

Abstract
Background
Hypothalamic‐pituitary‐adrenal (HPA) axis dysregulation has been implicated in the development and relapse of psychotic disorders. Elevated cortisol secretion has been positively linked with symptom severity in people with psychosis. Antiglucocorticoid and related drugs that target the HPA axis may be useful for the treatment of individuals with psychosis.
Objectives
1. To determine the effects of antiglucocorticoid and related drugs for the treatment of psychosis, when used alone or in combination with antipsychotic medication.
2. To determine whether the effects of these medications differs between those in a prodromal phase or first episode of psychosis, and those with more established illness.
Search methods
We searched the Cochrane Schizophrenia Group's Trials Register (August 2009 and April 2014).
Selection criteria
Randomised controlled trials (RCTs) comparing antiglucocorticoid and related drugs compared to placebo (either as a sole treatment or as an adjunct to atypical antipsychotics, typical antipsychotics, antidepressants or other combination treatment) for people with a primary diagnosis of a psychotic disorder, or for individuals at high risk of developing a psychotic disorder.
Data collection and analysis
Review authors independently selected trials, assessed methodological quality and extracted data. We used a fixed‐effect meta‐analysis. We calculated risk ratios (RRs) with 95% confidence intervals (CIs) for dichotomous outcomes, and mean differences (MDs) and standardised mean differences (SMDs) with 95% CIs for continuous measures. We assessed risk of bias for included studies and used GRADE (Grading of Recommendations Assessment, Development and Evaluation) to create a 'Summary of findings' table.
Main results
We included 11 studies that randomly assigned 509 people with schizophrenia, schizoaffective disorder or psychotic depression. No trials were conducted in patients at their first episode of psychotic illness and none included populations at high risk for developing psychosis. Our pre‐stated outcomes of interest were mental state, global state, general functioning, adverse effects and quality of life.
Two trials compared antiglucocorticoid drugs (mifepristone) versus placebo as sole treatment. Limited data from one trial showed no difference in the proportion responding to mifepristone when mental state was assessed immediately post intervention using the Brief Psychiatric Rating Scale (BPRS) (n = 5, 1 RCT, MD ‐5.20, 95% CI ‐17.91 to 7.51; very low‐quality evidence); depressive symptoms (Hamilton Rating Scale for Depression (HAMD) total) were also similar between groups (n = 5, 1 RCT, MD 1.67, 95% CI ‐16.44 to 19.78; very low‐quality evidence). However, a significant difference favoured treatment at short‐term follow‐up for global state (30% reduction in total BPRS, n = 221, 1 RCT, RR 0.58, 95% CI 0.38 to 0.89; low‐grade quality evidence). This effect was also seen for short‐term positive psychotic symptoms (50% reduction in BPRS positive symptom subscale, n = 221, 1 RCT, RR 0.60, 95% CI 0.43 to 0.84; low‐grade quality evidence). Participants receiving mifepristone experienced a similar overall number of adverse effects as those receiving placebo (n = 226, 2 RCTs, RR 0.92, 95% CI 0.77 to 1.09; moderate‐quality evidence). No data on general functioning or quality of life were available.
One trial compared an antiglucocorticoid, dehydroepiandrosterone (DHEA), as an adjunct to atypical antipsychotic treatment to adjunctive placebo. Data for main outcomes of interest were of low quality, and analysis of useable data showed no significant effects of treatment on mental state or adverse effects. Data on global state, general functioning and quality of life were not available.
Data from six trials comparing antiglucocorticoid drugs as an adjunct to combination treatment versus adjunctive placebo showed no significant differences between groups in mean endpoint scores for overall psychotic symptoms (n = 171, 6 RCTs, SMD 0.01, 95% CI ‐ 0.29 to 0.32) or positive psychotic symptoms (n = 151, 5 RCTs, SMD ‐0.07, 95% CI ‐ 0.40 to 0.25). Data from three trials showed no differences between groups in mean endpoint scores for negative symptoms (n = 94, 3 RCTs, MD 2.21, 95% CI ‐0.14 to 4.55). One study found improvements in global state that were similar between groups (n = 30, 1 RCT, RR 0.58, 95% CI 0.32 to 1.06; very low‐quality evidence). In this comparison, pooled results showed that antiglucorticoids caused a greater overall number of adverse events (n = 199, 7 RCTs, RR 2.66, 95% CI 1.33 to 5.32; moderate quality evidence), but no quality of life data were available.
Authors' conclusions
Good evidence is insufficient to conclude whether antiglucocorticoid drugs provide effective treatment for psychosis. Some global state findings suggest a favourable effect for mifepristone, and a few overall adverse effect findings favour placebo. Additional large randomised controlled trials are needed to justify findings.
Keywords: Humans, Dehydroepiandrosterone, Dehydroepiandrosterone/therapeutic use, Dexamethasone, Dexamethasone/therapeutic use, Glucocorticoids, Glucocorticoids/antagonists & inhibitors, Hypothalamo‐Hypophyseal System, Ketoconazole, Ketoconazole/therapeutic use, Mifepristone, Mifepristone/adverse effects, Mifepristone/therapeutic use, Pituitary‐Adrenal System, Psychotic Disorders, Psychotic Disorders/drug therapy, Randomized Controlled Trials as Topic
Plain language summary
Antiglucocorticoid and related treatments for psychosis
Psychosis is a broad term that includes several mental illnesses such as schizophrenia, schizophreniform disorder, schizoaffective disorder, psychotic depression and bipolar disorder with psychotic features. Psychotic disorders affect about 3% of the population and may cause high levels of disability, making it a significant public health problem both socially and economically.
Stress may result in the release of cortisol and has been linked with both onset and relapse of psychotic disorders. Elevated cortisol levels have been found in some people with psychosis, especially among those suffering with psychotic depression and those in earlier phases of psychosis. Antiglucocorticoid drugs have been reported to reduce the effects of cortisol and may be useful for people with psychotic depression and bipolar disorder. We reviewed all randomised trials comparing antiglucocorticoid and related drugs versus placebo in people with psychosis ‐ prodromal psychosis or first episode of psychosis.
Eleven studies (involving 509 participants) were included in this review. Several antiglucocorticoid‐related drugs were examined, including dehydroepiandrosterone (DHEA) (n = 5), mifepristone (n = 4), dexamethasone (n = 1) and ketoconazole (n = 1). All participants were adults with a diagnosis of schizophrenia, schizoaffective disorder or psychotic depression. Most trials examined giving antiglucocorticoid drugs as an additional part of regular treatment. Available data from these trials revealed no effects for overall psychotic symptoms, 'positive' symptoms or 'negative' symptoms. One large trial comparing mifepristone versus placebo as the sole treatment revealed a significant difference in the proportion of people responding to treatment with mifepristone versus placebo. This effect was not seen immediately but 21 days after the intervention was begun. Adverse effect data varied. When individual anticorticoids such as mifepristone and DHEA were compared with placebo, the incidence of side effects was similar between groups; however, pooled data on various antiglucorticoids given as an adjunct to combination treatment showed that antiglucocorticoids increased incidence of side effects than placebo. In summary, very few trials are under way, and most involve a small number of people. Limited available data do not provide enough evidence to support the use of antiglucocorticoid treatments for psychosis; additional trials are needed.
Summary of findings
Summary of findings for the main comparison. Any antiglucocorticoid compared with placebo as sole treatment (data only for mifepristone) for psychosis.
| Any antiglucocorticoid compared with placebo as sole treatment (data only for mifepristone) for psychosis | ||||||
| Patient or population: people with psychosis Settings: inpatient/outpatient Intervention: any antiglucocorticoid Comparison: placebo as sole treatment (data only for mifepristone) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as sole treatment (data only for mifepristone) | Any antiglucocorticoid | |||||
| Mental state: 1. General ‐ average endpoint score BPRS total scores Follow‐up: 4 days | Mean mental state: 1. General ‐ average endpoint score in intervention groups was 5.2 lower (17.91 lower to 7.51 higher) | 5 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |||
| Mental state: 2. Specific ‐ depression ‐ average endpoint score HAMD total Follow‐up: 4 days | Mean mental state: 2. Specific ‐ depression ‐ average endpoint score in intervention groups was 1.67 higher (16.44 lower to 19.78 higher) | 5 (1 study) | ⊕⊝⊝⊝ Very lowa,b,c | |||
| Global state: 1. General ‐ no clinically significant improvement ‐ short term < 30% improvement on BPRS Follow‐up: 7 days | Lowd | RR 0.58 (0.38 to 0.89) | 221 (1 study) | ⊕⊕⊝⊝ Lowe,f | ||
| 200 per 1000 | 116 per 1000 (76 to 178) | |||||
| Moderated | ||||||
| 400 per 1000 | 232 per 1000 (152 to 356) | |||||
| Highd | ||||||
| 600 per 1000 | 348 per 1000 (228 to 534) | |||||
| Global state: 2. Specific: positive ‐ no clinically significant improvement ‐ short term (< 50% improvement on BPRS PSS) Follow‐up: 7 days | Lowd | RR 0.6 (0.43 to 0.84) | 221 (1 study) | ⊕⊕⊝⊝ Lowe,f | ||
| 300 per 1000 | 180 per 1000 (129 to 252) | |||||
| Moderated | ||||||
| 500 per 1000 | 300 per 1000 (215 to 420) | |||||
| Highd | ||||||
| 700 per 1000 | 420 per 1000 (301 to 588) | |||||
| General functioning: improved to an important degree | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome |
| Adverse effects: general: overall number of events Follow‐up: 7 days | Lowd | RR 0.92 (0.77 to 1.09) | 226 (2 studies) | ⊕⊕⊕⊝ Moderatee | ||
| 500 per 1000 | 460 per 1000 (385 to 545) | |||||
| Moderated | ||||||
| 700 per 1000 | 644 per 1000 (539 to 763) | |||||
| Highd | ||||||
| 900 per 1000 | 828 per 1000 (693 to 981) | |||||
| Quality of life: improved to an important degree | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aRisk of bias: rated 'serious' ‐ unclear how undertaken, groups imbalanced. bImprecision: rated 'serious' ‐ small study. cPublication bias: rated 'strongly suspected' ‐ one very small trial, may well be other unpublished work. dModerate risk roughly equal to that of people in control group of trial. eRisk of bias: rated 'serious' ‐ allocation concealment unclear, blinding untested, study authors allied with relevant company. fIndirectness: rated 'serious' ‐ BPRS not direct measure of global state.
Summary of findings 2. Any antiglucocorticoid compared with placebo as adjunct to atypical antipsychotic treatment (data only for DHEA) for psychosis.
| Any antiglucocorticoid compared with placebo as adjunct to atypical antipsychotic treatment (data only for DHEA) for psychosis | ||||||
| Patient or population: people with psychosis Settings: inpatient/outpatient Intervention: any antiglucocorticoid Comparison: placebo as adjunct to atypical antipsychotic treatment (data only for DHEA) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as adjunct to atypical antipsychotic treatment (data only for DHEA) | Any antiglucocorticoid | |||||
| Mental state: 1. General ‐ average endpoint score PANSS total Follow‐up: 12 weeks | Mean mental state: 1. General ‐ average endpoint score in intervention groups was 1.7 lower (10.78 lower to 7.38 higher) | 40 (1 study) | ⊕⊕⊝⊝ Lowa,b | |||
| Mental state: 2. Specific ‐ negative symptoms ‐ average endpoint score PANSS subscale Follow‐up: 12 weeks | Mean mental state: 2. Specific ‐ negative symptoms ‐ average endpoint score in intervention groups was 0.7 higher (2.63 lower to 4.03 higher) | 40 (1 study) | ⊕⊕⊝⊝ Lowa,b | |||
| Global state: general ‐ no clinically significant improvement | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome |
| General functioning: improved to an important degree | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome |
| Adverse effects: specific: extrapyramidal symptoms ‐ average endpoint scores parkinsonism SAS total Follow‐up: 12 weeks | Mean adverse effects: extrapyramidal symptoms ‐ average endpoint scores parkinsonism in intervention groups was 0 higher (0.88 lower to 0.88 higher) | 40 (1 study) | ⊕⊕⊝⊝ Lowa,b | |||
| Quality of life: improved to an important degree | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aRisk of bias: rated 'serious' ‐ small study with some imbalance in groups. bImprecision: rated 'serious' ‐ small study.
Summary of findings 3. Any antiglucocorticoid compared with placebo as adjunct to combination treatment for psychosis.
| Any antiglucocorticoid compared with placebo as adjunct to combination treatment for psychosis | ||||||
| Patient or population: people with psychosis Settings: inpatient/outpatient Intervention: any antiglucocorticoid Comparison: placebo as adjunct to combination treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo as adjunct to combination treatment | Any antiglucocorticoid | |||||
| Mental state: 1. General ‐ average endpoint score ‐ short term ‐ only mifepristone BPRS total Follow‐up: 7 days | Mean mental state: 1. General ‐ average endpoint score ‐ short term ‐ only mifepristone in intervention groups was 2.1 higher (2.86 lower to 7.06 higher) | 20 (1 study) | ⊕⊕⊝⊝ Lowa,b | |||
| Mental state: 2. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate PANSS negative subscale | Mean mental state: 2. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate in intervention groups was 1.68 higher (0.93 lower to 4.3 higher) | 70 (2 studies) | ⊕⊕⊝⊝ Lowa,c | |||
| Global state: general ‐no clinically significant improvement ‐ data only for mifepristone < 30% improvement on BPRS Follow‐up: 6 weeks | Lowd | RR 0.58 (0.32 to 1.06) | 30 (1 study) | ⊕⊝⊝⊝ Very lowe,f | ||
| 700 per 1000 | 406 per 1000 (224 to 742) | |||||
| Moderated | ||||||
| 800 per 1000 | 464 per 1000 (256 to 848) | |||||
| Highd | ||||||
| 900 per 1000 | 522 per 1000 (288 to 954) | |||||
| General functioning: average endpoint scores ‐ data only for DHEA GAF/SOFA Follow‐up: 6 weeks | Mean functioning: average endpoint scores ‐ data only for DHEA in intervention groups was 4.4 higher (3.4 lower to 12.2 higher) | 30 (1 study) | ⊕⊕⊝⊝ Lowe | |||
| Adverse effects: 1. General ‐ overall number of events Follow‐up: 12 weeks | Lowd | RR 2.66 (1.33 to 5.32) | 199 (7 studies) | ⊕⊕⊕⊝ Moderatea | ||
| 50 per 1000 | 133 per 1000 (67 to 266) | |||||
| Moderated | ||||||
| 100 per 1000 | 266 per 1000 (133 to 532) | |||||
| Highd | ||||||
| 150 per 1000 | 399 per 1000 (200 to 798) | |||||
| Adverse events: 2. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA SHRS total Follow‐up: 6 weeks | Mean adverse events: 2. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA in intervention groups was 5 lower (8.85 to 1.15 lower) | 30 (1 study) | ⊕⊕⊝⊝ Lowe | |||
| Quality of life: improved to an important degree | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aRisk of bias: rated 'serious' ‐ small study or studies. bImprecision: rated 'serious' ‐ few data from small study or studies. cInconsistency: rated 'serious' ‐ I2 91%. dModerate risk roughly equates to that of control group. eRisk of bias: rated 'very serious' ‐ small trial, imbalanced groups. fIndirectness: rated 'serious' ‐ BPRS not direct measure of global state.
Background
Description of the condition
Psychosis is a generic term that encompasses a group of severe mental illnesses with considerable variation in prognosis, including schizophrenia, schizophreniform disorder, schizoaffective disorder, psychotic depression and bipolar disorder with psychotic features. The lifetime prevalence of psychotic disorders is relatively high at around 3%, with many sufferers having a high level of disability, making it a significant public health problem both socially and economically (Altindag 2007; Murray 1997; Perälä 2007). Antipsychotic medications are the primary treatment for psychosis. The newer atypical antipsychotics are preferable to typical antipsychotics, as they are associated with fewer extrapyramidal symptoms (Kerwin 2004). Atypical antipsychotics are generally effective in alleviating the 'positive' symptoms (e.g. hallucinations, delusions) but have only modest effects on 'negative' (e.g. anhedonia, withdrawal, flat affect) and cognitive symptoms (Keefe 1999; Leucht 1999). A significant proportion of patients are treatment resistant, and many do not achieve complete remission of symptoms. Although fewer extrapyramidal symptoms have been reported, significant adverse effects are associated with antipsychotic treatment, such as weight gain and diabetes mellitus, which can lead to increased risk of a range of co‐morbid medical conditions and medication non‐compliance (Alvarez‐Jimenez 2008; Newcomer 2005).
Description of the intervention
Psychosocial interventions (such as cognitive‐behavioural treatment) for psychosis have been associated with reasonable levels of efficacy (Pilling 2002). Shifting the focus of intervention from chronic illness to intervention at earlier stages of the illness has also resulted in better outcomes (Killackey 2007). An increase in interest has been noted in the delivery of treatments to young people at ultra‐high risk (UHR) of developing a psychotic disorder, or with subthreshold symptoms, to reduce the likelihood of this group transitioning to a full‐blown psychotic disorder (McGorry 2002).
The search for more effective and benign treatments for all phases of psychotic disorders is ongoing. These may involve alternative medications to atypical antipsychotics, or adjunctive treatments to augment symptom reduction or alleviate adverse effects. The search for more benign treatments is considered particularly important in the treatment of initial episodes of psychosis and for UHR patients as they are at an early stage of their illness, and the potential for positive outcomes is therefore greater (McGorry 2006). Many UHR individuals will not go on to have psychosis with a chronic deteriorating course (Yung 2007); therefore for these people, the risks associated with taking medications with serious adverse effects may outweigh the benefits.
Antiglucocorticoid and related drugs that target the hypothalamic‐pituitary‐adrenal (HPA) axis are increasingly investigated as treatment for psychosis, particularly for those at early stages of illness and/or as adjunctive treatment. Recent clinical trials suggest that the antiglucocorticoid drug mifepristone may be useful in the treatment of individuals with psychotic depression and bipolar disorder (DeBattista 2006; Young 2004).
How the intervention might work
The HPA axis has been implicated in the development and relapse of major psychiatric disorders, including psychosis (Phillips 2006). Several studies have identified abnormalities of HPA axis function in patients with schizophrenia (Lammers 1995; Sharma 1988), psychotic depression (Nelson 1997), bipolar disorder (Watson 2004) and first‐episode psychosis (Pariante 2004; Ryan 2004), and in those at prodromal stages of psychosis (Garner 2005; Thompson 2007).
Higher levels of circulating cortisol and impaired regulation of the HPA axis have been reported, particularly among patients with psychotic depression and those in the acute phase of psychosis. It has been suggested that HPA axis dysfunction may cause or exacerbate psychotic and depressive symptoms and neuropsychological dysfunction. Supporting this is the observation that corticosteroid therapy used for a variety of medical conditions can often induce symptoms of depression and psychosis, including hallucinations and delusions, as well as cognitive impairment (Brown 2001). In addition, Cushing's syndrome (a condition characterised by hypercortisolaemia) is associated with significant cognitive impairment, which improves when cortisol levels have returned to normal following treatment (Starkman 1999). Atypical antipsychotics have been shown to suppress HPA axis activity (Cohrs 2006), and some evidence suggests that normalisation of HPA axis activity correlates with improvement in clinical symptoms among patients with schizophrenia (Zhang 2005). As a result, the HPA axis is increasingly viewed as an important therapeutic target in psychosis.
Why it is important to do this review
A large body of evidence suggests that stress and HPA axis function are important factors in the development of psychosis. Given the need for more benign treatments, particularly at earlier stages of psychosis, a review of the efficacy of antiglucocorticoid and related medications will be useful. These medications are increasingly popular, but given the uncertain efficacy and side effects for those with psychosis, a systematic review is essential. A Cochrane systematic review that examined use of antiglucocorticoids for mood disorders included nine studies (Gallagher 2005), and given the increasing number of trials investigating these medications for psychosis, this review is timely.
Objectives
1. To determine the effects of antiglucocorticoid and related drugs for the treatment of psychosis, when used alone or in combination with antipsychotic medication.
2. To determine whether the effects of these medications differs between those in the prodromal phase or first episode of psychosis and those with more established illness.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCTs). When a trial was described as 'double‐blind', but it was only implied that the study was randomised, we included it in a sensitivity analysis. If we noted no substantive differences within primary outcomes (see Types of outcome measures) when these 'implied randomisation' studies were added, we included these in the final analysis. If we observed a substantive difference, we used only clearly randomised trials and described results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those in which investigators allocated by using alternate days of the week.
Types of participants
We included people with a primary diagnosis of a psychotic disorder (including schizophrenia, schizophreniform disorder, schizoaffective disorder, psychotic depression and bipolar disorder with psychotic features) diagnosed by a clinician using any diagnostic system, those determined to be at ultra‐high risk (UHR) for psychosis and those in the prodromal phase of psychosis. We included individuals at all stages of psychosis (e.g. prodromal through chronic psychosis) treated in an inpatient or outpatient setting with any length of untreated or treated illness of any severity. We also included those with co‐morbidity.
Types of interventions
1. Pharmacological treatments targeting components of the hypothalamic‐pituitary‐adrenal (HPA) axis including the following main categories:
1.1 Glucocorticoid receptor antagonists (e.g. mifepristone); 1.2 Mineralocorticoid receptor antagonists (e.g. spironolactone); 1.3 Glucocorticoid receptor/mineralocorticoid receptor agonists (e.g. hydrocortisone, dexamethasone); 1.4 Corticotrophin‐releasing hormone antagonists (e.g. R121919, ORG 34116); and 1.5 Steroid‐synthesis inhibitors (e.g. metyrapone, ketoconazole) used alone or as adjunctive treatment.
2. Neuroactive steroids considered to have antiglucocorticoid effects, such as dehydroepiandrosterone (DHEA)
3. Comparison interventions
3.1 Placebo 3.2 Atypical antipsychotic treatment 3.3 Typical antipsychotic treatment 3.4 Antidepressant treatment 3.5 Other combination treatment (e.g. atypical or typical antipsychotic or both, antidepressant)
Types of outcome measures
We grouped outcomes into immediate (under two weeks), short‐term (two to 12 weeks) and long‐term (over 12 weeks).
Primary outcomes
1. Mental state
1.1 Clinically significant change in mental state (as defined by individual studies) 1.2 Average change in total psychotic symptom scores 1.3 Average endpoint in total psychotic symptom scores 1.4 Average change in positive symptom scores 1.5 Average endpoint in positive symptom scores 1.6 Average change in negative symptom scores 1.7 Average endpoint in negative symptom scores
Secondary outcomes
1. Global state
1.1 Relapse 1.2 Remission rate 1.3 Transition rate or time to onset of psychosis 1.4 Clinically important change in response (as defined by individual studies) 1.5 Leaving the study early
2. Mental state
2.1 Average change in depressive symptom scores 2.2 Average endpoint in depressive symptom scores 2.3 Average change in anxiety symptom scores 2.4 Average endpoint in anxiety symptom scores
3. Cognitive functioning
3.1 Clinically important change in cognitive functioning in any of the following domains: executive functioning, working memory, declarative learning and memory, vigilance/attention or psychomotor speed 3.2 Average endpoint in cognitive functioning scores 3.3 Average change in cognitive functioning scores
4. General functioning
4.1 Clinically important change in general functioning 4.2 Average change in general functioning scores 4.3 Average endpoint in general functioning scores
5. Adverse effects
5.1 General adverse effects 5.2 Serious adverse effects 5.3 Extrapyramidal symptoms (EPS) 5.4 Weight gain
6. Quality of life
6.1 Clinically significant change in quality of life (as defined by individual studies) 6.2 Average change in quality of life scores 6.3 Average endpoint in quality of life scores
'Summary of findings' table
We used the GRADE (Grading of Recommendations Assessment, Development and Evaluation) approach to interpret findings (Schünemann 2008) and used the GRADE profiler (GRADEPRO) to import data from RevMan 5.1 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effects of interventions examined and the sum of available data on all outcomes rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Mental state
Global state
General functioning
Adverse effects
Quality of life
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group’s Trials Register
The Trials Search Co‐ordinator (TSC) searched the Registry of Trials of the Cochrane Schizophrenia Group (2014 April 28) using the following phrase:
(*acetoxyandrost* or *aminoglutethimide* or *corticotropin* or *corticoid* or *dexamethasone* or *etomidate* or *hydrocortisone* or *ketoconazole* or *metyrapone* or *mifepristone* or *mitot?ne* or *org?34116* or *r?121919* or *ru?486* or *spironolactone* or *steroid* or *dehydroepiandrosterone*):ti,ab in REFERENCE or (*acetoxyandrost* or *aminoglutethimide* or *corticotropin* or *corticoid* or *dexamethasone* or *etomidate* or *hydrocortisone* or *ketoconazole* or *metyrapone* or *mifepristone* or *mitot?ne* or *org?34116* or *r?121919* or *ru?486* or *spironolactone* or *steroid* or *dehydroepiandrosterone*):sin in STUDY
The Registry of Trials of the Cochrane Schizophrenia Group is compiled by systematic searches of major resources (including Allied and Complementary Medicine Database (AMED), BIOSIS, Cumulative Index to Nursing and Allied Health Literature (CINAHL), EMBASE, MEDLINE, PsycINFO, PubMed and registries of clinical trials) and their monthly updates, handsearches and searches of grey literature and conference proceedings (see Group Module). No language, date, document type or publication status limitations are applied when records are included in the Register.
For previous searches, see Appendix 1.
Searching other resources
1. Reference lists
We searched reference lists of articles and other reviews retrieved from the search for relevant studies.
2. Handsearching
We handsearched published abstracts from the following conferences: International Early Psychosis Conference, Birmingham, October 2006; International Early Psychosis Conference, Vancouver, October 2004; Schizophrenia Research, 13th Biennial Winter Workshop, Davos, February 2006; and Schizophrenia Bulletin, 10th International Congress on Schizophrenia Research, Savannah, April 2005.
3. Personal communication
We contacted the authors of included trials to ask about additional trials, published or unpublished. We noted their responses in the Characteristics of included studies and Description of studies sections.
Data collection and analysis
Since the protocol for this review was published, the Cochrane Schizophrenia Group has updated its template for Methods sections, and we have updated these sections of the text to reflect this. For previous text, see also Appendix 2.
Selection of studies
Two review authors (BG, SH or LP) independently inspected the title and abstract of all articles identified in the searches. When disagreement occurred, we attempted to resolve this by discussion, and when doubt remained, we acquired the full article for further inspection. Once we had obtained the full article, we (BG, LP, SH, SB) independently decided whether the study met the review criteria. If we could not resolve disagreement by discussion, we sought further information and added these trials to the list of those awaiting assessment.
Data extraction and management
1. Extraction
Four review authors (BG, SH, LP, SB) independently extracted data from all included studies. When disputes arose, we discussed and documented decisions made and, if necessary, contacted authors of studies for clarification. For remaining problems, a third review author (BG, SH, LP) helped clarify issues and documented decisions made. We extracted data presented only in graphs and figures when possible, but we include these data only when two review authors independently reported the same result. We attempted to contact study authors through an open‐ended request to obtain missing information or clarification when necessary. For multi‐centre studies, when possible, we separately extracted data relevant to each component centre.
2. Management
2.1 Forms
We extracted data onto standard, predesigned, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and
the measuring instrument had not been written or modified by one of the trialists for that particular trial.
The ideal measuring instrument should be a self report or a report completed by an independent rater or relative (not the therapist). We realise that often this is not reported clearly, and under Description of studies, we noted whether this was the case.
2.3 Endpoint versus change data
Both endpoint and change data offer advantages. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change requires two assessments (baseline and endpoint), which can be difficult to perform in unstable and difficult to measure conditions such as schizophrenia. We have decided to use primarily endpoint data and to use change data only when the former are not available. We combined endpoint and change data in the analysis if possible, as we preferred to use mean differences (MDs) rather than standardised mean differences (SMDs) (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes often are not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to data before inclusion.
We entered into the analysis data from studies of at least 200 participants, for example, irrespective of the following rules, because skewed data pose less of a problem in large studies. We also entered change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed. We presented and entered change data into the statistical analyses.
For endpoint data:
When a scale started from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. Values lower than 1 strongly suggest a skew, and we excluded these data. If this ratio was higher than one but lower than two, skew was suggested. We entered the data and tested whether inclusion or exclusion of data changed the results substantially. Finally, if the ratio was larger than two, we included these data because skew was less likely (Altman 1996; Higgins 2011).
When a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS); Kay 1986), which can provide values from 30 to 210, we modified the calculation described above to take the scale starting point into account. In these cases, skew was present if 2 standard deviations (SD) > (S ‐ S min), where 'S' was the mean score and 'S min' was the minimum score.
2.5 Common measure
To facilitate comparison between trials, we intended, if possible, to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month), to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
When possible, we made efforts to convert outcome measures to dichotomous data, which can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' and 'not clinically improved'. It was generally assumed that a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS; Overall 1962) or the Positive and Negative Syndrome Scale (PANSS; Kay 1986) could be considered a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original study authors.
2.7 Direction of graphs
When possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for antiglucocorticoids. When keeping to this makes it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we reported data when the left of the line indicates an unfavourable outcome. This is noted in the relevant graphs.
Assessment of risk of bias in included studies
Again review authors BG, SH, LP and SB worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimation of effect and high risk of bias of the study, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the two raters disagreed, we determined the final rating by consensus, with involvement of another member of the review group (BG, LP, SH or SB). When inadequate details of randomisation and other characteristics of trials were provided, we contacted study authors to obtain further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial was to be allocated, again, we resolved this matter by discussion.
We noted the level of risk of bias in both the text of the review and the 'Summary of findings' table.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RRs are more intuitive (Boissel 1999) than odds ratios, and that odds ratios tend to be interpreted as RRs by clinicians (Deeks 2000). The number needed to treat for an additional harmful outcome (NNTH) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and in its interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table/s, when possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes, we estimated mean differences (MDs) and 95% CIs between groups. When different measurement tools were used to measure the same outcome, we estimated standardised mean differences (SMDs) and 95% CIs between groups.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster‐randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data pose problems. First, study authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
We included no cluster trials in this review. If we had identified such studies, we would have applied the following approach: When clustering was not accounted for in primary studies, we would present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We would seek to contact first authors of studies to obtain intraclass correlation coefficients and to ask for clustered data; we would adjust for these by using accepted methods (Gulliford 1999). When clustering had been incorporated into the analysis of primary studies, we would have presented these data as if obtained from a non‐cluster‐randomised study but with adjustment for the clustering effect.
We sought statistical advice and have been advised that binary data as presented in a report should be divided by a 'design effect'. This is calculated by using the mean number of participants per cluster (m) and the intraclass correlation coefficient (ICC) [Design effect = 1 + (m ‐ 1) * ICC] (Donner 2002). If the ICC was not reported, it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed with ICCs considered and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect, which occurs when an effect (e.g. pharmacological, physiological, psychological) of treatment in the first phase is carried over to the second phase. As a consequence of entry into the second phase, participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate when the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we used data only from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
When a study had involved more than two treatment arms, if relevant, we would have presented additional treatment arms in comparisons. If data had been binary, we would have simply added these and combined them within the two‐by‐two table. If data had been continuous, we would have combined data by applying the formula provided in Section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011). When additional treatment arms were not relevant, we would not have used these data. One trial in this review included multiple (four) treatment arms, of which two were irrelevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data and would not use them within analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' table/s by downgrading quality. Finally, we also downgraded quality within 'Summary of findings' table/s when total loss was 25% to 50%.
2. Binary
When attrition for a binary outcome was between 0 and 50%, and when these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). We assumed that all those leaving the study early had the same rates of negative outcome as those who completed the study, with the exception of the outcomes of death and adverse effects (for these outcomes, we used the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ for those who did not). We undertook a sensitivity analysis by testing how prone primary outcomes were to change when data only from people who completed the study to that point were compared with the ITT analysis under the above assumptions.
3. Continuous
3.1 Attrition
When attrition for a continuous outcome was between 0 and 50%, and when data only from people who completed the study to that point were reported, we reproduced these.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain missing values from study authors. If these were not available, when measures of variance for continuous data were missing, but an exact standard error and confidence intervals were available for group means, and P value or 't' value was available for differences in means, we could calculate SDs by following the rules provided in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011): When only the standard error (SE) was reported, we calculated SDs by using the formula: SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011) presents detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated SDs according to a validated imputation method based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude outcomes of a given study and thus to lose information. We nevertheless examined the validity of the imputations by performing a sensitivity analysis that excluded imputed values.
3.3 Assumptions about participants who left trials early or were lost to follow‐up
Various methods are available to account for participants who leave trials early or are lost to follow‐up. Some trials present only the results of study completers, others use the method of last observation carried forward (LOCF) and more recently investigators have used methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) as the standard. Although the latter methods seem somewhat better than LOCF (Leon 2006), we believe that the high percentage of participants leaving studies early and differences between groups in reasons for leaving studies early often represent the core problem in randomised schizophrenia trials. Therefore we did not exclude studies on the basis of the statistical approach used. However, we preferred to use data from studies based on more sophisticated approaches. For example, we preferred MMRM or multiple imputation to LOCF, and we presented completer analyses only if no ITT data were available. Moreover, we addressed this issue in the item "Incomplete outcome data" for the risk of bias tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without knowledge of comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise. When such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without knowledge of comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise. When such methodological outliers arose, we fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on both magnitude and direction of effects and strength of evidence for heterogeneity (e.g. P value from Chi2 test, confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic was interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When we found substantial levels of heterogeneity in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when dissemination of research findings is influenced by the nature and direction of results. These are described in Section 10.1 of the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011). We attempted to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol versus those in the published report. If the protocol was not available, we compared outcomes listed in the Methods section of the trial report versus actually reported results.
2. Funnel plot
Reporting biases arise when dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful for investigating reporting biases but are of limited power for detecting small‐study effects. We did not use funnel plots for outcomes when we identified 10 or fewer studies, or when all studies were of similar size. In other cases, when use of funnel plots is possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that no closed argument can be presented for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that different studies are estimating different, yet related, intervention effects. This often seems to be true, and the random‐effects model takes into account differences between studies even when no statistically significant heterogeneity is observed. However, the random‐effects model does present a disadvantage. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can inflate or deflate effect size. We chose a fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
Diagnosis
'Prodrome' versus 'first‐onset' versus 'chronic'
Type of antiglucocorticoid drug
Adults versus adolescents
Length of untreated illness
1.2 Clinical state, stage or problem
We also proposed to undertake this review to provide an overview of the effects of antiglucocortoicoids for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state or stage and with similar problems.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed studies outside of the rest to see whether homogeneity was restored. For this review, we decided that should this occur when data contributed no more than around 10% of total weighting to the 'Summary of findings', we would present the data. If this was not the case, we would have pooled data and would have discussed issues. We know of no supporting research for this 10% cut‐off, but we are investigating the use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or other versions of this review. We did not undertake analyses of this kind.
Sensitivity analysis
We performed sensitivity analyses to investigate the degree to which effect sizes depended on assumptions made by review authors. Towards this end, we excluded trials with 'high risk' or 'unclear risk' for allocation concealment, trials with 'high risk' or 'unclear risk' for blinding of outcome assessment and trials with 'high risk' or 'unclear risk' for ITT analysis.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if in some way the study report implied randomisation. For primary outcomes, we included these studies, and if we observed no substantive differences when implied randomised studies were added to those with better descriptions of randomisation, we included all data from these studies.
2. Assumptions for lost binary data
When assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of primary outcomes when we used our assumption/s, and when we used only data from people who had completed the study to that point. If we noted a substantial difference, we reported results and discussed them but continued to employ our assumption.
When assumptions had to be made regarding missing SD data (see Dealing with missing data), we compared the findings of primary outcomes when we used our assumption/s, and when we used only data from people who had completed the study to that point. We undertook a sensitivity analysis to test how prone results were to change when completer‐only data were compared with imputed data on the basis of the above assumption. If we noted a substantial difference, we reported results and discussed them but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no additional details available), allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of effect estimates, we would have included in the analysis data from these trials.
4. Imputed values
We also undertook a sensitivity analysis to assess the effects of including data from trials for which we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from excluded trials with those of other trials contributing to the outcome but presented them separately.
5. Fixed‐effect and random‐effects
We synthesised all data using a fixed‐effect model; however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether this approach would alter the significance of the results.
Results
Description of studies
For substantive descriptions of studies, please see Characteristics of included studies and Characteristics of excluded studies.
Results of the search
In total, we identified 1847 publications through the electronic search strategy. We judged most of these (1785) to be irrelevant on the basis of information provided in the title and the abstract, leaving 62 reports of studies for possible inclusion in the review. After obtaining full publications of these studies, we excluded 25 additional publications (see Characteristics of excluded studies tables). Of the remaining 37 publications, nine are awaiting assessment. The remaining 28 publications described a total of 13 trials, which met the criteria for inclusion in the review. We classified two of these trials as ongoing, leaving 11 trials for analysis (Figure 1).
1.
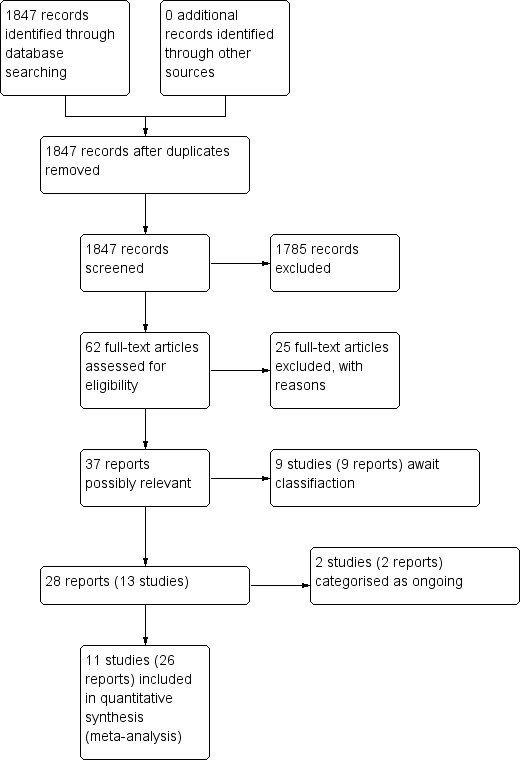
Study flow diagram 2009, 2014 searches.
Included studies
1. Setting
Five trials (Belanoff 2001; Nachshoni 2005; Newcomer 1998; Strous 2003; Strous 2007) were conducted in an inpatient setting, three trials (DeBattista 2006; Flores 2006; Ritsner 2006) consisted of both inpatients and outpatients and the remaining three trials (Gallagher 2005; Marco 2002; Ritsner 2010) took place in an outpatient setting.
2. Participants
All trials were conducted in an adult population. The youngest participant was 18 years of age and the oldest was 74 years.
Nine trials (DeBattista 2006; Flores 2006; Gallagher 2005; Marco 2002; Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007) used the Structured Clinical Interview for DSM Disorders (SCID) to determine the diagnosis. In Belanoff 2001 and Newcomer 1998, diagnoses were determined by clinician interview(s).
Eight trials required a diagnosis of schizophrenia or schizoaffective disorder based on the Diagnostic and Statistical Manual (DSM) III‐R (Marco 2002; Newcomer 1998) or IV (Gallagher 2005; Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007). Four of these trials (Strous 2003; Strous 2007; Ritsner 2006; Ritsner 2010) consisted of participants with chronic schizophrenia/schizoaffective disorder of at least two years' duration. In addition, Strous 2003 required participants to score 25 or higher on the Scale for the Assessment of Negative Symptoms (SANS) for inclusion in the trial. Marco 2002 and Gallagher 2005 required patients to be clinically stable but symptomatic (duration of illness not stated). In the trial by Nachshoni 2005, hospitalised patients with schizophrenia with any form of extrapyramidal symptoms (EPS) were referred for study recruitment (duration of illness not stated).
Three trials included participants with a diagnosis of psychotic major depression based on DSM IV (Belanoff 2001; DeBattista 2006; Flores 2006). DeBattista 2006 required participants to score 38 or higher on the BPRS and 20 or higher on the Hamilton Rating Scale for Depression (HAMD); Flores 2006 required participants to score five or higher on the BPRS four‐item positive symptom subscale and 21 or higher on the HAMD (21‐item) for inclusion in the trial.
3. Size
We included 11 trials involving 509 participants (Belanoff 2001; DeBattista 2006; Flores 2006; Gallagher 2005; Marco 2002; Nachshoni 2005; Newcomer 1998; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007). Three trials (Belanoff 2001; Gallagher 2005; Ritsner 2006) were of a cross‐over design. We used in the analysis only data from the first intervention phase of the cross‐over trial. The trial by Ritsner 2010 involved multiple treatment arms; we used in the analysis only data from DHEA and placebo treatment arms. Sample size varied widely between studies, ranging from five to 221 participants.
4. Interventions
All trials except for two (Belanoff 2001; DeBattista 2006) administered the intervention treatment as an adjunct to antipsychotic or regular treatment. In the case of Strous 2007, antipsychotic treatment consisted of stable doses of olanzapine only. In Newcomer 1998, participants received ongoing typical antipsychotic treatment, but anticholinergics were also administered. For all other trials, regular treatment involved a combination of atypical, typical and/or antidepressant or other psychotropic medication.
Four trials administered mifepristone (Belanoff 2001; DeBattista 2006; Flores 2006; Gallagher 2005) at a dose of 600 mg/d for between four and eight days. Five trials administered DHEA (Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007); doses ranged from 100 mg/d to 400 mg/d, and the intervention duration ranged from seven days to 12 weeks (see Characteristics of included studies for details). Ritsner 2010 included four treatment arms (pregnenolone ‐ 30 mg, pregnenolone ‐ 200 mg, DHEA ‐ 400 mg and placebo) and reported only data from DHEA and placebo arms. One trial (Newcomer 1998) administered successive doses (0.5, 1, 1, 1 mg) of dexamethasone over four days. Another trial (Marco 2002) administered ketoconazole titrated up to 800 mg/d over four weeks.
5. Outcomes
5.1 Assessment times
All trials conducted baseline and post‐treatment assessments (i.e. immediately following cessation of the intervention). Three trials (DeBattista 2006; Gallagher 2005; Newcomer 1998) conducted follow‐up assessments up to a maximum of 21 days following the end of the intervention.
5.2 Missing outcomes
None of the trials examined relapse or remission rates. No trials involved first‐episode psychosis patients or individuals at ultra‐high risk for psychosis; therefore we did not examine time to transition to psychosis or transition rates.
5.3 Outcome scales
5.3.1 Mental state
5.3.1.1 Brief Psychopathological Rating Scale ‐ BPRS (Overall 1962) This observer‐rated scale is used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. Items are rated from zero (absent) to six (severe) according to clearly defined anchor points. Scores can range from 0 to 126, with high scores indicating more severe symptoms. The BPRS has displayed good concurrent and discriminant validity in studies involving psychotic participants (Faustman 1994) and high inter‐rater reliability ranging from 0.67 to 0.88 for individual items and overall score (Hedlund 1980). The BPRS positive symptom subscale (as defined by studies included in this review) consists of the following four core psychotic items of the BPRS: suspiciousness, hallucinatory behaviour, disorganised thinking and unusual thought content.
5.3.1.2 Positive and Negative Symptom Scale ‐ PANSS (Kay 1986) This observer‐rated scale is used to evaluate positive, negative and other symptom dimensions in schizophrenia. This scale consists of 30 items, each of which can be defined on a seven‐point scoring system ranging from one (absent) to seven (extreme). This scale can be divided into three subscales for measuring the severity of general psychopathology (PANSS‐G), positive symptoms (PANSS‐P) and negative symptoms (PANSS‐N).
5.3.1.3 Scale for Assessment of Negative Symptoms (SANS) (Andreasen 1989) This observer‐rated scale is widely used to assess negative symptoms in psychotic illness. It includes 25 items that collapse to five subscales: affective flattening, alogia, avolition‐apathy, anhedonia‐asociality and attention. All items are rated from zero to five (absent to severe).
5.3.2.1 Global state
5.3.2.1.1 Response rates Two trials (DeBattista 2006; Flores 2006) used one or more of the following measures of efficacy based on BPRS or HAMD scale level of response: (1) 30% reduction in total BPRS score, (2) 50% reduction in BPRS positive symptoms subscale (four items of the BPRS); or (3) 50% reduction in HAMD.
5.3.2.2.2 Hamilton Rating Scale for Depression (HAMD) (Hamilton 1960) and Anxiety (HAMA) (Hamilton 1959) The HAMD is an observer‐rated scale that is used to assess the presence and severity of depressive states. This scale has 17‐item and 21‐item versions and is widely used for clinical assessment of depressive symptoms. The HAMA is widely used to assess the severity of anxiety symptoms. It consists of 14 items, each defined by a series of symptoms.
5.3.2.2.3 Calgary Depression Scale for Schizophrenia – CDSS (Addington 1997) This observer‐rated scale was specifically developed for assessment of the level of depression in schizophrenia. The scale is designed to assess the presence of depression separate from other dimensions of psychopathology in schizophrenia such as negative symptoms.
5.3.2.3 Cognitive functioning
Seven trials assessed neurocognitive functioning across several domains using a range of cognitive tests. Three trials (Gallagher 2005; Ritsner 2006; Ritsner 2010) used the Cambridge Neuropsychological Test Automated Battery (CANTAB). Available data were insufficient for meta‐analysis. We have presented data from only one trial.
5.3.2.4 General functioning
5.3.2.4.1 Global Assessment of Functioning ‐ GAF (American Psychiatric Association 2000) The GAF is a numerical observer‐rated scale (0 to 100) that is used to assess social, occupational and psychological functioning.
5.3.2.5 Adverse effects
5.3.2.5.1 Extrapyramidal Symptoms Rating Scale – ESRS (Chouinard 1980) This observer‐rated scale assesses parkinsonian symptoms, dyskinetic movements and tardive dyskinesia. High scores indicate severe levels of movement disorder.
5.3.2.5.2 The St. Hans Rating Scale ‐ SHRS (Gerlach 1993) This multi‐dimensional observer‐rated scale is used to evaluate neuroleptic‐induced hyperkinesia, parkinsonism, akathisia and dystonia.
5.3.2.5.3 Abnormal Involuntary Movement Scale – AIMS (Guy 1976) This observer‐rated scale is used to assess abnormal involuntary movements associated with antipsychotic drugs, such as tardive dyskinesia and chronic akathisia. Scoring consists of rating movement severity in anatomical areas (facial/oral, extremities and trunk) on a five‐point scale (zero to four). A low score indicates low levels of dyskinetic movement.
5.3.2.5.4 Barnes Akathisia Rating Scale – BARS (Barnes 1989) This four‐item observer‐rated scale is used to assess the presence and severity of drug‐induced akathisia. It is the most widely used comprehensive rating scale for akathisia and includes both objective items (e.g. observed restlessness) and subjective items (e.g. patient's awareness of restlessness and related distress), together with a global clinical assessment of akathisia.
5.3.2.5.5 Simpson and Angus Scale – SAS (Simpson 1970) This observer‐rated scale is used to evaluate the presence and severity of drug‐induced parkinsonian symptoms. It is a 10‐item rating scale, and each item is rated on a five‐point scale from zero (complete absence of the condition) to four (extreme presence of the condition).
5.3.2.5.6 Weight gain
Only one trial (Strous 2007) reported average pretreatment and post‐treatment patient weight. It was not possible to analyse weight gain.
5.3.2.6 Quality of life
5.3.2.4.1 Observer‐rated Quality of Life scale (QOL) (Heinrichs 1984)
5.3.2.4.2 The Social and Occupational Functioning Assessment Scale (SOFAS) (American Psychiatric Association 2000) This numerical observer‐rated scale (0 to 100) is used to assess social and occupational functioning.
Excluded studies
We excluded 25 reports of studies; 12 (Barkai 1985; Brambilla 1988; Katz 2002; Kim 1960; Kline 1968; Korsgaard 1981; Lembke 2013; Rees 1951; Rees 1956; Rothschild 2005; Schatzberg 2003; Simpson 2005) were not randomised, eight (Beasley 1998; David 1999; Iager 1986; Lane 2001; Loranger 1968; Miodownik 2011; Stein 1984; Tollefson 1998) did not involve an antiglucocorticoid or related treatment, two were not intervention studies (Harrigan 2004; Nihalani 2007), one did not involve patients with a psychotic disorder (Young 2004), one was not placebo controlled (Belanoff 2002) and one reported no data that could be included (Silbergeld 1973). Details can be found in the Characteristics of excluded studies tables.
Ongoing studies
Two trials (Jarskog 2009; Solvason 2008) were described as ongoing trials. We identified no subsequent publications from these trials in our search.
Studies awaiting assessment
Nine publications (Hardwick 1957; Kleiser 1984; Owen 1996; Pivac 2002; Sluchevskii 1986; Smidt 1988; Volk 1976; Volk 1977; Watson 2002 (N0573099798)) are awaiting assessment, as we were unable to obtain full details on them. One of these publications (Smidt 1988) is a description of a study registered with ClinicalTrials.gov in 2002, but no published reports have followed.
Risk of bias in included studies
For a summary of risk of bias across all trials, see Figure 2 and Figure 3.
2.
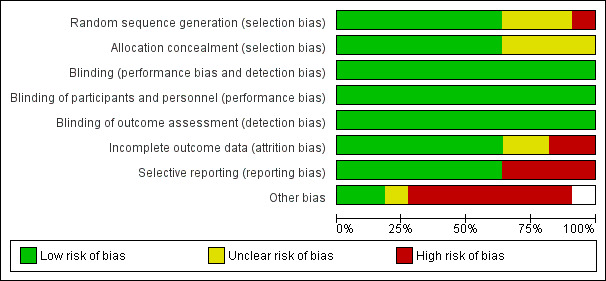
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
3.
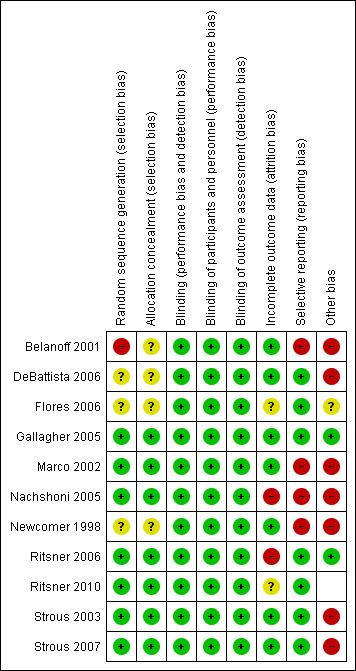
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All included trials were stated to be randomised. Seven trials (Gallagher 2005; Marco 2002; Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007) used a computer‐generated randomisation list or random number generation. These trials confirmed that allocation was concealed by a key‐based numerical code on envelopes (Marco 2002) or by an independent pharmacist (Gallagher 2005; Ritsner 2006; Ritsner 2010), or it was maintained under lock and key in a concealed fashion (Nachshoni 2005; Strous 2003; Strous 2007). These trials were rated as 'low risk'. Three trials (DeBattista 2006; Flores 2006; Newcomer 1998) provided no details of randomisation generation nor concealment of allocation and were rated as 'unclear risk'. The remaining trial (Belanoff 2001) likewise provided no details of randomisation generation nor concealment of allocation and reported an imbalance in length of illness between groups, which calls into question the success of randomisation. This trial was rated 'high risk'.
Blinding
All included trials were described as 'double‐blind'. When it was unclear whether the term 'double‐blind' referred to participants, providers and/or outcome assessors, we sought additional information from the study authors. Eight trials (Flores 2006; Gallagher 2005; Marco 2002; Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007) confirmed that participants, providers and outcome assessors were blind to the treatment condition.
Incomplete outcome data
Three trials reported no participants leaving the study early (Belanoff 2001; Gallagher 2005; Marco 2002). DeBattista 2006 performed efficacy analyses on the ITT sample (n = 221), which consisted of all randomly assigned participants who received at least one dose of study medication. Data were observed at day 28 for 170 of the 221 participants (77%). For the 51 participants with missing data at day 7 or day 28, BPRS and HAMD data were imputed by using a mixed‐effects model for repeated measurements. In Nachshoni 2005, of the 34 randomly assigned participants, four were withdrawn for failing to meet trial requirements because investigators altered medication dose during the study and conducted the analysis on 30 participants. In Flores 2006, one participant dropped out during the intervention and researchers conducted an observed case analysis. Ritsner 2006 randomly assigned 62 participants. Seven participants failed to complete the 12 weeks of the cross‐over randomisation phase because they withdrew study consent after randomisation but had previously received the first dose of study medication. Investigators conducted an analysis that included 55 participants. Strous 2003 randomly assigned 30 participants. Investigators eliminated three participants from study analysis after they failed to complete three weeks of the randomisation phase (selected a priori). They imputed missing data on four participants by using last observation carried forward (LOCF) (i.e. analysis was conducted on 27 participants). In Strous 2007, nine participants dropped out during the intervention and researchers conducted an ITT analysis. In Newcomer 1998, analyses excluded participants with missing data (n = 3); however, investigators also ran analyses that included all participants (n = 19) by applying conservative adjustments for missing data. Ritsner 2010 randomly assigned 58 participants to one of four treatment groups (pregnenolone ‐ 30 mg, pregnenolone ‐ 200 mg, DHEA ‐ 400 mg and placebo). Fourteen participants left the study early after completing at least four weeks of the trial. Investigators conducted analyses on 44 participants, allocated 16 to placebo (five of whom left the study early) and allocated 16 to DHEA (three of whom left the study early). They conducted analyses on 11 participants in the placebo group and in 13 in the DHEA group.
Selective reporting
Most trials reported all outcome measures. Belanoff 2001 did not report data from the Clinical Global Impression (CGI) scale and paragraph recall test. Marco 2002 stated that they found no significant differences, but investigators provided no data for the following outcome measures; PANSS, Beck Depression Inventory (BDI), Bunney‐Hamburg Global Rating Scale and neurocognitive tests. In the trial by Nachshoni 2005, which investigated effects of DHEA administration on medication‐induced EPS, the frequency of participants demonstrating tardive dyskinesia (10%) or dystonia (0%) was very low; therefore, researchers could not analyse these measures. Finally, Newcomer 1998 did not report symptomatic outcomes (e.g. BPRS/PANSS, SANS).
Other potential sources of bias
Other potential sources of bias included the small sample size in some trials (e.g. Belanoff 2001, n = 5). Baseline imbalances in gender (Marco 2002; Strous 2003), length of illness (Belanoff 2002) and symptom severity (Strous 2007) were evident between groups. In Marco 2002, participants receiving ketoconazole had higher baseline cortisol levels and appeared to be taking a greater number of concomitant medications, suggesting that they may have had a more severe or complex disorder. Some study authors in DeBattista 2006 were employed by or received funding from the sponsor, and in Flores 2006, one study author had a financial interest in the company that licensed IP (Intellectual Property) for use of mifepristone in the treatment of patients with psychotic depression.
Effects of interventions
See: Table 1; Table 2; Table 3
1. Any antiglucocorticoid vs placebo as sole treatment (data only for mifepristone)
Only Belanoff 2001 and DeBattista 2006, both providing mifepristone treatment, could be included in this comparison (total n = 226).
1.1 Mental state
1.1.1 General: average endpoint score (BPRS total scores, higher score = poor)
Only Belanoff 2001 (a four‐day trial) reported continuous data and observed no statistically significant differences between intervention and control (n = 5, 1 RCT, MD ‐5.20, 95% CI ‐17.91 to 7.51; Analysis 1.1).
1.1. Analysis.
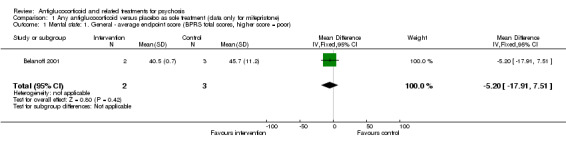
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 1 Mental state: 1. General ‐ average endpoint score (BPRS total scores, higher score = poor).
1.1.2 Specific: depression: average endpoint score (HAMD total, higher score = poor)
Belanoff 2001 also reported continuous data for this outcome and described no statistically significant differences between intervention and control (n = 5, 1 RCT, MD 1.67, 95% CI ‐16.44 to 19.78; Analysis 1.2).
1.2. Analysis.
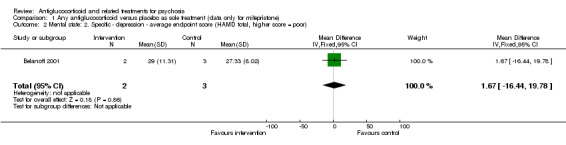
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 2 Mental state: 2. Specific ‐ depression ‐ average endpoint score (HAMD total, higher score = poor).
1.2 Global state
1.2.1 General ‐ no clinically significant improvement (< 30% improvement BPRS)
DeBattista 2006 reported very short‐term data (immediate) defined as at least a 30% reduction in BPRS scores. They noted no differences between intervention and control (n = 221, 1 RCT, RR 1.49, 95% CI 0.93 to 2.38). Twenty‐one days after cessation of the intervention (mifepristone, short‐term), DeBattista 2006 assessed this outcome and provided data revealing a difference in favour of treatment (n = 221, 1 RCT, RR 0.58, 95% CI 0.38 to 0.89; Analysis 1.3) with a number needed to treat for additional beneficial outcome (NNTB) value of seven.
1.3. Analysis.
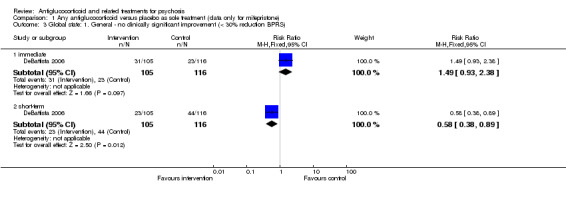
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 3 Global state: 1. General ‐ no clinically significant improvement (< 30% reduction BPRS).
1.2.2 Specific: positive symptoms ‐ no clinically significant improvement in BPRS positive symptoms (< 50% improvement BPRS, PSS)
For the very short term, DeBattista 2006 reported outcome data defined as at least a 50% reduction in BPRS positive symptom scores and observed no statistically significant differences between intervention and control (n = 221, 1 RCT, RR 1.49, 95% CI 0.85 to 2.64). After three weeks, the same study found a statistically significant difference in favour of treatment (n = 221, 1 RCT, RR 0.60, 95% CI 0.43 to 0.84; Analysis 1.4) with an NNTB value of five.
1.4. Analysis.
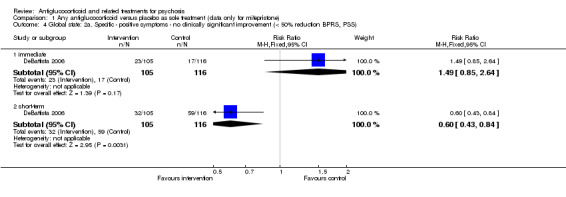
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 4 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement (< 50% reduction BPRS, PSS).
1.2.3 Specific: depressive symptoms ‐ no clinically significant improvement (< 50% improvement HAMD)
Very short‐term data from DeBattista 2006 defined at least a 50% reduction in HAMD scores as important and reported no statistically significant differences between intervention and control (n = 221, 1 RCT, RR 1.55, 95% CI 0.84 to 2.84). By three weeks, no effect was evident (n = 221, 1 RCT, RR 0.83, 95% CI 0.58 to 1.19; Analysis 1.5).
1.5. Analysis.
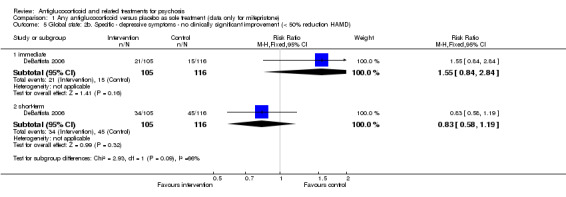
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 5 Global state: 2b. Specific ‐ depressive symptoms ‐ no clinically significant improvement (< 50% reduction HAMD).
1.2.4 Leaving the study early
Both trials recorded the number of participants leaving the study early and noted no clear differences between intervention and control (n = 226, 2 RCTs, RR 1.24, 95% CI 0.77 to 2.01; Analysis 1.6).
1.6. Analysis.
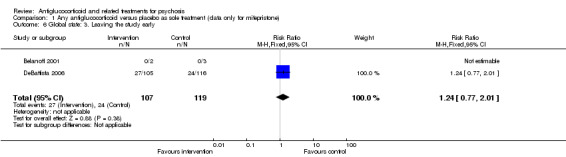
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 6 Global state: 3. Leaving the study early.
1.3 Adverse effects
1.3.1 General: overall number of events
Both trials reported the overall rate of adverse events and observed no significant differences between intervention and control (n = 226, 2 RCTs, RR 0.92, 95% CI 0.77 to 1.09; Analysis 1.7).
1.7. Analysis.
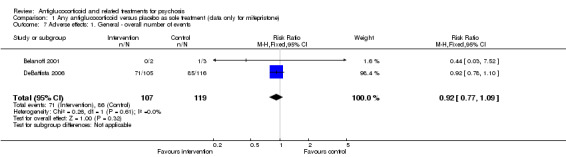
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 7 Adverse effects: 1. General ‐ overall number of events.
1.3.2 Specific: various effects
DeBattista 2006 reported a long series of adverse events including allergy, various cardiovascular difficulties, central nervous system problems, gastrointestinal complaints, metabolic issues and various pains. None were prevalent or more common in the mifepristone group (Analysis 1.8).
1.8. Analysis.
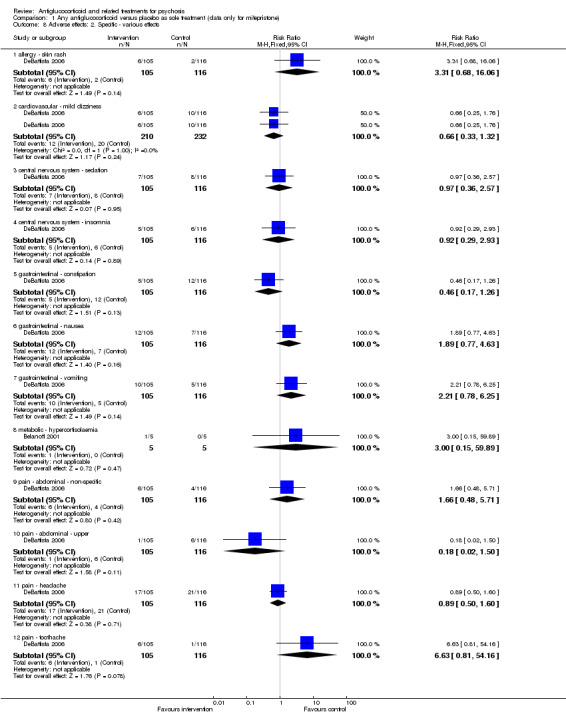
Comparison 1 Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone), Outcome 8 Adverse effects: 2. Specific ‐ various effects.
2. Any antiglucorticoid vs placebo as adjunct to atypical antipsychotic treatment (data only for DHEA)
Only Strous 2007, which provided DHEA treatment, could be included in this comparison (n = 40).
2.1 Mental state
2.1.1 General: average endpoint score (PANSS total, high score = poor)
Investigators reported no statistically significant differences between DHEA and control (n = 40, 1 RCT, MD ‐1.70, 95% CI ‐10.78 to 7.38; Analysis 2.1).
2.1. Analysis.
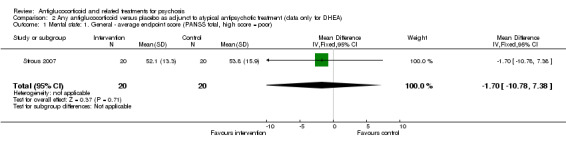
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 1 Mental state: 1. General ‐ average endpoint score (PANSS total, high score = poor).
2.1.2. Specific: positive and negative symptoms ‐ average endpoint score (PANSS subscales, high score = poor)
Data from the PANSS negative symptom subscale revealed no statistically significant differences between intervention and control (n = 40, 1 RCT, MD 0.70, 95% CI ‐2.63 to 4.03). This also applied to the positive subscale (n = 40, 1 RCT, MD ‐1.00, 95% CI ‐3.20 to 1.20; Analysis 2.2).
2.2. Analysis.
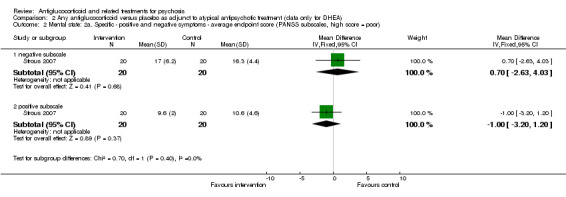
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 2 Mental state: 2a. Specific ‐ positive and negative symptoms ‐ average endpoint score (PANSS subscales, high score = poor).
2.1.3 Specific: negative symptoms and depression (high score = poor, data skewed)
Data for the SANS were skewed and are presented as 'Other data' in Analysis 2.3. They do not suggest a clear effect favouring either approach.
2.3. Analysis.
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 3 Mental state: 2b. Specific ‐ negative symptoms ‐ average endpoint scores (SANS negative subscale, high score = poor, data skewed).
| Mental state: 2b. Specific ‐ negative symptoms ‐ average endpoint scores (SANS negative subscale, high score = poor, data skewed) | ||||
|---|---|---|---|---|
| Study | Interventions | Mean | SD | N |
| Strous 2007 | DHEA | 34.2 | 20.2 | 20 |
| Strous 2007 | Placebo | 31.2 | 22.7 | 20 |
Average depression endpoint scores (CDSS total) were also skewed but did not clearly favour either group (Analysis 2.4).
2.4. Analysis.
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 4 Mental state: 2c. Specific ‐ depression ‐ average endpoint scores (CDSS total, high score = poor, skewed data).
| Mental state: 2c. Specific ‐ depression ‐ average endpoint scores (CDSS total, high score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Strous 2007 | DHEA | 2.0 | 2.7 | 20 |
| Strous 2007 | Placebo | 3.4 | 4.0 | 20 |
2.2 Global state: leaving the study early
Researchers noted no differences between intervention and control (n = 40, 1 RCT, RR 0.80, 95% CI 0.25 to 2.55; Analysis 2.5).
2.5. Analysis.
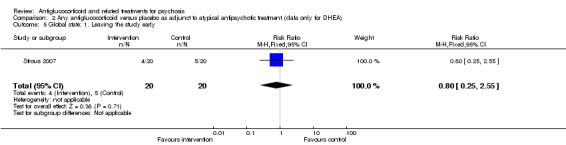
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 5 Global state: 1. Leaving the study early.
2.3 Adverse effects
2.3.1 Extrapyramidal symptoms (EPS) ‐ average endpoint scores (high scores = poor)
Investigators reported no statistically significant differences between intervention and control for parkinsonism when using the SAS (n = 40, 1 RCT, MD 0.00, 95% CI ‐0.88 to 0.88; Analysis 2.6).
2.6. Analysis.
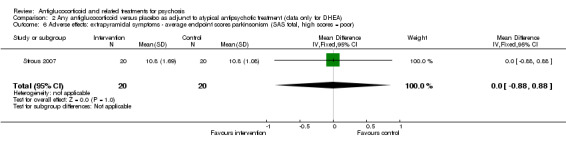
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 6 Adverse effects: extrapyramidal symptoms ‐ average endpoint scores parkinsonism (SAS total, high scores = poor).
They provided data for other EPS such as akathisia and tardive dyskinesia but only for the intervention groups (Table 4). They could undertake no analyses.
1. Adverse effect data for intervention group (control group not reported).
| Outcome | Measure | Mean | SD | N |
| Akathisia | BAS total | 0.05 | 0.24 | 20 |
| Tardive dyskinesia | AIMS total | 0.7 | 2.44 | 20 |
2.3.2 Weight gain ‐ average body weight endpoint
Study authors reported no differences between intervention and control (n = 40, 1 RCT, RR 5.20, 95% CI ‐4.51 to 14.91; Analysis 2.7).
2.7. Analysis.
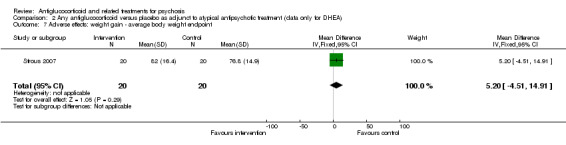
Comparison 2 Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA), Outcome 7 Adverse effects: weight gain ‐ average body weight endpoint.
3. Any antiglucocorticoid vs placebo as adjunct to combination treatment
Eight trials were included in this comparison involving the following intervention treatments: mifepristone (Flores 2006; Gallagher 2005), ketoconazole (Marco 2002), DHEA (Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003) and dexamethasone (Newcomer 1998).
3.1 Mental state
3.1.1 General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, higher score = poor)
All trials except Newcomer 1998 provided an overall psychotic symptom score. Data from Nachshoni 2005 were skewed and therefore are presented as 'Other data' in Analysis 3.2. For the remaining six trials including a total of 171 participants, researchers reported no overall statistically significant differences between intervention and control for immediate follow‐up (n = 171, 6 RCTs, SMD 0.01, 95% CI ‐0.29 to 0.32; Analysis 3.1). The test for subgroup differences was not statistically significant (Chi² = 4.58, df = 2, P value = 0.10, I² = 56.4%).
3.2. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 2 Mental state: 1b. General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, high score = poor, skewed data).
| Mental state: 1b. General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, high score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Nachshoni 2005 | DHEA | 21.0 | 12.5 | 15 |
| Nachshoni 2005 | Placebo | 22.2 | 8.3 | 15 |
3.1. Analysis.
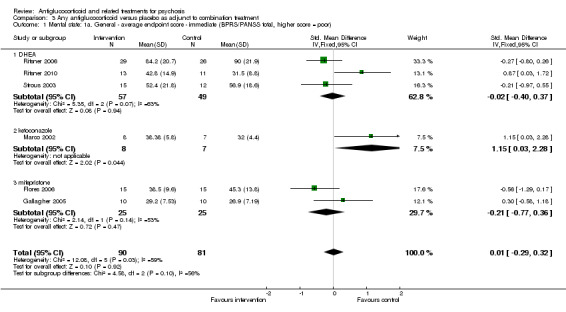
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 1 Mental state: 1a. General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, higher score = poor).
a. DHEA
Researchers observed no statistically significant differences between DHEA and control (n = 106, 3 RCTs, SMD ‐0.02, 95% CI ‐0.40 to 0.37).
b. Ketoconazole
The one trial involving ketoconazole (Marco 2002) showed a statistically significant difference in favour of control (n = 15, 1 RCT, SMD 1.15, 95% CI 0.03 to 2.28).
c. Mifepristone
Investigators noted no statistically significant differences between intervention and control (n = 50, 2 RCTs, SMD ‐0.21, 95% CI ‐0.77 to 0.36).
3.1.2 General: average endpoint score ‐ short term ‐ only mifepristone (BPRS total, higher score = poor)
Gallagher 2005 conducted follow‐up assessment 14 days after cessation of treatment. Study authors described no significant differences between intervention and control (n = 20, 1 RCT, MD 2.10, 95% CI ‐2.86 to 7.06; Analysis 3.3).
3.3. Analysis.
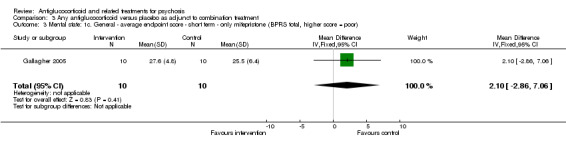
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 3 Mental state: 1c. General ‐ average endpoint score ‐ short term ‐ only mifepristone (BPRS total, higher score = poor).
3.1.3 Specific: positive symptoms ‐ average endpoint scores ‐ immediate (BPRS/PANSS positive subscale, higher score = poor)
Five of the eight trials provided a measure of positive psychotic symptoms (Flores 2006; Marco 2002; Ritsner 2006; Ritsner 2010; Strous 2003). Overall investigators described no statistically significant differences between intervention and control (n = 151, 5 RCTs, SMD ‐0.07, 95% CI ‐0.40 to 0.25) and no statistically significant subgroup differences (Analysis 3.4).
3.4. Analysis.
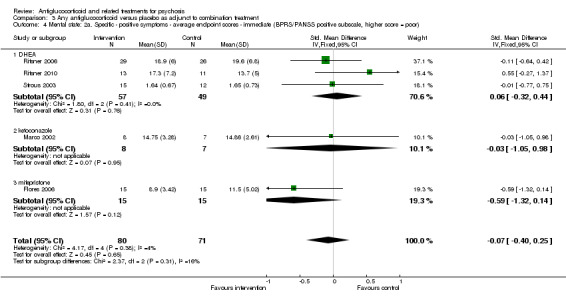
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 4 Mental state: 2a. Specific ‐ positive symptoms ‐ average endpoint scores ‐ immediate (BPRS/PANSS positive subscale, higher score = poor).
3.1.4 Specific: negative symptoms ‐ average endpoint scores ‐ immediate (PANSS negative subscale, higher score = poor)
Four of the eight trials reported a measure of negative symptoms (Marco 2002; Ritsner 2006; Ritsner 2010; Strous 2003). Overall, researchers observed no statistically significant differences between intervention and control (n = 94, 3 RCTs, MD 2.21, 95% CI ‐0.14 to 4.55), but the test for subgroup differences was statistically significant (Chi² = 7.64, df = 1, P value = 0.006, I² = 86.9%; Analysis 3.5). Data from Strous 2003 (SANS) were skewed and are presented as 'Other data' in Analysis 3.6.
3.5. Analysis.
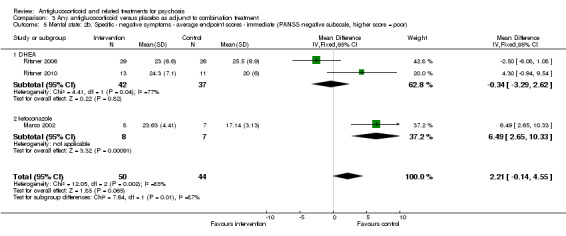
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 5 Mental state: 2b. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate (PANSS negative subscale, higher score = poor).
3.6. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 6 Mental state: 2c. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate ‐ only DHEA (SANS, high score = poor, skewed data).
| Mental state: 2c. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate ‐ only DHEA (SANS, high score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Strous 2003 | DHEA | 26.53 | 18.44 | 15 |
| Strous 2003 | Placebo | 38.42 | 18.51 | 15 |
a. DHEA
The two trials (Ritsner 2006; Ritsner 2010) reported no statistically significant differences between intervention and control (n = 79, 2 RCTs, MD ‐0.34, 95% CI ‐3.29 to 2.62), but the test for subgroup differences was statistically significant (Chi² = 4.41, df = 1, P value = 0.04, I² = 77%).
b. Ketoconazole
Marco 2002 observed a statistically significant difference in favour of control (n = 15, 1 RCT, MD 6.49, 95% CI 2.65 to 10.33).
3.1.5 Specific: anxiety symptoms ‐ average endpoint scores ‐ data only for DHEA (HAMA total, higher score = poor, skewed data)
Only Strous 2003 reported an anxiety symptom score, but the data were skewed and are presented as 'Other data' in Analysis 3.7.
3.7. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 7 Mental state: 2d. Specific ‐ anxiety symptoms ‐ average endpoint scores ‐ only DHEA (HAMA total, higher score = poor, skewed data).
| Mental state: 2d. Specific ‐ anxiety symptoms ‐ average endpoint scores ‐ only DHEA (HAMA total, higher score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Strous 2003 | DHEA | 2.67 | 2.64 | 15 |
| Strous 2003 | Placebo | 5.42 | 4.93 | 12 |
3.1.6 Specific: depression ‐ average endpoint scores ‐ data only for mifepristone (HAMD total, higher score = poor)
3.1.6.1. Immediate
Four trials provided a depression symptom score. However, data from three of these trials were skewed and are presented in Analysis 3.9 (Gallagher 2005; Marco 2002; Strous 2003).
3.9. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 9 Mental state: 2f. Specific ‐ depression ‐ average endpoint scores (CDSS/HAMD total, high score = poor, skewed data).
| Mental state: 2f. Specific ‐ depression ‐ average endpoint scores (CDSS/HAMD total, high score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| DHEA | ||||
| Strous 2003 | DHEA | 4.1 | 4.5 | 15 |
| Strous 2003 | Placebo | 5.6 | 4.5 | 12 |
| ketoconazole | ||||
| Marco 2002 | Ketoconazole | 16.0 | 9.2 | 8 |
| Marco 2002 | Placebo | 23.0 | 6.9 | 7 |
| mifepristone | ||||
| Gallagher 2005 | Mifepristone | 7.6 | 6.4 | 10 |
| Gallagher 2005 | Placebo | 8.7 | 9.7 | 10 |
In the remaining trial by Flores 2006, study authors noted no statistically significant differences between intervention and control (n = 30, 1 RCT, MD ‐3.20, 95% CI ‐9.12 to 2.72).
3.1.6.2. Short‐term
Gallagher 2005 reported no statistically significant differences between intervention and control (n = 20, 1 RCT, RR 0.90, 95% CI ‐3.75 to 5.55).
3.2 Global state
3.2.1 General ‐ no clinically significant improvement ‐ data only for mifepristone (< 30% improvement BPRS)
Flores 2006 recorded outcomes as at least a 30% reduction in BPRS scores and described no statistically significant differences between mifepristone and control (n = 30, 1 RCT, RR 0.58, 95% CI 0.32 to 1.06; Analysis 3.10).
3.10. Analysis.
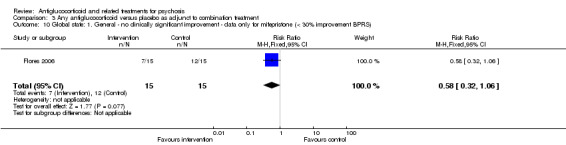
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 10 Global state: 1. General ‐ no clinically significant improvement ‐ data only for mifepristone (< 30% improvement BPRS).
3.2.2 Specific ‐ positive symptoms ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement BPRS PSS)
Flores 2006 recorded outcomes as at least a 50% reduction in BPRS positive symptoms subscale scores and described a statistically significant difference in favour of mifepristone (n = 30, 1 RCT, RR 0.38, 95% CI 0.18 to 0.81; Analysis 3.11) with an NNTB value of two.
3.11. Analysis.

Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 11 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement BPRS, PSS).
3.2.3 Depression ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement HAMD)
With regards to depression symptoms, and with outcome defined as at least a 50% reduction in HAMD scores, the short‐term trial by Flores 2006 showed no statistically significant differences between mifepristone and control (n = 30, 1 RCT, RR 0.85, 95% CI 0.59 to 1.22; Analysis 3.12).
3.12. Analysis.

Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 12 Global state: 2b. Specific ‐ depression ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement HAMD).
3.2.4 Leaving the study early
Seven trials reported these data (Flores 2006; Gallagher 2005; Marco 2002; Newcomer 1998; Ritsner 2006; Ritsner 2010; Strous 2003). Four (Gallagher 2005; Marco 2002; Newcomer 1998; Ritsner 2006) reported no participants leaving the study during the intervention period. Flores 2006 explained that one person in the intervention group left the study early, and Strous 2003 reported three people leaving early, all belonging to the placebo group. Ritsner 2010 observed that eight people left the study early ‐ five belonged to the placebo group and three to the DHEA group. Researchers noted no statistically significant differences between intervention and control (n = 201, 7 RCTs, RR 0.56, 95% CI 0.20 to 1.52; Analysis 3.13).
3.13. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 13 Global state: 3. Leaving the study early.
a. Dexamethasone
Newcomer 1998 reported that no participants left the study during the intervention period.
b. DHEA
Strous 2003 reported that three people left early, all of whom belonged to the placebo group. Ritsner 2006 reported that no participants left the study during the intervention period, and Ritsner 2010 indicated that five people belonging to the placebo group left early, along with three people belonging to the DHEA group.
c. Ketoconazole
Marco 2002 reported that no participants left the study during the intervention period.
d. Mifepristone
Flores 2006 reported that one person in the intervention group left the study early. Gallagher 2005 reported that no participants left the study during the intervention period.
3.3 Cognitive functioning: average endpoint scores, various tasks
3.3.1 Information processing and sustained attention
Newcomer 1998 observed no statistically significant differences between intervention and control (n = 19, 1 RCT, MD ‐0.50, 95% CI ‐1.87 to 0.87; Analysis 3.14).
3.14. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 14 Cognitive functioning: 1a. Average endpoint scores, various tasks.
3.3.2 Spatial thinking
Newcomer 1998 also noted no statistically significant differences between intervention and control (n = 19, 1 RCT, MD ‐0.10, 95% CI ‐1.63 to 1.43; Analysis 3.14).
3.3.3 Vigilance
Data from Newcomer 1998 were skewed and are presented as 'Other data' in Analysis 3.15.
3.15. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 15 Cognitive functioning: 1b. Vigilance task ‐ average endpoint scores ‐ skewed data.
| Cognitive functioning: 1b. Vigilance task ‐ average endpoint scores ‐ skewed data | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Vigilance | ||||
| Newcomer 1998 | Dexamethasone | 2.5 | 2.6 | 11 |
| Newcomer 1998 | Placebo | 2.6 | 1.4 | 8 |
3.4 General functioning
3.4.1 Average endpoint scores GAF/SOFAS (low = poor) ‐ DHEA only
Two trials (Nachshoni 2005; Ritsner 2010) reported these data and described no statistically significant differences between intervention and control (n = 54, 2 RCTs, MD 1.05, 95% CI ‐5.55 to 7.66; Analysis 3.16).
3.16. Analysis.
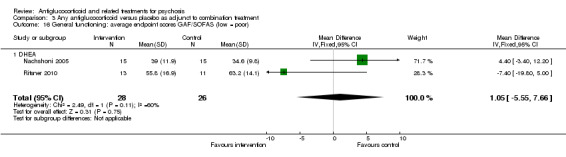
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 16 General functioning: average endpoint scores GAF/SOFAS (low = poor).
3.5 Adverse effects
3.5.1 General: overall number of events
All trials reported the number of general adverse events. Three trials (Newcomer 1998; Ritsner 2006; Ritsner 2010) reported no adverse events in the intervention group or in the placebo group. Overall a statistically significant difference favoured control (n = 223, 8 RCTs, RR 2.66, 95% CI 1.35 to 5.32; Analysis 3.17).
3.17. Analysis.
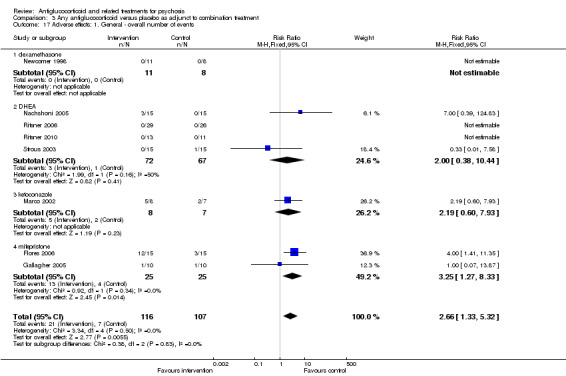
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 17 Adverse effects: 1. General ‐ overall number of events.
a. Dexamethasone
Newcomer 1998 reported no adverse events in the intervention group nor in the placebo group.
b. DHEA
Researchers noted no statistically significant differences between intervention and control (Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003) (n = 139, 4 RCTs, RR 2.00, 95% CI 0.38 to 10.44). Ritsner 2006 reported no adverse events in the intervention group and none in the placebo group.
c. Ketoconazole
Investigators observed no statistically significant differences between intervention and control (Marco 2002) (n = 15, 1 RCT, RR 2.19, 95% CI 0.60 to 7.93).
d. Mifepristone
Study authors described a statistically significant difference in favour of control (Flores 2006; Gallagher 2005) (n = 50, 2 RCTs, RR 3.25, 95% CI 1.27 to 8.33) with a number needed to treat for additional harmful outcome (NNTH) value of seven.
3.5.2 Specific: extrapyramidal symptoms ‐ only DHEA
Nachshoni 2005 reported a statistically significant difference in favour of DHEA (n = 30, 1 RCT, MD ‐5.00, 95% CI ‐8.85 to ‐1.15; Analysis 3.18). Similar data in two trials (Ritsner 2006; Ritsner 2010) were skewed and are presented as 'Other data' in Analysis 3.19.
3.18. Analysis.
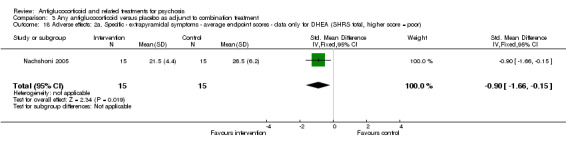
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 18 Adverse effects: 2a. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA (SHRS total, higher score = poor).
3.19. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 19 Adverse effects: 2b. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA (skewed data).
| Adverse effects: 2b. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| extrapyramidal symptoms (ESRS total, higher score = poor) | ||||
| Ritsner 2006 | DHEA | 4.0 | 6.8 | 29 |
| Ritsner 2006 | Placebo | 5.5 | 8.5 | 26 |
| Ritsner 2010 | DHEA | 2.9 | 4.8 | 13 |
| Ritsner 2010 | Placebo | 1.7 | 4.1 | 11 |
| Akathisia and tardive dyskinesia (AIMS/BARS total, higher score = poor) | ||||
| Ritsner 2006 | DHEA | 1.1 | 2.8 | 29 |
| Ritsner 2006 | Placebo | 0.8 | 2.3 | 26 |
| Ritsner 2010 | DHEA | 0.23 | 0.6 | 13 |
| Ritsner 2010 | Placebo | 0.18 | 0.4 | 11 |
3.5.3 Specific: various effects
Trials presented data for various effects including allergy (skin rash), blurred vision, dizziness, appetite increase/decrease, fatigue, irritability, constipation, nausea and dysmenorrhoea. Investigators described no differences between treatment groups for any of these adverse effects (Analysis 3.20).
3.20. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 20 Adverse effects: 2c. Specific: various effects.
3.6. Quality of life
3.6.1 Observer‐rated quality of life scale ‐ DHEA only
Ritsner 2006 assessed general functioning using an observer‐rated QOL scale and noted no statistically significant differences between intervention and control (n = 55, 1 RCT, MD 6.20, 95% CI ‐1.37 to 13.77; Analysis 3.21).
3.21. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 21 Quality of life: observer‐rated scale.
Subgroup analyses ‐ by diagnostic group
The purpose of the remaining comparisons was to assess the efficacy of each type of intervention treatment across different diagnoses. All included trials were conducted in people with schizophrenia/schizoaffective disorder or in individuals with psychotic depression. All five trials (Nachshoni 2005; Ritsner 2006; Ritsner 2010; Strous 2003; Strous 2007) involving DHEA as the intervention treatment were conducted in patients with schizophrenia/schizoaffective disorder; therefore, analysis by diagnostic group for DHEA was not possible.
4. Diagnostic group: mifepristone vs placebo as adjunct to combination treatment
The purpose of this remaining comparison was to assess the efficacy of each type of intervention treatment across different diagnoses. All included trials were conducted in people with schizophrenia/schizoaffective disorder or in individuals with psychotic depression.
Two trials are included in this comparison: Flores 2006 was conducted in people with psychotic depression, and Gallagher 2005 in people with schizophrenia.
4.1. Mental state
4.1.1 General ‐ average endpoint score (BPRS total scores, higher score = poor)
Both trials provided an overall psychotic symptom score and described no statistically significant differences between intervention and control (n = 50, 2 RCTs, MD ‐1.02, 95% CI ‐6.16 to 4.12), and the test for subgroup diagnostic differences was not statistically significant (Chi² = 2.79, df = 1, P value = 0.09, I² = 64.2%; Analysis 4.1).
4.1. Analysis.
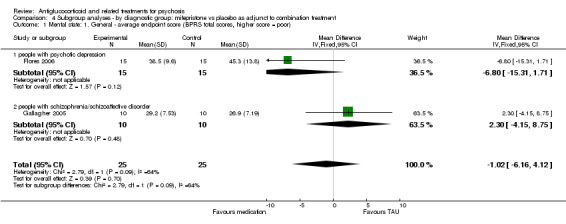
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 1 Mental state: 1. General ‐ average endpoint score (BPRS total scores, higher score = poor).
a. People with psychotic depression
Results show no statistically significant differences between mifepristone and control (n = 30, 1 RCT, MD ‐6.80, 95% CI ‐15.31 to 1.71).
b. People with schizophrenia/schizoaffective disorder
Study authors reported no statistically significant differences between mifepristone and control (n = 20, 1 RCT, MD 2.30, 95% CI ‐4.15 to 8.75).
4.1.2 Specific a. positive symptoms ‐ average endpoint scores ‐ people with psychotic depression (BPRS positive subscale, higher score = poor)
Flores 2006 was the only trial to provide a measure of positive psychotic symptoms and reported no statistically significant differences between intervention and control (n = 30, 1 RCT, MD ‐2.60, 95% CI ‐5.67 to 0.47; Analysis 4.2).
4.2. Analysis.
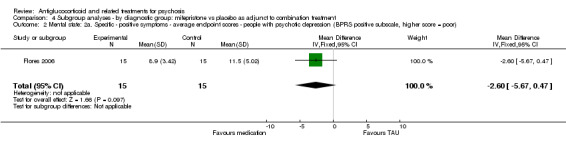
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 2 Mental state: 2a. Specific ‐ positive symptoms ‐ average endpoint scores ‐ people with psychotic depression (BPRS positive subscale, higher score = poor).
4.1.3 Specific b. depression ‐ average endpoint scores ‐ people with psychotic depression (HAMD total, higher score = poor)
Only Flores 2006 reported outcome data defined as a 50% or greater reduction on HAMD scores and no statistically significant differences between intervention and control (n = 30, 1 RCT, MD ‐3.20, 95% CI ‐9.12 to 2.72; Analysis 4.3).
4.3. Analysis.
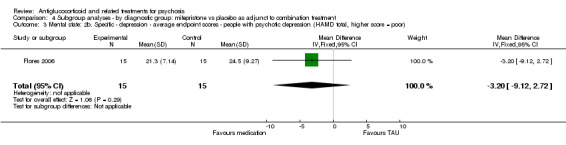
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 3 Mental state: 2b. Specific ‐ depression ‐ average endpoint scores ‐ people with psychotic depression (HAMD total, higher score = poor).
4.2 Global state
4.2.1 General ‐ no clinically significant improvement ‐ people with psychotic depression (< 30% improvement on BPRS)
Only Flores 2006 recorded outcome as at least a 30% reduction in BPRS scores and no statistically significant differences between intervention and control (n = 30, 1 RCT, RR 0.58, 95% CI 0.32 to 1.06; Analysis 4.4).
4.4. Analysis.
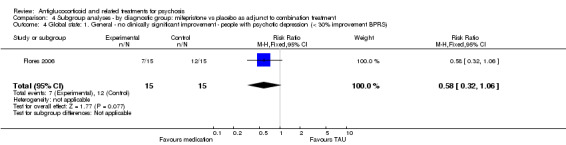
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 4 Global state: 1. General ‐ no clinically significant improvement ‐ people with psychotic depression (< 30% improvement BPRS).
4.2.2 Specific: positive symptoms ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement BPRS PSS)
Flores 2006 also recorded outcome as at least a 50% reduction in BPRS positive symptoms subscale scores and noted a statistically significant difference in favour of the intervention (n = 30, 1 RCT, RR 0.38, 95% CI 0.18 to 0.81; Analysis 4.5). with an NNTB value of two.
4.5. Analysis.
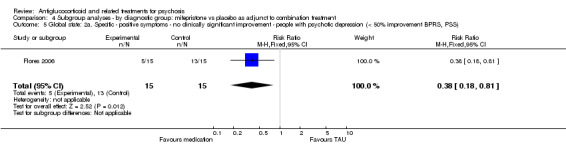
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 5 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement BPRS, PSS).
4.2.3 Specific: depression ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement on HAMD)
Only Flores 2006 provided outcome data defined as at least a 50% reduction in HAMD scores and showed no statistically significant differences between intervention and control (n = 30, 1 RCT, RR 0.85, 95% CI 0.59 to 1.22; Analysis 4.6).
4.6. Analysis.

Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 6 Global state: 2b. Specific ‐ depression ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement HAMD).
4.2.4 Leaving the study early
No participants left the study early in the trial by Gallagher 2005, and Flores 2006 reported was no statistically significant differences between intervention and control (n = 50, 2 RCTs, RR 3.00, 95% CI 0.13 to 68.26; Analysis 4.7).
4.7. Analysis.
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 7 Global state: 3. Leaving the study early.
a. People with psychotic depression
Researchers observed no statistically significant differences between intervention and control (n = 30, 1 RCT, RR 3.00, 95% CI 0.13 to 68.26).
b. People with schizophrenia/schizoaffective disorder
No participants left the study early in Gallagher 2005.
4.3 Adverse events
4.3.1 Overall number of events
Both trials reported adverse events and noted a statistically significant difference in favour of control (n = 50, 2 RCTs, RR 3.25, 95% CI 1.27 to 8.33; Analysis 4.8).
4.8. Analysis.
Comparison 4 Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment, Outcome 8 Adverse events: overall number of events.
a. People with psychotic depression
Study authors described a statistically significant difference in favour of control (n = 30, 1 RCT, RR 4.00, 95% CI 1.41 to 11.35).
b. People with schizophrenia/schizoaffective disorder
Investigators observed no statistically significant differences between intervention and control (n = 20, 1 RCT, RR 1.00, 95% CI 0.07 to 13.87).
We could not conduct subgroup analyses on the following because no or few trials were available: (1) prodrome versus 'first‐onset' versus 'chronic'; (2) type of antiglucocorticoid drug; (3) adults versus adolescents; and (4) duration of untreated illness.
Discussion
Summary of main results
The review includes data from 11 trials (involving 509 participants) that assessed the following antiglucocorticoid and related treatments: mifepristone, ketoconazole, dexamethasone and DHEA. The small number of trials in each comparison yielded insufficient evidence to permit definitive conclusions on the efficacy and safety of these treatments for psychosis as sole treatment or as an adjunct to regular treatment. Results for each antiglucocorticoid or related drug are summarised below. All findings are graded as of low or very low quality (Table 1; Table 2; Table 3) with the exception of adverse events, which are more convincingly in favour of placebo.
1. Mifepristone
Four trials assessed the efficacy of mifepristone as sole treatment (n = 225, DeBattista 2006; n = 5, Belanoff 2001) or as an adjunct to regular treatment (n = 20, Gallagher 2005; n = 30, Flores 2006) in patients diagnosed with psychotic depression or schizophrenia. Analysis of available continuous data showed no beneficial effect of mifepristone. For clinical response (dichotomous) variables, data from Flores 2006 revealed a favourable effect of mifepristone for clinically significant improvement in positive psychotic symptoms, but not in depression symptoms. Dichotomous data from DeBattista 2006 showed no differences between intervention and control immediately post intervention; however at 21 days' follow‐up, they revealed a beneficial effect of mifepristone for total and positive psychotic symptoms, but not for depression symptoms.
All trials reported the rate of general adverse events (refer to Table 5 for a detailed description) and provided evidence for a higher rate of general adverse events with treatment. Only Flores 2006 stated that researchers observed no serious adverse events.
2. Summary of included studies and baseline psychopathology.
| Study | Intervention | Dosage | Length | As adjunct (Y/N) | Assessment tool | BPRS/PANSS | HAMD/CDSS |
SANS/ PANSS negative |
SANS/ PANSS negative |
||
| Intervention | Control | Intervention | Control | Intervention | Control | ||||||
| Belanoff 2001 | Mifepristone | 600 mg/d | 4 days | N | BPRS, HAMD | 51.5 (3.5) | 45.3 (11.6) | 37.5 (12.0) | 29.0 (5.3) | ‐ | ‐ |
| DeBattista 2006 | Mifepristone | 600 mg/d | 7 days | N | BPRS, HAMD | 55.8 (11.6) | 55.7 (9.2) | 37.3 (8.4) | 37.3 (7.5) | ‐ | ‐ |
| Flores 2006 | Mifepristone | 600 mg/d | 8 days | Y | BPRS, HAMD | 46.9 (5.7) | 50.1 (8.5) | 29.3 (5.0) | 31.5 (5.1) | ‐ | ‐ |
| Gallagher 2005 | Mifepristone | 600 mg/d | 7 days | Y | BPRS, HAMD | 31.5 (10.1) | 27.8 (9.7) | 9.6 (8.8) | 11.7 (8.1) | ‐ | ‐ |
| Nachshoni 2005 | DHEA | 100 mg/d | 7 days | Y | BPRS | 23.8 (12.2) | 24.9 (9.6) | ‐ | ‐ | ‐ | ‐ |
| Ritsner 2006 | DHEA | 200 mg/d | 6 weeks | Y | PANSS | 91.6 (17.1) | 95.6 (17.2) | ‐ | ‐ | 25.3 (6.3) | 26.7 (6.2) |
| Strous 2003 | DHEA | Titrated up to 100 mg/d | 6 weeks | Y | PANSS, HAMD, SANS | 66.4 (20.9) | 63.8 (18.8) | 8.9 (5.3) | 7.0 (5.6) | 47.9 (13.7) | 40.8 (16.5) |
| Strous 2007 | DHEA | Titrated up to 150 mg/d | 8 weeks | Y | PANSS, CDSS, SANS | 65.5 (15.6) | 65.1 (18.8) | 3.0 (3.7) | 4.3 (3.2) | 52.8 (15.9) | 39.7 (23.9) |
| Ritsner 2010 | DHEA | DHEA 400 mg/d | 8 weeks | Y | PANSS | 46.2 (11.1) | 37.1 (8.8) | ‐ | ‐ | 25.8 (6.1) | 22.8 (5.8) |
| Marco 2002 | Ketoconazole | Titrated up to 800 mg/d | 4 weeks | Y | PANSS, HAMD | 37.5 (6.7) | 32.4 (6.3) | 20.0 (6.0) | 20.0 (5.1) | 21.6 (5.4) | 17.4 (5.1) |
| Newcomer 1998 | Dexamethasone | Successive doses of 0.5, 1, 1 and 1 mg | 4 days | Y | BPRS, SANS | No data reported | No data reported | ‐ | ‐ | No data reported | No data reported |
Too few trials were identified to investigate the efficacy of mifepristone treatment across diagnostic groups or the optimal method of treatment (e.g. single or adjunctive therapy, length of treatment).
2. Ketoconazole
One trial (n = 15, Marco 2002) assessed the efficacy of ketoconazole, a cortisol synthesis inhibitor, as an adjunct to regular treatment in patients with schizophrenia/schizoaffective disorder. Data from this small trial show no benefit from treatment and in some cases a more favourable outcome with placebo. Study authors described no adverse events in the intervention group and none in the placebo group.
3. DHEA
Five trials assessed the efficacy of DHEA as an adjunct to atypical antipsychotic (n = 40, Strous 2007) or combination treatment (n = 62, Ritsner 2006; n = 32, Ritsner 2010; n = 30, Strous 2003; n = 34, Nachshoni 2005) in patients with schizophrenia or schizoaffective disorder. A large proportion of the data were skewed and therefore could not be included in the meta‐analysis. Data available for analysis showed no evidence for an effect of treatment on clinical symptoms. The rate of participant attrition or general adverse events did not differ between DHEA treatment and placebo. With regards to extrapyramidal symptoms, Nachshoni 2005 revealed a beneficial effect of treatment, whereas Strous 2007 showed no effect of treatment.
4. Dexamethasone
No primary outcome data were provided in the trial involving dexamethasone treatment (Newcomer 1998), and no evidence suggested an effect of dexamethasone treatment on cognitive functioning.
Available data were insufficient for comparison of the effects of intervention treatment in the following subgroups: (1) 'prodrome' versus 'first‐onset' versus 'chronic'; (2) type of antiglucocorticoid drug; (3) adults versus adolescents; and (4) duration of untreated illness.
Overall completeness and applicability of evidence
Only short‐term outcomes (up to 12 weeks) were assessed, and most studies measured outcomes immediately following cessation of the intervention. Three trials conducted follow‐up assessments up to a maximum of 21 days post treatment and provided scant data on general functioning; however, this aspect would be more relevant to trials with longer follow‐up assessments.
All trials were conducted in adult populations, primarily with a diagnosis of schizophrenia or schizoaffective disorder (eight of 11 trials). The remaining three studies were conducted in people with psychotic depression, and all trials involved mifepristone treatment. Some trials required a specified level of symptom severity for entry into the trial. No trials were conducted in patients at their first episode of psychotic illness and none included populations at high risk for developing psychosis.
Quality of the evidence
The quality of data was variable. Most trials included fewer than 40 participants, with the exception of DeBattista 2006 (n = 225) and Ritsner 2006 (n = 62). Just over half of the trials (seven of 11) used adequate randomisation, allocation concealment and blinding. It should be noted however that in some of these trials, it was necessary to request this information from the study authors because it was not adequately described in the publication. The remaining four trials did not adequately describe the randomisation and allocation procedures and therefore have unclear risk of bias. Most participants were followed up, or intention‐to‐treat analysis was conducted. A large proportion of available data, particularly regarding depression symptoms, was skewed and therefore could not be included in the meta‐analysis. Other risks of bias involved small sample size, and in some trials baseline imbalances in clinical symptom scores were apparent between intervention and control groups (refer to Table 5). Analysis of final endpoint data in this review, as opposed to change scores, may have biased the results.
Potential biases in the review process
We tried to identify all relevant trials in our search. However, we may not have identified all studies. We are aware that the search date is old at the time of publication and new studies may be available. Our review has been limited to articles written in English, so bias may involve including no studies published in languages other than English.
Agreements and disagreements with other studies or reviews
Strous 2003 and Strous 2007 reported an improvement in negative symptoms and/or depression and in anxiety symptoms with dehydroepiandrosterone (DHEA) treatment. In contrast, results of this review show no evidence for a beneficial effect of DHEA treatment on clinical symptoms. In partial agreement with this review, the 12‐week cross‐over trial by Ritsner 2006 reported no significant clinical improvement with DHEA treatment, although the first six weeks of DHEA treatment was associated with significant improvement in Positive and Negative Syndrome Scale (PANSS) scores compared with placebo. These discrepancies are likely due to the type of data analysed (e.g. change scores vs final endpoint data) and the fact that data were skewed and were not included in the meta‐analyses.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
This review suggests that little trial‐based evidence shows the effectiveness of antiglucocorticoid and related medications in treating individuals with psychosis. People with psychosis should continue to look at new trials and reviews in this area, as this review suggests that mifepristone may be effective, and with more trials, its effectiveness may be established in the future.
2. For clinicians
Given the lack of data at this stage, antiglucocorticoid treatments cannot be recommended. Very few trials have been conducted, and most have involved a small sample. Limited available data do not provide enough evidence to support or refute the use of antiglucocorticoid treatment for psychotic disorders, although some findings suggest a favourable effect for mifepristone. Additional trials are needed.
3. For managers or policy makers
Managers and policy makers have little evidence upon which to base decisions about provision of these medications.
Implications for research.
1. General
Some trials appeared to follow the CONSORT statement and offered clear reporting of the conduct of the trial, but this was not the case in all studies, and clearer reporting would have allowed this review to be more informative. Particularly disappointing was the lack of reporting of useable outcome data.
2. Specific
2.1 Reviews
Suggestions for future reviews include the neurosteroid pregnenolone, which is synthesised from cholesterol and is a precursor to glucocorticoids. Pregnenolone enhances learning and memory in rodents, and clinical trials are now being conducted to investigate adjunctive pregnenolone for cognitive and negative symptoms in patients with schizophrenia or schizoaffective disorder. See also Table 6.
3. Suggestions for future reviews.
| Title | Possible included studies |
| Antiglucocorticoid dose for psychosis | Belanoff 2002 |
| Pregnenolone for psychosis | Savitz AJ, Silverstein SM, McGovern KC, Schenkel L, Grant L. The neurosteroid, pregnenolone, reduces negative symptoms in patients with schizophrenia: results of a preliminary double‐blind study.Schizophrenia Bulletin. Vol 33. 2007:489‐9 Marx CE, Keefe RSE, Buchanan RW, et al. Proof‐of‐concept trial with the neurosteroid pregnenolone targeting cognitive and negative symptoms in schizophrenia. Neuropsychopharmacology. Vol 34. Issue 8. 2009:1885‐903 |
2.2 Trials
Well‐designed, larger‐scale trials are needed across the range of psychotic disorders and at different stages of illness. In particular, there is a need to investigate these types of treatments in at‐risk and early psychosis populations. Treatments aimed at correcting HPA axis dysfunction might be more effective at early stages of illness, during which hyperactivity of the HPA axis is thought to be pronounced. These trials need to include outcome measures such as time to transition to psychosis and transition rate, along with assessment of clinical symptoms and general functioning. Neuroendocrine measurements and/or screening of patients with demonstrable HPA axis dysfunction for inclusion in the trial would be useful for delivering more targeted treatment. Trials with longer follow‐up assessments are needed to determine the short‐, medium‐ and long‐term outcomes of these treatments. It will be important to assess levels of social and occupational functioning, which often are considered more pertinent to the clinician and patient than symptom reduction alone. Finally, trials need to follow CONSORT guidelines for reporting data from clinical trials (Table 7).
4. Suggested design for future study.
| Methods | Allocation: randomised, clearly described. Blinding: double, tested. Duration: 1 week. Need short‐, medium‐ and long‐term follow‐up. |
| Participants | Diagnosis: people at ultra‐high risk of developing psychosis. N = 300.* Age: young adults. Sex: male only (due to mifepristone interaction with progesterone receptor). History: demonstrable HPA axis hyperactivity. |
| Intervention | 1. Mifepristone: flexible dose 400 to 600 mg/day. N = 150. 2. Placebo. N = 150. |
| Outcomes | Global state: relapse, time to transition to psychosis. Service outcomes: admission, time in hospital. Mental state: CGI, Total psychotic (BPRS) and depressive symptoms. General functioning: quality of life measures. Neuroendocrine measurements: cortisol awakening response and circadian rhythm measured on 2 consecutive days. |
| Notes | *Powered to be able to identify a difference of ˜ 20% between groups for primary outcome with adequate degree of certainty. |
History
Protocol first published: Issue 1, 2008 Review first published: Issue 1, 2016
| Date | Event | Description |
|---|---|---|
| 27 October 2008 | Amended | Minor amendments to protocol |
| 9 September 2008 | Amended | Converted to Rev Man 5 format |
Acknowledgements
The Cochrane Schizophrenia Group maintains a standard template for the Methods section, and we have used and adapted this for our requirements.
We would like to thank Muayad Alzuabi for peer review and Dolores Matthews for copy editing.
Appendices
Appendix 1. Previous search strategies
September 2007 search
1. Cochrane Schizophrenia Group Trials Register (see Electronic searches for strategy)
2. Cochrane Central Register of Controlled Trials (CENTRAL) and bibliographic databases, including MEDLINE, PsycINFO and EMBASE, were searched (OVID 1950 to November 2007). The search strategy used for these databases is included in an additional table (Table 12).
5. Search terms.
| PsycINFO (OVID 1950 to August 2009) | EMBASE (OVID 1950 to August 2009) | MEDLINE (OVID 1950 to August 2009) |
| 1. Psychosis/
2. exp Acute Psychosis/
3. Affective Psychosis/
4. exp Hallucinosis/
5. exp "Paranoia (Psychosis)"/
6. exp Schizophrenia/
7. #1 or #2 or #3 or #4 or #5 or #6
8. exp Adrenal Cortex Hormones/
9. exp Glucocorticoids/
10. Corticotropin releasing factor 11. Corticotropin 12. Vasopressin 13. Hypothalamic pituitary adrenal axis 14. Corticosteroids/ 15. R121919 16. ORG 34116 17. 3‐acetoxyandrost‐5‐ene‐7,17‐dione 18. dehydroepiandrosterone 19. mifepristone 20. mitotane 21. aminoglutethimide 22. spironolactone 23. ketoconazole 24. metyrapone 25. #8 or #9 or #10 or #11or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 26. #7 AND #25 27. Clinical Trials/ 28. Controlled trial$.tw 29. (controlled studies or controlled study).tw 30. Random$.tw 31. Random Sampling/ 32. ((singl$ or doubl$ or trebl$ or tripl$) adj5 (blind$ or dummy or mask$)).tw 33. Placebo$mp 34. #27 or #28 or # 29 or #30 or #31 or #32 or #32 or #33 35. #26 AND #34 |
1. Psychosis/ or psychotic disorder$.tw 2. exp Delusion/ 3. exp Hallucination/ 4. exp Paranoid Psychosis/ 5. exp Schizohprenia/ 6. #1 or #2 or #3 or #4 or #5 7. exp corticosteroid/ 8. exp corticosteroid receptor/ 9. Corticotropin releasing factor 10. Corticotropin Releasing Factor Receptor 11. Corticotropin Releasing Factor Receptor 1 12. Corticotropin Releasing Factor Receptor 2 13. Corticotropin 14. Vasopressin 15. Hypothalamus Hypophysis Adrenal System 16. R121919 17. ORG 34116 18. 3‐acetoxyandrost‐5‐ene‐7,17‐dione 19. Prasterone 20. Mifepristone 21. Mitotane 22. Aminoglutethimide 23. Spironolactone 24. Ketoconazole 25. Metyrapone 26. # 7 or #8 or #9 or #10 or #11or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 27. #6 AND #26 28. exp controlled study/ 29. (controlled trial$ or controlled study or controlled studies).tw 30. random$.tw 31. single blind procedure/ 32. double blind procedure/ 33. ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$ or dummy)).tw 34. placebo$.mp 35. #28 or #29 or #30 or #31 or #32 or #33 or #34 36. #27 AND #35 | 1. exp psychotic disorders/ 2. Delusions/ 3. Hallucinations/ 4. Paranoid Disorders/ 5. Schizophrenia/ 6. (psychotic disorder$ or psychoses or psychosis).tw. 7. (delusion$ or hallucination$ or paranoid$).tw. 8. (schizoaffective disorder$ or schizophreniform disorder$ or schizophrenia).tw. 9. exp mood disorders/ 10. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 11. Receptors, Steroid/ 12. Glucocorticoids/ 13. Receptors, Corticotropin‐Releasing Hormone/ 14. Receptors, Corticotropin/ 15. Dexamethasone/ 16. Hydrocortisone/ 17. Adrenocorticotropic Hormone/ 18. Corticotrophs/ 19. Hydroxycorticosteroids/ 20. R 121919.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 21. ORG 34116.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 22. 3‐acetoxyandrost‐5‐ene‐7,17‐dione.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 23. Dehydroepiandrosterone/ 24. Mifepristone/ 25. Mitotane/ 26. Aminoglutethimide/ 27. Spironolactone/ 28. Ketoconazole/ 29. Metyrapone/ 30. Corticotropin‐Releasing Hormone/ 31. 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 31. 10 and 31 32. clinical trial.pt 33. clinical trial$.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 34. random$.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 35. placebo.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 36. placebo.ti,ab 37. groups.ti,ab 38. dt.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 39. trial.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 40. groups.mp. [mp=title, original title, abstract, name of substance word, subject heading word] 41. 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 42. 31 and 41 |
3. We searched the National Research Register (http://www.updatesoftware.com/National/nrr‐frame.html), Clinical Trials (http://clincaltrials.gov/ct/gui/c/r), the Australian Clinical Trials Register (http://www.actr.org.au/trialSearch.aspx) and Current Controlled Trials (http://www.controlled‐trials.com) databases. Additionally, we searched the trial databases of pharmaceutical companies.
November 2009 search
We searched the Cochrane Schizophrenia Group Trials Register (September 2007, November 2009) using the phrase:
[(*Steroid* or *corticoid* or *cort?cotrop* or *dexamethasone* or *hydrocortisone* or *R?121919* or *ORG?34116* or *3‐acetoxyandrost* or dehydroepiandrosteron* or *mifepristone* or *mitot?ne* or *aminoglutethimide* or *spironolactone* or *ketoconazole* or *metyrapone* or *etomidate* or *RU‐486* in TI, AB or IN fields of REFERENCE) or (*steroids* or Hydrocortisone or Corticotropin or Mifepristone or Dehydroepiandrosterone or Etomidate or Ketoconazole or Glucocoticoid receptor antagonist or aminogluteth* or mitotane* or dexamethas* or metyrapon* in Intervention field of STUDY)] This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module).
Appendix 2. Previously published data extraction and methods
Authors BG, SH, LP and SB independently extracted data from selected trials. When disputes arose, we attempted to resolve these by discussion. When this was not possible and further information was necessary to resolve the dilemma, we did not enter data but added the trial to the list of those awaiting assessment. We collected information on participants (age, gender, ethnicity, diagnosis, diagnostic criteria and first‐episode/prodromal criteria used, setting of care, country, inclusion and exclusion criteria for the trial, duration of treated and untreated illness, previous treatment and psychiatric co‐morbidity); interventions (description of medication, method of delivery, dose, duration of treatment, actual dosage received) and other interventions used in intervention group; interventions in comparison group with similar detail; outcome measures (description of measures used, timing of administration) and results (point estimates and measures of variability, frequency counts for dichotomous variables) and methods (randomisation and allocation procedure, blinding, number of participants randomly assigned, withdrawn, dropped out, analysed, baseline comparability, intention‐to‐treat analysis, other problems).
Assessment of risk of bias in included studies
Authors BG, SH, LP and SB assessed the risk of bias of included studies using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2009). Risk of bias was assessed as 'yes', 'no' or 'unclear' on the following points. 1. Sequence generation 2. Allocation concealment 3. Blinding of participants, personnel and outcome assessors 4. Incomplete outcome data 5. Selective outcome reporting 6. Other sources of bias
All studies meeting inclusion criteria were included regardless of the outcome of the assessment of risk of bias. However, a sensitivity analysis was performed for the primary outcome excluding trials with 'no' or 'unclear' ratings for allocation concealment. Poor concealment has been associated with overestimation of treatment effect (Schulz 1995). All risk of bias items were summarised and described in the Risk of bias in included studies section.
When disputes arose as to which category a trial should be allocated, again resolution was attempted by discussion. When this was not possible, we did not enter the data, and we added the trial to the list of those awaiting assessment until further information could be obtained.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated the risk ratio (RR) and its 95% confidence interval (CI) based on the fixed‐effect model. Risks ratios are more intuitive (Boissel 1999) than odds ratios, and odds ratios tend to be interpreted as risk ratios by clinicians (Deeks 2000). This misinterpretation then leads to overestimation of the impression of the effect. When overall results were significant, we calculated the number needed to treat for additional beneficial outcome (NNTB) and the number needed to treat for additional harmful outcome (NNTH). When people were lost to follow‐up at the end of the study, we assumed that they had had a poor outcome, and that once they were randomly assigned, they would be included in the analysis (intention‐to‐treat/ITT analysis).
2. Continuous data
2.1 Rating scales
A wide range of instruments are available for measuring mental health outcomes. These instruments vary in quality; many are not valid and are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore, we included continuous data from rating scales only if the measuring instrument had been described in a peer‐reviewed journal.
2.2 Final endpoint value versus change data
When both final endpoint data and change data were available for the same outcome category, we presented only final endpoint data. We acknowledge that by doing this, we may have excluded a large portion of the published change data but argue that endpoint data are more clinically relevant, and that if change data were to be presented along with endpoint data, they would be given undeserved equal prominence. When studies reported only change data, we contacted study authors to ask for endpoint figures.
2.3 Multiple linear regression data
Many trials in psychiatry report estimates of treatment effects from multiple linear regression models. These models adjust for varying factors such as age, sex and baseline of the outcome. We pooled treatment estimates from these trials using fixed‐effect (inverse variance) meta‐analysis. We converted P values and confidence intervals for treatment effect to standard errors and entered them into RevMan using the generic inverse variance.
2.4 Skewed data
Continuous data on outcomes in trials relevant to mental health issues often are not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to continuous final value endpoint data before inclusion: (1) Standard deviations and means were reported in the paper or could be obtained from study authors; (2) when a scale started from zero, the standard deviation, when multiplied by two, should be less than the mean (otherwise, the mean is unlikely to be an appropriate measure of the centre of distribution (Altman 1996)); in cases with data that are greater than the mean, we entered data into the 'Other data' table as skewed data. If a scale starts from a positive value (such as PANSS, which can have values from 30 to 210), the calculation described above in (2) should be modified to take the scale starting point into account. In these cases, skewness is present if 2 SD > (S ‐ Smin), where S is the mean score and S min is the minimum score. We reported non‐normally distributed data (skewed) in the 'Other data types' tables.
For change data (mean change from baseline on a rating scale), it is impossible to tell whether or not data are non‐normally distributed (skewed) unless individual patient data are available. After consulting the ALLSTAT electronic statistics mailing list, we entered change data into RevMan analyses and reported the finding in the text to summarise available information. In doing this, we assumed that data were not skewed or that the analysis could cope with the unknown degree of skew.
2.5 Data synthesis
For continuous outcomes, we estimated a mean difference (MD) when the same measure was used and a standardised mean difference (SMD) when different measures were used to measure the same outcome with a fixed‐effect model.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ cluster‐randomisation (such as randomisation by clinician or practice), but analysis and pooling of clustered data pose problems. First, study authors often fail to account for intraclass correlation in clustered studies, leading to a unit of analysis error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999). When clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation co‐efficients of clustered data and will adjust for this by using accepted methods (Gulliford 1999). When clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster‐randomised study but will adjust for the clustering effect. We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a design effect, which is calculated by using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1 + (m ‐ 1) * ICC] (Donner 2002). If the ICC was not reported, it was assumed to be 0.1 (Ukoumunne 1999). If cluster studies had been appropriately analysed with consideration of intraclass correlation co‐efficients and relevant data documented in the report, we synthesised these with other studies using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological, psychological) of treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase, participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in psychosis, we have used only data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
When a study involved more than two treatment arms, if relevant, we presented additional treatment arms in comparisons. When additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
We excluded data from studies in which more than 50% of participants in any group were lost to follow‐up (this did not include the outcome of 'leaving the study early'). In studies with less than 50% attrition, we considered people leaving early to have had the negative outcome, except for the event of death. We analysed the impact of including studies with high attrition rates (25% to 50%) in a sensitivity analysis. If inclusion of data from this latter group resulted in a substantive change in the estimate of effect, we did not add the data to trials with less attrition but presented them separately.
Assessment of heterogeneity
First, we considered all included studies within any comparison to judge for clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. We supplemented this by using the I2 statistic, which provides an estimate of the percentage of variability due to heterogeneity rather than to chance alone. When the I2 estimate was greater than or equal to 50%, we interpreted this as indicating the presence of considerable levels of heterogeneity (Higgins 2003).
Assessment of reporting biases
We assessed reporting bias by assessing whether data appropriate for use in the meta‐analysis and/or results were reported for the main clinical outcomes of the review.
Data synthesis
When appropriate, we performed meta‐analysis and pooled effect estimates obtained by using the Review Manager statistical software programme. We used a fixed‐effect model. When possible, we entered data into RevMan in such a way that the area to the left of the 'line of no effect' indicated a 'favourable' outcome for the antiglucocorticoid interventions. Wem this was not possible, we labelled the graphs in RevMan analyses accordingly, so that the direction of any effects was clear.
Subgroup analysis and investigation of heterogeneity
Clinical homogeneity was satisfied when participants, interventions and outcome measures were considered to be similar. When we found heterogeneous results, we investigated the reasons for this; when heterogeneity substantially altered the results, we did not summarise these data but presented them separately along with reasons for heterogeneity investigated by the following subgroup and sensitivity analyses.
Subgroup analysis
Diagnosis
Prodrome versus first‐onset versus chronic
Type of antiglucocorticoid drug
Adults versus adolescents
Duration of untreated illness
Sensitivity analysis
We performed sensitivity analyses to investigate the degree to which effect sizes depend on assumptions made by review authors.
Sensitivity analysis
Excluding trials with 'high risk' or 'unclear risk' for allocation concealment
Excluding trials with 'high risk' or 'unclear risk' for blinding of outcome assessment
Excluding trials with 'high risk' or 'unclear risk' for intention‐to‐treat analysis
Data and analyses
Comparison 1. Any antiglucocorticoid versus placebo as sole treatment (data only for mifepristone).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. General ‐ average endpoint score (BPRS total scores, higher score = poor) | 1 | 5 | Mean Difference (IV, Fixed, 95% CI) | ‐5.20 [‐17.91, 7.51] |
| 2 Mental state: 2. Specific ‐ depression ‐ average endpoint score (HAMD total, higher score = poor) | 1 | 5 | Mean Difference (IV, Fixed, 95% CI) | 1.67 [‐16.44, 19.78] |
| 3 Global state: 1. General ‐ no clinically significant improvement (< 30% reduction BPRS) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 immediate | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.93, 2.38] |
| 3.2 short‐term | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.38, 0.89] |
| 4 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement (< 50% reduction BPRS, PSS) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 immediate | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.85, 2.64] |
| 4.2 short‐term | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.43, 0.84] |
| 5 Global state: 2b. Specific ‐ depressive symptoms ‐ no clinically significant improvement (< 50% reduction HAMD) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 immediate | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.84, 2.84] |
| 5.2 short‐term | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.58, 1.19] |
| 6 Global state: 3. Leaving the study early | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.77, 2.01] |
| 7 Adverse effects: 1. General ‐ overall number of events | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.77, 1.09] |
| 8 Adverse effects: 2. Specific ‐ various effects | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 allergy ‐ skin rash | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.31 [0.68, 16.06] |
| 8.2 cardiovascular ‐ mild dizziness | 1 | 442 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.33, 1.32] |
| 8.3 central nervous system ‐ sedation | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.36, 2.57] |
| 8.4 central nervous system ‐ insomnia | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.29, 2.93] |
| 8.5 gastrointestinal ‐ constipation | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.17, 1.26] |
| 8.6 gastrointestinal ‐ nausea | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.89 [0.77, 4.63] |
| 8.7 gastrointestinal ‐ vomiting | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [0.78, 6.25] |
| 8.8 metabolic ‐ hypercortisolaemia | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.15, 59.89] |
| 8.9 pain ‐ abdominal ‐ non‐specific | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [0.48, 5.71] |
| 8.10 pain ‐ abdominal ‐ upper | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.02, 1.50] |
| 8.11 pain ‐ headache | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.50, 1.60] |
| 8.12 pain ‐ toothache | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.63 [0.81, 54.16] |
Comparison 2. Any antiglucocorticoid versus placebo as adjunct to atypical antipsychotic treatment (data only for DHEA).
Comparison 3. Any antiglucocorticoid versus placebo as adjunct to combination treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1a. General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, higher score = poor) | 6 | 171 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.29, 0.32] |
| 1.1 DHEA | 3 | 106 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐0.40, 0.37] |
| 1.2 ketoconazole | 1 | 15 | Std. Mean Difference (IV, Fixed, 95% CI) | 1.15 [0.03, 2.28] |
| 1.3 mifepristone | 2 | 50 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐0.77, 0.36] |
| 2 Mental state: 1b. General ‐ average endpoint score ‐ immediate (BPRS/PANSS total, high score = poor, skewed data) | Other data | No numeric data | ||
| 3 Mental state: 1c. General ‐ average endpoint score ‐ short term ‐ only mifepristone (BPRS total, higher score = poor) | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 2.10 [‐2.86, 7.06] |
| 4 Mental state: 2a. Specific ‐ positive symptoms ‐ average endpoint scores ‐ immediate (BPRS/PANSS positive subscale, higher score = poor) | 5 | 151 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.40, 0.25] |
| 4.1 DHEA | 3 | 106 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐0.32, 0.44] |
| 4.2 ketoconazole | 1 | 15 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐1.05, 0.98] |
| 4.3 mifepristone | 1 | 30 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.59 [‐1.32, 0.14] |
| 5 Mental state: 2b. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate (PANSS negative subscale, higher score = poor) | 3 | 94 | Mean Difference (IV, Fixed, 95% CI) | 2.21 [‐0.14, 4.55] |
| 5.1 DHEA | 2 | 79 | Mean Difference (IV, Fixed, 95% CI) | ‐0.34 [‐3.29, 2.62] |
| 5.2 ketoconazole | 1 | 15 | Mean Difference (IV, Fixed, 95% CI) | 6.49 [2.65, 10.33] |
| 6 Mental state: 2c. Specific ‐ negative symptoms ‐ average endpoint scores ‐ immediate ‐ only DHEA (SANS, high score = poor, skewed data) | Other data | No numeric data | ||
| 7 Mental state: 2d. Specific ‐ anxiety symptoms ‐ average endpoint scores ‐ only DHEA (HAMA total, higher score = poor, skewed data) | Other data | No numeric data | ||
| 8 Mental state: 2e. Specific ‐ depression ‐ average endpoint scores ‐ only mifepristone (HAMD total, higher score = poor) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 immediate | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐3.20 [‐9.12, 2.72] |
| 8.2 short‐term | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐3.75, 5.55] |
| 9 Mental state: 2f. Specific ‐ depression ‐ average endpoint scores (CDSS/HAMD total, high score = poor, skewed data) | Other data | No numeric data | ||
| 9.1 DHEA | Other data | No numeric data | ||
| 9.2 ketoconazole | Other data | No numeric data | ||
| 9.3 mifepristone | Other data | No numeric data | ||
| 10 Global state: 1. General ‐ no clinically significant improvement ‐ data only for mifepristone (< 30% improvement BPRS) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.32, 1.06] |
| 11 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement BPRS, PSS) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.18, 0.81] |
| 12 Global state: 2b. Specific ‐ depression ‐ no clinically significant improvement ‐ data only for mifepristone (< 50% improvement HAMD) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.59, 1.22] |
| 13 Global state: 3. Leaving the study early | 7 | 201 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.20, 1.52] |
| 13.1 dexamethasone | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 DHEA | 3 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.13, 1.28] |
| 13.3 ketoconazole | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.4 mifepristone | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 14 Cognitive functioning: 1a. Average endpoint scores, various tasks | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1 Information processing and sustained attention (serial addition task) | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐1.87, 0.87] |
| 14.2 Spatial thinking (Benton Line Orientation task) | 1 | 19 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐1.63, 1.43] |
| 15 Cognitive functioning: 1b. Vigilance task ‐ average endpoint scores ‐ skewed data | Other data | No numeric data | ||
| 15.2 Vigilance | Other data | No numeric data | ||
| 16 General functioning: average endpoint scores GAF/SOFAS (low = poor) | 2 | 54 | Mean Difference (IV, Fixed, 95% CI) | 1.05 [‐5.55, 7.66] |
| 16.1 DHEA | 2 | 54 | Mean Difference (IV, Fixed, 95% CI) | 1.05 [‐5.55, 7.66] |
| 17 Adverse effects: 1. General ‐ overall number of events | 8 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.66 [1.33, 5.32] |
| 17.1 dexamethasone | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.2 DHEA | 4 | 139 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.38, 10.44] |
| 17.3 ketoconazole | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.19 [0.60, 7.93] |
| 17.4 mifepristone | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [1.27, 8.33] |
| 18 Adverse effects: 2a. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA (SHRS total, higher score = poor) | 1 | 30 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐1.66, ‐0.15] |
| 19 Adverse effects: 2b. Specific ‐ extrapyramidal symptoms ‐ average endpoint scores ‐ data only for DHEA (skewed data) | Other data | No numeric data | ||
| 19.1 extrapyramidal symptoms (ESRS total, higher score = poor) | Other data | No numeric data | ||
| 19.2 Akathisia and tardive dyskinesia (AIMS/BARS total, higher score = poor) | Other data | No numeric data | ||
| 20 Adverse effects: 2c. Specific: various effects | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 allergy ‐ skin rash ‐ data only for mifepristone | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.67 [0.64, 20.94] |
| 20.2 anticholinergic ‐ minor blurred vision ‐ data only for DHEA | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 20.3 cardiovascular ‐ mild dizziness ‐ data only for ketoconazole | 1 | 15 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.19 [0.60, 7.93] |
| 20.4 gastrointestinal ‐ appetite ‐ increase ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 20.5 central nervous system ‐ fatigue ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 19.78] |
| 20.6 central nervous system ‐ irritability/agitation ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 20.7 gastrointestinal ‐ appetite ‐ decrease ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 20.8 gastrointestinal ‐ constipation ‐ data only for DHEA | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 20.9 gastrointestinal ‐ constipation ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 20.10 gastrointestinal ‐ nausea ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 20.11 hormonal ‐ dysmenorrhoea ‐ data only for mifepristone | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 21 Quality of life: observer‐rated scale | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | 6.20 [‐1.37, 13.77] |
| 21.1 DHEA | 1 | 55 | Mean Difference (IV, Fixed, 95% CI) | 6.20 [‐1.37, 13.77] |
3.8. Analysis.
Comparison 3 Any antiglucocorticoid versus placebo as adjunct to combination treatment, Outcome 8 Mental state: 2e. Specific ‐ depression ‐ average endpoint scores ‐ only mifepristone (HAMD total, higher score = poor).
Comparison 4. Subgroup analyses ‐ by diagnostic group: mifepristone vs placebo as adjunct to combination treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. General ‐ average endpoint score (BPRS total scores, higher score = poor) | 2 | 50 | Mean Difference (IV, Fixed, 95% CI) | ‐1.02 [‐6.16, 4.12] |
| 1.1 people with psychotic depression | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐6.80 [‐15.31, 1.71] |
| 1.2 people with schizophrenia/schizoaffective disorder | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 2.30 [‐4.15, 8.75] |
| 2 Mental state: 2a. Specific ‐ positive symptoms ‐ average endpoint scores ‐ people with psychotic depression (BPRS positive subscale, higher score = poor) | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐2.60 [‐5.67, 0.47] |
| 3 Mental state: 2b. Specific ‐ depression ‐ average endpoint scores ‐ people with psychotic depression (HAMD total, higher score = poor) | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐3.20 [‐9.12, 2.72] |
| 4 Global state: 1. General ‐ no clinically significant improvement ‐ people with psychotic depression (< 30% improvement BPRS) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.32, 1.06] |
| 5 Global state: 2a. Specific ‐ positive symptoms ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement BPRS, PSS) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.18, 0.81] |
| 6 Global state: 2b. Specific ‐ depression ‐ no clinically significant improvement ‐ people with psychotic depression (< 50% improvement HAMD) | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.59, 1.22] |
| 7 Global state: 3. Leaving the study early | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 7.1 people with psychotic depression | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 7.2 people with schizophrenia/schizoaffective disorder | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Adverse events: overall number of events | 2 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [1.27, 8.33] |
| 8.1 people with psychotic depression | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.0 [1.41, 11.35] |
| 8.2 people with schizophrenia/schizoaffective disorder | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 13.87] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Belanoff 2001.
| Methods | Allocation: randomised.
Blindness: participant/personnel, unclear whether blinded at outcome.
Duration: 4 days. Design: cross‐over (× 1). |
|
| Participants | Diagnosis: major depression with psychotic features. DSM IV, clinician interview(s).
N = 5.
Age: range 44 to 67 years; average mifepristone ˜ 48 (SD 4), placebo 56 (SD ˜ 12) years (first arm)
Sex: 3 M, 2 F (first arm).
Setting: inpatient.
History: duration of illness ‐ mifepristone 4.5 months (SD ˜ 5), placebo 98 months (SD 123)
Excluded: any sign of Cushing syndrome apart from hypercortisolaemia, women of child‐bearing potential, patients using illicit drugs within a month before admission, patients consuming up to 2 ounces of alcohol daily. Country: USA. |
|
| Interventions | 1. Mifepristone: dose 600 mg/day. N = 2. 2. Placebo: N = 3. Other concurrent treatments: no antipsychotic medication for 3 days before entering study, no antidepressant upon entering study, no participants started on antidepressant medication while in study, benzodiazepines permitted for insomnia and acetaminophen for headaches. |
|
| Outcomes | Mental state: total psychotic symptoms (BPRS), depression (HAMD total score). Global state: leaving the study early. Adverse event: overall adverse event rate. Unable to use: cognition paragraph recall (not reported). |
|
| Notes | All means and SDs were calculated from individual data by BG. Funded by NARSAD Young Investigator Award, Pritzker Foundation, NIMH. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "Randomised"; no other statement provided. Imbalance in duration of illness between groups calls into question the success of randomisation. |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ no statement provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "..patients served as their own controls in a random‐assignment, double‐blind crossover design" (pg 517). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | High risk | Two outcome measures not reported in the results: Clinical Global Impression (CGI) scale, paragraph recall cognitive test. |
| Other bias | High risk | Small sample size (n = 5). |
DeBattista 2006.
| Methods | Allocation: randomised. Blindness: participant/personnel, unclear whether blinded at outcome. Duration: 7 days. Assessment points: baseline (day 0), daily during dosing (days 1 to 7), days 14 and 28. |
|
| Participants | Diagnosis: psychotic depression. Structured Clinical Interview for DSM Disorders (SCID), DSM IV, clinician interview(s).
N = 221.
Age: mifepristone group mean ˜ 41 years (SD ˜ 11), placebo group mean ˜ 42 years (SD 11).
Sex: 112 M, 109 F.
Setting: inpatient and outpatient.
History: unclear.
Excluded: unstable medication condition, use of systemic or inhaled corticosteroids, ECT in past 3 months, antidepressant and/or antipsychotic in past 7 days, history of illicit drug use in past month, alcohol or drug dependence in past 6 months. Country: USA (29 sites). |
|
| Interventions | 1. Mifepristone: dose 600 mg/day. N = 105. 2. Placebo: N = 116. Other concurrent treatments: antipsychotics and antidepressants not allowed for 7 days of study, thereafter any indicated treatment. |
|
| Outcomes | Global state: rapid response (> 30% reduction BPRS total at days 7 and 28), response (> 30% reduction BPRS total at day 28 but not at day 7), positive psychotic response (< 50% improvement BPRS PSS) and depression response (< 50% improvement HAMD). Leaving the study early. Adverse events: spontaneous report of adverse events. |
|
| Notes | Funded by: This work was sponsored by Corcept Therapeutics, Menlo Park, California. “We acknowledge the following disclosures; CD: Speakers Bureau, Wyeth, Cephalon, Pfizer, GSK, Lilly, BMS, Cyberonics. Grant support; Wyeth, GSK, Cephalon, Pritzker Foundation, NARSAD, NIMH, Neuronetics, Cyberonics. Consultant; Corcept Therapeutics, Wyeth, Lilly, Roche, BMS. Stock‐holder; Corcept Therapeutics. JB: CEO and equity‐holder; Corcept Therapeutics, Menlo Park, CA. CB: Statistical consultant; Corcept Therapeutics. LLC: Consultant; GlaxoSmithKline, Novartis, Pfizer, Johnson & Johnson, Sepracor, Cyberonics, Bristol‐Myers Squibb, Medronic, and Wyeth. Grants/Research support; NIH, US Dept of the Interior, Cyberonics, Pfizer, Corcept Therapeutics, Medtronic, and UCB Pharma. Speakers Bureau or has received Honoraria for speaking; Cyberonics, Pfizer, Wyeth, AstraZeneca, and Cephalon." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients who met the study criteria were randomised 1:1 to 7 days..." (pg 1344). |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ no statement provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "randomised 1:1 to 7 days of inpatient treatment in a double‐blind, placebo controlled, parallel group design" (pg 1344). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data imputed using appropriate methods (ITT analysis conducted). |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported. |
| Other bias | High risk | Study authors have conflict of interest with sponsor/funding source. |
Flores 2006.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 days. |
|
| Participants | Diagnosis: psychotic major depression, SCID, DSM IV and clinician interview(s).
N = 31.
Age: mean ‐ mifepristone group ˜ 36 years (SD ˜ 13), placebo group ˜ 39 years (SD ˜ 13).
Sex: 13 M, 17 F.
Setting: inpatient and outpatient.
History: unclear.
Excluded: pregnant/lactating women, major medical illnesses, history of seizures, major head trauma, abnormal clinical laboratory tests, those taking systemic steroids, people younger than 18 years, actively suicidal, obsessive‐compulsive disorder. Country: USA. |
|
| Interventions | 1. Mifepristone: dose 600 mg/day. N = 17. 2. Placebo: N = 15. Other concurrent treatments: remained on current medications during study (included antidepressants, antipsychotics, anxiolytics, mood stabilisers). |
|
| Outcomes | Mental state: total psychotic symptoms (BPRS), positive psychotic symptoms (BPRS positive symptoms subscale), depression (HAMD total score). Global state: response (30% reduction BPRS total and 50% reduction BPRS positive). Leaving the study early. Adverse events: overall rate, serious adverse outcomes. |
|
| Notes | Funded by National Institute of Mental Health and National Institutes of Health. "Corcept Therapeutics has licensed intellectual property for the use of mifepristone in the treatment of PMD. Dr Alan Schatzberg is cofounder of Corcept Therapeutics and is the only author involved in this submission who has any financial interest in this company. However, Dr Schatzberg continues to be full‐time faculty at Stanford University. In addition, Dr Schatzberg played no direct role in the recruitment, assessment, or follow‐up of subjects enrolled in this study. Dr Schatzberg was not directly involved in the analysis of data stemming from this research." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "..patients were randomised to..." (pg.630). |
| Allocation concealment (selection bias) | Unclear risk | No statement provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "Patients were randomised to either 600mg per day of double‐blind mifepristone or placebo for eight days" (pg 630). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients were randomised to either 600mg per day of double‐blind mifepristone or placebo for 8 days" (pg 630). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients were rated on the HDRS and BPRS by a blinded, trained clinical administrator..." (pg 630). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One participant dropped out during the intervention and was not included in the analysis. |
| Selective reporting (reporting bias) | Low risk | All outcome measures were reported. |
| Other bias | Unclear risk | One study author has financial interest in the company that has licensed IP for use of mifepristone in treatment of PMD. |
Gallagher 2005.
| Methods | Allocation: randomised. Blindness: double. Duration: 7 days. | |
| Participants | Diagnosis: schizophrenia, SCID, DSM IV.
N = 20.
Age: average 43 years (SD 9.6), range 27 to 61 years.
Sex: 18 M, 2 F.
Setting: outpatient.
History: unclear.
Excluded: women of child‐bearing potential, neuropsychological confounds of previous major head injury, current or previous neurological disease, co‐morbid medical condition. Country: UK. |
|
| Interventions | 1. Mifepristone: dose 600 mg/day. N = 10. 2. Placebo: N = 10. Other concurrent treatments: "Patients' medication had been unchanged for six weeks before participation and remained so throughout the study period. Two subjects had minor dose reductions during the previous eight weeks, but otherwise there had been no medication changes for at least eight months." All 20 participants were taking at least 1 antipsychotic (see details, p 157). In addition, 7 were taking anticholinergics, 1 carbamazepine, 6 an antidepressant, 1 regular paracetamol, 1 ibuprofen and 1 carbimazole. |
|
| Outcomes | Mental state: total psychotic symptoms (BPRS), depression post intervention, and depression at follow‐up (HDRS‐17). Global state: leaving the study early. Adverse events: general adverse events. Unable to use cognitive functioning: spatial working memory task (CANTAB), Rey‐Auditory Verbal Learning Test, short‐term memory span, visuospatial learning and memory, executive function and attention. Cross‐over study design: data not reported for the first phase of the study. Study authors reported no significant differences between groups in any cognitive measure. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (from correspondence): "Using a computerised randomiser for trial design." |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "It was held by pharmacy and was not known to anyone involved in the study. All medication was dispensed by pharmacy in identical packaging, and active/placebos were identical and produced by the manufacturer (Exelygen)." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Administration of medication was in a double‐blind design" (pg 156). Outcome assessors were blind (source: correspondence). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Administration of medication was in a double‐blind design" (pg 156). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blind (source: correspondence). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data for phase 1 of cross‐over trial. Only data from phase 1 included in the meta‐analysis. |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported. |
| Other bias | Low risk | Study appears to be free of other sources of bias. |
Marco 2002.
| Methods | Allocation: randomised.
Blindness: unclear.
Duration: 4 weeks. Funded by NARSAD, the National Alliance for the Mentally Ill, the Stanley Foundation. Active ketoconazole and placebo capsules were provided at no cost by Janssen Pharmaceuticals. |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder, SCID, DSM III‐R.
N = 19.
Age: average 48 year (SD 8.5), range 33 to 62 years.
Sex: 12 M, 3 F. Setting: outpatient. History: unclear. Excluded: no change in medication in the past 6 weeks, medically unhealthy, no use of other steroid‐containing medications. Country: USA. |
|
| Interventions | 1. Ketoconazole: started at 200 mg/day and advanced to maximum dose 800 mg/day. N = 8. 2. Placebo: N = 7. Other concurrent treatments: All participants were taking stable doses of antipsychotic medication (including risperidone, perphenazine, haloperidol decanoate and thioridazine) and, when applicable, antidepressant and/or mood stabiliser medication (including clonazepam, lithium, trazodone, benztropine mesylate, diphenhydramine, propranolol, carbamazepine, sucralfate, sertraline, lorazepam and trihexyphenidyl). One participant with schizophrenia was taking no concurrent treatment. As a criterion of clinical stability, all participants were well known to the outpatient psychiatry clinic and had not required any change in medication dosage for a minimum of 6 weeks. No changes in pre‐stabilised, open‐label antipsychotic, antidepressant and/or mood‐stabilising medication regimens were allowed during the study period. Other concurrent treatments (placebo group): All participants were taking stable doses of antipsychotic medication (including risperidone, haloperidol, perphenazine and trifluoperazine) plus, when applicable, antidepressant and/or mood stabiliser medication (including paroxetine, lorazepam, benztropine mesylate, diphenhydramine, fluoxetine, buspirone and methylphenidate). Two participants with schizophrenia were taking no concurrent treatment. |
|
| Outcomes | Mental state: total psychotic symptoms (PANSS), positive psychotic symptoms (PANSS), negative psychotic symptoms (PANSS), depression (HAMD). Global state: leaving the study early. Adverse events: general adverse events. Unable to use: cognition. California Verbal Learning Test, Trails A & B, FAS Verbal Fluency Test. Alternate versions of the CVLT and Verbal Fluency Test were used for baseline and week 4 assessments. Data were not provided. Study authors reported no significant differences between groups on cognitive measures. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (from correspondence): "by computer sequence." |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "Envelopes with key based on numeric code. Pharmacist had filled blinded Rx based on numeric code." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participant/providers: Yes. Quote: "Subjects were randomised to receive in a double‐blind manner..." (pg 157). Outcome assessors: Yes. Quote: "Subjects and blinded raters were not able to accurately guess treatment assignment based on side effects." (pg 159). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Subjects and blinded raters were not able to accurately guess treatment assignment based on side effects" (pg 159). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | High risk | Data reported only for HAMD and cortisol measures. For PANSS, BDI, Bunney‐Hamburg Global Rating Scale and neurocognitive function tests, it was stated that no significant differences were noted (data not reported). |
| Other bias | High risk | Baseline gender imbalance. Participants in the ketoconazole group seem to be taking a greater number of concomitant medications, suggesting that they may have more severe or complex illness. Those in the ketoconazole group had higher baseline cortisol levels. |
Nachshoni 2005.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 7 days. Funded by: no extramural funding (source: study author). |
|
| Participants | Diagnosis: schizophrenia, schizoaffective disorder. SCID, DSM III‐R.
N = 34.
Age: average ˜ 40 years (SD ˜ 12), range 19 to 64 years.
Sex: 23 M, 7 F.
Setting: inpatient.
History: unclear.
Excluded: receiving steroids, pregnant women, patients engaged in substance abuse, patients with significant medical or neurological illness. Country: Israel. |
|
| Interventions | 1. DHEA: dose 100 mg/day( 50 mg bid at 8am and 8pm). N = 18. 2. Placebo: N = 16. Other concurrent treatments: All participants had received fixed doses of antipsychotic medications for at least 3 weeks before study commencement, and no change in dosage or medication was permitted for the trial duration. Anti‐EPS agents were withdrawn with a 2‐day washout period before randomisation. |
|
| Outcomes | Mental state: total psychotic symptoms (BPRS). General functioning: GAF. Adverse events: general adverse events and general extrapyramidal symptoms (SHRS). Unable to use: adverse events ‐ extrapyramidal symptoms ‐ UKU (not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were randomised (by means of random number generation)..." (pg 252). |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "Randomization numbers provided by statistician to research assistant assigning study medication and maintained under lock and key in concealed fashion." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participant/providers: Yes. Quote: "...randomised to receive either DHEA at a fixed dose of 100 mg/day or placebo in double‐blind fashion..." (pg 252). Outcome assessors blind (from correspondence). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blind (from correspondence). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing outcome data for 4 participants not accounted for in analysis. |
| Selective reporting (reporting bias) | High risk | Study authors unable to analyse tardive dyskinesia or dystonia because few participants had tardive dyskinesia (10%) or dystonia (0%). UKU scores not reported. |
| Other bias | High risk | Only 11/30 participants exhibited akathisia, and slight differences in baseline akathisia symptoms were noted between groups. Sample size was small and the intervention was of short duration. |
Newcomer 1998.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: 4 days. Funded by NIMH Scientist Development Awards. |
|
| Participants | Diagnosis: schizophrenia. DMS III‐R and clinician interview.
N = 19.
Age: average ˜ 32 years (SD 8).
Sex: 9 M, 10 F.
Setting: inpatient.
History: unclear.
Excluded: history of DSM‐III‐R substance dependence or abuse within the past 6 months, current pregnancy, any current medical illness including trauma, fever or dehydration in the past month, neurological disorders (except possible tardive dyskinesia) including any history of significant head injury, defined as loss of consciousness for longer than 5 minutes and/or with neurological sequelae. Body weight < 80% of ideal body weight, treatment with narcotics in the past month, any treatment with corticosteroids or high‐dose oestrogens within past 6 months Country: USA. |
|
| Interventions | 1. Dexamethasone: successive doses of 0.5, 1, 1 and 1 mg at 2300 h on days 0 to 3. N = 11. 2. Placebo: N = 8. Other concurrent treatments: Participants were studied during ongoing 'typical' antipsychotic treatment, including haloperidol and adjunctive anticholinergics. They were also studied during ongoing nicotine use. |
|
| Outcomes | Global state: leaving the study early. Cognitive function: paced serial addition task, vigilance task, Benton line orientation task. Adverse events: spontaneous reporting of adverse events. Unable to use: mental state. Total psychotic symptoms (BPRS not reported), negative psychotic symptoms (SANS not reported). Cognitive functioning: paragraph recall test (data not available). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No statement provided. |
| Allocation concealment (selection bias) | Unclear risk | No statement provided. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "...after 4 days of double‐blind, placebo controlled treatment with DEX..." (pg 67). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analyses excluded participants with missing data (n = 3). However, Quote: "analyses were also run that included all subjects (n=19) by applying conservative adjustments for missing data (mean performance values for the other three test days as the washout (day 11) performance value)." |
| Selective reporting (reporting bias) | High risk | Only baseline BPRS and SANS scores reported. |
| Other bias | High risk | Participants in placebo group received on average lower dose of antipsychotic medication compared with intervention group. |
Ritsner 2006.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 6 weeks. Funded by Stanley Foundation. |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder. SCID, DSM IV.
N = 62.
Age: average ˜ 36 years (SD 10), range 20 to 53 years.
Sex: 41 M, 14 F. Setting: inpatient and outpatient. History: length of illness ‐ "chronic" average ˜ 14 years (SD ˜ 9). Excluded: evidence of organic brain damage, mental retardation, major medical illness, alcohol or drug abuse, prostate nodules or cancer; symptoms of benign prostatic hypertrophy, pregnant women, history of breast or uterine illness. Country: Israel. |
|
| Interventions | 1. DHEA first: 100 mg BID (total 200 mg/day). N = 29. 2. Placebo first: N = 26. Other concurrent treatments: Throughout duration of study, participants continued to receive regular antipsychotic medication, with antipsychotic medication dose kept constant for at least 2 weeks before study entry and throughout the study period. 24 participants received first‐generation antipsychotic medication: chlorpromazine equivalent mean dose of 660 mg/d (SD 53). 20 received second‐generation antipsychotic medication: chlorpromazine equivalent mean dose of 375 mg/d (SD 266). 11 received both types of antipsychotic medication: chlorpromazine equivalent mean dose of 1077 mg/d (SD 357). Participants allowed to receive benzodiazepine or antiparkinsonian medications as indicated. |
|
| Outcomes | Mental state: total psychotic symptoms (PANSS), positive psychotic symptoms (PANSS), negative psychotic symptoms (PANSS). Global state: leaving the study early. General functioning: observer‐rated QOL scale. Adverse events: extrapyramidal symptoms (ESRS, AIMS). Unable to use: cognitive functioning. CANTAB including visual and movement skills, attention and memory and executive function. Cross‐over study design: data not reported for first phase of the study. Study authors reported significant improvement in visual sustained attention, visual and movement skills following DHEA compared with placebo. They noted no significant differences between groups in the other cognitive domains. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (from correspondence): "Independent pharmacist dispensed either DHEA or placebo capsules according to a computer generated randomization list." |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "the allocation was done by a pharmacist." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participant/providers: participants, providers and key study personnel blinded (source: correspondence). Outcome assessors: yes (from correspondence). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants, providers and key study personnel blinded (source: correspondence). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blinded (source: correspondence). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number dropped out during intervention: 7. Quote: "..seven patients failed to complete the 12 weeks of the crossover randomisation phase because of withdrawal of study consent after randomisation, but previously have received the first dose of study medication" (pg 497). Analyses excluded participants with missing data (n = 7). |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported. |
| Other bias | Low risk | Study appears to be free of other sources of bias. |
Ritsner 2010.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 8 weeks. Funding: none reported. |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder. SCID, DSM IV.
N = 58.
Age: average 35.8 years (SD 8.3), range 23 to 55 years.
Sex: 32 M, 12 F.
Setting: outpatient.
History: average duration of illness (years): PREG30 = 15.1 (8.0); PREG200 = 11.7 (7.7); DHEA400 = 10.3 (7.3); placebo = 11.1 (6.5).
Excluded: unstable medical condition, any significant medical (including prostate illness) or neurological illness, pregnant women, receiving mood stabilisers or any steroid hormonal supplement (e.g. oestrogen). Country: Israel. |
|
| Interventions | 1. Pregnenolone (PREG30) 30 mg/day. yN = 16. 2. Pregnenolone (PREG200) 200 mg/day. N = 10. 3. DHEA 400 mg/d. N = 16. 4. Placebo identical capsules. N = 16. Other concurrent treatments: antipsychotic, anticholinergic, benzodiazepine medication continued from before the trial. PREG30: chlorpromazine equivalent 476.4 (337.6) mg/day. PREG200: chlorpromazine equivalent 585.0 (704.3) mg/day. DHEA: chlorpromazine equivalent 441.1 (276.2) mg/day. Placebo: chlorpromazine equivalent 621.3 (455.3) mg/day. |
|
| Outcomes | Mental state: total psychotic symptoms (PANSS), positive psychotic symptoms (PANSS), negative psychotic symptoms (PANSS). Global state: leaving the study early. General functioning: GAF. Adverse events: general adverse events, extrapyramidal symptoms (ESRS, BARS). Unable to use: cognitive functioning. CANTAB including matching to sample (MTS), delayed matching to sample (DMS), pattern recognition (PRM), rapid visual information processing (sustained attention) (RVP) and stockings of Cambridge (SOC). Data not provided. Study authors reported significant improvement in DMS and MTS with pregnenolone 30 mg/d treatment. No significant effects of DHEA on cognitive function reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomization procedure was performed using the Random Allocation Software, version 1.0" (pg 1353). |
| Allocation concealment (selection bias) | Low risk | "The pharmacist conducted randomization of participants by using a random and equal block size for placebo, DHEA and PREG30 arms (with ratio 1.5:1 for PREG30 and PREG200 arms, respectively) and conducted blinding of the trial. The patient allocation details were coded and kept confidential until the trial was completed" (pg 1353). |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Double‐blind study design" (pg 1352). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind study design" (pg 1352). Participants and study personnel blinded. "The patient allocation details were coded and kept confidential until the trial was completed" (pg 1353). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blinded (pg 1353). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | LOCF analysis described in methods (pg 1354): "Patients who completed the study (completers) were included in the statistical analysis. The LOCF procedure was used to analyze those subjects who completed at least four weeks (selected a priori) but failed to complete all eight weeks of the study (non‐completers)" (pg 1365): "Of the 58 patients randomly assigned to this trial, 14 patients dropped out." "More specifically 1,1,2, and 3 patients assigned to receive PREG‐30, PREG‐200, DHEA, and placebo, respectively, dropped out between four and six weeks, and seven patients dropped out between six and eight week." All 14 participants who left the the study early had completed at least 4 weeks of the trial. However, analysis was performed on 44, not LOCF (n = 58) Reasons for leaving the study early included lack of efficacy (n = 4), change in antipsychotic drugs (n = 3), loss to follow‐up (n = 5), non‐compliance (n = 2). |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
Strous 2003.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 6 weeks. Funded by NARSAD Young Investigator Award. |
|
| Participants | Diagnosis: schizophrenia. SCID, DSM III‐R.
N = 30
Age: average ˜ 37 years (SD 12), range 20 to 67 years.
Sex: 12 M, 15 F.
Setting: inpatient.
History: duration of illness "chronic", average ˜ 200 months (SD 130).
Excluded: any significant medical (including prostate illness) or neurological illness, pregnant women, patients administered mood stabilisers or any steroid or hormonal supplement (e.g. oestrogen) All participants entered a 1‐week, single‐blind, placebo lead‐in phase of the study. Participants completing the placebo lead‐in who continued to demonstrate the above criteria for negative symptoms, with no meaningful change in SANS score (defined by a change > 20% in clinical ratings score), qualified to enter study treatment phase. Country: Israel |
|
| Interventions | 1. DHEA: dose 25 mg/d for first 2 weeks (8am), 50 mg/day in equally divided doses for the following 2 weeks (8am and 8pm), then 100 mg/d in equally divided doses for the final 2 weeks (8am and 8pm). N = 15. 2. Placebo: N = 15. Other concurrent treatments: Participants were required to have been administered a stable dose of their current 'typical' or 'atypical' antipsychotic medication for at least a month before study commencement. Participants were required to continue taking their regular medications for the duration of the study. No change in dose or addition of any other psychoactive medication was permitted during the study. All participants entered a 1‐week, single‐blind, placebo lead‐in phase of the study. Five individuals were taking olanzapine, 4 clozapine, 3 haloperidol, 1 fluphenazine, 1 zuclophenthixol and 1 risperidone. Four participants were taking benzodiazepine medication, and 3 anticholingeric medication. |
|
| Outcomes | Mental state: total psychotic symptoms (PANSS), positive psychotic and negative symptoms (PANSS, SANS), depression (HAMD total score), anxiety (HAMA). Global state: leaving the study early. Adverse events: overall rate, monitored and assessed daily for any adverse events, formally assessed weekly by a physician for any DHEA medication adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were then randomised (by means of random number generation) to receive..." (pg 134). |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "Randomisation numbers provided by statistician to research assistant assigning study medication and maintained under lock and key in concealed fashion." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participant/providers: yes. Quote: "..to receive either DHEA or placebo, each for six weeks in a double‐blind manner (administered and monitored by a hospital pharmacist)" (pg 134). Outcome assessors blinded (Source: correspondence). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blinded (source: correspondence). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number leaving early during intervention: 3 participants (DHEA = 0; placebo = 3) eliminated from study analysis after failing to complete 3 weeks of randomisation phase (selected a priori). Missing data on 4 participants (DHEA = 2; placebo = 2) imputed using LOCF. Three participants eliminated from study analysis after failing to complete 3 weeks of randomisation phase (selected a priori). Missing data on 4 participants imputed using LOCF. |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported. |
| Other bias | High risk | Gender imbalance. No data given on number of individuals screened for study inclusion. |
Strous 2007.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 12 weeks. Funded by NARSAD Young Investigator Award. |
|
| Participants | Diagnosis: schizophrenia. DSM IV, SCID.
N = 40.
Age: average 34 years (SD ˜ 10), range 18 to 58 years.
Sex: 27 M 13 F.
Setting: inpatient.
History: duration of illness 'chronic', longer than 2 years.
Excluded: people with any significant medical (including prostate illness) or neurological illness, pregnant women, people who had been administered mood stabilisers or any steroid or hormonal supplement (e.g. oestrogen). Country: Israel. |
|
| Interventions | 1. DHEA: dose 50 mg/day for first 2 weeks, 100 mg/day for following 2 weeks, finally 150 mg/day for last 8 weeks (each administered in divided morning and evening doses). N = 20. 2. Placebo: N = 20. Other concurrent treatments: participants required to have been maintained on a stable dose of olanzapine for at least 1 month before study commencement. Participants were required to continue taking this dose of olanzapine throughout the duration of the study. Aside from olanzapine, concurrent medications allowed included medications that were clinically required before study recruitment to maintain and stabilise clinical status (e.g. benzodiazepines). Clinicians were requested to not change these ancillary medications over the course of the study. All participants entered a 1‐week, single‐blind, placebo lead‐in phase of the study. |
|
| Outcomes | Mental state: total psychotic symptoms (PANSS), positive psychotic symptoms (PANSS), negative psychotic symptoms (PANSS), depression (CDSS). Global state: leaving the study early. Adverse events: extrapyramidal symptoms (parkinsonism) (SAS), body weight Unable to use cognitive functioning: 'Mindstreams' cognitive test battery (Go‐NoGo Response Inhibition Test, Stroop Interference Test, Staged Information Processing Speed tests); test of verbal and non‐verbal memory. Data not provided. Study authors reported no significant differences in cognitive performance between intervention and control groups. Adverse events: extrapyramidal symptoms (tardive dyskinesia, akathisia) (BARS/AIMS). Only data for intervention groups reported (Table 4). No analyses could be undertaken. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients then were randomised (by means of random number generation)..." (pg 97). |
| Allocation concealment (selection bias) | Low risk | Quote (from correspondence): "Randomisation numbers provided by statistician to research assistant assigning study medication and maintained under lock and key in concealed fashion" |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participant/providers: Yes. Quote: "...to receive either DHEA or placebo, each for 12 weeks in a double‐blind manner" (pg 97). Outcome assessors blinded (source: correspondence). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind" ‐ probably undertaken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors blinded (source: correspondence). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Low risk | All outcome measures reported. |
| Other bias | High risk | No data given on numbers of individuals screened for study inclusion. Tendency towards baseline imbalance in SANS scores (P value = 0.051). |
AIMS ‐ Abnormal Involuntary Movement Scale BARS ‐ Barnes Akathisia Rating Scale BDI ‐ Beck Depression Inventory
BPRS ‐ Brief Psychaitric Rating Scale BPRS PSS ‐ BPRS Positive Symptom Subscale CANTAB ‐ Cambridge Neuropsychological Test Automated Battery CDSS ‐ Calgary Depression Scale for Schizophrenia CGI‐S ‐ Clinical Global Impressions Scale CPZ ‐ Chlorpromazine CVLT ‐ California Verbal Learning Test DHEA ‐ Dehydroepiandrosterone DSM ‐ Diagnostic and Statistical Manual of Mental Disorders ESRS ‐ Extrapyramidal Symptom Rating Scale F ‐ Female
GSK ‐ GlaxoSmithKline HAMA Hamilton Scale for Anxiety HAMD ‐ Hamilton Scale for Depression HDRS‐17 ‐ Hamilton Depression Rating Scale ‐ 17 item LOCF ‐ Last observation carried forward M ‐ Male MADRS ‐ Montgomery‐Asberg Depression Rating Scale
NARSAD ‐ National Alliance for Research on Schizophrenia and Depression
NIH ‐ National Institutes of Health
NIMH ‐ National Institute of Mental Health OAS ‐ Overt Aggression Scale PANSS ‐ Positive and Negative Syndrome Scale for Schizophrenia
PMD ‐ Psychotic major depression QOL ‐ Quality of life SANS ‐ Scale for the Assessment of Negative Symptoms SAS ‐ Simpson‐Angus Extrapyramidal Symptom Scale SHRS ‐ St Hans Rating Scale UKU ‐ Udvalg for Kliniske Undersøgelser side effects rating scale
USPHS ‐ United States Public Health Service YMRS ‐ Young Mania Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Barkai 1985 | Allocation: not randomised (not an intervention study, review article). |
| Beasley 1998 | Allocation: randomised. Participants: people with psychosis. Intervention: olanzapine vs haloperidol vs placebo, not antiglucocorticoid or related intervention. |
| Belanoff 2002 | Allocation: randomised. Participants: people with psychotic major depression. Intervention: dose finding study (50 mg to 400 mg of mifepristone), not a placebo‐controlled trial. |
| Brambilla 1988 | Allocation: not randomised (all participantts were allocated to receive placebo treatment first, followed by the intervention treatment). |
| David 1999 | Allocation: randomised. Participants: people with psychosis. Intervention: olanzapine vs risperidone, not antiglucocorticoid or related intervention. |
| Harrigan 2004 | Allocation: randomised.
Participants: people with a psychotic disorder. Interventions: not an intervention study (ketoconazole administered as CYP inhibitor to examine antipsychotic effects on cardiac functioning measures). |
| Iager 1986 | Allocation: randomised. Participants: people with psychosis. Intervention: 1‐desamino‐8‐D‐arginine vasopressin (DDAVP) vs placebo, not antiglucocorticoid or related intervention. |
| Katz 2002 | Allocation: not randomised (not an intervention study, review article). |
| Kim 1960 | Allocation: not randomised (case control study). |
| Kline 1968 | Allocation: not randomised. |
| Korsgaard 1981 | Allocation: not randomised (all participantts were allocated to receive placebo treatment first, followed by the intervention treatment). |
| Lane 2001 | Allocation: randomised. Participants: people with schizophrenia. Intervention: clozapine plus water vs clozapine plus super‐strength grapefruit juice, not antiglucocorticoid or related intervention. |
| Lembke 2013 | Allocation: not randomised (not an intervention study). |
| Loranger 1968 | Allocation: randomised. Participants: mixed diagnostic group (diagnoses: schizophrenic reaction (50%), psychoneurotic reaction (22%), manic‐depressive reaction (18%), sociopathic personality disturbance (5%), involutional psychotic reaction (3%), 'paranoid state' (2%). Interventions: cyclopregnol (6beta‐hydroxy‐3,5‐cyclopregnan‐20‐one) vs chlorpromazine vs placebo, not antiglucocorticoid or related intervention. |
| Miodownik 2011 | Allocation: randomised. Participants: schizophrenia, schizoaffective disorder. Intervention: L‐theanine vs placebo. Reason for exclusion: not an antiglucocorticoid treatment. |
| Nihalani 2007 | Allocation: not randomised (not an intervention study, review article). |
| Rees 1951 | Allocation: not randomised (case control study). |
| Rees 1956 | Allocation: not randomised (case control study). |
| Rothschild 2005 | Allocation: not randomised (not an intervention study, letter to the editor). |
| Schatzberg 2003 | Allocation: not randomised (not an intervention study, review article). |
| Silbergeld 1973 | Allocation: randomised. Participants: people with depression, schizophrenia, anxiety‐type reactions. Intervention: dexamethasone vs placebo. Reason for exclusion: no includable data. |
| Simpson 2005 | Allocation: not randomised (no comparison group, all participants received intervention treatment). |
| Stein 1984 | Allocation: randomised. Participants: people with schizophrenia. Intervention: vasopressin derivative vs placebo, not antiglucocorticoid or related intervention. |
| Tollefson 1998 | Allocation: randomised. Participants: people with schizophrenia. Intervention: olanzapine vs haloperidol, not antiglucocorticoid or related intervention. |
| Young 2004 | Allocation: randomised. Participants: people with bipolar disorder, not psychotic disorder. |
CYP ‐ Cytochrome P mg ‐ Milligrams
Characteristics of studies awaiting assessment [ordered by study ID]
Hardwick 1957.
| Methods | Allocation: unclear. Blindness: double (preliminary investigation only, 'main series' not an RCT). Duration: 15 days. Funded by: British Drug Houses Ltd. |
| Participants | Diagnosis: schizophrenia, dementia, 'anxiety hysteria', 'anxiety state', 'depressive state'. N = 10 (preliminary investigation) and N = 10 (main series). Age: 28 to 76 years. Sex (M:F): preliminary investigation 2:8; main series 3:7. Setting: preliminary investigation: unclear; main series: outpatients. History: preliminary investigation: 'chronic' average length of illness unclear; main series: varied, but predominantly described as 'longstanding'; average duration of illness unclear. Excluded: unclear. Country: United Kingdom. |
| Interventions | Preliminary investigation: 300 mg/day 6B‐hydroxy‐3:5‐cyclopregnan‐20‐one for 15 days, followed by placebo (15 days) (N unknown) or 15 days placebo, followed by 15 days 300 mg/day 6B‐hydroxy‐3:5‐cyclopregnan‐20‐one (N unknown). Main series: 300 mg/day 6B‐hydroxy‐3:5‐cyclopregnan‐20‐one. |
| Outcomes | Unclear (participant interview and subjective report only, no standardised measures used). |
| Notes | Awaiting assessment given lack of available information. |
Kleiser 1984.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: schizophrenia. Age: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Owen 1996.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: schizophrenia. Age: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Pivac 2002.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: schizophrenia. Age: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear |
| Notes | Awaiting assessment given lack of available information. |
Sluchevskii 1986.
| Methods | Allocation: unclear. Blindness: unclear. Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: alcohol abstinence syndrome, acute and chronic alcoholic psychoses; alcoholism. Age: unclear. Gender: unclear. |
| Interventions | 1. Vasopressin: dose unclear. 2. Corticotropin: dose unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Smidt 1988.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: 3 weeks. Funding: unclear. |
| Participants | Diagnosis: schizophrenia. Age: 22 to 39 years. N = 12. Age: unclear. Sex (M:F): unclear. Setting: unclear. History: unclear. Excluded: unclear. |
| Interventions | 1. Prednisolone: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. Other concurrent treatments: neuroleptic medication. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Volk 1976.
| Methods | Allocation: randomised. Blindness: unclear. Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: unclear N = unclear. Age: unclear. Sex (M:F): unclear. Setting: unclear. History: unclear. Excluded: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Volk 1977.
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: unclear. N = unclear. Age: unclear. Sex (M:F): unclear. Setting: unclear. History: unclear. Excluded: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
Watson 2002 (N0573099798).
| Methods | Allocation: unclear.
Blindness: unclear.
Duration: unclear. Funding: unclear. |
| Participants | Diagnosis: unclear. N = unclear. Age: unclear. Sex (M:F): unclear. Setting: unclear. History: unclear. Excluded: unclear. |
| Interventions | 1. Unclear: dose unclear. N = unclear. 2. Unclear: dose unclear. N = unclear. |
| Outcomes | Unclear. |
| Notes | Awaiting assessment given lack of available information. |
F ‐ Female M ‐ Male RCT ‐ Randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
Jarskog 2009.
| Trial name or title | Dehydroepiandrosterone (DHEA) for schizophrenia. |
| Methods | Allocation: randomised.
Blindness: double.
Duration: 6 weeks. Funded by Stanley Foundation. |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder.
N = 30.
Age: unclear. Sex (M:F): men only. Setting: unclear. History: persistent symptoms with prior adequate trials of antipsychotic drugs. Excluded: unclear. Country: USA. |
| Interventions | 1. DHEA: dose 400 mg/day. 2. Placebo. Other concurrent treatments: unclear, DHEA administered as adjunctive treatment. |
| Outcomes | Mental state: positive and negative symptoms, psychopathology (PANSS). Global state: CGI. Adverse effects: AIMS. |
| Starting date | Unclear. |
| Contact information | Dr L Fredrik Jarskog 1. Department of Psychiatry, University of North Carolina at Chapel Hill CB # 7160 2. Neurosciences Hospital, Chapel Hill, NC 27599 USA Email address: jarskog@med.unc.edu |
| Notes |
Solvason 2008.
| Trial name or title | Treatment of schizoaffective disorder using mifepristone. |
| Methods | Allocation: unclear (quote: "randomised").
Blindness: unclear (quote: "double‐blind").
Duration: unclear. Funded by Pritzker Foundation. |
| Participants | Diagnosis: schizoaffective disorder.
N = 30.
Age: 18 to 75 years.
Sex (M:F): mixed gender.
Setting: unclear.
History: unclear.
Excluded: Participants must be between the ages of 18 and 75 and must have no major medical problems. Country: USA. |
| Interventions | 1. mifepristone; 2. placebo. |
| Outcomes | Psychiatric symptomatology. |
| Starting date | April 1998. |
| Contact information | Dr Gregory H Cohen MSW Telephone: (650) 723‐3305 Email address: ghcohen@stanford.edu |
| Notes |
AIMS ‐ Abnormal Involuntary Movement Scale
DHEA ‐ Dehydroepiandrosterone
F ‐ Female
M ‐ Male
PANSS ‐ Positive and Negative Syndrome Scale for Schizophrenia
Differences between protocol and review
Since the protocol was published, we have updated the review in keeping with new RevMan 5 formatting. Main changes have involved identifying a primary outcome versus secondary outcomes and the approach to assessing and reporting on risk of bias in trials and in completing 'Summary of findings' tables. The Cochrane Schizophrenia Group has also updated its template for the Methods section, and we have updated these sections of the text to reflect this. For previous text, see Appendix 2.
As some trials were of very short duration, we have added a new category to the grouping of outcomes. Follow‐ups less than two weeks were considered to be 'immediate‐term', and 'short‐term' follow‐up was defined as two to 12 weeks post intervention.
Contributions of authors
Belinda Garner: co‐ordinated development of the protocol, trial selection, data extraction and write‐up of the review.
Sarah Hetrick: co‐ordinated development of the protocol, trial selection, data extraction and write‐up of the review.
Sarah Bendall: trial selection and data extraction.
Lisa Phillips: trial selection and data extraction.
Sources of support
Internal sources
ORYGEN Research Centre, University of Melbourne, Australia.
External sources
No sources of support supplied
Declarations of interest
Belinda Garner: no conflict of interest.
Sarah Hetrick: no conflict of interest.
Sarah Bendall: no conflict of interest.
Lisa Phillips: no conflict of interest.
New
References
References to studies included in this review
Belanoff 2001 {published data only}
DeBattista 2006 {published data only}
Flores 2006 {published data only}
- Carroll BJ, Rubin RT. Is mifepristone useful in psychotic depression? [comment]. Neuropsychophamacology 2006;12:2793‐4. [PUBMED: 17109015 ] [DOI] [PubMed] [Google Scholar]
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology 2006; Vol. 31, issue 3:628‐36. [DOI] [PubMed]
- Keller J, Schatzberg AF. Reply: Clinical and biological effects of mifepristone treatment for psychotic treatment. Neuropsychopharmacology 2006;3:628‐36. [PUBMED: 16160710] [DOI] [PubMed] [Google Scholar]
- NCT00048269. HPA axis/dopamine interactions in psychotic depression. https://www.clinicaltrials.gov/ct/show/NCT00048269 (accessed 28 February 2012).
Gallagher 2005 {published data only}
- Gallagher P, Watson S, Dye CE, Young AH, Ferrier IN. Persistent effects of mifepristone (RU‐486) on cortisol levels in bipolar disorder and schizophrenia. Journal of Psychiatric Research 2008;42:1037‐41. [DOI] [PubMed] [Google Scholar]
- Gallagher P, Watson S, Smith MS, Ferrier IN, Young AH. Effects of adjunctive mifepristone (RU‐486) administration on neurocognitive function and symptoms in schizophrenia. Biological Psychiatry 2005; Vol. 57, issue 2:155‐61. [DOI] [PubMed]
- Mackin P, Gallagher P, Watson S, Young AH, Ferrier IN. Changes in brain‐derived neurotrophic factor following treatment with mifepristone in bipolar disorder and schizophrenia. Australian and New Zealand Journal of Psychiatry 2007;41(4):321‐6. [DOI] [PubMed] [Google Scholar]
- Young A. Mifepristone for bipolar disorder and schizophrenia. Stanley Foundation Research Programs 2009.
Marco 2002 {published data only}
Nachshoni 2005 {published data only}
- Nachshoni T, Ebert T, Abramovitch Y, Asael‐Amir MM, Weizman A, Kotler M, et al. The effect of DHEA administration on extrapyramidal symptoms in schizophrenia: a randomized double blind placebo controlled trial. Journal of the European College of Neuropsychopharmacology 2005;15(Suppl 2):S135. [DOI] [PubMed] [Google Scholar]
- Nachshoni T, Ebert T, Abramovitch Y, Assael‐Amir M, Kotler M, Maayan R, et al. Improvement of extrapyramidal symptoms following dehydroepiandrosterone (DHEA) administration in antipsychotic treated schizophrenia patients: a randomized, double‐blind placebo controlled trial. Schizophrenia Research 2005; Vol. 79, issue 2‐3:251‐6. [DOI] [PubMed]
Newcomer 1998 {published data only}
Ritsner 2006 {published data only}
- Ritsner M. Randomized, double‐blind, crossover study of dehydroepiandrosterone (DHEA) for schizophrenia. Stanley Foundation Research Programs 2009.
- Ritsner MS, Gibel A, Ratner Y, Tsinovoy G, Strous RD. Improvement of sustained attention and visual and movement skills, but not clinical symptoms, after dehydroepiandrosterone augmentation in schizophrenia: a randomized, double‐blind, placebo‐controlled, crossover trial. Journal of Clinical Psychopharmacology 2006; Vol. 26, issue 5:495‐9. [DOI] [PubMed]
- Ritsner MS, Strous RD. Neurocognitive deficits in schizophrenia are associated with alterations in blood levels of neurosteroids: a multiple regression analysis of findings from a double‐blind, randomized, placebo‐controlled, crossover trial with DHEA. Journal of Psychiatric Research 2010;44:75‐80. [DOI] [PubMed] [Google Scholar]
- Strous RD, Gibel A, Maayan R, Weizman A, Ritsner MS. Hormonal response to dehydroepiandrosterone administration in schizophrenia: findings from a randomized, double‐blind, placebo‐controlled, crossover study. Journal of Clinical Psychopharmacology 2008; Vol. 28, issue 4:456‐9. [DOI] [PubMed]
Ritsner 2010 {published data only}
- NCT00174889. Efficacy and safety of pregnenolone augmentation in the management of schizophrenia patients: a randomised double‐blind placebo‐controlled trial. https://www.clinicaltrials.gov/ct/show/NCT00174889 (accessed 28 February 2012).
- Ritsner MS, Gibel A, Shleifer T, Boguslavsky I, Zayed A, Maayan R, et al. Pregnenolone and dehydroepiandrosterone as an adjunctive treatment in schizophrenia and schizoaffective disorder: an 8‐week, double‐blind, randomized, controlled, 2‐center, parallel‐group trial. Journal of Clinical Psychiatry 2010;71(10):1351‐62. [DOI] [PubMed] [Google Scholar]
Strous 2003 {published data only}
- Strous R, Maayan R, Lapidus R, Stryjer R, Lustig M, Kotler M, et al. Use of dehydro‐epiandrosterone in the management of negative symptoms of schizophrenia. Journal of the European College of Neuropsychopharmacology 2002;12(Suppl 3):S286. [Google Scholar]
- Strous RD. Dehydroepiandrosterone (DHEA) augmentation in the management of schizophrenia symptomatology. Essential Psychopharmacology 2005;6(3):141‐7. [PubMed] [Google Scholar]
- Strous RD, Maayan R, Kotler M, Weizman A. Hormonal profile effects following dehydroepiandrosterone (DHEA) administration to schizophrenic patients. Clinical Neuropharmacology 2005;28(6):265‐9. [DOI] [PubMed] [Google Scholar]
- Strous RD, Maayan R, Lapidus R, Stryjer R, Lustig M, Kotler M, et al. Dehydroepiandrosterone augmentation in the management of negative, depressive, and anxiety symptoms in schizophrenia. Archives of General Psychiatry 2003; Vol. 60, issue 2:133‐41. [DOI] [PubMed]
Strous 2007 {published data only}
- Strous RD, Stryjer R, Maayan R, Gal G, Eisner D, Weizman A. Amelioration of symptomatology, Parkinsonism and neurocognitive dysfunction following dehydroepiandrosterone (DHEA) administration in schizophrenia. Journal of the European College of Neuropsychopharmacology 2006;16(Suppl 4):S408. [Google Scholar]
- Strous RD, Stryjer R, Maayan R, Gal G, Viglin D, Katz E, et al. Analysis of clinical symptomatology, extrapyramidal symptoms and neurocognitive dysfunction following dehydroepiandrosterone (DHEA) administration in olanzapine treated schizophrenia patients: a randomized, double‐blind placebo controlled trial. Psychoneuroendocrinology 2007; Vol. 32, issue 2:96‐105. [DOI] [PubMed]
References to studies excluded from this review
Barkai 1985 {published data only}
- Barkai AI. Combined electroconvulsive and drug therapy. Comprehensive Therapy 1985; Vol. 11, issue 7:48‐53. [PubMed]
Beasley 1998 {published data only}
- Beasley CM, Sayler ME, Keisler GM, Potvin JH, Sanger TM, Tollefson GD. The influence of pharamacotherapy on self‐directed and externally‐directed aggression in schizophrenia. Schizophrenia Research 1998; Vol. 29, issue 1‐2:28.
Belanoff 2002 {published data only}
Brambilla 1988 {published data only}
David 1999 {published data only}
- David SR, Meehan KM, Sutton VK, Taylor CC. Treatment of negative symptoms with olanzapine in comparison with other novel antipsychotic agents. Journal of the European College of Neuropsychopharmacology 1999; Vol. 9:S292.
Harrigan 2004 {published data only}
- Harrigan EP, Miceli JJ, Anziano R, Watsky E, Reeves KR, Cutler NR, et al. A randomized evaluation of the effects of six antipsychotic agents on QTC, in the absence and presence of metabolic inhibition. Journal of Clinical Psychopharmacology. United States of America, 2004; Vol. 24, issue 1:62‐9. [DOI] [PubMed]
Iager 1986 {published data only}
Katz 2002 {published data only}
- Katz PR, Jeste DV, Tariot PN. Pharmacotherapy for the older patient with psychosis. Journal of the American Medical Directors Association 2002;3(4 Suppl 1):H34‐7. [DOI] [PubMed] [Google Scholar]
Kim 1960 {published data only}
Kline 1968 {published data only}
- Kline NS, Blair J, Cooper TB, Esser AH, Hackett E, Vestergaard P. A controlled seven year study of endocrine and other indices in drug treated chronic schizophrenics. Acta Psychiatrica Scandinavica Supplementum 1968; Vol. 206:7‐75. [MEDLINE: ] [PubMed]
Korsgaard 1981 {published data only}
Lane 2001 {published data only}
Lembke 2013 {published data only}
- Lembke A, Gomez R, Tenakoon L, Keller J, Cohen G, Williams GH, et al. The mineralocorticoid receptor agonist, fludrocortisone, differentially inhibits pituitary—adrenal activity in humans with psychotic major depression. Psychoneuroendocrinology 2013;38:115‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Loranger 1968 {published data only}
Miodownik 2011 {published data only}
- Miodownik C, Maayan R, Ratner Y, Lerner V, Pintov L, Mar M, et al. Serum levels of brain‐derived neurotrophic factor and cortisol to sulfate of dehydroepiandrosterone molar ratio associated with clinical response to L‐theanine as augmentation of antipsychotic therapy in schizophrenia and schizoaffective disorder patients. Clinical Neuropharmacology 2011;34(4):155‐60. [DOI] [PubMed] [Google Scholar]
Nihalani 2007 {published data only}
- Nihalani ND, Schwartz TL. Mifepristone, a glucocorticoid antagonist for the potential treatment of psychotic major depression. Current Opinion in Investigational Drugs 2007; Vol. 8, issue 7:563‐9. [PubMed]
Rees 1951 {published data only}
Rees 1956 {published data only}
Rothschild 2005 {published data only}
Schatzberg 2003 {published data only}
- Schatzberg AF. New approaches to managing psychotic depression. Journal of Clinical Psychiatry 2003; Vol. 64, issue Suppl 1:19‐23. [PubMed]
Silbergeld 1973 {published data only}
Simpson 2005 {published data only}
Stein 1984 {published data only}
Tollefson 1998 {published data only}
- Tollefson GD, Sanger TM. A blinded trial on the course and relationship of depressive symptoms in schizophrenia. Schizophrenia Research 1998; Vol. 29, issue 1,2:205.
Young 2004 {published data only}
References to studies awaiting assessment
Hardwick 1957 {published data only}
- Hardwick SW, Pearse JJ, Petrow V. 6β‐Hydroxy‐3:5‐Cyclopregnan‐20‐one in mental states. Journal of Mental Science 1957;103:835‐9. [DOI] [PubMed] [Google Scholar]
Kleiser 1984 {published data only}
- Kleiser B, Halberg F, Cornelissen G, Valkenburg C. Quantitative chronopharmacodynamic endpoint in health and schizophrenia: timing of plasma dehydroepiandrosterone (DHEA) versus DHEA‐sulfate. Annual Review of Chronopharmacology, Proceedings of the 1st International Montreux Conference of Biological Rhythms and Medications; 1984 Mar 26‐30; Montreux, Switzerland. Oxford: Pergamon Press, 1984:41‐4.
Owen 1996 {published data only}
- Owen M, Victor I, Sophia V, Theresa C, Francesca M, Elysa M, et al. Antiglucocorticoids in depression and schizophrenia. 149th Annual Meeting of the American Psychiatric Association; 1996 May 4‐9, New York, New York, USA. 1996.
Pivac 2002 {published data only}
- Pivac N, Muck‐Seler D, Jakovljevic M, Sagud M, Mihaljevic‐Peles A, Junaci S. The effects of olanzapine or fluphenazine on peripheral biochemical markers in schizophrenic patients. Proceedings of the 23rd Congress of the Collegium Internationale Neuro‐Psychopharmacologicum; 2002 Jun 23‐27; Montreal, Canada 2002.
Sluchevskii 1986 {published data only}
- Sluchevskii FI, Tikhomirov SM, Bakharev VD. Neuropeptides in the treatment of alcoholism and alcoholic psychoses. Zhurnal Nevropatologii i Psikhiatrii Imeni S. S. Korsakova 1986;86(2):244‐7. [PubMed] [Google Scholar]
Smidt 1988 {published data only}
- Smidt E, Axelsson R, Steen G. Treatment of chronic schizophrenia with glucocorticoids in combination with neuroleptic drugs: a pilot study. Current Therapeutic Research 1988;43:842‐50. [Google Scholar]
Volk 1976 {published data only}
- Volk W, Stoll KD. Double‐blind study on the therapy of postural hypotension in psychotic patients under psychotropic medication [Doppelblindversuchzur Therapie Orthostatischer Dysregulationserscheinungen bei Psychotikern unter Psychotroper Medikation]. Arzneimittel‐Forschung 1976;26(6):1188‐9. [PubMed] [Google Scholar]
Volk 1977 {published data only}
- Volk W. Hypotonic disorders of circulation regulation caused by therapy with psychotropic drugs. Treatment with 9alpha‐fluorhydrocortisone. Medizinische Welt 1977; Vol. 28, issue 45:1853‐4. [PubMed]
Watson 2002 (N0573099798) {published data only}
- Watson S. The effects of the glucocorticoid receptor antagonist RU‐486 in schizophrenia and bipolar disorder. National Research Register 2002; Vol. 1.
References to ongoing studies
Jarskog 2009 {published data only}
- Jarskog LF. Dehydroepiandrosterone (DHEA) for schizophrenia. Stanley Foundation Research Programs 2009.
Solvason 2008 {published data only}
- NCT00725270. Treatment of schizoaffective disorder using mifepristone. https://www.clinicaltrials.gov/ct/show/NCT00725270 (accessed 28 February 2012).
Additional references
Addington 1997
- Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophrenia Research 1997;3:247‐51. [DOI] [PubMed] [Google Scholar]
Altindag 2007
- Altindag A. Lifetime prevalence of psychotic disorders in Finland is 3.1%. Evidence‐Based Mental Health 2007;10(3):96. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Alvarez‐Jimenez 2008
- Alvarez‐Jimenez M, Gonzalez‐Blanch C, Crespo‐Facorro B, Hetrick S, Rodriguez‐Sanchez JM, Perez‐Iglesias R, et al. Antipsychotic‐induced weight gain in schizophrenia spectrum disorders: a systematic critical reappraisal. CNS Drugs 2008;22(7):547‐62. [DOI] [PubMed] [Google Scholar]
American Psychiatric Association 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth. Washington: American Psychiatric Association, 2000. [Google Scholar]
Andreasen 1989
- Andreasen NC. The Scale for the Assessment of Negative Symptoms (SANS): conceptual and theoretical foundations. British Journal of Psychiatry Supplements 1989;7:49‐58. [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Brown 2001
- Brown ES, Chandler PA. Mood and cognitive changes during systemic corticosteroid therapy. Primary Care Companion Journal of Clinical Psychiatry 2001;3(1):17‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chouinard 1980
- Chouinard G, Ross‐Chouinard A, Annable L. Extrapyramidal symptom rating scale. Canadian Journal of Neurological Science 1980;7:233. [Google Scholar]
Cohrs 2006
- Cohrs S, Röher C, Jordan W, Meier A, Huether G, Wuttke W, et al. The atypical antipsychotics olanzapine and quetiapine, but not haloperidol, reduce ACTH and cortisol secretion in healthy subjects. Psychopharmacology 2006;185(1):11‐8. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town, South Africa. 2000.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials:methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Faustman 1994
- Faustman WO. Brief Psychiatric Rating Scale. The Use of Psychological Testing for Treatment Planning and Outcome Assessment. Mahwah, NJ: Lawrence Erlbaum Associates, 1994:371‐401. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Garner 2005
- Garner B, Pariante CM, Wood SJ, Velakoulis D, Phillips L, Soulsby B, et al. Pituitary volume predicts future transition to psychosis in individuals at ultra‐high risk of developing psychosis. Biological Psychiatry 2005;58(5):417‐23. [DOI] [PubMed] [Google Scholar]
Gerlach 1993
- Gerlach J. The St. Hans Rating Scale for extrapyramidal syndromes: reliability and validity. Acta Psychiatrica Scandinavica 1993;87:244‐52. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Early Clinical Drug Evaluation (ECDEU) Assessment Manual for Psychopharmacology. Washington, DC: National Institute of Mental Health, 1976. [Google Scholar]
Hamilton 1959
- Hamilton M. The assessment of anxiety states by rating. British Journal of Psychiatry 1959;32:50‐5. [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology and Neurosurgery in Psychiatry 1960;23:56‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hedlund 1980
- Hedlund JL, Vieweg BW. The Brief Psychiatric Rating Scale (BPRS): a comprehensive review. Journal of Operational Psychiatry 1980;11:48‐65. [Google Scholar]
Heinrichs 1984
- Heinrichs DW, Hanlon TE, Carpenter WT Jr. The quality of life scale; an instrument for rating the schizophrenic deficit syndrome. Schizophrenia Bulletin 1984;10:388‐98. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. British Medical Journal 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2009. www.cochrane‐handbook.org.
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Keefe 1999
- Keefe RS, Silva SG, Perkins DO, Lieberman JA. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: a review and meta‐analysis. Schizophrenia Bulletin 1999;25(2):201‐22. [DOI] [PubMed] [Google Scholar]
Kerwin 2004
- Kerwin RW. The new atypical antipsychotics. A lack of extrapyramidal side‐effects and new routes in schizophrenia research. British Journal of Psychiatry 2004;164(2):141‐8. [DOI] [PubMed] [Google Scholar]
Killackey 2007
- Killackey E, Yung AR. Effectiveness of early intervention in psychosis. Current Opinion in Psychiatry 2007;20(2):121‐5. [DOI] [PubMed] [Google Scholar]
Lammers 1995
- Lammers CH, Garcia‐Borreguero D, Schmider J, Gotthardt U, Dettling M, Holsboer F, et al. Combined dexamethasone/corticotropin‐releasing hormone test in patients with schizophrenia and in normal controls: II. Biological Psychiatry 1995;38(12):803‐7. [DOI] [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 1999
- Leucht S, Pitschel‐Walz G, Abraham D, Kissling W. Efficacy and extrapyramidal side‐effects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta‐analysis of randomized controlled trials. Schizophrenia Research 1999;35(1):51‐68. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Adams C, Bradley C, Joy C, Fenton M. Unpublished rating scales ‐ a major source of bias in randomised controlled trials of treatments for schizophrenia?. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
McGorry 2002
- McGorry PD, Yung AR, Phillips LJ, Yuen HP, Francey S, Cosgrave EM, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first‐episode psychosis in a clinical sample with subthreshold symptoms. Archives of General Psychiatry 2002;59(10):921‐8. [DOI] [PubMed] [Google Scholar]
McGorry 2006
- McGorry PD, Hickie IB, Yung AR, Pantelis C, Jackson H. Clinical staging of psychiatric disorders: a heuristic framework for choosing earlier, safer and more effective interventions. Australian and New Zealand Journal of Psychiatry 2006;40:616‐22. [DOI] [PubMed] [Google Scholar]
Murray 1997
- Murray CJL, Lopez AD. Global mortality, disability, and the contribution of risk factors. Lancet 1997;349:1436‐42. [DOI] [PubMed] [Google Scholar]
Nelson 1997
- Nelson JC, Davis JM. DST studies in psychotic depression: a meta‐analysis. American Journal of Psychiatry 1997;154(11):1497‐503. [DOI] [PubMed] [Google Scholar]
Newcomer 2005
- Newcomer JW. Second‐generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005;19(Suppl 1):1‐93. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pariante 2004
- Pariante CM, Vassilopoulou K, Velakoulis D, Phillips L, Soulsby B, Wood SJ, et al. Abnormal pituitary volume in psychosis. British Journal of Psychiatry 2004;185:5‐10. [DOI] [PubMed] [Google Scholar]
Perälä 2007
- Perälä J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S, et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Archives of General Psychiatry 2007;64(1):19‐28. [DOI] [PubMed] [Google Scholar]
Phillips 2006
- Phillips LJ, McGorry PD, Garner B, Thompson KN, Pantelis C, Wood SJ, et al. Stress, the hippocampus and the HPA axis: implications for the development of psychotic disorders.. Austrialian and New Zealand Journal Psychiatry 2006;40:725‐41. [DOI] [PubMed] [Google Scholar]
Pilling 2002
- Pilling S, Bebbington P, Kuipers E, Garety P, Geddes J, Orbach G, et al. Psychological treatments in schizophrenia: I. Meta‐analysis of family intervention and cognitive behaviour therapy. Psychological Medicine 2002;32(5):763‐82. [DOI] [PubMed] [Google Scholar]
Ryan 2004
- Ryan MCM, Sharifi N, Condren R, Thakore JH. Evidence of basal pituitary and renal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology 2004;29(8):1065‐70. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Sharma 1988
- Sharma RP, Pandey GN, Janicak PG, Peterson J, Comaty JE, Davis JM. The effect of diagnosis and age on the DST ‐ a meta‐analytic approach. Biological Psychiatry 1988;24(5):555‐68. [DOI] [PubMed] [Google Scholar]
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica Supplementum 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Starkman 1999
- Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE. Decrease in cortisol reverses human hippocampal atrophy following treatment of cushing's disease. Biological Psychiatry 1999;46(12):1595‐602. [DOI] [PubMed] [Google Scholar]
Thompson 2007
- Thompson KN, Phillips LJ, Komesaroff P, Yuen HP, Wood SJ, Pantelis C, et al. Stress and HPA‐axis functioning in young people at ultra high risk for psychosis. Journal of Psychiatric Research 2007;41(7):561‐9. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Watson 2004
- Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH. Hypothalamic‐pituitary‐adrenal axis function in patients with bipolar disorder. British Journal of Psychiatry 2004;184:496‐502. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Yung 2007
- Yung AR, Yuen HP, Berger G, Francey S, Hung T, Nelson B, et al. Declining transition rate in ultra high risk (prodromal) services: dilution or reduction of risk?. Schizophrenia Bulletin 2007;33:673‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2005
- Zhang XY, Zhou DF, Cao LY, Wu GY, Shen YC. Cortisol and cytokines in chronic and treatment‐resistant patients with schizophrenia: association with psychopathology and response to antipsychotics. Neuropsychopharmacology 2005;30(8):1532‐8. [DOI] [PubMed] [Google Scholar]