

Key Clinical Message
Lethal multiple pterygium syndrome is a very rare genetic disorder. The manifestations of this condition include growth deficiency of the fetus, craniofacial anomalies, joint contracture, and skin webbing (pterygia). This disorder is fatal before birth or shortly after birth. We reported a case of lethal multiple pterygium syndrome with multiple anomalies including pterygia involving the axilla, bilateral antecubital fossa, and groin. Arthrogryposis involving multiple lower and upper extremities joints. Cleft palate, microstomia and limitation of mouth opening, webbed neck, asymmetric small and narrow chest, ambiguous genitalia, depressed and wide nasal bridge, antemongoloid slant, low‐set, malformed, and posteriorly rotated ears, pterygia, syndactyly and camptodactyly of hands and rocket bottom feet. LMPS is a congenital genetic disease with multiple anomalies that is fatal in the second and third trimesters of pregnancy or shortly after birth. With genetic testing and counseling, it can be prevented from recurring in subsequent pregnancies.
Keywords: congenital disorders, genetic counseling, genetic screening, multiple pterygium syndrome, musculoskeletal anomaly
A newborn with pterygium underwent advanced resuscitation in the operating room.

1. INTRODUCTION
Multiple pterygium syndrome (MPS) is a rare genetic disorder that is characterized by arthrogryposis (joint contractures), skin webs on the joints (pterygia), short stature, kyphoscoliosis, craniofacial anomalies and other anomalies of bones, joints, and limb. 1 , 2 MPS includes two major forms: lethal and nonlethal (also called Escobar syndrome). The lethal form causes death during infancy (in the second or third trimester) or shortly after birth. 3 , 4 , 5 The main inheritance pattern of the disease is autosomal recessive) autosomal dominant and X‐linked have also been reported). 6 , 7 , 8
This syndrome is generally diagnosed during the prenatal period by fetal sonography, which shows evidence of oligohydramnios, arthrogryposis, and reduced fetal movements. 5 Due to the rare prevalence of this disease worldwide and in Iran, we describe a newborn diagnosed with this syndrome at our hospital.
2. CASE REPORT
A 26‐year‐old Afghan woman, G3P3L1IUFD1, gestational age: 38+5 weeks went to the emergency department complaining of labor pain. She had no underlying disease, and did not mention medication use, with routine pregnancy care. She had a nonconsanguineous marriage. Her first pregnancy was terminated at 24 weeks of gestational weeks due intrauterine fetal demise (IUFD) to severe oligohydramnios and fetal limb and renal anomalies. The parents did not perform a further evaluation to determine the underlying causes of this disorder. The second pregnancy lead to a term female newborn, without any medical problems, weighing 3400 g.
She did not performed aneuploidy screening tests. Ultrasound examination at 13 weeks of gestation manifested a severe oligohydramniosis that there was no evidence of rupture of the membrane in the amniosure test and the vaginal examination. The subsequent sonographic evaluation at 20 weeks of gestation for anomalies showed a single fetus with severe amniotic fluid reduction that caused complete evaluation impossible. The kidneys were echogenic, and the stomach and bladder were not seen. At this time, doctors had recommended pregnancy termination due to poor prognosis, but the parents had declined and did not accept further amniocentesis.
The last two‐dimensional (2D) ultrasound at 32 weeks of gestation showed an IUGR (Intrauterine growth restriction) fetus (weight and other parameters less than 2 percent of normal range) with absent umbilical artery diastolic flow. The bladder and stomach were not seen as before. In 38 weeks due to fetal distress shown in NST (nonstress test) as recurrent late deceleration, about 2 hours after admission with cervical dilatation of 2 cm and effacement of 20%, the patient underwent a cesarean section.
A dysmorphic, apneic baby boy weighing 900 grams, was born with Apgar 6 in the first minutes and the following anomalies: pterygia involving the axilla, bilateral antecubital fossa, and groin. Arthrogryposis involving multiple lower and upper extremities joints. Cleft palate, microstomia and limitation of mouth opening, webbed neck, asymmetric small and narrow chest, ambiguous genitalia, depressed and wide nasal bridge, antemongoloid slant, low‐set, malformed, and posteriorly rotated ears, pterygia, syndactyly and camptodactyly of hands and rocket bottom feet (Figure 1A–D).
FIGURE 1.
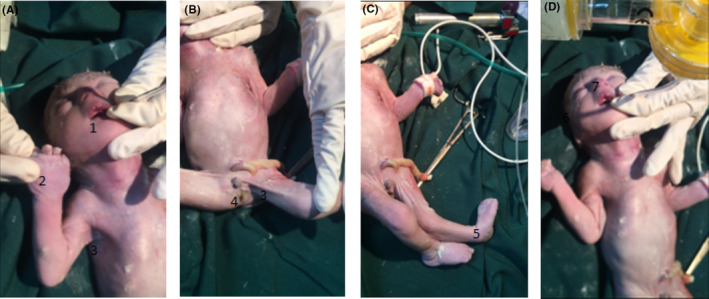
(A–D): Anomalies features that are shown with numbers: 1‐ cleft palate, 2‐ camptodactyly, 3‐ pterygia, 4‐ ambiguous genitalia, 5‐ club foot, 6‐ depressed nasal ridge, and 7‐ low‐set ear.
The ABG (atrial blood gas) was 7.25. He required extensive resuscitation at birth, including full cardiopulmonary resuscitation and intubation. After intubation, the chest did not expand properly, and breathing sounds were not heard well. Despite advanced resuscitation, the baby expired about 10 minutes after birth. Whole exome sequencing (WES) showed a homozygous mutation of the CHRNG gene.
3. DISCUSSION
LMPS (lethal multiple pterygium syndrome) is characterized by arthrogrypotic contractures of the limbs, pterygia (webbing of the neck, popliteal, and antecubital fossa), lack of muscle movement (akinesia), fetal hydrops with cystic hygroma, short stature, congenital vertical talus, camptodactyly, hip dislocation, facial anomalies, cryptorchidism, pulmonary, cardiac and brain hypoplasia, congenital diaphragmatic hernia, kidney abnormalities, progressive scoliosis, intrauterine growth restriction, and severe oligohydramnios. 1 , 2 , 9
Facial anomalies include depressed nasal bone, hypertelorism, microretrognathism and microstomia, epicanthal folds, low‐set malformed ears, and cleft palate, 4 cryptorchidism and underdeveloped phallus may be seen in male patients, and females may have missing or underdeveloped labia majora. 5 In this case, we could observe a large number of these anomalies, as mentioned above.
The main inheritance pattern of the disease is autosomal recessive, which is caused by a mutation in the gamma subunit of the cholinergic receptor (CHRNG) on chromosome 2, as we saw in our case genetic examination. Still, cases of autosomal dominant and X‐linked have also been reported. 6 , 7 , 8 The gamma subunit is expressed in fetal muscles and helps develop neuromuscular junctions. According to studies, it has been determined that there is no relationship between phenotype and genotype, which means that the same mutations can cause both lethal and nonlethal forms. 10 , 11 , 12
The differential diagnosis includes Bartsocas–Papas, FADS (fetal akinesia deformation sequence, also known as Pena–Shokeir syndrome type I), Neu–Laxova, and arthrogryposis multiplex; Congenital restrictive myopathy and Bruck syndrome. And the diagnosis is confirmed with genetic examinations. 5 , 13
The early diagnosis of MPS is difficult because the only sonographic features present in the first trimester are increased nuchal translucency and fetal hydrops. But we did not find this presentation in second trimester sonography. MPS is often diagnosed in the last months of pregnancy with the manifestations mentioned above, and genetic counseling should be done before the next pregnancy. 14 LMPS is lethal during pregnancy or soon after birth 4 , 5 ; as we see in our case, that expired about 10 minutes after birth. Knowing how to diagnose and deal with this disorder prevents additional evaluations and investigations and reduces parents' worries. Depending on the inheritance pattern, which is often autosomal recessive, parents should have genetic counseling and can be offered genetic testing and preimplantation genetic tests for future pregnancies.
4. CONCLUSION
LMPS is a congenital genetic disease with multiple anomalies that is fatal in the second and third trimesters of pregnancy or shortly after birth. With genetic testing and counseling, it can be prevented from recurring in subsequent pregnancies.
AUTHOR CONTRIBUTIONS
Parvaneh Sadeghimoghadam: Conceptualization. Saeedeh Shirdel: Writing – original draft. Sedigheh Hantoushzadeh: Supervision. Zeinab Hashemi: Writing – review and editing. Marjan Ghaemi: Conceptualization.
FUNDING INFORMATION
None.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to declare.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
The authors would like to acknowledge the case parents to allow the publishing and genetic examination of this case.
Sadeghimoghadam P, Shirdel S, Hantoushzadeh S, Hashemi Z, Ghaemi M. Lethal multiple pterygium syndrome in a newborn, a case report. Clin Case Rep. 2023;11:e7678. doi: 10.1002/ccr3.7678
DATA AVAILABILITY STATEMENT
Data is available upon request.
REFERENCES
- 1. Escobar V, Bixler D, Gleiser S, Weaver DD, Gibbs T. Multiple pterygium syndrome. Am J Dis Child. 1978;132(6):609‐611. [DOI] [PubMed] [Google Scholar]
- 2. Teebi AS, Daoud AS. Multiple pterygium syndrome: a relatively common disorder among Arabs. J Med Genet. 1990;27(12):791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hall JG, Reed SD, Rosenbaum KN, Gershanik J, Chen H, Wilson KM. Limb pterygium syndromes: a review and report of eleven patients. Am J Med Genet. 1982;12(4):377‐409. [DOI] [PubMed] [Google Scholar]
- 4. Hall JG. The lethal multiple pterygium syndromes. Am J Med Genet. 1984;17(4):803‐807. [DOI] [PubMed] [Google Scholar]
- 5. Kariminejad A, Ghaderi‐Sohi S, Hossein‐Nejad Nedai H, Varasteh V, Moslemi AR, Tajsharghi H. Lethal multiple pterygium syndrome, the extreme end of the RYR1 spectrum. BMC Musculoskelet Disord. 2016;17:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tolmie JL, Patrick A, Yates JR. A lethal multiple pterygium syndrome with apparent X‐linked recessive inheritance. Am J Med Genet. 1987;27(4):913‐919. [DOI] [PubMed] [Google Scholar]
- 7. Meyer‐Cohen J, Dillon A, Pai GS, Conradi S. Lethal multiple pterygium syndrome in four male fetuses in a family: evidence for an X‐linked recessive subtype? Am J Med Genet. 1999;82(1):97‐99. [DOI] [PubMed] [Google Scholar]
- 8. Chong JX, Burrage LC, Beck AE, et al. Autosomal‐dominant multiple Pterygium syndrome is caused by mutations in MYH3. Am J Hum Genet. 2015;96(5):841‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Gennaro GL, Greggi T, Parisini P. Scoliosis in Escobar syndrome (multiple pterygium syndrome). Description of two cases. Chir Organi Mov. 1996;81(3):317‐323. [PubMed] [Google Scholar]
- 10. Kariminejad A, Almadani N, Khoshaeen A, Olsson B, Moslemi AR, Tajsharghi H. Truncating CHRNG mutations associated with interfamilial variability of the severity of the Escobar variant of multiple pterygium syndrome. BMC Genet. 2016;17(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vogt J, Morgan NV, Rehal P, et al. CHRNG genotype‐phenotype correlations in the multiple pterygium syndromes. J Med Genet. 2012;49(1):21‐26. [DOI] [PubMed] [Google Scholar]
- 12. Missias AC, Chu GC, Klocke BJ, Sanes JR, Merlie JP. Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR gamma‐to‐epsilon switch. Dev Biol. 1996;179(1):223‐238. [DOI] [PubMed] [Google Scholar]
- 13. Mohtisham FS, Sallam A, Shawli A. Lethal multiple pterygium syndrome. BMJ Case Rep. 2019;12(5):e229045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joshi T, Noor NN, Kural M, Tripathi A. Lethal multiple pterygium syndrome. J Family Med Prim Care. 2016;5(2):477‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available upon request.