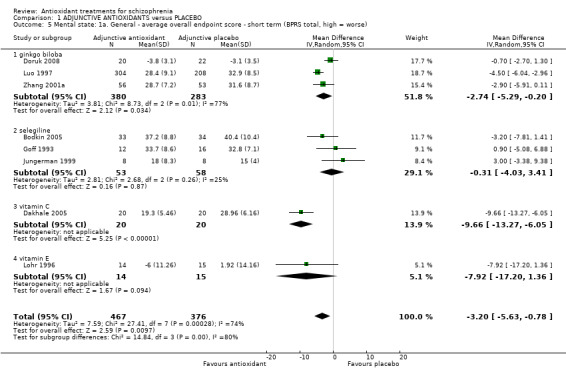
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 5 Mental state: 1a. General ‐ average overall endpoint score ‐ short term (BPRS total, high = worse).
Overall, data were favourable, however, when data were subgrouped into individual antioxidant treatments, the results varied.
1.3.1.1 Ginkgo biloba
Data from three short‐term studies showed a favourable effect for Ginkgo biloba (3 RCTS, n = 663, MD ‐2.74, 95% CI ‐5.29 to ‐0.20). This subgroup had important levels of heterogeneity (Chi2 = 8.73; df = 2; P = 0.013; I2 = 77%)
1.3.1.2 Selegiline
No effect was found for selegiline (3 RCTS, n = 111, MD ‐0.31, 95% CI ‐4.03 to 3.41)
1.3.1.3 Vitamin C
One small study showed a favourable effect for vitamin C (n = 40, MD ‐9.66, 95% CI ‐ 13.27 to ‐6.05)
1.3.1.4 Vitamin E
A favourable effect was not found for vitamin E (n = 29, MD ‐7.92, 95% CI ‐17.20 to 1.36)
1.3.2 Mental state: 1b. General ‐ short‐term PANSS
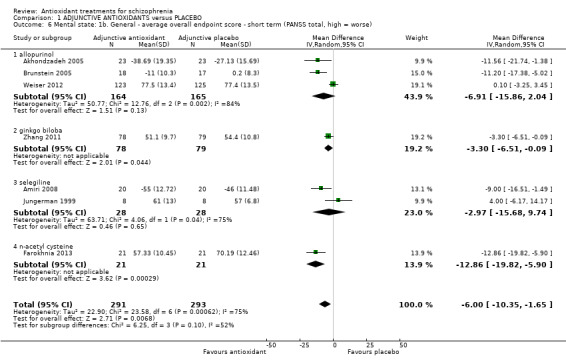
Overall, there was substantial heterogeneity regarding differences of any antioxidant and placebo in mental state scores for the PANSS (Chi² = 16.96, df = 5 (P = 0.005); I² = 71%). Data showed a favourable effect for antioxidants (7 RCTS, n = 584, MD ‐6.00, 95% CI ‐10.35 to ‐1.65) (Analysis 1.6). Data could be subgrouped into individual antioxidants.
1.6. Analysis.
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 6 Mental state: 1b. General ‐ average overall endpoint score ‐ short term (PANSS total, high = worse).
1.3.2.1 Allopurinol
In this subgroup we found three relevant trials (n = 329). There was no significant difference between adjunctive antioxidants and placebo (MD ‐6.91 95% CI ‐15.86 to 2.04). This subgroup had important levels of heterogeneity (Chi2 = 12.76; df = 2; P = 0.002; I2 = 84%).
1.3.2.2 Ginkgo biloba
In this subgroup we only found one relevant trial (n = 157). There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐3.30 95% CI ‐6.51 to ‐0.09).
1.3.2.3 Selegiline
In this subgroup we found two relevant trials (n = 56). There was no significant difference between adjunctive antioxidants and placebo (MD ‐2.97 95% CI ‐15.68 to 9.74, Analysis 1.6). This subgroup had important levels of heterogeneity (Chi2 = 4.06; df = 1; P = 0.044; I2 = 75%).
1.3.2.4 NAC
In this subgroup we only found one relevant trial (n = 42). There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐12.86 95% CI ‐19.82 to ‐5.90).
1.3.3 Mental state: 1c. General ‐ medium‐term PANSS
Only one relevant trial (randomising NAC) presented medium‐term mental state data (n = 135). There was no significant difference between adjunctive antioxidants and placebo (MD ‐1.71 95% CI ‐7.55 to 4.13, Analysis 1.7).
1.7. Analysis.
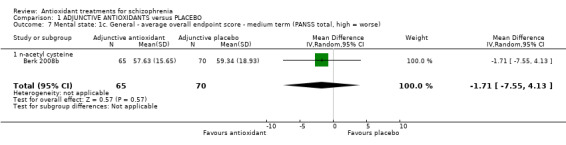
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 7 Mental state: 1c. General ‐ average overall endpoint score ‐ medium term (PANSS total, high = worse).
1.3.4 Mental state: 1d. General ‐ long‐term BPRS
Again, only one trial (randomising vitamin E) presented long‐term mental state data. There was no significant difference between adjunctive antioxidants and placebo (n = 104, MD 1.20 95% CI ‐2.47 to 4.87, Analysis 1.8).
1.8. Analysis.
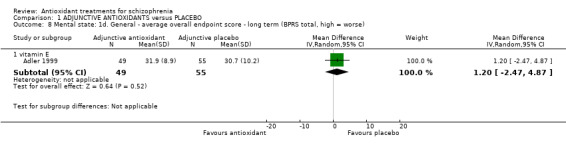
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 8 Mental state: 1d. General ‐ average overall endpoint score ‐ long term (BPRS total, high = worse).
1.3.5 Mental state: 2a. Specific ‐ short‐term PANSS negative symptoms
Nine studies presented specific short‐term scores for negative symptoms using the PANSS. Overall, results showed no effect for antioxidants over placebo (9 RCTs, n = 653, MD ‐0.38, 95% CI ‐0.77 to 0.00) (Analysis 1.9). Significant levels of heterogeneity were found (i2 = 79%)
1.9. Analysis.
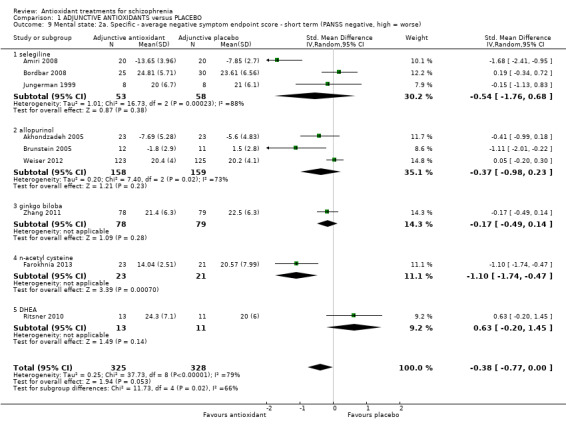
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 9 Mental state: 2a. Specific ‐ average negative symptom endpoint score ‐ short term (PANSS negative, high = worse).
Data could be subgrouped into individual antioxidants.
1.3.5.1 Selegiline
In this subgroup we found three relevant trials (n = 111). There was a statistically significant difference between adjunctive antioxidants and placebo (standardised mean difference (SMD) ‐0.54 95% CI ‐1.76 to 0.68). This subgroup had important levels of heterogeneity (Chi2 = 16.73; df = 2; P = 0.0; I2 = 88%).
1.3.5.2 Allopurinol
In this subgroup we found three relevant trials (n = 317). There was a statistically significant difference between adjunctive antioxidants and placebo (SMD ‐0.37 95% CI ‐0.98 to 0.23). This subgroup had important levels of heterogeneity (Chi2 = 7.4; df = 2; P = 0.025; I2 = 73%).
1.3.5.3 Ginkgo biloba
In this subgroup we only found one relevant trial (n = 157) (Zhang 2011). There was a statistically significant difference between adjunctive antioxidants and placebo (SMD ‐0.17 95% CI ‐0.49 to 0.14).
1.3.5.4 NAC
In this subgroup we only found one relevant trial (n = 44) (Farokhnia 2013). There was a statistically significant difference between adjunctive antioxidants and placebo (SMD ‐1.10 95% CI ‐1.74 to ‐0.47).
1.3.5.5 DHEA
In this subgroup we only found one relevant trial (n = 24) (Ritsner 2010). There was no significant difference between adjunctive antioxidants and placebo (SMD 0.63 95% CI ‐ 0.20 to 1.45,Analysis 1.9).
1.3.6. Mental state: 2b. Specific ‐ short‐term SANS negative
Three studies presented short‐term data for negative symptoms using the SANS scale. All three studies were assessing Ginkgo biloba compared with placebo. There was a statistically significant difference between the antioxidant Ginkgo biloba and placebo (3 RCTs, n = 667, MD ‐7.46 95% CI ‐12.46 to ‐2.46, Analysis 1.10). This subgroup had important levels of heterogeneity (Chi2 = 8.32, df = 2 (P = 0.02); I² = 76%).
1.10. Analysis.
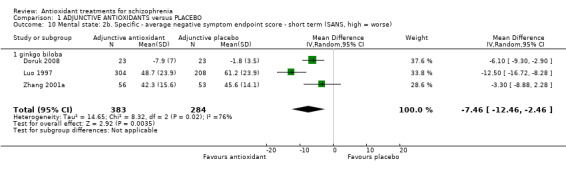
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 10 Mental state: 2b. Specific ‐ average negative symptom endpoint score ‐ short term (SANS, high = worse).
1.3.7 Mental state: 2c. Specific ‐ medium‐term PANSS negative
1.11.1 NAC
Only one NAC trial (n = 135) presented medium‐term data for negative symptoms. There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐2.33 95% CI ‐4.24 to ‐0.42, Analysis 1.11).
1.11. Analysis.
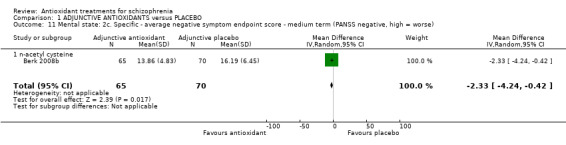
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 11 Mental state: 2c. Specific ‐ average negative symptom endpoint score ‐ medium term (PANSS negative, high = worse).
1.3.8 Mental state: 2d. Specific ‐ short‐term PANSS positive
Eight trials presented short‐term data for positive symptoms using the PANSS scale. No effect was found (8 RCTs, n = 611, MD ‐0.96 95% CI ‐2.50 to 0.58). Substantial levels of heterogeneity were found (I2 = 83 %)(Analysis 1.12).
1.12. Analysis.
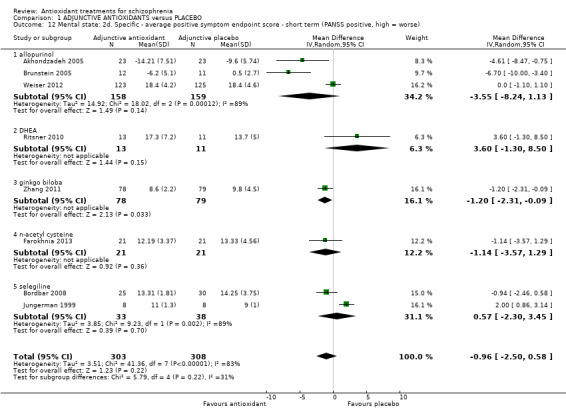
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 12 Mental state: 2d. Specific ‐ average positive symptom endpoint score ‐ short term (PANSS positive, high = worse).
Data could be subgrouped into individual antioxidants
1.3.8.1 Selegiline
In this subgroup we found two relevant trials (n = 71). There was no significant difference between adjunctive antioxidants and placebo (MD 0.57 95% CI ‐2.30 to 3.45). This subgroup had important levels of heterogeneity (Chi2 = 9.23; df = 1; P = 0.002; I2 = 89%).
1.3.8.2 Allopurinol
In this subgroup we found three relevant trials (n = 317). There was no significant difference between adjunctive antioxidants and placebo (MD ‐3.55 95% CI ‐8.24 to 1.13). This subgroup had important levels of heterogeneity (Chi2 = 18.02; df = 2; P = 0.0; I2 = 89%).
1.3.8.3 Ginkgo biloba
In this subgroup we only found one relevant trial (n = 157) (Zhang 2011). There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐1.20 95% CI ‐2.31 to ‐0.09).
1.3.8.4 NAC
In this subgroup we only found one relevant trial (n = 42) (Farokhnia 2013). There was no significant difference between adjunctive antioxidants and placebo (MD ‐1.14 95% CI ‐ 3.57 to 1.29).
1.3.8.5 DHEA
In this subgroup we only found one relevant trial (n = 24) (Ritsner 2010). There was no significant difference between adjunctive antioxidants and placebo (MD 3.60 95% CI ‐ 1.30 to 8.50 ).
1.3.9 Mental state: 2e. Specific ‐ short‐term SAPS positive
Two Ginkgo biloba trials (n = 151) presented short‐term data using the SAPS positive. There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐5.06 95% CI ‐7.52 to ‐2.59, Analysis 1.13).
1.13. Analysis.
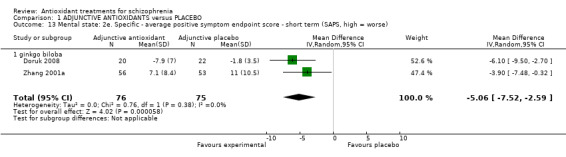
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 13 Mental state: 2e. Specific ‐ average positive symptom endpoint score ‐ short term (SAPS, high = worse).
1.3.10 Mental state: 2f. Specific ‐ medium‐term PANSS positive
1.3.10.1 NAC
Only one NAC trial (n = 135) presented medium‐term positive mental state data (Berk 2008b). There was no significant difference between adjunctive antioxidants and placebo (MD 0.47 95% CI ‐1.43 to 2.37, Analysis 1.14).
1.14. Analysis.
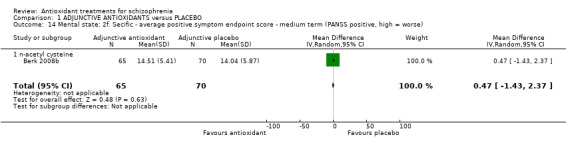
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 14 Mental state: 2f. Secific ‐ average positive symptom endpoint score ‐ medium term (PANSS positive, high = worse).
1.4 General functioning
1.4.1 General functioning: 1a. General ‐ short‐term GAS total endpoint
Two small studies presented equivocal short‐term data for general functioning (2 RCTs, n = 52, MD ‐1.11 95% CI ‐8.07 to 5.86) (Analysis 1.15).
1.15. Analysis.
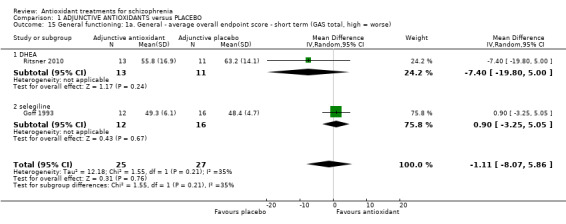
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 15 General functioning: 1a. General ‐ average overall endpoint score ‐ short term (GAS total, high = worse).
Data could be subgrouped into individual antioxidants.
1.4.1.1 Selegiline
In this subgroup we only found one relevant trial (n = 28) (Goff 1993). There was no significant difference between the antioxidant selegiline and placebo (MD 0.90 95% CI ‐3.25 to 5.05).
1.4.1.2 DHEA Again, equivocal data were found for the antioxidant DHEA in one trial (n = 24, MD ‐7.40 95% CI ‐19.80 to 5.00)
1.4.2 General functioning: 1b. General ‐ medium‐term GAS total endpoint
One trial comparing NAC with placebo presented medium‐term general functioning data(Berk 2008b). There was no significant difference between adjunctive NAC and placebo (n = 135, MD 2.84 95% CI ‐2.09 to 7.77 ) (Analysis 1.16).
1.16. Analysis.
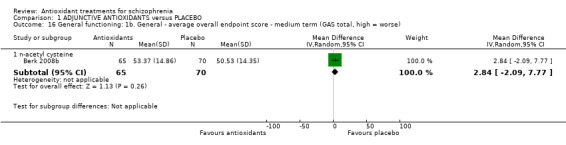
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 16 General functioning: 1b. General ‐ average overall endpoint score ‐ medium term (GAS total, high = worse).
1.4.3 General functioning; 1c. General ‐ long‐term GAS total endpoint
In this subgroup we only found one relevant trial (n = 104) (Adler 1999). There was no significant difference between adjunctive vitamin E and placebo (MD 0.10 95% CI ‐5.60 to 5.80) (Analysis 1.17).
1.17. Analysis.
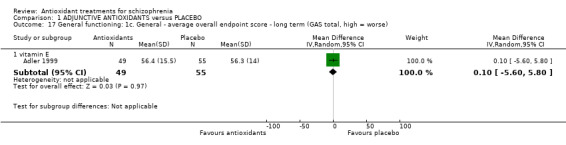
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 17 General functioning: 1c. General ‐ average overall endpoint score ‐ long term (GAS total, high = worse).
1.5 Adverse effects:
Adverse effects were usually not reported in a usable way in most trials, with the exceptions of Akhondzadeh 2005; Amiri 2008; Berk 2008b; Brunstein 2005; Dickerson 2009; and Zhang 2001a. These trials reported different measures of adverse effects, including specific adverse effects, serious adverse effects and scale data. See Analysis 1.18; Analysis 1.19; Analysis 1.20.
1.18. Analysis.
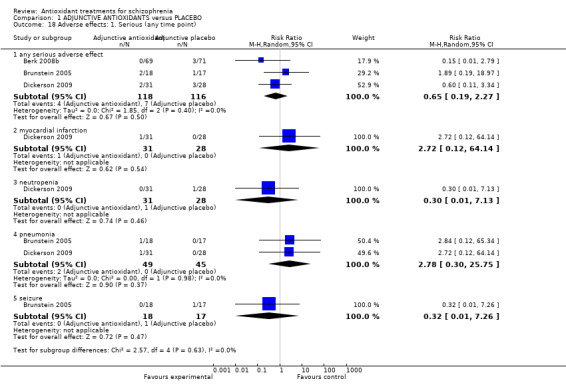
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 18 Adverse effects: 1. Serious (any time point).
1.19. Analysis.
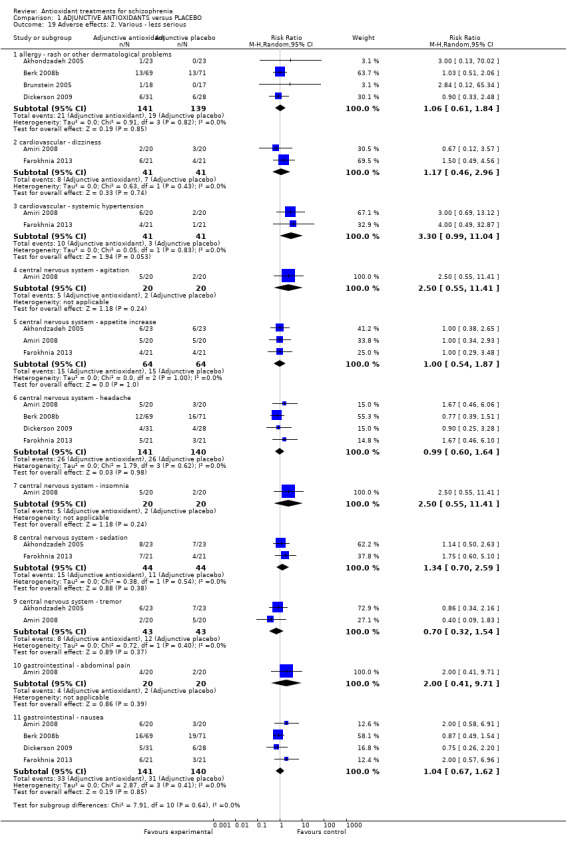
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 19 Adverse effects: 2. Various ‐ less serious.
1.20. Analysis.
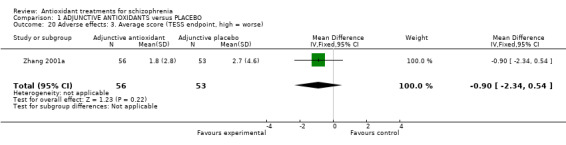
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 20 Adverse effects: 3. Average score (TESS endpoint, high = worse).
1.5.1 Serious adverse effects (any time point)
1.5.1.1 Any 'serious' adverse effect
Three relevant trials recorded 'any serious adverse effect) (n = 234). There was no significant difference between adjunctive antioxidants and placebo (3 RCTS, n = 234, RR 0.65 95% CI 0.19 to 2.27).
1.5.1.2 Myocardial infarction
In this subgroup we only found one relevant trial (n = 59) (Dickerson 2009). There was no significant difference between adjunctive antioxidants and placebo (RR 2.72 95% CI 0.12 to 64.14).
1.5.1.3 Neutropenia
In this subgroup we only found one relevant trial (n = 59) (Dickerson 2009). There was no significant difference between adjunctive antioxidants and placebo (RR 0.30 95% CI 0.01 to 7.13).
1.5.1.4 Pneumonia
In this subgroup we found two relevant trials (n = 94). There was no significant difference between adjunctive antioxidants and placebo (RR 2.78 95% CI 0.30 to 25.75)
1.5.1.5 Seizure
In this subgroup we only found one relevant trial (n = 35) (Brunstein 2005). There was no significant difference between adjunctive antioxidants and placebo (RR 0.32 95% CI 0.01 to 7.26, Analysis 1.18).
1.5.2 Adverse effects 2. Various ‐ less 'serious'
1.5.2.1 Allergy ‐ rash or other dermatological problems
In this subgroup we found four relevant trials (n = 280). There was no significant difference between adjunctive antioxidants and placebo (RR 1.06 95% CI 0.61 to 1.84).
1.5.2.2 Cardiovascular ‐ dizziness
In this subgroup we found two relevant trials (n = 82) (Amiri 2008; Farokhnia 2013). There was no significant difference between adjunctive antioxidants and placebo (RR 1.17 95% CI 0.46 to 2.96).
1.5.2.3 Cardiovascular ‐ systemic hypertension
IIn this subgroup we found two relevant trials (n = 82) (Amiri 2008; Farokhnia 2013). There was no significant difference between adjunctive antioxidants and placebo (RR 3.30 95% CI 0.99 to 11.04)
1.5.2.4 Central nervous system ‐ agitation
In this subgroup we only found one relevant trial (n = 40) (Amiri 2008). There was no significant difference between adjunctive antioxidants and placebo (RR 2.50 95% CI 0.55 to 11.41, Analysis 1.19).
1.5.2.5 Central nervous system ‐ appetite increase
In this subgroup we found three relevant trials (n = 128). There was no significant difference between adjunctive antioxidants and placebo (RR 1.00 95% CI 0.54 to 1.87, Analysis 1.19).
1.5.2.6 Central nervous system ‐ headache
In this subgroup we found four relevant trials (n = 281). There was no significant difference between adjunctive antioxidants and placebo (RR 0.99 95% CI 0.60 to 1.64, Analysis 1.19).
1.5.2.7 Central nervous system ‐ insomnia
In this subgroup we only found one relevant trial (n = 40) (Amiri 2008). There was no significant difference between adjunctive antioxidants and placebo (RR 2.50 95% CI 0.55 to 11.41, Analysis 1.19).
1.5.2.8 Central nervous system ‐ sedation
In this subgroup we only found two relevant trials (n = 88) (Akhondzadeh 2005; Farokhnia 2013). There was no significant difference between adjunctive antioxidants and placebo (RR 1.34 95% CI 0.70 to 2.59, Analysis 1.19).
1.5.2.9 Central nervous system ‐ tremor
In this subgroup we found two relevant trials (n = 86). There was no significant difference between adjunctive antioxidants and placebo (RR 0.70 95% CI 0.32 to 1.54, Analysis 1.19).
1.5.2.10 Gastrointestinal ‐ abdominal pain
In this subgroup we only found one relevant trial (n = 40) (Amiri 2008). There was no significant difference between adjunctive antioxidants and placebo (RR 2.00 95% CI 0.41 to 9.71).
1.5.2.11 Gastrointestinal ‐ nausea
In this subgroup we found four relevant trials (n = 281). There was no significant difference between adjunctive antioxidants and placebo (RR 1.04 95% CI 0.67 to 1.62).
1.5.3 Adverse effects: 3. Average scores ‐ endpoint total TESS
For this outcome, we only found one relevant trial (n = 109) (Zhang 2001a). There was a statistically significant difference between adjunctive antioxidants and placebo (MD ‐0.90 95% CI ‐2.34 to 0.54, Analysis 1.20).
1.6 Laboratory data (serum tests)
1.6.1 MDA levels
In this subgroup we only found one relevant trial (n = 40) (Dakhale 2005). There was a statistically significant difference between adjunctive antioxidants and placebo (SMD ‐2.18 95% CI ‐2.98 to ‐1.38, Analysis 1.21).
1.21. Analysis.
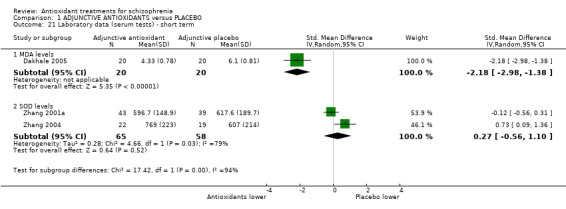
Comparison 1 ADJUNCTIVE ANTIOXIDANTS versus PLACEBO, Outcome 21 Laboratory data (serum tests) ‐ short term.
1.6.2 SOD levels
In this subgroup we found two relevant trials (n = 123). There was no significant difference between adjunctive antioxidants and placebo (SMD 0.27 95% CI ‐0.56 to 1.10, Analysis 1.21). This subgroup had important levels of heterogeneity (Chi2 = 4.66; df = 1; P = 0.031; I2 = 79%).
Discussion
Summary of main results
With 22 trials, including over 2000 participants, studies testing adjunctive drugs with antioxidant potential are mostly preliminary. They are characterised by a short follow‐up period, mostly underpowered, do not offer data on the most clinically meaningful outcomes and are of a relatively low quality. Efficacy data on psychopathology scale scores is highly heterogenous, which can be explained by the use of drugs with a different pharmacological profile. Safety appeared not to be a concern for the studies, with most studies not reporting relevant adverse events at all. All these issues make it difficult to interpret the effect of adding an antioxidant to the standard treatment of schizophrenia.
Regarding particular drugs, Ginkgo biloba is the most studied agent, overwhelmingly in Chinese participants, who were all inpatients. In these studies, ginkgo significantly reduced rating scale scores in the short term, mostly for positive symptoms. In the Chinese trials, there was also a lower rate of patients leaving the study early when compared with placebo.
A very recent independently run, high quality and adequately powered trial comparing allopurinol with placebo (Weiser 2012) changed an initial impression of benefit given by three previous small trials (Akhondzadeh 2005; Brunstein 2005; Dickerson 2009). Although allopurinol was as tolerable as placebo, at present there is no evidence for benefit in terms of improvement, total scale scores or positive or negative symptoms of schizophrenia.
For selegiline, data from four fairly small trials and of low quality do not suggest any benefit in terms of scale scores. One study suggested worsening of positive symptoms (Jungerman 1999), and pooled data showed a trend towards lower tolerability for adjunctive selegiline.
We identified two studies comparing adjunctive N‐acetyl cysteine (NAC) with placebo, one short term (Farokhnia 2013) and one medium term (Berk 2008b). The data indicate a benefit in terms of negative symptoms, with no difference in positive symptoms, functioning or tolerability. One small short‐term trial and two long‐term trials investigated vitamin E effects, albeit they were mostly interested in tardive dyskinesia outcomes (Adler 1993a; Adler 1999; Dorevitch 1997a). They were not able to establish meaningful differences between adjunctive vitamin E and placebo in terms of symptoms of tolerability. One small trial showed a very large benefit for adjunctive vitamin C over placebo in terms of total psychopathology scores (Dakhale 2005).
Overall completeness and applicability of evidence
1. Completeness
Albeit we were able to identify a number of trials, with two trials being appreciably larger than the rest (Luo 1997; Weiser 2012), we were not able to identify our primary outcomes of interest. Most data pertained to rating scale scores, with hardly any measure of clinical improvement, functioning or quality of life.
2. Applicability
Trials recruited both inpatients and outpatients who would be recognisable in everyday care. All trials in China using adjunctive ginkgo, however, were conducted exclusively in inpatients. The interventions are accessible. Outcomes, nonetheless, were wanting in clinical applicability.
Quality of the evidence
Most trials included were very small and underpowered, with an overall poor quality of reporting. Some data were highly heterogenous. There was a recent trend, however, for better standards of reporting (for instance, see Berk 2008b; Zhang 2004; Zhang 2011). The largest trial by far (Luo 1997), however, did not report allocation concealment. Grading of the quality of evidence can be seen in the Table 1.
Potential biases in the review process
1. Missing studies
We made a significant effort to identify relevant trials. However, these are small studies generally and it is likely that we have failed to identify other studies of limited power. A few studies were reported only in abstract form and we were not able to obtain data from the authors. We were able to obtain unpublished data for two studies (Berk 2008b; Bordbar 2008), and for two Chinese papers (Luo 1997; Xu 2002); we were also able to obtain help from a native speaker.
Agreements and disagreements with other studies or reviews
There is a published Cochrane review on vitamin E for tardive dyskinesia (TD) (Soares‐Weiser 2011). This was mostly interested in TD, not schizophrenia, and only on vitamin E, so there is no particular disagreement or overlap between the two reviews.
One recent systematic review investigated the use of Ginkgo biloba for schizophrenia (Singh 2010). This review had a more liberal approach to inclusion (it included one trial that did not use a placebo comparison and an open‐label trial). The direction of results are generally the same and the authors conclude more data are needed to disentangle ginkgo's pharmacological properties.
Authors' conclusions
Implications for practice.
1. For clinicians
The many difficulties with included studies, amongst them low quality and sample size and a lack of clinically meaningful outcomes, preclude us from giving general suggestions regarding the use of adjunctive substances with antioxidant potential in people with schizophrenia. Another concern was the poor reporting of adverse events throughout the trials.
Specifically, however, the data indicate the adjunctive use of allopurinol should not be encouraged at this point, since an adequately powered trial failed to show any benefit
2. For people with schizophrenia
Although there is preliminary data that suggests substances with antioxidant potential may be of use, definitive studies are lacking. This means there is substantial uncertainty on the potential benefit and risks these different compounds carry.
3. For policy makers
Currently,there is not enough good‐quality evidence to suggest the use of allopurinol for schizophrenia is appropriate.
Implications for research.
As different substances have different pharmacological properties and clinical effects, it is more useful to think about these medications not as antioxidants, but as distinct drugs with antioxidant potential. As such, it is preferable to discuss implications for each drug, and not for antioxidants in general. There is medium‐quality evidence for the adjunctive efficacy of Ginkgo biloba, with relatively large numbers of people randomised to receive it, mostly in China. These studies are also short‐term ones and lack measures that are most useful to patients and clinicians. The only trial we located on adjunctive N‐acetyl cysteine (NAC), on the other hand, was of high quality and measured outcomes over a longer period of time and suggested benefit for negative symptoms of schizophrenia.
Overall, available data suggest that real‐world studies, adequately powered with more substantial follow‐up periods need to be conducted if clinicians and people with schizophrenia are to adopt adjunctive antioxidants in everyday practice. Table 2 suggests a design for such a study. Outcomes should be meaningful for those with schizophrenia, and include measures of improvement and relapse (not just rating scale scores), functioning and quality of life and acceptability and, importantly, safety data. Because of the various features discussed, ginkgo and NAC emerged from the trials located as the most promising interventions, and should have priority in the design of further trials.
1. Suggested design of future trial.
| Methods | Allocation: centralised sequence generation with table of random numbers or computer‐generated code, stratified by severity of illness, sequence concealed till interventions assigned. Blinding: those recruiting and assigning participants, those assessing outcomes, all blind to allocated group, blinding could be tested. Duration: minimum of 24 weeks |
| Participants | Diagnosis: schizophrenia, if operational criteria used these should be in the context of routine care. N = 450*. Age: adults. Sex: men and women. Setting: anywhere. |
| Interventions | 1. Adjunctive placebo 2. Adjunctive antioxidant (preferably Ginkgo biloba EGb or NAC) |
| Outcomes | Quality of life: healthy days,** SF‐36***. Service outcomes: days in hospital, time attending psychiatric outpatient clinic. Satisfaction with care: patients/carers. Global state: CGI.*** Mental state: CGI. Social functioning: to include occupational status. Adverse effects: including mortality. Economic data. |
| Notes | * size of study to detect a 10% difference in improvement with 80% certainty. ** Primary outcome. *** If scales are used to measure outcome then there should be binary cut off points, defined before study starts, of clinically important improvement. |
CGI ‐ Clinical Global Impression; NAC ‐ N‐acetyl cysteine; SF 36 ‐ Short form 36
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods sections of their reviews. We have used this text as the basis of what appears here and adapted it as required.
Appendices
Appendix 1. Previous searches
1.1 Search in 2012
1.1.1 Electronic searches
1.1.1.1 Cochrane Schizophrenia Group Trials Register (November 2010)
The register was searched by the Trial Search co‐ordinator of the Cochrane Schizophrenia Group using the phrase
[(*vitamin C* OR *ascorbic acid* OR *vitamin E* OR *alpha‐tocopherol* OR *selegiline* OR *deprenyl* OR *n‐acetyl cysteine* OR *n‐acetyl‐l‐cysteine* OR *n‐acetylcysteine* OR *acetylcysteine* OR *superoxide dismutase* OR *SOD * OR *dehydroepiandrosterone* OR *antioxidant*) in title, abstract and index terms of REFERENCE and Intervention of STUDY]
The Cochrane Schizophrenia Group’s Registry of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, EMBASE, MEDLINE, PsycINFO, PubMed, and registries of Clinical Trials) and their monthly updates, handsearches, grey literature, and conference proceedings. The searches do not have language limitation (for details of databases searched by the Schizophreia Group please see their Group Module).
1.1.2 Searching other resources
1.1.2.1 Reference searching
We inspected the reference lists of all retrieved articles, previous reviews and major text books of schizophrenia were inspected for additional trials.
1.1.2.2 Personal contact
If necessary we contacted the authors of significant papers, as well as other experts in the field, and asked for their knowledge of further studies, published or unpublished, relevant to the review. We noted any responses in the Characteristics of included studies
1.2 Search in 2012
1.2.1 CENTRAL 2012
Review authors ran an additional search through the CENTRAL database in November 2012 using the above search term.
Data and analyses
Comparison 1. ADJUNCTIVE ANTIOXIDANTS versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: No clinically important response (20% improvement PANSS) | 3 | 229 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.53, 1.12] |
| 1.1 allopurinol ‐ short term | 2 | 94 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.36, 1.32] |
| 1.2 n‐acetyl cysteine ‐ medium term | 1 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.92] |
| 2 Leaving the study early: 1a. Short term | 16 | 1584 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.48, 1.11] |
| 2.1 allopurinol | 4 | 388 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.55, 1.24] |
| 2.2 DHEA | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.17, 2.10] |
| 2.3 ginkgo biloba | 4 | 857 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.17, 1.03] |
| 2.4 n‐acetyl cysteine | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.51] |
| 2.5 selegiline | 3 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 5.22 [0.90, 30.31] |
| 2.6 vitamin C | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.03, 2.05] |
| 2.7 vitamin E | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.06, 14.48] |
| 3 Leaving the study early: 1b. Medium term | 1 | 140 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.67, 1.48] |
| 3.1 n‐acetyl cysteine | 1 | 140 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.67, 1.48] |
| 4 Leaving the study early: 1c. Long term | 2 | 195 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.59, 1.35] |
| 4.1 vitamin E | 2 | 195 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.59, 1.35] |
| 5 Mental state: 1a. General ‐ average overall endpoint score ‐ short term (BPRS total, high = worse) | 8 | 843 | Mean Difference (IV, Random, 95% CI) | ‐3.20 [‐5.63, ‐0.78] |
| 5.1 ginkgo biloba | 3 | 663 | Mean Difference (IV, Random, 95% CI) | ‐2.74 [‐5.29, ‐0.20] |
| 5.2 selegiline | 3 | 111 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐4.03, 3.41] |
| 5.3 vitamin C | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐9.66 [‐13.27, ‐6.05] |
| 5.4 vitamin E | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐7.92 [‐17.20, 1.36] |
| 6 Mental state: 1b. General ‐ average overall endpoint score ‐ short term (PANSS total, high = worse) | 7 | 584 | Mean Difference (IV, Random, 95% CI) | ‐6.00 [‐10.35, ‐1.65] |
| 6.1 allopurinol | 3 | 329 | Mean Difference (IV, Random, 95% CI) | ‐6.91 [‐15.86, 2.04] |
| 6.2 ginkgo biloba | 1 | 157 | Mean Difference (IV, Random, 95% CI) | ‐3.30 [‐6.51, ‐0.09] |
| 6.3 selegiline | 2 | 56 | Mean Difference (IV, Random, 95% CI) | ‐2.97 [‐15.68, 9.74] |
| 6.4 n‐acetyl cysteine | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐12.86 [‐19.82, ‐5.90] |
| 7 Mental state: 1c. General ‐ average overall endpoint score ‐ medium term (PANSS total, high = worse) | 1 | 135 | Mean Difference (IV, Random, 95% CI) | ‐1.71 [‐7.55, 4.13] |
| 7.1 n‐acetyl cysteine | 1 | 135 | Mean Difference (IV, Random, 95% CI) | ‐1.71 [‐7.55, 4.13] |
| 8 Mental state: 1d. General ‐ average overall endpoint score ‐ long term (BPRS total, high = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 vitamin E | 1 | 104 | Mean Difference (IV, Random, 95% CI) | 1.20 [‐2.47, 4.87] |
| 9 Mental state: 2a. Specific ‐ average negative symptom endpoint score ‐ short term (PANSS negative, high = worse) | 9 | 653 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.77, 0.00] |
| 9.1 selegiline | 3 | 111 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐1.76, 0.68] |
| 9.2 allopurinol | 3 | 317 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.98, 0.23] |
| 9.3 ginkgo biloba | 1 | 157 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.49, 0.14] |
| 9.4 n‐acetyl cysteine | 1 | 44 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐1.74, ‐0.47] |
| 9.5 DHEA | 1 | 24 | Std. Mean Difference (IV, Random, 95% CI) | 0.63 [‐0.20, 1.45] |
| 10 Mental state: 2b. Specific ‐ average negative symptom endpoint score ‐ short term (SANS, high = worse) | 3 | 667 | Mean Difference (IV, Random, 95% CI) | ‐7.46 [‐12.46, ‐2.46] |
| 10.1 ginkgo biloba | 3 | 667 | Mean Difference (IV, Random, 95% CI) | ‐7.46 [‐12.46, ‐2.46] |
| 11 Mental state: 2c. Specific ‐ average negative symptom endpoint score ‐ medium term (PANSS negative, high = worse) | 1 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.33 [‐4.24, ‐0.42] |
| 11.1 n‐acetyl cysteine | 1 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.33 [‐4.24, ‐0.42] |
| 12 Mental state: 2d. Specific ‐ average positive symptom endpoint score ‐ short term (PANSS positive, high = worse) | 8 | 611 | Mean Difference (IV, Random, 95% CI) | ‐0.96 [‐2.50, 0.58] |
| 12.1 allopurinol | 3 | 317 | Mean Difference (IV, Random, 95% CI) | ‐3.55 [‐8.24, 1.13] |
| 12.2 DHEA | 1 | 24 | Mean Difference (IV, Random, 95% CI) | 3.60 [‐1.30, 8.50] |
| 12.3 ginkgo biloba | 1 | 157 | Mean Difference (IV, Random, 95% CI) | ‐1.20 [‐2.31, ‐0.09] |
| 12.4 n‐acetyl cysteine | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐1.14 [‐3.57, 1.29] |
| 12.5 selegiline | 2 | 71 | Mean Difference (IV, Random, 95% CI) | 0.57 [‐2.30, 3.45] |
| 13 Mental state: 2e. Specific ‐ average positive symptom endpoint score ‐ short term (SAPS, high = worse) | 2 | 151 | Mean Difference (IV, Random, 95% CI) | ‐5.06 [‐7.52, ‐2.59] |
| 13.1 ginkgo biloba | 2 | 151 | Mean Difference (IV, Random, 95% CI) | ‐5.06 [‐7.52, ‐2.59] |
| 14 Mental state: 2f. Secific ‐ average positive symptom endpoint score ‐ medium term (PANSS positive, high = worse) | 1 | 135 | Mean Difference (IV, Random, 95% CI) | 0.47 [‐1.43, 2.37] |
| 14.1 n‐acetyl cysteine | 1 | 135 | Mean Difference (IV, Random, 95% CI) | 0.47 [‐1.43, 2.37] |
| 15 General functioning: 1a. General ‐ average overall endpoint score ‐ short term (GAS total, high = worse) | 2 | 52 | Mean Difference (IV, Random, 95% CI) | ‐1.11 [‐8.07, 5.86] |
| 15.1 DHEA | 1 | 24 | Mean Difference (IV, Random, 95% CI) | ‐7.40 [‐19.80, 5.00] |
| 15.2 selegiline | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐3.25, 5.05] |
| 16 General functioning: 1b. General ‐ average overall endpoint score ‐ medium term (GAS total, high = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 16.1 n‐acetyl cysteine | 1 | 135 | Mean Difference (IV, Random, 95% CI) | 2.84 [‐2.09, 7.77] |
| 17 General functioning: 1c. General ‐ average overall endpoint score ‐ long term (GAS total, high = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 17.1 vitamin E | 1 | 104 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐5.60, 5.80] |
| 18 Adverse effects: 1. Serious (any time point) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 any serious adverse effect | 3 | 234 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.19, 2.27] |
| 18.2 myocardial infarction | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 2.72 [0.12, 64.14] |
| 18.3 neutropenia | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.01, 7.13] |
| 18.4 pneumonia | 2 | 94 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [0.30, 25.75] |
| 18.5 seizure | 1 | 35 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 7.26] |
| 19 Adverse effects: 2. Various ‐ less serious | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 allergy ‐ rash or other dermatological problems | 4 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.61, 1.84] |
| 19.2 cardiovascular ‐ dizziness | 2 | 82 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.96] |
| 19.3 cardiovascular ‐ systemic hypertension | 2 | 82 | Risk Ratio (M‐H, Random, 95% CI) | 3.30 [0.99, 11.04] |
| 19.4 central nervous system ‐ agitation | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.55, 11.41] |
| 19.5 central nervous system ‐ appetite increase | 3 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.54, 1.87] |
| 19.6 central nervous system ‐ headache | 4 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.60, 1.64] |
| 19.7 central nervous system ‐ insomnia | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.55, 11.41] |
| 19.8 central nervous system ‐ sedation | 2 | 88 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.70, 2.59] |
| 19.9 central nervous system ‐ tremor | 2 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.32, 1.54] |
| 19.10 gastrointestinal ‐ abdominal pain | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.41, 9.71] |
| 19.11 gastrointestinal ‐ nausea | 4 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.67, 1.62] |
| 20 Adverse effects: 3. Average score (TESS endpoint, high = worse) | 1 | 109 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐2.34, 0.54] |
| 21 Laboratory data (serum tests) ‐ short term | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 21.1 MDA levels | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | ‐2.18 [‐2.98, ‐1.38] |
| 21.2 SOD levels | 2 | 123 | Std. Mean Difference (IV, Random, 95% CI) | 0.27 [‐0.56, 1.10] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adler 1993a.
| Methods | Allocation: randomised. Blindess: double blind: no further details. Duration: 12 weeks (36 week extension). Setting: outpatients, USA. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (36 of 37 at 36 weeks) (Diagnostic Criteria, Schooler and Kane). N = 28 (12 weeks), 40 (36 weeks). Sex: 2 female, 27 male. Age: average ˜ 60 yrs (SD ˜ 9.5). | |
| Interventions | 1. Vitamin E: dose increasing over 3 weeks to 1600 IU/day. N = 22.* 2. Placebo. N = 15.* Stable neuroleptic medication: dose average (CPE) vitamin E = 536 mg/day (SD 642); placebo = 921 mg/day (SD 1026). Compliance assessed by pill counts. | |
| Outcomes | Leaving the study early (at 12 weeks and at 36 weeks) Unable to use: adverse effects (safety data ‐ not reported) |
|
| Notes | No usable safety outcomes * 37 participants by 36 weeks |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random allocation, no further detail provided. |
| Allocation concealment (selection bias) | Unclear risk | Random allocation, no further detail provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Both rater and participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One participant left early and is not accounted for. |
| Selective reporting (reporting bias) | Unclear risk | Unclear since focuses on TD. |
| Other bias | Low risk | None obvious. |
Adler 1999.
| Methods | Allocation: randomised, co‐ordinated centrally, allocation with “biased coin” method, stratified by site, age, and baseline TD. Blindness: double‐blind, no further details. Duration: 1 year. Setting: outpatients, USA. Design: parallel. | |
| Participants | Diagnosis: schizophrenia, schizoaffective (DSM‐IV), and neuroleptic‐induced TD (Research Diagnostic Criteria). N = 158. Sex: 5F, 153M. Age: average ˜ 50 years (SD ˜ 10). | |
| Interventions | 1. Vitamin E: 1600 IU/day. N = 73.
2. Placebo. N = 85. Neuroleptic medication: not stable dose, average (CPE) vitamin E 380 mg/day (SD ˜110); placebo 458 mg/day (SD ˜433). |
|
| Outcomes | Leaving the study early. Mental state: BPRS endpoint score. General functioning: GAF endpoint score. Unable to use: safety data (not reported). |
|
| Notes | Not all patients leaving study early accounted for. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation. |
| Allocation concealment (selection bias) | Low risk | "Biased coin” method, stratified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis reported. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Akhondzadeh 2005.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 8 weeks. Setting: hospital, Iran. Design: parallel groups. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV) and PANSS > 59 criteria). N = 46. Sex: 13 female, 33 male. Age: average 33.8 (selegiline), 35.0 (placebo) | |
| Interventions | 1. Allopurinol: 300mg. N = 23. 2. Placebo. N = 23. Stable neuroleptic medication: haloperidol 15 mg. | |
| Outcomes | Leaving the study early. Mental state: PANSS change score. Adverse events: various specific effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | LOCF analysis reported. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Amiri 2008.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 8 weeks. Setting: in hospital, Iran. Design: parallel groups. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV) and PANSS > 59, PANSS negative >14 criteria). N = 40. Sex: 11F, 29M. Age: average 32.7 years (selegiline), 33.7 years (placebo). | |
| Interventions | 1. Selegiline: 10 mg/day. N = 20.
2. Placebo. N = 20. Stable risperidone dose: 6 mg/day. |
|
| Outcomes | Leaving the study early. Mental state: change scores in PANSS and PANSS negative subscale. Adverse events: various specific events. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated code. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical tablets. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "ITT performed". |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Berk 2008b.
| Methods | Allocation: randomised. Blindness: double‐blind: no further details. Duration: 24 weeks. Setting: mixed, but 95% were outpatients. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV) plus PANSS > 54. N = 140 Sex: 42F, 98M. Age: average ˜ 37 years. | |
| Interventions | 1. NAC (2 g/day). N = 69.
2. Placebo. N = 71. Stable neuroleptic medication: "usual antipsychotics medication". |
|
| Outcomes | Leaving the study early Mental state: PANSS (total, positive, negative, general endpoint score) Functioning: GAF endpoint score. Adverse events: various specific events. |
|
| Notes | Unpublished data obtained from authors | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple randomisation. |
| Allocation concealment (selection bias) | Low risk | Independent co‐ordinator generated sequence. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Raters blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 135 of 140 patients available for LOCF analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Bodkin 2005.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 12 weeks. Setting: outpatients, USA. Design: parallel groups. | |
| Participants | Diagnosis: schizophrenia (DSM‐III‐R and SANS >11 criteria). N = 67. Sex: 11F, 56M. Age: average 38.0 years (selegiline), 39.9 years (placebo). | |
| Interventions | 1. Selegiline (deprenyl): 10.mg/day. N = 33.
2. Placebo. N = 34. Stable neuroleptic medication: dose average (CPE) 727 (selegiline), 570 (placebo). |
|
| Outcomes | Leaving the study early. Mental state: SANS and BPRS endpoint scores. Unable to use: Adverse events: no usable data (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised no further details. |
| Allocation concealment (selection bias) | Low risk | "centrally based". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "drug and placebo in matched capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Investigators as well as subjects were completely blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Authors used random regression for main analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Bordbar 2008.
| Methods | Allocation: randomised, no further details Blindness: no further details. Duration: six weeks Setting: inpatients, Iran Design: parallel, three groups | |
| Participants | Diagnosis: schizophrenia (DSM‐IV) + PANSS negative >14. N = 80 (10 excluded). Sex: 45M, 45 F. Age: average ˜ 48 yrs | |
| Interventions | 1. Selegiline: 5 mg / day. N = 25 2. Selegiline: 10 mg/day. N = 25. 3. Placebo. N = 30. Stable neuroleptic medication: |
|
| Outcomes | Leaving the study early. Mental state: PANSS (negative and positive endpoint scores). Unable to use: safety (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Table of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | No further information. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Identical placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further information. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completer analysis, 10 patients excluded. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Brunstein 2005.
| Methods | Allocation: randomised, no further details. Blindness: double‐blind. Duration: six weeks. Setting: Brazil (mixed inpatients and outpatients) Design: cross‐over (but able to use data from first arm). |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV) and "moderately refractory". N = 35 (N = 23 for completer analysis). Sex: 9F, 14M (12 unclear because of completer analysis). Age: average 35.3 years (allopurinol), 42.3 years (placebo). | |
| Interventions | 1. Allopurinol: 600mg/day. N = 18. 2. Placebo. N = 17. Stable neuroleptic medication: dose average (CPE) 550 (allopurinol), 545(placebo). | |
| Outcomes | Global state: response (20% improvement in the PANSS). Leaving the study early. Mental state: PANSS change score. Adverse events: various and serious events. |
|
| Notes | Able to use LOCF analysis for total PANSS scores and response. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | LOCF analysis reported. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Dakhale 2005.
| Methods | Allocation: randomised. Blindness: double‐blind: no further details. Duration: 8 weeks Setting: outpatients, India. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 40. Sex: not reported. Age: average ˜ 38 yrs. | |
| Interventions | 1. Vitamin C (500 mg/day), N = 20. 2. Placebo. N = 20 Stable neuroleptic medication: atypical antipsychotics. | |
| Outcomes | Leaving the study early. Mental state: BPRS endpoint score. Laboratory data: serum MDA. Unable to use: adverse events (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Medicines identical in formulation. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis reported. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Dickerson 2009.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 8 weeks. Setting: outpatients, USA. Design: parallel groups. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV) and at least "moderate symptoms" criteria). N = 59. Sex: 20F, 39M. Age: average 43.1 years. | |
| Interventions | 1. Allopurinol: 600mg/day. N = 31. 2. Placebo. N = 28. Stable neuroleptic medication: antipsychotics as prescribed. | |
| Outcomes | Global state: response (at least 20% reduction in total PANSS score). Leaving the study early. Adverse events: serious and various events. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | LOCF used as imputation method. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Dorevitch 1997a.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 20 weeks total with cross‐over. Parallel period of 8 weeks. Setting: inpatients, Israel. Design: cross‐over study. | |
| Participants | Diagnosis: schizophrenia or schizoaffective (10%) (DSM‐IIR) and TD criteria). N = 40. Sex: 17F 23M. Age: average 64.4 years. | |
| Interventions | 1. Vitamin E: 1600 IU. N = 18. 2. Placebo. N = 22. Stable neuroleptic medication: antipsychotics as prescribed. | |
| Outcomes | Leaving the study early. Unable to use: adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Raters blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Two patients did not complete the study and no further details are given. |
| Selective reporting (reporting bias) | High risk | Not all expected outcomes reported. |
| Other bias | Low risk | None obvious. |
Doruk 2008.
| Methods | Allocation: randomised. Blindness: double‐blind Duration: 12 weeks. Setting: outpatients, Turkey. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (SCID), treatment‐resistant. N = 46. Sex: 17F, 29M. Age: average 30 years (Ginko EGb), 32 years (placebo). | |
| Interventions | 1. Ginkgo EGb, 120 mg/day. N = 23. 2. Placebo. N = 23. Stable neuroleptic medication: clozapine. | |
| Outcomes | Leaving the study early. Mental state: BRPS, SAPS, SANS change scores. Unable to use: adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random allocation, no details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinded rater. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Four people who left early were excluded from analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Farokhnia 2013.
| Methods | Allocation: randomised. Blindness: double‐blind Duration: 8 weeks. Setting: inpatients, Iran. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (SCID), PANSS >=60 and PANSS negative >=20. N = 46. Sex: 22F, 20M (4 unknown). Age: average 32 years (NAC), 33 years (placebo). | |
| Interventions | 1. NAC (2 g/day). N = 23.
2. Placebo. N = 23 Stable neuroleptic medication: risperidone (2‐6 mg/day). |
|
| Outcomes | Leaving the study early. Mental state: PANSS (total, negative, positive). Adverse events: various events. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Excel generated" |
| Allocation concealment (selection bias) | Low risk | "sealed envelope" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical containers" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Four people who left early were excluded from analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Goff 1993.
| Methods | Allocation: random, no further details. Blindess: double‐blind. Duration: six weeks. Setting: outpatients, USA. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (31/33), TD (Schooler & Kane). N = 33. Sex: 8F, 20M (5 unclear because of completer analysis). Age: average 48.7 (selegiline), 47.1 (placebo). | |
| Interventions | 1. Selegiline (deprenyl): 10 mg/day. N = 17. 2. Placebo. N = 16. Stable neuroleptic medication: dose average (CPE) 549 (selegiline), 683 (placebo). | |
| Outcomes | Leaving the study early. Mental state: BPRS endpoint score. General functioning: GAS endpoint score. Unable to use: adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical capsules given. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completer analysis used. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Jungerman 1999.
| Methods | Allocation: randomised, no further details Blindness: double‐blind Duration: eight weeks. Setting: outpatients, Israel. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 16. Sex: 8F, 8M. Age: average 36 years (SD 6 years). History: moderate residual symptoms. | |
| Interventions | 1. Selegiline (deprenyl): 15 mg/day. N = 8. 2. Placebo. N = 8. Stable neuroleptic medication: dose average (CPE) 360 (SD 180). | |
| Outcomes | Mental state: PANSS total endpoint score, PANSS positive endpoint score, PANSS negative endpoint score, BPRS total endpoint score Unable to use: adverse effects (not reported). |
|
| Notes | Information on allocation concealment and sequence generation obtained from authors. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "A randomization list prepared by a person who was not involvement in patient treatment or assessment". |
| Allocation concealment (selection bias) | Low risk | Identical drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Identical appearance". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Rater had no contact with medication preparation and allocation process". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participants left the study early. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Lohr 1996.
| Methods | Allocation: randomised, no further details. Double blind: no further details. Duration: eight weeks. Setting: outpatients, USA. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (29/35), bipolar disorder, unipolar depression (no specified criteria) and neuroleptic‐induced TD (Schooler and Kane criteria). N = 55 (20 patients left early, no data informed). Sex: 2F, 33M, 20 not informed. Age: average ˜ 50 years (SD ˜ 12). | |
| Interventions | 1. Vitamin E: 1600 IU/day. N = 27. 2. Placebo. N = 28. Stable neuroleptic medication for 2 weeks: dose average (CPE) vitamin E = 706 mg/day. (SD 680); placebo = 376 mg/day (SD 242). | |
| Outcomes | Mental state: BPRS endpoint score. Unable to use: leaving the study early (no group information reported). |
|
| Notes | Subgroup data for schizophrenia available for the BPRS. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completer analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Luo 1997.
| Methods | Allocation: randomised. Blindness: double‐blind, no further details. Duration: 16 weeks (36 week extension). Setting: inpatients, China. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (ICD‐10) plus SANS > 50 plus BPRS > 30. N = 545. Sex: 98F, 447M. Age: average ˜ 37 years. | |
| Interventions | 1. Ginkgo (EGb 360 mg/day), N = 315 2. Placebo. N = 230. Stable neuroleptic medication: mixed antipsychotics | |
| Outcomes | Leaving the study early. Mental state: BPRS endpoint score, SANS endpoint score. Unable to use: adverse effects (not reported). |
|
| Notes | Translation obtained from Prof. Yiming Wang | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Those who left early (33/545) were from excluded from analysis. |
| Selective reporting (reporting bias) | Unclear risk | No further details. |
| Other bias | Low risk | None obvious. |
Ritsner 2010.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 8 weeks. Setting: outpatients, Israel Design: parallel. | |
| Participants | Diagnosis: schizophrenia (n = 19) or schizoaffective disorder (n = 5) (DSM‐IV). N = 32. Sex: 6F, 18M (8 excluded not reported). Age: average ˜ 36 years. | |
| Interventions | 1. DHEA (400 mg/day), N =13 2. Placebo. N =11 . Stable neuroleptic medication: mixed antipsychotics. | |
| Outcomes | Leaving the study early. Mental state: PANSS. General functioning: GAF endpoint score. Unable to use: adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "random number generation" |
| Allocation concealment (selection bias) | Low risk | "allocation details coded and kept confidential" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Eight patients who discontinued the protocol followed up, but excluded from efficacy analysis. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Weiser 2012.
| Methods | Allocation: randomised. Blindess: double‐blind. Duration: 8 weeks. Setting: outpatients or inpatients, Romania. Design: parallel groups. | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (SCID). N = 248. Sex: 120 female, 128 male. Age: average 42.5 years. History: three psychotic episodes or continuously ill. | |
| Interventions | 1. Allopurinol: 600 mg/day. N = 123. 2. Placebo. N = 125. Stable neuroleptic medication: antipsychotics as prescribed. | |
| Outcomes | Leaving the study early. Mental state: PANSS endpoint score, PANSS positive endpoint, PANSS negative endpoint score. Unable to use: global state, adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Low risk | Central randomisation, individual bottles. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT and LOCF used. |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported. |
| Other bias | Low risk | None obvious. |
Zhang 2001a.
| Methods | Allocation: randomised, no further details. Blindess: double‐blind. Duration: 12 weeks. Setting: inpatients, China. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (SCID) and treatment resistant. N = 109 (82 for SOD analysis). Sex: 43F, 66M. Age: average 45 years. | |
| Interventions | 1. Ginkgo EGb, 360 mg/day. N = 56. 2. Placebo. N = 53. Stable neuroleptic medication: haloperidol, 16.8 mg/day (EGb), 16.5 mg/day (placebo). | |
| Outcomes | Leaving the study early. Mental state: BPRS endpoint score, SAPS endpoint score, SANS endpoint score. Adverse effects: TESS endpoint score. Laboratory data: SOD levels. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis reported. |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
Zhang 2004.
| Methods | Allocation: randomised, no further details. Blindness: double‐blind, no further details. Duration: 12 weeks. Setting: inpatients, China. Design: parallel. | |
| Participants | Diagnosis: schizophrenia (SCID) and neuroleptic‐induced TD (Schooler and Kane criteria). N = 41. Sex: 18F, 23M. Age: average ˜ 54 years (SD ˜ 10). | |
| Interventions | 1. Vitamin E: 1200 IU/day. N = 22. 2. Placebo. N = 19. Stable neuroleptic medication: dose average (CPE) vitamin E = 409.1 mg/day; placebo = 386.4 mg/day. | |
| Outcomes | Laboratory data: endpoint SOD. Unable to use: leaving the study early (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information on completion rate. |
| Selective reporting (reporting bias) | High risk | Not all expected outcomes reported. |
| Other bias | Low risk | None obvious. |
Zhang 2011.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 12 weeks. Setting: inpatients, China. Design: parallel. |
|
| Participants | Diagnosis: schizophrenia (SCID) and TD. N = 157. Sex: not reported. Age: average 45 years. | |
| Interventions | 1. Ginkgo EGb, 240 mg/day. N = 78. 2. Placebo. N = 79. Stable neuroleptic medication: most participants on clozapine, almost all on atypical antipsychotics (dose unclear). | |
| Outcomes | Leaving the study early. Mental state: PANSS (total, positive, negative) endpoint score. Unable to use: adverse effects (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer ‐ generated sequence". |
| Allocation concealment (selection bias) | Low risk | "Third ‐ party conducted allocation". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Identically appearing placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Raters blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "ITT analysis". |
| Selective reporting (reporting bias) | Low risk | Main outcomes reported. |
| Other bias | Low risk | None obvious. |
BPRS ‐ Brief Psychiatric Assessment Scale; CPE ‐ ;DHEA – dehydroepiandrosterone; DSM ‐ Diagnostic and Statistical Manual; EGb ‐ extract of Ginkgo biloba; GAF ‐ Global Assessment of Function; ICD ‐ International Classification of Diseases; ITT ‐ intention‐to‐treat; IU ‐ international units; LOCF ‐ last observation carried forward; MDA – malondialdehyde; NAC ‐ N‐acetyl cysteine; PANSS ‐ Positive and Negative Symptoms Scale; SANS ‐ Scale for the Assessment of Negative Symptoms; SCID – structured clinical interview for DSM disorders; SD – standard deviation; SOD ‐ superoxide dysmutase; TD ‐ tardive dyskinesia.
*
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bhavani 1962 | Allocation: randomised. Participants: inpatients, with schizophrenia. Intervention: vitamin C as monotherapy, not adjuvant. |
| Dorevitch 1997 | Allocation: randomised.
Participants: people with schizophrenia and tardive dyskinesia. Intervention: vitamin E (1200 IU) or placebo. Outcomes: no outcomes of interest (only tardive dyskinesia reported, which is not part of this review). |
| Elkashef 1990 | Allocation: randomised.
Participants: tardive dyskinesia. Intervention: vitamin E (1200 IU) or placebo. Outcomes: no outcomes of interest (only tardive dyskinesia reported). |
| Eranti 1998 | Allocation: randomised. Participants: not diagnosed with schizophrenia, but with an "acute transient psychosis". |
| Kapur 1991 | Allocation: randomised. Participants: chronic schizophrenia. Intervention: no parallel placebo group, riboflavin cross‐over trial. |
| Rees 1951 | Allocation: not randomised, matched pairs design. |
| Suresh 2007 | Allocation: randomised. Participants: schizophrenia (DSM‐IV) plus initial insomnia. Intervention: adjunctive melatonin (3‐12 mg) or placebo. Outcomes: no outcomes of interest evaluated (only effect on sleep outcomes reported). |
DSM ‐ Diagnostic and Statistical Manual
Characteristics of studies awaiting assessment [ordered by study ID]
Akhtar 1993.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: Four weeks. Setting: inpatients, India. Design: parallel groups. |
| Participants | Diagnosis: unclear, awaiting more information. N = 32. Sex: 14F, 18M. Age: average 53 (vitamin E), 57 (placebo) |
| Interventions | 1. Vitamin E. N = 17. 2. Placebo. N = 15. |
| Outcomes | Leaving the study early. |
| Notes |
Altman 1973.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 4 weeks. Setting: inpatients, USA. Design: parallel groups. |
| Participants | Diagnosis: unclear, awaiting more information. N = 151. Sex: unclear, awaiting more information. Age: unclear, awaiting more information. |
| Interventions | 1. Vitamin E. N = 75. 2. Placebo. N = 76. |
| Outcomes | Leaving the study early. |
| Notes |
Atmaca 2005.
| Methods | Allocation: randomised. Blindness: single‐blind. Duration: Eight weeks. Setting: outpatients, Turkey Design: parallel groups. |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 29. Sex: unclear, awaiting more information. Age: unclear, awaiting more information. |
| Interventions | 1. Ginkgo biloba. N = 15. 2. No Ginkgo (placebo?) : N = 14, awaiting more information. Antipsychotic medication: olanzapine ˜15 mg/day. |
| Outcomes | Leaving the study early. |
| Notes |
Ben‐Dor 1998.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: Six weeks. Setting: inpatients. Design: parallel groups. |
| Participants | Diagnosis: schizophrenia. N = 30. Sex: 14 female, 16 male. Age: average 53 years. History: chronic illness. |
| Interventions | 1. Vitamin E (2000 IU/day). N = unclear, awaiting more information.
2. Placebo. N = unclear, awaiting more information. Stable antipsychotic: haloperidol. |
| Outcomes | Leaving the study early. |
| Notes |
Egan 1992.
| Methods | Allocation: randomised. Blindess: double‐blind. Duration: Six weeks. Setting: mixed, USA. Design: cross‐over. |
| Participants | Diagnosis: schizophrenia, other psychoses. N = 21. Sex: unclear, awaiting more information. Age: average 54 years. |
| Interventions | 1. Vitamin E (1600 IU/day). N = 10 2. Placebo. N = 11. |
| Outcomes | Leaving the study early. Psychiatric Symptom Assessment Scale. Negative Symptom Rating Scale. |
| Notes |
Junker 1992.
| Methods | Allocation: randomised. Blindness; double‐blind. Duration: Four weeks. Setting: unclear, Germany. Design: parallel groups. |
| Participants | Diagnosis: unclear. N = 16. Sex: 6 M, 10 F. Age: average ˜ 52. History: tardive dyskinesia. |
| Interventions | 1. Vitamin E (1200 IU/day). N = unclear. 2. Placebo. N = unclear. |
| Outcomes | |
| Notes |
Kodesh 2003.
| Methods | Allocation: randomised. Blindess: double‐blind. Duration: three weeks. Setting: outpatients, Israel Design: cross‐over |
| Participants | Diagnosis: schizophrenia. N = 10. Sex: 10M. Age: unclear. History: sexual dysfunction. |
| Interventions | 1. Selegiline (15 mg/day). N = unclear.
2. Placebo. N = unclear. Stable antipsychotic: "typical neuroleptic agents". |
| Outcomes | Leaving the study early. PANSS. |
| Notes |
Lam 1994.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: six weeks. Setting: inpatients, Hong Kong. Design: cross‐over. |
| Participants | Diagnosis: schizophrenia. N = 16. Sex: unclear. Age: unclear. |
| Interventions | 1. Vitamin E (1200 mg/day). N = unclear.
2. Placebo. N = unclear. Stable antipsychotic: "typical neuroleptic agents". |
| Outcomes | Leaving the study early. BPRS. |
| Notes |
Meng 1996.
| Methods | Allocation: randomised. Blindness: double ‐ blind. Duration: eight weeks. Setting: inpatients, China. Design: parallel groups. |
| Participants | Diagnosis: schizophrenia. N = 40. Sex: unclear. Age: unclear. |
| Interventions | 1. Ginkgo biloba (240 mg/day). N = 21.
2. Placebo. N = 19. Stable antipsychotic: antipsychotics, no further description. |
| Outcomes | Leaving the study early. BPRS. SANS. |
| Notes | Very probably a subsample of Luo 1997 |
Milner 1963.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: three weeks. Setting: inpatients, UK. Design: parallel groups. |
| Participants | Diagnosis: "chronic psychiatric patients". N = 40. Sex: unclear. Age: unclear. |
| Interventions | 1. Vitamin C (1000 mg/day). N = 20.
2. Placebo. N = 20. Stable antipsychotic: unclear |
| Outcomes | Leaving the study early. Self‐rated symptoms. |
| Notes |
Salmasi 2009.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: eight weeks. Setting: unclear, Iran. Design: parallel groups. |
| Participants | Diagnosis: schizophrenia. N = 32. Sex: unclear. Age: unclear. |
| Interventions | 1. Vitamin E (800 IU/day). N = unclear.
2. Placebo. N = unclear. Stable antipsychotic: olanzapine. |
| Outcomes | No outcomes of interest reported. |
| Notes |
Schmidt 1991.
| Methods | Allocation: randomised. Blindess: double‐blind. Duration: two weeks. Setting: inpatients, Switzerland. Design: cross‐‐over. |
| Participants | Diagnosis: schizophrenia, major depression, schizoaffective disorder. N = 23. Sex: 9 F, 10 M (4 unclear) Age: average 45 years. History: tardive dyskinesia |
| Interventions | 1. Vitamin E (1600 IU /day). N = 13.
2. Placebo. N = 10. Stable antipsychotic: typical antipsychotics. |
| Outcomes | Leaving the study early. |
| Notes |
Shamir 2000.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: four weeks. Setting: inpatients, Israel. Design: cross‐over. |
| Participants | Diagnosis: schizophrenia. N = 19. Sex: 11 F, 18 M. Age: average 74 years. History: tardive dyskinesia |
| Interventions | 1. Melatonin (2 mg/day). N = 9.
2. Placebo. N = 10. Stable antipsychotic: "regular regimen". |
| Outcomes | Unable to use any data (only cross‐over reported) |
| Notes |
Shamir 2001a.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: six weeks. Setting: inpatients, Israel. Design: cross‐over. |
| Participants | Diagnosis: schizophrenia. N = 22. Sex: 11 F, 11 M. Age: average 64 years. History: tardive dyskinesia. |
| Interventions | 1. Melatonin (10 mg/day). N = 10.
2. Placebo. N = 12. Stable antipsychotic: "regular regimen". |
| Outcomes | Unable to use any data (only cross‐over reported) |
| Notes |
Shriqui 1992.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: four weeks. Setting: outpatients, Canada. Design: cross‐over. |
| Participants | Diagnosis: schizophrenia, bipolar disorder. N = 27. Sex: unclear. Age: unclear. History: tardive dyskinesia. |
| Interventions | 1. Vitamin E (1200 IU/day). N = unclear.
2. Placebo. N = unclear. Stable antipsychotic: "psychotropic medication". |
| Outcomes | Leaving the study early. |
| Notes |
Stearns 1996.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 16 weeks. Setting: unclear Design: cross‐over |
| Participants | Diagnosis: schizophrenia. N = 14. Sex: 3F, 11M. Age: unclear. History: tardive dyskinesia. |
| Interventions | 1. Selegiline (unclear dose). N = unclear.
2. Placebo. N = unclear. Stable antipsychotic: unclear. |
| Outcomes | Unable to use any data (only cross‐over reported) |
| Notes |
Xu 2002.
| Methods | Allocation: randomised. Blindess: unclear if single‐blind or open‐label. Duration: eight weeks Setting: inpatients, China Design: parallel |
| Participants | Diagnosis: schizophrenia (CCMD‐2‐R). N = 100. Sex: 21% female.. Age: average ˜ 46 years. |
| Interventions | 1. Ginkgo (EGb 49.6 mg three times daily), n = 50 2. Placebo. N = 50 Stable neuroleptic medication: chlorpromazine |
| Outcomes | SAPS. SANS. |
| Notes | Translation obtained from Prof. Yiming Wang. |
BPRS ‐ Brief Psychiatric Assessment Scale; CCMD‐2‐R ‐ Chinese Classification of Mental Disorders; DSM ‐ Diagnostic and Statistical Manual; IU ‐ international units; PANSS ‐ Positive and Negative Symptoms Scale; SANS ‐ Scale for the Assessment of Negative Symptoms; SAPS ‐ Schedule for the Assessment of Positive Symptoms
Differences between protocol and review
As stated in the "Duration" section, we planned to divide the studies into immediate (four weeks or less), short term (4‐12 weeks), medium term (13‐26 weeks) and long term (over 26 weeks) However, Luo 1997 had a follow‐up period of 16 weeks and was analysed in the short‐term subgroup as it was designed as an acute study and much more similar to the other Ginkgo biloba studies, four weeks or less), short term (4‐12 weeks), medium term (13‐26 weeks) and long term (over 26 weeks).
We prestated that we would include the following outcomes in a 'Summary of findings' table. Global state: clinical response and relapse, mental state: clinical response, service use: hospital admission and days in hospital, adverse effects and quality of life.
This has not been possible. Relapse data service use and quality of life were not available. We thus opted to include, alongside improvement, endpoint psychopathology and risk of leaving the study before termination, as proxies of improvement and treatment acceptability. We noted the lack of available data for our prestated outcomes of interest in the abstract and main text.
Contributions of authors
Pedro Magalhães and Flávio Kapczinski drafted the original idea. Pedro Magalhães and Michael Berk were responsible for trial selection. Olivia Dean and Pedro Magalhães were responsible for data extraction and data management. All authors were responsible for the final drafting of the protocol.
Sources of support
Internal sources
-
Universidade Federal do Rio Grande do Sul, Brazil.
Employs Profs. Magalhaes and Kapczinski
-
Deakin University, Australia.
Employs Prof. Berk
-
University of Toronto, Canada.
Employs Prof. Andreazza
External sources
No sources of support supplied
Declarations of interest
Pedro Magalhães has no conflicts of interest. Olivia Dean has received grant support from the Brain and Behavior Foundation, Simons Autism Foundation, Stanley Medical Research Institute, Lilly, NHMRC and an ASBD/Servier grant. Anna Andreazza has no conflicts of interest. Michael Berk was the lead investigator for one potentially relevant study for the review (Berk 2008; Berk 2008a) , but did not extract data from these trials for this review. He has received grant/research support from the Stanley Medical Research Foundation, MBF, NHMRC, beyondblue, Geelong Medical Research Foundation, Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Organon, Novartis, Mayne Pharma, Servier; has been a consultant/speaker for: Astra Zeneca, Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Janssen Cilag, Lundbeck, Pfizer, Sanofi Synthelabo, Servier, Solvay, Wyeth. Professor Berk is co‐inventor on two provisional patents regarding the use of NAC and related compounds for psychiatric indications, which, whilst assigned to the Mental Health Research Institute, could lead to personal remuneration upon a commercialisation event. Flávio Kapczinski has received grant/research support from Astra‐Zeneca, Eli Lilly, the Janssen‐Cilag, Servier, CNPq, CAPES, NARSAD and the Stanley Medical Research Institute; has been a member of the speakers’ boards for Astra‐Zeneca, Eli Lilly, Janssen and Servier; and has served as a consultant for Servier.
New
References
References to studies included in this review
Adler 1993a {published data only}
- Adler LA, Peselow E, Rotrosen J, Duncan E, Lee M, Rosenthal M, et al. Vitamin E treatment of tardive dyskinesia. American Journal of Psychiatry 1993;150(9):1405‐7. [DOI] [PubMed] [Google Scholar]
- Adler LA, Edson R, Lavori P, Peselow E, Duncan E, Rosenthal M, et al. Long‐term treatment effects of vitamin E for tardive dyskinesia. Biological Psychiatry 1998;15:868‐72. [DOI] [PubMed] [Google Scholar]
Adler 1999 {published data only}
- Adler LA, Rotrosen J, Edson R, Lavori P, Lohr J, Hitzemann R, et al. Vitamin E treatment for tardive dyskinesia. Archives of General Psychiatry 1999;56(9):836‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Akhondzadeh 2005 {published data only}
- Akhondzadeh S, Safarcherati A, Amini H. Beneficial antipsychotic effects of allopurinol as add‐on therapy for schizophrenia: a double blind, randomized and placebo controlled trial. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2005;29(2):253‐9. [PUBMED: 15694232] [DOI] [PubMed] [Google Scholar]
Amiri 2008 {published data only}
- Amiri A, Noorbala AA, Nejatisafa AA, Ghoreishi A, Derakhshan MK, Khodaie‐Ardakani MR, et al. Efficacy of selegiline add on therapy to risperidone in the treatment of the negative symptoms of schizophrenia: a double‐blind randomized placebo‐controlled study. Human Psychopharmacology 2008;23(2):79‐86. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Berk 2008b {published data only}
- Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N‐acetyl cysteine as a glutathione precursor for schizophrenia‐a double‐blind, randomized, placebo‐controlled trial. Biological Psychiatry 2008;64(5):361‐8. [DOI] [PubMed] [Google Scholar]
Bodkin 2005 {published data only}
- Bodkin JA, Siris SG, Bermanzohn PC, Hennen J, Cole JO. Double‐blind, placebo‐controlled, multicenter trial of selegiline augmentation of antipsychotic medication to treat negative symptoms in outpatients with schizophrenia. American Journal of Psychiatry 2005;162(2):388‐90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bordbar 2008 {published data only}
- Bordbar MR, Abdollahian E, Hojjat K, Samaric A. Effects of selegiline on negative symptoms in schizophrenia: A double‐blind clinical trial. Iranian Journal of Psychiatry and Clinical Psychology 2008;14(2):131‐9. [Google Scholar]
Brunstein 2005 {published data only}
- Brunstein MG, Ghisolfi ES, Ramos FL, Lara DR. A clinical trial of adjuvant allopurinol therapy for moderately refractory schizophrenia. Journal of Clinical Psychiatry 2005;66(2):213‐9. [PUBMED: 15705007] [DOI] [PubMed] [Google Scholar]
Dakhale 2005 {published data only}
- Dakhale GN, Khanzode SD, Khanzode SS, Saoji A. Supplementation of vitamin C with atypical antipsychotics reduces oxidative stress and improves the outcome of schizophrenia. Psychopharmacology 2005;182(4):494‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Dickerson 2009 {published data only}
- Dickerson FB, Stallings CR, Origoni AE, Sullens A, Khushalani S, Sandson N, et al. A double‐blind trial of adjunctive allopurinol for schizophrenia. Schizophrenia Research 2009;109(1‐3):66‐9. [PUBMED: 19195842] [DOI] [PubMed] [Google Scholar]
Dorevitch 1997a {published data only}
- Dorevitch A, Kalian M, Shlafman M, Lerner V. Treatment of long‐term tardive dyskinesia with vitamin E. Biological Psychiatry 1997;41(1):114‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Doruk 2008 {published data only}
- Doruk A, Uzun O, Ozsahin A. A placebo‐controlled study of extract of ginkgo biloba added to clozapine in patients with treatment‐resistant schizophrenia. International Clinical Psychopharmacology 2008;23(4):223‐7. [PUBMED: 18545061] [DOI] [PubMed] [Google Scholar]
Farokhnia 2013 {published data only}
- Farokhnia M, Azarkolah A, Adinehfar F, Khodaie‐Ardakani MR, Hosseini SM, Yekehtaz H, et al. N‐acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double‐blind, placebo‐controlled study. Clinical Neuropharmacology 2013;36(6):185‐92. [PUBMED: 24201233] [DOI] [PubMed] [Google Scholar]
Goff 1993 {published data only}
- Goff DC, Renshaw PF, SaridSegal O, Dreyfuss DA, Amico ET, Ciraulo DA. A placebo‐controlled trial of selegiline (L‐deprenyl) in the treatment of tardive dyskinesia. Biological Psychiatry 1993;33(10):700‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jungerman 1999 {published data only}
- Jungerman T, Rabinowitz D, Klein E. Deprenyl augmentation for treating negative symptoms of schizophrenia: a double blind, controlled study. Journal of Clinical Psychopharmacology 1999;19(6):522‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lohr 1996 {published data only}
- Lohr JB, Caligiuri MP. A double‐blind placebo‐controlled study of vitamin E treatment of tardive dyskinesia. Journal of Clinical Psychiatry 1996;57(4):167‐73. [PubMed] [Google Scholar]
Luo 1997 {published data only}
- Luo HC, Shen YC, Meng FQ. Therapeutic effect of shuxuening combining neuroleptics for the treatment of chronic schizophrenia‐‐a double blind study. Zhongguo Zhong xi yi jie he za zhi Zhongguo Zhongxiyi jiehe zazhi = Chinese Journal of Integrated Traditional and Western Medicine / Zhongguo Zhong xi yi jie he xue hui, Zhongguo Zhong yi yan jiu yuan zhu ban 1997;17(3):139‐42. [PubMed] [Google Scholar]
Ritsner 2010 {published data only}
- Ritsner MS, Gibel A, Shleifer T, Boguslavsky I, Zayed A, Maayan R, et al. Pregnenolone and dehydroepiandrosterone as an adjunctive treatment in schizophrenia and schizoaffective disorder: an 8‐week, double‐blind, randomized, controlled, 2‐center, parallel‐group trial. The Journal of Clinical Psychiatry 2010;71(10):1351‐62. [PUBMED: 20584515] [DOI] [PubMed] [Google Scholar]
Weiser 2012 {published data only}
- Weiser M, Gershon AA, Rubinstein K, Petcu C, Ladea M, Sima D, et al. A randomized controlled trial of allopurinol vs. placebo added on to antipsychotics in patients with schizophrenia or schizoaffective disorder. Schizophrenia Research 2012;138(1):35‐8. [PUBMED: 22483162] [DOI] [PubMed] [Google Scholar]
Zhang 2001a {published data only}
- Zhang XY, Zhou DF, Zhang PY, Wu GY, Su JM, Cao LY. A double‐blind, placebo‐controlled trial of extract of Ginkgo biloba added to haloperidol in treatment‐resistant patients with schizophrenia. Journal of Clinical Psychiatry 2001;62(11):878‐83. [PUBMED: 11775047] [DOI] [PubMed] [Google Scholar]
Zhang 2004 {published data only}
- Zhang XY, Zhou DF, Cao LY, Xu CQ, Chen da C, Wu GY. The effect of vitamin e treatment on tardive dyskinesia and blood superoxide dismutase: a double‐blind placebo‐controlled trial. Journal of Clinical Psychopharmacology 2004;24(1):83‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Zhang 2011 {published data only}
- Zhang WF, Tan YL, Zhang XY, Chan RC, Wu HR, Zhou DF. Extract of Ginkgo biloba treatment for tardive dyskinesia in schizophrenia: a randomized, double‐blind, placebo‐controlled trial. Journal of Clinical Psychiatry 2011;72(5):615‐21. [PUBMED: 20868638] [DOI] [PubMed] [Google Scholar]
- Zhang XY, Zhou DF, Su JM, Zhang PY. The effects of extract of ginkgo biloba added to haloperidol on superoxide dismutase in inpatients with chronic schizophrenia. Journal of Clinical Psychopharmacology 2001;21(1):85‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bhavani 1962 {published data only}
- Bhavani AD, Sen NN, Punekar BD. Impact of ascorbic acid administration on memory, attention, intelligence and motor functions in schizophrenics. Transactions of all India Institute of Mental Health 1962;3:80‐8. [Google Scholar]
Dorevitch 1997 {published data only}
- Dorevitch A, Lerner V, Shalfman M, Kalian M. Lack of effect of vitamin E on serum creatine phosphokinase in patients with long‐term tardive dyskinesia. International Clinical Psychopharmacology 1997;12(3):171‐3. [PUBMED: 9248874] [DOI] [PubMed] [Google Scholar]
Elkashef 1990 {published data only}
- Elkashef AM, Ruskin PE, Bacher N, Barrett D. Vitamin E in the treatment of tardive dyskinesia. American Journal of Psychiatry 1990;147(4):505‐6. [DOI] [PubMed] [Google Scholar]
Eranti 1998 {published data only}
- Eranti VS, Gangadhar BN, Janakiramaiah N. Haloperidol induced extrapyramidal reaction: lack of protective effect by vitamin E. Psychopharmacology 1998;140(4):418‐20. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kapur 1991 {published data only}
- Kapur S, Ganguli R, Ulrich R, Raghu U. Use of random‐sequence riboflavin as a marker of medication compliance in chronic schizophrenics. Schizophrenia Research 1991;6(1):49‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rees 1951 {published data only}
- Rees L, King GM. Desoxycortone acetate and ascorbic acid in the treatment of schizophrenia. Journal of Mental Science 1951;97:376‐80. [DOI] [PubMed] [Google Scholar]
Suresh 2007 {published data only}
- Suresh Kumar PN, Andrade C, Bhakta SG, Singh NM. Melatonin in schizophrenic outpatients with insomnia: a double‐blind, placebo‐controlled study. Journal of Clinical Psychiatry 2007;68(2):237‐41. [PUBMED: 17335321] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Akhtar 1993 {published data only}
- Akhtar S, Jajor TR, Kumar S. Vitamin E in the treatment of tardive dyskinesia. Journal of Postgraduate Medicine 1993;39(3):124‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Altman 1973 {published data only}
- Altman H, Mehta D, Evenson RC, Sletten IW. Behavioral effects of drug therapy on psychogeriatric inpatients: II. multivitamin supplement. Journal of the American Geriatrics Society 1973;21(6):249‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Atmaca 2005 {published data only}
- Atmaca M, Tezcan E, Kuloglu M, Ustundag B, Kirtas O. The effect of extract of ginkgo biloba addition to olanzapine on therapeutic effect and antioxidant enzyme levels in patients with schizophrenia. Psychiatry and Clinical Neurosciences 2005;59(6):652‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ben‐Dor 1998 {published data only}
- Ben‐Dor A, Gelkoph M. Vitamin E: an alternative to anticholinergic drugs?. Proceedings of the 9th Congress of the Association of European Psychiatrists; 1998 Sep 20‐24; Copenhagen, Denmark. 1998.
Egan 1992 {published data only}
- Egan MF, Hyde TM, Albers GW, Elkashef A, Alexander RC, Reeve A, et al. Treatment of tardive dyskinesia with vitamin E. American Journal of Psychiatry 1992;149(6):773‐7. [DOI] [PubMed] [Google Scholar]
Junker 1992 {published data only}
- Junker D, Steigleider P, Gattaz WF. Alpha‐tocopherol in the treatment of tardive dyskinesia. Proceedings of the 7th Biennial Winter Workshop on Schizophrenia; 1994 Jan 23‐28; Les Diablerets, Switzerland. 1992; Vol. 6, issue 2:123.
Kodesh 2003 {published data only}
- Kodesh A, Weizman A, Aizenberg D, Hermesh H, Gelkopf M, Zemishlany Z. Selegiline in the treatment of sexual dysfunction in schizophrenic patients maintained on neuroleptics: a pilot study. Clinical Neuropharmacology 2003;26(4):193‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lam 1994 {published data only}
- Lam LCW, Chiu HFK, Hung SF. Vitamin E in the treatment of tardive dyskinesia: A replication study. Journal of Nervous and Mental Disease 1994;182(2):113‐4. [DOI] [PubMed] [Google Scholar]
Meng 1996 {published data only}
- Meng Fan Qiang, Cui Yu Hua, Wang Shan Hui. A double‐blind placebo controlled study of EGb761 in the treatment of chronic schizophrenia. Jounal of Clinical Psychological Medicine 1996;6:339‐341. [Google Scholar]
Milner 1963 {published data only}
- Milner G. Ascorbic acid in chronic psychiatric patients ‐ a controlled trial. British Journal of Psychiatry 1963;109:294‐9. [Google Scholar]
Salmasi 2009 {published data only}
- Salmasi FB, Jazayeri M, Ghaeli P, Hashemian F, Akhondzadeh S, Raisi F, et al. Comparing the effects of high‐dose vitamin E with those of placebo on insulin resistance in patients with schizophrenia treated with olanzapine. Journal of Clinical Psychopharmacology 2009;29(2):182‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Schmidt 1991 {published data only}
- Schmidt M, Meister P, Baumann P. Treatment of tardive dyskinesias with vitamin E. European Psychiatry 1991;6(4):201‐7. [Google Scholar]
Shamir 2000 {published data only}
- Shamir E, Barak Y, Plopsky I, Zisapel N, Elizur A, Weizman A. Is melatonin treatment effective for tardive dyskinesia?. Journal of Clinical Psychiatry 2000;61(8):556‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shamir 2001a {published data only}
- Shamir E, Barak Y, Shalman I, Laudon M, Zisapel N, Tarrasch R, et al. Melatonin treatment for tardive dyskinesia: a double‐blind, placebo‐controlled, crossover study. Archives of General Psychiatry 2001;58:1049‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shriqui 1992 {published data only}
- Shriqui CL, Bradwejn J, Annable L, Jones BD. Vitamin E in the treatment of tardive dyskinesia: a double‐blind placebo‐ controlled study. American Journal of Psychiatry 1992;149(3):391‐3. [DOI] [PubMed] [Google Scholar]
Stearns 1996 {published data only}
- Stearns AI, Sambunaris A, Elkashef AM, Issa F, Egan MF, Wyatt RJ. Selegiline for negative symptoms and tardive dyskinesia. Proceedings of the 149th Annual Meeting of the American Psychiatric Association; 1996 May 4‐9; New York, New York, USA. 1996.
Xu 2002 {published data only}
- Xu X, Ji Y, He M. The efficacy of Ginkgo biloba tablet inadjuvant treatment of schizophrenia. Shanghai Medical andPharmaceutical Journal 2002;23:456‐7. [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1990
- Andreasen NC. Methods for assessing positive and negative symptoms. Modern problems of pharmacopsychiatry 1990;24:73‐88. [PUBMED: 2336066] [DOI] [PubMed] [Google Scholar]
Berk 2008
- Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N‐acetyl cysteine for depressive symptoms in bipolar disorder ‐ a double‐blind randomized placebo‐controlled trial. Biological Psychiatry 2008;64(6):468‐75. [PUBMED: 18534556] [DOI] [PubMed] [Google Scholar]
Berk 2008a
- Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N‐acetyl cysteine as a glutathione precursor for schizophrenia ‐ a double‐blind, randomized, placebo‐controlled trial. Biological Psychiatry 2008;64(5):361‐8. [PUBMED: 18436195] [DOI] [PubMed] [Google Scholar]
Berk 2009
- Berk M. Neuroprogression: pathways to progressive brain changes in bipolar disorder. International Journal of Neuropsychopharmacology 2009;12(4):441‐5. [PUBMED: 18922203] [DOI] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT Jr, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;33(10):681‐90. [DOI] [PubMed] [Google Scholar]
Dean 2009
- Dean OM, Buuse M, Bush AI, Copolov DL, Ng F, Dodd S, et al. A role for glutathione in the pathophysiology of bipolar disorder and schizophrenia? Animal models and relevance to clinical practice. Current Medicinal Chemistry 2009;16(23):2965‐76. [PUBMED: 19689277] [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape town. Cape Town: The Cochrane Collaboration, 2000.
DeLisi 2008
- DeLisi LE. The concept of progressive brain change in schizophrenia: implications for understanding schizophrenia. Schizophrenia Bulletin 2008;34(2):312‐21. [PUBMED: 18263882] [DOI] [PMC free article] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [PUBMED: 3802833] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Endicott 1976
- Endicott J, Spitzer RL, Fleiss JL, Cohen J. The global assessment scale. A procedure for measuring overall severity of psychiatric disturbance. Archives of General Psychiatry 1976;33(6):766‐71. [PUBMED: 938196] [DOI] [PubMed] [Google Scholar]
Freemantle 1999
- Freemantle N, Mason J, Eccles M. Deriving treatment recommendations from evidence within randomized trials. The role and limitation of meta‐analysis. International Journal of Technology Assessment in Health Care 1999;15(2):304‐15. [PUBMED: 10507190] [PubMed] [Google Scholar]
Guy 1991
- Guy W. Treatment Emergent Symptom Scale. ECDEU Assessment Manual for Psychopharmacology. National Institutes for Mental Health, 1991.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Jones 1995
- Jones SH, Thornicroft G, Coffey M, Dunn G. A brief mental health outcome scale‐reliability and validity of the Global Assessment of Functioning (GAF). British Journal of Psychiatry: the journal of mental science 1995;166(5):654‐9. [PUBMED: 7620753] [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and negative syndrome scale (PANSS) manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Tollefson GD, Charles C, Zipursky R, Sharma T, Kahn RS, Keefe RS, Green AI, Gur RE, McEvoy J, Perkins D, Hamer RM, Gu H, Tohen M, HGDH Study Group. Antipsychotic drug effects on brain morphology in first‐episode psychosis. Archives of General Psychiatry 2005;62(4):361‐70. [PUBMED: 15809403] [DOI] [PubMed] [Google Scholar]
Lieberman 2006
- Lieberman JA, Malaspina D, Jarskog LF. Preventing clinical deterioration in the course of schizophrenia: the potential for neuroprotection. CNS Spectrums 2006;11(4):Suppl 1‐13; Quiz Suppl 14‐5. [PUBMED: 16641837] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [PUBMED: 10755072] [DOI] [PubMed] [Google Scholar]
Ng 2008
- Ng F, Berk M, Dean O, Bush AI. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. International Journal of Neuropsychopharmacology 2008;11(6):851‐76. [PUBMED: 18205981] [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Padurariu 2010
- Padurariu M, Ciobica A, Dobrin I, Stefanescu C. Evaluation of antioxidant enzymes activities and lipid peroxidation in schizophrenic patients treated with typical and atypical antipsychotics. Neuroscience Letters 2010;479(3):317‐20. [PUBMED: 20561936] [DOI] [PubMed] [Google Scholar]
Petiti 2000
- Petiti DB. Meta‐Analysis, Decision Analysis, and Cost‐Effectiveness Analysis: Methods for Quantitative Synthesis in Medicine. 2nd Edition. New York: Oxford University Press, 2000. [Google Scholar]
Rossler 2005
- Rossler W, Salize HJ, Os J, Riecher‐Rossler A. Size of burden of schizophrenia and psychotic disorders. European Neuropsychopharmacology 2005;15(4):399‐409. [PUBMED: 15925493] [DOI] [PubMed] [Google Scholar]
Satoh 1978
- Satoh K. Serum lipid peroxide in cerebrovascular disorders determined by a new colorimetric method. Clinica Chimica Acta; international journal of clinical chemistry 1978;90(1):37‐43. [PUBMED: 719890] [DOI] [PubMed] [Google Scholar]
Singh 2010
- Singh V, Singh SP, Chan K. Review and meta‐analysis of usage of ginkgo as an adjunct therapy in chronic schizophrenia. International Journal of Neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum (CINP) 2010;13(2):257‐71. [PUBMED: 19775502] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2011
- Soares‐Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD000209.pub2; PUBMED: 21328246] [DOI] [PubMed] [Google Scholar]
Uttara 2009
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current Neuropharmacology 2009;7(1):65‐74. [PUBMED: 19721819] [DOI] [PMC free article] [PubMed] [Google Scholar]
Valko 2007
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International journal of Biochemistry & Cell Biology 2007;39(1):44‐84. [PUBMED: 16978905] [DOI] [PubMed] [Google Scholar]
Wang 2008
- Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology 2008;33(6):1276‐86. [PUBMED: 17687264] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wood 2009
- Wood SJ, Yucel M, Pantelis C, Berk M. Neurobiology of schizophrenia spectrum disorders: the role of oxidative stress. Annals of the Academy of Medicine 2009;38(5):396‐6. [PUBMED: 19521638] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]