

Abstract
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths in the United States. Despite ongoing efforts aimed at increasing screening for CRC and early detection, and development of more effective therapeutic regimens, the overall morbidity and mortality from this malignancy remains a clinical challenge. Therefore, identifying and developing genomic and epigenomic biomarkers that can improve CRC diagnosis and help predict response to current therapies are of paramount importance for improving survival outcomes in CRC patients, sparing patients from toxicity associated with current regimens, and reducing the economic burden associated with these treatments. Although efforts to develop biomarkers over the past decades have achieved some success, the recent availability of high-throughput analytical tools, together with the use of machine learning algorithms, will likely hasten the development of more robust diagnostic biomarkers and improved guidance for clinical decision-making in the coming years. In this article, we provide a systematic and comprehensive overview on the current status of genomic and epigenomic biomarkers in CRC, and comment on their potential clinical significance in the management of patients with this fatal malignancy, including in the context of precision medicine.
Keywords: biomarkers, colorectal cancer, epigenomics, genomics, liquid biopsy
INTRODUCTION
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths in the western world. Despite ongoing efforts aimed at increasing screening of individuals and improving strategies for early detection, ~20% of patients still present with metastatic disease at diagnosis, and ~35% of patients who undergo surgery for primary disease experience relapse (Siegel et al., 2020). Over the past three decades, the median overall survival (OS) of patients with metastatic CRC (mCRC) has gradually increased because of the implementation of combined chemotherapy regimens as well as the introduction of targeted molecular therapies against epidermal growth factor receptor (EGFR), angiogenic factors, and, more recently, BRAF. In addition, the discovery that immunotherapy is effective for patients with microsatellite instability-high (MSI-H) or deficient DNA mismatch-repair (dMMR) mCRC has introduced a new therapeutic option for this subset of patients (Ruiz-Bañobre, Kandimalla, & Goel, 2019).
CRC develops through sequential accumulation of genetic and epigenetic alterations in precursor lesions (mainly adenomas and serrated polyps). These precursor lesions progressively develop a more aggressive phenotype, acquiring a range of dysplastic features and eventually progressing to adenocarcinoma, the most malignant stage (Jung, Hernández-Illán, Moreira, Balaguer, & Goel, 2020). Greater understanding of the molecular pathways involved in carcinogenesis of CRC is needed to improve the detection of therapeutically targetable vulnerabilities and to develop more effective early diagnostic strategies in order to detect disease before it progresses to malignancy. To date, three pathways have been well established in CRC carcinogenesis: chromosome instability (CIN), microsatellite instability (MSI), and the serrated pathway. These pathways are characterized by multistep genetic and epigenetic alterations, involving several oncogene and tumor suppressor genes, as described below.
CIN pathway
The CIN phenotype is observed in as many as 80% of sporadic CRCs, and is characterized by chromosome changes such as somatic copy number variations, deletions, insertions, amplifications, or loss of heterozygosity (Grady & Carethers, 2008). Mutation of the APC gene is a key event in CRC tumors that exhibit CIN, and is considered the earliest genomic event in CRC carcinogenesis through this pathway (Powell et al., 1992). Loss of APC activity results in translocation of beta-catenin to the nucleus and subsequent activation of the WNT signaling pathway, which leads to constitutive activation of other relevant genes such as MYC, CCND1, VEGF, and PPARD (Mann et al., 1999). The WNT signaling pathway is activated in nearly all CRCs that exhibit CIN, and APC mutations have been identified in more than 80% of these cancers (Guinney et al., 2015). Later mutations that are characteristic of CRC arise in a sequential manner after mutation of APC as lesions progress toward adenocarcinoma. Activating mutations in KRAS often arise following acquisition of mutations in APC, and are present in 40–50% of CRCs (Santini et al., 2008). Later in the adenoma–carcinoma sequence, activating mutations in PIK3CA and inactivating mutations in P53 arise in ~15% and 60% of CRCs, respectively (Baker et al., 1990; Liao et al., 2012). Loss of heterozygosity at chromosome 18q, which harbors various tumor suppressor genes from the TGF-beta pathway, such as SMAD2 and SMAD4, is also found in more than 70% of CRCs (Fearon & Vogelstein, 1990).
MSI pathway
MSI is the primary mechanism for the development of colorectal tumors with a hypermutable phenotype. CRC that arises due to MSI develops more rapidly than CRC arising from CIN, having an estimated tumor development time of a few years as compared to approximately two decades for the CIN pathway (Aust, Sommer, & Baretton, 2012). CRCs arising due to MSI are often characterized by the presence of single nucleotide substitutions or insertions and deletions within microsatellite repeats. Microsatellites are short tandem repeat DNA sequences spread throughout the human genome. Because they are formed of highly repetitive sequences, they have a higher propensity for acquiring mutations than do other regions of the genome. The process of repairing errors in microsatellite repeats is tightly governed by an intact MMR system (Ruiz-Bañobre & Goel, 2019). MMR deficiency resulting from somatic or germline DNA alterations in any of the key MMR genes (MLH1, MSH2, MSH6, and PMS2), or from deletions in the EPCAM gene (which leads to constitutional repression of MSH2 gene expression through promoter methylation), is the initiating event for MSI-driven carcinogenesis. The loss of MLH1 expression due to biallelic MLH1 promoter methylation is the key somatic event responsible for ~75%–80% of sporadic cancers with MSI. Overall, MSI is observed in ~12–15% of sporadic CRCs and in the majority of CRCs from patients with Lynch syndrome and its variants (Muir–Torre or Turcot’s syndromes) (Ruiz-Bañobre & Goel, 2019). Moreover, sporadic CRCs with MSI frequently exhibit a CpG island methylator phenotype (CIMP) and harbor BRAF V600E mutations, while APC and P53 mutations are found in a smaller proportion of MSI-H CRCs compared with MSS CRCs (Nguyen, Goel, & Chung, 2020; Rajagopalan et al., 2002; TCGA Research Network, 2012).
Serrated pathway
The serrated pathway represents the origin of ~15% of CRCs, and describes the progression of precursor malignant lesions that present as serrated polyps (Toyota et al., 1999). The term “serrated polyp” encompasses a heterogeneous spectrum of lesions that include, in decreasing order of prevalence, benign hyperplastic polyps (the most frequent subtype), sessile serrated adenomas (SSAs), and traditional serrated adenomas (TSAs; the less frequent subtype) (McCarthy, Serra, & Chetty, 2019). SSAs and TSAs are more advanced lesions that have a higher propensity for malignant progression. The earliest driving molecular event in the serrated pathway appears to be BRAF V600E mutation. This mutation is present in a large proportion of microvesicular hyperplastic polyps, which subsequently progress to SSA, then to SSA with dysplasia, and finally to CRC (Davies et al., 2002). Following the appearance of the BRAF V600E mutation, serrated tumors can follow two different routes and develop either into MSI CRC through acquisition of CIMP and methylation of the MLH1 promoter, or into microsatellite stable (MSS) CRCs (Guinney et al., 2015). In addition, RNF43 mutation constitutes a subsequent important genomic event for the serrated pathway in both MSI and MSS CRCs (Yan et al., 2017). TSAs are less prevalent than SSAs (TSAs represent 1–7% of all serrated lesions) and therefore are not characterized as well, but they often exhibit three somewhat distinct molecular patterns: BRAF-mutated and CIMP-high (CIMP-H), KRAS-mutated and CIMP-low, or BRAF and KRAS wild-type. In addition, TSAs almost never display an MSI phenotype (McCarthy et al., 2019).
Although differences and similarities in the molecular profiles for these three pathways are well defined, a deeper temporospatial understanding of these carcinogenesis pathways will help for more rational biomarker development. Conceptually, events that occur early during carcinogenesis will serve better as potential biomarkers for the early detection of precursor lesions. In contrast, molecular profiles from both early and later lesions will be useful for identifying biomarkers for prognostication, and therapeutic benefit prediction. However, as we will discuss in this chapter, the biomarker field is complex, and it is not simple to identify robust biomarkers, thus reinforcing the importance of comprehensive molecular profiling in a clearly defined clinical context (Figure 1).
Figure 1. Schematic and general overview of the different biospecimen sources and biomarkers involved in diagnosis, prognosis, and prediction of clinical benefit of therapies for CRC.
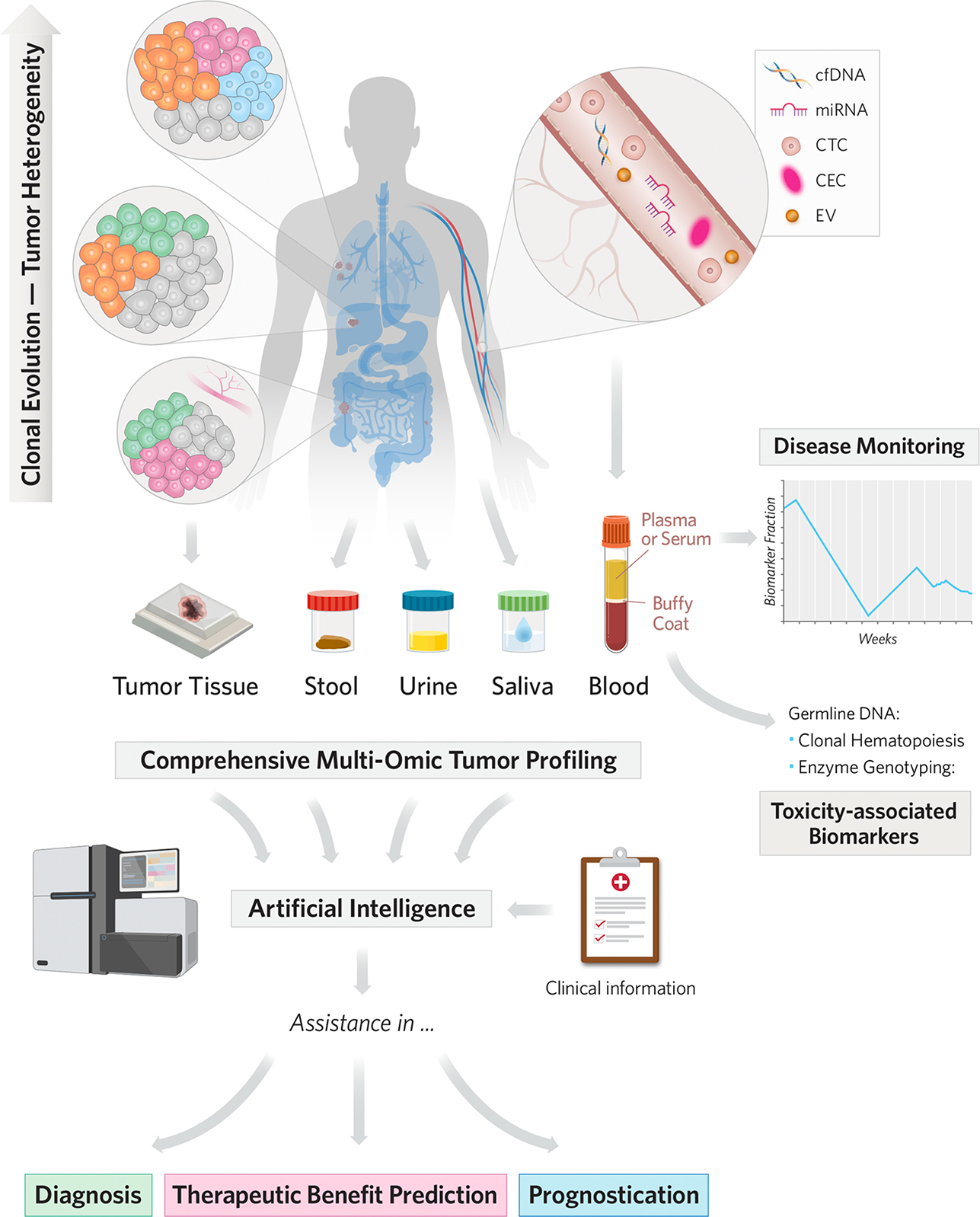
Starting from a single ancestral clone, cancer cells undergo different routes of clonal evolution, which are responsible for temporospatial tumor heterogeneity. Although a single tumor tissue biopsy cannot capture all of the tumor variability, it is postulated that different types of liquid biopsies and biospecimens can capture this heterogeneity over time. In addition, there are many sources and types of biomarkers that can be detected in tumor tissue and other biospecimens, with blood, stool, saliva, and urine being the most promising. To date, for CRC early-diagnosis, the most frequently studied biospecimens have been stool and blood and for prognosis and clinical benefit prediction tumor tissue and blood have been most frequently studied. Moreover, because of their capacity to recapitulate tumor heterogeneity, liquid biopsies can facilitate disease monitoring and therapeutic response, especially through serial blood analyses. Identification of genotypic markers encoding drug-metabolizing enzymes is also possible by analyzing germline DNA from peripheral blood mononuclear cells. Last, implementation of novel high-throughput molecular analytic techniques, together with integration of multi-omic approaches from multiple sources with clinical and epidemiologic data using artificial intelligence, will accelerate biomarker development and assist in the clinical decision-making process.
Abbreviations: CEC, circulating endothelial cells; cfDNA, cell-free DNA; CRC, colorectal cancer; CTC, circulating tumor cell; EV, extracellular vesicle; miRNA, microRNA.
DIAGNOSTIC BIOMARKERS
Recently, the incidence of CRC has been rising in adults under the age of 50, and such CRC is referred to as early-onset CRC. This increasing incidence suggests that CRC screening programs for early detection should be expanded to a younger, asymptomatic, average-risk population. Although there is a lack of consensus within the medical community regarding this issue, some medical societies are discussing implementing currently available screening strategies across a wider range of ages, including younger individuals (USPSTF, 2020). Regarding screening strategies, these can be prioritized into several tiers: the first tier could include colonoscopy, fecal immunochemical test (FIT), and fecal occult blood test (FOBT); the second tier computed tomographic colonography, FIT-DNA, and flexible sigmoidoscopy; and the third tier capsule endoscopy. Although colonoscopy is considered the gold standard for CRC screening, it has several limitations, including invasiveness, cost, and low compliance. In contrast, FOBT and FIT, which are the most commonly used non-invasive screening tests in Europe and other Western countries, lack sensitivity and specificity compared to colonoscopy, especially for precursor lesions (Quintero et al., 2012). Given the challenges associated with these commonly used screening approaches, there is a clear need for developing more robust, non-invasive strategies to diagnose premalignant and malignant colorectal lesions.
These strategies are likely to include novel biomarkers for early detection and improved, clinically useful devices and kits that leverage existing and new biomarkers. Some molecular alterations are particularly appealing for potential use as non-invasive biomarkers, especially alterations that can be detected in stool or blood. For example, epigenetic changes, either on their own or, more recently, in combination with DNA mutations and circulating cells, have emerged as front runners for the development of early detection biomarkers in colorectal neoplasia (which includes both precursor lesions and CRC). In this section we provide a summary of genomic and epigenomic diagnostic biomarkers for CRC (Table 1), highlighting well-established assays and describing some of the most promising and novel potential biomarkers in the field.
Table 1.
Established and potential biomarkers in CRC diagnosis.
| References | Biomarker | Specimen | Test | Accuracy | Regulatory status |
|---|---|---|---|---|---|
| miRNAs | |||||
|
Ng et al. (2009)
Uratani et al. (2016) Yau, Tang, Harriss, Dickins, and Polytarchou (2019) |
miR-92a | Blood | NA | CRC: AUROC: 0.89 (plasma) Sensitivity: 0.89 Specificity: 0.70 A: AUROC: 0.75 (serum) Sensitivity: 0.65 Specificity: 0.79 AA: AUROC: 0.84 (serum) |
– |
| Stool | NA | CRC: AUROC: 0.80 Sensitivity: 0.47 Specificity: 0.91 A: AUROC: 0.64 Sensitivity: 0.43 Specificity: 0.91 |
– | ||
|
Peng et al. (2017)
Toiyama et al. (2013) Yau et al. (2019) |
miR-21 | Blood | NA | CRC: AUROC: 0.85 (plasma)/0.87 (serum) Sensitivity: 0.69/0.75 Specificity: 0.86/0.84 A: AUROC: 0.80 (serum) Sensitivity: 0.77 Specificity: 0.81 |
– |
| Stool | NA | CRC: AUROC: 0.84 Sensitivity: 0.60 Specificity: 0.86 A: AUROC: 0.77 Sensitivity: 0.60 Specificity: 0.83 |
– | ||
| Yau et al. (2019) | 2-miRNA panel: miR-92 miR-21 |
Stool | NA | CRC: AUROC: 0.84 Sensitivity: 0.54 Specificity: 0.88 |
– |
| Duran-Sanchon et al. (2020) | GBM modela: miR-421 miR-27a-3p |
Stool | NA | CRC and AA: AUROC: 0.74 (training set)/0.63 (test set) Sensitivity: 0.74/0.67 Specificity: 0.63/0.60 CRC: AUROC: 0.86/0.74 Sensitivity: 0.96/0.96 Specificity: 0.36/0.33 AA: AUROC: 0.71/0.64 Sensitivity: 0.61/0.59 Specificity: 0.71/0.69 |
– |
| Liu, Klein, et al. (2018) and Liu, Liu, et al. (2018) | 4-miRNA panel: miR-21 miR-92a miR-29a miR-125b |
Blood (serum) | NA | CRC: AUROC: 0.95 Sensitivity: 0.85 Specificity: 0.99 |
– |
| Chang et al. (2016) | 2-miRNA panel: miR-223 miR-92a |
Blood (plasma) | NA | CRC: AUROC: 0.91 Sensitivity: 0.97 Specificity: 0.75 |
– |
| Herreros-Villanueva et al. (2019) | 6-miRNA panel: miR-19a miR-19b miR-15b miR-29a miR-335 miR-18a |
Blood (plasma) | NA | CRC and AA: AUROC: 0.92 Sensitivity: 0.85 Specificity: 0.90 |
– |
| Sazanov, Kiselyova, Zakharenko, Romanov, and Zaraysky (2017) | 2-miRNA panel: miR-21 miR-92a |
Saliva | NA | CRC: Sensitivity: 0.97 Specificity: 0.91 |
– |
| Rapado-González et al. (2019) | 5-miRNA panel: miR-186-5p miR-29a-3p miR-29c-3p miR-766-3p miR-491-5p |
Saliva | NA | CRC: AUROC: 0.75 Sensitivity: 0.72 Specificity: 0.67 |
– |
| DNA methylation | |||||
| Church et al. (2014) | SEPT9 | Blood (plasma) | Epi proColon | CRC: Sensitivity: 0.68 Specificity: 0.79 AN: Sensitivity: 0.25 Specificity: 0.79 AA: Sensitivity: 0.22 Specificity: 0.79 |
Registered trademark. FDA approval: alternative for CRC screeningb |
|
Baek et al. (2009)
Carmona et al. (2013) Chen et al. (2005) Fu et al. (2018) Itzkowitz et al. (2007) Kisiel et al. (2013) Li et al. (2009) Lu et al. (2014) |
VIM | Blood (plasma) | NA | CRC: Sensitivity: 0.59 Specificity: 0.93 |
– |
| Stool | ColoSure | CRC: Sensitivity: 0.81 Specificity: 0.95 |
Trademark. Not FDA approval | ||
| Jung et al. (2020) | SFRP2 | Blood (plasma and serum) | NA | CRC: Sensitivity: 0.64–0.67 Specificity: 0.97–1.00 A: Sensitivity: 0.06–0.81 Specificity: 0.73–1.00 |
– |
| Stool | NA | CRC: Sensitivity: 0.56–0.94 Specificity: 0.77–0.97 A: Sensitivity: 0.28–0.76 Specificity: 0.55–1.00 |
– | ||
| Luo et al. (2020) | cd-score (multi-region panel) | Blood (plasma) | NA | CRC: AUROC: 0.96 Sensitivity: 0.88 Specificity: 0.90 |
– |
| Luo et al. (2020) | cg10673833 (single probe) | Blood (plasma) | NA | CRC: AUROC: 0.90 Sensitivity: 0.90 Specificity: 0.87 |
– |
| Kandimalla et al. (2020) | Multi-region panel | Blood (plasma) | EpiPanGI-Dx | CRC: AUROC: 0.98 |
– |
| ColonES Product Sheet (2020) | Multi-region panel | Blood (plasma) | ColonEs | CRC and A: Sensitivity: 0.91 (A)/0.97 (I)/0.94 (II–IV) Specificity: 0.99 |
– |
| Multi-modal | |||||
| USPSTF. US Preventive Services Task Force et al. (2020) | Multi-modal panel: KRAS mutations (×7) NDRG4 m.s. BMP3 m.s. FIT |
Stool | Cologuard | CRC: Sensitivity: 0.93 Specificity: 0.85 AN: Sensitivity: 0.47 Specificity: 0.89 AA: Sensitivity: 0.43 Specificity: 0.89 |
Registered trademark. FDA approval: CRC screeningc |
| Friedland et al. (2020) | Multi-modal panel: Circulating GI endothelial cells Somatic mutationsd SEPT9 m.s. |
Blood | FirstSight | CRC: Sensitivity: 1.00 Specificity: 0.90 AA: Sensitivity: 0.80 |
Trademark |
| Cohen et al. (2018) | Multi-modal panele: 8 proteins Mutations |
Blood (plasma) | CancerSEEK | CRC: AUROC: 0.85 |
– |
| Duran-Sanchon et al. (2020) | GBM modela: miR-421 miR-27a-3p FIT |
Stool | NA | CRC and AA: AUROC: 0.72 (training set)/0.70 (test set) Sensitivity: 0.74/0.68 Specificity: 0.63/0.64 CRC: AUROC: 0.90/0.93 Sensitivity: 0.96/0.97 Specificity: 0.36/0.43 AA: AUROC: 0.70/0.64 Sensitivity: 0.61/0.49 Specificity: 0.71/0.71 |
– |
| Metabolomic | |||||
| Deng et al. (2017) | Metabolites: Succinic acid Ascorbic acid Carnitine |
Urine | PolypDx | HP and A: Sensitivity: 0.43 Specificity: 0.91 |
Trademark. Available for CLIA-certified laboratories |
miRNA-based predictive model included miR-421 and miR-27a-3p, along with age and sex.
Alternative in average-risk individuals who refuse other screening modalities.
CRC screening in average-risk population.
Somatic mutations in oncogenes and tumor suppressor genes.
8 proteins (cancer antigen 125, carcinoembryonic antigen, cancer antigen 19–9, proactin, hepatocyte growth factor, osteopontin, myeloperoxidase, and tissue inhibitor of metalloproteinases 1) and mutations (SNVs or INDELs) in 1933 distinct genomic positions.
Abbreviations: A, adenoma; AA, advanced adenoma; CRC, colorectal cancer; AN, advanced neoplasia; AUROC, area under the receiver operating characteristic curve; CLIA, Clinical Laboratory Improvement Amendments; FIT, fecal immunochemical test; GBM, gradient boosting machine; GI, gastrointestinal; HP, hyperplastic polyp; m.s., methylation status; NA, not available.
MicroRNAs (miRNAs)
The potential of miRNAs as biomarker candidates lies in their small size, relatively limited numbers as compared to protein-coding genes/mRNAs, and stability in a variety of biological specimens, such as tissue, blood, and stool (Bovell et al., 2012; Cortez et al., 2011; Esteller, 2011; J. Lu et al., 2005; Volinia et al., 2006). In addition, there is a wide variety of routine laboratory techniques for identifying and quantifying miRNAs (e.g., microarrays, quantitative reverse transcription PCR) in virtually all specimen types, which would further support application of miRNAs as biomarkers. Over the past decade, the number of studies investigating miRNAs in CRC has increased exponentially, and efforts to evaluate their potential as biomarkers have particularly increased. Despite this enthusiasm, only a few well-designed studies have thus far been conducted that include large patient cohorts, well-defined patient populations, and analysis of independent validation cohorts. None of the biomarkers evaluated in these studies have yet been adopted in clinical practice because of the lack of prospective randomized studies comparing them with current gold standard screening methods. Below, we discuss some of the most promising miRNAs for diagnostic purposes.
miR-92a –
The presence of miR-92a has shown remarkable utility for discriminating patients with CRC from healthy controls when measured in blood or stool samples. In plasma samples, miR-92a showed good performance for detecting CRC, with an area under the receiver operating characteristic curve (AUROC), sensitivity, and specificity of 0.89, 89%, and 70%, respectively (Ng et al., 2009). In the context of adenoma detection, serum miR-92a levels yielded an AUROC of 0.75, and corresponding sensitivity and specificity of 65% and 79%, respectively (Uratani et al., 2016). Recently, Yau et al. (Yau, Tang, Harriss, Dickins, & Polytarchou, 2019) reported the results of a meta-analysis involving 17 studies evaluating the diagnostic potential of several miRNAs, specifically in stool samples. Expression of miR-92a in stool had an AUROC, sensitivity, and specificity of 0.80, 47%, and 91%, respectively, for detecting CRC (Yau et al., 2019), and of 0.64, 43%, and 91%, respectively, for detecting adenoma (Yau et al., 2019). Intriguingly, the diagnostic accuracy of miR-92a for adenoma detection was higher using blood than stool samples (Uratani et al., 2016; Yau et al., 2019).
miR-21 –
The presence of miR-21 has demonstrated high sensitivity and specificity for detecting CRC in both blood and stool samples, although results from stool samples showed greater variability. In a meta-analysis of 11 studies conducted between 2012 and 2014 and involving almost 800 patients, the overall AUROC, sensitivity, and specificity of circulating miR-21 for blood-based early detection of CRC were 0.86, 72%, and 84%, respectively (Peng et al., 2017). For the diagnosis of colorectal adenomas, miR-21 expression in serum showed an AUROC of 0.80 with a sensitivity and specificity of 76.8% and 81.1%, respectively (Toiyama et al., 2013). In a recent meta-analysis, miR-21 expression in stool samples yielded an AUROC, sensitivity, and specificity of 0.84, 60%, and 86%, respectively for CRC (Yau et al., 2019). In the same study, the overall AUROC, sensitivity, and specificity for adenoma detection were 0.77, 60%, and 83%, respectively (Yau et al., 2019).
miRNA panels –
Although miRNA panels have been suggested as a potentially more accurate and robust diagnostic approach than the use of a single miRNA biomarker, these approaches have not yielded consistent findings among studies. For example, in some studies, the analysis of miR-21 and miR-92a in blood samples for the non-invasive diagnosis of CRC yielded sensitivities and specificities ranging from 90% to 89%, and 90% to 96%, respectively, whereas in another cohort, the combination of these two biomarkers yielded a sensitivity of 68% and a specificity of 91% (Liu et al., 2013). In contrast, in another study, a serum-based four-miRNA panel (miR-21, miR-29a, miR-92a and miR-125b) showed an excellent performance for CRC diagnosis, with an AUROC of 0.95, sensitivity of 85%, and specificity of 99% (Liu et al., 2018). Along similar lines, a two-miRNA panel (miR-223 and miR-92a) exhibited an AUROC of 0.91, sensitivity of 97%, and specificity of 75% for detection of CRC in blood specimens from a cohort of 291 patients with CRC and 452 self-reported healthy controls (Chang et al., 2016). On the other hand, a recent meta-analysis evaluating the potential for CRC diagnosis using a combination panel of miR-21 and miR-92a detected in stool samples yielded an AUROC of 0.84, sensitivity of 0.54, and specificity of 0.88 (Yau et al., 2019). A plasma-based six-miRNA panel (miR-19a, miR-19b, miR-15b, miR-29a, miR-335, and miR-18a), when analyzed in a cohort of 300 individuals, could accurately identify healthy controls from patients with advanced colorectal neoplasms (CRC and advanced adenoma), with an AUROC of 0.92, sensitivity of 85%, and specificity of 90% (Herreros-Villanueva et al., 2019).
Recently, Duran-Sanchon et al. (Duran-Sanchon et al., 2020) proposed a new miRNA-based model to discriminate patients with advanced neoplasms (CRC and advanced adenomas) from those with non-relevant findings at colonoscopy. The authors conducted a study composed of four stages (discovery in tissue samples, technical validation in a subset of stool samples from patients of the discovery phase, clinical validation in an independent set of stool samples, and predictive modeling) and identified a panel of 3 miRNAs (miR-421, miR-130b-3p, and miR-27a-3p) that were significantly up-regulated in fecal samples obtained from participants with advanced neoplasms. Subsequently, they developed a predictive model by combining stool expression levels of miR-421, miR27a-3p, and hemoglobin. This model could identify patients with CRC with an AUROC of 0.93, sensitivity of 97%, and specificity of 43%, as compared with an AUROC of 0.67, sensitivity of 100%, and a specificity of 31% for fecal hemoglobin concentration alone. However, the accuracy of this model for identifying patients with advanced neoplasms was significantly lower when compared with participants who had non-relevant findings at colonoscopy (AUROC of 0.62, sensitivity of 62%, specificity of 58%) or normal colonoscopy findings (AUROC of 0.59, sensitivity of 43%, specificity of 63%). One of the major limitations of this study, together with the small sample size of the validation set (n = 189), was the fact that it included only individuals with a positive result from a FIT, and therefore, as the authors stated, further studies are needed in FIT-naive subjects.
Although saliva is a less explored bodily fluid for CRC diagnosis, it has recently gained attention as a potential biospecimen for non-invasive liquid biopsy because it is easy and safe to collect (Malathi, Mythili, & Vasanthi, 2014). Several studies have reported the existence of various salivary molecular indicators of local and systemic disorders, including cancer (Malathi et al., 2014). In this context, Sazanov et al. recently evaluated the expression levels of miR-21 in plasma and saliva samples from CRC patients (Sazanov, Kiselyova, Zakharenko, Romanov, & Zaraysky, 2017). miR-21 showed a plasma sensitivity and specificity of 65% and 85%, respectively, for discriminating between CRC patients (n = 34) and healthy controls (n = 34), and even greater sensitivity (97%) and specificity (91%) in saliva. The high sensitivity and specificity values for salivary miR-21 reflect the potential of this saliva-based test for CRC diagnostic purposes. Another study recently identified a set of five miRNAs (miR-186-5p, miR-29a-3p, miR-29c-3p, miR-766-3p, and miR-491-5p) that were significantly upregulated in saliva from CRC patients (Rapado-González et al., 2019). Interestingly, the combined analysis of these five miRNAs yielded an AUROC of 0.754, sensitivity of 72%, and specificity of 67% for differentiating CRC patients (n = 51) from healthy individuals (n = 37). Unfortunately, and in line with Sazanov et al. study (Sazanov et al., 2017), the authors did not explore the accuracy of the panel for identifying adenoma or adenoma or CRC. Furthermore, although both saliva-based studies showed promising results, they must still be evaluated in validation trials in larger retrospective and prospective cohorts.
In general, studies conducted to date on potential miRNA biomarkers share two main characteristics that limit their external validity—relatively small sample size and the lack of detailed clinical-pathological information on the CRC population evaluated in the study. These limitations must be addressed, and the miRNA biomarkers or panels evaluated in randomized prospective studies before they can be translated to the clinic.
Long non-coding RNAs (lncRNAs)
In recent years, lncRNAs have gained increasing attention as potential biomarkers for CRC. In contrast to miRNAs, the precise number of functional lncRNAs remains unclear, because of ongoing discovery of new lncRNAs. Despite promising early results as biomarkers, research on lncRNAs still focuses mostly on discovery of new lncRNAs and their functions. Studies exploring the potential of lncRNAs as biomarkers in large patient cohorts are still elusive. Although tissue-derived lncRNAs have been the primary analyte in the most studies, a few studies have also analyzed blood-derived lncRNAs to determine their potential as non-invasive biomarkers. The lncRNA HOTAIR has been analyzed in both serum and tissues and is upregulated in early stages of CRC (Svoboda et al., 2014; Zhao, Song, Zhang, Kuerban, & Wang, 2015). Along similar lines, upregulation of the lncRNA colon cancer-associated transcript 1 (CCAT1) in tumor tissue and blood also seems to be an early event during colorectal carcinogenesis (Alaiyan et al., 2013; X. He et al., 2014; Nissan et al., 2012; Ozawa et al., 2017). Evaluation of HOTAIR and CCAT1 in a panel combining both markers revealed that upregulated expression of both markers had greater sensitivity and specificity for diagnosing CRC in plasma samples than of either lncRNA separately (Zhao et al., 2015). These two lncRNAs were also recently evaluated in a stool-based lncRNA extended panel (Gharib et al., 2020). The study population consisted of 150 participants, including a training and a validation set, and a group of 30 subjects with colon polyps. Expression levels of lncRNAs were evaluated by quantitative real-time PCR. To design a predictive panel, the investigators selected 10 significantly dysregulated lncRNAs, including CCAT1, CCAT2, H19, HOTAIR, HULC, MALAT1, PCAT1, MEG3, PTENP1, and TUSC7. The diagnostic performance of the panel in terms of distinguishing CRC as compared to healthy tissue yielded an AUROC of 0.8554 in the training set and 0.8465 in the validation set. The panel’s AUROC for early-stage CRC (I-II TNM stages) was 0.7871 in the training set and 0.8121 in the validation set, and for advanced CRC (III-IV TNM stages) the AUROC was 0.9281 in the training set and 0.9236 in the validation set. The corresponding AUROC values for CRC vs. colon polyp were 0.9228 (I-IV TNM stages), 0.9042 (I-II TNM stages), and 0.9362 (III-IV TNM stages). Again, in addition to the small sample size of the cohort included in the study, there is a lack of information regarding the clinical-pathological characteristics of the polyps analyzed in the study beyond the sex and age of the corresponding subjects.
Histone modifications
Although some studies have evaluated histone modifications as non-invasive diagnostic biomarkers, the data are still preliminary and further studies are required to determine the feasibility and utility of this approach. Methylation levels of histone 3 at lysine 4 are higher in CRC and in adenomas than in normal colonic mucosa, and acetylation levels of histone 3 at lysine 27 and histone 4 at lysine 12 are markedly greater in CRC compared to normal colonic mucosa (Ashktorab et al., 2009; Karczmarski et al., 2014; Nakazawa et al., 2012). In an attempt to identify non-invasive biomarkers and to take advantage of the stability of nucleosomes within the circulation, some studies have demonstrated the potential of using histone modifications in circulating nucleosomes as CRC diagnostic biomarkers. Chromatin immunoprecipitation studies revealed that reduced levels of trimethylation of histone 3 at lysine 9 and of histone 4 at lysine 20 in circulating nucleosomes were present in patients with CRC as compared with healthy control individuals (Gezer et al., 2013, 2015). Although histone modifications have a central role in cancer pathogenesis, their lack of cancer-specificity and various technical limitations associated with their use as quantitative analytes are, to date, some of the major obstacles to their use as non-invasive biomarkers.
DNA methylation
One of the first epigenetic alterations to be discovered in human cancer was the low level of DNA methylation in tumors as compared to normal tissue (Feinberg & Vogelstein, 1983). The loss of methylation is primarily due to hypomethylation of repetitive DNA sequences (e.g., LINE, SINE and Alu elements) and demethylation of coding regions and introns (Feinberg & Tycko, 2004). During carcinogenesis, the degree of hypomethylation of genomic DNA increases as a lesion progresses from a benign proliferation of cells to an invasive cancer (Fraga et al., 2004). In addition to hypomethylation of genomic DNA, hypermethylation of specific CpG islands in the promoter regions of tumor-suppressor genes is also a major event in the origin of many cancers. All of these changes in DNA methylation modulate gene expression: gene silencing via hypermethylation of CpG islands in promoters, gene activation via hypomethylation of CpG-poor gene promoters, and oncogene overexpression via hypermethylation of gene bodies (Yang et al., 2014).
DNA methylation profiling provides several advantages over somatic mutation analysis for detecting cancer, including greater clinical sensitivity and dynamic range, multiple detectable methylation target regions, greater prevalence in precancerous lesions and early stage cancers, and presence of multiple altered CpG sites within each targeted genomic region (Heyn & Esteller, 2012; Laird, 2003). Importantly, altered methylation is also seen as a field effect in colonic mucosa that predisposes normal tissue to neoplastic transformation (Luo, Yu, & Grady, 2014). In addition, the alterations in CpG methylation are relatively constant in each type of cancer, whereas there are usually no predominant somatic mutations. Despite the relatively high frequency of somatic mutations in cancers, the patterns of mutations are highly heterogeneous in individual patients, making somatic mutations less than ideal markers for early detection of cancers (Kandoth et al., 2013). Given favorable characteristics of DNA methylation markers and strong supporting evidence of their utility as biomarkers, several assays using DNA methylation biomarkers have been commercialized during the last decade, are used in current clinical practice, or have even entered clinical guidelines.
One of the most widely studied non-invasive DNA methylation biomarkers for CRC diagnosis is methylation of the SEPT9 gene in plasma. SEPT9 encodes septin 9, a GTP-binding protein involved in actin dynamics, cytoskeletal remodeling, vesicle trafficking, and exocytosis. Multiple studies have evaluated the diagnostic accuracy of this methylation biomarker in large cohorts of patients with CRC, and in these studies sensitivity and specificity ranged from 48% to 90% and 73% to 97%, respectively (Bergheim et al., 2018; Church et al., 2014; Fu et al., 2018; Song, Jia, Peng, Xiao, & Li, 2017; Song, Peng, et al., 2017; Tänzer et al., 2010; Wu et al., 2016). In 2016, this biomarker (commercialized as the Epi proColon test by Epigenomics) was approved by the US FDA as the first molecular blood-based assay for CRC screening. This test is not recommended for routine CRC screening, and represents an alternative only for individuals who refuse other screening modalities such as FOBT, FIT, flexible sigmoidoscopy, or colonoscopy. In one of the largest studies in which SEPT9 plasma methylation was analyzed using Epi proColon test 2.0 methodology, this biomarker yielded an overall sensitivity of 73.7% and specificity of 97% in a large cohort (300 patients with CRC and 568 healthy control individuals) (He et al., 2018). However, findings of both this study and a 2017 meta-analysis agree that this test performed statistically significantly better in patients with advanced stage (III-IV) CRC than in those with early-stage (I–II) CRC (He et al., 2018; Song, Jia, et al., 2017). The Epi proColon test requires only a 10 ml blood sample, out of which ~3.5 ml of plasma is used to obtain free circulating DNA. Methylated SEPT9 DNA within the plasma is amplified via PCR, along with β-actin as an internal control. Because this screening test only requires a blood draw, which can be performed in conjunction with other routine laboratory exams, this test is attractive to patients and has high compliance rates.
In an effort to further improve the diagnostic accuracy of Epi proColon test, which, as noted above, is suboptimal for early-stage CRC, other groups have attempted to combine SEPT9 plasma methylation levels with additional biomarkers, such as FIT or other plasma-based methylated genes (for example, SHOX2 and ALX4) (Bergheim et al., 2018; Tänzer et al., 2010; Wu et al., 2016). Wu et al. evaluated a combination of SEPT9 plasma methylation, FIT and serum carcinoembryonic antigen (CEA) levels for CRC screening that included all CRC stages, and achieved an overall sensitivity of 97.2% (Wu et al., 2016). Although the majority of the data for this test were obtained from case–control or cohort studies, SEPT9 plasma methylation has also been analyzed in asymptomatic, intermediate-risk populations of healthy individuals to further assess its diagnostic potential as an alternative to FIT or colonoscopy (Church et al., 2014). However, when Song et al. (Song, Jia, et al., 2017) compared the data from this study with that from a meta-analysis of FIT in a similar population, they found lower sensitivity (68% vs. 79%) and specificity (80% vs. 94%) for SEPT9 plasma methylation than for FIT. One of the major limitations of the SEPT9 plasma methylation biomarker is its poor sensitivity (ranges from 7.9% to 38.7%) for identifying precursor lesions (adenomas) (Fu et al., 2018; He et al., 2018; Song, Peng, et al., 2017; Wu et al., 2016). SEPT9 plasma methylation levels exhibited the highest sensitivity (83.3%) in a subgroup of patients with villous adenoma, suggesting this biomarker has potential for identifying advanced adenomas (Song, Peng, et al., 2017). However, the number of patients included in this study was small (n = 18), highlighting the need for the development of more ambitious approaches for detecting precancerous lesions and early-stage CRC (Song, Peng, et al., 2017).
Methylation of the VIM gene has also been evaluated as a non-invasive biomarker for CRC diagnosis. VIM encodes the intermediate filament protein vimentin, which, together with microtubules and actin microfilaments, constitutes the cytoskeleton. The diagnostic accuracy of VIM methylation for CRC appears be greater for stool samples than for blood specimens. For example, the sensitivity and specificity of VIM methylation were 81% and 95%, respectively, in stool samples, with similar values seen across CRC stages (Baek et al., 2009; Carmona et al., 2013; W.-D. Chen et al., 2005; Fu et al., 2018; Itzkowitz et al., 2007; Kisiel et al., 2013; Li et al., 2009; H. Lu et al., 2014). In contrast, in plasma samples, VIM methylation had a sensitivity and specificity of 59% and 93%, respectively, with substantially greater sensitivity in advanced disease stages(Li et al., 2009). Given the performance of VIM methylation in stool samples, this biomarker has been commercialized as the ColoSure test (LabCorp) (Ned, Melillo, & Marrone, 2011), but has not yet obtained US FDA clearance or approval for use as a CRC screening test.
In contrast to VIM methylation, methylation of secreted frizzled-related protein 2 (SFRP2) has shown more promising results for adenomas. SFRP2 methylation has been studied for detection of precancerous lesions (both adenomas and hyperplastic polyps). The sensitivity of plasma-based SFRP2 methylation for detecting adenomas ranged from 6.4% to 81.1%, and the specificity from 73% to 100% (Barták et al., 2017; Tang et al., 2011), while the sensitivity and specificity of stool-based SFRP2 methylation ranged from 27.8% to 76% and from 55% to 100%, respectively (Glöckner et al., 2009; Park et al., 2017; Zhang, Zhu, Wu, Zhang, & Qi, 2014).
As high-throughput tools for molecular development continue to improve, new studies are emerging to evaluate the potential of DNA methylation biomarkers through the use of more comprehensive approaches. In 2020, Luo et al (Luo et al., 2020) identified CRC-specific methylation signatures by comparing CRC tissues to normal blood leukocytes. Using machine learning algorithms, the authors developed a predictive diagnostic model (cd-score) by using cell-free DNA (cfDNA) samples from a cohort of 801 patients with CRC and 1021 normal controls. In the discovery dataset, the cd-score discriminated patients with CRC (n = 528) from normal controls (n = 674) with an AUROC of 0.96, sensitivity of 87.5%, and specificity of 89.9%. This accuracy was confirmed in a validation dataset of 273 CRC patients and 347 normal controls, yielding an AUROC of 0.96, sensitivity of 87.9%, and specificity of 89.6%. With the goal of making screening methods for cancer as simple as possible, the authors prospectively investigated the efficiency of using the methylation status of CpG site cg10673833 for detecting CRC as well as precancerous lesions in high-risk populations. To identify an appropriate population, all enrolled participants (n = 16,890) were invited to take a cancer risk assessment using an established Clinical Cancer Risk Score System (H. Chen et al., 2019). This identified 1493 participants (ages range: 45–75 years) who were considered to be at high risk for CRC. These high-risk subjects were subsequently scheduled to undergo screening colonoscopy and were recruited into the study to undergo methylation profiling at the time of the screening. The cg10673833 methylation test identified 19 of 21 participants with CRC and 7 of 8 participants with CRC in situ (diagnosis as high-grade dysplasia), with an AUROC of 0.90, sensitivity of 89.7%, and specificity of 86.8%. The positive predictive value and negative predictive value were 0.118 (95% CI, 0.101 to 0.138) and 0.998 (95% CI, 0.993 to 0.999), respectively. For advanced precancerous lesions, the sensitivity was 33.3%, much higher than the positivity rate for subjects who did not have CRC or advanced precancerous lesions (12.1%).
More recently in 2020, Liu et al. (Liu et al., 2020) reported the results of a prospective case-controlled substudy from the Circulating Cell-free Genome Atlas (CCGA; NCT02889978) and the STRIVE (NCT03085888) projects. This substudy assessed the ability of targeted methylation analysis of circulating cfDNA to detect and localize multiple cancer types across all stages at high specificity. The CCGA study was designed to determine if genome-wide cfDNA sequencing in combination with machine learning algorithms could detect and localize a large number of cancer types at sufficiently high specificity to be considered for a general population-based cancer screening program. In previous discovery work in a first CCGA substudy, whole-genome bisulfite sequencing to interrogate genome-wide methylation patterns outperformed whole-genome sequencing and targeted sequencing approaches to interrogate copy-number variants and single-nucleotide variants (SNVs)/small insertions and deletions, respectively) (Liu et al., 2018; Oxnard et al., 2019). In addition, targeted sequencing with SNV-based classification was significantly confounded by clonal hematopoiesis of indeterminate potential (Swanton et al., 2018), suggesting that such a test would require concurrent sequencing of white blood cells to return accurate results. The cfDNA methylation-based classifier achieved consistently high overall specificity between the cross-validated training and independent validation sets (>99%) across all cancer types (>50) with a sensitivity of ~55%. Incorporation of clinical baseline demographic information and blood sample quality metrics to the model did not sufficiently improve sensitivity (<10%). Although this study did not evaluate the accuracy specifically across CRC stages, both the training and validation cohorts included patients with CRC (training, n = 122 with CRC; validation, n = 53 with CRC) among all the cancer types included (Liu et al., 2020). Because not all participants with cancer were asymptomatic, additional studies in an asymptomatic screening population are currently ongoing.
Similar efforts to identify methylation-based biomarkers are ongoing that are specifically focused on gastrointestinal (GI) cancers. One of the most promising tests is the EpiPanGI-Dx (Kandimalla et al., 2020), a cfDNA methylation fingerprint for the early detection of several GI cancers, including CRC, esophageal squamous cell and adenocarcinoma (ESCC and EAC), gastric cancer (GC), hepatocellular carcinoma (HCC), and pancreatic ductal adenocarcinoma (PDAC). Using a tissue-based genome-wide DNA methylation analysis of these GI cancers to select the most informative differentially methylated regions (DMRs), Kandimalla et al. (Kandimalla et al., 2020) developed a novel three distinct categories of DMR panels and sequenced 300 plasma specimens from all GI cancers, as well as age-matched healthy controls, with 40X coverage. They found 1) cancer-specific biomarker panels with AUROC values of 0.98 (CRC), 0.94 (ESCC), 0.90 (EAC), 0.90 (GC), 0.98 (HCC), and 0.85 (PDAC); 2) a pan-GI cancer biomarker panel that detected all GI cancers with an AUROC of 0.90; and 3) a multi-cancer prediction panel, EpiPanGI Dx, with a prediction accuracy around 0.85 for most GI cancers. All three groups of DMR panels, after being trained and tested using the cfDNA cohorts, achieved excellent diagnostic accuracy as indicated by AUROC values ranging from 0.74 to 0.98, even for each of the early-stage GI cancers. This study represents the first specific GI cancer cfDNA methylation test that has potential to be applied in the clinic for the early detection of all GI cancers.
Recently, Chen et al. (Chen et al., 2020) described a new blood-based cancer diagnostic test based on cfDNA methylation analysis, the PanSeer assay. Using publicly available microarray and whole genome bisulfite sequencing data from The TCGA and genomic regions known to be cancer-related in the literature, as well as internal reduced representation bisulfite sequencing data from a variety of cancer tissues, the authors compiled a targeted panel of 595 genomic regions for further interrogation in plasma samples. Because tumor DNA tends to be rare in plasma, especially in patients with early stage cancer, and because conventional methods for constructing sequencing libraries that incorporate bisulfite conversion and double-stranded ligation typically have a high DNA loss rate (Aigrain, Gu, & Quail, 2016), the authors used the Singlera library construction method. This method uses semi-targeted PCR, which requires only a single ligation event and a single PCR primer per amplicon, allowing single-molecule counting at a higher molecular recovery rate than conventional methods (Gansauge & Meyer, 2013; Zheng et al., 2014). Preliminary results of PanSeer on plasma samples from 605 asymptomatic individuals, 191 of whom were later diagnosed with stomach, esophageal, colorectal, lung, or liver cancer within four years of blood draw, demonstrated an accuracy of cancer diagnosis of 95%. Using a similar approach, another plasma-based assay of circulating tumor methylated DNA (ColonES, Singlera Genomics) showed impressive preliminary results, with sensitivities of 91%, 97%, and 94% for detecting adenoma, stage I CRC, and stage II-IV CRC, respectively (Singlera Genomics, 2020). The reported preliminary specificity of the test is 99%. Although additional studies are needed in larger population-based cohorts, DNA methylation genome-wide approaches are leading candidates for biomarker panels and seem to be the future for molecular blood-based diagnostics for CRC and many other tumor types.
Multi-modal approach
Although epigenetic alterations are among the most well-characterized CRC diagnostic biomarkers, considering that CRC evolves through acquisition of many genetic and epigenetic alterations, combinations of multiple types of molecular and cellular biomarkers have also been evaluated to see if they enable more accurate detection of colorectal polyps and CRC. Indeed, several promising combinations have been proposed to improve diagnostic performance. Among the tests that combine different biological approaches, currently, only Cologuard (Exact Sciences), a stool-based multi-target panel, is approved by the US FDA for CRC screening. Cologuard tests three genomic biomarkers (seven point mutations in KRAS and methylation status of NDRG4 and BMP3) and includes a quantitative enzyme-linked immunosorbent assay for the presence of hemoglobin. In a study of ~10,000 intermediate-risk individuals, Imperiale et al. (Imperiale et al., 2014) found that Cologuard had a significantly higher sensitivity for CRC detection compared to FIT (92.3% vs. 73.8%). More importantly, the sensitivity for detecting advanced precancerous lesions (advanced adenomas or sessile serrated lesions ≥1 cm) was almost two-fold higher for Cologuard than for FIT (42.4% vs. 23.8%). However, this increased sensitivity came at the cost of lower specificity (89.8% [Cologuard] vs. 96.4% [FIT]) in patients with negative colonoscopy (Imperiale et al., 2014). Given these promising results, Cologuard has been included in the United States Preventive Services Task Force and National Comprehensive Cancer Network (NCCN) guidelines as a CRC screening option at 3-year intervals, which is on equal standing with the other traditional screening options (colonoscopy, FIT, and FOBT) (USPSTF). Ongoing trials are prospectively evaluating the utility of Cologuard in other scenarios, such as longitudinal testing in an average-risk population at 3-year intervals (NCT02419716), in an average-risk population aged 45 to 49 (NCT03728348), and with FIT to model stool-based molecular surveillance approaches to inform health policy decisions (NCT02715141).
At the 2020 ASCO Virtual Scientific Program, Friedland et al. (Friedland et al., 2020) presented interim results of a study of a new promising diagnostic test, the FirstSight assay. FirstSight is a blood-based assay that evaluates three biomarkers: 1) circulating GI epithelial cells, 2) validated somatic oncogene and tumor suppressor mutations, and 3) methylation of SEPT9 in cfDNA. Based on results for these three biomarkers, the assay calculates a diagnostic score (CMx Score). FirstSight was evaluated in 354 patients who had no prior history of CRC and were scheduled to undergo a colonoscopy. Eighty-six percent of the patients were asymptomatic and 14% had reported symptoms or a positive-FIT result. Prior to colonoscopy, patient blood samples were analyzed using the FirstSight assay. In this study, FirstSight achieved specificity of 90% and sensitivity of 100% for detecting CRC and sensitivity of 75.5% for detecting advanced adenomas. Overall, the test had a sensitivity of 79.7% for detecting advanced adenomas and CRC. In addition, there was a significant association between CMx score and polyp size (F value = 5.80, p-value = 0.017), but not for DNA mutation (F value = 1.29, p-value = 0.263) or methylation status (F value = 0.34, p-value = 0.560) and polyp size. Similarly, there was a significant association between CMx score and the number of polyps (F value = 23.71, p-value < 0.0001), but again not for DNA mutation (F value = 1.57, p-value = 0.210) or methylation status (F value = 1.34, p-value = 0.248) and the number of polyps. These results suggest that CMx score, which incorporates circulating epithelial cells, provided predictive information for polyp sizes and number above and beyond DNA mutation and methylation status alone.
Analysis of cancer-associated mutations in cfDNA has been also explored in conjunction with plasma protein analysis. In 2018, Cohen et al. (Cohen et al., 2018) reported on a new blood test, called CancerSEEK, for cancer diagnosis. CancerSEEK evaluates plasma levels of eight proteins (cancer antigen 125, CEA, cancer antigen 19-9, proactin, hepatocyte growth factor, osteopontin, myeloperoxidase, and tissue inhibitor of metalloproteinases) together with the presence of mutations (SNVs or INDELs) in 1933 distinct genomic sites. The presence of a mutation in an assayed gene or elevated levels of any of these proteins would classify a patient as positive. In this study, 1005 patients with nonmetastatic, clinically detected cancers of the ovary, liver, stomach, pancreas, esophagus, colorectum, lung, or breast were evaluated using CancerSEEK. The median sensitivity of CancerSEEK among the eight cancer types evaluated was 70% (78% for stage III, 73% for stage II, and 43% for stage I cancers). The specificity of CancerSEEK was >99% across cancers of all stages. Moreover, the test could localize the anatomic site of origin of the cancer in a median of 83% of the patients. Given that driver gene mutations are not usually tissue-specific, the vast majority of the localization information was derived from protein markers. The accuracy of prediction for primary cancer prediction varied by tumor type, but the test was most accurate for CRC (84%).
Although it is beyond the scope of this article, many other approaches are being evaluated for diagnosing polyps and CRC in various bodily fluids. For example, PolypDx (Deng et al., 2017), is a metabolomic-based urine test for detecting adenomas. Although PolypDx was originally designed on a nuclear magnetic resonance platform, it was subsequently developed as a mass spectrometry-based urine metabolomic test to detect three metabolites: succinic acid, ascorbic acid, and carnitine. Detection of these metabolites used in combination with three clinical features (age, sex, and smoking status) yielded higher sensitivity (43%) and similar specificity (91%) to two different FITs (sensitivities of 18% and 21%, and specificities of 97% and 92%, respectively) for detecting hyperplastic polyps and adenomas (Deng et al., 2017).
Primary tumor of origin
Tissue-based markers are also being developed to guide CRC diagnosis, and there are various molecular platforms that can help define the primary tumor of origin in cases classified as cancer of unknown primary (CUP). Initial work to identify original tumor sites was performed using algorithms based on genome-wide gene expression profiles. This work achieved an accuracy of 88% for primary tumors within 56 categories and of 78% for CUP (Ojala, Kilpinen, & Kallioniemi, 2011). However, despite an accuracy rate of 78.6% for site prediction in silico, results suggested that comprehensive genome-wide profiling of gene expression by microarray analysis might not yet be suitable for clinical application in patients with CUP. Currently, two assays, a 92 gene RT- PCR assay and a 64 tissue-specific miRNA assay are commercially available for prediction of tumor sites of origin (Erlander et al., 2011; Meiri et al., 2012). Although comprehensive gene expression profiling can be influenced by irrelevant variants, and dimension reduction is necessary because of the large number of variables, the 92 gene RT-PCR assay might be more informative as a result of the limited number of biologically relevant genes on which it is based (Ma et al., 2006). Other methods that are based on miRNAs, which can regulate the expression of large numbers of protein-coding genes, also have achieved a high accuracy rate (Rosenfeld et al., 2008; Varadhachary et al., 2011). In this regard, recently a miRNA-based tissue signature has been proposed for the specific diagnosis of CRC with mucinous differentiation (Ruiz-Bañobre et al., 2020). Although data are still very preliminary and validation studies are needed, this signature could be especially useful for classifying CRC in those cases where a small tumor biopsy is not able to capture the whole histological nature of the tumor (Moran et al., 2016).
Classifiers of cancer type based on DNA methylation profiles, which can overcome the inherent limitations of working with RNA in formalin-fixed paraffin-embedded (FFPE) tissue samples, are also being developed. One such platform, the EPICUP, showed a specificity of 99.6%, a sensitivity of 97.7%, a positive predictive value of 88.6%, and a negative predictive value of 99.9% in a validation set of 7691 tumors (Moran et al., 2016). DNA methylation profiling predicted a primary cancer of origin in 188 (87%) of 216 patients with CUP. In addition, patients who received a tumor type-specific therapy consistent with their EPICUP diagnoses showed improved OS compared with those who received empiric therapy. Although this approach shows promise for achieving a real impact in daily clinical practice, the efficacy of any given assay must be demonstrated in randomized clinical trials, a scenario that has been a serious limitation in the majority of previous studies (Hainsworth et al., 2012).
PROGNOSTIC AND PREDICTIVE BIOMARKERS
During recent decades, in parallel with the development of anti-neoplastic strategies, substantial efforts have been devoted to identifying biomarkers that can help in decision-making in the clinic. Although biomarkers likely have complex associations with patient outcomes, robust discrimination between prognostic and predictive biomarkers is challenging in clinical studies that aim to evaluate therapeutic benefits. To demonstrate that a biomarker is predictive of treatment benefit, biomarker status for all patients must be obtained, including patients who were treated with the agent of interest and untreated patients, preferably in the context of a randomized study. A formal statistical test of the treatment-by-biomarker interaction should be significant. To establish whether a marker is purely prognostic, it must be shown that there is significant association between the biomarker and outcome, regardless of treatment, and that treatment effects do not depend on the biomarker. Finally, a biomarker may have both predictive and prognostic implications (Ballman, 2015). This prognostic-predictive complexity is partly driven by the search for more effective therapies for patients who have a poor prognosis if treated with standard therapies (Sveen, Kopetz, & Lothe, 2020). Therefore, genetic alterations that are classically associated with CRC that has a poor prognosis are now targets of some of the most promising targeted-therapies and, consequently, are also considered response prediction biomarkers for these cancers. Because the number of prognostic and/or predictive biomarkers is rising, in this article we have chosen to focus on biomarkers that are already established in clinical practice or those with a very promising future. Our discussion is structured to follow the typical clinical path, from the adjuvant to the palliative setting.
Adjuvant setting
Regarding biomarkers used in the adjuvant setting, only MSI-H/dMMR is thus far established as a useful prognostic indicator. The first reports of a favorable prognostic association of MSI in CRC were published in 1993 (Lothe et al., 1993; Thibodeau, Bren, & Schaid, 1993). This association has since been confirmed for OS and disease-free survival by metanalyses comparing data from 1277 (Popat, 2004) and 12782 (Guastadisegni, Colafranceschi, Ottini, & Dogliotti, 2010) patients with primary MSI-H CRCs to those with MSS CRCs. Retrospective analyses of data from randomized trials showed this association also applies to patients with stage II and III colon cancer patients who have not received adjuvant chemotherapy (Hutchins et al., 2011; Sargent et al., 2010). A corresponding association of MSI with clinicopathological features has also been reported for prognosis; patients with proximal tumors have a greater favorable prognostic association of MSI than those with distal tumors (Sinicrope et al., 2013). Various studies report stronger effects of MSI in stage II CRC (Klingbiel et al., 2015; Merok et al., 2013), similar prognostic associations across stages II and III CRC (Benatti et al., 2005; Sinicrope et al., 2011), or an even a stronger effect in stage III CRC (Samowitz et al., 2001). However, no statistically significant interactions with clinicopathological features, including tumor stage, were observed in a pooled analysis of data from 7,326 stage II or III colon cancers (Dienstmann et al., 2017), suggesting that the prognostic value of MSI is independent of disease stage.
The prognostic implications of MSI after adjuvant treatment might be confounded by also having a predictive value for 5-FU-based chemotherapy, thus illustrating prognostic–predictive biomarker complexity (Vilar & Gruber, 2010). Loss of MMR function could result in failure to recognize and respond to incorporation of 5-FU into tumor DNA (Jo & Carethers, 2006). However, many retrospective analyses have shown inconsistent results regarding the effects of 5-FU-based regimens in patients with MSI-H tumors (Vilar & Gruber, 2010). Most studies that compared the effects of 5-FU-based chemotherapy to no treatment failed to show significant improvements in OS or disease-free survival (DFS) in patients with MSI-H/dMMR tumors, even though 5-FU-based chemotherapy significantly improved the outcomes of patients with MSS or MMR-proficient colon cancers or CRCs (Benatti et al., 2005; Carethers et al., 2004; Hutchins et al., 2011; Lanza et al., 2006; Sargent et al., 2010). Only two of the seven studies revealed a statistically significant interaction between MSI status and the response to chemotherapy (Jover et al., 2009; Ribic et al., 2003). A meta-analysis including data from 396 patients with MSI-H CRCs revealed a substantial degree of heterogeneity with respect to the effects of 5-FU-based chemotherapy, and a lack of benefit could not be definitively confirmed (Guastadisegni et al., 2010).
MSI status is not predictive of a lack of benefit from the combination chemotherapies typically used in patients with CRC. The addition of oxaliplatin to 5-FU-based adjuvant chemotherapy regimens improves patient survival (André et al., 2004), and this effect is also seen among patients with dMMR stage II or III CRC (Cohen et al., 2020; Flejou et al., 2013; Tougeron et al., 2016). Furthermore, patients with dMMR tumors have better survival outcomes after treatment with FOLFOX compared with those with MMR-proficient tumors (in one study the association was tumor location dependent) (Gavin et al., 2012; Sinicrope et al., 2013, 2014; Zaanan et al., 2011), consistent with the favorable prognostic effect of MSI. Paradoxically, metastatic MSI-H tumors are aggressive and are associated with inferior PFS and OS outcomes relative to metastatic tumors for MSS CRC (Heinemann et al., 2018; Kim et al., 2016; Tran et al., 2011; Venderbosch et al., 2014). Although other reports indicate that no prognostic associations exist and additional data are needed, the survival benefits associated with primary MSI-H CRCs seem to be lost in the metastatic setting (Jin et al., 2018; Margonis et al., 2018). On the other hand, although neoadjuvant therapy is not currently a standard therapeutic strategy in CRC, the combination of nivolumab plus ipilimumab has yielded encouraging results in CRC patients in this setting, especially among those with MSI-H/dMMR tumors. In a recent study reporting the results of a phase II NICHE trial (NCT03026140) (Chalabi et al., 2020), among 35 patients who received both nivolumab and ipilimumab and were evaluable for efficacy (two patients with pMMR CRCs were deemed ineligible post-surgery), 100% of those with dMMR CRC and 27% with pMMR CRC had pathologically-relevant responses. The majority of responders (19/20 with dMMR CRC and 3/15 with pMMR CRC) had major pathologically relevant responses, including 12 complete responses. Subsequent organoid-based investigations suggested that the lower response rates of patients with pMMR CRCs reflected a lack of highly immunogenic T cell antigens, as opposed to other tumor-intrinsic factors (Chalabi et al., 2020).
In 2012, Ebert et al. (Ebert et al., 2012) proposed the epigenetic biomarker methylation of TFAP2E in tumor tissue specimens as a promising negative predictive biomarker for 5-FU-based chemotherapy. The authors suggested the lack of response to 5-FU was probably mediated by DKK4, a downstream effector of TFAP2E that is implicated in chemoresistance to 5-FU in CRC cell lines (Xi, Formentini, Nakajima, Kornmann, & Ju, 2008; Xi, Nakajima, Schmitz, Chu, & Ju, 2006). Although the expectations were high, this study presented various important limitations for its clinical usefulness. Primarily, it interrogated a relatively small cohort of patients with CRC (n = 220), which was actually a combined collection of patients with non-metastatic and mCRC from four different prospective trials that were analyzed together as one large cohort. Second, only a very small subset of the entire cohort was analyzed for methylation and expression of TFAP2E, as well as expression of the DKK4 protein. Third, although all patients received 5-FU–based chemotherapy, their regimens differed and also included either oxaliplatin, irinotecan and/or cetuximab, or radiotherapy (chemoradiotherapy). Unfortunately, in a subsequent study the negative predictive potential of TFAP2E was not confirmed (Murcia et al., 2018). Although methylation of TFAP2E intron 3 is tumor-related, it did not correlate with loss of TFAP2E protein expression, and, more importantly, TFAP2E methylation did not play any role in predicting response to 5-FU-based chemotherapy in patients with CRC.
In January 2016, Dalerba et al. (Dalerba et al., 2016) reported a novel approach to the problem of identifying patients with CRC who might benefit from adjuvant chemotherapy. They reasoned that the presence of a stem cell-like state could be associated with more aggressive tumors, and performed a bioinformatic search for a gene-expression signature obtained from populations of stem cells and progenitor cells. By mining a large, preexisting database of CRCs, the authors identified a panel of 16 genes for which expression was inversely related to the stem cell-like state. CDX2 was the most clinically actionable of these genes because it could be detected using immunohistochemical analysis. The investigators performed a series of validation analyses involving multiple independent data sets, which is a necessary approach for data-mining research. The first analysis confirmed an inverse relationship between CDX2 expression and patient outcomes in which CDX2-negative tumors (present in 6.9% of patients in the discovery data set) were associated with significantly lower rates of 5-year DFS compared to CDX2-positive tumors (41% vs. 74%). A validation dataset was created by immunohistochemical analysis to confirm CDX2 protein expression. Analysis of the validation dataset, in which 13% of the patients had CDX2-negative tumors, confirmed 5-year survival rates of 48% among patients with CDX2-negative tumors vs. 71% among patients with CDX2-positive tumors. However, this finding was not sufficient to prove that the subgroup of patients with a worse natural history would benefit from adjuvant chemotherapy; instead they might be less responsive to treatment. To address this, the investigators focused on patients with stage II CRC and confirmed that CDX2-negative cancers were associated with significantly lower rates of survival than were CDX2-positive cancers (48–51% vs. 80–87%). Finally, they used an expanded database to demonstrate that the benefit observed with the administration of chemotherapy in terms of disease-free survival in CDX2-negative cohorts was superior to that observed in CDX2-positive cohorts in both the stage II subgroup and the stage III subgroup. However, despite the rigorous bioinformatics analysis, the number of patients who had stage II CRC and CDX2-negative tumors was small, and so this result is not definitive. This retrospective study requires prospective confirmation with uniform interventions. In addition, the immunohistochemical analysis was performed on tissue microarrays, which facilitated rapid throughput but may have underestimated the heterogeneity of CDX2 expression throughout the tumor. Furthermore, these findings raise the question of what mechanism might be at work in silencing CDX2; the answer to this question could lead to the discovery of new approaches to treating the fundamental problem. Meanwhile, given these limitations, use of CDX2 has not entered clinical practice to indicate (or not) if adjuvant chemotherapy should be given to stage II CRC patients.
On the other hand, based on the cfDNA methylation analysis, Luo et al. (Luo et al., 2020) recently developed a combined score (cp-score) for the prognostic purposes in CRC. This score also includes clinical characteristics such as age, gender, primary tumor site, and TNM stage. Using machine learning algorithms and proportional hazards regression methods, the authors conducted a variable selection on the training set (n = 528, events = 157) and built a composite score on the validation set (n = 273, events = 77). Uni-Cox and LASSO-Cox methods were implemented to reduce the dimensionality, and a Cox-model was constructed for prognostication using a five-marker panel, which dichotomized the patients into high-risk and low-risk groups. To characterize the discrimination potential of the composite score, TNM stage, CEA levels, primary tumor location, and the combination of all the existing biomarkers, the investigators applied time-dependent receiver operating characteristic curves. Multivariate Cox regression analysis indicated that the cp-score was highly correlated with the risk of death and was an independent prognostic factor in both the training and validation sets. As expected, TNM stage, CEA status, and primary tumor location were also prognostic factors for survival of patients with CRC. An integrative model combining the cp-score and clinical characteristics demonstrated greater prognostic accuracy in both the training (AUROC of 0.82) and validation (AUROC of 0.87) cohorts. Finally, in an attempt to develop an easier-to-use tool, the authors constructed a nomogram with a point scale of the next four variables: cp-score, CEA concentration, TNM stage, and primary tumor location.
The possibility that ctDNA could be a useful prognostic marker for minimal residual disease was also been suggested for a small series of advanced CRC patients who underwent resection of liver metastases (Diehl et al., 2008). This approach has been subsequently demonstrated in localized CRC (Reinert et al., 2019; Jeanne Tie et al., 2020, 2016; Jeanne Tie, Cohen, Wang, Christie, et al., 2019; Jeanne Tie, Cohen, Wang, Li, et al., 2019), and many ongoing clinical trials are evaluating the role of ctDNA status in adjuvant therapy decision-making and the impact on patient survival outcomes (Coakley, Garcia-Murillas, & Turner, 2019).
Finally, although it is not a genomic or epigenomic biomarker itself, Immunoscore, deserves specific attention in this section. Proposed for the first time in 2006 by Jerome Galon (Galon et al., 2006), Immunoscore was recently recognized in the current Localised Colon Cancer ESMO Clinical Guidelines as a useful tool for prognostication in the non-metastatic colon cancer (CC) setting. Immunoscore has been validated in a huge prospective cohort of 2,681 stage I-III CC patients, and was a strong predictor for time to recurrence, OS, and DFS, independent of patient age, sex, MSI, and other relevant prognostic factors. Moreover, Immunoscore showed the highest relative contribution to the risk of all clinical parameters, including the UICC TNM staging system (Pagès et al., 2018). Based on this data, the ESMO Clinical Guidelines concluded that Immunoscore could help refine the prognosis for colon cancer patients with early stage disease in conjunction with the TNM staging system (level of evidence III – prospective cohort studies, grade of recommendation C – insufficient evidence for efficacy or benefit does not outweigh the risk or the disadvantages, optional). Oppositely, the NCCN Colon Guidelines, to date, do not even mention Immunoscore, probably because of unclear evidence for its prognostic value for predicting the risk of recurrence and death in stage II or III CC separately or for its predictive role in predicting adjuvant chemotherapy benefit. In an attempt to clarify these questions specifically in stage III CC, the International Society for Immunotherapy of Cancer Immunoscore Consortium conducted a retrospective study to evaluate Immunoscore in 763 patients with TNM stage III CC from two retrospective cohorts: cohort 1 (Canada/United States) and cohort 2 (Europe/Asia) (Mlecnik et al., 2020). In this analysis, patients with a high Immunoscore also had the lowest risk of recurrence, and a significantly prolonged TTR, OS, and DFS. The association of Immunoscore with TTR was independent of major prognostic covariates such as sex, T stage, N stage, primary tumor location, and MSI status. Moreover, Immunoscore had the strongest contribution to survival risk for TTR and OS. Importantly, chemotherapy was significantly associated with survival in the high-Immunoscore group for patients with either low-risk or high-risk stage II CC as compared to the low-Immunoscore group (Mlecnik et al., 2020). These studies have paved the path for investigating the prognostic role of Immunoscore in larger prospective studies, and, even more importantly, in randomized clinical trials to evaluate its predictive potential for chemotherapy benefit.
Palliative setting
Although the backbone of treatment in patients with mCRC has historically been chemotherapy, over the last few decades targeted molecular therapies against EGFR and angiogenic factors have been introduced into daily clinical practice. Furthermore, new treatment options have recently been added to the mCRC armamentarium, including BRAF inhibitors (BRAFi) and anti-programmed cell death 1 (PD-1) antibodies. Historically, and in parallel with drug development, multiple research efforts have been undertaken to discover and implement molecular biomarkers to guide therapeutic strategies. This becomes even more important in today’s clinical scenario in which multiple therapeutic options are available, and therefore treatment selection aims not only to improve patient survival, but also to spare patients from unnecessary toxicity and reduce the economic burden of expensive treatments. In this section, we summarize the most relevant milestones achieved in the field of biomarkers for various treatments in mCRC (Figure 2) and provide insights into some of the important clinical and methodological aspects.
Figure 2. Predictive biomarkers for metastatic CRC treatment.
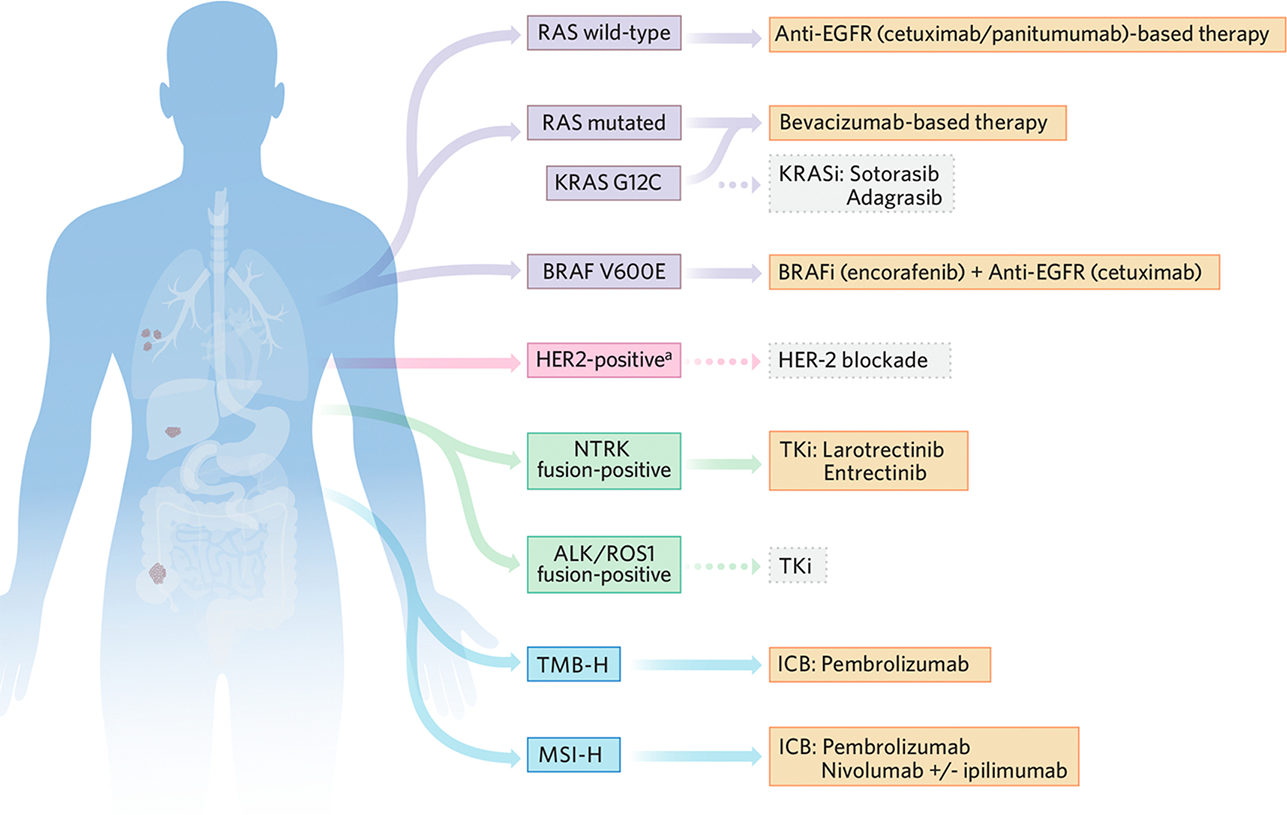
Summary of currently known molecular tumor alterations that enable improved clinical decision-making regarding use of therapies that are tailored to the metastatic CRC (mCRC) patient. The therapies shown go beyond classical chemotherapeutic agents. Excluding RAS mutations for EGFR-targeted therapies, which are considered a negative predictive biomarker, the molecular alterations shown represent positive predictors of benefit with the indicated targeted therapies. Although bevacizumab is considered a useful therapeutic option in combination with chemotherapy in mCRC independent of any particular molecular alteration, currently there is no consistent predictive biomarker to guide bevacizumab use. Although many of these molecular alterations are applicable only to a minority of mCRC patients, collectively these low-prevalence actionable characteristics support a new targeted therapeutic horizon for many patients. Color-coded boxes: yellow, US FDA-approved therapy; grey, not US FDA approved therapy.
aHER2 amplification or overexpression.
Abbreviations: BRAFi, BRAF inhibitors; CRC, colorectal cancer; ICB, immune checkpoint blockade; KRASi, KRAS inhibitors; MSI-H, microsatellite instability high; TKi, tyrosine kinase inhibitors; TMB-H, tumor mutation burden high.
Conventional Chemotherapy
The backbone of treatment in patients with mCRC has historically been chemotherapy, and several chemotherapeutic agents are now approved in this setting: fluoropyrimidines (fluorouracil [FU] and capecitabine), oxaliplatin, irinotecan, and, since 2015, trifluridine/tipiracil (TAS-102).
*Fluoropyrimidine-based chemotherapy and TAS-102 –
Several studies have investigated the predictive role of fluoropyrimidine metabolic pathway enzymes in response to FU-based therapies. Various retrospective and prospective studies of the role of thymidylate synthase (TS) in fluoropyrimidine-based therapy (primarily FU plus leucovorin) have yielded discordant results. In this regard, multiple studies have shown that low levels of TS expression in metastatic tumor tissues generally correlate with higher overall response rate (ORR) (Aschele et al., 2002; Aschele, Debernardis, Tunesi, Maley, & Sobrero, 2000; Cascinu et al., 2000; Etienne-Grimaldi et al., 2008). Surprisingly, such a correlation was not evident when TS levels were measured in primary tumor tissues (Aschele et al., 2000; Johnston et al., 2003). Similarly, low levels of TS and dihydropyrimidine dehydrogenase (DPD) in metastatic tumor tissues were associated with a favorable response to FU in patients with mCRC (Salonga et al., 2000); however, a subsequent report in 2006 failed to validate these findings (Smorenburg et al., 2006). Likewise, the role of thymidine phosphorylase as a predictive biomarker was investigated, but the results remain inconclusive (Gustavsson et al., 2009; Lindskog, Derwinger, Gustavsson, Falk, & Wettergren, 2014).
In 2009, a meta-analysis of five studies examining a total of 861 patients with mCRC concluded that, compared with MSS patients, MSI-H patients did not achieve a statistically significant better response rate to FU-based chemotherapy (Des Guetz, Uzzan, Nicolas, Schischmanoff, & Morere, 2009). Similarly, a study of the relationship between MSH2 gene expression and capecitabine efficacy in patients with mCRC revealed that observed that higher MSH2 expression was associated with a better response (Jensen, Danenberg, Danenberg, & Jakobsen, 2007).
In an attempt to identify noncoding RNA-based predictive biomarkers, a low expression of miR-143 was shown to be associated with improved ORR and progression-free survival (PFS) in patients treated with capecitabine (Simmer et al., 2015). Likewise, low expression of miR-429 correlated with improved 5-year disease-free survival and OS in patients with mCRC treated with FU-based chemotherapy (Dong, Cai, & Li, 2016).
On the basis of the results of the phase III RECOURSE (Study of TAS-102 in Patients With Metastatic Colorectal Cancer Refractory to Standard Chemotherapies) trial, the US FDA approved TAS-102 for patients with mCRC. In 2017, Suenaga et al. (Suenaga et al., 2017) analyzed genomic DNA extracted from 233 samples from three cohorts: an evaluation cohort of 52 patients who received TAS-102, a validation cohort of 129 patients who received TAS-102, and a control cohort of 52 patients who were treated with regorafenib. Single nucleotide polymorphisms of genes involved in homologous recombination repair (ATM, BRCA1, BRCA2, XRCC3, FANCD2, H2AX, and RAD51), and cell cycle checkpoints (ATR, CHEK1, CHEK2, CDKN1A, TP53, CHE1, PIN1, and PCNA) were analyzed by PCR-based direct sequencing. Interestingly, patients who harbored any G allele at the ATM rs609429 locus exhibited improved OS rates when compared with patients with a C/C variant (Suenaga et al., 2017).
*Oxaliplatin-based chemotherapy –
Several key genes involved in the nucleotide excision repair pathway have been explored as potential predictive biomarkers for treatment with oxaliplatin. The most notable studies in this area have been on the ERCC1 gene, but as reported based on results from the MAVERICC (Marker Evaluation for Avastin Research in CRC) trial (NCT01374425), intratumoral ERCC1 expression failed to predict response to oxaliplatin treatment (Parikh et al., 2019). The XRCC1 gene, a base excision repair modulator, has also been evaluated as a possible predictive biomarker for oxaliplatin treatment, and a polymorphism in this gene (XRCC1-839 Arg/Gln or Gln/Gln) correlated with worse ORR to treatment with FU/oxaliplatin (Stoehlmacher et al., 2001).
Several microRNAs (miRNAs) have also been assessed for predictive response potential to FU, leucovorin, and oxaliplatin (FOLFOX)- or capecitabine plus oxaliplatin (CAPEOX)-based regimens. In this treatment setting, high miR-625-3p and low miR-148a expression were associated with poor response (Rasmussen et al., 2013; Takahashi et al., 2012), whereas high miR- 126 microvessel density was associated with improved PFS (Torben Frostrup Hansen, Nielsen, Jakobsen, & Sorensen, 2013).
*Irinotecan-based chemotherapy –
The most notable marker studied as a potential predictive biomarker for irinotecan treatment is topoisomerase 1 (TOP1). The first large study in which the predictive power of TOP1 was evaluated used tumor tissue samples from the FOCUS (Fluorouracil, Oxaliplatin, CPT-11: Use and Sequencing; NCT00008060) trial and reported that moderate or high expression of TOP1 was associated with a significant gain in survival after irinotecan-based therapy (Braun et al., 2008). Unfortunately, these findings were not subsequently confirmed in an analysis of samples from 545 patients involved in the CAIRO (Capecitabine, Irinotecan, Oxaliplatin; NCT00312000) study, even though patients received similar treatment regimens and similar analytical approaches were used (Koopman et al., 2009).
A study of the role of genetic polymorphisms in the TDP1 and XRCC1 genes in response to irinotecan-based regimens, showed a positive correlation with improved ORR in patients with the TDP1 IVS12+79G.T and XRCC1 GGCC-G/GGCC-G genotypes (Hoskins et al., 2008). Aprataxin, a protein member of the histidine triad superfamily, has also shown potential ability to be used to discriminate responders from nonresponders. In a retrospective cohort of 128 patients treated with irinotecan-based chemotherapy, low expression of aprataxin correlated with improved disease control, PFS, and OS (Dopeso et al., 2010). Although studies of UGT1A gene polymorphisms and their predictive value for response to irinotecan treatment have yielded conflicting results (Cecchin et al., 2009; Dias, McKinnon, & Sorich, 2012), higher methylation levels of the BNIP3 gene in patients treated with irinotecan plus S1 correlated with lower response rates (Hiraki et al., 2010).
Anti-angiogenic Drugs
*Bevacizumab –
Since the introduction of bevacizumab for the management of patients with mCRC, substantial efforts have been made to discover predictive biomarkers for this anti-angiogenic drug. To date, the most promising approach evaluated the correlation between VEGF-A levels and clinical benefit from bevacizumab-based chemotherapy. A 2014 study reported higher ORR and improved PFS in patients with a low expression of VEGF-A in primary tumor specimens (Bruhn et al., 2014). Unfortunately, these encouraging results were not confirmed by the MAVERICC trial (Parikh et al., 2019). The possible predictive role of RAS was evaluated in a subset of 230 patients with mCRC treated with irinotecan, FU, and leucovorin (IFL) plus placebo, or IFL plus bevacizumab, in a phase III randomized clinical trial. In this study, only the subset of patients who had wild-type KRAS showed a significantly higher response rate in the bevacizumab arm (Hurwitz, Yi, Ince, Novotny, & Rosen, 2009).
*Regorafenib –
While most of the studies have explored biomarkers from a radiomic perspective, others have used tissue- and plasma-based approaches. As a potential tissue-based predictive molecular marker for regorafenib, Wong et al. (Wong et al., 2015) described a correlation between downregulation of p53 and phosphorylated-proline-rich AKT substrate and higher PFS and metabolic response, respectively. The authors also evaluated the association between total plasma circulating cfDNA at baseline and major efficacy outcomes and found a significant inverse correlation with PFS (p = 0.048). Moreover, patients with detectable KRAS mutation in plasma presented a worse PFS (p = 0.04).
Regarding RAS gene evaluated in circulating ccfDNA, a small retrospective study involving 21 CRC patients showed a clinically significant association between reduced rates of RAS mutation in plasma, as detected by digital droplet PCR, and improved PFS (p = 0.01) and OS (p = 0.06)(Khan et al., 2017). A prognostic score for regorafenib has also been developed based on combinatorial clinico-molecular models and the REBECCA (Regorafenib in Metastatic Colorectal Cancer: A Cohort Study in the Real-Life Setting) study. This prognostic score included the following parameters that were independently associated with poorer OS: high Eastern Cooperative Oncology Group performance status, a shorter time from initial diagnosis of metastases, an initial regorafenib dose of 160 mg, more than three metastatic sites, presence of liver metastases, and KRAS mutations (Adenis et al., 2016).
*Aflibercept and ramucirumab –
A 2015 study of 87 patients with mCRC who were enrolled on the phase II AFFIRM (Study of Aflibercept and Modified FOLFOX6 as First-Line Treatment in Patients With Metastatic Colorectal Cancer) trial and were treated with aflibercept plus mFOLFOX6 reported that high plasma levels of interleukin-8 at baseline, together with increase in these plasma levels during the course of treatment, correlated with shorter PFS (Lambrechts et al., 2015). In 2017, Tabernero et al. (Tabernero et al., 2018) evaluated the predictive potential of VEGF-D in a translational research study based on the RAISE (Ramucirumab Versus Placebo in Combination With Second-Line FOLFIRI in Patients With Metastatic Colorectal Carcinoma That Progressed During or After First-Line Therapy With Bevacizumab, Oxaliplatin, and a Fluoropyrimidine) trial. Using an adaptive analysis design, the researchers analyzed plasma proteins from two sets of patients, one exploratory (n = 294) and one confirmatory (n = 590), and found that high VEGF-D basal levels correlated with a better PFS (p = 0.0013) and OS (p = 0.0107) in patients treated in the ramucirumab arm. Although VEGF-D is not a genomic or epigenomic biomarker, it is the most compelling study to date in a ramucirumab-treated CRC cohort.
Anti-EGFR Drugs: Cetuximab and Panitumumab
KRAS mutations in tumor tissues were the first predictive biomarker approved to guide decision-making and determine eligibility for anti-EGFR therapy in patients with mCRC. One of the first studies of KRAS mutations as predictive biomarkers comprised 30 patients with mCRC treated with cetuximab-based regimens. This study showed a significant correlation between the presence of KRAS mutations and the lack of response to anti-EGFR therapy (Lievre et al., 2006), an observation that was subsequently validated in a cohort of 427 patients with mCRC treated with panitumumab (Amado et al., 2008). Similarly, the potential of NRAS mutation status as a negative response predictor for panitumumab and cetuximab was later confirmed in several clinical trials and meta-analyses (De Roock, Claes, et al., 2010; De Roock, Jonker, et al., 2010; Douillard et al., 2013; Peeters M, Oliner KS, 2010; Sorich M J, Wiese M D, Rowland A, Kichenadasse G, McKinnon R A, 2015). Interestingly, in addition to the lack of efficacy in patients with RAS-mutated mCRC, panitumumab or cetuximab also showed reduced efficacy when given in combination with FOLFOX in a retrospective data analysis from two phase III clinical trials (Bokemeyer et al., 2017; Douillard et al., 2013). Such a negative effect for panitumumab or cetuximab when given in conjunction with FU, leucovorin, and irinotecan (FOLFIRI) has not been confirmed to date (Stintzing et al., 2017). In an attempt to translate these tissue-based predictive biomarkers into circulating tumor DNA (ctDNA)-based liquid biopsy assays, two 2017 studies used an innovative beads, emulsions, amplification, and magnetics (BEAMing) assay and reported promising agreement rates of 89.7% and 93% for the mutational status of RAS between tissue and ctDNA (Grasselli et al., 2017; Vidal et al., 2017).
BRAF mutations have also been evaluated as a predictive biomarker for anti-EGR therapies. The presence of V600E mutation within BRAF often reflects a poor prognosis in patients with CRC (Yuan et al., 2013). In addition, two meta-analyses reported a lack of benefit in terms of PFS, OS, and ORR when anti-EGFR therapies were combined with standard chemotherapy in a subset of patients harboring BRAF mutations (Pietrantonio et al., 2015; Rowland et al., 2015). Despite these early data suggesting that the presence of the BRAF V600E mutation may suggest a lack of response to anti-EGFR-based therapies, there is not yet enough clinical evidence to consider BRAF mutational status as a negative predictive biomarker in patients with advanced disease. Moreover, emerging therapeutic strategies that combine anti-EGFR drugs with BRAF inhibitors have recently changed the paradigm of how we must understand these molecular alterations from a therapeutic perspective.
In 2014, Manceau et al. (Manceau et al., 2014) investigated the roles of different miRNAs in response to anti-EGFR therapies. They conducted miRNA profiling in a training set of tissue specimens (33 fresh-frozen [FF] and 35 FFPE samples that were retrospectively collected and 19 prospectively collected FF samples) from 87 patients with mCRC refractory to chemotherapy and treated with anti-EGFR antibodies, and determined that hsa-miR-31-3p expression levels were significantly associated with PFS. They subsequently validated these results in an independent validation cohort consisting of 19 FF and 26 FFPE prospectively collected samples. The percentage of size variation of target lesions by RECIST criteria in the validation series and risk status were significantly associated with expression levels of hsa-miR-31-3p. In addition, the investigators built and validated nomograms to predict PFS based on hsa-miR-31-3p expression. In the same context, Miller-Phillips et al. (L Miller-Phillips et al., 2018) evaluated the role of miR-21 as a predictive biomarker for anti-EGFR therapies. Using an RT-qPCR assay, they quantified miR-21 expression in FFPE tumor samples from patients enrolled on the FIRE-3 trial (NCT00433927) patients. The median miR-21 expression within the FIRE-3 population was determined and subsequently used to segment the population into low and high miR-21 expression groups. Patients who had wild-type RAS wild-type and low miR-21 expression showed significantly improved ORR and OS when cetuximab, but not bevacizumab, was added to FOLFIRI chemotherapy. In contrast, patients who had wild-type RAS but high miR-21 expression showed no significant difference in ORR and OS between the cetuximab and bevacizumab treatment groups. Similarly, patients with mutated RAS and either high or low miR-21 expression showed no significant difference in ORR between cetuximab or bevacizumab added to FOLFIRI. Unfortunately, other correlations between groups and PFS or OS were not significant. In a later study to determine the biological changes underlying these differences in efficacy outcomes, Miller-Phillips et al. (Lisa Miller-Phillips et al., 2019) used normalized mRNA microarray data to compare gene expression in the miR-21 low and high groups. This analysis identified 538 genes that were significantly and differentially expressed in patients with wild-type RAS after adjustment for multiple testing. Including data from the two groups into single-sample Gene Set Enrichment Analysis showed 23 hallmarks of cancer gene sets significantly differentially enriched, being KRAS-signaling higher in the miR-21 high group.
Anti–PD-1 Drugs: Pembrolizumab and Nivolumab
In May and July of 2017, the US FDA approved the anti-PD-1 therapies pembrolizumab and nivolumab for the treatment of patients with MSI-H mCRC for whom the disease has progressed after treatment with fluoropyrimidine, oxaliplatin, and irinotecan. Almost a year later, in July 2018, a nivolumab plus ipilimumab combination regimen was approved, which opened three novel treatment options for patients with MSI-H or dMMR mCRC (patients with MSI-H or dMMR mCRC represent approximately 5% of all patients with mCRC) (Braun et al., 2017). Although patients with MSI-positive mCRC have worse prognosis, it is thought that they derive clinical benefit from anti-PD-1 therapy because of a large proportion of lymphocytic infiltration and the presence of mutation-associated neoantigens (Giannakis et al., 2016) (Le et al., 2017, 2015; Overman et al., 2017, 2018). This exciting discovery has led to universal MSI testing for the management of patients with mCRC.
Not surprisingly, in May 2020 the US FDA approved pembrolizumab as first-line therapy for patients with MSI-H/dMMR mCRC. This approval was based on the results of the KEYNOTE-177 study (NCT02563002), a multicenter, international, open-label, active-controlled, randomized trial that compared first-line therapy with pembrolizumab vs. chemotherapy in 307 patients with MSI-H/dMMR mCRC. This study demonstrated a statistically significant improvement in PFS, with a median PFS of 16.5 months vs. 8.2 months for pembrolizumab compared to chemotherapy standard-of-care. Longer-term analysis is needed to assess the effect on OS. Moreover, in June 2020 the US FDA granted accelerated approval to pembrolizumab for the treatment of patients with any unresectable or metastatic solid tumor with high mutational burden (as determined by the FDA-approved test, the FoundationOneCDx assay) whose cancer has progressed after previous treatment and has no satisfactory alternative treatment options (Bersanelli, 2020). Several clinical trials evaluating the combination of anti-PD-1 therapy with chemotherapy are ongoing for previously untreated MSI-H/dMMR mCRC patients with the goal of improving on results from previous studies and further extending survival of these patients. Meanwhile, other different immunotherapeutic approaches are being evaluated for treatment of MSS CRC, which is less responsive to immune-checkpoint inhibition than MSI-H mCRC. Although all of these results represent substantial therapeutic advances in the treatment of mCRC, they also emphasize the growing need for more precise predictive biomarkers to support more rational development of immunotherapies. A more comprehensive understanding of the intersection between genomics, epigenomics, and immunology in mCRC seems essential for meeting this need for new strategies.
Recently, Grasso et al. (Grasso et al., 2018) reported the results of a large-scale genomic analysis (TCGA, Nurses’ Health Study, and Health Professionals Follow-up Study cohorts) involving 1211 primary CRC tumor specimens. Mutations in genes involved in immune modulatory pathways, as well as in the neoantigen-presentation machinery (mainly B2M and HLA), significantly correlated with MSI-H. Along with JAK1/ 2 and IFN-gamma receptor 1 mutations, similar alterations have been observed in melanoma, non-small cell lung cancer, and CRC and deemed to be genetic drivers of primary or acquired resistance to immune-checkpoint blockade, reflecting their role as a mechanism of adaptive resistance against T-cell tumor infiltration (Gao et al., 2016; Giannakis et al., 2016; Grasso et al., 2018; Gurjao et al., 2019; Le et al., 2017; Zaretsky et al., 2016). The interaction between somatic alterations and the immune system is complex, as indicated by a recent study in which 11 out of 13 B2M-mutant CRC patients achieved mCRC control with anti-PD-1 or anti-PD-L1 agents, despite the presence of a mutation that, theoretically, conferred primary resistance to ICI (Middha et al., 2019). On the other hand, for both MSS and MSI-H tumors, active WNT/β-catenin signaling was inversely associated with tumor T-cell infiltration, providing evidence of the existence of an anti-immune response mechanism beyond the MSI profile (Grasso et al., 2018).
BRAF Inhibitors: Vemurafenib and Encorafenib
BRAF mutations occur in 10–15% of all CRCs and in ~7% of all mCRC (Clarke & Kopetz, 2015; Davies et al., 2002). Although most BRAF mutations occur in codon 600 (mainly BRAF V600E), which leads to constitutive BRAF kinase activity and sustained MAPK pathway signaling, 2% of mCRCs have atypical BRAF mutations that are outside of codon 600, usually in codon 594 (Jones et al., 2017). Surprisingly, although monotherapy with BRAFi has proven effective in the treatment of BRAF-mutant melanoma, it was ineffective in BRAF V600E-mutant CRCs. Preclinical evidence demonstrated that despite transient inhibition of pERK by BRAFi such as vemurafenib, rapid ERK reactivation occurs through EGFR-mediated activation of RAS and CRAF (Corcoran et al., 2012). Furthermore, the fact that BRAF V600E-mutant CRCs express higher levels of pEGFR than do BRAF-mutant melanomas, positions them for EGFR-mediated resistance (Corcoran et al., 2012). Collectively, these findings provided rationale to test dual BRAF and EGFR blockade. Results from preclinical studies and early phase clinical trials, have demonstrated this strategy is feasible and safe, and can potentially improve therapeutic efficacy of BRAFi. Moreover, preclinical studies have suggested that combined inhibition of BRAF and MEK was more effective than dual BRAF and EGFR blockade. This strategy was tested in subsequent phase 1 and phase 2 clinical trials that combined BRAF inhibitors with both anti-EGFR monoclonal antibodies and MEK inhibitors (Corcoran et al., 2018, 2010, 2012). Results of these trials led to US FDA approval (in April 2020) of encorafenib, a BRAF tyrosine kinase inhibitor, used in combination with cetuximab for the treatment of adult patients with BRAF V600E-mutated mCRC. The efficacy of this combination of drugs was evaluated in the BEACON CRC study (Kopetz et al., 2019), a phase 3 randomized, active-controlled, open-label, multicenter trial (NCT02928224). In this trial, encorafenib plus cetuximab demonstrated a clinical and statistically significant OS and PFS benefit compared to the control arm of either irinotecan or FOLFIRI plus cetuximab in patients with BRAF V600E-mutated mCRC who had progressed on one or two prior regimens. This trial also evaluated the efficacy of triple-therapy with encorafenib, binimetinib (a MEK inhibitor [MEKi]), and cetuximab in a second experimental arm, but although this regimen showed an improved OS and PFS compared to the control arm, it was more toxic than the dual BRAF and EGFR blockade and had similar efficacy.
Another BRAFi, vemurafenib, which has more modest clinical activity, was recently included in the NCCN guidelines as a treatment option for patients with BRAF V600E-mutated mCRC when used in combination with cetuximab/panitumumab plus irinotecan (Kopetz et al., 2020; “National Comprehensive Cancer Network. Colon Cancer (Version 4.2020). https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed December 6, 2020.,” n.d.). Inclusion in the guidelines was based on results of the randomized phase 2 Southwest Oncology Group (SWOG) 1406 trial, in which the triple-therapy (vemurafenib, cetuximab, and irinotecan) demonstrated improved PFS and ORR as compared with cetuximab plus irinotecan (Kopetz et al., 2020). In addition to the previously described regimens, based on the results of a phase 1 study (Corcoran et al., 2018), the NCCN Panel has recommend the combination of dabrafenib (BRAFi) plus trametinib (MEKi) plus either cetuximab or panitumumab as another treatment option beyond the first line setting for BRAF V600E-mutated mCRC (NCCN. Colon Cancer version 4, 2020).
HER-2 Blockade
A large body of evidence, accrued primarily from breast and gastric cancer patients, supports the role of HER-2 amplification or overexpression as a predictive biomarker for anti-HER-2-based therapies. Therefore, there is renewed interest in evaluating HER-2 as a clinically actionable target in mCRC. Although initial mCRC clinical trials interrogating the anti-HER-2 monoclonal antibody trastuzumab in combination with other chemotherapeutic agents (either FOLFOX or irinotecan) closed early due to lack of patient accrual, mechanistic insights gained from preclinical analyses of HER-2-amplified mCRC patient-derived xenografts has led to improved design of new clinical trials (Bertotti et al., 2011; Clark JW, Niedzwiecki D, Hollis D, 2003; Ramanathan et al., 2004). Three phase 2 clinical trials evaluated dual HER-2 blockade in a biomarker-selected subset of heavily pretreated mCRC patients. Study treatment included trastuzumab plus lapatinib (HERACLES trial, NCT03225937), pertuzumab and trastuzumab (MyPathway trial, NCT02091141), or the antibody-drug conjugate trastuzumab deruxtecan (DESTINY-CRC01, NCT03384940). Results of these studies demonstrated an impressive ORR of ~30–45% (Hainsworth et al., 2018; Sartore-Bianchi et al., 2017; Siena et al., 2020). These data have paved the way for development of ongoing phase 2 clinical trials evaluating the efficacy of new anti-HER-2 agents, such as S1613 (NCT03365882), trastuzumab-emtansine (NCT03418558), or tucatinib (NCT03043313) in this clinical scenario comprising ~5% of RAS wild-type mCRC patients (Valtorta et al., 2015). Furthermore, determining the utility of ctDNA analyses in monitoring therapeutic efficacy and in identifying mechanisms of resistance to dual HER-2 blockade is also an attractive area of study (Siravegna et al., 2018).
Tyrosine Kinase Inhibitors
New drugs that target tyrosine kinase (TK) fusions in genes such as NRTK1/2/3, RET, ALK, and ROS1 are showing promising preliminary results in phase 1 and 2 clinical trials that include patients with CRC. One agent, LOXO 101 (larotrectinib), is a selective tropomyosin receptor kinase (TRK) inhibitor that demonstrated tumor-agnostic efficacy in patients with NTRK fusion-positive malignancies (including four patients with CRC who achieved a partial response) (Drilon et al., 2018). A second agent, entrectinib, an ALK, ROS1, TRKA, TRKB, and TRKC selective inhibitor, demonstrated clinical activity in patients who had fusions in the previously described TK genes (Drilon, Siena, et al., 2017). Patients who responded to entrectinib included two patients whose mCRC harbored CAD-ALK or LMNA-NTRK1 gene fusions (Amatu et al., 2015; Sartore-Bianchi et al., 2016). Anticipating potential resistance mechanisms to larotrectinib based on evidence from other pan-TK inhibitors, Drilon et al. (Drilon, Nagasubramanian, et al., 2017) developed LOXO-195 (selitrectinib), a potent and selective TRK kinase inhibitor designed to have a molecular structure that would overcome typical TRK resistance mutations. LOXO-195 was initially evaluated in a mCRC patient whose cancer had an LMNA-NTRK1 rearrangement with a G595R larotrectinib-resistance mutation. This patient successfully achieved a durable partial response (Drilon, Nagasubramanian, et al., 2017). Although the prevalence of rearrangements in TK genes in mCRC patients may be as low as 1.5%, the accelerated development of TK inhibitors offers new hope for some heavily pretreated mCRC patients who have no other therapeutic options (Pietrantonio et al., 2017). Given these promising results, the US FDA granted accelerated approval to larotrectinib (November 2018) and entrectinib (August 2019) for patients with NTRK gene fusion-positive solid tumors without a known acquired resistance mutation. The Committee for Medicinal Products for Human Use of the European Medicines Agency has also recommended the granting of a conditional marketing authorization for larotrectinib (July 2019) and entrectinib (May 2020) for the same indication.
KRAS Inhibitors
KRAS is one of the most commonly altered oncogenes in human cancers, and was long considered an undruggable target because of the small size of abnormal KRAS proteins, the presence of few binding sites, and the rapid, tight binding of active KRAS to GTP. However, recent data have suggested that KRAS may be targetable. For example, preliminary data on the activity of AMG510 (sotorasib), a small covalent inhibitor, have shown that it rapidly and irreversibly occupies KRAS G12C and extinguishes its activity through a unique interaction with the P2 pocket (Janes et al., 2018). The KRAS G12C mutation occurs in ~4% of CRC (Neumann, Zeindl-Eberhart, Kirchner, & Jung, 2009). In a recent phase 1 trial, sotorasib showed encouraging anti-tumor activity in heavily pretreated patients who had advanced, KRAS G12C-mutated solid tumors (Hong et al., 2020). A total of 129 patients were included in this study, 42 of whom had CRC. Within CRC patients, sotorasib treatment yielded an ORR and disease control rate (DCR) of 7.1% and 73.8%, respectively. The median duration of stable disease was 5.4 months and the median PFS was 4.0 months. Although sotorasib showed promising anticancer activity in patients with heavily pre-treated solid tumors bearing the KRAS G12C mutation, inconsistency was seen in tumor response between patients with non-small cell lung cancer and those with CRC, which the authors suggested indicated either that KRAS G12C is not the dominant oncogenic driver for CRC or that other pathways, such as the WNT or EGFR pathways, mediate oncogenic signaling beyond KRAS. These hypotheses are supported by solid preclinical evidence (Amodio et al., 2020; Lee et al., 2018; Xue et al., 2020), and therefore, clinical trials that combine sotorasib with other agents that block additional pathways have already been initiated (i.e., NCT04185883 and NCT04303780). Although many KRAS G12C inhibitors in addition to sotorasib are under development, to date only adagrasib, an irreversible covalent inhibitor, has shown promising antitumor activity in KRAS G12C-mutated CRC. Furthermore, inhibitors for mutations other than KRAS G12C are being developed. For example, initial preclinical data for MRTX1133, a new, first-in-class KRAS G12D inhibitor, have demonstrated significant tumor regression in preclinical animal models (Mirati Therapeutics, 2020). Thus, through development of a range of inhibitors, effective means of targeting KRAS are emerging.
Disease Monitoring by Liquid Biopsies
Liquid biopsies have recently emerged as powerful tools for monitoring disease evolution and therapeutic response through the analysis of cfDNA and RNA biomarkers in bodily fluids. One of the basis for this concept of liquid biopsies came from a study that reported that a gradual decrease in CEA levels during chemotherapy was significantly associated with better survival rates (Al-Sarraf, Baker, Talley, Kithier, & Vaitkevicius, 1979). Such a correlation between CEA flare and improved PFS and OS was confirmed a few years later in a subset of 670 patients with mCRC being treated with first-line chemotherapy (Strimpakos et al., 2010). Although CEA is not a CRC-specific biomarker, CEA monitoring in blood, alone or in addition to CA19-9 monitoring, is still commonly performed in routine clinical practice (Stiksma, Grootendorst, & van der Linden, 2014). In addition to this strategy, analyses of circulating tumor cells and endothelial cells in mCRC have been undertaken by several groups (S. J. Cohen et al., 2008; Ronzoni et al., 2010). In 2015, Hansen et al. (T F Hansen, Carlsen, Heegaard, Sørensen, & Jakobsen, 2015) reported that circulating levels of miRNA-126 in a subset of 68 patients with mCRC were predictive of tumor response to bevacizumab-based chemotherapy. On the other hand, the role of ctDNA in genotyping CRCs and tracking clonal evolution of CRC during and after treatment with anti-EGFR–based regimens has been evaluated (Siravegna et al., 2015). Multiple studies have since reported distinct genetic alterations in ctDNA from patients with primary or acquired resistance to anti-EGFR–based therapies. These alterations were seen in genes such as KRAS, NRAS, MET, ERBB2, FLT3, EGFR, and MAP2K1 and detected by approaches including droplet digital PCR, BEAMing, and next-generation sequencing methodologies (Siravegna et al., 2015; Van Emburgh et al., 2016). Further demonstrating the potential utility of changes in ctDNA, a massively parallel sequencing-based assay of ctDNA from a prospective cohort of 53 patients with mCRC showed that early changes in ctDNA during first-line standard chemotherapy can predict subsequent radiologic response (J Tie et al., 2015). Similarly, a 2017 study demonstrated a significant correlation between a decrease in RAS mutant clones in blood after 8 weeks of therapy and improved PFS and OS in a cohort of patients treated with regorafenib (Khan et al., 2017).
Intriguingly, although clonal evolution is dynamic, the emergence of drug-resistant clones in circulation increases during treatment and decreases after drug withdrawal. Understanding of this fact has led to novel treatment strategies that are being evaluated in studies such as the RASINTRO (RAS Mutations in ctDNA and Anti-EGFR Reintroduction in mCRC) study (NCT03259009) and the CHRONOS (Rechallenge With Panitumumab Driven by RAS Dynamic of Resistance) trial (NCT03227926). Both of these studies are evaluating the predictive impact of ctDNA RAS mutations on the efficacy of anti-EGFR monotherapy rechallenge in patients with mCRC with wild-type RAS whose disease has progressed after anti-EGFR–free chemotherapy. Another recently described approach involved a five-gene methylation panel for monitoring tumor burden in liquid biopsies using a methyl-BEAMing assay (Barault et al., 2017). Using this method to analyze plasma samples from 182 patients with mCRC treated with chemotherapy and/or targeted therapy, revealed a significant correlation between the dynamics of circulating cfDNA methylation markers and ORR and PFS. Using a similar approach, as part of studies presented in previous sections of this chapter, Luo et al. (Luo et al., 2020) evaluated the utility of ddPCR analysis of cg10673833 methylation for monitoring disease evolution. Dynamic changes in cg10673833 methylation were consistent with treatment response, and these changes were more pronounced than changes in CEA. Patients who had positive responses to treatment had parallel decreases in cg10673833 methylation as compared with untreated patients, and a further reduction in cg10673833 methylation was observed in patients after surgery. In contrast, patients who had progressive or recurrent disease showed increased methylation of cg10673833.
TOXICITY-ASSOCIATED BIOMARKERS
DPYD gene
Severe adverse events and side effects as a result of treatment-related toxicity are a major concern for any cancer therapy. One particular concern in the context of CRC is DPD deficiency, an inherited defect in the enzyme that catabolizes fluoropyrimidines and thus increases the risk of toxicity secondary to fluoropyrimidine-based therapies (Diasio, Beavers, & Carpenter, 1988; Tuchman et al., 1985; Amstutz et al., 2018; Mattison, Soong, & Diasio, 2002). DPD deficiency causes a deficit in the metabolism of thymine and uracil, resulting in excessive amounts of uracil and thymine in the blood, urine, and cerebrospinal fluid (Innocenti et al., 2020). It can be inherited as an autosomal recessive trait, such as homozygosity for deleterious DPYD variants. Patients who are homozygous for DPYD variants can develop severe DPD deficiency, which can result in neurologic and other severe defects during infancy. However, patients with homozygous variants may also be asymptomatic. The prevalence of DPD deficiency in the population ranges from 2% to 8% depending on ethnicity (Innocenti et al., 2020). The Clinical Pharmacogenetics Implementation Consortium selected 4 DPYD variants that confer DPD deficiency and for which recommendations exist to aid fluoropyrimidine treatment and dosing decisions: c.1905*1G>A (*2A), c.2846A>T, c.1679T>G, and c.1129-5923C>G (haplotype B3). These patients could be offered alternative regimens or receive dose reductions (Amstutz et al., 2018). Two prospective studies have shown DPYD genotyping followed by tailoring of the fluoropyrimidine dose to be feasible in clinical practice and to improve patient safety and cost effectiveness (Deenen et al., 2016; Henricks et al., 2018, 2019). We note that universal DPYD genotyping before starting fluoropyrimidine treatment remains controversial. For example the NCCN Panel does not currently support this practice, while the ESMO Clinical Guidelines, in accordance to the Pharmacovigilance Risk Assessment Committee of the European Medicines Agency statement (March 2020) (“EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine. https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine. Accessed Novem,” n.d.), strongly recommend DPYD genotyping or DPD phenotyping before initiating fluoropyrimidine-based therapy in the adjuvant setting (Argilés et al., 2020).
UGT1A1
The UDP-glucuronosyltransferase (UGT) enzyme encoded by UGT1A1 is important in the metabolism of irinotecan, a pro-drug that is widely used in the treatment of mCRC. Irinotecan is converted by carboxylesterase enzymes into the active metabolite SN-38. SN-38 produces severe treatment-related toxicities, including early and late forms of diarrhea, dehydration, and severe neutropenia (Innocenti et al., 2004; “Package Insert. Camptosar® (Irinotecan) Injection, intravenous infusion. 2019. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020571s050lbl.pdf. Accessed November 16, 2020.,” n.d.). SN-38 is inactivated and detoxified by the UGT1A1 enzyme. UGT enzymes are responsible for glucuronidation, which transforms lipophilic metabolites into hydrophilic substances that can be easily excreted from the body. Deficiencies in UGT1A1 can be caused by certain genetic polymorphisms and can result in conditions associated with accumulation of unconjugated hyperbilirubinemias, such as types I and II of the Crigler-Najjar and Gilbert syndromes. Moreover, certain genetic polymorphisms UGT1A1 can result in a decreased glucuronidation of SN-38, resulting in increased drug levels and thus increased risk for toxicity, although severe irinotecan-related toxicity is not experienced by all patients who have these polymorphisms (Innocenti et al., 2004; X. Liu, Cheng, Kuang, Liu, & Xu, 2014; O’Dwyer & Catalano, 2006). UGT1A is located on chromosome 2q37, which encodes for multiple genes and pseudogenes. Moreover it exists in many alternatively spliced isoforms (van Es et al., 1993). Variants of UGT1A1 that decrease UGT1A1 enzyme activity can lead to jaundice. Up to 135 genetic variants of UGT1A1 have been reported; UGT1A1*1 is the wild-type allele associated with normal enzyme activity (Strassburg, 2008). The most common variant allele is UGT1A1*28, which is also associated with drug toxicity. Current literature suggests that one copy of the UGTA1A*28 allele results in an approximately 35% decrease in transcriptional activity, and 2 copies (*28/*28, homozygous) result in approximately a 70% decrease (Barbarino, Haidar, Klein, & Altman, 2014; Lam, 2019). Approximately 10% of the North American population is homozygous for the *28 allele (*28/*28 genotype, also known as UGT1A1 7/7 genotype) and are at an increased risk of neutropenia following intravenous irinotecan therapy (Hall, Ybazeta, Destro-Bisol, Petzl-Erler, & Di Rienzo, 1999). The rate of severe neutropenia in *28/*28 homozygous patients is as high as 36%, and is strongly associated with a higher hospitalization rate (Etienne-Grimaldi et al., 2015; Shulman et al., 2011). There is less evidence to support a link between UGT1A1 genotype and irinotecan treatment-related diarrhea, and data is conflicting regarding the influence of UGT1A genotype on response to irinotecan therapy (Dias et al., 2012; “Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan?,” 2009).
Individuals homozygous for another variant allele, UGT1A1*6, which is more prevalent in Asian populations, have reduced UGT1A1 enzyme activity and homozygosity appears to be an important predictor of severe toxicity to irinotecan therapy in this population (Dean, 2012).
Moreover, many other genetic markers, including genetic variations in ABCC1 and ABCB2, SLC transporter genes, TGFB, and NR1I2 may influence the risk of irinotecan toxicity (Dean, 2012). On the other hand, emerging data suggests that other UGT1 variant alleles may have a protective effect. This could be the case for UGT1A1 c.-1068A>G, which seems to have a lower risk of irinotecan-induced neutropenia, probably due to an enhanced capacity for glucuronidation (S. Chen et al., 2015).
FUTURE PERSPECTIVES AND CONCLUSION
Over the past several years, many different approaches have been developed and tested for diagnosing and treating CRC. The rising incidence of early-onset CRC, coupled with the typical screening and diagnosis of the disease in individuals of older ages, represents a tremendous challenge for healthcare systems worldwide (Akimoto et al., 2020). Given current evidence and recent advances, the future of CRC early-diagnosis would appear to be in multimodal liquid-biopsy approaches that include robust DNA methylation analyses. However, much additional study is needed to identify the ideal diagnostic test as well as a better understanding of the molecular characteristics of the various subsets of CRC and premalignant lesions.
Beyond diagnosis, a detailed understanding of CRC from a multi-omic perspective seems necessary to improve clinical outcomes in the adjuvant and palliative settings. Although risk stratification of CRC based on multi-omic molecular analyses has not yet been implements in the clinic, such a strategy together with in-depth knowledge of the interaction among (epi)genetic somatic and germline alterations and the immune system could have an important role in patient selection for adjuvant therapy. Moreover, although current studies have involved only a small number of patients, the growing evidence of the utility of various target genomic alterations such as specific gene mutations and fusions, together with specific genomic phenotypes, are laying the groundwork for developing more precise therapeutic approaches. This is exemplified by, for example, the emerging importance of NRTK fusions and KRAS mutations as targets for TK and RAS inhibitors, respectively. Moreover, many alterations that were previously identified in other tumor types such as HER2 amplification in breast cancer, are also showing promise for CRC.
Finally, an understanding of CRC from a phylogenetic perspective and a better characterization of tumor heterogeneity and evolution should be incorporated into clinical guidelines for future CRC screening and therapeutic strategies. Cancer is a complex disease, and using rational tissue sampling, detailed clinical phenotype annotation, and integrative genomics coupled with comprehensive mathematical models of cancer evolution and biomarker shedding should become the basis for data generation and biomarker development (Avanzini et al., 2020; Wheeler et al., 2020). Implementing all of these approaches will require making use of novel high-throughput molecular analytic techniques for comprehensive molecular profiling as well as rethinking and adapting the design of the next generation of clinical trials. Integrating multi-omic approaches with clinical and epidemiologic data using machine-learning algorithms will hasten biomarker development, and, most importantly, the decision-making process (Ruiz-Bañobre et al., 2019). To make this feasible in daily clinical practice, it will be necessary to implement user-friendly decision support tools that foster interactive treatment planning based on streamlined, expert-driven genomic-data interpretation and reporting (Tamborero et al., 2020).
Gradually, CRC management is moving from a “one-size-fits-all” approach to a more personalized medicine strategy in which increasing numbers of subsets of patients can obtain long-term survival benefits targeting low prevalence driver molecular alterations. With this concept in mind, biomarker research is moving forward, bringing together many types of “omics” data and establishing novel liquid biopsy-based strategies to capture temporo-spatial tumor heterogeneity. The future of CRC diagnosis and treatment is promising as many scientific disciplines come together, generating new knowledge and strategies that will improve patient care and outcomes.
ACKNOWLEDGEMENTS
We thank Raquel Varela-Fernández and Jared Millar for their assistance in medical illustration.
FUNDING INFORMATION
This work was supported by CA72851, CA181572, CA184792 and CA227602 grants from the National Cancer Institute, National Institutes of Health to AG. In addition, JR-B is supported by Río Hortega fellowship from the Institute of Health Carlos III (CM19/00087) and 2020 TTD Research Grant from the Spanish Cooperative Group for the Treatment of Digestive Tumors (TTD).
References
- Adenis A, de la Fouchardiere C, Paule B, Burtin P, Tougeron D, Wallet J, … Andre T (2016). Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer, 16(1), 412. 10.1186/s12885-016-2440-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigrain L, Gu Y, & Quail MA (2016). Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays - a systematic comparison of DNA library preparation kits for Illumina sequencing. BMC Genomics, 17(1), 458. 10.1186/s12864-016-2757-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto N, Ugai T, Zhong R, Hamada T, Fujiyoshi K, Giannakis M, … Ogino S (2020). Rising incidence of early-onset colorectal cancer — a call to action. Nature Reviews Clinical Oncology. 10.1038/s41571-020-00445-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sarraf M, Baker L, Talley RW, Kithier K, & Vaitkevicius VK (1979). The value of serial carcinoembryonic antigen (CEA) in predicting response rate and survival of patients with gastrointestinal cancer treated with chemotherapy. Cancer, 44(4), 1222–1225. [DOI] [PubMed] [Google Scholar]
- Alaiyan B, Ilyayev N, Stojadinovic A, Izadjoo M, Roistacher M, Pavlov V, … Nissan A (2013). Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer, 13, 196. 10.1186/1471-2407-13-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, … Chang DD (2008). Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 26(10), 1626–1634. 10.1200/JCO.2007.14.7116 [DOI] [PubMed] [Google Scholar]
- Amatu A, Somaschini A, Cerea G, Bosotti R, Valtorta E, Buonandi P, … Siena S (2015). Novel CAD-ALK gene rearrangement is drugable by entrectinib in colorectal cancer. British Journal Of Cancer, 113, 1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, … Misale S (2020). EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discovery, 10(8), 1129 LP–1139. 10.1158/2159-8290.CD-20-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstutz U, Henricks LM, Offer SM, Barbarino J, Schellens JHM, Swen JJ, … Schwab M (2018). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clinical Pharmacology and Therapeutics, 103(2), 210–216. 10.1002/cpt.911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, … de Gramont A (2004). Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment for Colon Cancer. New England Journal of Medicine, 350(23), 2343–2351. 10.1056/NEJMoa032709 [DOI] [PubMed] [Google Scholar]
- Argilés G, Tabernero J, Labianca R, Hochhauser D, Salazar R, Iveson T, … Arnold D (2020). Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up† Annals of Oncology, 31(10), 1291–1305. 10.1016/j.annonc.2020.06.022 [DOI] [PubMed] [Google Scholar]
- Aschele C, Debernardis D, Bandelloni R, Cascinu S, Catalano V, Giordani P, … Monfardini S (2002). Thymidylate synthase protein expression in colorectal cancer metastases predicts for clinical outcome to leucovorin-modulated bolus or infusional 5-fluorouracil but not methotrexate-modulated bolus 5-fluorouracil. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 13(12), 1882–1892. [DOI] [PubMed] [Google Scholar]
- Aschele C, Debernardis D, Tunesi G, Maley F, & Sobrero A (2000). Thymidylate synthase protein expression in primary colorectal cancer compared with the corresponding distant metastases and relationship with the clinical response to 5-fluorouracil. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 6(12), 4797–4802. [PubMed] [Google Scholar]
- Ashktorab H, Belgrave K, Hosseinkhah F, Brim H, Nouraie M, Takkikto M, … Smoot D (2009). Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Digestive Diseases and Sciences, 54(10), 2109–2117. 10.1007/s10620-008-0601-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aust DE, Sommer U, & Baretton GB (2012, September). What can molecular pathology offer for optimal decision making? Annals of Oncology : Official Journal of the European Society for Medical Oncology. England. 10.1093/annonc/mds346 [DOI] [PubMed] [Google Scholar]
- Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, … Reiter JG (2020). A mathematical model of ctDNA shedding predicts tumor detection size. Science Advances, 6(50), eabc4308. 10.1126/sciadv.abc4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek YH, Chang E, Kim YJ, Kim BK, Sohn JH, & Park D Il. (2009). Stool methylation-specific polymerase chain reaction assay for the detection of colorectal neoplasia in Korean patients. Diseases of the Colon and Rectum, 52(8), 1452–1463. 10.1007/DCR.0b013e3181a79533 [DOI] [PubMed] [Google Scholar]
- Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, … Vogelstein B (1990). p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Research, 50(23), 7717–7722. [PubMed] [Google Scholar]
- Ballman KV (2015). Biomarker: Predictive or Prognostic? Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 33(33), 3968–3971. 10.1200/JCO.2015.63.3651 [DOI] [PubMed] [Google Scholar]
- Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, … Di Nicolantonio F (2017). Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarino JM, Haidar CE, Klein TE, & Altman RB (2014). PharmGKB summary: very important pharmacogene information for UGT1A1. Pharmacogenetics and Genomics, 24(3), 177–183. 10.1097/FPC.0000000000000024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barták BK, Kalmár A, Péterfia B, Patai ÁV, Galamb O, Valcz G, … Molnár B (2017). Colorectal adenoma and cancer detection based on altered methylation pattern of SFRP1, SFRP2, SDC2, and PRIMA1 in plasma samples. Epigenetics, 12(9), 751–763. 10.1080/15592294.2017.1356957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti P, Gafa R, Barana D, Marino M, Scarselli A, Pedroni M, … Lanza G (2005). Microsatellite instability and colorectal cancer prognosis. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 11(23), 8332–8340. 10.1158/1078-0432.CCR-05-1030 [DOI] [PubMed] [Google Scholar]
- Bergheim J, Semaan A, Gevensleben H, Groening S, Knoblich A, Dietrich J, … Dietrich D (2018). Potential of quantitative SEPT9 and SHOX2 methylation in plasmatic circulating cell-free DNA as auxiliary staging parameter in colorectal cancer: a prospective observational cohort study. British Journal of Cancer, 118(9), 1217–1228. 10.1038/s41416-018-0035-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersanelli M (2020). Tumour mutational burden as a driver for treatment choice in resistant tumours (and beyond). The Lancet Oncology, 21(10), 1255–1257. 10.1016/S1470-2045(20)30433-2 [DOI] [PubMed] [Google Scholar]
- Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, … Trusolino L (2011). A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discovery, 1(6), 508–523. 10.1158/2159-8290.CD-11-0109 [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Köhne C-H, Ciardiello F, Lenz H-J, Heinemann V, Klinkhardt U, … Tejpar S (2017). FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. European Journal of Cancer, 51(10), 1243–1252. 10.1016/j.ejca.2015.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovell L, Shanmugam C, Katkoori VR, Zhang B, Vogtmann E, Grizzle WE, & Manne U (2012). miRNAs are stable in colorectal cancer archival tissue blocks. Frontiers in Bioscience (Elite Edition), 4, 1937–1940. 10.2741/514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, … Seymour MT (2008). Predictive Biomarkers of Chemotherapy Efficacy in Colorectal Cancer: Results From the UK MRC FOCUS Trial. Journal of Clinical Oncology, 26(16), 2690–2698. 10.1200/JCO.2007.15.5580 [DOI] [PubMed] [Google Scholar]
- Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, … Seymour MT (2017). Predictive Biomarkers of Chemotherapy Efficacy in Colorectal Cancer : Results From the UK MRC FOCUS Trial, 26(16). 10.1200/JCO.2007.15.5580 [DOI] [PubMed] [Google Scholar]
- Bruhn MA, Townsend AR, Khoon Lee C, Shivasami A, Price TJ, Wrin J, … Hardingham JE (2014). Proangiogenic tumor proteins as potential predictive or prognostic biomarkers for bevacizumab therapy in metastatic colorectal cancer. International Journal of Cancer, 135(3), 731–741. 10.1002/ijc.28698 [DOI] [PubMed] [Google Scholar]
- Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, … Boland CR (2004). Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology, 126(2), 394–401. 10.1053/j.gastro.2003.12.023 [DOI] [PubMed] [Google Scholar]
- Carmona FJ, Azuara D, Berenguer-Llergo A, Fernández AF, Biondo S, de Oca J, … Moreno V (2013). DNA methylation biomarkers for noninvasive diagnosis of colorectal cancer. Cancer Prevention Research (Philadelphia, Pa.), 6(7), 656–665. 10.1158/1940-6207.CAPR-12-0501 [DOI] [PubMed] [Google Scholar]
- Cascinu S, Catalano V, Aschele C, Barni S, Debernardis D, Gallo L, … Catalano G (2000). Immunohistochemical determination of p53 protein does not predict clinical response in advanced colorectal cancer with low thymidylate synthase expression receiving a bolus 5-fluorouracil-leucovorin combination. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 11(8), 1053–1056. [DOI] [PubMed] [Google Scholar]
- Cecchin E, Innocenti F, D’Andrea M, Corona G, De Mattia E, Biason P, … Toffoli G (2009). Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 27(15), 2457–2465. 10.1200/JCO.2008.19.0314 [DOI] [PubMed] [Google Scholar]
- Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, … Haanen JB (2020). Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nature Medicine. 10.1038/s41591-020-0805-8 [DOI] [PubMed] [Google Scholar]
- Chang P-Y, Chen C-C, Chang Y-S, Tsai W-S, You J-F, Lin G-P, … Chan E-C (2016). MicroRNA-223 and microRNA-92a in stool and plasma samples act as complementary biomarkers to increase colorectal cancer detection. Oncotarget, 7(9), 10663–10675. 10.18632/oncotarget.7119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li N, Ren J, Feng X, Lyu Z, Wei L, … He J (2019). Participation and yield of a population-based colorectal cancer screening programme in China. Gut, 68(8), 1450–1457. 10.1136/gutjnl-2018-317124 [DOI] [PubMed] [Google Scholar]
- Chen S, Laverdiere I, Tourancheau A, Jonker D, Couture F, Cecchin E, … Guillemette C (2015). A novel UGT1 marker associated with better tolerance against irinotecan-induced severe neutropenia in metastatic colorectal cancer patients. The Pharmacogenomics Journal, 15(6), 513–520. 10.1038/tpj.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-D, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L, … Markowitz SD (2005). Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. Journal of the National Cancer Institute, 97(15), 1124–1132. 10.1093/jnci/dji204 [DOI] [PubMed] [Google Scholar]
- Chen X, Gole J, Gore A, He Q, Lu M, Min J, … Jin L (2020). Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nature Communications, 11(1), 3475. 10.1038/s41467-020-17316-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, … Ransohoff DF (2014). Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut, 63(2), 317–325. 10.1136/gutjnl-2012-304149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JW, Niedzwiecki D, Hollis D,P (2003). Phase-II trial of 5-fluororuacil (5-FU), leucovorin (LV), oxaliplatin (Ox), and trastuzumab (T) for patients with metastatic colorectal cancer (CRC) refractory to initial therapy. Onkologie, 26, 13–46.14605451 [Google Scholar]
- Clarke CN, & Kopetz ES (2015). BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. Journal of Gastrointestinal Oncology, 6(6), 660–667. 10.3978/j.issn.2078-6891.2015.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coakley M, Garcia-Murillas I, & Turner NC (2019). Molecular Residual Disease and Adjuvant Trial Design in Solid Tumors. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 25(20), 6026–6034. 10.1158/1078-0432.CCR-19-0152 [DOI] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, … Papadopoulos N (2018). Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science, 359(6378), 926 LP–930. 10.1126/science.aar3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R, Taieb J, Fiskum J, Yothers G, Goldberg R, Yoshino T, … Shi Q (2020). Microsatellite Instability in Patients With Stage III Colon Cancer Receiving Fluoropyrimidine With or Without Oxaliplatin: An ACCENT Pooled Analysis of 12 Adjuvant Trials. Journal of Clinical Oncology, JCO.20.01600. 10.1200/JCO.20.01600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, … Meropol NJ (2008). Relationship of Circulating Tumor Cells to Tumor Response, Progression-Free Survival, and Overall Survival in Patients With Metastatic Colorectal Cancer. Journal of Clinical Oncology, 26(19), 3213–3221. 10.1200/JCO.2007.15.8923 [DOI] [PubMed] [Google Scholar]
- ColonES Product Sheet. Singlera Genomics. https://singleraoncology.com/wp-content/uploads/2018/10/Singlera-ColonES-Product-Sheet-V2018OCT.pdf. Accesed on 6 December 2020. (n.d.).
- Corcoran RB, Andre T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, … Van Cutsem E (2018). Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, & Engelman JA (2010). BRAF Gene Amplification Can Promote Acquired Resistance to MEK Inhibitors in Cancer Cells Harboring the BRAF V600E Mutation. Science Signaling, 3(149), ra84 LP–ra84. 10.1126/scisignal.2001148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, … Engelman JA (2012). EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF-Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discovery, 2(3), 227 LP–235. 10.1158/2159-8290.CD-11-0341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, & Calin GA (2011). MicroRNAs in body fluids--the mix of hormones and biomarkers. Nature Reviews. Clinical Oncology, 8(8), 467–477. 10.1038/nrclinonc.2011.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P, Sahoo D, Paik S, Guo X, Yothers G, Song N, … Clarke MF (2016). CDX2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer. New England Journal of Medicine, 374(3), 211–222. 10.1056/NEJMoa1506597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, … Futreal PA (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949–954. 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, … Tejpar S (2010). Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet. Oncology, 11(8), 753–762. 10.1016/S1470-2045(10)70130-3 [DOI] [PubMed] [Google Scholar]
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, … Tejpar S (2010). Association of KRAS p.G13D Mutation With Outcome in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer Treated With Cetuximab. JAMA, 304(16), 1812–1820. 10.1001/jama.2010.1535 [DOI] [PubMed] [Google Scholar]
- Dean L (2012). Irinotecan Therapy and UGT1A1 Genotype. In Pratt VM, McLeod HL, Rubinstein WS, Scott SA, Dean LC, Kattman BL, & Malheiro AJ (Eds.). Bethesda (MD). [PubMed] [Google Scholar]
- Deenen MJ, Meulendijks D, Cats A, Sechterberger MK, Severens JL, Boot H, … Schellens JHM (2016). Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 34(3), 227–234. 10.1200/JCO.2015.63.1325 [DOI] [PubMed] [Google Scholar]
- Deng L, Chang D, Foshaug RR, Eisner R, Tso VK, Wishart DS, & Fedorak RN (2017). Development and Validation of a High-Throughput Mass Spectrometry Based Urine Metabolomic Test for the Detection of Colonic Adenomatous Polyps. Metabolites, 7(3), 32. 10.3390/metabo7030032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Guetz G, Uzzan B, Nicolas P, Schischmanoff O, & Morere J-F (2009). Microsatellite instability: a predictive marker in metastatic colorectal cancer? Targeted Oncology, 4(1), 57–62. 10.1007/s11523-008-0103-8 [DOI] [PubMed] [Google Scholar]
- Dias MM, McKinnon RA, & Sorich MJ (2012). Impact of the UGT1A1*28 allele on response to irinotecan: a systematic review and meta-analysis. Pharmacogenomics, 13(8), 889–899. 10.2217/pgs.12.68 [DOI] [PubMed] [Google Scholar]
- Diasio RB, Beavers TL, & Carpenter JT (1988). Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. The Journal of Clinical Investigation, 81(1), 47–51. 10.1172/JCI113308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, … Diaz LAJ (2008). Circulating mutant DNA to assess tumor dynamics. Nature Medicine, 14(9), 985–990. 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Mason MJ, Sinicrope FA, Phipps AI, Tejpar S, Nesbakken A, … Guinney J (2017). Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 28(5), 1023–1031. 10.1093/annonc/mdx052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S-J, Cai X-J, & Li S-J (2016). The Clinical Significance of MiR-429 as a Predictive Biomarker in Colorectal Cancer Patients Receiving 5-Fluorouracil Treatment. Medical Science Monitor : International Medical Journal of Experimental and Clinical Research, 22, 3352–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopeso H, Mateo-Lozano S, Elez E, Landolfi S, Ramos Pascual FJ, Hernandez-Losa J, … Arango D (2010). Aprataxin tumor levels predict response of colorectal cancer patients to irinotecan-based treatment. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 16(8), 2375–2382. 10.1158/1078-0432.CCR-09-3275 [DOI] [PubMed] [Google Scholar]
- Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, … Patterson SD (2013). Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. The New England Journal of Medicine, 369(11), 1023–1034. 10.1056/NEJMoa1305275 [DOI] [PubMed] [Google Scholar]
- Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, … Hyman DM (2018). Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. New England Journal of Medicine, 378(8), 731–739. 10.1056/NEJMoa1714448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, … Hyman DM (2017). A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discovery, 7(9), 63–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Siena S, Ou S-HI, Patel M, Ahn MJ, Lee J, … De Braud FG (2017). Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discovery, 7(4), 400 LP–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Sanchon S, Moreno L, Augé JM, Serra-Burriel M, Cuatrecasas M, Moreira L, … Castells A (2020). Identification and Validation of MicroRNA Profiles in Fecal Samples for Detection of Colorectal Cancer. Gastroenterology, 158(4), 947–957.e4. 10.1053/j.gastro.2019.10.005 [DOI] [PubMed] [Google Scholar]
- Ebert MPA, Tänzer M, Balluff B, Burgermeister E, Kretzschmar AK, Hughes DJ, … Schmid RM (2012). TFAP2E-DKK4 and chemoresistance in colorectal cancer. The New England Journal of Medicine, 366(1), 44–53. 10.1056/NEJMoa1009473 [DOI] [PubMed] [Google Scholar]
- EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine. https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine. Accessed Novem. (n.d.).
- Erlander MG, Ma X-J, Kesty NC, Bao L, Salunga R, & Schnabel CA (2011). Performance and clinical evaluation of the 92-gene real-time PCR assay for tumor classification. The Journal of Molecular Diagnostics : JMD, 13(5), 493–503. 10.1016/j.jmoldx.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M (2011). Non-coding RNAs in human disease. Nature Reviews Genetics, 12(12), 861–874. 10.1038/nrg3074 [DOI] [PubMed] [Google Scholar]
- Etienne-Grimaldi M-C, Boyer J-C, Thomas F, Quaranta S, Picard N, Loriot M-A, … Le Guellec C (2015). UGT1A1 genotype and irinotecan therapy: general review and implementation in routine practice. Fundamental & Clinical Pharmacology, 29(3), 219–237. 10.1111/fcp.12117 [DOI] [PubMed] [Google Scholar]
- Etienne-Grimaldi M-C, Formento J-L, Francoual M, Francois E, Formento P, Renee N, … Milano G (2008). K-Ras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 14(15), 4830–4835. 10.1158/1078-0432.CCR-07-4906 [DOI] [PubMed] [Google Scholar]
- Fearon ER, & Vogelstein B (1990). A genetic model for colorectal tumorigenesis. Cell, 61(5), 759–767. 10.1016/0092-8674(90)90186-i [DOI] [PubMed] [Google Scholar]
- Feinberg AP, & Tycko B (2004, February). The history of cancer epigenetics. Nature Reviews. Cancer. England. 10.1038/nrc1279 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, & Vogelstein B (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301(5895), 89–92. 10.1038/301089a0 [DOI] [PubMed] [Google Scholar]
- Flejou J-F, André T, Chibaudel B, Scriva A, Hickish T, Tabernero J, … De Gramont A (2013). Effect of adding oxaliplatin to adjuvant 5-fluorouracil/leucovorin (5FU/LV) in patients with defective mismatch repair (dMMR) colon cancer stage II and III included in the MOSIAC study. Journal of Clinical Oncology, 31(15_suppl), 3524. 10.1200/jco.2013.31.15_suppl.3524 [DOI] [Google Scholar]
- Fraga MF, Herranz M, Espada J, Ballestar E, Paz MF, Ropero S, … Esteller M (2004). A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Research, 64(16), 5527–5534. 10.1158/0008-5472.CAN-03-4061 [DOI] [PubMed] [Google Scholar]
- Friedland S, Pan JY, Watson D, Chen Y, Nimgaonkar A, Gulzar Z, … Mei R (2020). A sensitive and quantitative multimodal blood test for the detection of colorectal adenomas and cancer: Correlation with size and number of polyps. Journal of Clinical Oncology, 38(15_suppl), 1555. 10.1200/JCO.2020.38.15_suppl.1555 [DOI] [Google Scholar]
- Fu B, Yan P, Zhang S, Lu Y, Pan L, Tang W, … Liu W (2018). Cell-Free Circulating Methylated SEPT9 for Noninvasive Diagnosis and Monitoring of Colorectal Cancer. Disease Markers, 2018, 6437104. 10.1155/2018/6437104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Galon J, Costes A, Costes A, Sanchez-Cabo F, Sanchez-Cabo F, … Pagès F (2006). Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science, 313(September), 1960–1964. 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- Gansauge M-T, & Meyer M (2013). Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nature Protocols, 8(4), 737–748. 10.1038/nprot.2013.038 [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, … Sharma P (2016). Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell, 167(2), 397–404.e9. 10.1016/j.cell.2016.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, … Pogue-Geile KL (2012). Mutation Profiling and Microsatellite Instability in Stage II and III Colon Cancer: An Assessment of Their Prognostic and Oxaliplatin Predictive Value. Clinical Cancer Research, 18(23), 6531 LP–6541. 10.1158/1078-0432.CCR-12-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gezer U, Ustek D, Yörüker EE, Cakiris A, Abaci N, Leszinski G, … Holdenrieder S (2013). Characterization of H3K9me3- and H4K20me3-associated circulating nucleosomal DNA by high-throughput sequencing in colorectal cancer. Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology and Medicine, 34(1), 329–336. 10.1007/s13277-012-0554-5 [DOI] [PubMed] [Google Scholar]
- Gezer U, Yörüker EE, Keskin M, Kulle CB, Dharuman Y, & Holdenrieder S (2015). Histone Methylation Marks on Circulating Nucleosomes as Novel Blood-Based Biomarker in Colorectal Cancer. International Journal of Molecular Sciences, 16(12), 29654–29662. 10.3390/ijms161226180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharib E, Nazemalhosseini-Mojarad E, Baghdar K, Nayeri Z, Sadeghi H, Rezasoltani S, … Asadzadeh Aghdaei H (2020). Identification of a stool long non-coding RNAs panel as a potential biomarker for early detection of colorectal cancer. Journal of Clinical Laboratory Analysis, n/a(n/a), e23601. 10.1002/jcla.23601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, … Garraway LA (2016). Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Reports, 15(4), 857–865. 10.1016/j.celrep.2016.03.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckner SC, Dhir M, Yi JM, McGarvey KE, Van Neste L, Louwagie J, … Ahuja N (2009). Methylation of <em>TFPI2</em> in Stool DNA: A Potential Novel Biomarker for the Detection of Colorectal Cancer. Cancer Research, 69(11), 4691 LP–4699. 10.1158/0008-5472.CAN-08-0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, & Carethers JM (2008). Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology, 135(4), 1079–1099. 10.1053/j.gastro.2008.07.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli J, Elez E, Caratù G, Matito J, Santos C, Macarulla T, … Vivancos A (2017). Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Annals of Oncology, 28(6), 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, … Peters U (2018). Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discovery. [Google Scholar]
- Guastadisegni C, Colafranceschi M, Ottini L, & Dogliotti E (2010). Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. European Journal of Cancer (Oxford, England : 1990), 46(15), 2788–2798. 10.1016/j.ejca.2010.05.009 [DOI] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, … Tejpar S (2015). The consensus molecular subtypes of colorectal cancer. Nature Medicine, 21(11), 1350–1356. 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurjao C, Liu D, Hofree M, AlDubayan SH, Wakiro I, Su M-J, … Giannakis M (2019). Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair-Deficient Colorectal Cancer. Cancer Immunology Research, 7(8), 1230–1236. 10.1158/2326-6066.CIR-18-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson B, Kaiser C, Carlsson G, Wettergren Y, Odin E, Lindskog EB, … Ma D (2009). Molecular determinants of efficacy for 5-FU-based treatments in advanced colorectal cancer: mRNA expression for 18 chemotherapy-related genes. International Journal of Cancer, 124(5), 1220–1226. 10.1002/ijc.23852 [DOI] [PubMed] [Google Scholar]
- Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, … Kurzrock R (2018). Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. Journal of Clinical Oncology, 36(6), 536–542. 10.1200/JCO.2017.75.3780 [DOI] [PubMed] [Google Scholar]
- Hainsworth JD, Rubin MS, Spigel DR, Boccia RV, Raby S, Quinn R, & Greco FA (2012). Molecular Gene Expression Profiling to Predict the Tissue of Origin and Direct Site-Specific Therapy in Patients With Carcinoma of Unknown Primary Site: A Prospective Trial of the Sarah Cannon Research Institute. Journal of Clinical Oncology, 31(2), 217–223. 10.1200/JCO.2012.43.3755 [DOI] [PubMed] [Google Scholar]
- Hall D, Ybazeta G, Destro-Bisol G, Petzl-Erler ML, & Di Rienzo A (1999). Variability at the uridine diphosphate glucuronosyltransferase 1A1 promoter in human populations and primates. Pharmacogenetics, 9(5), 591–599. [PubMed] [Google Scholar]
- Hansen TPTF, Carlsen a L., Heegaard NHH, Sørensen FB, Jakobsen A, Zhang JJ, … Toffoli G (2014). TS gene polymorphisms are not good markers of response to 5-FU therapy in stage III colon cancer patients. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 14(1), 128–135. 10.1158/1078-0432.CCR-13-3271 [DOI] [Google Scholar]
- Hansen TF, Carlsen a L., Heegaard NHH, Sørensen FB, & Jakobsen a. (2015). Changes in circulating microRNA-126 during treatment with chemotherapy and bevacizumab predicts treatment response in patients with metastatic colorectal cancer. British Journal of Cancer, (October 2014), 1–6. 10.1038/bjc.2014.652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen Torben Frostrup, Nielsen BS, Jakobsen A, & Sorensen FB (2013). Visualising and quantifying angiogenesis in metastatic colorectal cancer : A comparison of methods and their predictive value for chemotherapy response. Cellular Oncology (Dordrecht), 36(4), 341–350. 10.1007/s13402-013-0139-3 [DOI] [PubMed] [Google Scholar]
- He N, Song L, Kang Q, Jin P, Cai G, Zhou J, … Wu K (2018). The Pathological Features of Colorectal Cancer Determine the Detection Performance on Blood ctDNA. Technology in Cancer Research & Treatment, 17, 1533033818791794. 10.1177/1533033818791794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Tan X, Wang X, Jin H, Liu L, Ma L, … Fan Z (2014). C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology and Medicine, 35(12), 12181–12188. 10.1007/s13277-014-2526-4 [DOI] [PubMed] [Google Scholar]
- Heinemann V, Kraemer N, Buchner H, Fischer von Weikersthal L, Decker T, Kiani A, … Stintzing S (2018). Somatic DNA mutations, tumor mutational burden (TMB), and MSI Status: Association with efficacy in patients (pts) with metastatic colorectal cancer (mCRC) of FIRE-3 (AIO KRK-0306). Journal of Clinical Oncology, 36(15_suppl), 3591. 10.1200/JCO.2018.36.15_suppl.359130372390 [DOI] [Google Scholar]
- Henricks LM, Lunenburg CATC, de Man FM, Meulendijks D, Frederix GWJ, Kienhuis E, … Schellens JHM (2018). DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. The Lancet. Oncology, 19(11), 1459–1467. 10.1016/S1470-2045(18)30686-7 [DOI] [PubMed] [Google Scholar]
- Henricks LM, Lunenburg CATC, de Man FM, Meulendijks D, Frederix GWJ, Kienhuis E, … Schellens JHM (2019). A cost analysis of upfront DPYD genotype-guided dose individualisation in fluoropyrimidine-based anticancer therapy. European Journal of Cancer (Oxford, England : 1990), 107, 60–67. 10.1016/j.ejca.2018.11.010 [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Duran-Sanchon S, Martín AC, Pérez-Palacios R, Vila-Navarro E, Marcuello M, … Arroyo R (2019). Plasma MicroRNA Signature Validation for Early Detection of Colorectal Cancer. Clinical and Translational Gastroenterology, 10(1), e00003. 10.14309/ctg.0000000000000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, & Esteller M (2012). DNA methylation profiling in the clinic: applications and challenges. Nature Reviews. Genetics, 13(10), 679–692. 10.1038/nrg3270 [DOI] [PubMed] [Google Scholar]
- Hiraki M, Kitajima Y, Nakafusa Y, Nakamura J, Hashiguchi K, Sumi K, … Miyazaki K (2010). CpG island methylation of BNIP3 predicts resistance against S-1/CPT-11 combined therapy in colorectal cancer patients. Oncology Reports, 23(1), 191–197. [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, … Li BT (2020). KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. New England Journal of Medicine, 383(13), 1207–1217. 10.1056/NEJMoa1917239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins JM, Marcuello E, Altes A, Marsh S, Maxwell T, Van Booven DJ, … Baiget M (2008). Irinotecan pharmacogenetics: Influence of pharmacodynamic genes. Clinical Cancer Research, 14(6), 1788–1796. 10.1158/1078-0432.CCR-07-1472 [DOI] [PubMed] [Google Scholar]
- https://ir.mirati.com/news-releases/news-details/2020/Mirati-Therapeutics-Reports-Investigational-Adagrasib-MRTX849-Preliminary-Data-Demonstrating-Tolerability-and-Durable-Anti-Tumor-Activity-as-well-as-Initial-MRTX1133-Preclinical-Data/default.aspx.
- Hurwitz HI, Yi J, Ince W, Novotny WF, & Rosen O (2009). The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. The Oncologist, 14(1), 22–28. 10.1634/theoncologist.2008-0213 [DOI] [PubMed] [Google Scholar]
- Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, … Quirke P (2011). Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 29(10), 1261–1270. 10.1200/JCO.2010.30.1366 [DOI] [PubMed] [Google Scholar]
- Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, … Berger BM (2014). Multitarget stool DNA testing for colorectal-cancer screening. The New England Journal of Medicine, 370(14), 1287–1297. 10.1056/NEJMoa1311194 [DOI] [PubMed] [Google Scholar]
- Innocenti F, Mills SC, Sanoff H, Ciccolini J, Lenz H-J, & Milano G (2020). All You Need to Know About DPYD Genetic Testing for Patients Treated With Fluorouracil and Capecitabine: A Practitioner-Friendly Guide. JCO Oncology Practice, OP.20.00553. 10.1200/OP.20.00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, … Ratain MJ (2004). Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 22(8), 1382–1388. 10.1200/JCO.2004.07.173 [DOI] [PubMed] [Google Scholar]
- Itzkowitz SH, Jandorf L, Brand R, Rabeneck L, Schroy PC 3rd, Sontag S, … Shuber A (2007). Improved fecal DNA test for colorectal cancer screening. Clinical Gastroenterology and Hepatology : The Official Clinical Practice Journal of the American Gastroenterological Association, 5(1), 111–117. 10.1016/j.cgh.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, … Liu Y (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell, 172(3), 578–589.e17. 10.1016/j.cell.2018.01.006 [DOI] [PubMed] [Google Scholar]
- Jensen LH, Danenberg KD, Danenberg PV, & Jakobsen A (2007). Predictive value of MSH2 gene expression in colorectal cancer treated with capecitabine. Clinical Colorectal Cancer, 6(6), 433–435. 10.3816/CCC.2007.n.012 [DOI] [PubMed] [Google Scholar]
- Jin Z, Sanhueza CT, Johnson B, Nagorney DM, Larson DW, Mara KC, … Hubbard JM (2018). Outcome of Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Mayo Clinic Experience. The Oncologist, 23(9), 1083–1091. 10.1634/theoncologist.2017-0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo W-S, & Carethers JM (2006). Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomarkers : Section A of Disease Markers, 2(1–2), 51–60. 10.3233/cbm-2006-21-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston PG, Benson AB 3rd, Catalano P, Rao MS, O’Dwyer PJ, & Allegra CJ (2003). Thymidylate synthase protein expression in primary colorectal cancer: lack of correlation with outcome and response to fluorouracil in metastatic disease sites. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 21(5), 815–819. 10.1200/JCO.2003.07.039 [DOI] [PubMed] [Google Scholar]
- Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, … Grothey A (2017). (Non-V600) BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 35(23), 2624–2630. 10.1200/JCO.2016.71.4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, … Alenda C (2009). The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. European Journal of Cancer, 45(3), 365–373. 10.1016/j.ejca.2008.07.016 [DOI] [PubMed] [Google Scholar]
- Jung G, Hernández-Illán E, Moreira L, Balaguer F, & Goel A (2020). Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nature Reviews Gastroenterology & Hepatology, 17(2), 111–130. 10.1038/s41575-019-0230-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandimalla R, Xu J, Link A, Matsuyama T, Yamamura K, Parker I, … Goel A (2020). Abstract 1084: EpiPanGI-Dx: A cell-free DNA methylation fingerprint for the early detection of gastrointestinal cancers. Cancer Research, 80(16 Supplement), 1084 LP–1084. 10.1158/1538-7445.AM2020-1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, … Ding L (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502(7471), 333–339. 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczmarski J, Rubel T, Paziewska A, Mikula M, Bujko M, Kober P, … Ostrowski J (2014). Histone H3 lysine 27 acetylation is altered in colon cancer. Clinical Proteomics, 11(1), 24. 10.1186/1559-0275-11-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K, Rata M, Cunningham D, Koh D-M, Tunariu N, Hahne JC, … Valeri N (2017). Functional imaging and circulating biomarkers of response to regorafenib in treatment-refractory metastatic colorectal cancer patients in a prospective phase II study. Gut. 10.1136/gutjnl-2017-314178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CG, Ahn JB, Jung M, Beom SH, Kim C, Kim JH, … Shin SJ (2016). Effects of microsatellite instability on recurrence patterns and outcomes in colorectal cancers. British Journal of Cancer, 115(1), 25–33. 10.1038/bjc.2016.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisiel JB, Yab TC, Nazer Hussain FT, Taylor WR, Garrity-Park MM, Sandborn WJ, … Ahlquist DA (2013). Stool DNA testing for the detection of colorectal neoplasia in patients with inflammatory bowel disease. Alimentary Pharmacology & Therapeutics, 37(5), 546–554. 10.1111/apt.12218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingbiel D, Saridaki Z, Roth AD, Bosman FT, Delorenzi M, & Tejpar S (2015). Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: results of the PETACC-3 trial. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 26(1), 126–132. 10.1093/annonc/mdu499 [DOI] [PubMed] [Google Scholar]
- Koopman M, Knijn N, Richman S, Seymour M, Quirke P, van Tinteren H, … Nagtegaal ID (2009). The correlation between Topoisomerase-I (Topo1) expression and outcome of treatment with capecitabine and irinotecan in advanced colorectal cancer (ACC) patients (pts) treated in the CAIRO study of the Dutch Colorectal Cancer Group (DCCG). EJC Supplements, 7(2), 321–322. 10.1016/S1359-6349(09)71098-5 [DOI] [Google Scholar]
- Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, … Tabernero J (2019). Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. New England Journal of Medicine, 381(17), 1632–1643. 10.1056/NEJMoa1908075 [DOI] [PubMed] [Google Scholar]
- Kopetz S, Guthrie KA, Morris VK, Lenz H-J, Magliocco AM, Maru D, … Hochster HS (2020). Randomized Trial of Irinotecan and Cetuximab With or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). Journal of Clinical Oncology, JCO.20.01994. 10.1200/JCO.20.01994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW (2003). The power and the promise of DNA methylation markers. Nature Reviews. Cancer, 3(4), 253–266. 10.1038/nrc1045 [DOI] [PubMed] [Google Scholar]
- Lam YWF (2019). Chapter 1 - Principles of Pharmacogenomics: Pharmacokinetic, Pharmacodynamic, and Clinical Implications. In Lam YWF & S. A. B. T.-P. (Scott Second E. (Eds.) (pp. 1–53). Academic Press. 10.1016/B978-0-12-812626-4.00001-2 [DOI] [Google Scholar]
- Lambrechts D, Thienpont B, Thuillier V, Sagaert X, Moisse M, Peuteman G, … Van Cutsem E (2015). Evaluation of efficacy and safety markers in a phase II study of metastatic colorectal cancer treated with aflibercept in the first-line setting. British Journal of Cancer, 113(7), 1027–1034. 10.1038/bjc.2015.329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza G, Gafà R, Santini A, Maestri I, Guerzoni L, & Cavazzini L (2006). Immunohistochemical test for MLH1 and MSH2 expression predicts clinical outcome in stage II and III colorectal cancer patients. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 24(15), 2359–2367. 10.1200/JCO.2005.03.2433 [DOI] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, … Diaz LAJ (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, N.Y.), 357(6349), 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, … Diaz LA (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine, 372(26), 2509–2520. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-K, Jeong W-J, Cho Y-H, Cha P-H, Yoon J-S, Ro EJ, … Choi K-Y (2018). β-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3β. EMBO Reports, 19(12). 10.15252/embr.201846060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Chen W-D, Papadopoulos N, Goodman SN, Bjerregaard NC, Laurberg S, … Vogelstein B (2009). Sensitive digital quantification of DNA methylation in clinical samples. Nature Biotechnology, 27(9), 858–863. 10.1038/nbt.1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, … Ogino S (2012). Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 18(8), 2257–2268. 10.1158/1078-0432.CCR-11-2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, … Laurent-Puig P (2006). KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Research, 66(8), 3992–3995. 10.1158/0008-5472.CAN-06-0191 [DOI] [PubMed] [Google Scholar]
- Lindskog EB, Derwinger K, Gustavsson B, Falk P, & Wettergren Y (2014). Thymidine phosphorylase expression is associated with time to progression in patients with metastatic colorectal cancer. BMC Clinical Pathology, 14, 25. 10.1186/1472-6890-14-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G-H, Zhou Z-G, Chen R, Wang M-J, Zhou B, Li Y, & Sun X-F (2013). Serum miR-21 and miR-92a as biomarkers in the diagnosis and prognosis of colorectal cancer. Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology and Medicine, 34(4), 2175–2181. 10.1007/s13277-013-0753-8 [DOI] [PubMed] [Google Scholar]
- Liu H-N, Liu T-T, Wu H, Chen Y-J, Tseng Y-J, Yao C, … Shen X-Z (2018). Serum microRNA signatures and metabolomics have high diagnostic value in colorectal cancer using two novel methods. Cancer Science, 109(4), 1185–1194. 10.1111/cas.13514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Klein E, Hubbell E, Maddala T, Aravanis AM, Beausang JF, … Oxnard G (2018). Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Annals of Oncology, 29, viii14. 10.1093/annonc/mdy269.048 [DOI] [Google Scholar]
- Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, … Berry DA (2020). Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology, 31(6), 745–759. 10.1016/j.annonc.2020.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cheng D, Kuang Q, Liu G, & Xu W (2014). Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. The Pharmacogenomics Journal, 14(2), 120–129. 10.1038/tpj.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothe RA, Peltomäki P, Meling GI, Aaltonen LA, Nyström-Lahti M, Pylkkänen L, … Børresen A-L (1993). Genomic Instability in Colorectal Cancer: Relationship to Clinicopathological Variables and Family History. Cancer Research, 53(24), 5849 LP–5852. [PubMed] [Google Scholar]
- Lu H, Huang S, Zhang X, Wang D, Zhang X, Yuan X, … Huang Z (2014). DNA methylation analysis of SFRP2, GATA4/5, NDRG4 and VIM for the detection of colorectal cancer in fecal DNA. Oncology Letters, 8(4), 1751–1756. 10.3892/ol.2014.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, … Golub TR (2005). MicroRNA expression profiles classify human cancers. Nature, 435(7043), 834–838. 10.1038/nature03702 [DOI] [PubMed] [Google Scholar]
- Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, … Xu R (2020). Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Science Translational Medicine, 12(524), eaax7533. 10.1126/scitranslmed.aax7533 [DOI] [PubMed] [Google Scholar]
- Luo Y, Yu M, & Grady WM (2014). Field cancerization in the colon: a role for aberrant DNA methylation? Gastroenterology Report, 2(1), 16–20. 10.1093/gastro/got039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X-J, Patel R, Wang X, Salunga R, Murage J, Desai R, … Erlander M (2006). Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Archives of Pathology & Laboratory Medicine, 130(4), 465–473. 10.1043/1543-2165(2006)130[465:MCOHCU]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Malathi N, Mythili S, & Vasanthi HR (2014). Salivary diagnostics: a brief review. ISRN Dentistry, 2014, 158786. 10.1155/2014/158786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau G, Imbeaud S, Thiébaut R, Liébaert F, Fontaine K, Rousseau F, … Laurent-Puig P (2014). Hsa-miR-31-3p expression is linked to progression-free survival in patients with KRAS wild-type metastatic colorectal cancer treated with anti-EGFR therapy. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 20(12), 3338–3347. 10.1158/1078-0432.CCR-13-2750 [DOI] [PubMed] [Google Scholar]
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, … Hanski C (1999). Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proceedings of the National Academy of Sciences of the United States of America, 96(4), 1603–1608. 10.1073/pnas.96.4.1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margonis GA, Buettner S, Wagner D, McVey J, Andreatos N, Beer A, … Weiss MJ (2018). Microsatellite instability in resectable colorectal liver metastasis: An international multi-institutional analysis. Journal of Clinical Oncology, 36(4_suppl), 220. 10.1200/JCO.2018.36.4_suppl.22029206554 [DOI] [Google Scholar]
- Mattison LK, Soong R, & Diasio RB (2002). Implications of dihydropyrimidine dehydrogenase on 5-fluorouracil pharmacogenetics and pharmacogenomics. Pharmacogenomics, 3(4), 485–492. 10.1517/14622416.3.4.485 [DOI] [PubMed] [Google Scholar]
- McCarthy AJ, Serra S, & Chetty R (2019). Traditional serrated adenoma: an overview of pathology and emphasis on molecular pathogenesis. BMJ Open Gastroenterology, 6(1), e000317. 10.1136/bmjgast-2019-000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiri E, Mueller WC, Rosenwald S, Zepeniuk M, Klinke E, Edmonston TB, … Aharonov R (2012). A second-generation microRNA-based assay for diagnosing tumor tissue origin. The Oncologist, 17(6), 801–812. 10.1634/theoncologist.2011-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merok MA, Ahlquist T, Røyrvik EC, Tufteland KF, Hektoen M, Sjo OH, … Nesbakken A (2013). Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 24(5), 1274–1282. 10.1093/annonc/mds614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middha S, Yaeger R, Shia J, Stadler ZK, King S, Guercio S, … Hechtman JF (2019). Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and Are Microsatellite Instability-High. JCO Precision Oncology, (3), 1–14. 10.1200/PO.18.00321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Phillips L, Fischer von Weikersthal L, Kaiser F, Al-Batran S-E, Heintges T, Neureiter D, … Stintzing S (2018). Association of microRNA-21 (miR-21) with efficacy of cetuximab (cet) and bevacizumab (bev) in patients with metastatic colorectal cancer (mCRC) within the FIRE-3 study (AIO KRK-0306). Annals of Oncology, 29, viii39. 10.1093/annonc/mdy269.122 [DOI] [Google Scholar]
- Miller-Phillips Lisa, Heinemann V, Stahler A, Fischer von Weikersthal L, Kaiser F, Al-Batran S-E, … Stintzing S (2019). Association of microRNA-21 with efficacy of cetuximab in RAS wild-type patients in the FIRE-3 study (AIO KRK-0306) and microRNA-21’s influence on gene expression in the EGFR signaling pathway. Journal of Clinical Oncology, 37(15_suppl), 3593. 10.1200/JCO.2019.37.15_suppl.3593 [DOI] [Google Scholar]
- Mlecnik B, Bifulco C, Bindea G, Marliot F, Lugli A, Lee JJ, … Galon J (2020). Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. Journal of Clinical Oncology, 38(31), 3638–3651. 10.1200/JCO.19.03205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Martinez-Cardus A, Sayols S, Musulen E, Balana C, Estival-Gonzalez A, … Esteller M (2016). Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. The Lancet. Oncology, 17(10), 1386–1395. 10.1016/S1470-2045(16)30297-2 [DOI] [PubMed] [Google Scholar]
- Murcia O, Jover R, Egoavil C, Perez-Carbonell L, Juárez M, Hernández-Illán E, … Goel A (2018). TFAP2E Methylation and Expression Status Does Not Predict Response to 5-FU-based Chemotherapy in Colorectal Cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 24(12), 2820–2827. 10.1158/1078-0432.CCR-17-2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Kondo T, Ma D, Niu D, Mochizuki K, Kawasaki T, … Katoh R (2012). Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Human Pathology, 43(6), 834–842. 10.1016/j.humpath.2011.07.009 [DOI] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network. Colon Cancer (Version 4.2020). https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed December 6, 2020. (n.d.).
- Ned RM, Melillo S, & Marrone M (2011). Fecal DNA testing for Colorectal Cancer Screening: the ColoSureTM test. PLoS Currents, 3, RRN1220. 10.1371/currents.RRN1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Zeindl-Eberhart E, Kirchner T, & Jung A (2009). Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathology, Research and Practice, 205(12), 858–862. 10.1016/j.prp.2009.07.010 [DOI] [PubMed] [Google Scholar]
- Ng EKO, Chong WWS, Jin H, Lam EKY, Shin VY, Yu J, … Sung JJY (2009). Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut, 58(10), 1375–1381. 10.1136/gut.2008.167817 [DOI] [PubMed] [Google Scholar]
- Nguyen LH, Goel A, & Chung DC (2020). Pathways of Colorectal Carcinogenesis. Gastroenterology, 158(2), 291–302. 10.1053/j.gastro.2019.08.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissan A, Stojadinovic A, Mitrani-Rosenbaum S, Halle D, Grinbaum R, Roistacher M, … Gure AO (2012). Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. International Journal of Cancer, 130(7), 1598–1606. 10.1002/ijc.26170 [DOI] [PubMed] [Google Scholar]
- O’Dwyer PJ, & Catalano RB (2006). Uridine diphosphate glucuronosyltransferase (UGT) 1A1 and irinotecan: practical pharmacogenomics arrives in cancer therapy. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 24(28), 4534–4538. 10.1200/JCO.2006.07.3031 [DOI] [PubMed] [Google Scholar]
- Ojala KA, Kilpinen SK, & Kallioniemi OP (2011). Classification of unknown primary tumors with a data-driven method based on a large microarray reference database. Genome Medicine, 3(9), 63. 10.1186/gm279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman MJ, Lonardi S, Leone F, McDermott RS, Morse MA, Wong KYM, … Andre T (2017). Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: Update from CheckMate 142. Journal of Clinical Oncology, 35(4_suppl), 519. 10.1200/JCO.2017.35.4_suppl.519 [DOI] [Google Scholar]
- Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, … André T (2018). Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. Journal of Clinical Oncology, 36(8), 773–779. 10.1200/JCO.2017.76.9901 [DOI] [PubMed] [Google Scholar]
- Oxnard GR, Klein EA, Seiden MV, Hubbell E, Venn O, Jamshidi A, … Liu MC (2019). LBA77 - Simultaneous multi-cancer detection and tissue of origin (TOO) localization using targeted bisulfite sequencing of plasma cell-free DNA (cfDNA). Annals of Oncology, 30, v912. 10.1093/annonc/mdz394.074 [DOI] [Google Scholar]
- Ozawa T, Matsuyama T, Toiyama Y, Takahashi N, Ishikawa T, Uetake H, … Goel A (2017). CCAT1 and CCAT2 long noncoding RNAs, located within the 8q.24.21 “gene desert”, serve as important prognostic biomarkers in colorectal cancer. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 28(8), 1882–1888. 10.1093/annonc/mdx248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insert Package. Camptosar® (Irinotecan) Injection, intravenous infusion. 2019. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020571s050lbl.pdf. Accessed November 16, 2020. (n.d.).
- Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, … Galon J (2018). International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. The Lancet, 391(10135), 2128–2139. 10.1016/S0140-6736(18)30789-X [DOI] [PubMed] [Google Scholar]
- Parikh AR, Lee F-C, Yau L, Koh H, Knost J, Mitchell EP, … Lenz H-J (2019). MAVERICC, a Randomized, Biomarker-stratified, Phase II Study of mFOLFOX6-Bevacizumab versus FOLFIRI-Bevacizumab as First-line Chemotherapy in Metastatic Colorectal Cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 25(10), 2988–2995. 10.1158/1078-0432.CCR-18-1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-K, Baek HL, Yu J, Kim JY, Yang H-J, Jung YS, … Park D Il (2017). Is methylation analysis of SFRP2, TFPI2, NDRG4, and BMP3 promoters suitable for colorectal cancer screening in the Korean population? Intestinal Research, 15(4), 495–501. 10.5217/ir.2017.15.4.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters M, Oliner KS, P. A. (2010). Use of massively parallel, next-generation sequencing to identify gene mutations beyond KRAS that predict response to panitumumab in a randomized, phase 3, monotherapy study of metastatic colorectal cancer (mCRC). American Association for Cancer Research 101st Annual Meeting. Washington, DC, USA., April 17–2. [Google Scholar]
- Peng Q, Zhang X, Min M, Zou L, Shen P, & Zhu Y (2017). The clinical role of microRNA-21 as a promising biomarker in the diagnosis and prognosis of colorectal cancer: a systematic review and meta-analysis. Oncotarget, 8(27), 44893–44909. 10.18632/oncotarget.16488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Tejpar S, Sartore-Bianchi A, … Cremolini C (2017). ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. JNCI: Journal of the National Cancer Institute, 109(12), djx089–djx089. [DOI] [PubMed] [Google Scholar]
- Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, … Barni S (2015). Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. European Journal of Cancer (Oxford, England : 1990), 51(5), 587–594. 10.1016/j.ejca.2015.01.054 [DOI] [PubMed] [Google Scholar]
- Popat S (2004). Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. Journal of Clinical Oncology, 23(3), 609–618. 10.1200/JCO.2005.01.086 [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, … Kinzler KW (1992). APC mutations occur early during colorectal tumorigenesis. Nature, 359(6392), 235–237. 10.1038/359235a0 [DOI] [PubMed] [Google Scholar]
- Quintero E, Castells A, Bujanda L, Cubiella J, Salas D, Lanas Á, … González-Navarro A (2012). Colonoscopy versus Fecal Immunochemical Testing in Colorectal-Cancer Screening. New England Journal of Medicine, 366(8), 697–706. 10.1056/NEJMoa1108895 [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, & Velculescu VE (2002). Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature, 418(6901), 934. 10.1038/418934a [DOI] [PubMed] [Google Scholar]
- Ramanathan RK, Hwang JJ, Zamboni WC, Sinicrope FA, Safran H, Wong MK, … Finkelstein S (2004). Low overexpression of HER-2/neu in advanced colorectal cancer limits the usefulness of trastuzumab (Herceptin) and irinotecan as therapy. A phase II trial. Cancer Investigation, 22(6), 858–865. [DOI] [PubMed] [Google Scholar]
- Rapado-González Ó, Majem B, Álvarez-Castro A, Díaz-Peña R, Abalo A, Suárez-Cabrera L, … Suarez-Cunqueiro MM (2019). A Novel Saliva-Based miRNA Signature for Colorectal Cancer Diagnosis. Journal of Clinical Medicine. 10.3390/jcm8122029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MH, Jensen NF, Tarpgaard LS, Qvortrup C, Romer MU, Stenvang J, … Andersen CL (2013). High expression of microRNA-625-3p is associated with poor response to first-line oxaliplatin based treatment of metastatic colorectal cancer. Molecular Oncology, 7(3), 637–646. 10.1016/j.molonc.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? (2009). Genetics in Medicine : Official Journal of the American College of Medical Genetics, 11(1), 15–20. 10.1097/GIM.0b013e31818efd9d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, … Lindbjerg Andersen C (2019). Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncology, 5(8), 1124–1131. 10.1001/jamaoncol.2019.0528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, … Gallinger S (2003). Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. The New England Journal of Medicine, 349(3), 247–257. 10.1056/NEJMoa022289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzoni M, Manzoni M, Mariucci S, Loupakis F, Brugnatelli S, Bencardino K, … Danova M (2010). Circulating endothelial cells and endothelial progenitors as predictive markers of clinical response to bevacizumab-based first-line treatment in advanced colorectal cancer patients. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 21(12), 2382–2389. 10.1093/annonc/mdq261 [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, … Barshack I (2008). MicroRNAs accurately identify cancer tissue origin. Nature Biotechnology, 26(4), 462–469. 10.1038/nbt1392 [DOI] [PubMed] [Google Scholar]
- Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, & Sorich MJ (2015). Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. British Journal of Cancer, 112(12), 1888–1894. 10.1038/bjc.2015.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Bañobre J, & Goel A (2019). DNA Mismatch Repair Deficiency and Immune Checkpoint Inhibitors in Gastrointestinal Cancers. Gastroenterology. 10.1053/j.gastro.2018.11.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Bañobre J, Kandimalla R, & Goel A (2019). Predictive Biomarkers in Metastatic Colorectal Cancer: A Systematic Review. JCO Precision Oncology, (3), 1–17. 10.1200/PO.18.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Bañobre J, Roy R, Alustiza Fernández M, Murcia Ó, Jover R, Pera M, … Goel A (2020). Clinical significance of a microRNA signature for the identification and predicting prognosis in colorectal cancers with mucinous differentiation. Carcinogenesis, 41(11), 1498–1506. 10.1093/carcin/bgaa097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, … Danenberg PV (2000). Colorectal Tumors Responding to 5-Fluorouracil Have Low Gene Expression Levels of Dihydropyrimidine Dehydrogenase, Thymidylate Synthase, and Thymidine Phosphorylase. Clinical Cancer Research, 6(4), 1322 LP–1327. [PubMed] [Google Scholar]
- Samowitz WS, Curtin K, Ma KN, Schaffer D, Coleman LW, Leppert M, & Slattery ML (2001). Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiology, Biomarkers & Prevention : A Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology, 10(9), 917–923. [PubMed] [Google Scholar]
- Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, … Ruzzo A (2008). High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. The Oncologist, 13(12), 1270–1275. 10.1634/theoncologist.2008-0181 [DOI] [PubMed] [Google Scholar]
- Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, … Gallinger S (2010). Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 28(20), 3219–3226. 10.1200/JCO.2009.27.1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A, … Siena S (2016). Sensitivity to Entrectinib Associated With a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. JNCI: Journal of the National Cancer Institute, 108(1), djv306–djv306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, … Siena S (2017). Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. The Lancet Oncology, 17(6), 738–746. 10.1016/S1470-2045(16)00150-9 [DOI] [PubMed] [Google Scholar]
- Sazanov AA, Kiselyova EV, Zakharenko AA, Romanov MN, & Zaraysky MI (2017). Plasma and saliva miR-21 expression in colorectal cancer patients. Journal of Applied Genetics, 58(2), 231–237. 10.1007/s13353-016-0379-9 [DOI] [PubMed] [Google Scholar]
- Shulman K, Cohen I, Barnett-Griness O, Kuten A, Gruber SB, Lejbkowicz F, & Rennert G (2011). Clinical implications of UGT1A1*28 genotype testing in colorectal cancer patients. Cancer, 117(14), 3156–3162. 10.1002/cncr.25735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, … Jemal A (2020). Colorectal cancer statistics, 2020. CA: A Cancer Journal for Clinicians, 70(3), 145–164. 10.3322/caac.21601 [DOI] [PubMed] [Google Scholar]
- Siena S, Di Bartolomeo M, Raghav KPS, Masuishi T, Loupakis F, Kawakami H, … Yoshino T (2020). A phase II, multicenter, open-label study of trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing metastatic colorectal cancer (mCRC): DESTINY-CRC01. Journal of Clinical Oncology, 38(15_suppl), 4000. 10.1200/JCO.2020.38.15_suppl.4000 [DOI] [Google Scholar]
- Simmer F, Venderbosch S, Dijkstra JR, Vink-Borger EM, Faber C, Mekenkamp LJ, … Nagtegaal ID (2015). MicroRNA-143 is a putative predictive factor for the response to fluoropyrimidine-based chemotherapy in patients with metastatic colorectal cancer. Oncotarget, 6(26), 22996–23007. 10.18632/oncotarget.4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinicrope FA, Foster NR, Thibodeau SN, Marsoni S, Monges G, Labianca R, … Sargent DJ (2011). DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. Journal of the National Cancer Institute, 103(11), 863–875. 10.1093/jnci/djr153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinicrope FA, Mahoney MR, Smyrk TC, Thibodeau SN, Warren RS, Bertagnolli MM, … Alberts SR (2013). Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 31(29), 3664–3672. 10.1200/JCO.2013.48.9591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinicrope FA, Yoon HH, Mahoney MR, Nelson GD, Thibodeau SN, Goldberg RM, … Alberts SR (2014). Overall survival result and outcomes by KRAS, BRAF, and DNA mismatch repair in relation to primary tumor site in colon cancers from a randomized trial of adjuvant chemotherapy: NCCTG (Alliance) N0147. Journal of Clinical Oncology, 32(15_suppl), 3525. 10.1200/jco.2014.32.15_suppl.3525 [DOI] [Google Scholar]
- Siravegna G, Lazzari L, Crisafulli G, Sartore-Bianchi A, Mussolin B, Cassingena A, … Bardelli A (2018). Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell, 34(1), 148–162.e7. 10.1016/j.ccell.2018.06.004 [DOI] [PubMed] [Google Scholar]
- Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, … Bardelli A (2015). Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nature Medicine, 21, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smorenburg CH, Peters GJ, van Groeningen CJ, Noordhuis P, Smid K, van Riel AMGH, … Giaccone G (2006). Phase II study of tailored chemotherapy for advanced colorectal cancer with either 5-fluouracil and leucovorin or oxaliplatin and irinotecan based on the expression of thymidylate synthase and dihydropyrimidine dehydrogenase. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 17(1), 35–42. 10.1093/annonc/mdj046 [DOI] [PubMed] [Google Scholar]
- Song L, Jia J, Peng X, Xiao W, & Li Y (2017). The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Scientific Reports, 7(1), 3032. 10.1038/s41598-017-03321-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Peng X, Li Y, Xiao W, Jia J, Dong C, … Han X (2017). The SEPT9 gene methylation assay is capable of detecting colorectal adenoma in opportunistic screening. Epigenomics, 9(5), 599–610. 10.2217/epi-2016-0146 [DOI] [PubMed] [Google Scholar]
- Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, K. C. S. (2015). Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 26(1), 13–21. 10.1126/scitranslmed.3002442 [DOI] [PubMed] [Google Scholar]
- Stiksma J, Grootendorst DC, & van der Linden PWG (2014). CA 19-9 As a Marker in Addition to CEA to Monitor Colorectal Cancer. Clinical Colorectal Cancer, 13(4), 239–244. 10.1016/j.clcc.2014.09.004 [DOI] [PubMed] [Google Scholar]
- Stintzing S, Miller-Phillips L, Modest DP, Fischer von Weikersthal L, Decker T, Kiani A, … Heinemann V (2017). Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: analysis of the FIRE-3 (AIO KRK-0306) study. European Journal of Cancer, 79, 50–60. 10.1016/j.ejca.2017.03.023 [DOI] [PubMed] [Google Scholar]
- Stoehlmacher J, Ghaderi V, Iobal S, Groshen S, Tsao-Wei D, Park D, & Lenz HJ (2001). A polymorphism of the XRCC1 gene predicts for response to platinum based treatment in advanced colorectal cancer. Anticancer Research, 21(4B), 3075–3079. [PubMed] [Google Scholar]
- Strassburg CP (2008). Pharmacogenetics of Gilbert’s syndrome. Pharmacogenomics, 9(6), 703–715. 10.2217/14622416.9.6.703 [DOI] [PubMed] [Google Scholar]
- Strimpakos AS, Cunningham D, Mikropoulos C, Petkar I, Barbachano Y, & Chau I (2010). The impact of carcinoembryonic antigen flare in patients with advanced colorectal cancer receiving first-line chemotherapy. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 21(5), 1013–1019. 10.1093/annonc/mdp449 [DOI] [PubMed] [Google Scholar]
- Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Murgioni S, … Marmorino F (2017). Genetic variants of DNA repair-related genes predict efficacy of TAS-102 in patients with refractory metastatic colorectal cancer, (February), 1–8. 10.1093/annonc/mdx035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveen A, Kopetz S, & Lothe RA (2020). Biomarker-guided therapy for colorectal cancer: strength in complexity. Nature Reviews Clinical Oncology, 17(1), 11–32. 10.1038/s41571-019-0241-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda M, Slyskova J, Schneiderova M, Makovicky P, Bielik L, Levy M, … Vodicka P (2014). HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis, 35(7), 1510–1515. 10.1093/carcin/bgu055 [DOI] [PubMed] [Google Scholar]
- Swanton C, Venn O, Aravanis A, Hubbell E, Maddala T, Beausang JF, … Baselga J (2018). Prevalence of clonal hematopoiesis of indeterminate potential (CHIP) measured by an ultra-sensitive sequencing assay: Exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. Journal of Clinical Oncology, 36(15_suppl), 12003. 10.1200/JCO.2018.36.15_suppl.12003 [DOI] [Google Scholar]
- Tabernero J, Hozak RR, Yoshino T, Cohn AL, Obermannova R, Bodoky G, … Van Cutsem E (2018). Analysis of angiogenesis biomarkers for ramucirumab efficacy in patients with metastatic colorectal cancer from RAISE, a global, randomized, double-blind, phase III study. Annals of Oncology, 29(3), 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Cuatrecasas M, Balaguer F, Hur K, Toiyama Y, Castells A, … Goel A (2012). The clinical significance of MiR-148a as a predictive biomarker in patients with advanced colorectal cancer. PloS One, 7(10), e46684. 10.1371/journal.pone.0046684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Dienstmann R, Rachid MH, Boekel J, Baird R, Braña I, … consortium CCE (2020). Support systems to guide clinical decision-making in precision oncology: The Cancer Core Europe Molecular Tumor Board Portal. Nature Medicine, 26(7), 992–994. 10.1038/s41591-020-0969-2 [DOI] [PubMed] [Google Scholar]
- Tang D, Liu J, Wang D, Yu H, Li Y, & Zhang J (2011). Diagnostic and prognostic value of the methylation status of secreted frizzled-related protein 2 in colorectal cancer. Clinical and Investigative Medicine. Medecine Clinique et Experimentale, 34(2), E88–95. 10.25011/cim.v34i1.15105 [DOI] [PubMed] [Google Scholar]
- Tänzer M, Balluff B, Distler J, Hale K, Leodolter A, Röcken C, … Ebert MPA (2010). Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PloS One, 5(2), e9061. 10.1371/journal.pone.0009061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau SN, Bren G, & Schaid D (1993). Microsatellite instability in cancer of the proximal colon. Science (New York, N.Y.), 260(5109), 816–819. 10.1126/science.8484122 [DOI] [PubMed] [Google Scholar]
- Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, … Gibbs P (2015). Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 26(8), 1715–1722. 10.1093/annonc/mdv177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Jeanne, Cohen JD, Lo SN, Wang Y, Li L, Christie M, … Gibbs P (2020). Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. International Journal of Cancer. 10.1002/ijc.33312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Jeanne, Cohen JD, Wang Y, Christie M, Simons K, Lee M, … Gibbs P (2019). Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncology, 5(12), 1710–1717. 10.1001/jamaoncol.2019.3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Jeanne, Cohen JD, Wang Y, Li L, Christie M, Simons K, … Gibbs P (2019). Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut, 68(4), 663–671. 10.1136/gutjnl-2017-315852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Jeanne, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, … Gibbs P (2016). Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Science Translational Medicine, 8(346), 346ra92. 10.1126/scitranslmed.aaf6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiyama Y, Takahashi M, Hur K, Nagasaka T, Tanaka K, Inoue Y, … Goel A (2013). Serum miR-21 as a diagnostic and prognostic biomarker in colorectal cancer. Journal of the National Cancer Institute, 105(12), 849–859. 10.1093/jnci/djt101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tougeron D, Mouillet G, Trouilloud I, Lecomte T, Coriat R, Aparicio T, … Zaanan A (2016). Efficacy of Adjuvant Chemotherapy in Colon Cancer With Microsatellite Instability: A Large Multicenter AGEO Study. Journal of the National Cancer Institute, 108(7). 10.1093/jnci/djv438 [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, & Issa JP (1999). CpG island methylator phenotype in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America, 96(15), 8681–8686. 10.1073/pnas.96.15.8681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran B, Kopetz S, Tie J, Gibbs P, Jiang Z-Q, Lieu CH, … Desai J (2011). Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer, 117(20), 4623–4632. 10.1002/cncr.26086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, Stoeckeler JS, Kiang DT, O’Dea RF, Ramnaraine ML, & Mirkin BL (1985). Familial Pyrimidinemia and Pyrimidinuria Associated with Severe Fluorouracil Toxicity. New England Journal of Medicine, 313(4), 245–249. 10.1056/NEJM198507253130407 [DOI] [PubMed] [Google Scholar]
- Uratani R, Toiyama Y, Kitajima T, Kawamura M, Hiro J, Kobayashi M, … Kusunoki M (2016). Diagnostic Potential of Cell-Free and Exosomal MicroRNAs in the Identification of Patients with High-Risk Colorectal Adenomas. PloS One, 11(10), e0160722. 10.1371/journal.pone.0160722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- USPSTF. US Preventive Services Task Force et al. Screening for colorectal cancer: US preventive services task force recommendation statement (October 27, 2020). https://uspreventiveservicestaskforce.org/uspstf/draft-recommendation/colorectal-cancer-screening3#ful. [Google Scholar]
- Valtorta E, Martino C, Sartore-Bianchi A, Penaullt-Llorca F, Viale G, Risio M, … Gambacorta M (2015). Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Modern Pathology : An Official Journal of the United States and Canadian Academy of Pathology, Inc, 28(11), 1481–1491. 10.1038/modpathol.2015.98 [DOI] [PubMed] [Google Scholar]
- Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, … Bardelli A (2016). Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nature Communications, 7, 13665. 10.1038/ncomms13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es HH, Bout A, Liu J, Anderson L, Duncan AM, Bosma P, … Schurr E (1993). Assignment of the human UDP glucuronosyltransferase gene (UGT1A1) to chromosome region 2q37. Cytogenetics and Cell Genetics, 63(2), 114–116. 10.1159/000133513 [DOI] [PubMed] [Google Scholar]
- Varadhachary GR, Spector Y, Abbruzzese JL, Rosenwald S, Wang H, Aharonov R, … Raber MN (2011). Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 17(12), 4063–4070. 10.1158/1078-0432.CCR-10-2599 [DOI] [PubMed] [Google Scholar]
- Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, … Koopman M (2014). Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 20(20), 5322–5330. 10.1158/1078-0432.CCR-14-0332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal J, Muinelo L, Dalmases A, Jones F, Edelstein D, Iglesias M, … Montagut C (2017). Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Annals of Oncology. 10.1093/annonc/mdx125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar E, & Gruber SB (2010). Microsatellite instability in colorectal cancer-the stable evidence. Nature Reviews. Clinical Oncology, 7(3), 153–162. 10.1038/nrclinonc.2009.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu C-G, Ambs S, Cimmino A, Petrocca F, … Croce CM (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America, 103(7), 2257–2261. 10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Takebe N, Hinoue T, Hoadley KA, Cardenas MF, Hamilton AM, … Staudt LM (2020). Molecular Features of Cancers Exhibiting Exceptional Responses to Treatment. Cancer Cell. 10.1016/j.ccell.2020.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ALA, Lim JSJ, Sinha A, Gopinathan A, Lim R, Tan C-S, … Goh B-C (2015). Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. Journal of Translational Medicine, 13, 57. 10.1186/s12967-015-0405-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Zhou G, Jin P, Zhu J, Li S, Wu Q, … Qian J (2016). Detection of Colorectal Cancer Using a Simplified SEPT9 Gene Methylation Assay Is a Reliable Method for Opportunistic Screening. The Journal of Molecular Diagnostics : JMD, 18(4), 535–545. 10.1016/j.jmoldx.2016.02.005 [DOI] [PubMed] [Google Scholar]
- Xi Y, Formentini A, Nakajima G, Kornmann M, & Ju J (2008). Validation of biomarkers associated with 5-fluorouracil and thymidylate synthase in colorectal cancer. Oncol Rep, 19(1), 257–262. 10.3892/or.19.1.257 [DOI] [PubMed] [Google Scholar]
- Xi Y, Nakajima G, Schmitz JC, Chu E, & Ju J (2006). Multi-level gene expression profiles affected by thymidylate synthase and 5-fluorouracil in colon cancer. BMC Genomics, 7, 68. 10.1186/1471-2164-7-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, … Lito P (2020). Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature, 577(7790), 421–425. 10.1038/s41586-019-1884-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HHN, Lai JCW, Ho SL, Leung WK, Law WL, Lee JFY, … Leung SY (2017). RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut, 66(9), 1645–1656. 10.1136/gutjnl-2016-311849 [DOI] [PubMed] [Google Scholar]
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, & Liang G (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell, 26(4), 577–590. 10.1016/j.ccr.2014.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau TO, Tang C-M, Harriss EK, Dickins B, & Polytarchou C (2019). Faecal microRNAs as a non-invasive tool in the diagnosis of colonic adenomas and colorectal cancer: A meta-analysis. Scientific Reports, 9(1), 9491. 10.1038/s41598-019-45570-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z-X, Wang X-Y, Qin Q-Y, Chen D-F, Zhong Q-H, Wang L, & Wang J-P (2013). The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PloS One, 8(6), e65995. 10.1371/journal.pone.0065995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaanan A, Fléjou J-F, Emile J-F, Des GG, Cuilliere-Dartigues P, Malka D, … Taïeb J (2011). Defective mismatch repair status as a prognostic biomarker of disease-free survival in stage III colon cancer patients treated with adjuvant FOLFOX chemotherapy. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 17(23), 7470–7478. 10.1158/1078-0432.CCR-11-1048 [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, … Ribas A (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. New England Journal of Medicine, 375(9), 819–829. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhu Y-Q, Wu Y-Q, Zhang P, & Qi J (2014). Detection of promoter hypermethylation of Wnt antagonist genes in fecal samples for diagnosis of early colorectal cancer. World Journal of Gastroenterology, 20(20), 6329–6335. 10.3748/wjg.v20.i20.6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Song M, Zhang J, Kuerban M, & Wang H (2015). Combined identification of long non-coding RNA CCAT1 and HOTAIR in serum as an effective screening for colorectal carcinoma. International Journal of Clinical and Experimental Pathology, 8(11), 14131–14140. [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, … Le LP (2014). Anchored multiplex PCR for targeted next-generation sequencing. Nature Medicine, 20(12), 1479–1484. 10.1038/nm.3729 [DOI] [PubMed] [Google Scholar]