

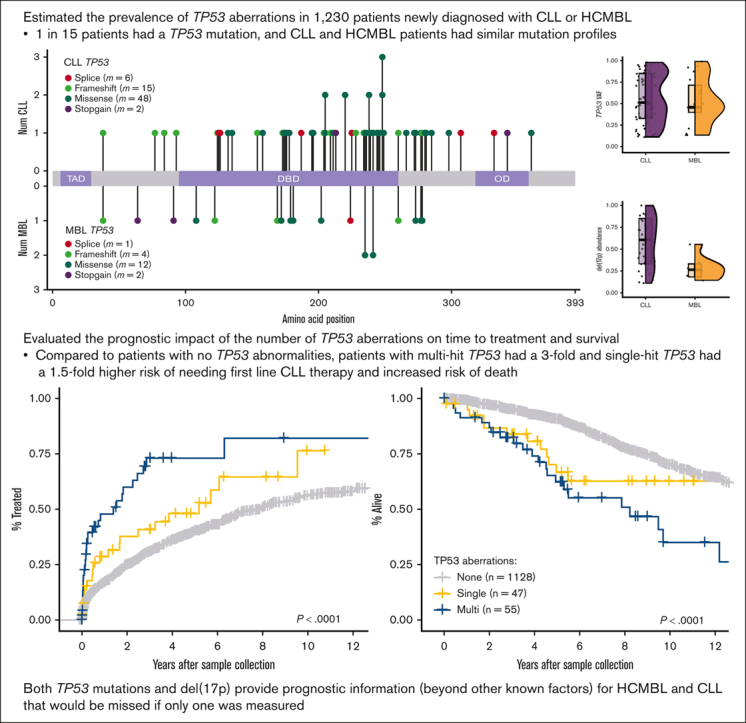
Key Points
-
•
Similar mutation frequency, type, and location in TP53 between 849 patients newly diagnosed with CLL and 381 patients with HCMBL.
-
•
Having multiple TP53 abnormalities increased the risk of progression to therapy and shortened the overall survival.
Visual Abstract
Abstract
TP53 aberrations, including mutations and deletion of 17p13, are important adverse prognostic markers in chronic lymphocytic leukemia (CLL) but are less studied in high count monoclonal B-cell lymphocytosis (HCMBL), an asymptomatic pre-malignant stage of CLL. Here we estimated the prevalence and impact of TP53 aberrations in 1,230 newly diagnosed treatment-naïve individuals (849 CLL, 381 HCMBL). We defined TP53 state as: wild-type (no TP53 mutations and normal 17p), single-hit (del(17p) or one TP53 mutation), or multi-hit (TP53 mutation and del(17p), TP53 mutation and loss of heterozygosity, or multiple TP53 mutations). Cox regression was used to estimate hazard ratios (HR) and 95% confidence intervals (CI) for time to first treatment and overall survival by TP53 state. We found 64 (7.5%) CLL patients and 17 (4.5%) HCMBL individuals had TP53 mutations with variant allele fraction >10%. Del(17p) was present in 58 (6.8%) of CLL and 11 (2.9%) of HCMBL cases. Most individuals had wild-type (N=1,128, 91.7%) TP53 state, followed by multi-hit (N=55, 4.5%) and then single-hit (N=47, 3.8%) TP53 state. The risk of shorter time to therapy and death increased with the number of TP53 abnormalities. Compared to wild-type patients, multi-hit patients had 3-fold and single-hit patients had 1.5-fold increased risk of requiring therapy. Multi-hit patients also had 2.9-fold increased risk of death compared to wild-type. These results remained stable after accounting for other known poor prognostic factors. Both TP53 mutations and del(17p) may provide important prognostic information for HCMBL and CLL that would be missed if only one were measured.
Introduction
Chronic lymphocytic leukemia (CLL), a neoplasm of B-cell lymphocytes, is characterized by clinical and biologic heterogeneity. Distinct genetic profiles exist in CLL, including recurrent cytogenetic abnormalities and somatic mutations that influence disease aggressiveness.1, 2, 3 One of the most well-known and clinically important prognostic markers in CLL would be abnormalities in TP53. Somatic aberrations include TP53 mutations and interstitial or complete deletions of the short arm of chromosome 17, del(17p). Prevalence of TP53 mutations (median variant allele fraction [VAF], 0.6%)4 ranges from 7% to 11% and that of del(17p) ranges from 4% to 8% in untreated CLL.3,5, 6, 7 Of patients with a TP53 mutation, ∼62% to 76% also had a del(17p).5,7,8 TP53 abnormalities increase with disease progression and relapse after treatment.6
Patients with CLL and TP53 abnormalities have a shorter progression-free survival, event-free survival, and overall survival (OS) when treated with chemoimmunotherapy and are less responsive to chemoimmunotherapy than patients with no TP53 abnormalities.6,7,9, 10, 11, 12 However, the prognostic importance of TP53 mutations with a low VAF and that of 17p deletions in few cells remain ambiguous. Prior studies have compared the OS of patients with wild-type vs mutated TP53; some studies13, 14, 15 found shorter survival among patients carrying TP53 mutations regardless of VAF, but another study16 found shorter survival only in patients carrying TP53 mutations with VAFs ≥12%. Currently, the TP53 Network of the European Research Initiative on Chronic Lymphocytic Leukemia (ERIC) recommends evaluating TP53 mutations with VAFs >10% because the clinical utility of low-VAF mutations remains unclear.17 In a study of the effect of 17p deletion clone size identified via fluorescence in situ hybridization (FISH) analysis,18 survival was most favorable among patients with less than 25% deleted nuclei (3-year survival rate, 92%), intermediate among patients with between 25% and 74% deleted nuclei (3-year survival rate, 67%), and least favorable among patients with 75% or more deleted nuclei (3-year survival rate, 40%; P < .001).
In addition, data from prior studies are inconclusive as to whether a single TP53 abnormality vs multiple TP53 abnormalities affects outcomes.3,19,20 In the chemoimmunotherapy era, survival was significantly shorter among patients with a single TP53 hit and further shortened among patients with multiple TP53 hits.20 More recent data indicate that patients with a single TP53 hit treated with ibrutinib had better OS than patients with multiple TP53 hits (5-year OS, 100% in single-hit vs 69% in multihit).19 Although these studies have shown the impact of TP53 aberrations in patients with CLL who have been treated, there are limited data on the prognostic impact of TP53 mutations, in particular the role of single vs multiple TP53 abnormalities in patients with newly diagnosed treatment-naive CLL.
Most CLL cases are preceded by a premalignant condition of circulating clonal B cells between 0.5 × 109/L and 4.9 × 109/L, called high-count monoclonal B-cell lymphocytosis (HCMBL) with a CLL-like immunophenotype.21,22 TP53 mutations (3.0%; 2/66) and del(17p)s (3.8%; 4/105) have been observed in HCMBL at a lower rate than in patients with CLL.23 Prior work by our group demonstrated the ability to predict time to first treatment (TTFT) using the CLL International Prognostic Index and found TP53 disruption, either via del(17p) FISH or TP53 mutation, among 4.8% (20/415) of individuals with HCMBL.24 However, in this study, TP53 mutations were detected via Sanger sequencing, limiting the ability to detect low-VAF mutations.24 Little is known about the impact of TP53 abnormalities on prognosis among individuals with HCMBL.
We aim to (a) examine whether TP53 aberrations occur less frequently in HCMBL cases than in CLL cases and to (b) evaluate the impact of single vs multiple aberrations compared with that of no TP53 aberrations on TTFT and OS in HCMBL and CLL. We estimated the prevalence and impact of TP53 abnormalities in a large cohort of individuals with HCMBL and those with newly diagnosed, treatment-naive CLL.
Methods
Participants
This research was approved by the Mayo Clinic institutional review board, and all participants provided written informed consent. TP53 was characterized in individuals with CLL or HCMBL that was diagnosed between 2000 and 2019, in accordance with the Mayo Clinic CLL resource, using pretreatment peripheral blood mononuclear cells (PBMCs) collected on average within 12 days of initial diagnosis. CLL diagnosis was made based on the 2008 International Workshop CLL criteria25 and updated to the 2018 International Workshop CLL criteria whenever possible.22 Flow cytometry was performed for samples from all patients for diagnosis and identification of CD19+/CD5+ tumor cells. All HCMBLs had clonal B-cell counts between 0.5 × 109/L and <5 × 109/L.
DNA sequencing
DNA was extracted from either PBMCs with >80% CD5+/CD19+ clonal B cells or those enriched for CD5+/CD19+ clonal B cells. The sequencing of the entire coding regions and intron-exon junctions of TP53 was part of a larger panel of 59 genes previously implicated in CLL.26 The median coverage depth per sample across the 59 genes was 1799 (range, 65-3675) with >83% of the samples having a median coverage depth of >1000 per nucleotide, allowing for detection of mutations with VAFs as low as 1%. For TP53 specifically, the median coverage was 1917 (range, 23-6004), with 83% of samples having a depth >1000, and 99% of samples having a depth of >100. Only mutations with at least 100 reads and 10 reads with the alternate allele were considered. Somatic mutations were called using MuTect2 in tumor-only mode. We included frameshift and in-frame deletions and insertions as well as nonsense, missense, and splicing mutations. Following the TP53 ERIC-updated recommendations,17 we evaluated TP53 mutations with VAFs >10% in our primary analyses. In secondary analyses, we evaluated the role of TP53 mutations with VAFs >1%.
Del(17p) status at time of sample collection or at diagnosis was available and extracted from the medical records for 82% (n = 1012) of the participants; individuals with >9.5% defective cells were considered to be del(17p)+ using FISH. The number of defective nuclei detected via FISH was also deduced and used to evaluate the mean del(17p) abundance in our cohort of patients with CLL and HCMBL. FISH data were missing in 218 participants (18%), and we inferred del(17p) copy number variations from targeted DNA sequencing using PatternCNV.27,28 First batch effects were adjusted by (a) quantifying the exon coverage of chromosomes without an established cytogenetic abnormality in CLL (ie, chromosomes 11, 12, 13, and 17 were excluded), (b) grouping patients into clusters with similar exon coverage patterns using principal component analysis and correlation matrixes, and then (c) rerunning PatternCNV on each cluster of patients to detect copy number variations on chromosomes 11, 12, 13, and 17. Del(17p) was inferred when the log ratio of coverage decreased by −0.25 from the log2 normalized median coverage in the genomic region (hg19) from 17:7134079 to 7722415. Calls were 98.62% concordant between PatternCNV and FISH, when available.
TP53 state
Somatic mutations and del(17p) status were used to define TP53 state for each patient based on the following criteria: (a) wild-type were patients with no TP53 mutations and normal 17p; (b) single-hit were those with either 1 TP53 heterozygous mutation (VAF <60%) or del(17p); and (c) multihit were those with TP53 mutation and copy neutral loss of heterozygosity (cnLOH), multiple TP53 mutations, or the combination of TP53 mutations and del(17p) (supplemental Figure 1). A full list of TP53 mutations and del(17p) status among patients with single-hit and multihit abnormalities is given in supplemental Table 1.
CLL and HCMBL outcomes
TTFT and OS were analyzed based on the TP53 state. TTFT was defined as the time from sample collection to either first treatment, death, or last follow-up date, whichever occurred first. OS was defined as the time from sample collection to either death or last follow-up date, whichever occurred first. Median and 5-year TTFT and OS were estimated using the Kaplan-Meier method. We used Cox regression to estimate hazard ratios (HRs) and 95% confidence intervals (CIs) for TTFT and OS associations. To investigate the sensitivity of our findings, we stratified the Kaplan-Meier analyses and adjusted the Cox regressions based on the type of TP53 and other known adverse prognostic factors, including age of >65 years, Rai stage I-IV, β 2-microglobulin (β2M) > 3.5 mg/L, or unmutated IGHV. For OS, we examined the effect of treatment by (a) evaluating OS based on the TP53 state when OS is defined as time from treatment to either death or last follow-up date, (b) censoring OS at the time of treatment, and (c) subsetting of patients who received the most used inhibitors: B-cell lymphoma 2 or Bruton tyrosine kinase (all treatments administered are described in supplemental Table 2.)
Tumor characteristics based on the TP53 state
Tumor mutational load (TML) was calculated as the number of mutated CLL driver genes (out of 59 total), as previously described.26 We then compared TML and mutation status of individual genes based on single-hit and multihit TP53 carriers.
Results
Patient characteristics
We characterized TP53 in 1230 individuals: 849 with CLL and 381 with HCMBL (Table 1). Most participants were male (69.6%, CLL; 63.8%, HCMBL). The median age at diagnosis was 61 years for CLL and 67 years for HCMBL (Table 1). Most of the patients with CLL were diagnosed at Rai stage 0 (54.9%) and had a median β2M level of 2.4 mg/L, and 51.1% had unmutated IGHV. Chromosomal aberrations in the participants of the study included del(13q) (50.8%, CLL; 42%, HCMBL), tri(12) (13%, CLL; 12.6%, HCMBL), del(11q) (10.5%, CLL; 3.9%, HCMBL), and del(17p) (6.8%, CLL; 2.9%, HCMBL) (Table 1). Samples were collected within 2 years of diagnosis for most participants (85%). Median follow-up after sample collection was 6.3 years.
Table 1.
Characteristics of individuals with CLL and HCMBL
| CLL (n = 849) | MBL (n = 381) | Overall (n = 1230) | ||||
|---|---|---|---|---|---|---|
| Female, n, % | 258 | 30.4 | 138 | 36.2 | 396 | 32.2 |
| Male | 591 | 69.6 | 243 | 63.8 | 834 | 67.8 |
| Median age (IQR), y | 61 (46-76) | 67 (52-82) | 63 (47-79) | |||
| Rai stage 0, n, % | 456 | 54.9 | ||||
| Rai stage 1-4, n, % | 374 | 45.1 | ||||
| Median β2M (IQR), mg/L | 2.4 (1.1-3.8) | 2.1 (1.4-2.9) | 2.3 (1.1-3.6) | |||
| IGHV unmutated, n, % | 406 | 51.1 | 71 | 24.6 | 477 | 44.0 |
| IGHV mutated, n, % | 389 | 48.9 | 218 | 75.4 | 607 | 56.0 |
| FISH del(11q), n, % | 89 | 10.5 | 15 | 3.9 | 104 | 8.5 |
| FISH del(13q), n, % | 431 | 50.8 | 160 | 42.0 | 591 | 48.0 |
| FISH tri(12), n, % | 110 | 13.0 | 48 | 12.6 | 158 | 12.8 |
| FISH or CNV del(17p), n, % | 58 | 6.8 | 11 | 2.9 | 69 | 5.6 |
| No TP53 mutations, n, % | 748 | 88.1 | 348 | 91.3 | 1096 | 89.1 |
| TP53 mutation(s) VAF >10%, n, % | 64 | 7.5 | 17 | 4.5 | 81 | 6.6 |
| TP53 state, wild-type, n, % | 768 | 90.5 | 360 | 94.5 | 1128 | 91.7 |
| TP53 state, single-hit, n, % | 35 | 4.1 | 12 | 3.1 | 47 | 3.8 |
| TP53 state, multihit, n, % | 46 | 5.4 | 9 | 2.4 | 55 | 4.5 |
| Single-hit TP53 type, n | 35 | 12 | 47 | |||
| No TP53 mutation; del(17p), n, % | 17 | 48.6 | 4 | 33.3 | 21 | 44.7 |
| Single TP53 mutation; normal 17p, n, % | 18 | 51.4 | 8 | 66.7 | 26 | 55.3 |
| Multihit TP53 type, n | 46 | 9 | 55 | |||
| TP53 LOH mutation, n, % | 3 | 6.5 | 1 | 11.1 | 4 | 7.3 |
| TP53 mutation(s); del(17p), n, % | 37 | 80.4 | 6 | 66.7 | 43 | 78.2 |
| Multiple TP53 mutations, n, % | 6 | 13.0 | 2 | 22.2 | 8 | 14.5 |
| Median follow-up, y | 5.95 | 7.27 | 6.27 | |||
CNV, copy number variation.
TP53 mutations
We found TP53 mutations with VAFs >10% in 64 patients with CLL (7.5%) and 17 individuals with HCMBL (4.5%) (Table 2). Patients with TP53 mutations were more likely to be older at diagnosis and have unmutated IGHV (Table 2). A total of 71 TP53 mutations were identified in 64 patients with CLL (Figure 1A). In individuals with HCMBL, 19 TP53 mutations were identified in 17 individuals (Figure 1B). For mutations with VAFs of >10%, the median TP53 VAF was not significantly different between individuals with CLL and those with HCMBL (Figure 1C) (median VAF, 51% in CLL and 46% in HCMBL; Kruskal-Wallis P > .68). Most of the TP53 mutations were missense, and we did not observe different patterns in the type or location of TP53 mutations between HCMBL and CLL cases (Figure 1E).
Table 2.
CLL and HCMBL by TP53 mutation status
| CLL | No TP53 mutation (n = 748) | VAF ≤10% (n = 37) | VAF >10% (n = 64) | Overall (n = 849) | ||||
|---|---|---|---|---|---|---|---|---|
| Female, n, % | 223 | 29.8 | 13 | 5.0 | 22 | 8.5 | 258 | 30.4 |
| Male, n, % | 525 | 70.2 | 24 | 4.1 | 42 | 7.1 | 591 | 69.6 |
| Median age (IQR), y | 61 (46-76) | 65 (55-75) | 66.5 (50-83) | 61 (46-76) | ||||
| Rai stage 0, n, % | 405 | 55.4 | 20 | 55.6 | 31 | 49.2 | 456 | 54.9 |
| Rai stage 1-4, n, % | 326 | 44.6 | 16 | 44.4 | 32 | 50.8 | 374 | 45.1 |
| Median β2M (IQR), mg/L | 2.4 (1.1-3.7) | 2.9 (1.1-4.8) | 2.9 (1.1-4.6) | 2.4 (1.1-3.8) | ||||
| IGHV unmutated, n, % | 343 | 49.0 | 24 | 68.6 | 39 | 65.0 | 406 | 51.1 |
| IGHV mutated, n, % | 357 | 51.0 | 11 | 31.4 | 21 | 35.0 | 389 | 48.9 |
| FISH del(11q), n, % | 84 | 11.2 | 2 | 5.4 | 3 | 4.7 | 89 | 10.5 |
| FISH del(13q), n, % | 370 | 49.5 | 19 | 51.4 | 42 | 65.6 | 431 | 50.8 |
| FISH tri(12), n, % | 103 | 13.8 | 4 | 10.8 | 3 | 4.7 | 110 | 13.0 |
| FISH/CNV del(17p), n, % | 10 | 1.3 | 7 | 18.9 | 41 | 64.1 | 58 | 6.8 |
| Median follow-up, y | 6.21 | 5.49 | 4.54 | 5.95 | ||||
| HCMBL | No TP53 mutation (n = 348) | VAF ≤10% (n = 16) | VAF >10% (n = 17) | Overall (n = 381) | ||||
|---|---|---|---|---|---|---|---|---|
| Female, n, % | 130 | 37.4 | 1 | 6.3 | 7 | 41.2 | 138 | 36.2 |
| Male, n, % | 218 | 62.6 | 15 | 93.8 | 10 | 58.8 | 243 | 63.8 |
| Median age (IQR), y | 67 (53-81) | 74 (63-85) | 70 (57-83) | 67 (52-82) | ||||
| Median β2M (IQR), mg/L | 2.1 (1.4-2.8) | 2.2 (1.5-2.9) | 2.2 (0.6-3.9) | 2.1 (1.4-2.8) | ||||
| IGHV unmutated, n, % | 66 | 25.0 | 1 | 8.3 | 4 | 30.8 | 71 | 24.6 |
| IGHV mutated, n, % | 198 | 75.0 | 11 | 91.7 | 9 | 69.2 | 218 | 75.4 |
| FISH del(11q), n, % | 13 | 3.7 | 1 | 6.3 | 1 | 5.9 | 15 | 3.9 |
| FISH del(13q), n, % | 145 | 41.7 | 8 | 50.0 | 7 | 41.2 | 160 | 42.0 |
| FISH tri(12), n, % | 47 | 13.5 | 1 | 6.3 | 0 | 0.0 | 48 | 12.6 |
| FISH/CNV del(17p), n, % | 3 | 0.9 | 1 | 6.3 | 7 | 41.2 | 11 | 2.9 |
| Median follow-up, y | 7.18 | 7.65 | 6.2 | 7.27 | ||||
Figure 1.

TP53 mutations and del(17p) based on CLL and HCMBL. Note that only TP53 mutations with VAF >10% were considered. Rate of mutations and del(17p) in CLL (A) and HCMBL (B) cases. Gold indicates single-hit (∗ denotes multihit based on cnLOH), and blue indicates multihit TP53. (C) VAF of TP53 mutations. Box plot (quartiles and median shown) and violin plots show distribution. (D) Percent of cells with del(17p) by FISH. (E) Location, type, and counts of TP53 mutations. Num, number.
Del(17p) was present in 58 patients with CLL (6.8%) and 11 individuals with HCMBL (2.9%) (Table 1; Figure 1A-B). Of those individuals with a FISH del(17p), the deletion occurred significantly more in leukemic cells in individuals with CLL (mean abundance, 58% of cells) compared with those in individuals with HCMBL (mean abundance, 28% of cells; Figure 1D; Kruskal-Wallis P = .0069). Most cases with del(17p) also had a TP53 mutation, 41 CLL (70.7%) and 7 HCMBL (63.6%) cases (Table 2).
Single-hit vs multihit TP53
Most of the individuals in our HCMBL/CLL cohort (91.7%; n = 1128) had no TP53 mutations with VAFs >10% and had normal 17p (wild-type TP53; supplemental Figure 1). The 47 individuals (3.8% of our cohort) with single-hit TP53 mutations included 21 individuals with del(17p) but no TP53 mutations, and 26 individuals with a single heterozygous TP53 mutation and normal 17p. The 55 (4.5%) individuals with multihit TP53 included 43 patients with both TP53 mutation(s) and del(17p), 8 with normal 17p but multiple TP53 mutations, and 4 with TP53 mutation and cnLOH (supplemental Figure 1). Among those with an TP53 aberration, CLL cases were more often multihit (56.8%; 46/81) than single-hit (43.2%; 35/81), and HCMBL cases were more often single-hit (57.1%; 12/21) than multihit (42.9%; 9/21), although these differences were not significant (Figure 1A-B). Because we did not observe significant differences between HCMBL and CLL, we evaluated TP53 state based on the TTFT and OS in the entire cohort, with HCMBL and CLL combined.
TTFT
In the entire cohort of individuals with treatment data (n = 1120), 417 patients subsequently received treatment: 58% untreated at 5 years (95% CI, 54.6%-61.6%). Patients with any TP53 abnormality had shorter TTFT than patients with wild-type TP53 (39% vs 60% untreated, respectively, at 5 years; P < .0001). Dividing the patients based on the TP53 state, we observed shorter TTFT among patients with multihit TP53 (n = 53; 29 events; 27% untreated at 5 years) than patients with single-hit TP53 (n = 42; 21 events; 52% untreated at 5 years; Figure 2A). In Cox regression, patients with multihit TP53 (HR, 3.05; 95% CI, 2.08-4.46) and those with single-hit TP53 (HR, 1.56; 95% CI, 1.00-2.42) had significantly shorter TTFTs compared with individuals with wild-type TP53 (Figure 2B), with a trend toward increased risk with increased number of TP53 abnormalities (likelihood ratio test, P = 1.25 × 10−6). These results did not change when restricted to individuals from whom a sample was collected within 2 years of diagnosis (supplemental Figure 2A-B) nor when we removed 4 potentially benign mutations (p.Asn235Ser, p.Glu298Lys, p.Arg202Cys, and p.Gly360Val) during sensitivity analyses (supplemental Figure 13).
Figure 2.

Kaplan-Meier curves and Cox regression forest plots. Based on TP53 normal, single-hit, and multihit state for TTFT (A-B) and OS (C-D). Note that only TP53 mutations with VAF >10% were considered.
Shorter TTFT was most pronounced among individuals with both del(17p) and a TP53 mutation (supplemental Figure 3B), with a median TTFT of 1.8 years among those with del(17p) plus a single TP53 mutation (n = 41; 21 treated) and a median of 0.1 year among those with del(17p) plus multiple TP53 mutations (n = 5; 4 treated). In comparison, patients with wild-type TP53 had a median TTFT of 7.9 years (n = 1025; 367 treated). In Cox regression, patients with both del(17p) plus TP53 mutation(s) had a shorter TTFT compared with patients with wild-type TP53 (HRs, 2.72 vs 9.02; supplemental Figure 3C). In sensitivity analyses, we analyzed these data among patients stratified based on other known prognostic factors, including (a) age, (b) Rai stage, (c) β2M, or (d) IGHV mutation status (supplemental Figures 4 and 5). Individuals with multihit TP53 had significantly shorter TTFT regardless of age, Rai stage, or β2M risk group. Patients with multihit TP53 had significantly shorter TTFT when IGHV was unmutated but not when IGHV was mutated (supplemental Figures 4D and 5D), as previously observed.29 In multivariate Cox regression, multihit TP53 remained a significant risk factor for shorter TTFT (HR, 2.00; 95% CI, 1.31-3.06; P = .001) after adjusting for age, Rai stage, β2M, and IGHV mutation status (Figure 3A). These data support that multiple hits to TP53 reduces the time to treatment beyond other clinical factors.
Figure 3.

Multivariate Cox regression. (A) TTFT and (B) OS. Note that only TP53 mutations with VAF >10% were considered.
OS
In the cohort of 1122 individuals (285 deaths) with survival data, 5-year survival was 88.4% (95% CI, 86.3%-90.5%). Patients with multihit TP53 abnormalities (n = 49; 23 deaths) had shorter OS (5-year survival 62.3%; 95% CI, 48.6%-79.8%) compared with patients with wild-type TP53 (n = 1032; 250 deaths; 5-year survival 90.5%; 95% CI, 88.5%-92.5%), and those with single-hit TP53 abnormalities (n = 41; 12 deaths; 5-year survival 66.6%; 95% CI, 52%-85.3%; Figure 2C). In Cox regression, patients with multihit abnormalities had a 2.89-fold increased risk of death compared with those with wild-type TP53 (95% CI, 1.88-4.43), and those with single-hit abnormalities had a 1.63-fold increased risk of death (95% CI, 0.91-2.91) compared with those with wild-type TP53 (Figure 2D), with a significant trend in the HR as the number of TP53 abnormalities increased (likelihood ratio test, P = 5.42 × 10−5). Results stayed consistent when restricted to individuals from whom a sample was collected within 2 years of diagnosis (supplemental Figure 2C-D).
In our cohort, 417 individuals received treatment. To address possible OS variability based on treatment type, we censored at the time of treatment and still observed shorter OS among patients with multihit TP53 compared with those with single-hit or wild-type TP53, but the effects were attenuated (supplemental Figure 6A). In addition, we stratified the data of 93 patients who received first-line B-cell lymphoma 2 or Bruton tyrosine kinase inhibitors and still observed significantly shorter OS among the patients with multihit TP53 compared with those with wild-type TP53 (supplemental Figure 6B).
Next, to test robustness of our findings further, we limited our OS analyses to patients with well-known higher risk characteristics (supplemental Figures 7 and 8). In each of these high-risk groups, we observed significantly shorter OS among patients with multiple hits (supplemental Figure 7). After adjusting for these factors in multivariate analysis (Figure 3B), multihit TP53 remained a significant risk factor for shorter OS (HR, 2.37; 95% CI, 1.52-3.70; P < .001). These data suggest that multihit TP53 remains an important prognostic marker.
Tumor differences by TP53 state
To investigate potential functional mechanisms underlying the differences in outcomes by TP53 state, we explored global differences in CLL driver mutations (supplemental Figure 9). TML did not significantly differ based on the TP53 state (Kruskal-Wallis, P = .56). Among the patients with single-hit TP53, NOTCH1 and SF3B1 were mostly commonly mutated, whereas among those with multihit TP53, MGA and NOTCH1 were mostly commonly mutated (supplemental Figure 9B). However, these genes (and other driver genes) were not mutated at significantly different rates in the 2 groups (Fisher exact tests, all P > .13).
TP53 mutations with VAFs ≤10%
Given the depth of sequencing data, we were able to detect TP53 mutations as low as VAFs >1%, which is well below the ERIC17-recommended VAF threshold of 10%. In our cohort, 37 patients with CLL (4.4%) and 16 with HCMBL (4.2%) had a TP53 mutation with a VAF ≤10% (Table 2; supplemental Figure 10A-B). Eleven patients with CLL and 2 with HCMBL had both a mutation with a VAF >10% and a mutation with a VAF ≤10% (supplemental Figure 10D). Low-VAF mutations were split between Rai stages and were slightly more common among patients with IGHV-unmutated CLL (Table 2). Co-occurrence of del(17p) was much less frequent with mutations with VAFs ≤10% (12.1%, CLL; 9.1%, HCMBL) than mutations with VAFs >10% (70.7%, CLL; 63.6%, HCMBL; Table 2). The low-VAF mutations were most often missense mutations, and no distinct mutation patterns were observed between patients with CLL and those with HCMBL (supplemental Figure 10E).
Considering all TP53 mutations (VAF > 1%), 1089 individuals had wild-type TP53, 74 individuals had a single-hit TP53 [54 single heterozygous mutation, 20 del(17p)], and 67 individuals had multihit TP53 [14 multiple mutations, 4 mutations and cnLOH, 37 single mutation and del(17p), and 12 multiple mutations and del(17p)]. TTFT remained shorter in patients with multihit TP53 than those with single-hit or wild-type TP53 (supplemental Figure 11A-B). Individuals with both a mutation and del(17p) had the shortest TTFTs (supplemental Figure 11C-E). These trends were the same for OS (supplemental Figure 12).
Discussion
The results of our study of newly diagnosed, treatment-naive CLL and HCMBL, which is the largest to date, to our knowledge, show that (a) 1 in 15 patients have a TP53 mutation (65% missense) at the time of initial diagnosis, with an average VAF of 50%; (b) 1 in 3 patients with a TP53 aberration have a TP53 mutation identified using sequencing studies in the absence of del(17p) via FISH testing; (c) compared with patients with no TP53 abnormalities, those with multihit TP53 have a threefold higher risk of needing first-line CLL therapy and increased risk of death; and (d) these results are true even after adjusting for other known prognostic factors, such as IGHV mutation status (for TTFT) and receipt of novel agent treatment (for OS). Collectively, these data have important implications in the counseling and management of patients with newly diagnosed CLL and HCMBL.
Overall, patients with HCMBL had lower VAF TP53 mutations and fewer del(17p) leukemic cells than patients with CLL. Regardless of HCMBL/CLL status, multiple TP53 abnormalities led to shorter TTFT and OS than single-hit TP53 or wild-type TP53. As in prior studies, most of the TP53 mutations were missense, and we did not observe different patterns in the type or location of TP53 mutations between HCMBL and CLL cases. Co-occurrence of TP53 mutations and del(17p) had the poorest outcomes, even after accounting for other known prognostic factors. These findings remained consistent even when low-VAF mutations were included. Thus, an increasing number of TP53 abnormalities may lead to worse prognosis for both patients with HCMBL and those with newly diagnosed CLL.
In CLL, many prior studies have focused on profiling TP53 at the time of treatment iniatition.6,8,16,19,20 Our study evaluated CLL at the time of diagnosis and found a similar prevalence of TP53 aberrations as prior studies that evaluated patients with early-stage disease.4,7,8,13, 14, 15,30 Together, these studies support evaluating TP53 status at the time of CLL diagnosis to gain prognostic information.
Current clinical practice usually does not consider single vs multihit TP53 abnormalities; however, our study supports that collectively the number of TP53 abnormalities lead to different outcomes. Individuals with multiple TP53 abnormalities had shorter TTFTs and OSs than those with a single-hit TP53, and individuals with both a TP53 mutation(s) and del(17p) had the poorest outcomes. These findings are consistent with prior studies among patients with CLL who were symptomatic.4,6,13,20 Thus, we found that having 1 TP53 mutation only increases risk of poor outcomes when del(17p) is also present. Some prior studies also observed this finding in that TP53 mutations in the absence of del(17p) were not prognostic for TTFT4 or OS.6,15 However, other studies reported significantly shorter OS7,8,13,14,30 in patients with only TP53 mutations. These discrepancies may be because of underlying differences in patient populations (considering that we enrolled only newly diagnosed untreated CLL), type of treatment, or TP53 sequencing and mutation detection strategies. A large, retrospective registry study (informCLL) reported that among 840 patients with CLL treated largely at community centers, only a third underwent testing for FISH, and only 1 in 10 patients underwent testing for TP53 mutation before treatment.31 Given that our study shows that patients with multihit TP53 abnormalities experience poor outcomes, additional outreach efforts to community physicians need to occur for optimal outcomes of patients with CLL.
Although limited by sample size, we observed shorter OS in individuals with multihit TP53 when restricted to the 93 patients who were treated with novel therapy in the front-line setting, patients with multihit TP53 had 16-fold increased risk of death (P = .003) whereas those with single-hit TP53 had a sixfold increased risk of death (P = .15) compared with patients with wild-type TP53. These results are in line with recent work showing that the concomitant presence of TP53 mutations and del(17p) is an independent negative prognostic factor for OS among patients with CLL on ibrutinib treatment.32 Screening for multihit TP53 among patients receiving novel therapy may be an important prognostic factor for OS in CLL and may have prognostic and counseling impact on HCMBL.
To our knowledge, this is currently the largest study of TP53 of HCMBL, and we found that TP53 may be important for evaluating HCMBL clinical course. In our cohort, 9.4% of patients with HCMBL had a TP53 abnormality, with 3.1% of these abnormalities being multihit (including all VAF mutations). TP53 mutations and del(17p) were less common, and del(17p) abundance was significantly lower in HCMBL than in CLL; these observations were expected, given that HCMBL is a precursor disease of CLL. Patients with HCMBL with multihit TP53 did, however, have an elevated risk of shorter TTFT and OS; multihit TP53 may already play a role in HCMBL clinical course and might be a prognostic factor in progression.
Following the ERIC TP53 mutation calling standards in CLL,17 our primary analyses defined TP53 mutations with VAFs >10%. However, we had the depth of sequencing to evaluate mutations with VAFs >1%. When all mutations with VAFs >1% were considered, we still observed shorter TTFT and OS in patients with multihit TP53. We also observed that patients with a single TP53 hit also had significantly worse outcomes than those with wild-type TP53, suggesting that any TP53 abnormality (single or multiple and mutation or deletion) is a significant prognostic factor. However, prior studies have found mixed results regarding the prognostic value of low-VAF TP53 mutation. Some prior studies observed significantly shorter OS in patients with low-VAF TP53 mutation, even when del(17p) status was included as a covariate.13,14,30 Conversely, other studies did not find prognostic value of low-VAF TP53 mutation in the absence of del(17p) within the context of treatment.15,16,33 Our study, which includes highly robust clinical information on our patient cohorts, supports the importance of screening small subclones with TP53 mutations at the time of diagnosis.
Our study is strengthened by deep targeted sequencing of TP53 in a large cohort of individuals with CLL and HCMBL from a single institution in which the clinical courses are robustly annotated. We followed a systematic approach to call and screen TP53 mutations to address the limitation of paired germ line sequencing not being available. Our findings are limited to the observational and retrospective nature of the study. Because of the relatively low frequency of TP53 abnormalities in CLL/HCMBL, stratification of results is also limited by small sample sizes; replication in an independent data set would strengthen our findings. However, our results are largely in line with prior studies of mostly symptomatic CLL and add evidence of the importance of screening for TP53 abnormalities in addition to measuring del(17p) at diagnosis in individuals with HCMBL and CLL.
In summary, we characterized TP53 in 849 individuals with CLL and 381 with HCMBL and observed mutations or 17p deletions in 141 individuals with similar mutation frequencies, types, and locations between CLL and HCMBL. Multiple TP53 hits reduced TTFT and OS, even after accounting for other clinical prognostic factors, primarily driven by individuals with both TP53 mutations and del(17p). Many recent studies have identified other markers of high-risk disease (eg, complex karyotype,34 subset #235). Similarly, patients with multiple TP53 aberrations may constitute an ultrahigh-risk group of patients with CLL. With many novel drugs being tested in early intervention trials, the accurate identification of high-risk patients may allow for an improved risk stratification.
Conflict-of-interest disclosure: N.E.K. serves on the advisory board for AbbVie, AstraZeneca, Beigene, Behring, Boehringer Ingelheim Pharmaceuticals Inc, Cytomx Therapy, Dava Oncology, Janssen, Juno Therapeutics, Oncotracker, Pharmacyclics, and Targeted Oncology; serves on the data safety monitoring committee for Agios Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, Celgene, CytomX Therapeutics, Dren Bio, Janssen, MorphoSys, and Rigel; and has received research funding from AbbVie, Acerta Pharma, Bristol Myers Squibb, Celgene, Genentech, MEI Pharma, Pharmacyclics, Sunesis, TG Therapeutics, and Tolero Pharmaceuticals. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the individuals who participated in our studies for their time and contribution.
This study was supported by awards from the National Cancer Institute, National Institutes of Health (K00CA234943) (R.G.), (R01CA235026 and R01CA254951) (S.L.S. and E.B.), (R21CA256648) (S.L.S. and G.K.), and (P50CA097274). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: S.L.S. and E.B. conceived and designed the study; S.L.S., E.B., S.A.P., and J.R.C. acquired the data; R.G., J.E.W.-N., C.E.M., and D.R.O. analyzed the data; R.G., J.E.W.-N., S.L.S., and E.B. drafted the manuscript; and all authors interpreted the data and reviewed the manuscript.
Footnotes
∗R.G. and J.E.W.-N. are joint first authors.
†S.L.S. and E.B. contributed equally to this study.
Supplemental Table 1 provides details on all TP53 mutations and 17p deletions observed in our participants.
Data are available on request from the corresponding authors.
The full-text version of this article contains a data supplement.
Contributor Information
Susan L. Slager, Email: slager.susan@mayo.edu.
Esteban Braggio, Email: braggio.esteban@mayo.edu.
Supplementary Material
References
- 1.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 2.Puente XS, Bea S, Valdes-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 3.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monti P, Lionetti M, De Luca G, et al. Time to first treatment and P53 dysfunction in chronic lymphocytic leukaemia: results of the O-CLL1 study in early stage patients. Sci Rep. 2020;10(1) doi: 10.1038/s41598-020-75364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dicker F, Herholz H, Schnittger S, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009;23(1):117–124. doi: 10.1038/leu.2008.274. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez D, Martinez P, Wade R, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29(16):2223–2229. doi: 10.1200/JCO.2010.32.0838. [DOI] [PubMed] [Google Scholar]
- 7.Rossi D, Cerri M, Deambrogi C, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15(3):995–1004. doi: 10.1158/1078-0432.CCR-08-1630. [DOI] [PubMed] [Google Scholar]
- 8.Zenz T, Eichhorst B, Busch R, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–4479. doi: 10.1200/JCO.2009.27.8762. [DOI] [PubMed] [Google Scholar]
- 9.Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2012;30(26):3209–3216. doi: 10.1200/JCO.2011.39.2688. [DOI] [PubMed] [Google Scholar]
- 10.Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematology Am Soc Hematol Educ Program. 2017;2017(1):329–337. doi: 10.1182/asheducation-2017.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 12.Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247–3254. doi: 10.1182/blood-2014-01-546150. [DOI] [PubMed] [Google Scholar]
- 13.Bomben R, Rossi FM, Vit F, et al. TP53 mutations with low variant allele frequency predict short survival in chronic lymphocytic leukemia. Clin Cancer Res. 2021;27(20):5566–5575. doi: 10.1158/1078-0432.CCR-21-0701. [DOI] [PubMed] [Google Scholar]
- 14.Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122–2130. doi: 10.1182/blood-2015-07-659144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brieghel C, Kinalis S, Yde CW, et al. Deep targeted sequencing of TP53 in chronic lymphocytic leukemia: clinical impact at diagnosis and at time of treatment. Haematologica. 2019;104(4):789–796. doi: 10.3324/haematol.2018.195818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blakemore SJ, Clifford R, Parker H, et al. Clinical significance of TP53, BIRC3, ATM and MAPK-ERK genes in chronic lymphocytic leukaemia: data from the randomised UK LRF CLL4 trial. Leukemia. 2020;34(7):1760–1774. doi: 10.1038/s41375-020-0723-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia. 2018;32(5):1070–1080. doi: 10.1038/s41375-017-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114(5):957–964. doi: 10.1182/blood-2009-03-210591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brieghel C, Aarup K, Torp MH, et al. Clinical outcomes in patients with multi-hit TP53 chronic lymphocytic leukemia treated with ibrutinib. Clin Cancer Res. 2021;27(16):4531–4538. doi: 10.1158/1078-0432.CCR-20-4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malcikova J, Smardova J, Rocnova L, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114(26):5307–5314. doi: 10.1182/blood-2009-07-234708. [DOI] [PubMed] [Google Scholar]
- 21.Marti GE, Rawstron AC, Ghia P, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol. 2005;130(3):325–332. doi: 10.1111/j.1365-2141.2005.05550.x. [DOI] [PubMed] [Google Scholar]
- 22.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760. doi: 10.1182/blood-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- 23.Rossi D, Sozzi E, Puma A, et al. The prognosis of clinical monoclonal B cell lymphocytosis differs from prognosis of Rai 0 chronic lymphocytic leukaemia and is recapitulated by biological risk factors. Br J Haematol. 2009;146(1):64–75. doi: 10.1111/j.1365-2141.2009.07711.x. [DOI] [PubMed] [Google Scholar]
- 24.Parikh SA, Rabe KG, Kay NE, et al. The CLL international prognostic index predicts outcomes in monoclonal B-cell lymphocytosis and Rai 0 CLL. Blood. 2021;138(2):149–159. doi: 10.1182/blood.2020009813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinstern G, O'Brien DR, Li X, et al. Tumor mutational load predicts time to first treatment in chronic lymphocytic leukemia (CLL) and monoclonal B-cell lymphocytosis beyond the CLL international prognostic index. Am J Hematol. 2020;95(8):906–917. doi: 10.1002/ajh.25831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Evans JM, Bhagwate AV, et al. PatternCNV: a versatile tool for detecting copy number changes from exome sequencing data. Bioinformatics. 2014;30(18):2678–2680. doi: 10.1093/bioinformatics/btu363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCabe CE, Jessen E, O'Brien DR, Wiedmeier-Nutor JE, Slager SL, Braggio E. Proceedings of the 113th Annual Meeting of the American Association for Cancer Research. 2022 April 8-13; New Orleans LA. AACR; 2022. Identifying copy number variations in chronic lymphocytic leukemia using targeted next generation sequencing [abstract] Abstract 3352. [Google Scholar]
- 29.Mansouri L, Thorvaldsdottir B, Sutton L-A, et al. Different prognostic impact of recurrent gene mutations in IGHV-mutated and IGHV-unmutated chronic lymphocytic leukemia: a retrospective, multi-center cohort study by Eric, the European Research Initiative on CLL, in harmony. Blood. 2021;138(suppl 1):2617. [Google Scholar]
- 30.Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–2147. doi: 10.1182/blood-2013-11-539726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mato AR, Barrientos JC, Ghosh N, et al. Prognostic testing and treatment patterns in chronic lymphocytic leukemia in the era of novel targeted therapies: results from the informCLL registry. Clin Lymphoma Myeloma Leuk. 2020;20(3):174–183.e3. doi: 10.1016/j.clml.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bomben R, Rossi FM, Vit F, et al. P596: clinical impact of TP53 disruption in chronic lymphocytic leukemia patients treated with a BCR inhibitor. A campus CLL experience. HemaSphere. 2022;6:495–496. [Google Scholar]
- 33.Malcikova J, Pavlova S, Kunt Vonkova B, et al. Low-burden TP53 mutations in CLL: clinical impact and clonal evolution within the context of different treatment options. Blood. 2021;138(25):2670–2685. doi: 10.1182/blood.2020009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baliakas P, Jeromin S, Iskas M, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133(11):1205–1216. doi: 10.1182/blood-2018-09-873083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaramillo S, Agathangelidis A, Schneider C, et al. Prognostic impact of prevalent chronic lymphocytic leukemia stereotyped subsets: analysis within prospective clinical trials of the German CLL Study Group (GCLLSG) Haematologica. 2020;105(11):2598–2607. doi: 10.3324/haematol.2019.231027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.