

Summary
The human genome comprises approximately 3% of tandem repeats with variable length (VNTR), a few of which have been linked to human rare diseases. Autosomal dominant tubulointerstitial kidney disease—MUC1 (ADTKD-MUC1) is caused by specific frameshift variants in the coding VNTR of the MUC1 gene. Calling variants from VNTR using short-read sequencing (SRS) is challenging due to poor read mappability. We developed a computational pipeline, VNtyper, for reliable detection of MUC1 VNTR pathogenic variants and demonstrated its clinical utility in two distinct cohorts: (1) a historical cohort including 108 families with ADTKD and (2) a replication naive cohort comprising 2,910 patients previously tested on a panel of genes involved in monogenic renal diseases. In the historical cohort all cases known to carry pathogenic MUC1 variants were re-identified, and a new 25bp-frameshift insertion in an additional mislaid family was detected. In the replication cohort, we discovered and validated 30 new patients.
Subject area(s): Genetics, Genomics, Techniques in genetics, Genotyping
Graphical abstract
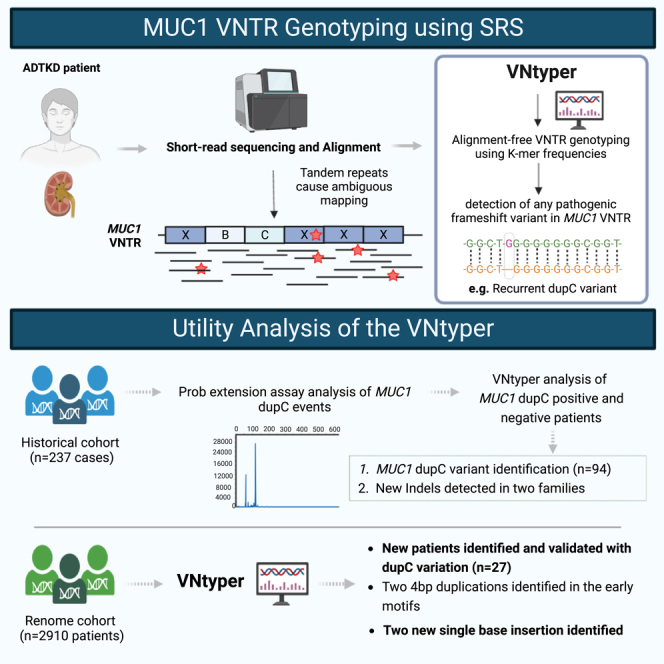
Highlights
-
•
Detecting pathogenic variants in VNTRs is challenging due to poor read mappability
-
•
VNtyper detects VNTR pathogenic variants in the MUC1 gene using Kestrel algorithm
-
•
VNtyper improves ADTKD-MUC1 diagnosis by accurate genotyping of MUC1 coding VNTR
-
•
Integration of VNtyper with SRS gene panel testing identified new overlooked patients
Genetics; Genomics; Techniques in genetics; Genotyping
Introduction
Autosomal dominant tubulointerstitial kidney disease (ADTKD, OMIM: 174000) is a hereditary condition characterized by progressive tubulointerstitial fibrosis with or without tubular dilation and atrophy, eventually leading to end-stage kidney failure.1 The affected individuals present mild-to-negative proteinuria and bland urinary sediment abnormalities, with normal kidney size.2 A positive family history is commonly reported for this disease. Molecular genetics play a key role in diagnosing and classifying ADTKD given its non-specific presentation. Pathogenic variants in different genes, including MUC1, UMOD, HNF1B, REN, and SEC61A1, are known to be responsible for ADTKD.1,3,4 The absence of a mutation does not rule out ADTKD as other loci remain to be identified.2 The recent advancements in high-throughput sequencing technology and the pan-genome graph pipelines have improved the diagnostic rate of Mendelian and multigenic complex diseases.5,6,7 However, some challenges remain for accurately detecting pathogenic variants in genes with complex structures. For instance, the detection of variants in the coding variable number of tandem repeat (VNTR) region of the MUC1 gene (encoding the mucin-1 protein) responsible for ADTKD-MUC1 (OMIM: 174000) is a real issue.
Mucin-1 is a transmembrane glycoprotein widely expressed in different segments of the nephron in the kidney, from the thick ascending limb of the loop of Henle to the collecting duct.8 The MUC1 gene maps to chromosome 1 and exhibits a large coding VNTR in exon 2, which embraces the combination of at least 34 different motifs of 60-mer that repeat 20 to 125 times with distinct random patterns for each allele. Each 60-mer motif encodes a highly glycosylated 20 amino acids (aa) block (see Figure 1). The initial five motifs (1-2-3-4(or 4p)-5(or 5C)) and the final four motifs (6(or 6p)-7-8-9) have likewise unique sequence. Any variant in these motifs except motif 7 could be found with short-read sequencing (SRS) and conventional pipelines. Between motifs 5(or 5C) and 6(or 6p), any combination of the remaining 22 known motifs is conceivable.9,10 Detailed information regarding the published motif sequences and their orientation is highlighted in Figure S1. Therefore, due to the complexity of the variation hotspot, finding pathogenic variants using SRS has proven to be challenging.
Figure 1.

Mucin-1 structural domains and recurrent MUC1 dupC variation hotspot
(A) Mucin-1 is a transmembrane glycoprotein widely expressed in different segments of the nephron in the kidney. Protein domains of the full-length mucin-1 protein are shown: N-terminus signal sequence, VNTR regions (variable repetition of 20aa blocks), SEA cleavage domain (is a highly conserved domain that undergoes an autocatalytic cleavage during folding in the endoplasmic reticulum), transmembrane domain (TM), and cytoplasmic domain.
(B and C) The frameshift caused by the insertion of a C in the VNTR motifs (blue boxes) with stretch of seven C (GCCCCCCCAGC) creates a new stop codon shortly (85aa) beyond the VNTR domain. This variation could be studied by the SNaPshot method using MwoI restriction enzyme. The 20 amino acid (aa) repeat blocks for wild-type (WT) and mutant (MT) protein are shown. The mutant protein harbors novel aa repeats (yellow boxes) and lacks the C-terminal domain.
Indeed, the insertion of a single nucleotide C in a motif with a stretch of seven C is a known recurrent variation in MUC1 VNTR.9,11 It has been established that the insertion of one or 3n+1 bases, or the deletion of two or 3n+2 nucleotides in the MUC1 VNTR (all variants leading to the same frameshift), is associated with ADTKD-MUC19,11,12,13 due to the production of toxic neo-protein.14 In all cases, the variant creates a new frame in the repeat and it alters the translational pattern of the 20 aa block. The mutant frame reaches a stop codon 85 amino acid after the last motif. The pathogenic frameshift variants in MUC1 VNTRs result in the production of a mucin 1 neo-protein containing many copies of a novel repeat sequence and lacking a C-terminal domain, which accumulates in the cytoplasm and activates unfolded protein response14 (Figure 1B).
Former studies have used alternative techniques, including probe extension assays followed by mass spectrometry9,15 or ddNTP (dideoxynucleotide) sequencing after Mwo1 digestion (SNaPshot),10 targeted analysis of one VNTR repeat with the use of Illumina system,16 or long-read sequencing (SMRT: single molecule real-time sequencing), to find causal variations; however, these approaches are either technically demanding or expensive.13,17,18 Using the SNaPshot approach,10 only the known single nucleotide insertion variant (dupC) could be investigated (Figure 1C). Other pathogenic variations than dupC or variants not affecting the Mwo1 restriction site in the non-conserved motifs could not be found or validated experimentally without the utilization of long-read sequencing technology.
A new method named code-adVNTR19 developed by Park et al. has recently been published for genotyping indels in the coding VNTRs. The utility of this method has been tested in a small cohort with three MUC1 dupC-positive individuals and 271 SNaPshot-negative individuals.19,20 Here we present a new pipeline named VNtyper with a genotyping algorithm based on k-mer approach21 to call variants from the MUC1 coding VNTR region using SRS data. Our first goal was to evaluate our pipeline in a well-described historical cohort before applying it to almost 3,000 patients with kidney disease (renome cohort) to identify new MUC1-positive patients.
Results
Historical cohort, SNaPshot results
The SNaPshot approach was used to study index cases (n = 108) and their relatives (symptomatic and asymptomatic individuals) within the historical cohort (Figure 2B). It allowed to detect 31 index cases (from 31 families) with dupC variation so classified as “MUC1 positive”. Beside index cases, 62/63 symptomatic relatives displayed a positive SNaPshot assay. In one symptomatic relative (NTIH_140) with atypical SNaPshot signal (very high undigested motifs) from a “MUC1 positive” family, we confirmed the bearing of the risk haplotype in the symptomatic individual by linkage analysis and concluded to an uninterpretable SNaPshot. All 24 asymptomatic relatives of the “MUC1 positive” index cases were negative by SNaPshot. In an additional index case, an abnormal SNaPshot signal (strong result of 7C + A and a shorter PCR product) led to the identification of a 5bp deletion in the restriction site of the MwoI enzyme. We confirmed the 5bp deletion event by subcloning and sequencing the PCR product (Figure 4E). This event was also detected in two symptomatic relatives in this family. Altogether, this investigation led to the identification of 32 families with confirmed MUC1-ADTKD and to the identification of 1/97 uninterpretable SNaPshot event.
Figure 2.

Historical cohort description flowchart
(A) The inclusion and exclusion criteria for MUC1-ADTKD are shown. In this cohort, 108 families with 237 individuals were studied by short-read sequencing. No pathogenic variant was found with standard pipeline in any gene related to ADTKD.
(B) The SNaPshot assay was performed to detect the known and recurrent MUC1 dupC variation. This method identified the dupC pathogenic variant in 31 index cases and their 62 symptomatic relatives. In one family with three affected members, our modified SNaPshot approach detected a 5bp deletion in the MwoI site.
(C) The VNtyper pipeline was applied to all individuals in the historical cohort. VNtyper re-identified all MUC1 dupC and 5bp deletion events. In one symptomatic case from MUC1-positive family (NTIH_140), linkage analysis validated the VNtyper results by confirming the segregation of the risk allele. In one family including six symptomatic members and negative SNaPshot, the pipeline detected a 25bp insertion.
Figure 4.

Modified SNaPshot method explaining new events at the Mwo1site
(A–C) Examples of the SNaPshot PCR product migration from symptomatic and asymptomatic individuals are shown. (B) In case of positive dupC event (REN6122000742), a 7C + C and 8C + A signals are present. With our modified protocol direct sequencing of the PCR product is feasible, which confirms the SNaPshot results. (C) A very strong 7C + A signal could be observed in some SNaPshots if Mwo1 digestion failed at one of the sites (due to a Mwo1 restriction site variants). In this instance, sequencing the PCR product could aid in the identification of potentially pathogenic (NTI6120004559) or polymorphic variants (HYP2316, NTI195).
(D and E) In one index patient with the clinics of ADTKD, a very strong 7C + A signal led to the detection of a 5bp deletion in the Mwo1 site and was confirmed by subcloning and sequencing the SNaPshot PCR product.
In 76 additional index cases and their relatives (109 symptomatic/116 individuals), the SNaPshot assay was negative. Such families were labeled as “MUC1 negative families” (Figure 2B).
Historical cohort, VNtyper-Kestrel results
We applied VNtyper-Kestrel on all 237 individuals from the historical cohort to re-identify positive cases and to determine if any MUC1-positive family members were overlooked (considered either as false negatives of the SNaPshot or false positives of the VNtyper).
VNtyper-Kestrel successfully re-identified MUC1 pathogenic variants in all symptomatic individuals from the MUC1 families (n = 97) (Figure 2C). In one previously described symptomatic relative with uninterpretable SNaPshots (NTIH_140), VNtyper reported a dupC, confirming the segregation of the risk allele.
In the MUC1-negative group (76 index cases; 116 individuals), VNtyper-Kestrel identified an additional family with six affected members, all bearing a 25bp insertion in the MUC1 VNTR, which could not be detected by the initial SNaPshot investigation. This variant was absent in their five asymptomatic relatives. Altogether, these results showed the applicability of the VNtyper-Kestrel pipeline to detect all MUC1 events in our cohort. It detected not only known cases with dupC but also new cases that cannot be detected by SNaPshot method.
Beside concordant true-positive and true-negative cases, VNtyper-Kestrel identified an independent cluster of 32 discordant individuals (Figure 5A – red dots below the dotted line). While all of them were negative by SNaPshot, VNtyper-Kestrel identified a MUC1 dupC with a statistically lower depth score than that in patients with relevant events and positive SNaPshot (respective mean ± SD: 0.0025 ± 0.0005 vs. 0.013 ± 0.007, p = 2.10−15).
Figure 5.

Historical cohort characterization
VNtyper analysis of the historical cohort identified MUC1 pathogenic variants in 33/108 families (30.6%).
(A) The VNtyper results of the historical cohort based on the depth score (log10 ratio) and the estimated depth of the alternate variant (AltDepth) are shown. Independent clustering of the MUC1 SNaPshot positive and negatives cases allowed the definition of a threshold to filter out variants with low support. All SNaPshot negative cases except (NTIH_140, marked with $) clustered together below the threshold (0.00469, in red), one patient was localized in the 10% above the threshold, and all SNaPshot positive cases (in green) as well as six cases with 25bp insertion (marked with #, in lightblue) were located above the 10% of the threshold (0.00515).
(B) In this section (magnification of the framed zone in (A)), the y axis scale was adjusted, and two plots were merged. Variants with depth scores below the threshold were filtered out and classified as false positive. Any variant with a depth score between the threshold and 10% above [0.00469–0.00515] or with AltDepth less than 20 is deemed low-confidence variants (L shape in gray), whereas any variant with a depth score above 0.00515 and AltDepth above 20 is considered to be high-confidence results.
(C) The analysis of the pipeline’s sensitivity and specificity with different depth score-based thresholds identified 0.00469 as the optimal threshold for distinguishing true positives from false positives.
(D) Among MUC1-positive families in this cohort, 31 had MUC1 dupC (94%), one had a 5bp deletion, and another had a 25bp insertion event (both 3%).
Altogether, we calculated a sensitivity (ability to detect all true positives) of 100% and a specificity (ability to exclude all true negatives) of 76.11% for the VNtyper-Kestrel. To determine the best depth-score threshold to detect 100% of true positives (sensitivity) while excluding the 32 aforementioned false positives (specificity), we calculated sensitivity and specificity at different depth-score points (Figure 5C). We determined the depth score of 0.00469 to be the optimal threshold because the sensitivity and specificity were both 100% at this point (Figure 5C). All individuals with a depth score below 0.00469 had a negative SNaPshot (red dots in Figure 5A).
Since there was no true positive in the historical cohort with AltDepth below 20, we decided to further study this zone in the renome cohort with caution by labeling variant with AltDepth <20 as low confidence. Also, as we identified few variants with a depth score within the zone of the 10% above the threshold [0.00469–0.00515], we decided to also label variants from this zone as low confidence for further analysis. Taken together, the analysis of the historical cohort allowed us to set up parameters to filter out false-positive VNtyper patients.
Comparing results from VNtyper-Kestrel and VNtyper-code-adVNTR within the historical cohort
We then compared the VNtyper-Kestrel to the recently published code-adVNTR method, included in our tool. While the VNtyper-Kestrel re-called all MUC1 SNaPshot-positive cases, the code-adVNTR missed five cases with MUC1 dupC events (NTIH155, NTIH153, NTIH377, NTI383, and NTI188). In contrast, code-adVNTR called no false-positive cases since it evaluates the likelihood of true calls and false calls owing to sequencing errors and filters out variants based on the p value >0.001 obtained from the statistical test. Another discrepancy between VNtyper-Kestrel and VNtyper-code-adVNTR was for the family with the 25bp insertion. The code-adVNTR result for affected members of this family was a 23bp deletion with the number of supporting reads larger than mean coverage (p value = 0), while VNtyper-Kestrel reported a 25bp insertion. By extracting reads containing the indicated variant from the fastq files, we confirmed the insertion of 25bp as predicted by Kestrel. No asymptomatic relative or affected MUC1 dupC patient had reads with this insertion pattern. The VNtyper-Kestrel and code-adVNTR results for all MUC1-positive cases from the historical cohort are shown in Table S3. Altogether, we report herein that code-adVNTR exhibits a lower sensitivity than VNtyper-Kestrel for the detection of MUC1 variants but is able to detect variants other than dupC that SNaPshot cannot detect.
Renome cohort, VNtyper-Kestrel results
Using this extensive cohort, our goal was to apply our VNtyper-Kestrel pipeline to previously obtained sequencing data to identify undiagnosed cases of ADTKD-MUC1 and estimate the specificity of VNtyper. The renome cohort had been previously studied for a panel of genes involved in monogenic renal diseases including the MUC1 gene. Figure 6C displays the distribution of different patient groups in this cohort according to their initial diagnosis.
Figure 6.

Characterization of the renome cohort. VNtyper analysis of the renome cohort identified 30 MUC1-positive patients
(A and B) The depth score-adapted threshold revealed from the historical cohort was applied to the naive renome cohort. Forty cases were reported above the threshold (dashed line), and 662 instances were below the threshold. To confirm the reliability of the threshold, we analyzed all cases with Mwo1 site variations (dupC or dupA) reported above (n = 35) the threshold. We extended the SNaPshot analysis to the 11 individuals that clustered just below the threshold (i.e., between the threshold and 10% below [0.004176–0.00469]) to ensure that no true positive were missed. All cases tested below the threshold and 9/12 of the cases reported with low confidence were SNaPshot negative (red points). SNaPshot verified MUC1 dupC events in 26 cases (25 high-confidence and 1 from low-confidence cases) as well as a case with MUC1 dupA event (green points). With a second high depth NGS, we verified a patient with low confidence single base insertion as true positive. Variations identified in the early conserved motifs were validated using IGV, see Figure S5. In section B (magnification of the framed zone in A), the y axis scale was adjusted, and the plots were merged.
(C) Patients from this cohort were assigned to different groups based on the initial clinical diagnosis.
(D) MUC1 pathogenic variants were discovered in various patient groups, particularly NTI. Several cases were also identified in other cohort groups like NPH, REN, NCR, and CAKUT.
(E) The description flowchart of this cohort is shown. Note: $Individuals with variants that cannot be confirmed by SNaPshot. ∗High confidence: patients with AltDepth above 20 or depth score above 0.00515. #Low confidence: patients with AltDepth below 20 or depth score between 0.00469 and 0.00515.
In two unrelated patients (REN_6122GM002870 and NTI_6121GM005428) a 4bp duplication had been previously identified in motifs 1 and 4, respectively, and a dupC variation was detected in three other independent cases (NTI1129, NTI_6121GM003097, and NPH1908593) in the early conserved motif 5. The IGV22 visualization of the bam files for these patients are shown in Figure S5. No MUC1-related pathogenic variant had been identified in other patients in this cohort before this investigation.
VNtyper-Kestrel was applied to the already available bam files from all patients of the renome cohort. Among the 2,910 cases, Kestrel-VNtyper reported a negative result for 2,208 patients. Among the 702 remaining cases, the depth score was below the threshold (0.00469) for 662 individuals, and the samples were thus filtered out as false positives. In 40 patients, the depth score was above the threshold (Figures 6A–6E).
To ensure the validity of our results, we categorized variants into two groups. The first group contained variants with a depth score above 0.00515 (the threshold +10%) and an AltDepth above 20, which were considered true positives. The second group included variants with an AltDepth below 20 or variants with a depth score within the zone of 10% above the threshold [0.00469–0.00515]. This separation allowed us to differentiate variants with high-confidence label from those with lower confidence (Figures 6A and 6B - gray band), which requires validation by other assays.
Among the 40 cases with a depth score above the threshold, 28 cases were reported with a high-confidence label and 12 were labeled as low-confidence cases due to AltDepth <20 and/or depth score below 10% of the threshold.
Among the 28 patients with a high-confidence positive result, 25 had a MUC1 dupC variant, one had a single nucleotide insertion other than C (an A insertion in NTI_6120004559), and two had 4bp duplication events in early conserved motifs (Figure 6E). These two 4bp duplications as well as three other dupC insertions in motif 5, all located in the early repeats and previously identified by NGS, were successfully re-identified by our pipeline with high confidence (see Figure S5). As expected, since variants were located in an early conserved motif, the variant depth was around half the total depth. SNaPshot analyses of all dupC-positive cases were conducted. SNaPshot was positive in 25/25 high-confidence cases. In one patient (NTI_6120004559), VNtyper-Kestrel identified a MUC1 dupA pathogenic variant with high confidence that we validated by our modified SNaPshot (Figure 4C).
Among the 12 patients with low-confidence label, one patient was found to carry a deletion of two nucleotides (CC) in the early motif 3, where a depth-score ratio of 50% is expected. This case exhibited a lower depth score than expected and had therefore been classified as false positive (Figure 6E). Regarding the 11 remaining low-confidence cases, only 2/11 were validated: HYP4100 (by SNaPshot) and NTI1179 (with second NGS). HYP4100 had a depth score above 0.00515 but a low AltDepth. NTI1179 had an AltDepth of 40 but a depth score of 0.00437. This particular case was validated with the second NGS (AltDepth equal to 537 and yielded a depth score of 0.00612). Two patients, NTI1168 and PK432, exhibited positive VNtyper (performed twice, but low confidence) but atypical SNaPshots. NTI1168 had a personal clinical history compatible with MUC1-related ADTKD, and no familial history was reported (Table S5). PK432 was a 6-year-old patient with a heterozygous deletion of the HNF1B gene and several microcysts. As we were unable to obtain new DNA samples from these patients and their parents, we have classified these cases as uninterpretable results. SNaPshot was negative in seven remaining patients with dupC variation. In addition, to confirm the reliability of the threshold (0.00469) to exclude false positives, we extended the SNaPshot analysis to the 11 individuals that clustered just below the threshold (i.e., between the threshold and 10% below [0.00411–0.00469]) to ensure that no true positive was missed. All 11 individuals with a depth score below the threshold were negative.
Altogether, in this replication cohort, we confirmed a high sensitivity of 100% for detecting MUC1-ADTKD individuals using VNtyper-Kestrel applied to SRS and showed that our pipeline can identify pathogenic variants in the early motifs and small indels other than dupC. However, its specificity decreased to 75.0% since we also detected false positive above the threshold of 0.00469. The specificity increases to 100% in the high-confidence group.
Method comparison within the renome cohort
In the second part of the investigation, code-adVNTR was utilized to compare findings for all 2,910 cases. The code-adVNTR method reported variants in 44 cases. Among these 44 cases, 30 were concordant with VNtyper-Kestrel, whereas 14 were not found by VNtyper-Kestrel (Table S6). The SNaPshot analysis was done on all reported positive code-adVNTR cases. Concordant cases were validated except two cases (PK423 and NTI1168) which were already classified as uninterpretable cases, while all 14 discordant cases were not confirmed, thus considered as false positive of the code-adVNTR. From 30 concordant cases, 28 were MUC1 dupC, one displayed an insertion of a T, and another an insertion of an A. Two out of 14 discordant cases had MUC1 dupC variation, and 12/14 had various insertions and deletions (Table S6). The SNaPshot and VNtyper-Kestrel were negative for all discordant dupC cases that all had unrelated clinics (Table S5).
The code-adVNTR method was able to detect not only dupC but also other variations such as small deletions but unable to detect pathogenic variants in the early conserved motifs (motifs 1–4). The final results for all cases identified by both methods in the renome cohort are shown in Table S4.
Data from both historical and renome cohorts were used for benchmarking the VNtyper-Kestrel and VNtyper-code-adVNTR methods within the pipeline. The median run time for mapping-free genotyping with the Kestrel algorithm was significantly lower than that for code-adVNTR within both cohorts (p value <0.0001). Figure S6 shows the mean speed of both Kestrel and code-adVNTR on small and large panels.
Discussion
We provide herein an accurate tool to detect pathogenic variations in the VNTR of the MUC1 gene (Figure 3), a genomic coding-region precedently unreachable by SRS due to poor read mappability and unpredictable sequence of 25–120 highly homologous 60-mer repeats motif (VNTR). Using our pipeline called VNtyper, we were able to re-identify 97 patients with a known MUC1-ADTKD and to identify 36 unrelated patients with unknown MUC1-ADTKD.
Figure 3.

Schematic overview of the VNtyper pipeline
The pipeline of short-read sequencing (SRS)-based deleterious variant detection in the coding VNTR of the gene MUC1 in ADTKD is shown.
Current methods for analyzing ADTKD-MUC1 patients include probe extension assays followed by mass spectrometry,9,15 SNaPshot,10 and long-read sequencing (SMRT).13,18 These approaches are either technically demanding or expensive13,17,18 and need the use of a specific processing other than SRS which is regularly used in the genetic laboratories. The issue of detecting MUC1 events using SRS data has been addressed in a more recent bioinformatical method called code-adVNTR, which utilizes multi-motif HMM (Hidden Markov Models) profiles to statistically detect frameshifts in coding VNTRs, as demonstrated in three independent individuals19,23 and 271 SNaPshot-negative individuals.19 In the present study, we implemented a pipeline including alignment-free genotyping algorithm as well as code-adVNTR method to identify variants in the VNTR region independently. First, we used the alignment-free genotyping algorithm based on haplotype reconstruction from k-mer frequencies.21 To bypass the issue of unknown sequence of reference for the MUC1 VNTR region, we developed a homemade motif dictionary as a reference. In the event of inconsistencies between the genotyping result and clinics, the code-adVNTR method was used to compare findings. We showed herein the superiority our VNtyper-Kestrel tool, since while code-adVNTR exhibits a good specificity (a few false positives in the renome cohort, Table S6), it also showed a lower sensitivity (93.3%) than VNtyper-Kestrel as it missed five cases whereas VNtyper-Kestrel detected all MUC1 cases.
The fact that the SNaPshot approach can only detect the variants affecting Mwo1 restriction site is a significant limitation. Using a specific MUC1 VNTR sequencing approach coupled with a spectrometry-based probe extension assay, Olinger et al. reported a prevalence of the dupC variant of 93.5% among MUC1-ADTKD in a cohort of 93 families.11 In our study, we used SRS coupled with alignment-free genotyping approach and detected a pathogenic frameshift MUC1 variant in 62 families in our two cohorts (historical and renome), including 90% of dupC variants that is consistent with the report by Olinger et al.11 However, there were a few discrepancies between VNtyper (both algorithms) and the SNaPshot in our cohorts. These cases (PK423 and NTI1168 from renome and NTIH_140 from historical) were identified by both algorithms but with atypical SNaPshot. The segregation analysis helped us consider NTIH_140 as true positive; however, for PK423 and NTI1168 due to lack of new samples from the patient and relatives, we considered these two results as uninterpretable. This underscores the importance of utilizing multiple methods to achieve accurate interpretation of ADTKD patients.
VNtyper pipeline effectively excluded false-positive cases due to technical errors. By applying VNtyper-Kestrel on all SNaPshot-negative cases in the historical cohort, we determined a depth score-adapted threshold to distinguish true positives from false positives. We tested the reliability of this threshold in a naive renome cohort of 2,910 individuals with likely hereditary kidney disease, in which MUC1 has been captured but unexplored due to technical issues. To investigate the possibility of false negatives in the zone of the threshold minus 10%, we used the SNaPshot method to analyze all individuals. However, no false negatives were detected in this zone, which confirms the high sensitivity of VNtyper-Kestrel in detecting true positives. Furthermore, when analyzing the distribution of 662 individuals with a depth score below the threshold of 0.00469 (considered as negatives), we have not identified an enrichment in the patients of NTI group but similar proportions of all disease groups. Due to the adjustment of the threshold specifically for dupC variations and the limited number of cases available for testing the threshold’s validity with other types of variations, we opted not to apply the threshold to variants other than dupC. Altogether, these findings support the applicability of the VNtyper-Kestrel to determine pathogenic frameshift variations in a single fast and accurate analysis. Taken together, our tool offers a new diagnostic perspective with high sensitivity and specificity compared to SNaPshot and to code-adVNTR alone.
One strength of our study is the capability to retrospectively re-analyze SRS data from 2,910 individuals with likely hereditary kidney diseases in an unbiased manner, thanks to the addition, in our library preparation kit, of probes to capture all exons and intron 2 of the MUC1 gene. By contrast, previously published cohorts have focused solely on patients with ADTKD symptoms that are compatible with a MUC1-related disease, likely due to technical limitations in analyzing MUC1 VNTR with standard SRS data. In the MUC1-positive cases originated from the naive renome cohort, only 74% of patients were referred for ADTKD (23/31) (see Figure 6). The remaining cases were identified in NPH (4/31), REN (2/31), NCR (1/31), and CAKUT (1/31). This supports the importance of applying MUC1 molecular diagnosis to the regular diagnosis of patients with likely hereditary kidney diseases with a tool like VNtyper that can be incorporated into the ADTKD genetic diagnosis using routine SRS.
We also conducted an analysis of MUC1 coverage and applied VNtyper on 2,328 high-coverage whole-genome sequencing files from the 1000 Genomes Project.24 The aim was to further examine MUC1 coverage in these cases to check the possibility of identifying any false positives. The mean and median coverage of the MUC1 gene in this project was 36.47 and 35.72, respectively. The obtained coverage was even lower than the mean coverage identified with the 5% downsampled read depth in the renome cohort (equal to 69.04), indicating that this depth may not be sufficient for detecting true positives or possible false positives.
The targeted approach used for MUC1 indel genotyping has the potential to be applied to other genes with disease-associated VNTRs. Protein-coding VNTRs have been identified in genes including, LPA,25 ACAN,26,27 TENT5A,26 MUC1,9 TCHH,26 PER3,28 MUC21,29 CEL,30 DRD4,31 ZFHX3,32 GP1BA,33 and MMP9.34 The repeat-unit size varies from 12bp in MMP9 to several kilobases in LPA, and the repeat count ranges from two in LPA and TENT5A to 125 in MUC1. The impact of coding-VNTRs on human phenotypes is determined by the length of the repeats (length polymorphisim) or the presence of deleterious variants (especially indels). Some deleterious frameshift variants have been reported in the VNTR of several genes including, MUC1 (single insertion leading to specific frame),9 CEL (single deletion),30 and MUC21 (4bp deletion).29 This goal could be achieved by creating a gene-specific VNTR motif reference file and applying disease-specific filtration steps to effectively report informative variations.
With our pipeline, we propose a diagnostic algorithm for ADTKD-MUC1. This algorithm is shown in Figure S7 and could be plugged to the standard pipeline analyzing data from panel sequencing. Briefly, only patients with a depth score above the threshold of 0.00469 should be considered for being true positives. Among them, patients labeled as low confidence (patients with a depth score within the 10% zone above the threshold and patients with AltDepth <20) should be confirmed by another assay since the specificity is not 100% in this zone. For other patients labeled high confidence, the sensitivity and specificity of VNtyper are 100% and in case of concordant clinic could be considered as true positives.
The recent development of innovative therapies for proteinopathies, including ADTKD-MUC1,14 compels us to diagnose ADTKD patients with a reliable routine diagnosis pipeline. Overall, this study offers a novel and accurate way to genotype coding VNTR in the MUC1 gene that can be incorporated to standard SRS regularly used in genetics laboratories. This will contribute to improve the identification of patients with ADTKD-MUC1.
Limitations of the study
VNtyper has limitations that must be mentioned and addressed in future works. The Kestrel and code-adVNTR are sensitive to the quality and coverage of the input data. When repeating NGS on patients with low confidence-labeled variants, it is recommended to use fresh and high-quality (not fragmented) DNA samples. Low-quality sequencing reads, such as short read length, very low-quality base calls, and very low depth, may increase the likelihood of false negatives, which might be a problem for using this pipeline with exome or genome sequencing data. When the sequencing depth is very low, due to the large VNTR length, a mutation dilution effect occurs, and the chance of missing variants may increase, secondary to the unsuccessful active region detection and the likelihood that the pipeline ignores positive cases. Further research involving whole-genome sequencing combined with VNtyper analysis of MUC1-positive patients is essential to determine the optimal depth in which we could identify all true positives. Code-adVNTR, on the other hand, appears to filter out several positive cases (five cases from historical cohort) that we found to be positive with VNtyper-Kestrel and SNaPshot. This should be due to the hard filtration that this method applies on reads harboring the mutation. The likelihood of missing potential variations in motifs not present in our motif dictionary is a further consideration. This could be remedied by incorporating newly described motifs into the reference file.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| VNtyper | This paper | https://github.com/hassansaei/VNtyper |
| Kestrel version 1.0.1 | Audano, P.A. et al.21 | https://github.com/paudano/kestrel |
| Code-adVNTR version 1.3.3 | Park, J. et al.19 | https://github.com/mehrdadbakhtiari/adVNTR |
| GATK version 4.2.5 | McKenna, A. et al.35 | https://gatk.broadinstitute.org/hc/en-us |
| BWA version v0.7.17-r1188 | Li, H. et al.36 | https://bio-bwa.sourceforge.net/ |
| Sambamba version 0.6.8 | Tarasov, A. et al.37 | https://lomereiter.github.io/sambamba/ |
| Samtools version 1.11 | Li, H. et al.38 | http://www.htslib.org/ |
| Fastp version 0.23.2 | Chen, S. et al.39 | https://github.com/OpenGene/fastp |
| IGV version 2.16.1 | Robinson, J.T. et al.22 | https://software.broadinstitute.org/software/igv/ |
| Python versions 3.9 and 2.7 | Python Software Foundation | https://www.python.org |
| Java version 8 | Orcale | https://www.java.com/en/ |
| Critical commercial assays | ||
| SureSelectXT Reagent kits | Agilent | #G9642C |
| Twist Library Preparation kit | Twist Bioscience | #100572 |
| Twist Hybridization and wash Kit | Twist Bioscience | #101025 |
| SNaPshot Multiplexing kit | ThermoFisher Scientific | #4323159 |
| MwoI restriction enzyme | New England BioLabs | #R0573 |
| ExoSAP_ITTM | ThermoFisher Scientific | #78201 |
| Ampure XP beads | Beckman Coulter | #A63882 |
| BigDyeTM Terminator v3.1 Cycle Sequencing Kit | ThermoFisher Scientific | #4337456 |
| Deposited data | ||
| Read-name replaced Bam files from MUC1 dupC positive patients | This paper | https://github.com/hassansaei/VNtyper |
| 1000 Genomes high coverage Project | Byrska-Bishop, M. et al.24 | https://www.internationalgenome.org/data-portal/data-collection/30x-grch38 |
| Oligonucleotides | ||
| SNaPshot amplification primer forward: actgtaaa acgacggccagtCTGGGAATCGCACCAGCGTGTG GCCCCGGGCTCCACC |
This paper | N/A |
| SNaPshot amplification primer reverse-Fluo: accag gaaacagctatgaccCGTGGATGAGGAGCCGCAGTG TCCGGGGCCGAGGTGACA |
This paper | N/A |
| Prob 7C: CGGGCTCCACCGCCCCCCC |
This paper | N/A |
| Prob 8C: gagagagagaCGGGCTCCACCGCCC CCCCc |
This paper | N/A |
| Other | ||
| 3500 Genetic Analyzer | ThermoFisher scientific | #4406017 |
| NextSeq 500 sequencer | Illumina | https://emea.illumina.com/systems/sequencing-platforms/nextseq.html |
| MiSeq sequencers | Illumina | https://emea.illumina.com/systems/sequencing-platforms/nextseq.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be promptly fulfilled by the lead contact Hassan Saei (hassan.saei@etu.u-paris.fr and Hassan.saei@inserm.fr).
Materials availability
This study did not generate new unique reagent.
Experimental model and participant details
Two patient cohorts were included and studied: the historical cohort and the renome cohort. All subjects or their legal representatives gave written informed consent to the molecular genetic analysis. The research was performed in accordance with the Declaration of Helsinki on human experimentation of the World Medical Association, and it was conducted with the approval of the Comité de Protection des Personnes pour la recherche biomédicale Ile de France. Information regarding each cohort is present in the “study cohorts” section in the method details. In brief, the historical cohort composed of 237 individuals belonging to 108 families (206 with ADTKD and 31 asymptomatic relatives). The renome cohort composed of 2910 patients with renal symptom tested on a panel of genes involved in monogenic renal diseases, from 2017 to 2022 in the Molecular Genetics Department of Necker University Hospital (Paris, France). Information regarding the ADTKD inclusion and exclusion criteria is present in Figure 2.
Method details
Study cohorts
Two distinct cohorts were analyzed with our pipeline. The first cohort was a historical cohort described in Figure 2, composed of 237 individuals belonging to 108 families with DNA available (206 with ADTKD and 31 asymptomatic at risk people). All these cases fulfilled the ADTKD-MUC1 inclusion criteria depicted in Figure 2A. Only index cases who had previously been tested for variations in the known ADTKD-related genes (MUC1, UMOD, HNF1b, DNAJB11, SEC61A1, and REN) were considered for the study. We enrolled individuals either tested negative on the gene panel or with a pathogenic variant in MUC1 (i.e in the early conserved motifs reachable by SRS). DNA from all individuals (108 index cases and 98 symptomatic/31 asymptomatic related) were screened for MUC1 pathogenic variations (1) by applying the VNtyper pipeline and (2) by probe extension assay method (SNaPshot) to detect the well-described cytosine duplication in the VNTR (Figure 2B).
The second cohort (named “renome”) comprises 2,910 patients with renal symptoms, studied from 2017 to 2022 in the Molecular Genetics Department of Necker University Hospital (Paris, France), each assigned to a group of hereditary renal diseases based on the initial presentation described as follows: Alport syndrome (ALP), congenital anomalies of the kidney and urinary tract (CAKUT), steroid-resistant nephrotic syndrome (NCR), autosomal recessive polycystic kidney disease (PK), renal ciliopathy not resembling autosomal recessive polycystic kidney disease (NPH), chronic tubulointerstitial nephropathy (NTI) and kidney failure of unknown origin (REN/CKD). Each patient in this cohort was tested for variants in a panel of genes listed in the renome including MUC1 gene. Of note, even if MUC1 gene was in the panel, only variants in the conserved early and late motifs except motif 7 were reachable with our regular pipeline. The complete list of genes studied in our panel is disclosed in Table S1.
Targeted Illumina panel sequencing
For all NGS analyses performed during the time or previously to the study, we used the following protocol. DNA was extracted from blood cells using standard procedures. During the study, the DNA fragmentation, libraries construction, and capture process were switched from the SureSelectXT (Agilent technology, Custom DNA Target Enrichment Probes) to the TWIST® technology (Twist Custom Panels). On the renome cohort, 1922 cases were prepared with TWIST and 988 cases were prepared with Agilent library preparation kit. All samples from NTI cohort were prepared using the TWIST kit. Using specific probes, we captured all exons and intron 2 of the MUC1 gene in our NTI (panel of six genes MUC1, UMOD, HNF1b, DNAJB11, SEC61A1, and REN) and renome panel (229 genes, Table S1). We applied an increased tiling density (X3) along MUC1 intron 2. Sequencing was performed paired-end 2∗150bp using Illumina NextSeq 500 or MiSeq sequencers.
After demultiplexing, the sequences were aligned to the human genome reference sequence (GRCh37, UCSC Genome Browser) using BWA36 software (v0.7.17). Variant calls were made with the GATK35 haplotypecaller (v4.2.5). Variants were annotated and filtered using the Polyweb software interface designed by the Bioinformatics platform of the Université de Paris Cité.
VNtyper design
The pipeline was developed utilizing the Python programming language and third-party tools, such as Kestrel21(v1.0.1), Sambamba37(v0.6.8), BWA36(v0.7.17-r1188), Samtools38(v1.11), Fastp39(v0.23.2), and code-adVNTR19,32(v1.3.3). VNtyper uses the alignment file or paired-end short-read sequencing data for fast and accurate genotyping of MUC1 coding-VNTR in ADTKD. It employs alignment-free genotyping of MUC1 VNTR using k-mer frequencies. Very briefly, Kestrel algorithm takes raw reads and our MUC1-specific reference file (MUC1-VNTR motif dictionary) and converts them to the IKC file (indexed k-mer count file) and to an array of k-mers respectively. The k-mer frequencies from the sequence reads are assigned to the ordered k-mers of the reference. A decline in frequency represents an active region where one or more variants are present. Following haplotype reconstruction and alignment, variants are retrieved from haplotypes.
The tool takes the following files as input: (i) the alignment and its index file (.bam and.bai) or Illumina Short-read sequencing file (.fastq), (ii) MUC1-VNTR motif dictionary (.fa), (iii) the reference sequence for chr1 from UCSC genome browser and (iv) VNTR database file for the code-adVNTR algorithm. In order to employ the Kestrel method, a 120-mer MUC1-VNTR motif dictionary comprised of known 34 motifs (Figure S1) was built. When the user provides an alignment file for analysis, the tool extracts reads aligned to the MUC1 gene on chromosome 1 (chr1:155158000-155163000) using Sambamba and also retains unaligned reads from the bam file. After removing duplicates, the tool converts the smaller bam files to fastq files.
After processing the input file, the pipeline runs the Kestrel toolkit. A complete workflow for the Kestrel algorithm is shown in Figure S2. The k-mer size into which the reads and references are split up is the most critical parameter for the algorithm. Typically, the length of a k-mer is between fifty percent and two-thirds of the read length. This size may not, however, be applicable to all conditions. Too-short k-mers produce short contigs (haplotypes in Kestrel), while too-long k-mers produce few but longer contigs. Since error-free k-mers must cover each other at every place inside a contig, insufficient coverage and sequencing errors result in few contigs. Shorter k-mers allow us to construct more haplotypes; nonetheless, this raises the possibility of incorrect variant calls. In our case, due to the small reference size, it is preferred to choose a shorter k-mer size. Therefore, the ideal approach was to conduct multiple trials with various k-mer sizes to minimize the chance of missing any case. After analyzing MUC1 positive cases with various K-mer sizes, we identified the k-mer size of 20 as the most suitable for our analysis using the default parameters of the Kestrel method. However, we did miss a few cases with this k-mer size and with the default parameters. To address this, we updated some parameters in the Kestrel algorithm, such as --maxalignstates (maximum number of alignments, to 30) and --maxhapstates (maximum number of haplotypes, to 30) to ensure that all variants could be called with this k-mer size. The vcf output contains several SNPs and indels, along with estimated depths for the variant. For each reported variant, Kestrel provides the estimated depth of the haplotypes with alternate variant (AltDepth) and the estimated depth of the haplotypes in the active region. We calculated and used a depth-score defined by the ratio of estimated depth of the haplotype with alternate variant over estimated depth of the haplotypes in the active region to filter out dupC variants with low support.
To prioritize variations in the pathogenic frame, VNtyper takes the vcf output and performs variant processing, which includes adding annotations to variants based on their type (SNV, Indel), separating tolerated frameshift variants from pathogenic frameshift variants based on calculated frame score. The frame score corresponds to the number of bases inserted or deleted that VNtyper calculates for each reported variant to be able to separate pathogenic frame from tolerated frame. The tool also distinguishes true calls from false calls based on the depth-score-adapted threshold. In addition, it labels each reported variant according to the level of confidence based on both the haplotype depth-score and the haplotype AltDepth. The motif sequence and variant position are added to the final result and the variation with the maximum depth-score is kept. This step generates a single result file containing motif data, variation type, variant position, estimated depths, depth-score, and confidence. If the user chooses to utilize both Kestrel and code-adVNTR methods (Figure S3), the VNtyper will additionally process the code-adVNTR result, which includes inframe variant filtering and frame score computation for frameshift variants, while maintaining the disease-associated indels. Ultimately, the pipeline combines the processed results from Kestrel and code-adVNTR into a single report. The complete overview of the VNtyper pipeline is shown in Figure 3. Of note, VNtyper referred to the pipeline with Kestrel as the main genotyping algorithm, and code-adVNTR as an optional step. In general, we utilized VNtyper for the combination of both methods. When referring to a specific method, we used VNtyper-Kestrel or VNtyper-code-adVNTR.
Implementation of a MUC1-specific motif dictionary
As previously noted, the coding VNTR of MUC1 consists of 34 distinct 60-mer motifs (encoding 20 amino acids) reported up to now.9,18 There could be additional motifs that are not yet characterized. Since the Kestrel algorithm failed to use a conventional human reference sequence in our case, a MUC1-specific reference sequence was required. We built a 120-mer motif dictionary containing all conceivable motif shufflings and labeled them according to their origin and order (n = 1156). Only 558 of these combinations are expected in the real life and were included in the motif dictionary. To generate the combinations, we used the Biopython package (v1.76) to create a list of motif sequences and used nested loops to concatenate the motifs in pairs and store the resulting 120-mer sequences and motif names in a dictionary. The dictionary has been sorted, indexed, and prepared for use with the Kestrel algorithm. In the event that additional motifs are discovered, they could be added to the dictionary. The common motifs observed within (non-conserved) and adjacent (conserved early motifs) to the MUC1 VNTR are depicted in Figure S1.
Experimental validation of MUC1 events
We used the Ekici et al.’s SNaPshot minisequencing (ThermoFisher Scientific) protocol adapted from the Kirby et al.’s method9 to screen the cytosine duplication in the VNTR of MUC1.10 Adding an M13 tag to the amplification primers allowed us to sequence the undigested PCR products, revealing the sequence of the undigested MwoI site. In brief, 100 ng of genomic DNA is digested once with MwoI. Remaining intact VNTRs are amplified using the extended primers tagged with M13 sequences to increase the PCR size and allow Sanger sequencing of the PCR products. A second digestion with MwoI is then performed, followed by a two-step purification: ExoSAPTM, to remove primers, single stranded PCR product, and dNTP not incorporated, and Ampure XP beads, for size selection. The SNaPshot probe extension kit is used and the products analyzed on a sequencer (ABI Genetic Analyzer 3500). A detailed stepwise protocol is described in Figure S4. The amplification primers and probe sequences are shown in Table S2.
The segregation of the risk allele in ADTKD-MUC1 positive families has been studied by linkage analysis to characterize affected family members in cases of negative SNaPshot result. The polymorphic markers used for linkage analysis in MUC1 positive families are shown in Figure S4.
Quantification and statistical analysis
To compare the runtime of two algorithms within VNtyper and analyze the mean coverage between the introduced cohorts in this study, we conducted statistical analysis using the Fisher’s exact test. The significance level was set to 0.05. Additionally, we devised a depth-score adapted threshold to filter out dupC variations in the MUC1 VNTR with low support. Detailed information regarding the calculation and threshold can be found in the method details section.
Acknowledgments
First, we thank all patients and their family members that participated in this study. We thank the bioinformatics platform of the Université Paris Cite at Imagine Institute, Paris, France, for providing access to the computational resources necessary for the setup and wide-scale analysis of the patient’s data. This work was supported by state funding from the “Agence Nationale de la Recherche” under “Investissements d’avenir” program (ANR-10-IAHU-01). Hassan Saei is a scholar of the PPU-Imagine International Doctoral Program, supported by the Institut Imagine. We extend our gratitude to all the clinicians listed below who have contributed as collaborators in this study: Asma Alla, Philippe Vanhille, Elodie Bailly, Stanislas Bataille, Julie Belliere, Sophie Blesson, Franck Bridoux, Guillaume Canaud, Christophe Charasse, Christian Combe, Mohamed Said Dahmoune, Myriam Dao, Nadege Devillard, Elsa Ferriere, Valérie Garrigue, Aurélie Hummel, Laurent Juilliard, Alexandre Karras, David Larmet, Alice Le Clech, Christophe Legendre, Charlene Levi, Ingrid Masson, Claire Maynard, Olivier Moranne, Othmane Mohib, Karine Moreau, Christiane Mousson, Mathilde Nizon, Laure Patrier, Marie-Noëlle Peraldi, Jean-Baptiste Philit, Romain Pszczolinski, Philippe Remy, Benjamin Savenkoff.
Author contributions
H.S. wrote the source code and developed the VNtyper pipeline. H.S., G.D., and C.A. designed the project and wrote the manuscript. L.H., C.A., G.D., and P.N. edited the manuscript. The experiments were planned and conducted by V.M., O.G., M.M., and S.L. P.N. did the VNtyper analysis and with F.T. provided the bioinformatics resources and supported the design of the VNtyper. B.K., S.B., P.V., L.H., and V.V. contributed to patient diagnosis and cohort description. G.D. and C.A. supervised the work.
Declaration of interests
The authors declare no competing interests.
Published: June 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107171.
Supplemental information
Data and code availability
-
•
The original code for VNtyper generated and used during this study are publicly available at GitHub (https://github.com/hassansaei/VNtyper). The docker image and configuration generated in this study are available in the docker hub (https://hub.docker.com/r/saei/VNtyper) and the GitHub page.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. We have included several read name-replaced bam files in the GitHub repository for the purpose of testing the setup.
-
•
This paper analyzes existing publicly available data, with information in the key resources table.
References
- 1.Eckardt K.-U., Alper S.L., Antignac C., Bleyer A.J., Chauveau D., Dahan K., Deltas C., Hosking A., Kmoch S., Rampoldi L., et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015;88:676–683. doi: 10.1038/ki.2015.28. [DOI] [PubMed] [Google Scholar]
- 2.Devuyst O., Olinger E., Weber S., Eckardt K.-U., Kmoch S., Rampoldi L., Bleyer A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Primers. 2019;5:60. doi: 10.1038/s41572-019-0109-9. [DOI] [PubMed] [Google Scholar]
- 3.Bolar N.A., Golzio C., Živná M., Hayot G., Van Hemelrijk C., Schepers D., Vandeweyer G., Hoischen A., Huyghe J.R., Raes A., et al. Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am. J. Hum. Genet. 2016;99:174–187. doi: 10.1016/j.ajhg.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayasreh N., Bullich G., Miquel R., Furlano M., Ruiz P., Lorente L., Valero O., García-González M.A., Arhda N., Garin I., et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1. Am. J. Kidney Dis. 2018;72:411–418. doi: 10.1053/j.ajkd.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Markello C., Huang C., Rodriguez A., Carroll A., Chang P.-C., Eizenga J., Markello T., Haussler D., Paten B. A complete pedigree-based graph workflow for rare candidate variant analysis. Genome Res. 2022;32:893–903. doi: 10.1101/gr.276387.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hickey G., Heller D., Monlong J., Sibbesen J.A., Sirén J., Eizenga J., Dawson E.T., Garrison E., Novak A.M., Paten B. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 2020;21:35. doi: 10.1186/s13059-020-1941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eggertsson H.P., Kristmundsdottir S., Beyter D., Jonsson H., Skuladottir A., Hardarson M.T., Gudbjartsson D.F., Stefansson K., Halldorsson B.V., Melsted P. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun. 2019;10:5402. doi: 10.1038/s41467-019-13341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patton S., Gendler S.J., Spicer A.P. The epithelial mucin, MUC1, of milk, mammary gland and other tissues. Biochim. Biophys. Acta. 1995;1241:407–423. doi: 10.1016/0304-4157(95)00014-3. [DOI] [PubMed] [Google Scholar]
- 9.Kirby A., Gnirke A., Jaffe D.B., Barešová V., Pochet N., Blumenstiel B., Ye C., Aird D., Stevens C., Robinson J.T., et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat. Genet. 2013;45:299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekici A.B., Hackenbeck T., Morinière V., Pannes A., Buettner M., Uebe S., Janka R., Wiesener A., Hermann I., Grupp S., et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int. 2014;86:589–599. doi: 10.1038/ki.2014.72. [DOI] [PubMed] [Google Scholar]
- 11.Olinger E., Hofmann P., Kidd K., Dufour I., Belge H., Schaeffer C., Kipp A., Bonny O., Deltas C., Demoulin N., et al. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int. 2020;98:717–731. doi: 10.1016/j.kint.2020.04.038. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto S., Kaimori J.-Y., Yoshimura T., Namba T., Imai A., Kobayashi K., Imamura R., Ichimaru N., Kato K., Nakaya A., et al. Analysis of an ADTKD family with a novel frameshift mutation in MUC1 reveals characteristic features of mutant MUC1 protein. Nephrol. Dial. Transplant. 2017;32:2010–2017. doi: 10.1093/ndt/gfx083. [DOI] [PubMed] [Google Scholar]
- 13.Okada E., Morisada N., Horinouchi T., Fujii H., Tsuji T., Miura M., Katori H., Kitagawa M., Morozumi K., Toriyama T., et al. Detecting MUC1 Variants in Patients Clinicopathologically Diagnosed With Having Autosomal Dominant Tubulointerstitial Kidney Disease. Kidney Int. Rep. 2022;7:857–866. doi: 10.1016/j.ekir.2021.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dvela-Levitt M., Kost-Alimova M., Emani M., Kohnert E., Thompson R., Sidhom E.-H., Rivadeneira A., Sahakian N., Roignot J., Papagregoriou G., et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell. 2019;178:521–535.e23. doi: 10.1016/j.cell.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Blumenstiel B., DeFelice M., Birsoy O., Bleyer A.J., Kmoch S., Carter T.A., Gnirke A., Kidd K., Rehm H.L., Ronco L., et al. Development and Validation of a Mass Spectrometry–Based Assay for the Molecular Diagnosis of Mucin-1 Kidney Disease. J. Mol. Diagn. 2016;18:566–571. doi: 10.1016/j.jmoldx.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Živná M., Kidd K., Přistoupilová A., Barešová V., DeFelice M., Blumenstiel B., Harden M., Conlon P., Lavin P., Connaughton D.M., et al. Noninvasive Immunohistochemical Diagnosis and Novel MUC1 Mutations Causing Autosomal Dominant Tubulointerstitial Kidney Disease. J. Am. Soc. Nephrol. 2018;29:2418–2431. doi: 10.1681/ASN.2018020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantere T., Kersten S., Hoischen A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019;10:426. doi: 10.3389/fgene.2019.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wenzel A., Altmueller J., Ekici A.B., Popp B., Stueber K., Thiele H., Pannes A., Staubach S., Salido E., Nuernberg P., et al. Single molecule real time sequencing in ADTKD-MUC1 allows complete assembly of the VNTR and exact positioning of causative mutations. Sci. Rep. 2018;8:4170. doi: 10.1038/s41598-018-22428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J., Bakhtiari M., Popp B., Wiesener M., Bafna V. Detecting tandem repeat variants in coding regions using code-adVNTR. iScience. 2022;25 doi: 10.1016/j.isci.2022.104785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popp B., Ekici A.B., Knaup K.X., Schneider K., Uebe S., Park J., Bafna V., Meiselbach H., Eckardt K.-U., Schiffer M., et al. Prevalence of hereditary tubulointerstitial kidney diseases in the German Chronic Kidney Disease study. Eur. J. Hum. Genet. 2022;30:1413–1422. doi: 10.1038/s41431-022-01177-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Audano P.A., Ravishankar S., Vannberg F.O. Mapping-free variant calling using haplotype reconstruction from k-mer frequencies. Bioinformatics. 2018;34:1659–1665. doi: 10.1093/bioinformatics/btx753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakhtiari M., Park J., Ding Y.-C., Shleizer-Burko S., Neuhausen S.L., Halldórsson B.V., Stefánsson K., Gymrek M., Bafna V. Variable number tandem repeats mediate the expression of proximal genes. Nat. Commun. 2021;12:2075. doi: 10.1038/s41467-021-22206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K., et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. 2022;185:3426–3440.e19. doi: 10.1016/j.cell.2022.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulovari A., Li R., Audano P.A., Porubsky D., Vollger M.R., Logsdon G.A., Chaisson M.J.P., Eichler E.E., Human Genome Structural Variation Consortium. Warren W.C. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. USA. 2019;116:23243–23253. doi: 10.1073/pnas.1912175116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukamel R.E., Handsaker R.E., Sherman M.A., Barton A.R., Zheng Y., McCarroll S.A., Loh P.-R. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. bioRxiv. 2021 doi: 10.1101/2021.01.19.427332. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stacey M.W., Neumann S.A., Dooley A., Segna K., Kelly R.E., Nuss D., Kuhn A.M., Goretsky M.J., Fecteau A.H., Pastor A., et al. Variable number of tandem repeat polymorphisms (VNTRs) in the ACAN gene associated with pectus excavatum. Clin. Genet. 2010;78:502–504. doi: 10.1111/j.1399-0004.2010.01492.x. [DOI] [PubMed] [Google Scholar]
- 28.Benedetti F., Dallaspezia S., Colombo C., Pirovano A., Marino E., Smeraldi E. A length polymorphism in the circadian clock gene Per3 influences age at onset of bipolar disorder. Neurosci. Lett. 2008;445:184–187. doi: 10.1016/j.neulet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Hijikata M., Matsushita I., Tanaka G., Tsuchiya T., Ito H., Tokunaga K., Ohashi J., Homma S., Kobashi Y., Taguchi Y., et al. Molecular cloning of two novel mucin-like genes in the disease-susceptibility locus for diffuse panbronchiolitis. Hum. Genet. 2011;129:117–128. doi: 10.1007/s00439-010-0906-4. [DOI] [PubMed] [Google Scholar]
- 30.Ræder H., Johansson S., Holm P.I., Haldorsen I.S., Mas E., Sbarra V., Nermoen I., Eide S.Å., Grevle L., Bjørkhaug L., et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat. Genet. 2006;38:54–62. doi: 10.1038/ng1708. [DOI] [PubMed] [Google Scholar]
- 31.LaHoste G.J., Swanson J.M., Wigal S.B., Glabe C., Wigal T., King N., Kennedy J.L. Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol. Psychiatry. 1996;1:121–124. [PubMed] [Google Scholar]
- 32.Bakhtiari M., Shleizer-Burko S., Gymrek M., Bansal V., Bafna V. Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res. 2018;28:1709–1719. doi: 10.1101/gr.235119.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cervera A., Tàssies D., Obach V., Amaro S., Reverter J.C., Chamorro A. The BC Genotype of the VNTR Polymorphism of Platelet Glycoprotein Ibα Is Overrepresented in Patients with Recurrent Stroke Regardless of Aspirin Therapy. Cerebrovasc. Dis. 2007;24:242–246. doi: 10.1159/000104485. [DOI] [PubMed] [Google Scholar]
- 34.Gremlich S., Nguyen D., Reymondin D., Hohlfeld P., Vial Y., Witkin S.S., Gerber S. Fetal MMP2/MMP9 polymorphisms and intrauterine growth restriction risk. J. Reprod. Immunol. 2007;74:143–151. doi: 10.1016/j.jri.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 35.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The original code for VNtyper generated and used during this study are publicly available at GitHub (https://github.com/hassansaei/VNtyper). The docker image and configuration generated in this study are available in the docker hub (https://hub.docker.com/r/saei/VNtyper) and the GitHub page.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. We have included several read name-replaced bam files in the GitHub repository for the purpose of testing the setup.
-
•
This paper analyzes existing publicly available data, with information in the key resources table.