

Abstract
Circulating tumor DNA (ctDNA) sequencing guides therapy decisions but has been studied mostly in small cohorts without sufficient follow-up to determine its influence on overall survival. We prospectively followed an international cohort of 1,127 patients with non-small-cell lung cancer and ctDNA-guided therapy. ctDNA detection was associated with shorter survival (hazard ratio (HR), 2.05; 95% confidence interval (CI), 1.74–2.42; P < 0.001) independently of clinicopathologic features and metabolic tumor volume. Among the 722 (64%) patients with detectable ctDNA, 255 (23%) matched to targeted therapy by ctDNA sequencing had longer survival than those not treated with targeted therapy (HR, 0.63; 95% CI, 0.52–0.76; P < 0.001). Genomic alterations in ctDNA not detected by time-matched tissue sequencing were found in 25% of the patients. These ctDNA-only alterations disproportionately featured subclonal drivers of resistance, including RICTOR and PIK3CA alterations, and were associated with short survival. Minimally invasive ctDNA profiling can identify heterogeneous drivers not captured in tissue sequencing and expand community access to life-prolonging therapy.
Multiplex tissue sequencing can match patients with non-small-cell lung cancer (NSCLC) to a growing number of life-prolonging targeted therapies1–3. In many real-world settings, however, patients do not receive multiplex tumor sequencing due to an inability to acquire tissue, sequencing failure or lack of access4–6. Sequencing of circulating tumor DNA (ctDNA) from plasma (‘liquid biopsies’) may similarly identify targetable alterations. Community adoption of such methods is rising7,8, although the effectiveness of ctDNA for targeted therapy matching has been studied mostly in small cohorts with limited follow-up9–16.
Short-term studies have suggested that ctDNA-matched targeted therapy produces equivalent rates of radiologic response to tissue-matched therapy12,17, but whether it translates to equal overall survival (OS) benefit is unknown. ctDNA detection is also thought to reflect aggressive tumor biology and disease burden18–23, but whether its prognostic value is independent of tumor volume or clinical factors is not yet defined. Some ctDNA alterations may be observed in plasma but not in tissue24. This genomic diversity between plasma and tissue may represent a mixture of artifacts25, clonal hematopoiesis (CH)26 and spatial tumor heterogeneity27. Whether alterations corresponding to tumor heterogeneity have prognostic relevance or common genomic features is unclear. Despite increasing commercial use of ctDNA detection, clinical interpretation is challenging without adequately powered studies to address such biological questions.
Recent reviews have highlighted the need for large prospective studies confirming the clinical utility of liquid biopsies28,29. To define the usefulness of ctDNA as a genomic biomarker to guide treatments and its association with and impact on survival, we conducted a prospective international cohort study of patients with metastatic NSCLC undergoing plasma ctDNA sequencing.
Results
Patient characteristics
Patients were enrolled at Memorial Sloan Kettering Cancer Center (MSK, New York, NY, USA), an academic cancer center, and GenesisCare (Sydney, Australia), a community-based oncology practice including physicians affiliated with the University of Sydney. The study included adults with stage IV or recurrent NSCLC and either no known driver mutation before enrollment or progression of disease following targeted therapy. Of the 1,357 patients initially enrolled in the study, 1,127 were included in the analysis (Fig. 1 and Table 1).
Fig. 1 |. Status of enrolled patients.
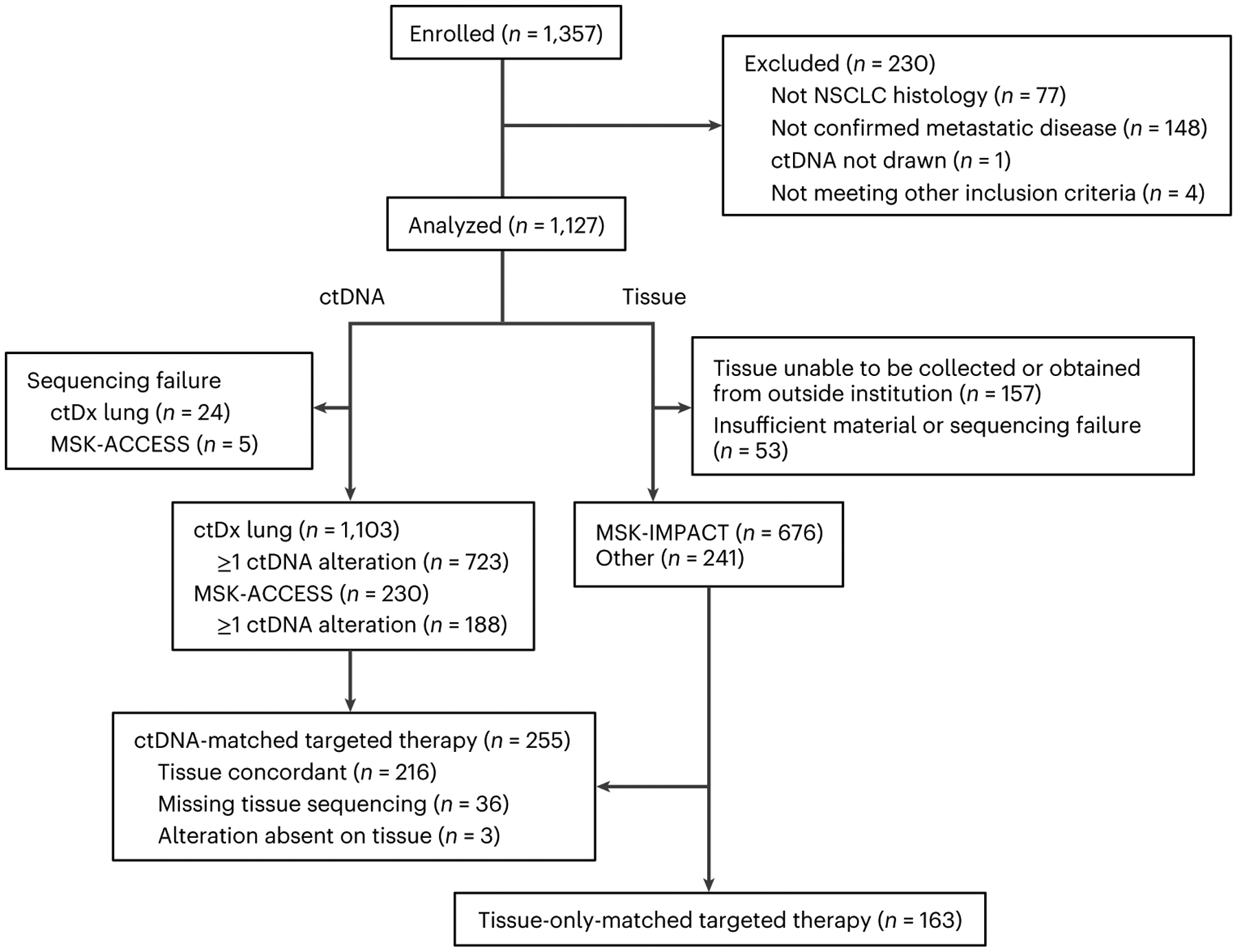
CONSORT diagram showing excluded patients as well as status of ctDNA/tissue sequencing and targeted therapy during the study for included patients.
Table 1 |.
Baseline patient characteristics
| Characteristic | MSK (n=1,002) | Sydney (n=125) | Total (n=1,127) |
|---|---|---|---|
| Age, median (IQR), years | 68 (59–75) | 67 (57–74) | 68 (59–75) |
| Sex, n (% total) | |||
| Men | 432 (43%) | 48 (38%) | 480 (43%) |
| Women | 570 (57%) | 77 (62%) | 647 (57%) |
| Race, n (% total) | |||
| White | 730 (73%) | 37 (30%) | 767 (68%) |
| Asian | 164 (16%) | 34 (27%) | 196 (17%) |
| Black | 53 (5%) | 0 (0%) | 53 (5%) |
| Other | 20 (2%) | 0 (0%) | 20 (2%) |
| Unknown | 35 (3%) | 54 (43%) | 89 (8%) |
| Histology, n (% total) | |||
| Adenocarcinoma | 849 (85%) | 120 (96%) | 969 (86%) |
| Squamous | 84 (8%) | 4 (3%) | 88 (8%) |
| Othera | 69 (7%) | 1 (1%) | 70 (6%) |
| Smoking history, n (% total) | |||
| Current/former | 553 (55%) | 42 (34%) | 595 (53%) |
| Never | 449 (45%) | 66 (53%) | 515 (46%) |
| Unknown | 0 (0%) | 17 (14%) | 17 (2%) |
| Treatment history, n (% total) | |||
| Treatment naive | 677 (68%) | 51 (41%) | 728 (65%) |
| Previous treatment | 325 (32%) | 74 (59%) | 399 (35%) |
| Previous targeted therapy | 152 (15%) | 49 (39%) | 201 (18%) |
| Previous nontargeted therapy | 173 (17%) | 25 (20%) | 198 (17%) |
| Additional sequencing, n (% total) | |||
| MSK-ACCESS | 230 (23%) | NA | 230 (20%) |
| Within 30 days | 163 (16%) | NA | 163 (14%) |
| MSK-IMPACT | 676 (67%) | NA | 676 (60%) |
| Within 30 days | 429 (43%) | NA | 429 (38%) |
| Other tissue sequencing | 129 (13%) | 112 (90%) | 241 (21%) |
| No tissue sequencingb | 197 (20%) | 12 (10%) | 209 (19%) |
| Concurrent PET, n (% total) | MSK (n=337) | Sydney (n=120) | Total (n=457) |
| Extrapulmonary disease | 252 (75%) | 83 (69%) | 335 (73%) |
| No extrapulmonary disease | 85 (25%) | 37 (31%) | 122 (27%) |
Includes dedifferentiated, neuroendocrine and mixed histologic subtypes.
Reasons included an inability to collect samples from the patient or an outside institution, insufficient material or sequencing failure.
Sequencing overview
All patients received ctDNA sequencing via the Resolution Bioscience ctDx Lung platform, a targeted next-generation sequencing panel including known molecular drivers of NSCLC (Supplementary Table 1)10,30–32. Plasma was drawn on the day of study enrollment and could be redrawn subsequently at the provider’s discretion. Most patients (n = 1,003, 89%) provided a single sample. Only mutations passing a filter for germline variants were included in the analysis (Methods and Supplementary Fig. 1). A summary of the pathogenic alterations detected on first ctDx Lung sequencing is shown in Supplementary Table 2. All patients enrolled at MSK consented to tissue next-generation sequencing with Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a targeted capture sequencing assay cleared by the Food and Drug Administration and approved by the New York State Department33,34. From 10 June 2019, patients were eligible for additional plasma sequencing with Analysis of Circulating cfDNA to Evaluate Somatic Status (MSK-ACCESS)35 at the provider’s discretion. Both MSK assays use matched white blood cell (WBC) sequencing to remove CH and germline variants.
We confirmed lower failure rates and faster turnaround time with ctDNA compared to tissue; 164 of 1,219 (13%) tissue sequencing attempts failed after sample receipt due to insufficient material or sequencing failure. By contrast, 37 of 1,919 (2%) ctDx Lung or MSK-ACCESS samples failed. Only two patients did not have successful molecular profiling of either plasma or tissue. Time from blood draw (that is, ctDNA sample collection or matched WBC collection for MSK-IMPACT) to sequencing report was shorter for plasma than for tissue sequencing: 33 days (interquartile range (IQR), 25–41) for MSK-IMPACT versus 11 days (IQR, 9–14) for MSK-ACCESS or ctDx Lung (Extended Data Fig. 1). Mutations detected in time-matched MSK-ACCESS and ctDx Lung assays were highly concordant (Methods (“ctDx Lung versus MSK-ACCESS” and Supplementary Fig. 2).
Independent prognostic value of ctDNA alterations
To test whether ctDNA alterations are prognostic, we compared OS among patients with versus without detectable ctDNA on the earliest ctDx Lung sequencing, correcting for the timing of sample collection (Methods). Because the majority of plasma DNA is derived from non-tumor tissue14,26,36, we define ctDNA detection here as the identification of at least one mutation or copy number change, in keeping with other studies using DNA sequencing-based liquid biopsies25,37.
Of the 1,127 patients, 722 (64%) had at least one detectable ctDNA alteration by ctDx Lung. Patients with ctDNA alterations had shorter survival (hazard ratio (HR), 2.05; 95% CI, 1.74–2.42; P < 0.001; Fig. 2a). This observation could be generalized to the MSK and Sydney cohorts and in treatment-naive and post-treatment settings (Fig. 2b).
Fig. 2 |. ctDNA alterations and OS.
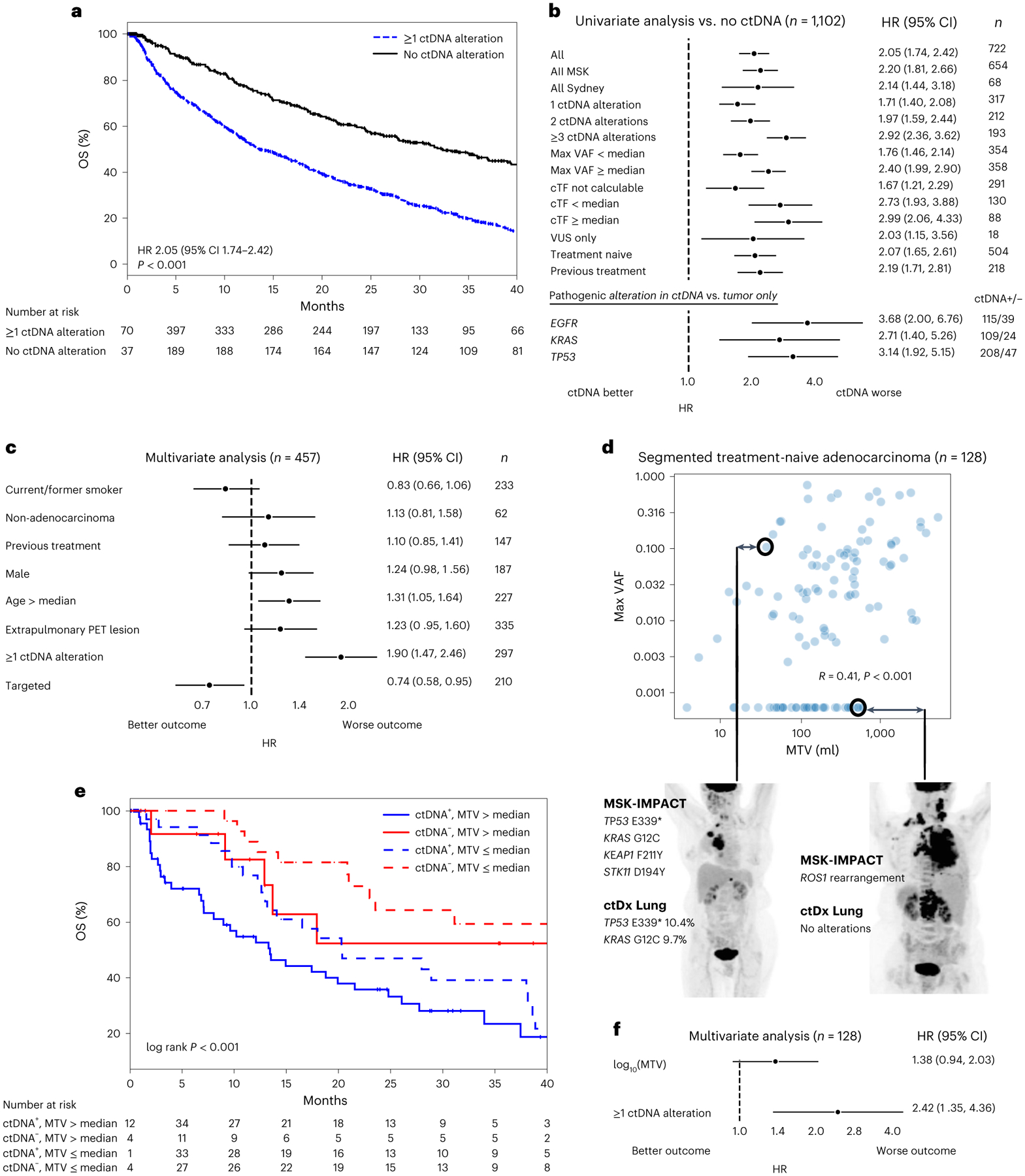
a, Kaplan–Meier survival curves for patients with versus without ctDNA detected (defined by alteration presence). Number at risk adjusted for study/variable entry time. b, Forest plots comparing patients with versus without ctDNA alterations in the listed subgroups (median OS and the number of independent patients in both the ctDNA+ and ctDNA− arms are given in Supplementary Table 3). Error bars represent 95% CI. EGFR, KRAS and TP53 subgroups compare patients with alterations in these genes in ctDNA to those with alterations in these genes in tumor tissue only. c, Multivariate Cox proportional hazards model with the listed variables in patients with time-matched PET imaging. ‘Targeted’, treated with targeted therapy. Error bars represent 95% CI. Total cohort size and number of independent patients in each variable arm are listed in the figure (n). d, Scatterplot showing the relationship between ctDNA maximum VAF and MTV among segmented patients with treatment-naive adenocarcinoma. Values of zero are set to the minimum of the respective log axes. R and P values from two-sided Spearman’s correlation coefficient (P = 2 × 10−6). Representative examples of discordance between MTV and VAF are shown. e. Kaplan–Meier survival curves for patients stratified by MTV and ctDNA detection. Number at risk adjusted for study/variable entry time. f, Multivariate Cox proportional hazards model with log10(MTV) as a continuous variable and ctDNA detection. Error bars represent 95% CI. n, number of independent patients in the total analysis.
Previous studies conflict as to whether ctDNA burden does38 or does not17 associate with survival. In our large NSCLC cohort, higher numbers of ctDNA alterations and higher maximum variant allele frequency (VAF) were associated with shorter survival (Fig. 2b and Supplementary Table 3). We sought to assess whether circulating tumor fraction (cTF), that is, an estimate of tumor burden in plasma based on clonal mutations that is corrected for copy number changes (Methods), might provide additional prognostic stratification. Higher cTF itself was not associated with shorter survival, possibly because the requirement that clonal mutations be present in plasma was itself a strong prognostic sign. cTF could not be calculated for patients without clonal alterations in ctDNA; patients in this group had better survival than those with calculable cTF (Fig. 2b).
Some patients had only variants of unknown significance (VUS; that is, pathogenic status unknown32) in ctDNA. These patients also had worse survival than those without ctDNA alterations, suggesting that ctDNA portends worse outcomes even in the absence of pathogenic alterations. Among patients with known pathogenic alterations (in TP53, EGFR or KRAS), we tested whether it is the presence of those alterations themselves, rather than their detection in ctDNA, that is associated with worse prognosis. In these subgroups, detection of the pathogenic alteration in ctDNA was associated with worse prognosis than detection in tissue only (Fig. 2b).
We tested whether ctDNA is more likely to be detected in patients with higher tumor mutational burden (TMB) and whether this might affect our conclusions regarding survival. TMB on MSK-IMPACT sequencing was not associated with greater ctDNA burden, possibly due to the small footprint of the ctDx Lung assay (Supplementary Fig. 3a). Among patients with TMB of <10 mutations per Mb39, those with ctDNA detection had shorter survival; among those with TMB of ≥10 mutations per Mb, ctDNA detection was associated with a trend toward worse survival (Supplementary Fig. 3b). Finally, it is possible that ctDNA’s prognostic value may vary based on treatment patterns. In subgroup analyses limited to patients treated with targeted therapy, immunotherapy or chemotherapy only, ctDNA detection remained an independent predictor of worse outcome. Among patients treated with immunotherapy, our findings of ctDNA detection as a poor prognostic marker held true regardless of TMB stratification (Supplementary Fig. 3c). In summary, we found that ctDNA is prognostic in a manner that scales with ctDNA burden but is independent of treatment patterns and tumor genomics.
Multivariate and radiologic analysis
ctDNA detection is influenced by histology27, disease extent and clinical factors38, findings that were confirmed in this cohort (Extended Data Fig. 2). Of 335 patients with extrapulmonary disease on time-matched positron emission tomography (PET) imaging (Supplementary Table 4), 248 (74%) had detectable ctDNA; by contrast, 49 of 122 (40%) patients with intrapulmonary disease had detectable ctDNA (chi-squared test, P < 0.001). In a multivariate model including extrapulmonary disease and clinical features as variables, ctDNA detection independently predicted worse survival (Fig. 2c). In addition, advanced age was associated with worse survival, whereas receipt of targeted therapy was associated with better survival.
Previous studies differ in whether ctDNA levels correlate with metabolic tumor volume (MTV)40,41. It is also unknown whether the prognostic value of ctDNA reflects tumor burden or another aspect of tumor biology not captured by metabolic imaging. To examine the relationships among ctDNA levels, MTV and survival, we computed MTV for treatment-naive patients with adenocarcinoma who had time-matched PET imaging and MSK-IMPACT sequencing (Methods).
MTV was correlated with ctDNA VAF, although there were cases in which high ctDNA levels were observed with low MTV and vice versa (Fig. 2d). Among patients with high or low MTV (relative to the median), ctDNA remained independently prognostic (Fig. 2e). It is possible that ctDNA may appear independently prognostic among patients with high or low MTV because it reflects relative tumor burden within those discrete cohorts. In multivariate analyses treating MTV as a continuous variable, higher MTV was associated with a trend toward worse survival, while ctDNA detection was significantly associated with worse survival (Fig. 2f). We repeated these analyses with other continuous radiologic features, namely, maximum standardized uptake value (SUV), SUVmean, the product of MTV and SUVmean (total lesion glycolysis) and the number of lesions. In all cases, ctDNA levels were correlated with radiologic features but remained independently prognostic (Supplementary Fig. 4).
Patients treated at MSK had higher ctDNA burden than those treated at Sydney (Supplementary Fig. 5b). In multivariate analysis including treatment site as a variable, ctDNA detection remained prognostic, but treatment site was not (Supplementary Fig. 5c). In summary, all multivariate analyses confirmed the independent prognostic value of ctDNA detection.
ctDNA matching to targeted therapy
We examined the clinical utility of ctDNA-matched targeted therapy and its impact on survival, a subject of equivocal results in previous smaller short-term studies12,17,23. A total of 418 (37%) patients were treated with targeted therapy (Supplementary Table 5) after study entry; 255 (23%) patients had molecular targets identified on ctDNA (Supplementary Table 6), and 163 (14%) patients were matched based on tissue analysis only (Fig. 1). The odds of matching to targeted therapy by ctDNA were lower in smokers, patients with non-adenocarcinoma histology and patients with intrapulmonary disease only (Supplementary Table 7).
Among patients treated with targeted therapy, those with detectable ctDNA had worse survival (Supplementary Fig. 3c and Fig. 3a). Among patients with detectable ctDNA, those matched to targeted therapy by liquid biopsy had longer OS than those not receiving targeted therapy (HR, 0.63; 95% CI, 0.52–0.76; P < 0.001; Fig. 3a). Among patients without detectable ctDNA, those treated with targeted therapy did not have longer survival than those not treated with targeted therapy to a statistically significant extent (HR, 0.92; 95% CI, 0.7–1.2; P > 0.1), suggesting greater relative benefit to targeted therapy among those patients with detectable ctDNA. Genes targeted in the ctDNA-matched and tissue-only-matched groups are shown in Fig. 3b. OS varied by gene target; trends toward shorter OS among patients with ctDNA were observed in all genes (Supplementary Fig. 6).
Fig. 3 |. Clinical utility of ctDNA and tissue sequencing.
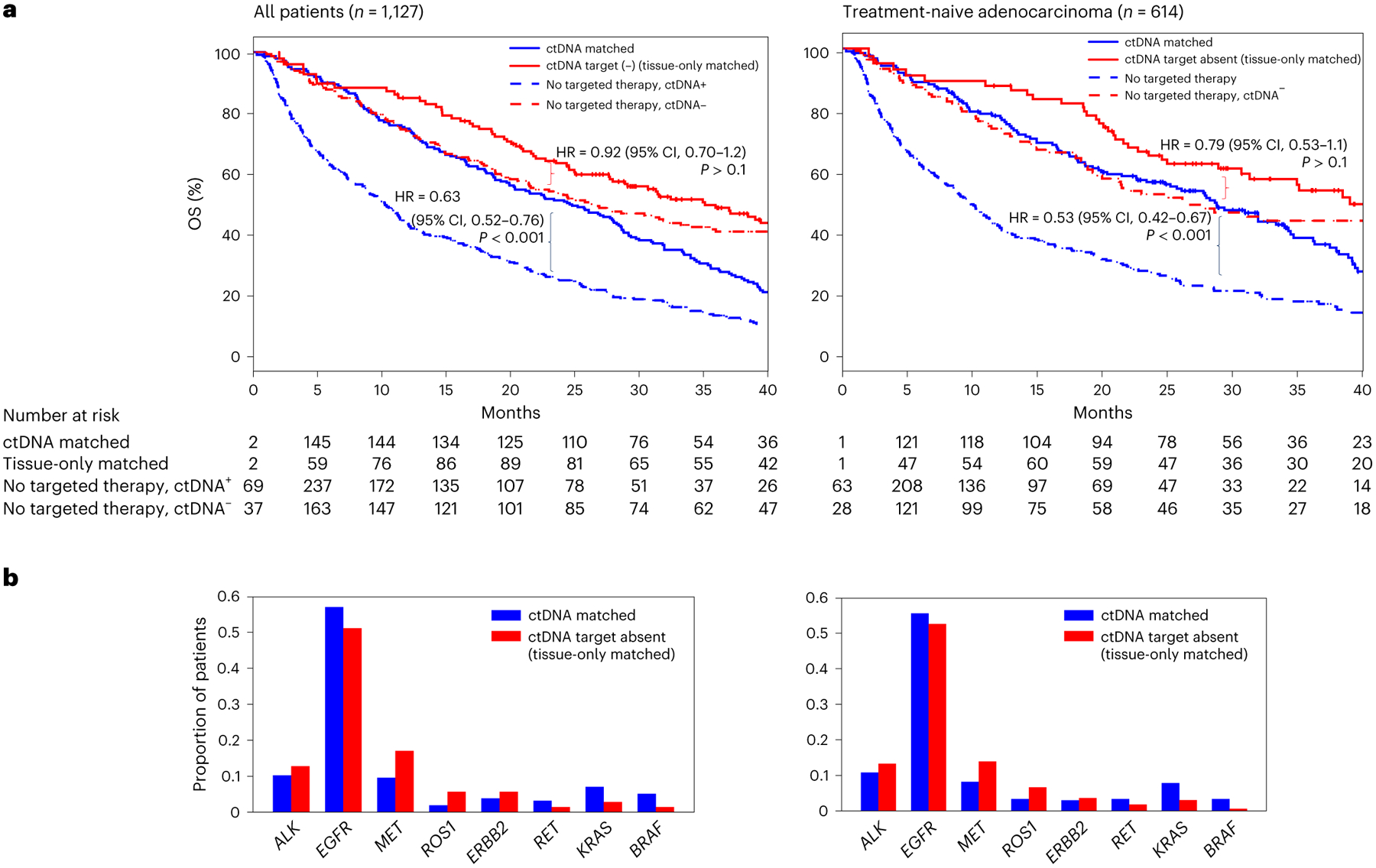
a, Kaplan–Meier survival curves for patients matched to targeted therapy by ctDNA, not treated with targeted therapy but ctDNA+, matched to targeted therapy by tissue only and not treated with targeted therapy and ctDNA−. Number at risk adjusted for study/variable entry time. P values are from a two-sided Cox proportional hazards model and were not adjusted for multiple hypotheses. b, Number of patients matched to a given molecular target by ctDNA and tissue sequencing or tissue sequencing only.
To test whether our findings regarding ctDNA-matched targeted therapy might be confounded by the histopathologic and treatment diversity of our cohort, we repeated these analyses restricted to treatment-naive patients with adenocarcinoma (Fig. 3) and in multivariate analyses including clinical and radiologic variables (Supplementary Fig. 7). The relationships for patients treated or not treated with targeted therapy, stratified by ctDNA presence, were similar.
In summary, despite the lower baseline survival of patients with detectable ctDNA, among those patients, ctDNA-matched targeted therapy was associated with survival benefit, to an even greater extent than with tissue-matched therapy among those without detectable ctDNA. Previous studies have suggested that ctDNA presence does not affect radiologic response to targeted therapy12,17; however, we found that ctDNA-matched targeted therapy was associated with shorter OS than tissue-only-matched therapy, emphasizing the importance of long-term follow-up in evaluating the full benefits of any biomarker-driven therapy.
ctDNA may match patients to targeted therapy when tissue sequencing is unavailable6. Among the patients without tissue sequencing, ctDNA-matched targeted therapy was associated with better survival than no targeted therapy (Extended Data Fig. 3). ctDNA may also be useful for repeat sampling that is impractical with tissue. Here, ctDNA matched 11 patients to a second targeted therapy based on detection of an additional oncogenic driver at progression (Supplementary Fig. 8), although it should be noted that the evidence supporting targeted therapy directed against a second oncogene ranges considerably42–44.
Plasma–tissue divergent genomics
It is established that tumor mutations may be absent in ctDNA due to lower rates of DNA shedding or limits of plasma assay sensitivity8,10,12; these tumor-only mutations are associated with better survival (Fig. 2b). By contrast, the clinical relevance of alterations found on plasma but not clinical tissue sequencing is less clear. Of 429 patients with tissue sequencing (MSK-IMPACT) within 30 days of ctDx Lung sequencing, 109 (25%) patients had mutations or copy number alterations (CNAs) detected in ctDNA that were absent on tumor sequencing (‘ctDNA-only’ alterations). Three of these patients had EGFR T790M mutations, leading to changes in treatment. Additional potentially actionable ctDNA-only mutations included one KRAS G12C mutation, one BRAF V600E mutation and one RET fusion; none of these led to changes in therapy due to their absence in tissue.
We sought to evaluate whether ctDNA-only alterations associate with survival. The subgroup of patients with ctDNA-only alterations had worse survival than those with tissue-matched ctDNA alterations (Fig. 4a). These findings were consistent when the ctDNA-only alteration was either a mutation or CNA (Fig. 4b and Supplementary Table 8).
Fig. 4 |. ctDNA mutations not detected on time-matched tissue sequencing (MSK-IMPACT).
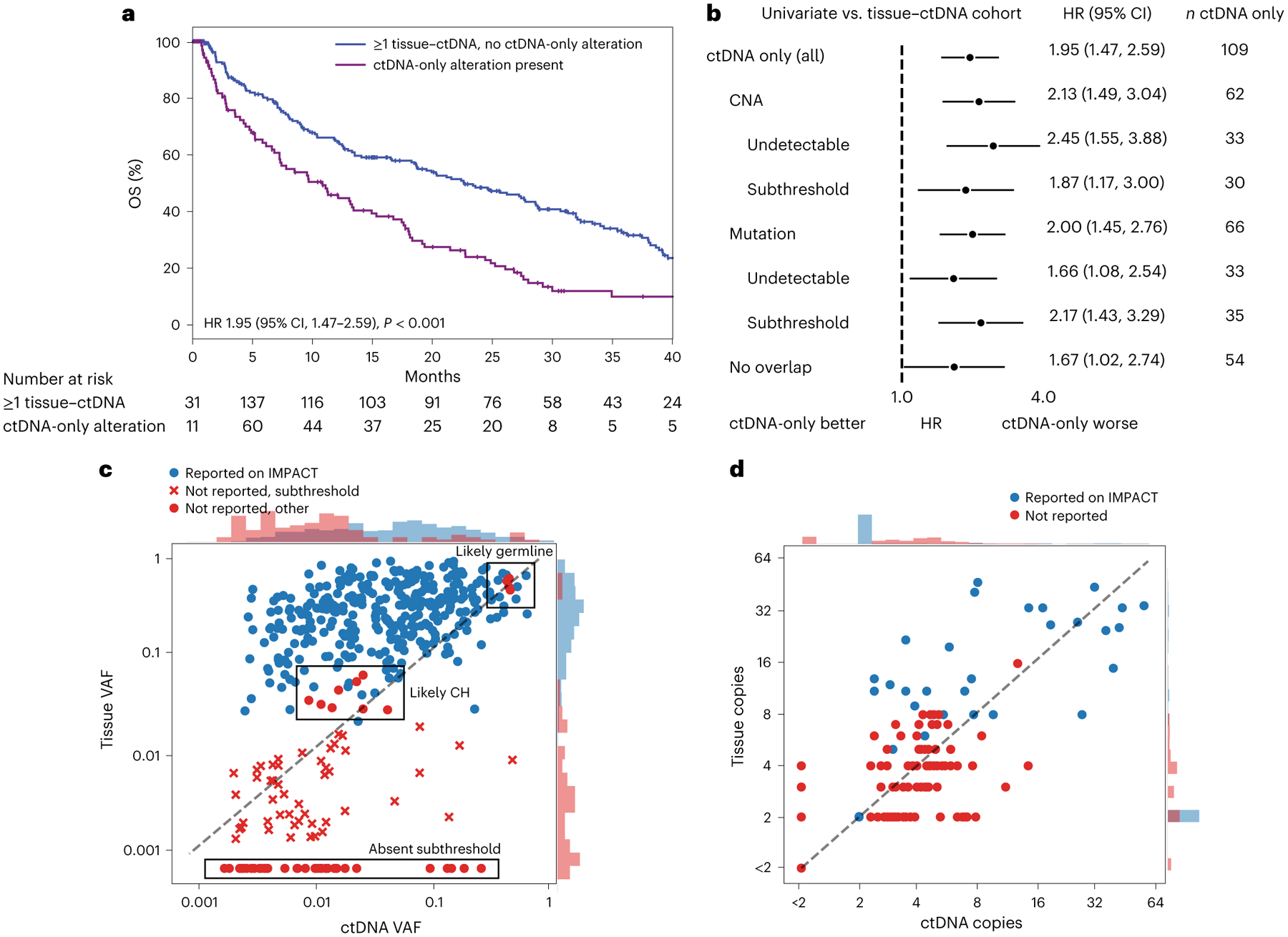
a, Kaplan–Meier survival curves for patients without ctDNA alterations (black) or with ctDNA-only alterations (purple), compared by two-sided Cox proportional hazards model (P = 1 × 10−5). b, Forest plots comparing patients with ctDNA-only alterations versus those with matched ctDNA–tissue alterations only in the listed subgroups (the median OS and number of independent patients in each arm are given in Supplementary Table 8). Error bars represent 95% CI. c,d, Scatterplots of mutations (c) and CNAs (d) detected in ctDNA. Red dots correspond to ctDNA-only alterations. The y axis in d shows CNA levels calculated algorithmically from tissue MSK-IMPACT. Histograms at the top and right show the density of points of each category along the respective axis. Raw values of zero were set to the minimum value of the log axis.
Using MSK-IMPACT, we quantified the proportion of ctDNA-only mutations attributable to CH or germline variation and tumor mutations missed because of stringent base-calling thresholds. Of the 109 patients with ctDNA-only alterations, 66 patients had ctDNA-only mutations. Among these 66 patients, 92 ctDNA-only mutations were detected. Of these 92 mutations, 11 (12%) were filtered out in MSK-IMPACT based on WBC sequencing suggesting a CH or germline mutation (Fig. 4c). Of the remaining 81 mutations, 44 (54%) were detectable in tissue sequenced by MSK-IMPACT at subthreshold levels (Fig. 4c). Similarly, of the 110 ctDNA-only CNAs, 56 (51%) were detectable at subclinical reporting levels in tissue MSK-IMPACT (Fig. 4d). Both called and subthreshold MSK-IMPACT mutations could be detected in WBC controls at low frequencies (Supplementary Fig. 9); it is unclear whether these mutations represent artifacts, low-level CH or tumor DNA in buffy coat. We investigated whether subthreshold detectability in MSK-IMPACT had any bearing on OS. Patients with ctDNA-only alterations had shorter survival than those with tissue-only-matched ctDNA alterations whether or not the alterations were at a detectable subthreshold level (Fig. 4b).
In summary, we found that ctDNA-only alterations are common and have significant prognostic value. A subgroup of 26 patients had no common alterations observed between ctDNA and tissue. These patients also had worse survival than those with tissue-matched ctDNA alterations (Fig. 4b).
Correlates of ctDNA-only alterations
We sought to assess whether ctDNA-only alterations might reflect higher cTF, tumor burden or other clinical variables. Patients with ctDNA-only alterations did not have higher average cTF (Fig. 5a). Patients without ctDNA alterations had smaller MTV (median, 122 mL; IQR, 58–284 mL) than those with tissue-matched ctDNA alterations (median, 252 mL; IQR, 109–484 mL; P < 0.001; Fig. 5b). The latter, in turn, had smaller MTV than patients with ctDNA-only alterations (median, 737 mL; IQR, 125–1,598 mL; P < 0.001; Fig. 5b). Patients with extrapulmonary disease were more likely to have ctDNA-only alterations, but patients with advanced age, male sex, smoking history, previous treatment and non-adenocarcinoma histologies were not more likely to have these alterations (Supplementary Fig. 10a). ctDNA-only alterations were not associated with TMB, tumor purity, sequencing depth (Supplementary Fig. 10b) or sequencing site (Supplementary Fig. 10c). Together, these findings suggest that genomic divergence between liquid and tumor biopsies is most related to tumor burden.
Fig. 5 |. Correlates of plasma–tissue divergence.
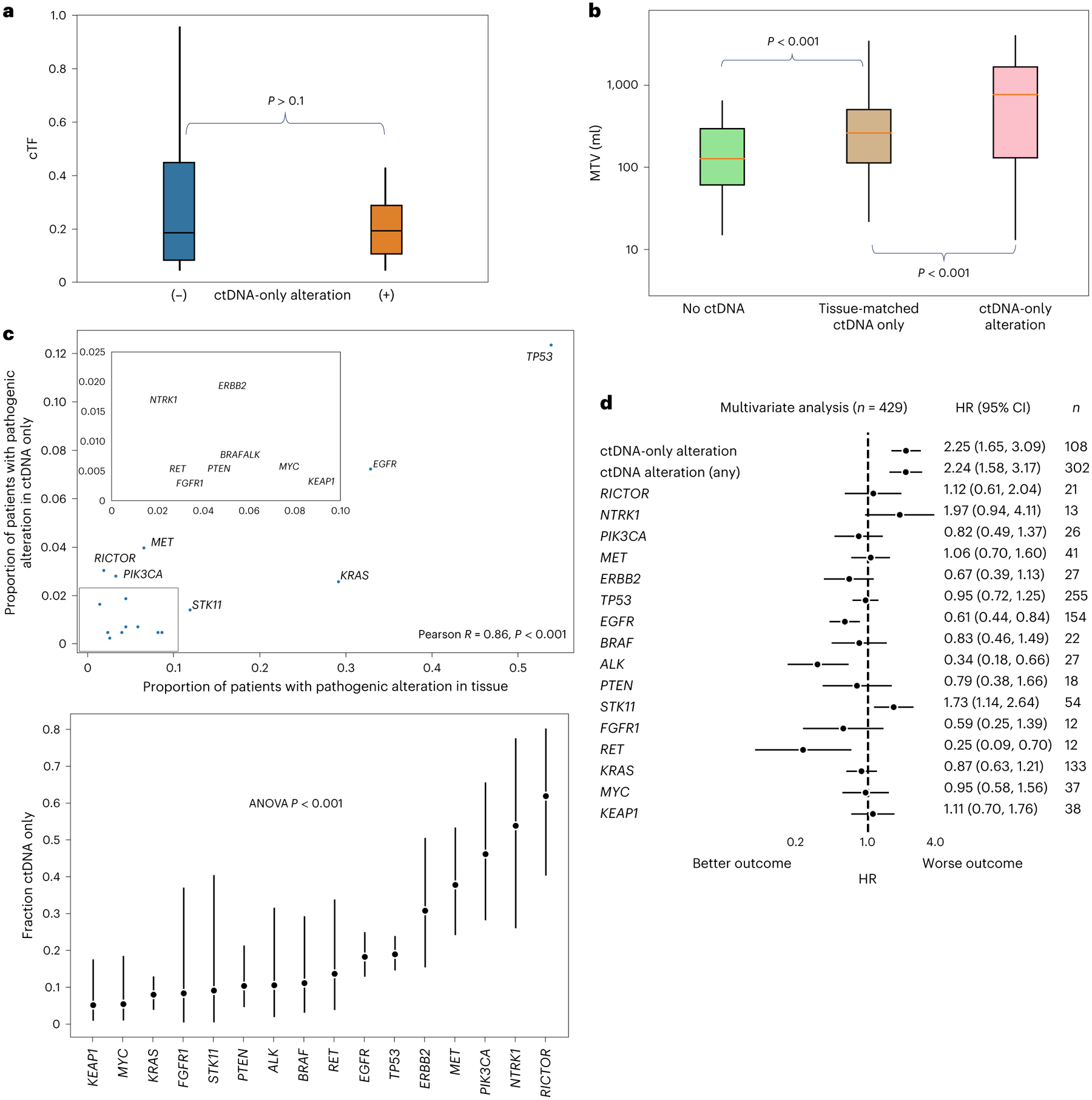
a,b, Boxplots showing cTF for patients with (n = 102 independent patients) and without (n = 30 independent patients) ctDNA-only alterations (two-sided Mann–Whitney U test, P = 0.5) (a) and MTV on time-matched PET imaging (segmented independent patients only, n = 129) for patients without ctDNA (n = 41), with tissue-matched ctDNA only (n = 62) and with ctDNA-only alterations (n = 25) (b). Boxes denote medians ±IQRs. Whiskers denote 1.5 times IQRs. P values are from two-sided Mann–Whitney U tests (P value for tissue-matched versus no ctDNA, 8 × 10−10; ctDNA-only versus tissue-matched, 1 × 10−5). c, Top, scatterplot showing the proportion of patients with an oncogenic driver in a given gene as detected in either tissue (±ctDNA) or in ctDNA only (that is, not in tissue). Pearson’s two-sided P value = 1 × 10−5. Bottom, fraction of the total number of patients with an oncogenic driver in the indicated gene detected only in ctDNA. Center points are fractions; whiskers are binomial 95% CI. Only genes with at least 10 patients possessing such a driver by either method are shown. ANOVA, P value = 4 × 10−13. d, Multivariate Cox proportional hazards model with the listed variables. Oncogenic driver gene alterations refer to those found in either tissue or ctDNA.
We sought to study the genomic composition of ctDNA-only alterations. The gene-level distribution of pathogenic alterations32 in ctDNA-only versus tissue alterations is shown in Fig. 5c. The altered genes more commonly seen in tissue were also more commonly seen in only ctDNA (Pearson’s R = 0.86, P < 0.001; Fig. 5c). The relative proportions of ctDNA-only alterations to tissue alterations, however, showed over-representation of commonly subclonal alterations in secondary resistance genes45,46, in particular, RICTOR, NTRK1, MET and ERBB2 amplifications and PIK3CA mutations (Fig. 5c). These genes remained over-represented in alterations absent or at subthreshold levels in MSK-IMPACT and in treatment-naive patients (Supplementary Fig. 11). In patients without overlap in genomic alterations between ctDNA and tissue, no significant over-representation was observed, leaving open the possibility that these cases may indicate a second primary rather than a single heterogeneous disease.
The presence of the aforementioned alterations in secondary resistance genes, even in tissue, might confer worse prognosis. To test whether the worse survival seen in patients with ctDNA-only alterations is a result of underlying tumor genetics, we performed multivariate survival analysis including the presence of any ctDNA alteration, the presence of a ctDNA-only alteration and the presence of specific pathogenic gene alterations in either ctDNA or tissue. As expected based on previous tissue-based analyses47, patients with ALK, RET and EGFR alterations had better survival, as expected given frequent treatment with targeted therapy among this group, while the presence of STK11 alterations was associated with worse survival. The presence of RICTOR, NTRK, MET, ERBB2 and PIK3CA alterations was not associated with worse survival. Conversely, the presence of ctDNA alterations and the presence of ctDNA-only alterations were both independently associated with worse survival (Fig. 5d). These findings suggest that ctDNA–tissue divergence, and not the genomic changes themselves, is prognostic and highlight the utility of liquid biopsies for detecting meaningful spatial genomic heterogeneity when paired with tissue sequencing.
We studied whether CH database filtering or serial sampling might differentiate whether ctDNA-only alterations are related to the tumor or CH. Mutations commonly seen in NSCLC (that is, ‘hotspots’) were more likely to be recovered as ctDNA mutations; however, mutations could not be discriminated from CH on this basis because these mutations were also likely to be CH ‘hotspots’ (Supplementary Fig. 12)48. CH mutations were not associated with statistically significantly worse survival in this cohort (Supplementary Fig. 13). Although CH mutations are associated with subtle differences in survival48–50, the worse survival seen in patients with ctDNA-only alterations here was evidently not attributable to CH. Among the seven patients with ctDNA-only mutations and multiple ctDNA samples, ctDNA-only mutations qualitatively appeared in multiple samples or followed VAF trends of analogous tissue-matched ctDNA mutations (Supplementary Fig. 14).
Discussion
Despite the increased attention received in the technology industry7 and community adoption of liquid biopsy as a promising means of monitoring treatment response and selecting targeted therapy51, robust studies on its survival impact are lacking. We present the results of the largest prospective international cohort (n = 1,127) of patients with advanced NSCLC who underwent ctDNA-guided therapy with longitudinal follow-up for survival.
We find that ctDNA levels are correlated with radiologic features but have independent prognostic power in the general metastatic setting. The robust, independent prognostic utility of ctDNA suggests that liquid biopsies are a marker for increased micrometastatic or especially aggressive tumor biology and should supplement radiologic reports when assessing disease burden and chance of relapse following systemic therapy.
Overall, 23% of the patients were matched to targeted therapy by ctDNA testing. Targeted therapy was associated with greater relative survival benefit in patients with detectable ctDNA than in those without, highlighting the potentially greater survival gains with liquid biopsy-matched therapy. Future studies that use ctDNA for patient selection51 should account for the lower baseline OS of patients with detectable ctDNA to avoid underestimating the effects of therapy. At the same time, even though patients without detectable ctDNA have better prognosis overall, those patients may still further benefit from tissue-matched targeted therapy. Patients treated with targeted therapy, such as those with EGFR-mutant lung cancer, may not derive the same benefit from nontargeted treatments such as immunotherapy as those without driver mutations52, who comprise the control arm here. Randomized trials of novel therapies should be adequately stratified and powered to determine the relative survival benefits of particular targeted agents in patients with and without detectable ctDNA.
Plasma sequencing without paired WBC sequencing is common among commercial liquid biopsy assays7. We found that appropriate germline filters and a focus on NSCLC drivers may greatly reduce but not eliminate inclusion of CH and germline mutations. Even with WBC pairing, methods such as fragment-length analysis53 may be helpful in differentiating tumor DNA from CH at especially low VAF.
Of the patients with time-matched clinical tissue sequencing, 25% had alterations in ctDNA not detected in tissue. These ctDNA-only alterations disproportionately featured commonly subclonal drivers of resistance such as RICTOR amplification and PIK3CA mutation45,46 even in the treatment-naive setting. Patients with ctDNA-only alterations had worse survival and greater MTV than even those with tumor-matched ctDNA alterations, which suggests that ctDNA-only alterations are related to tumor heterogeneity missed by spatially restricted tissue sequencing54. Genomic heterogeneity in lung cancer is widespread and may be associated with worse prognosis55. The presence of ctDNA-only alterations did not increase with age, unlike CH, and was not commonly found in matched WBCs. Plasma may have higher specificity for tumor mutations than previously reported10 and may be a valuable means of detecting subclonal genomic diversity when paired with tissue sequencing.
This study has a number of limitations. The nonrandom, real-world nature of this study makes it challenging to assess the survival benefit of any single targeted therapy with ctDNA matching. We find that ctDNA-matched therapy, like tissue-matched therapy1, is associated with longer survival; however, the baseline of patients not treated with targeted therapy is heterogeneous and consists of driver-negative and nontargetable driver groups treated with a variety of therapies, as expected given the evolution of NSCLC treatments over the past decade2,56. Patients with previous treatment reflect a particularly heterogeneous genomic landscape, and further studies on the interaction between specific resistance alterations and ctDNA detection are warranted. Despite our attempts to correct for immortality bias using variable entry times, randomized trials are necessary to confirm the benefits of particular targeted therapies. Reassuringly, our findings align with historic randomized trials and propensity-matched analyses, suggesting that targeted therapy matched by either ctDNA or tissue is associated with survival benefit1,57. Our cohort was predominantly recruited from an academic cancer center in New York; although findings were validated in a community oncology practice in Sydney, further exploration of ctDNA use in community settings is warranted. Indeed, our findings suggest that treatment patterns differ between primary and referral settings, as do levels of ctDNA, likely due to differences in disease burden at presentation. Both cohorts also had lower smoking rates than expected from a typical population of patients with lung cancer. Generalization of our results to cohorts should be performed with caution. Both tumor and ctDNA sequencing were performed using targeted panels; estimates of tumor fraction, clonality and copy number changes will likely improve with broader sequencing approaches22,40. Of the 230 patients with plasma sequenced by the 129-gene MSK-ACCESS panel, 188 (82%) had at least one detectable ctDNA alteration. The higher proportion of patients with detectable alterations using a broader panel aligns with other studies, suggesting that the rate of ctDNA detection is higher with such panels35,58. The focused ctDx Lung panel was centered on therapeutic actionability. The calculation of cTF itself is challenging, particularly with sparse sequencing panels, and methods for cTF calculation vary considerably; our findings in this respect should be interpreted with caution.
Nonetheless, given the growing number of targeted therapies for NSCLC2, next-generation sequencing of tumor DNA is crucial. Liquid biopsy-matched therapy provided survival benefit in patients with failed or missing tissue sequencing for practical reasons. Even in an academic setting in which consent to tissue sequencing was routine, tissue sequencing was absent in 20% of the patients. In real-world settings, the prevalence of incomplete tissue sequencing may exceed 50% and is particularly low in disadvantaged groups4,5,59. Although tumor sequencing remains valuable, our study suggests that in both academic and community settings, ctDNA-guided therapy may increase survival. ctDNA sequencing may increase clinical trial enrollment60, which is also lower in disadvantaged patients4,61, owing to fast turnaround of results37. Lower failure rates and faster turnaround time with liquid versus tissue biopsies here corroborate this notion, especially after accounting for the time required to obtain tissue from external institutions, schedule biopsies or process samples. The minimally invasive nature of ctDNA collection may also increase clinical trial participation among older patients also underrepresented due to comorbidities and time demands on both the patient and provider61–63. Owing to their low failure rate coupled with a fast turnaround time, liquid biopsies are well suited to expansion into retail clinics within pharmacies and home settings, further lowering barriers to precision cancer medicine for communities at large64,65. Greater availability of these assays will in turn provide an invaluable tool for monitoring disease relapse19,20,66, emergence of secondary actionable drivers67–69 and tumor evolution70. As technologies continue to improve, liquid biopsies may ultimately offer similar benefits in the early detection of cancer40,71. By helping to overcome barriers to sequencing, liquid biopsies provide an opportunity to match patients to life-prolonging therapies on a previously unseen scale.
Methods
Statistics and reproducibility
The study included adults with stage IV or recurrent NSCLC and either no known driver mutation before enrollment or progression of disease following targeted therapy. The enrollment began on 21 October 2016; this analysis included patients enrolled until 1 November 2020 with follow-up until 31 December 2021. Patients were followed for a median of 445 (IQR, 171–826) days following study enrollment. Patients provided written informed consent and were enrolled in a continuous, nonrandom fashion. Patients were not compensated for participation in the study. The study was independently approved by the institutional review board of each site. Patients in MSK were enrolled as part of a prospective sequencing protocol (NCT01775072). No statistical method was used to predetermine sample size. Only patients not meeting the criteria for study inclusion (Fig. 1) were excluded from the analysis; no data were otherwise excluded unless they did not meet the criteria for particular analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Germline variant filtering and benchmarking
To reduce inclusion of incidental germline mutations, we applied two filters to variant calls from the plasma-only ctDx Lung assay. Filter 1 removed variant calls present in gnomAD72 at greater than 0.5% population frequency, a threshold with high specificity for germline mutations73 that is standard for ctDx Lung clinical reporting. Filter 2 removed mutations present in gnomAD at any population frequency with a VAF of 35–65%, a range in which germline mutations are frequently recovered in plasma35, but retained known oncogenic driver mutations32. There were 10,410 ctDx Lung unfiltered ctDNA mutations identified across all 1,631 samples; of these, 7,165 (69%) mutations were removed by filter 1 and an additional 156 (1%) were removed by filter 2. The remaining 3,089 mutations were analyzed (Supplementary Fig. 1).
To examine the accuracy of this filtering method, we examined filtered mutations in samples with time-matched WBC filtering (MSK-IMPACT or MSK-ACCESS), which served as a gold-standard control for the detection of germline mutations. A total of 555 ctDx Lung samples had time-matched MSK-IMPACT or MSK-ACCESS sequencing. Among these samples, 1,616 plasma mutations with a VAF of 35–65% were filtered out and had WBC concentrations with a VAF of 35–65% (true negatives), 31 were unfiltered and had WBC VAF < 35% (true positives), 4 were unfiltered and had WBC VAF > 35% (false positives) and one was excluded but had WBC VAF < 35% (false negative). The positive predictive value for tumor mutations with a VAF of 35–65% was 89%, and the negative predictive value was >99%.
Survival analyses
All OS analyses measured time from diagnosis of stage IV or recurrent NSCLC to the time of death, right-censored at the date of the last follow-up. To mitigate immortality bias, left truncation at the time of cohort entry was applied, and targeted therapy treatment was encoded as a time-dependent variable. HRs, CIs and P values were calculated using Cox proportional hazards models. P values <0.05 were considered significant, although in the setting of multiple hypotheses, this should be interpreted as exploratory. Analyses were conducted in Python 3.7.4 using lifeline package 0.25.7. In the analyses of the prognostic value of ctDNA, those with failed ctDx Lung testing (n = 24) were excluded. In multivariate analyses including radiologic features as variables, those without time-matched PET scans were excluded.
Prognostic value and correlates of ctDNA
We compared the survival of patients with any mutation or CNA on their first ctDx Lung test (that is, those with ‘detectable’ ctDNA) to those without any alteration. Patients with ctDNA testing failure (n = 24) were excluded from this analysis.
We assessed the relationships between age (greater or less than median), smoking status (current/former versus never smoker), sex, histologic subtype (adenocarcinoma, squamous cell carcinoma or other), previous NSCLC systemic therapy and radiologic extrapulmonary disease (at least one SUV avid extrapulmonary lesion on [18F]fluorodeoxyglucose ([18F]FDG) PET within 30 days of ctDNA draw) with ctDNA alteration number and VAF using histograms and Mann–Whitney U or Kruskal–Wallis tests. We evaluated the prognostic value of ctDNA alterations in a multivariate model, including the aforementioned clinical features and receipt of targeted therapy as variables.
Targeted therapy
We compared OS for patients matched to targeted therapy following study entry to that for patients treated with other therapies; patients were further grouped based on matching (ctDNA-only versus tissue-only). Only targeted therapies initiated after study enrollment were considered. Patients were matched to targeted therapy based on OncoKB level 1 or 2 alterations, with the majority being matched based on level 1 alterations (Supplementary Table 6). Patients with ctDNA testing failure were treated as lacking ctDNA alterations (either mutations or CNAs) for this analysis.
ctDNA–tissue discordance
We measured the incidence of ctDNA alterations absent on time-matched (from a specimen obtained within 30 days of plasma draw) tissue sequencing with MSK-IMPACT. Only loci overlapping between panels from the earliest sample pair were considered. Mutations present in ctDNA that were absent in MSK-IMPACT were genotyped for subthreshold evidence of mutation from MSK-IMPACT Binary Alignment Map files34; CNAs were similarly reevaluated using a previously validated algorithm74. Among the 429 patients with time-matched ctDx Lung and MSK-IMPACT sequencing, 131 CNAs were detected across the 11 genes in both panels by the MSK-IMPACT clinical pipeline. Of these, 31 (24%) were detected by ctDx Lung. An additional 110 CNAs were detected by ctDx Lung but not MSK-IMPACT. Among the 290 MSK-IMPACT mutations in regions covered by ctDx Lung, 178 (61%) mutations were detected by ctDx Lung. An additional 96 mutations were detected by ctDx Lung but not MSK-IMPACT. We compared the OS of patients who had ctDNA-only alterations to those who did not.
cTF calculation
The relationship between VAF and cTF was assumed as
where TCN is the tumor copy number at a given allele and BAF is the B allele frequency or proportion of TCN assumed to be mutant. cTF in ctDNA was thus calculated as follows.
The cTF of a sample was taken as the maximum cTF of all mutations. Given the small panel size used for ctDNA sequencing, TCN and BAF were calculated using Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) applied to time-matched tumor IMPACT sequences. Only clonal mutations (that is, those with either cancer cell fraction >0.8 or cancer cell fraction >0.7 and upper 95% CI of cancer cell fraction >0.9 according to FACETS) were considered. Patients in whom at least one clonal tissue mutation did not appear in ctDNA or for whom FACETS did not converge were labeled as having incalculable cTF.
PET–CT imaging analysis
Time-matched FDG PET–computed tomography (CT) scan images were retrieved from the picture archiving and communication system. The MTV of all tumor lesions on PET–CT was semiautomatically contoured using the Beth Israel PET–CT viewer plugin for FIJI (ImageJ version 1.53q)75. MTV was calculated using the recommended 41% SUVmax threshold76. Based on the MTVs, additional PET metrics of SUVs (SUVmax and SUVmean) and total lesion glycolysis (SUVmean x MTV) were calculated. For MTV and total lesion glycolysis, the sum of all lesions was used for analysis.
MSK-ACCESS pipeline
MSK-ACCESS is a hybrid-capture, duplex barcoded sequencing panel that allows for two bioinformatic methods of calling mutations: (1) de novo base calling, requiring a given mutation to be present in at least three separate duplex consensus reads for recurrently observed ‘hotspot’ mutations and at least five separate duplex consensus reads for non-hotspot mutations, and (2) ‘genotyping’, in which a variant previously observed in a given patient (in either MSK-IMPACT or MSK-ACCESS samples with de novo base calling) is called as a mutation with a lower threshold (at least one duplex consensus read or two simplex (single-stranded) consensus reads)35. In this study, a total of four actionable mutations resulting in therapy matching were identified in MSK-ACCESS ctDNA samples using genotyping alone (that is, these would not have been discovered were they not previously discovered in a given patient’s tissue sample): one EGFR L858R mutation, one EGFR exon 19 deletion and two KRAS G12C mutations. Because this is the default base calling algorithm for MSK-ACCESS and because it is possible for driver mutation profiles to change across time independently of base calling methods (that is, for biological reasons), these patients were treated as being matched to targeted therapy by ctDNA for the purposes of analysis.
ctDx Lung versus MSK-ACCESS
Among the 163 patients with ctDx Lung and MSK-ACCESS tests within 30 days of each other, 54 mutations were present on ctDx Lung testing only, while 55 mutations were present on MSK-ACCESS only. Of the 54 unique ctDx Lung mutations, 30 (56%) of them were filtered out based on WBC sequencing (Extended Data Fig. 3). The 24 (44%) remaining mutations were below the subthreshold or not detected at any frequency in MSK-ACCESS. A total of 216 mutations were detected on both assays. The concordance correlation coefficient of all mutations together was 0.98; the Pearson’s R for these same mutations was also 0.98, P < 0.001. Of the 230 patients with both ctDx Lung and MSK-ACCESS testing, 13 were matched to targeted therapy based on a ctDNA alteration not detected on ctDx Lung testing, while 3 patients were matched based on a ctDNA alteration not detected on MSK-ACCESS.
Extended Data
Extended Data Fig. 1 |. Turnaround time of plasma and tissue sequencing.
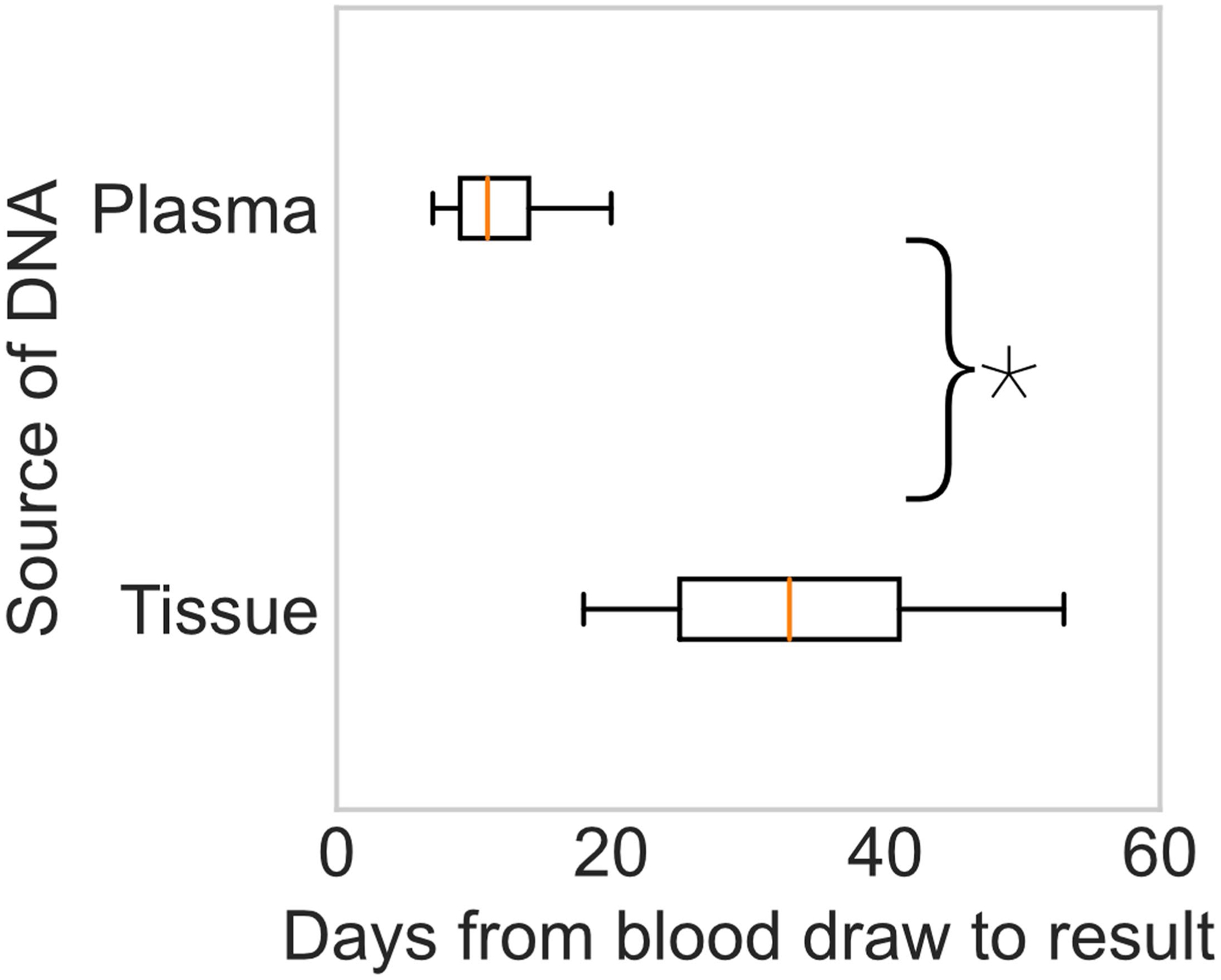
Boxplots showing median (center) +/−25%ile (boxes) and 95%ile (whiskers) of turnaround time for liquid biopsy (MSK-ACCESS and ctDx Lung, N independent samples = 2,162) and tissue (MSK-IMPACT, N independent samples = 612) sequencing from date of blood collection. Tissue start time is the date of white blood cell control collection. The turnaround time for plasma ctDNA sequencing was significantly faster than for tissue sequencing (*two-sided Mann-Whitney U, p < 0.001).
Extended Data Fig. 2 |. Correlates of ctDNA alteration levels.
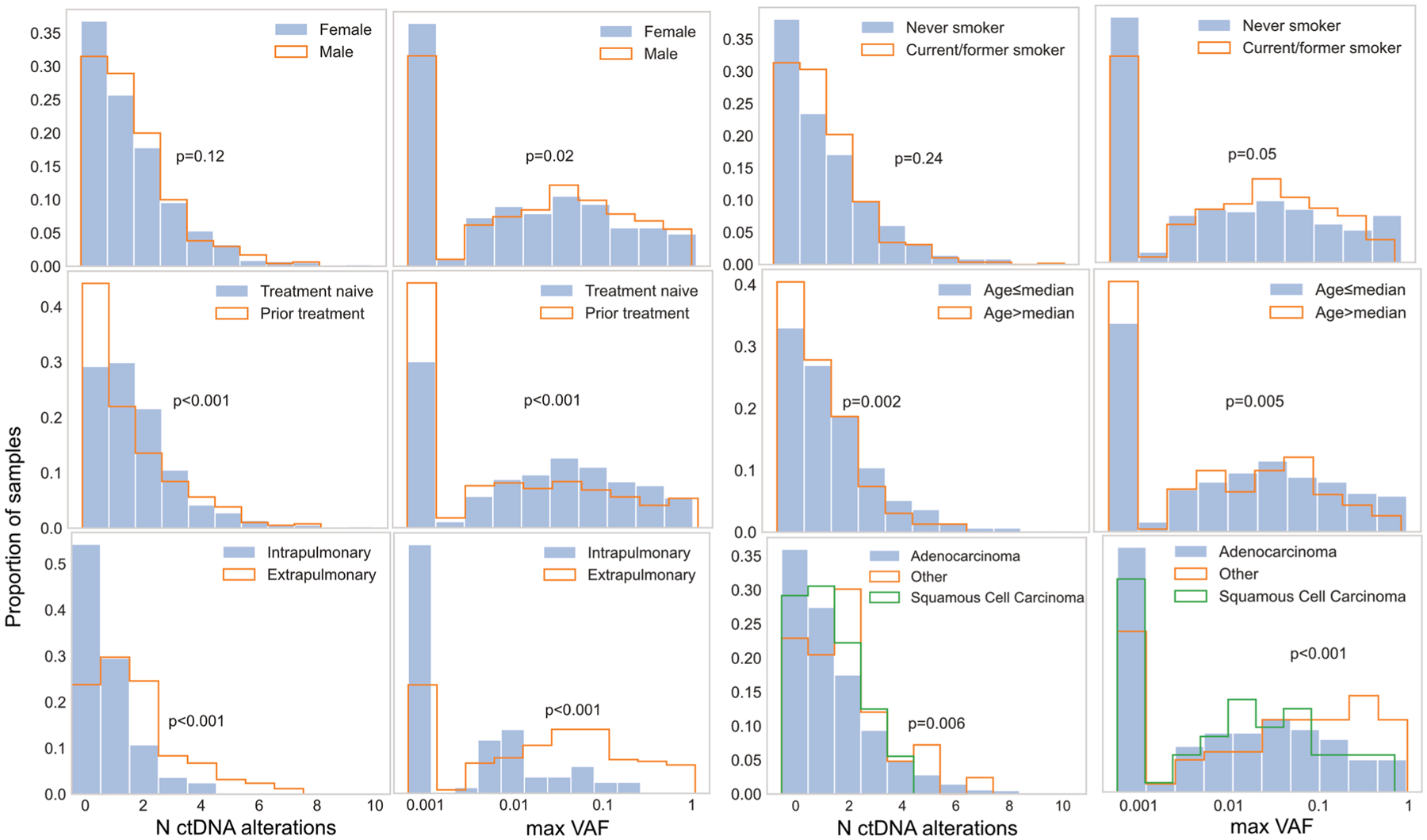
Histograms showing the proportion of patients with either number of ctDNA alterations or maximum mutation variant allele frequency (VAF) per sample. P-values are from Mann-Whitney U tests or Kruskal-Wallis tests for histologic subgroups. Raw VAFs of zero are set to the minimum value of the log axis.
Extended Data Fig. 3 |. Survival of patients without tissue sequencing matched to targeted therapy by ctDNA.
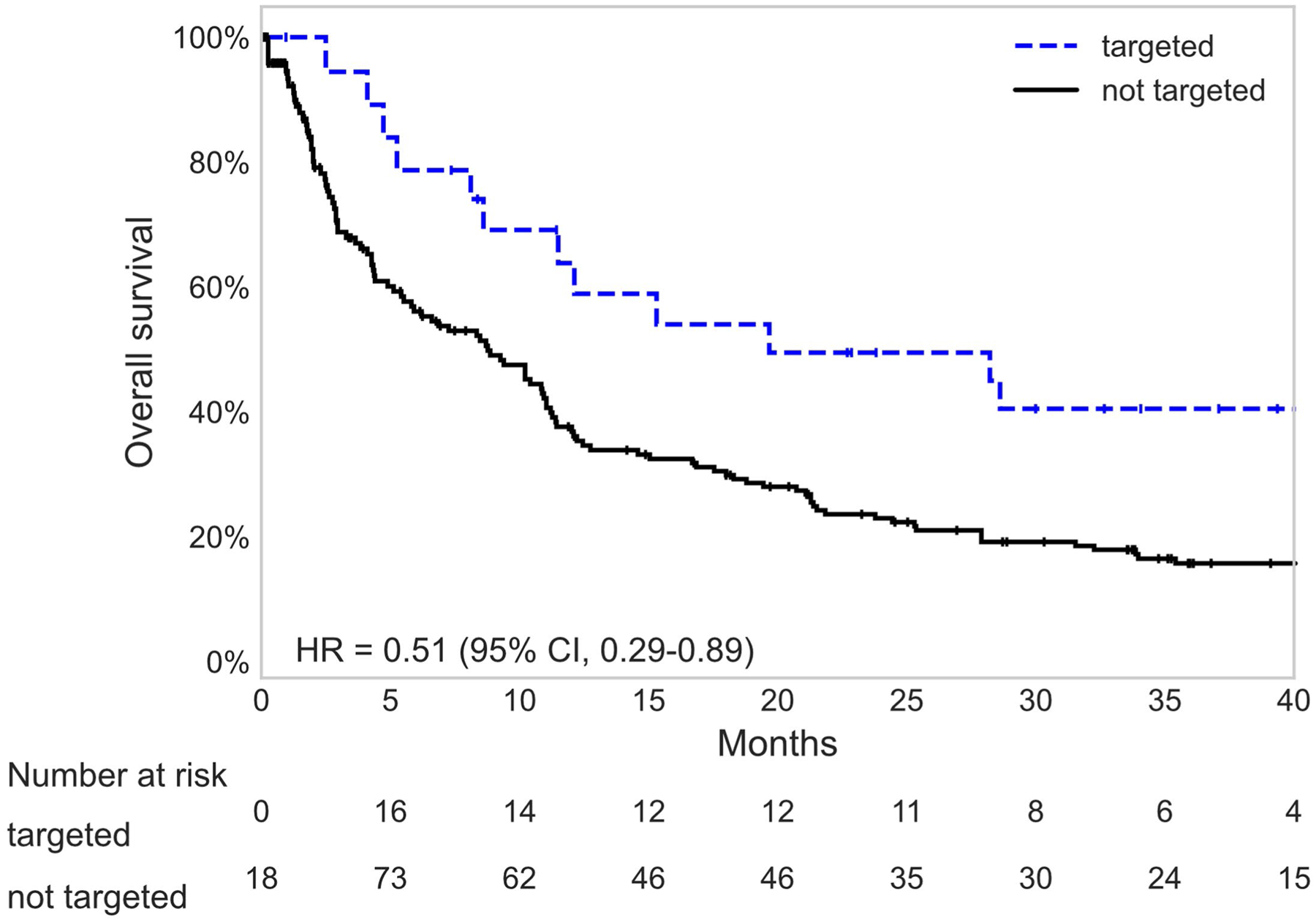
Kaplan-Meier survival curves for patients without tissue sequencing matched or not matched to targeted therapy. Number at risk in each category is adjusted for left truncation and time-dependent nature of targeted therapy variables.
Supplementary Material
Acknowledgements
This work was supported by a grant from the Antidote Health Foundation for Cure of Cancer (BTL), the National Institutes of Health (T32-CA009207 (J.J.), CTSA UL1TR00457 (M.R.M.-G.), P50 CA24774901 and P30 CA008748), the Molecular Diagnostics Service in the Department of Pathology, and the Marie-Josee and Henry R. Kravis Center for Molecular Oncology.
Competing interests
J.J. has a patent licensed by MDSeq. A.N. reports serving as a one-time paid consultant for Bayer. P.K.P. receives compensation for consulting or advisory board participation from Bicara Therapeutics, Boehringer Ingelheim, EMD Serono, GlaxoSmithKline, Takeda Pharmaceuticals, WC Communications and Xencor and receives honoraria for participation in CME educational programs from PeerVoice, ACE Oncology Research to Practice, Clinical Care Options, Spring to Life and Touch Independent Medical Education. J.E.C. has served as a consultant for Astra Zeneca, Bristol-Myers Squib, Genentech, Merck, Flame Biosciences, Novartis, Regeneron-Sanofi, Guardant Health and Janssen and has received research funding to her institution from Astra Zeneca, Bristol-Myers Squib, Genentech and Merck. A.R.B. has stock ownership in Johnson & Johnson. R.B. reports a grant from Archer, honoraria for advisory board participation from Loxo oncology and speaking fees from Illumina. A.Z. has received speaking fees from Illumina. D.C. has consulted with/received honoraria from Pfizer, Loxo/Lilly Oncology, BridgeBio, FORE Therapeutics, Scorpion Therapeutics and Vividion Therapeutics. Y.R.M.-G. acknowledges receipt of training through an institutional K30 grant from the NIH (CTSA UL1TR00457). She has received funding from a Kristina M. Day Young Investigator Award from Conquer Cancer, the ASCO Foundation, funded by Charles M. Baum and Carol A. Baum. She reports travel, accommodation and expenses from AstraZeneca and honoraria from Virology Education. She acknowledges research funding to the institution from Loxo Oncology at Eli Lilly, Elucida Oncology, Taiho Oncology, Hengrui USA/Jiangsu Hengrui Pharmaceuticals and Endeavor Biomedicines. She acknowledges royalties from Rutgers University Press and Wolters Kluwer. H.-Y.T. has received academic travel support from Resolution Bioscience. C.X. has received honoraria from AstraZeneca, BeiGene, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Merck & Co., Novartis, Pfizer and Roche; has received research support from BeiGene and Hengrui Pharmaceutical; and has received reimbursement for travel and accommodation expenses from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Merck & Co., Novartis, Pfizer and Roche. B.D. reports equity interest in Eli Lilly and Company, Roche and CVS Health and family member involvement with Eli Lilly and Company and CVS Health. H.A.Y. has consulted for AstraZeneca, Blueprint Medicine, Janssen Oncology, Cullinan and Daiichi. Her institution has received research funding for clinical trials from AstraZeneca, Daiichi, Pfizer, Novartis, Cullinan Oncology and Lilly. M.O. reports personal fees from PharmaMar, personal fees from Novartis, personal fees from Targeted Oncology, personal fees from Bristol-Myers Squibb, personal fees from Merck Sharp & Dohme, personal fees from Jazz Pharmaceuticals and personal fees from Astro Pharmaceuticals, outside the submitted work. M.D.H. reports grants from BMS and personal fees from Achilles, Adagene, Adicet, Arcus, AstraZeneca, Blueprint, BMS, DaVolterra, Eli Lilly, Genentech/Roche, Genzyme/Sanofi, Janssen, Immunai, Instil Bio, Mana Therapeutics, Merck, Mirati, Natera, Pact Pharma, Shattuck Labs and Regeneron, as well as equity options from Factorial, Immunai, Shattuck Labs and Arcus. A patent filed by Memorial Sloan Kettering related to the use of TMB to predict response to immunotherapy (PCT/US2015/062208) is pending and licensed by PGDx. P.L. is listed as an inventor on patent applications filed by MSKCC that describe approaches to treat KRAS or BRAF-mutant tumors. K.C.A. reports personal fees from AstraZeneca and nonfinancial support from Takeda and Novartis outside the submitted work. In the last 3 years, M.G.Z. has received consulting fees from Takeda, GlaxoSmithKline, Expert Connect, Aldeyra Therapeutics, Novocure and Atara and honoraria from Research to Practice, Medical Learning Institute and OncLive. Memorial Sloan Kettering receives research funding from the Department of Defense, the National Institutes of Health, Precog, GlaxoSmithKline, Epizyme, Polaris, Sellas Life Sciences, Bristol Myers Squibb, Millenium/Takeda, Curis and Atara for research conducted by M.G.Z. M.G.Z. serves as chair of the board of directors of the Mesothelioma Applied Research Foundation, uncompensated. M.G.K. receives personal fees from Novartis, Sanofi-Genzyme, AstraZeneca, Pfizer, Janssen and Daiichi-Sankyo; received honoraria for participation in educational programs from WebMD, OncLive, Physicians Education Resources, Prime Oncology, Intellisphere, Creative Educational Concepts, Peerview, i3 Health, Paradigm Medical Communications, AXIS, Carvive Systems and AstraZeneca; received travel support from AstraZeneca, Pfizer and Genentech; and received editorial support from Hoffman La-Roche. Memorial Sloan Kettering has received research funding from the National Cancer Institute (USA), the Lung Cancer Research Foundation and Genentech Roche for research conducted by M.G.K. I.P. has consulted or served on the advisory boards of Pfizer, AstraZeneca, Blueprint Medicine, DavaOncology, Eli Lilly and Curio Science. W.V.L. receives institutional research funding from Daiichi Sankyo, Amgen and Abbvie and has been a compensated consultant for PharmaMar, G1 Therapeutics, AstraZeneca and Jazz Pharmaceuticals. D.M. is a consultant for AstraZeneca, Johnson & Johnson, Boston Scientific, Bristol Myers Squibb and Merck. G.R. receives royalties from Scanlan International. B.J.P. is a consultant for AstraZeneca. L.P.L. is an employee and shareholder of Agilent Technologies. M.L. is an employee and shareholder of Agilent Technologies. C.I.D. reports serving on the advisory board for Amgen and Ipsen, honoraria for Merck and academic travel support from Roche. M.I. reports serving on the advisory boards of Pfizer and Takeda; receiving honoraria from Roche, AstraZeneca, MSD, Bristol Myers Squibb, Pfizer, Takeda and Novartis; and travel support from Roche, AstraZeneca and MSD. S.C. reports advisory board fees from Roche and Astra Zeneca and travel support from Bristol Myers Squibb, outside the submitted work. N.P. has served on advisory boards or received personal honoraria from Boehringer Ingelheim, MSD, Merck, Bristol-Myers Squib, Astra Zeneca, Takeda, Pfizer, Roche, Novartis, Ipsen and Bayer and has received research funding to his institution from Bayer, Pfizer and Roche. A.L. reports personal fees and travel funding from Mundipharma/Helsinn, personal fees from Bayer and personal fees from Eisai. G.J.R. reports grants from National Institutes of Health/National Cancer Institute, has been an uncompensated consultant to Daiichi, Pfizer and Mirati and has institutional research support from Mirati, Takeda, Merck, Roche, Pfizer and Novartis. In addition, G.J.R. has pending patents US20170273982A1 and WO2017164887A8. D.B.S. has served as a consultant for/received honoraria from Loxo Oncology, Lilly Oncology, Pfizer, QED Therapeutics, Vivideon Therapeutics and Illumina. M.L. has received honoraria for advisory board participation from Merck, Astra-Zeneca, Bristol Myers Squibb, Blueprint Medicines, Janssen Pharmaceuticals, Takeda Pharmaceuticals, Lilly Oncology, LOXO Oncology, Bayer, ADC Therapeutics, Riken Genesis and Paige AI and research support from LOXO Oncology, Merus, and Helsinn Therapeutics. Marc Ladanyi reports honoraria for ad-hoc advisory board participation from Merck, AstraZeneca, Bristol Myers Squibb, Takeda, Bayer, and Lilly Oncology; and research support from LOXO Oncology, Merus and Helsinn Therapeutics. V.W.R. reports grants from Genelux, grants from Genentech, other from DaVinci Surgery, nonfinancial support from Bristol Myers Squibb and personal fees from NIH/Coordinating Center for Clinical Trials, outside the submitted work. A.R. reports grants from Varian Medical Systems, grants and personal fees from AstraZeneca, grants and personal fees from Merck, grants and personal fees from Boehringer Ingelheim, grants from Pfizer, personal fees from Research to Practice, personal fees from Cybrexa, personal fees from More Health and nonfinancial support from Philips/Elekta, outside the submitted work. D.G. reports grants from Varian, AstraZeneca, Merck and Bristol Myers Squibb and personal fees from Varia, AstraZeneca, Merck, US Oncology, Bristol Myers Squibb, Relfexion, WebMD, Vindico and Medscape and has served on the advisory board for AstraZeneca. A.D. has received honoraria or worked on the advisory boards of Ignyta/Genentech/Roche, Loxo/Bayer/Lilly, Takeda/Ariad/Millenium, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Tyra Biosciences, Verastem, MORE Health, Abbvie, 14ner/Elevation Oncology, Remedica, ArcherDX, Monopteros, Novartis, EMD Serono, Melendi, Liberum, Repare RX, Nuvalent, Merus, AXIS, Chugai Pharm and EPG Health; has received funding through his institution from Pfizer, Exelixis, GlaxoSmithKline, Teva, Taiho and PharmaMar; has received research support from Foundation Medicine; receives royalties from Wolters Kluwer; has received other support from Boehringer Ingelheim; has received food/beverage from Merck, Puma and Merus; and has received CME honoraria from Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis, Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences, Med Learning, Imedex, Answers in CME and Clinical Care Options. H.I.S. reports the following support: compensated consultant/advisor to Ambry Genetics, Konica Minolta, Bayer, Pfizer, Sun Pharmaceuticals and WCG Oncology; uncompensated consultant/advisory to Amgen, ESSA Pharma, Janssen Research & Development, Janssen Biotech and Sanofi Aventis; he has received research funding (to his institution) from Epic Sciences, Illumina, Janssen, Menarini Silicon Biosystems, Prostate Cancer Foundation and ThermoFisher; intellectual property rights from BioNTech, Elucida Oncology, MaBVAX and Y-mAbs Therapeutics; and nonfinancial support from Amgen, Asterias Biotherapeutics, Bayer, ESSA Pharma, Menarini Silicon Biosystems, Phosplatin, Pfizer, Prostate Cancer Foundation and WCG Oncology. S.P.S. is a shareholder and consultant of Canesia Health. M.F.B. reports a consulting/advisory role with PetDx and Eli Lilly; research support from Grail; and a patent pending related to cfDNA profiling. R.L. is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Syndax. Zentalis, Ajax, Bakx, Auron, Prelude, C4 Therapeutics and Isoplexis for which he receives equity support. He receives research support from Ajax and Abbvie and has consulted for Incyte, Janssen, Morphosys and Novartis. He has received honoraria from Astra Zeneca and Kura for invited lectures and from Gilead for grant reviews. P.R. received institutional grant/funding from Grail, Illumina, Novartis, Epic Sciences and ArcherDx and consultation/ad board/honoraria from Novartis, Foundation Medicine, AstraZeneca, Epic Sciences, Inivata, Natera and Tempus. J.R.-F. is a paid consultant of Goldman Sachs, Paige.AI and REPARE Therapeutics, a member of the scientific advisory board of Goldman Sachs, Paige.AI and Volition RX, and an ad hoc member of the scientific advisory board of Roche, Genentech, Roche Tissue Diagnostics, Ventana, Novartis, InVicro and GRAIL. D.R.J. serves as a consultant for AstraZeneca and Merck. C.M.R. reports personal fees from AbbVie, Amgen, Ascentage, AstraZeneca, Bicycle, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Jansen, Jazz, Lilly/Loxo, Pfizer, PharmaMar, Syros, Vavotek, Bridge Medicines and Harpoon Therapeutics, outside the submitted work. J M.I. reports equity in LumaCyte and has served as an uncompensated member of a steering committee for Genentech. B.T.L. has served as an uncompensated advisor and consultant to Amgen, Genentech, Boehringer Ingelheim, Lilly, AstraZeneca and Daiichi Sankyo. He has received research grants to his institution from Amgen, Genentech, AstraZeneca, Daiichi Sankyo, Lilly, Illumina, GRAIL, Guardant Health, Hengrui Therapeutics, MORE Health and Bolt Biotherapeutics. He has received academic travel support from MORE Health and Jiangsu Hengrui Medicine. He is an inventor on two institutional patents at Memorial Sloan Kettering (US62/685,057, US62/514,661) and has intellectual property rights as a book author at Karger Publishers and Shanghai Jiao Tong University Press. The remaining authors declare no competing interests.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-022-02047-z.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
No software was used for data collection.
Extended data is available for this paper at https://doi.org/10.1038/s41591-022-02047-z.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-022-02047-z.
Data availability
Genomic and clinical data are available on cBioPortal (https://www.cbioportal.org/study/summary?id=nsclc_ctdx_msk_2022). The raw sequencing data for MSK-IMPACT and MSK-ACCESS are protected and are not broadly available due to privacy laws. Researchers at MSK with appropriate IRB permission may request the data from the Center for Molecular Oncology (skicmopm@mskcc.org).
References
- 1.Kris MG et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311, 1998–2006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howlader N et al. The effect of advances in lung-cancer treatment on population mortality. N. Engl. J. Med 383, 640–649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasche B & Grant SC Non-small cell lung cancer and precision medicine a model for the incorporation of genomic features into clinical trial design. JAMA 311, 1975 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Bruno DS et al. Racial disparities in biomarker testing and clinical trial enrollment in non-small cell lung cancer (NSCLC). J. Clin. Oncol 39, 9005 (2021). [Google Scholar]
- 5.Robert NJ et al. Biomarker tissue journey among patients (pts) with untreated metastatic non-small cell lung cancer (mNSCLC) in the U.S. Oncology Network community practices. J. Clin. Oncol. 39, 9004 (2021). [Google Scholar]
- 6.Zugazagoitia J et al. Clinical utility of plasma-based digital next-generation sequencing in patients with advance-stage lung adenocarcinomas with insufficient tumor samples for tissue genotyping. Ann. Oncol 30, 290–296 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Sheridan C Investors keep the faith in cancer liquid biopsies. Nat. Biotechnol 37, 972–974 (2019) [DOI] [PubMed] [Google Scholar]
- 8.Rolfo C et al. Liquid biopsy for advanced non-small cell lung cancer: a consensus statement from the International Association for the Study of Lung Cancer (IASLC). J. Thorac. Oncol 16, 1647–1662 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Heitzer E, Haque IS, Roberts CES & Speicher MR Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet 20, 71–88 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Sabari JK et al. A prosspective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J. Natl Cancer Inst 111, 575–583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oxnard GR et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J. Clin. Oncol 34, 3375–3382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal C et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol. 5, 173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sacher AG et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2, 1014–1022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bettegowda C et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci. Transl. Med 6, 224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leighl NB et al. Clinical utility of comprehensive cell-free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non-small cell lung cancer. Clin. Cancer Res 25, 4691–4700 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Remon J et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann. Oncol 28, 784–790 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Tran HT et al. Clinical outcomes in non-small cell lung cancer patients treated with EGFR-tyrosine kinase inhibitors and other targeted therapies based on tumor versus plasma genomic profiling. JCO Precis. Oncol 5, 1241–1249 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang M et al. Incorporating blood-based liquid biopsy information into cancer staging: time for a TNMB system? Ann. Oncol 29, 311–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhuri AA et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 7, 1394–1403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nabet BY et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 183, 363–376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg SB et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin. Cancer Res 24, 1872–1880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandara DR et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small cell lung cancer patients treated with atezolizumab. Nat. Med 24, 1441–1448 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Karachaliou N et al. Association of EGFR L858R mutation in circulating free DNA with survival in the EURTAC trial. JAMA Oncol. 1, 149–157 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Mack PC et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: analysis of over 8000 cases. Cancer 126, 3219–3228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razavi P et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med 25, 1928–1937 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merker JD et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J. Clin. Oncol 36, 1631–1641 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Alix-Panabières C & Pantel K Liquid biopsy: from discovery to clinical application. Cancer Discov. 11, 858–873 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Paweletz CP et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin. Cancer Res 22, 915–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Supplee JG et al. Sensitivity of next-generation sequencing assays detecting oncogenic fusions in plasma cell-free DNA. Lung Cancer 134, 96–99 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Chakravarty D et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol 2017, 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandelker D et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA 318, 825–835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brannon AR et al. Enhanced specificity of high sensitivity somatic variant profiling in cell-free DNA via paired normal sequencing: design, validation, and clinical experience of the MSK-ACCESS liquid biopsy assay. Nat. Comm 12, 3770 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li BT et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: results from the Actionable Genome Consortium. Ann. Oncol. J. Eur. Soc. Med. Oncol 30, 597–603 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura Y et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med 26, 1859–1864 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Pairawan S et al. Cell-free circulating tumor DNA variant allele frequency associates with survival in metastatic cancer. Clin. Cancer Res 26, 1924–1931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramalingam SS et al. Abstract CT078: tumor mutational burden (TMB) as a biomarker for clinical benefit from dual immune check point blockade with nivolumab (nivo) + ipilimumab (ipi) in first-line (1L) non-small cell lung cancer (NSCLC): identification of TMB cutoff from CheckMate 568. Cancer Research 78, CT078 (2018). [Google Scholar]
- 40.Chabon JJ et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morbelli S et al. Circulating tumor DNA reflects tumor metabolism rather than tumor burden in chemotherapy-naive patients with advanced non-small cell lung cancer (NSCLC): 18F-FDG PET/CT study. J. Nucl. Med 58, 1764–1769 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Piotrowska Z et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 8, 1529–1539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Offin M et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis. Oncol 2, PO.18.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piper-Vallillo AJ, Sequist LV & Piotrowska Z Emerging treatment paradigms for EGFR-mutant lung cancers progressing on osimertinib: a review. J. Clin. Oncol 38, 2926–2936 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Tan AC Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 11, 511–518 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicholas M et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med 7, 283ra54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen R et al. Harnessing clinical sequencing data for survival stratification of patients with metastatic lung adenocarcinomas. JCO Precis. Oncol 10.1200/po.18.00307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolton KL et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet 52, 1219–1226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coombs CC et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolton KL et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat. Commun 12, 5975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ignatiadis M, Sledge GW & Jeffrey SS Liquid biopsy enters the clinic – implementation issues and future challenges. Nat. Rev. Clin. Oncol 18, 297–312 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Lisberg A et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor (TKI) naive patients with advanced NSCLC. J. Thorac. Oncol 13, 1138–1145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cristiano S et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parikh AR et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 25, 1415–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamal-Hanjani M et al. Tracking the evolution of non-small cell lung cancer. N. Engl. J. Med 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Mateo J et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT). Ann. Oncol. J. Eur. Soc. Med. Oncol 29, 1895–1902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramalingam SS et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med 382, 41–50 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Presley CJ et al. Association of broad-based genomic sequencing with survival among patients with advanced non-small cell lung cancer in the community oncology setting. JAMA 320, 469–477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rothwell DG et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat. Med 25, 738–743 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Murthy VH, Krumholz HM & Gross CP Participation in cancer clinical trials: race-, sex-, and age-based disparities. JAMA 291, 2720–2726 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Scher KS & Hurria A Under-representation of older adults in cancer registration trials: known problem, little progress. J. Clin. Oncol 30, 2036–2038 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Sedrak MS et al. Older adult participation in cancer clinical trials: a systematic review of barriers and interventions. CA Cancer J. Clin 71, 78–92 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rolfo C, Russo A & de Miguel-Pérez D Speeding tumor genotyping during the SARS-CoV-2 outbreak through liquid biopsy. Cancer 126, 4089–4091 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li BT et al. Reimagining patient-centric cancer clinical trials: a multi-stakeholder international coalition. Nat. Med 28, 620–626 (2022). [DOI] [PubMed] [Google Scholar]
- 66.Syeda MM et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: a clinical validation study. Lancet Oncol. 22, 370–380 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turner NC et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 21, 1296–1308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chabon JJ et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun 7, 11815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bardelli A & Pantel K Liquid biopsies, what we do not know (yet). Cancer Cell. 31, 172–179 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Siravegna G et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med 21, 795–801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lennon AM et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 369, 6499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNulty SN, Parikh BA, Duncavage EJ, Heusel JW & Pfeifer JD Optimization of population frequency cutoffs for filtering common germline polymorphisms from tumor-only next-generation sequencing data. J. Mol. Diagn 21, 903–912 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Shen R & Seshan VE FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanoun S et al. Influence of software tool and methodological aspects of total metabolic tumor volume calculation on baseline [18F]FDG PET to predict survival in Hodgkin lymphoma. PLoS ONE 10, e0140830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boellaard R et al. FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging: version 1.0. Eur. J. Nucl. Med. Mol. Imaging 37, 181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic and clinical data are available on cBioPortal (https://www.cbioportal.org/study/summary?id=nsclc_ctdx_msk_2022). The raw sequencing data for MSK-IMPACT and MSK-ACCESS are protected and are not broadly available due to privacy laws. Researchers at MSK with appropriate IRB permission may request the data from the Center for Molecular Oncology (skicmopm@mskcc.org).