

Abstract
Granzyme A from killer lymphocytes cleaves gasdermin B (GSDMB) and triggers pyroptosis in targeted human tumor cells, eliciting anti-tumor immunity. However, GSDMB has a controversial role in pyroptosis and has been linked to both anti- and pro-tumor functions. Here we found that GSDMB splicing variants are functionally distinct. Cleaved N-terminal (NT) fragments of GSDMB isoforms 3 and 4 caused pyroptosis, but isoforms 1, 2 and 5 did not. The nonfunctional isoforms have a deleted or modified exon 6 and therefore lack a stable belt motif. The belt likely contributes to the insertion of oligomeric GSDMB-NTs into the membrane. Consistently, noncytotoxic GSDMB-NTs blocked pyroptosis caused by cytotoxic GSDMB-NTs in a dominant negative manner. Upon natural killer (NK) cell attack, GSDMB3-expressing cells died by pyroptosis, whereas GSDMB4-expressing cells died by mixed pyroptosis and apoptosis, and GSDMB1/2-expressing cells died only by apoptosis. GSDMB4 partially resisted NK cell-triggered cleavage, suggesting that only GSDMB3 is fully functional. GSDMB1-3 were the most abundant isoforms in tested tumor cell lines and were similarly induced by IFNγ and the chemotherapy drug methotrexate. Expression of cytotoxic GSDMB3/4 isoforms, but not GSDMB1/2 isoforms that are frequently upregulated in tumors, was associated with better outcomes in bladder and cervical cancers, suggesting GSDMB3/4-mediated pyroptosis was protective in those tumors. Our study indicates that tumors may block and evade killer cell-triggered pyroptosis by generating noncytotoxic GSDMB isoforms. Therefore, therapeutics that favor the production of cytotoxic GSDMB isoforms by alternative splicing may improve anti-tumor immunity.
One-Sentence Summary:
GSDMB splicing isoforms play distinct roles in killer lymphocyte-mediated pyroptosis of human tumor cells and anti-tumor immunity.
Introduction
Pyroptosis, a lytic form of programmed necrotic cell death that releases inflammatory mediators to activate immune responses, is mediated by pore-forming gasdermins (GSDMs). The human GSDM family consists of five active members: GSDMA, GSDMB, GSDMC, GSDMD and GSDME (1). After the C-terminal (CT) auto-inhibitory domains are removed by proteolysis, the N-terminal (NT) domains of these GSDMs bind lipids, oligomerize, and form pores in the plasma membrane to induce pyroptosis (2–9). Granzyme B (GzmB) from killer lymphocytes causes cancer-associated pyroptosis (CAP) by cleaving GSDME in tumor cells (10). CAP increases immune cell infiltration into the tumor and elicits anti-tumor protection (11, 12). Similarly, killer cell granzyme A (GzmA) cleaves GSDMB to trigger CAP (13). GSDMB expression, which can be induced by TNF-α or IFN-γ, dramatically increases the efficacy of immune checkpoint blockade in mouse tumor models (13).
Nevertheless, the role of GSDMB in CAP is controversial. The pore-forming activity of GSDMB-NT was verified in some studies (13, 14) but disputed in others (15–17). The concept that GSDMB triggers CAP and anti-tumor immunity has also been challenged in studies showing that GSDMB expression in gastric and breast cancers correlates with poor clinical outcomes (18–20) and that GSDMB promotes cell proliferation and migration during epithelial repair in a pyroptosis-independent manner (15). Five GSDMB isoforms are generated by alternative splicing of exons 6-7, leading to different GSDMB-NTs upon cleavage by GzmA. Most of the previous studies focused on one isoform without considering that distinct isoforms might have different functions. Here we investigate the functions of GSDMB splicing variants and find that only isoforms 3 and 4, which contain an intact exon 6, trigger killer cell-mediated pyroptosis. Our study systematically defines the functional landscape of GSDMB isoforms and reveals a role of alternative splicing in modulating cancer-associated pyroptosis.
Results
GSDMB isoforms differ in pore-forming activity
The conflicting GSDMB literature prompted us to investigate whether GSDMB isoforms exhibit different pore-forming activities. GSDMB isoforms are generated by alternative splicing of exons 6-7. According to previous studies (13, 21), we designated the five previously reported isoforms as gasdermin B1 (exon 6 skipping, Δ6; 403 amino acids (aa)), B2 (Δ6, Δ7; 394 aa), B3 (416 aa, no exon skipping), B4 (Δ7; 407 aa) and B5 (Δ7 and a 12-nucleotide insertion in exon 6 due to an alternative splicing acceptor; 411 aa), respectively (Fig. 1A). Additionally, in this study, we identified a new isoform that we named GSDMB6.
Fig. 1. N-terminal fragments of GSDMB splicing isoforms exhibit different pore-forming activity.
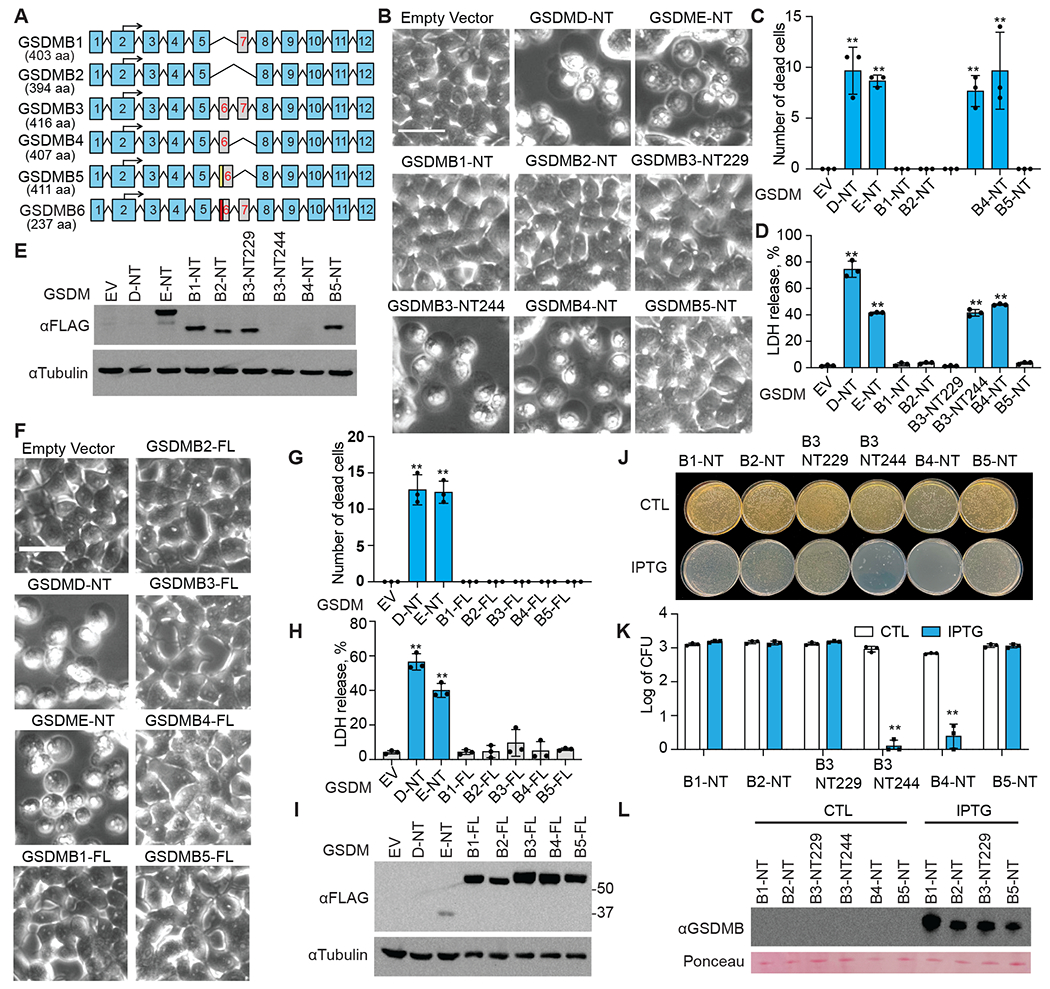
A, Schematic of six alternative splicing variants of GSDMB. Numbered blue boxes represent exons. An alternative splicing acceptor results in an insertion (yellow box) in exon 6 of GSDMB5. The red box in GSDMB6 indicates a 13-nucleotide deletion in exon 6. B-E, The effect of overexpressing GSDMB-NTs on HEK293T cell death, assessed by morphology using microscopy (B and C) and by LDH release (D). Dead cells were counted and quantified using three images (C). Expression of indicated FLAG-tagged GSDMB-NTs was determined by anti-FLAG immunoblot (E). F-I, The effects of overexpressing full-length GSDMBs on HEK293T cell death, assessed by morphology using microscopy (F and G) and by LDH release (H). Dead cells were counted and quantified using three images (C). Expression of indicated FLAG-tagged full-length GSDMBs was assessed by anti-FLAG immunoblots (I). J-L, The effect of overexpressing N-terminal GSDMBs on E. coli cell death, accessed by colony formation on LB plates without (CTL, non-treatment) or with IPTG (J and K). CFU, colony-forming units. Expression of indicated N-terminal GSDMBs was assessed by anti-GSDMB immunoblots (L). Ponceau S stained bands were used as loading controls. Data are mean ± s.d. of biological triplicates and are representative of three independent experiments. Statistical analysis was performed using the two-tailed Student’s t-test. **P < 0.01. Scale bar, 20 μm
The original study identifying the GzmA-GSDMB pathway examined GSDMB3 (13), while two recent studies, which found no pore-forming activity, tested GSDMB5 (15, 17). To determine if GSDMB-NTs, generated by GzmA cleavage of different GSDMB isoforms (Fig. S1), exhibit pore-forming activity, we ectopically expressed the NTs of GSDMB1-5 in HEK293T cells. As previously reported, GSDMD-NT and GSDME-NT induced pyroptosis (2, 3, 22, 23), while only the GSDMB3- and B4-NTs caused pyroptosis, indicated by cell membrane ballooning (Fig. 1B, C) and LDH release (Fig. 1D). The other GSDMB isoforms were noncytotoxic even though the NT fragments were well expressed, indicated by immunoblotting (Fig. 1E). Cytotoxic NT fragments of GSDMB3 and GSDMB4 were barely detected due to cell death. Full-length GSDMB has been shown to bind membrane lipids (24). To test if full-length (FL) GSDMBs also induce pyroptosis, we expressed full-length GSDMB1-5 isoforms in HEK293T cells (Fig. 1I). None of them were cytotoxic, as indicated by normal cell morphology (Fig. 1F and G) and background LDH release (Fig. 1H). GzmA was shown to cleave GSDMB3 at two sites, a major site K244 and a minor site K229 (Fig. S1), generating a long (GSDMB3-NT244) and a short NT (GSDMB3-NT229), respectively (13). Only the long GSDMB3-NT244, but not the short cleavage fragment GSDMB3-NT229, induced pyroptosis when ectopically expressed in HEK293T cells (Fig. 1B–E).
GSDMB-NT has also been shown to bind to bacterial membranes and kill bacteria (17). To test if GSDMB-NTs kill bacteria, NTs of GSDMB1-5 were ectopically expressed in BL21 E. coli through IPTG induction. Only GSDMB3-NT244 and GSDMB4-NT killed bacteria (Fig. 1J, K), while the other isoforms were noncytotoxic even though the NT fragments were well expressed, indicated by immunoblotting (Fig.1L). Thus, GzmA only generates GSDMB pores when it cuts GSDMB3 at the major cleavage site and GSDMB4, but not the other isoforms, reconciling the seemingly conflicting observations in the literature.
A belt motif in GSDM-NT promotes pore formation
To investigate why GSDMB isoforms have contrasting pore-forming activities, we compared the predicted structural models of GSDMB-NT isoforms generated by AlphaFold (25) to structures of human GSDMD-NT and mouse GSDMA3-NT obtained by cryo-electron microscopy and X-ray crystallography (5, 9, 26–28). Previous truncation experiments identified residues 1-243 as the minimal GSDMD-NT fragment capable of inducing pyroptosis (2, 29). Further scanning mutagenesis demonstrated that Thr239 and Phe240 are necessary for GSDMD-NT activity (29). Thr239 and Phe240 in GSDMD, and the corresponding residues in GSDMA3 (Thr231 and Phe232), are visible in both membrane-inserted and auto-inhibited conformations (5, 9, 26, 27) (Fig. 2A–C). In these structures, the β9 and β11 strands form a hairpin that is stabilized by a belt motif that wraps around the hairpin and ends with the critical Thr and Phe residues just near the GSDM-NT C-terminus. The ThrPhe residues nest inside a cavity formed by the α3-β6-β9 region by forming hydrogen bonding and hydrophobic interactions (Fig. 2C) (9, 29). Therefore, the belt is not part of the disordered linker between the NT and CT domains of GSDMs, but rather an ordered, integral structure within the active NT domain. Cryo-EM structures of membrane-inserted GSDMB (30, 31) published during the revision of this manuscript confirmed the orderedness of the belt.
Fig. 2, Structural and functional analysis of N-terminal GSDMB isoforms.
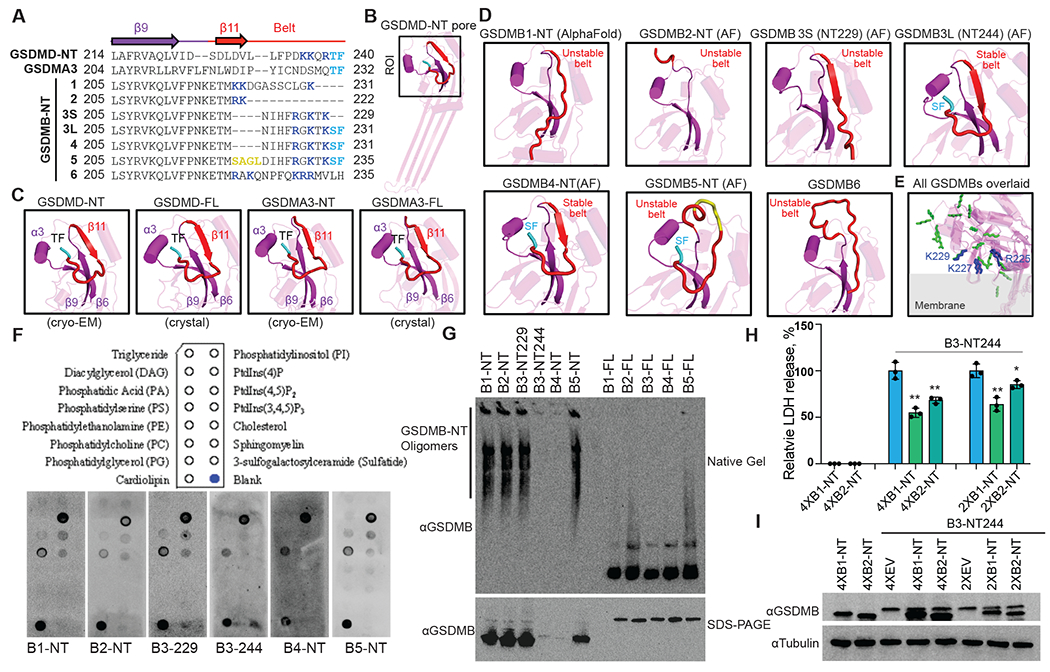
A, Sequence alignment of the N-terminal domains (NTs) of human GSDMD, mouse GSDMA3, and human GSDMB variants near the belt motif (red). Arrows: β strands. Yellow: Extra residues in the belt in GSDMB5. Blue: Positively charged residues in the belt. Cyan: ThePhe or SerPhe residues necessary for GSDM-NT activity. Dashes: Missing residues. B, Cryo-EM structure of the human GSDMD-NT pore (PDB: 6VFE) showing a single subunit. The region of interest (ROI) – near the belt – is boxed and enlarged in panel C. C, Zoomed-in views of the belt region of GSDMD and GSDMA3 in both membrane-inserted (cryo-EM structures, PDB: 6VFE, 6CB8) and auto-inhibited (crystal structures, PDB: 6N9O, 5B5R) conformations. For clarity, residues after the belt, including the linker and GSDM-CTs, are not shown. 3S, GSDMB3-NT229; 3L, GSDMB3-NT244. D, AlphaFold structural models of the GSDMB-NT variants, zoomed-in at the belt region (red). For clarity, residues after the belt are hidden. E, Positions and orientations of positively charged residues in the belt based on AlphaFold models of GSDMB-NTs. A hypothetical membrane is shown in gray. The positively charged residues are shown as blue sticks in GSDMB3-NT244 and GSDMB4-NT, and as green sticks in other isoforms. F, Membrane lipid strips (upper panel) were incubated with indicated proteins, and binding was assessed by blotting for GSDMB (lower panel). G, HEK293T cells, transfected with indicated plasmids, were lysed and resolved on a native gel (upper panel) and an SDS-PAGE (lower panel), immunoblotted for GSDMB. H and I, HEK293T cells, transiently transfected with the indicated amount of plasmids (2X, 2 times of plasmid compared to GSDMB3-NT; 4X, four times; EV, empty vector), were assessed 24 h after transfection for pyroptosis by LDH release (H) and GSDMB expression by immunoblots (I). Data are mean ± s.d. of biological triplicates and are representative of at least two independent experiments. Comparisons were calculated by two-tailed Student’s t-test. *P < 0.05, **P < 0.01.
The GSDMB-NT splicing variants only differ in their sequences in the belt motif (Fig. 2A). In GSDMB-NT, the critical Thr in the ThrPhe residues is replaced by a conservative Ser→Thr substitution. The noncytotoxic NTs of GSDMB isoforms 1, 2 and the short form of 3 (3S, NT229) all lack the critical SerPhe residues (Fig. 2A). The sequences of the pore-forming GSDMB4-NT and long form of GSDMB3-NT (3L, NT244) are identical in this region, while GSDMB5 has a 4-residue insertion, potentially disrupting the belt and shifting residues within the belt (Fig. 2A), in line with a recent cryo-EM analysis of GSDMB5 (30). Structural modeling of GSDMB-NT isoforms indicated that the belt is unstable in isoforms 1, 2, 3S (NT229), and 5, which either lack the β6-β11 hairpin, or have the C-terminus hanging far from the α3-β6-β9 cavity due to the lack of SerPhe, or both (Fig. 2D). Consistently, crystal structures of GSDMB5 showed that the belt is disordered in this isoform (30–32). Therefore, the belt motif in isoforms 3L and 4, which resembles the stabilizing structures formed in GSDMD and GSDMA3, is necessary for pore formation and unstable in the noncytotoxic isoforms.
Noncytotoxic GSDMB-NTs are incapable of membrane insertion
To examine the mechanism by which the belt motif promotes the pore-forming activity of GSDMB-NTs, we first investigated whether GSDMB-NTs differ in lipid binding. There are positively charged residues within the variable belt region (Fig. 2A). According to predicted structures, the positively charged residues (R225, K227, and K229) in GSDMB3-NT244 and GSDMB4-NT are clustered and point towards the membrane. The unstable belts in the NTs of GSDMB1, 2, 3S and 5 alter the positions and orientations of the positively charged residues, potentially hindering lipid binding by these isoforms (Fig. 2E). However, when we incubated purified GSDMB-NTs with strips dotted with different lipids, all GSDMB-NTs similarly bound to cardiolipin, PtdIns(4)P, and phosphatidylserine (PS), suggesting that the belt motif does not markedly influence lipid binding of GSDMB-NTs (Fig. 2F). It is possible that there are subtle lipid binding differences under the detection limit of our assay, as triple charge-reversal mutations of R225-K227-K229 modestly lowered the activity of GSDMB3 in liposome-based experiments (30). Nonetheless, less aggressive double mutations of R225-K227 to alanines did not affect GSDMB3 activity (31), supporting our postulation that the belt promotes pore formation mainly by an alternative mechanism. Besides the lipid strip results, all GSDMB-NTs contain the hydrophobic anchor and three positively charged patches previously identified as crucial for membrane binding by GSDM-NTs (9, 26, 32). Additionally, the positively charged residues within the GSDMB belt are not conserved among GSDMs (Fig. 2A).
Next, we tested whether GSDMB-NTs differ in oligomerization, as the belt is located near an inter-subunit contact site (9, 26). We expressed NT fragments or FL GSDMB splicing isoforms in HEK293T cells and analyzed the lysates by immunoblotting following denaturing or native gels (Fig. 2G). Under denaturing conditions, all proteins migrated mostly as monomers. Under native conditions, high molecular weight oligomers were visible in cells transfected with GSDMB-NTs but not GSDMB-FLs. Oligomers were barely detectable for cytotoxic GSDMB3-NT244 and GSDMB4-NT due to cell death. Surprisingly, oligomers were detected in all noncytotoxic GSDMB-NTs including B1, B2, B3S and B5, suggesting that the belt does not markedly contribute to GSDMB-NT oligomerization. Our results contrast with mutagenesis experiments and molecular dynamics simulations pointing towards the involvement of the belt in GSDMB-NT oligomerization (30, 31, 33).
Because noncytotoxic GSDMB-NTs still bind lipids and oligomerize (Fig. 2F, G), we reasoned that the pore formation process is arrested at the insertion step. Prior to membrane insertion, GSDM-NTs oligomerize into prepores, arcs, and slits (9, 26, 34, 35). The belt likely contributes to the insertion of the oligomeric GSDMB-NTs. If so, one would expect that noncytotoxic GSDMB-NTs impair the pore-forming abilities of cytotoxic GSDMB-NTs. Indeed, when we co-expressed cytotoxic GSDMB3-NT244 with either GSDMB1-NT or GSDMB2-NT in HEK293 cells, pyroptosis was dampened in a dose-dependent, dominant-negative manner, manifested by significantly reduced LDH release (P < 0.01) (Fig. 2H). The blockade effect was not due to decreased GSDMB3-NT244 expression, given consistent expression levels shown by immunoblotting (Fig. 2I). Likewise, flow cytometry experiments using mCherry as an indicator of GSDMB3-NT244 expression similarly showed dampened SYTOX uptake at the same levels of GSDMB3-NT244 in the presence of noncytotoxic isoforms (Fig. 2H, Fig. S2). Together, our data indicate that the belt facilitates the membrane insertion of membrane-bound, oligomeric GSDMB-NTs. Exact mechanisms of blockade by noncytotoxic GSDMBs await further investigation. Indirect mechanisms may exist such as competition among GSDMBs for activating proteases and membrane lipids.
GSDMB isoforms are heterogeneously expressed
To better understand the functional importance of GSDMB splicing, we profiled the expression of endogenous GSDMB isoforms in a panel of cell lines including cancer lines. One pair of primers (F1/R1) was designed to detect total GSDMB expression and another pair (F2/R2) flanking exons 6-7 was used to detect GSDMB variants (Fig. 3A). Total GSDMB expression was analyzed by qRT-PCR using F1/R1 primers (Fig. 3B). All these cell lines expressed GSDMB to different degrees. Colorectal adenocarcinoma SW837, SW1116 and HT29 showed the highest endogenous GSDMB expression. Hepatocellular carcinoma HepG2, neuroblastoma SH-SY5Y and lung carcinoma A549 cells showed moderate expression, while the other cells had minimal expression. HeLa had the lowest expression, about 3-fold lower than the low-expressing cell lines. Using flanking primers F2/R2 to probe cDNAs from these cells, we detected five bands at different molecular weights corresponding to GSDMB isoforms 1-5 by size (Fig. 3C). Sanger sequencing of these bands in SW1116 and HepG2 cells confirmed that GSDMB isoforms 1-4 were expressed in these cells (Fig. S3). However, instead of GSDMB5, an out-of-frame variant (named GSDMB6 hereafter) containing a 13-nucleotide deletion in exon 6 was identified. GSDMB6 is a 237-amino acid truncated fragment, which lacks a stable belt and should therefore be noncytotoxic like GSDMB5 (Fig. 2A, D). Among the six variants, GSDMB1/2/3 were abundant isoforms, and GSDMB4/6 were much less abundant, while GSDMB5 was not detected in our experiments. We therefore focused on the two abundant noncytotoxic isoforms GSDMB1/2 and two cytotoxic isoforms GSDMB3/4 hereafter.
Fig. 3. Expression of GSDMB isoforms.
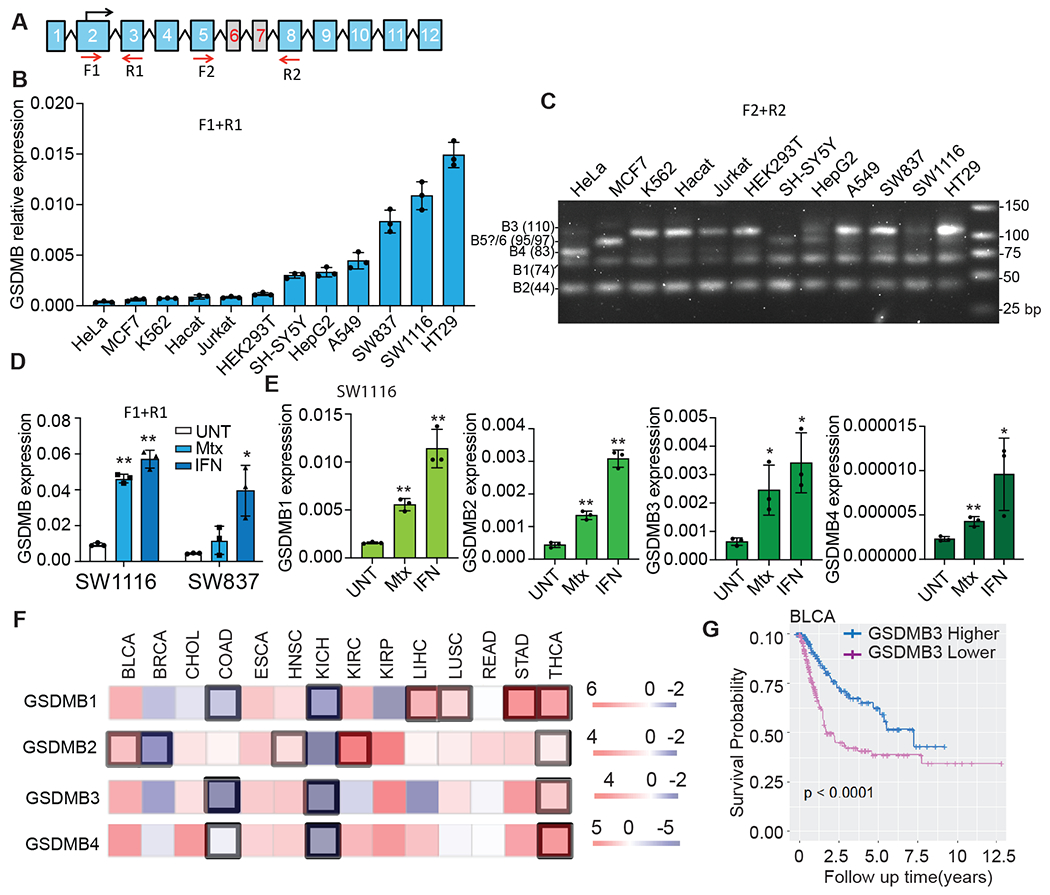
A, Schematic of the PCR primers used to detect total GSDMB (F1/R1) or specific isoforms (F2/R2). B, Expression of GSDMB in indicated cell lines, assessed by qRT-PCR using F1/R1 primers, relative to GAPDH. C, PCR products of GSDMB isoforms, resolved on 3% agarose gels, were amplified using F2/R2 primers. D, Effect of IFNγ and methotrexate (Mtx) on total GSDMB expression in SW837 and SW1116 cells, assessed by qRT-PCR with F1/R1 primers. E, Effect of IFNγ and Mtx on the expression of GSDMB isoforms in SW1116 cells, assessed by qRT-PCR with isoform-specific primers. F, Expression of GSDMB isoforms in primary tumors compared to adjacent normal tissue in the Cancer DEIso and TCGA database. Comparisons were calculated by paired Wilcoxon test and P<0.01 was considered significant (highlighted in black boxes). G, Correlation of GSDMB3 expression and survival in BLCA patients. Data in B and D are mean ± s.d. of biological triplicates and are representative of three independent experiments. Statistical analysis was performed using the two-tailed Student’s t-test. **P < 0.01. *P < 0.05.
Because HeLa cells barely express endogenous GSDMB (Fig. 3B), they are an ideal model for studying GSDMB isoforms in an overexpression setting. We stably overexpressed GSDMB isoforms 1-4 in HeLa cells individually, using an empty vector (EV) construct as a control. Anti-GSDMB immunoblotting showed that those isoforms were comparably expressed (Fig. S4A). Isoform-specific primers detected the expected GSDMB isoforms in HeLa GSDMB1-4 cells, but not in the EV control (Fig. S4B).
GSDMBs are differentially regulated in cancers and linked to survival outcomes
GSDMB is highly expressed in SW837 and SW1116 colorectal carcinoma cells. Its expression can be transcriptionally upregulated by IFNγ in those cells (13). Methotrexate (Mtx) treatment also induces GSDMB expression (15). GSDMB proteins induced by IFNγ and Mtx have been reported to concentrate in different cellular compartments and regulate cell proliferation and migration (15). We confirmed that GSDMB could be significantly (P < 0.01) induced by both IFNγ and Mtx in SW1116 cells but only by IFNγ in SW837 cells (Fig. 3D). To test if these two stimuli upregulate different GSDMB isoforms in SW1116 cells, we performed qRT-PCR using isoform-specific primers on treated SW1116 cells. All four isoforms were induced by both IFNγ and Mtx in SW1116 cells (Fig. 3E), although less by Mtx, suggesting their expression is similarly regulated.
Nonetheless, when comparing GSDMB isoform expression in tumors to adjacent normal tissues using the Cancer DEIso and TCGA databases (Fig. 3F), we found that the expression of GSDMB isoforms is differentially regulated in tumors. Our bioinformatics analysis showed that noncytotoxic GSDMB2 expression is significantly upregulated in multiple tumor types (P < 0.01), including bladder urothelial carcinoma (BLCA), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC) and thyroid carcinoma (THCA), but considerably reduced in breast invasive carcinoma (BRCA) compared to adjacent normal tissues. Similarly, GSDMB1 is upregulated considerably in four tumor types but suppressed in two other tumor types (Fig. 3F). Cytotoxic GSDMB3/4 are simultaneously downregulated in colon adenocarcinoma (COAD) and kidney chromophobe tumors (KICH), but upregulated only in THCA (Fig. 3F). These data indicate that noncytotoxic GSDMB1/2 may be more likely to be overexpressed while cytotoxic GSDMB3/4 is suppressed in primary tumors, suggesting opposite roles in tumor development. We also verified the expression of GSDMB isoforms in some TCGA primary tumors compared to the GTEX normal tissue in the UCSC Xena database (Fig. S5). We found that most of our expression data can be reproduced, except that GSDMB1 and GSDMB4 are not differentially expressed in KICH and THCA, respectively, and that GSDMB2 is reduced in THCA compared to normal tissues. However, these results do not contradict the observed expression trends of GSDMB isoforms in primary tumors. When analyzing the effect of tumor expression of GSDMB on patient survival, significant differences in survival (P < 0.01) were observed only in BLCA and KIRC patients but not in other patients. In BLCA, increased expression of cytotoxic GSDMB3 was associated with better survival (Fig. 3G) while expression of the other isoforms has little effect on patient survival (Fig. S6A). In KIRC patients, higher expression of any of the four GSDMB isoforms, among which only GSDMB2 was significantly upregulated, was associated with worse survival (P < 0.01) (Fig. S6B).
Some genomic single nucleotide polymorphism (SNP) variants also differentially regulate GSDMB isoform expression. Homozygotes for the minor allele of GSDMB rs11078928 showed almost no expression of exon 6-containing cytotoxic GSDMB3 and 4 isoforms but expressed the noncytotoxic GSDMB1/2 isoforms (36). Another minor variant, GSDMB rs8067378 has been associated with increased cervical cancer risk in East Asians (37–40). We analyzed those two minor alleles and found they are closely linked (R2=0.9951, P < 0.0001 in East Asians), meaning most people carry both or neither of these two minor alleles. This observation suggests that loss of cytotoxic GSDMB isoforms in rs11078928 is associated with higher cervical cancer risk. These data together suggest that expression of cytotoxic GSDMB3/4 may correlate to better clinical outcomes and reduced tumor risk, while noncytotoxic GSDMB1/2 is frequently overexpressed in tumors and may exert functions that promote tumor development.
Pyroptosis-independent functions of GSDMB may be context-specific
We next investigated previously described pyroptosis-independent functions of GSMDB. GSDMB1 has been reported to be nuclear localized and function as a transcription activator (21), although other studies reported cytosolic localization of GSDMB (15). To assess cellular localization, C-terminal GFP-tagged GSDMB isoforms were expressed in HeLa cells, and their localization was determined by confocal microscopy (Fig. S7A). All the ectopically expressed GSDMB isoforms were distributed to both the cytoplasm and the nucleus of HeLa cells and none were exclusively in the nucleus. We further tested whether GSDMBs function as transcription activators in GSDMB-expressing HeLa cells by examining the expression of 5-lipoxygenase (5-LO) and TGF-β1, which have been shown to be upregulated by GSDMB in human bronchial epithelial cells (21). Compared to HeLa cells expressing empty vector, HeLa cells overexpressing GSDMB isoforms did not show different TGF-β1 expression but showed markedly reduced 5-LO expression in GSDMB-expressing cells (Fig. S7B). We next tested whether GSDMBs promote tumor cell proliferation and migration as reported (15, 19, 20). In HeLa cells, proliferation rates, colony formation and cell migration were unchanged by ectopic GSDMB1-4 expression compared to cells transfected with an empty vector control (Fig. S7C–F). These data suggest that the reported non-pyroptotic roles of GSDMB in transcriptional activation, cell proliferation and migration may be limited to certain cells and contexts.
GSDMB isoforms 3 and 4 induce pyroptosis during killer lymphocyte attacks
Killer lymphocytes activate pyroptosis in GSDMB-expressing target cells by GzmA release, but activate caspase-independent noninflammatory cell death, apoptosis, in targets that do not express GSDMB (13). To test whether GSDMB isoforms are differentially activated by killer cells, the human NK cell line NK-92 MI was incubated with target HeLa cells stably expressing GSDMB1-4 or EV (Fig. S4) and loaded with calcein AM (acetoxymethyl), a cell membrane-permeable dye released only when the cell membrane is disrupted (41) (Fig. 4A–E). Pyroptosis causes rapid membrane permeabilization with characteristic cell membrane ballooning, while apoptosis features cell shrinking, DNA condensation and cell membrane blebbing, with delayed cell membrane disruption. After 2.5 hours, NK-92 MI cocultures with HeLa cells expressing EV, GSDMB1 and GSDMB2 contained dying HeLa cells with apoptotic features and bright calcein staining, indicating that isoforms 1 and 2 did not trigger pyroptosis (Fig. 4A, B). In contrast, there were many pyroptotic HeLa cells expressing GSDMB3, which had cell membrane balloons and released calcein. Both apoptotic and pyroptotic cells were observed in GSDMB4-expressing HeLa cells. Quantification confirmed that only GSDMB3- and GSDMB4-expressing HeLa cells released calcein into the supernatant above background (Fig. 4C). To confirm these results, Vybrant DiD, a cell membrane dye, was used to label NK-92 MI-targeted GSDMB-expressing HeLa cells. Giant membrane balloons were again observed in dying GSDMB3-expressing HeLa cells and to a less extent in GSDMB4-expressing HeLa cells, but not in cells expressing the other isoforms, which showed shrunken cells dying an apoptotic cell death (Fig. S8).
Fig. 4. GSDMB3 and 4 mediate GzmA-triggered pyroptosis.
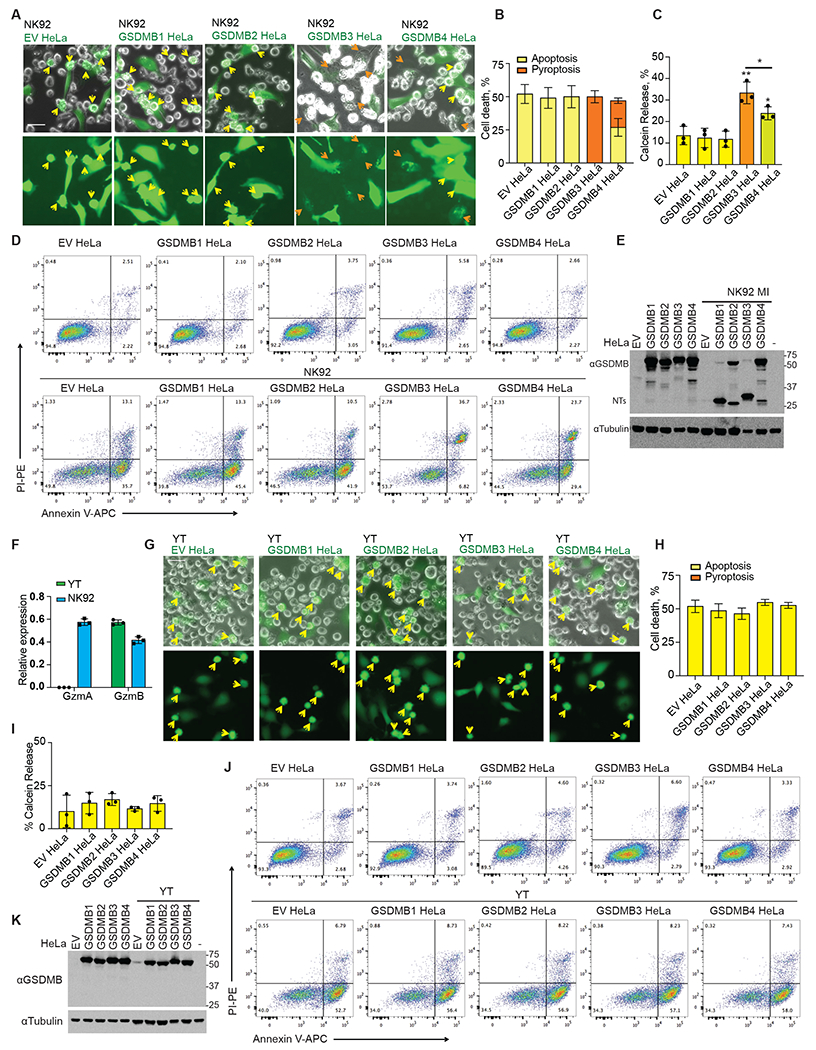
A and B, Representative fluorescent microscopy images of cocultures of NK-92 MI cells with calcein-labeled (green) empty vector and GSDMB-expressing HeLa cells (A). Yellow arrows indicate apoptotic cells and orange arrows indicate pyroptotic cells. The percentage of dead cells was counted and quantified using three images (B). C, Calcein release of empty vector and GSDMB-expressing HeLa cells induced by NK-92 MI cells (E/T ratio=3/1; 2h). D, GSDMB cleavage in GSDMB-expressing HeLa cells incubated with NK-92 MI cells at the E/T ratio of 3/1 for 2.5 h, assessed by immunoblotting. E, Indicated HeLa cells were co-incubated with NK-92 MI cells at the E/T ratio of 3/1 for 2.5 h. Cell death was assessed by Annexin V/ PI staining and flow cytometry. F, Expression of GzmA and GzmB in YT-INDY and NK-92 MI cells, assessed by qRT-PCR. G and H, Representative fluorescent microscopy images of cocultures of YT NK cells with calcein-labelled (green) empty vector and GSDMB-expressing HeLa cells (G). Yellow arrows indicate apoptotic cells. The percentage of dead cells was counted and quantified from three images (H). I, Calcein release of empty vector and GSDMB-expressing HeLa cells induced by YT cells (E/T ratio=3/1; 2h). J, GSDMB cleavage in GSDMB HeLa cells incubated with YT cells at the E/T ratios=3/1 for 2.5 h, assessed by immunoblotting. K, Indicated HeLa cells were co-incubated with YT cells at the E/T ratio of 3/1 for 2.5 h. Cell death was assessed by Annexin V/ PI staining and flow cytometry. Data in bar graphs are mean ± s.d. of biological triplicates and are representative of three independent experiments. Comparisons were calculated by two-tailed Student’s t-test. **P < 0.01. *P < 0.05 Scale bar, 20 μm.
Annexin V/ propidium iodide (PI) staining has been widely used to characterize apoptotic cell death. Early apoptotic cells can be stained by Annexin V conjugates but not by PI (Annexin V+/PI−), and will eventually become Annexin V+/PI+ and Annexin V−/PI+ when the cell membrane is disrupted by secondary necrosis. By contrast, pyroptotic cells are Annexin V+/PI+ from the beginning of cell death (13). We characterized NK-92 MI-targeted GSDMB-expressing HeLa cells by Annexin V/ PI staining and flow cytometry analysis by 2.5 hours after coculture, when secondary necrosis rarely occurs (Fig. 4D). Our data indicate that in empty vector control HeLa or HeLa cells expressing noncytotoxic GSDMB1 and GSDMB2, the majority of the dead cells stained Annexin V+/PI−, an indicator of early apoptosis. While in GSDMB3-expressing HeLa cells, most of the dead cells were Annexin V+/PI+, supporting that these cells mainly underwent pyroptosis. GSDMB4-expressing cells contained both Annexin V+/PI− and Annexin V+/PI+ populations, agreeing with our observation that GSDMB4 cells underwent both apoptosis and pyroptosis. A small Annexin V+/PI+ cell population was observed in control or GSDMB1/2 HeLa cells, presumably because of the secondary necrosis or membrane damage of cells during sample handling.
Next, we assessed GSDMB isoform cleavage in NK-92 MI-treated HeLa cells by immunoblotting (Fig. 4E). A GSDMB-NT band of the expected size was generated after NK attack of the HeLa cells expressing each of the GSDMB isoforms, but uncleaved, full-length GSDMB2 and GSDMB4 were prominent in cells expressing those isoforms, suggesting that isoforms 2 and 4 were partially resistant to GzmA cleavage. Therefore, although both GSDMB3-NT244 and GSDMB4-NT are cytotoxic, GSDMB4 is less likely to trigger pyroptosis due to resistance to cleavage by GzmA. To further confirm the NK cell killing results, we treated GSDMB-expressing HeLa cells with primary human NK (hNK) cells isolated from peripheral blood mononuclear cells (Fig. S9A) and assessed cell death by Annexin V/ PI staining and flow cytometry (Fig. S9B). hNK cells rapidly killed ~85% of the target cells in 2.5 hours. Like NK-92 MI cells, hNK cells triggered similar types of cell death in HeLa cells expressing four GSDMBs, respectively. Thus, GSDMB3 and, to a lesser extent, GSDMB4, but not GSDMB1 or GSDM2, trigger killer lymphocyte-mediated pyroptosis.
Killer cell-triggered GSDMB-induced pyroptosis depends on GzmA
Previous work suggested that GzmA is responsible for GSDMB cleavage and target cell pyroptosis after killer cell attacks (13). To confirm this finding, GSDMB1-4-expressing HeLa cells were challenged with YT-INDY cells, a human NK cell line expressing GzmB but not GzmA (42) (Fig. 4F). 2.5 hours after YT challenge, apoptosis was induced in all GSDMB1-4-expressing and EV control HeLa cells, indicated by cell shrinkage, membrane blebbing, bright calcein staining and the absence of membrane balloons (Fig. 4G, H). Moreover, GSDMB expression did not increase calcein in culture supernatants above background (Fig. 4I) and did not increase Annexin V+/PI+ dead cells in flow cytometry analyses (Fig. 4J). GZMA−/− YT attack did not lead to GSDMB cleavage, as assessed by immunoblotting (Fig. 4K). Thus, GzmA is responsible for cleaving GSDMB and triggering pyroptosis in target cells subjected to killer lymphocyte attack.
Discussion
Here we showed that GSDMB splicing variants differ in pore-forming activity and susceptibility to GzmA cleavage. Only GzmA-activated GSDMB3 and GSDMB4 isoforms form cell membrane pores. Therefore, splicing isoforms of GSDMB play distinct roles in killer cell-triggered pyroptosis. These data explain the conflicting reports about GSDMB activity. In our study, GSDMB2 and GSDMB4, both of which lack the exon 7-encoded region, were partially resistant to killer cell GzmA-mediated cleavage, in contrast with uniform cleavage of purified recombinant GSDMBs in vitro (13). GzmA primarily cleaves after a conserved lysine residue in all GSDMB isoforms (Fig S1), but the sequences preceding this residue vary among isoforms. The different results observed in vitro and in cells may reflect distinct protein conformations, post-translational modifications, and concentrations in cells. It is also possible that GSDMB isoforms complex differently with other cellular proteins, which alters their cleavage efficiency in cells.
While we studied six GSDMB isoforms, other isoforms have been reported, which encode short truncations or generate a circular RNA (43, 44). Out-of-frame isoforms are generally susceptible to nonsense-mediated RNA decay, a mechanism that prevents the production of truncated proteins in cells (44), and therefore their RNAs are unstable in cells. The out-of-frame variant GSDMB6 we identified encodes a 237-amino acid truncated protein and has very low expression in cells. GSDMB5 (411 aa) has been widely used in GSDMB studies. However, GSDMB5 is not a major GSDMB isoform in the cells tested in our study.
GSDMB is the only GSDM that lacks a mouse counterpart, which hampers the utilization of mouse tumor models to dissect GSDMB function in vivo. A recent study used a knock-in strategy to express GSDMB2 in mice ubiquitously (45). Although expression of GSDMB2 did not affect the overall frequency of spontaneous neoplasia, coexpression of GSDMB2 and HER2/NEU increased the incidence of breast cancer in mice, suggesting that pro-tumor functions of GSDMB2 could be context-dependent. When overexpressing GSDMB isoforms in HeLa cells, we did not observe any change in cell proliferation and migration, which could be attributed to a cell type-dependent effect.
Our study suggested that noncytotoxic GSDMB isoforms (like GSDMB1/2) function as negative regulators of GSDMB-mediated pyroptosis. The NT fragments of noncytotoxic and cytotoxic isoforms bound lipids and oligomerized similarly, but coexpression of noncytotoxic isoforms (GSDMB1/2) dampened GSDMB3-NT244-mediated pyroptosis. Noncytotoxic isoforms (1/2) might oligomerize with the cytotoxic isoforms (3/4), forming heterooligomers to block the concerted conformational transition from intermediate assemblies to membrane pores. Therefore, noncytotoxic isoforms could be employed by tumors to downregulate the pore-forming abilities of cytotoxic isoforms.
Aberrant alternative splicing has been implicated in many aspects of cancer, including angiogenesis, cell proliferation, invasion, metastasis and generation of neoantigens (46). In primary tumors, cytotoxic GSMDB3/4 are prone to be downregulated, but noncytotoxic GSDMB1/2 are frequently upregulated, suggesting tumors may employ alternative splicing to differentially regulate GSDMB isoforms to avoid GSDMB pyroptosis. The splicing alterations of GSDMB in tumors could be caused by the occurrence of cancer driver mutations in genes encoding components of the splicing machinery, or genetic mutations at the splicing sites. The minor allele of GSDMB rs11078928 alters a splicing site and leads to downregulation of cytotoxic GSDMB isoforms. Interestingly, the rs11078928 allele is associated with a higher risk of cervical cancer, suggesting that cervical cancer cells may employ the GSDMB splicing to promote tumorigenesis. Additionally, the minor allele of rs11078928 is associated with a lower risk of asthma (14), suggesting that GSDMB-mediated pyroptosis might also contribute to the pathogenesis of asthma.
Expression of the fully functional GSDMB3 is associated with better patient survival rates in BLCA cancer patients. However, the survival of KIRC patients negatively correlated with the expression of any GSDMB isoform. Our and other studies suggest that whether GSDMB3 expression correlates with better survival in patients is not only determined by GSDMB3 alone, but also by other GSDMB isoforms. Although GSDMB3 mediates killer lymphocyte-triggered pyroptosis that enhances anti-tumor immunity (13), other studies also suggested that noncytotoxic GSDMBs, particularly GSDMB2, promoted tumorigenesis by promoting tumor cell proliferation and migration (19, 20). Furthermore, our study revealed that noncytotoxic GSDMBs could block anti-tumor GSDMB pyroptosis by interfering with cytotoxic GSDMBs. Therefore, the clinical outcomes could be determined by the tug-of-war between the pro- and anti-tumor functions of different GSDMB isoforms. In this case, the fact that GSDMB3 expression correlates with better survival in BLCA but not KIRC could be due to the different expression levels of other GSDMB isoforms in these two tumors. Broadly speaking, because of the heterogeneity of cancers on many levels, including their diverse mechanisms of tumorigenesis and evasion of anti-tumor immunity, the expression of GSDMB3 may be anti-tumor in a cancer type-dependent manner.
From a therapeutic perspective, our data suggest that tumors may manipulate GSDMB splicing to avoid pyroptosis. Modulating GSDMB splicing to increase cytotoxic GSDMB isoforms and suppress noncytotoxic isoforms could improve anti-tumor immunity and enhance immunotherapy.
Materials and Methods
Study design.
The objective of this study was to determine if GSDMB splicing variants play distinct roles in killer lymphocyte-mediated pyroptosis and anti-tumor immunity. To achieve this goal, we have used molecular biological, biochemical and cellular experiments to show that GSDMB isoforms exhibit distinct pore-forming activity and differentially mediate NK cell-triggered pyroptosis. We dissected the underlying molecular mechanism by a combination of structural modeling and biochemical and cellular experiments. Bioinformatics analysis further showed that GSDMBs are differentially regulated in cancers and linked to survival outcomes. All experiments were independently repeated at least two or three times, as indicated in figure legends. This study was not blinded.
Cell lines.
SW837 (RRID: CVCL_1729) and SW1116 (RRID:CVCL_0544) are from Nidhi Sahni lab and all other cells used in this study are from Judy Lieberman lab. HeLa (RRID:CVCL_0030), HEK293T (RRID:CVCL_0063), SW837, HaCaT (RRID:CVCL_0038) and MCF7 (RRID:CVCL_0031) were cultured in complete DMEM medium as previously reported (10). SW1116, NK-92 MI (RRID:CVCL_3755), YT-Indy (RRID:CVCL_1797), K562 (RRID:CVCL_0004), Jurkat (RRID:CVCL_0065), SH-SY5Y (RRID:CVCL_0019), HepG2 (RRID:CVCL_0027), A549 (RRID:CVCL_0023) and HT29 (RRID:CVCL_0320) cells were cultured in complete RPMI medium as reported (10). Primary human NK cells were cultured in MACS NK medium with 1% MACS NK supplement (Miltenyi Biotec), 5% serum AB (Sigma) and 500 IU/ml IL-2 (Miltenyi Biotec) for 1-2 weeks before being used for NK killing assay. All cell lines were verified to be free of mycoplasma by PCR.
Plasmids.
pLVX-puro GSDMB 1-5 plasmids were synthesized by GenScript. GSDMB 1-5 NTs were amplified by PCR from mentioned plasmids and cloned into c-FLAG pcDNA3 (Addgene) using XhoI and BamHI. A GFP fragment was amplified from pWPI vector (Addgene) and inserted into pLVX-puro GSDMB 1-4 plasmids using XhoI and NEBuilder HiFi DNA Assembly Master Mix. To express GSDMB-NT proteins in E. Coli, GSDMB1-5 NTs were cloned into the pET28a vector using XhoI and BamHI. A mutation was introduced into those pET28a-GSDMB-NT plasmids to inactivate the N-terminal His tags by quick change PCR using primers 5’-actggtggacagcaataaggtcgcggatccatgttc-3’ and 5’-gaacatggatccgcgaccttattgctgtccaccagt-3’. All plasmids were verified by Sanger sequencing.
Generation of stable cell lines.
For lentivirus generation, pLVX-Puro GSDMB plasmids were transfected into HEK293T cells with pSPAX2 and pCMV-VSV-G at a 2:2:1 ratio. Supernatants collected 2 days later were used to transduce HeLa for 48 hours. Puromycin (Sigma, 2 μg/ml) was then added to select GSDMB-positive cells. pLVX-Puro empty vector was used to generate the control cells.
Semi-quantitative and quantitative RT-PCR.
For GSDMB induction, cells were treated with 10 ng/ml IFNγ or 10 μM Mtx for 48 hr. RNA was extracted using TRIzol reagent (Life Technologies) according to the manufacturer’s instructions and was subject to reverse transcription using M-MLV reverse transcriptase (Invitrogen). GSDMB, TGF-β1 and 5-LO expression were determined by qRT-PCT using iTaq Universal SYBER Green Supermix (Bio-Rad).
Sanger sequencing for GSDMB isoforms.
F2/R2 primer pair was used to amplify the different GSDMB isoforms from SW1116 and HepG2 cDNA. Then gel-purification was performed using QIAquick Gel Purification Kit (Qiagen 28706) for the PCR products (40-120 bp mixture) on agarose gel. Purified PCR products were cloned into pCR2.1-TOPO vector (Invitrogen 45-0245) and plasmid DNA were extracted for Sanger sequencing.
Cytotoxicity assay in HEK293T cells.
The transfection and LDH release in HEK293T was performed as reported (10). Pyroptotic cells were imaged using EVOS FL cell imaging system. Alternatively, cells were stained with SYTOX Green (Invitrogen, R37168) and analyzed on a CytoFLEX flow cytometer (Beckman Coulter).
SDS-PAGE and native gel Immunoblot.
SDS-PAGE and native gel Immunoblot were performed as previously reported (4). Antibodies used were α-GSDMB (ab215729, Abcam, RRID:AB_2909483), α-FLAG (F1804, Sigma, RRID:AB_262044), and α-tubulin (T5168, Sigma, RRID:AB_477579).
Immunoprecipitation and lipid binding assay.
For the purification of C-terminal FLAG-tagged GSDMB-NT proteins by immunoprecipitation, HEK293T cells transiently expressing GSDMB-NT were lysed in lysis buffer containing complete protease inhibitor cocktail (Roche). Lysates were incubated with anti-FLAG M2 beads (Sigma) for 3 h at 4 °C. Beads were washed three times with lysis buffer and proteins were eluted with lysis buffer containing 100 μg/ml 3xFLAG peptides (Sigma). For lipid binding assay, precipitates were spotted on Membrane Lipid Strips (Echelon Biosciences) according to the manufacturer’s instructions.
Isolation of primary NK cells.
Human NK cells were isolated from human peripheral blood mononuclear cells (PBMC) using the human NK Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Isolated NK cells were accessed using anti-CD56 (BioLegend, cat# 362507) and anti-CD3 (BioLegend, cat# 300306) staining and flow cytometry.
NK cell killing assay.
For NK cell-induced cell death, HeLa cells were seeded in 24-well plates overnight. Cells were loaded with 5 μM calcein AM (eBioscience) before co-incubating with NK-92 MI, YT or primary NK cells at E: T ratios = 3:1. 2 or 3 hours after incubation, calcein release was determined by recording fluorescence at 528 nM after excitation at 485 nM using a BioTek Synergy plate reader. For flow cytometry analysis, cells were collected by trypsinization and stained with annexin V conjugates (ThermoFisher A23204) and PI (Abcam). Samples were immediately analyzed by flow cytometry. For Vybrant DiD staining, HeLa cells were seeded overnight and stained with Vybrant DiD dye (ThermoFisher) according to the manufacturer’s instructions, and then NK-92 MI cells were added at an E: T ratio of 3:1. Pyroptotic cells were imaged using EVOS FL cell imaging system.
Bacterial killing assay.
For the cytotoxicity of N termini of GSDMB isoforms in E. Coli, 50 μl BL21 cells (DE3, Thermo Scientific, EC0114) were transformed with a pET28a-GSDMB-NT plasmid (100 ng) and plated equally on Kanamycin-containing LB plates with or without 0.1 M IPTG. The plates were incubated overnight at 37°C before the colony-forming units were determined.
GSDMB localization.
HeLa cells stably expressing GSDMB-GFP were seeded in 12-well plates. The next day cells were washed with PBS twice and fixed with cold 100% methanol for 10 minutes at −20°C. The cells were washed twice with PBS and stained with 1 μg/ml DAPI (Abcam) for 10 minutes at room temperature, then washed with PBS, and mounted with Vectashield antifade mounting medium (Vector Laboratories, cat# H-1200-10). Z-stack images (63×) were acquired with Zeiss LSM 880 confocal microscope and processed with Fiji software.
Proliferation, colony formation and wound healing assays.
Proliferation was measured using CellTiter-Glo ATP viability kit (Promega) according to the manufacturer’s instructions. Colony formation assay was performed using Crystal Violet staining. Wound healing assay (Abcam) was performed according to the manufacturer’s instructions. Images were taken by EVOS FL cell imaging system and analyzed by Image J.
Bioinformatics analysis.
Isoform-level expression information based on FPKM regarding four GSDMB isoforms (NM_001042471, NM_001165958, NM_001165959, and NM_018530) were obtained from Cancer DEIso (47). We got 19 tumor samples and 19 matched normal samples in TCGA-BLCA (T19 N19), 111 paired samples in TCGA-BRCA, 9 paired samples in TCGA-CHOL, 40 paired samples in TCGA-COAD, 6 paired samples in TCGA-ESCA, 42 paired samples in TCGA-HNSC, 23 paired samples in TCGA-KICH, 72 paired samples in TCGA-KIRC, 31 paired samples in TCGA-KIRP, 42 paired samples in TCGA-LIHC, 49 paired samples in TCGA-LUSC, 9 paired samples in TCGA-READ, 26 paired samples in TCGA-STAD, 58 paired samples in TCGA-THCA. To identify differentially expressed isoforms between tumors and adjacent normal tissues in these cancers, we performed paired Wilcoxon test for each isoform of GSDMB. P values<0.01 was considered significant.
To verify the expression of GSDMB isoforms in BLCA, KICH and THCA, we used TCGA and GTEX samples in the UCSC Xena. Besides samples in TCGA, normal tissue samples whose sources are “Bladder”, “Kidney” and “Thyroid” in GTEX were used as additional TCGA-BLCA, TCGA-KICH and TCGA-THCA normal samples, respectively. We got 406 tumor and 27 normal samples in TCGA-BLCA, 65 tumor and 24 normal samples in TCGA-KICH, and 510 tumor and 337 normal samples in TVGA-THCA. Processed RSEM expected count of transcript expression across samples was recomputed using UCSC TOIL RNA-seq pipeline and extracted from the Xena browser. Unpaired Wilcoxon test was performed on TCGA tumors (TCGA sample size is more), TCGA normal and GTX normal tissues.
For survival analysis, we collected tumor samples from 406 BLCA patients and tumor samples from 527 KIRC patients and defined those samples in which isoform expression is greater than median expression across samples as a higher group and otherwise lower group. We did the survival analysis between the higher and lower group across cancers based on log-rank test. P values <0.01 was considered significant.
Statistics.
Student’s t-test (two-tailed), paired samples Wilcoxon signed-rank test, unpaired Wilcoxon rank sum test or log-rank test were used to determine differences between 2 groups as indicated in the figure captions. P values <0.05 were considered significant (unless stated otherwise) and shown in figures.
Supplementary Material
Fig. S1. Cleavage sites of GzmA in GSDMB isoforms.
Fig. S2. Blockade of cytotoxic GSDMB-NT by noncytotoxic isoforms shown by flow cytometry.
Fig. S3. Sequencing validation of GSDMB isoforms.
Fig. S4. Expression of GSDMB isoforms in stably transfected HeLa cells.
Fig. S5. Expression of GSDMB isoforms in TCGA primary tumors compared to GTEX normal tissue in the UCSC Xena database.
Fig. S6. Effects of GSDMB isoforms’ expression on patient survival.
Fig. S7. The role of GSDMB variants in tumor cell growth and migration.
Fig. S8. Visualization of pyroptosis in membrane-stained HeLa cells.
Fig. S9. Cell death induced by human primary NK cells in GSDMB-expressing HeLa cells.
Table S1. Primers used for semi-qPCR or qRT-PCR.
Reproducibility Checklist
Data file S1. Raw data file.
Acknowledgments:
We thank MD Anderson Sequencing and Microarray Facility (SMF) for Sanger sequencing. We thank Dr. Xiyu Ma at Boston Children’s Hospital, Dr. Shengqing Gu at MD Anderson Cancer Center and Dr. Jianbin Ruan at University of Connecticut Health Center for their helpful discussion.
Funding:
This study was supported by the University of Texas Rising STARs Award and Elsa U. Pardee Research Grant to ZZ; National Institutes of Health grant R01CA240955 to JL; Howard Hughes Medical Institute funding and National Institutes of Health grant R01EB030015 to MBE; National Institutes of Health grant R01AI139914 to HW; Ovarian Cancer Research Alliance Early Career Investigator Grant 649968 and Department of Defense grant W81XWH-18-PRCRP-CDA CA181455 to NS; National Institutes of Health grant R35GM133658, Komen Foundation grant CCR19609287, and the 2022-2023 program in Oncological Data and Computational Sciences sponsored by The Joint Center for Computational Oncology between the Oden Institute, MD Anderson and TACC to SSY; Postdoctoral fellowship from Jane Coffin Childs Memorial Fund for Medical Research to SX; and National Institutes of Health grant P30CA016672 to MD Anderson SMF.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are in the main text or the Supplementary Materials.
References and Notes:
- 1.Liu X, Xia S, Zhang Z, Wu H, Lieberman J, Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov 20, 384–405 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Sborgi L et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO journal 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell research 26, 1007–1020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aglietti RA et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–7863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia S et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Zhang Y, Lieberman J, Lighting a Fire: Can We Harness Pyroptosis to Ignite Antitumor Immunity? Cancer Immunol Res 9, 2–7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong Q, Zhang Z, Cancer-associated pyroptosis: A new license to kill tumor. Frontiers in immunology 14, 1082165 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Z et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science, (2020). [DOI] [PubMed] [Google Scholar]
- 14.Panganiban RA et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. The Journal of allergy and clinical immunology 142, 1469–1478.e1462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rana N et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell 185, 283–298 e217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. Journal of molecular cell biology 11, 496–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen JM et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 184, 3178–3191 e3118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komiyama H et al. Alu-derived cis-element regulates tumorigenesis-dependent gastric expression of GASDERMIN B (GSDMB). Genes & genetic systems 85, 75–83 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Hergueta-Redondo M et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 7, 56295–56308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hergueta-Redondo M et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PloS one 9, e90099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das S et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci U S A 113, 13132–13137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Rogers C et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nature communications 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao KL, Kulakova L, Herzberg O, Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci U S A 114, E1128–e1137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirdita M et al. ColabFold: making protein folding accessible to all. Nature methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruan J, Xia S, Liu X, Lieberman J, Wu H, Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z et al. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 51, 43–49 e44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z et al. Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei X et al. Enterovirus 71 Inhibits Pyroptosis through Cleavage of Gasdermin D. J Virol 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C et al. Structural basis for GSDMB pore formation and its targeting by IpaH7.8. Nature, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong X et al. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature, (2023). [DOI] [PubMed] [Google Scholar]
- 32.Yin H et al. Insights into the GSDMB-mediated cellular lysis and its targeting by IpaH7.8. Nature communications 14, 61 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oltra SS et al. Distinct GSDMB protein isoforms and protease cleavage processes differentially control pyroptotic cell death and mitochondrial damage in cancer cells. Cell Death Differ, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulvihill E et al. Mechanism of membrane pore formation by human gasdermin-D. The EMBO journal 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mari SA et al. Gasdermin-A3 pore formation propagates along variable pathways. Nature communications 13, 2609 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison FS et al. The splice site variant rs11078928 may be associated with a genotype-dependent alteration in expression of GSDMB transcripts. BMC genomics 14, 627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miura K et al. A significant association between rs8067378 at 17q12 and invasive cervical cancer originally identified by a genome-wide association study in Han Chinese is replicated in a Japanese population. Journal of human genetics 61, 793–796 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Shi Y et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nature genetics 45, 918–922 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Yang YC et al. Replication of results from a cervical cancer genome-wide association study in Taiwanese women. Scientific reports 8, 15319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S et al. Association Between GSDMB Gene Polymorphism and Cervical Cancer in the Northeast Chinese Han Population. Front Genet 13, 860727 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Somanchi SS, McCulley KJ, Somanchi A, Chan LL, Lee DA, A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PloS one 10, e0141074 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suck G et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol 33, 1160–1171 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Sun Q et al. Expression of GSDML Associates with Tumor Progression in Uterine Cervix Cancer. Translational oncology 1, 73–83 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardamone G et al. The Characterization of GSDMB Splicing and Backsplicing Profiles Identifies Novel Isoforms and a Circular RNA That Are Dysregulated in Multiple Sclerosis. Int J Mol Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarrio D et al. Gasdermin-B Pro-Tumor Function in Novel Knock-in Mouse Models Depends on the in vivo Biological Context. Front Cell Dev Biol 10, 813929 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Qian J, Gu C, Yang Y, Alternative splicing and cancer: a systematic review. Signal Transduct Target Ther 6, 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang TH et al. Cancer DEIso: An integrative analysis platform for investigating differentially expressed gene-level and isoform-level human cancer markers. Comput Struct Biotechnol J 19, 5149–5159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cleavage sites of GzmA in GSDMB isoforms.
Fig. S2. Blockade of cytotoxic GSDMB-NT by noncytotoxic isoforms shown by flow cytometry.
Fig. S3. Sequencing validation of GSDMB isoforms.
Fig. S4. Expression of GSDMB isoforms in stably transfected HeLa cells.
Fig. S5. Expression of GSDMB isoforms in TCGA primary tumors compared to GTEX normal tissue in the UCSC Xena database.
Fig. S6. Effects of GSDMB isoforms’ expression on patient survival.
Fig. S7. The role of GSDMB variants in tumor cell growth and migration.
Fig. S8. Visualization of pyroptosis in membrane-stained HeLa cells.
Fig. S9. Cell death induced by human primary NK cells in GSDMB-expressing HeLa cells.
Table S1. Primers used for semi-qPCR or qRT-PCR.
Reproducibility Checklist
Data file S1. Raw data file.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are in the main text or the Supplementary Materials.