

Abstract
Background
Large cell lung carcinoma (LCLC) is an exceptionally aggressive disease with a poor prognosis. At present, little is known about the molecular pathology of LCLC.
Methods
Ultra-deep sequencing of cancer-related genes and exome sequencing were used to detect the LCLC mutational in 118 tumor-normal pairs. The cell function test was employed to confirm the potential carcinogenic mutation of PI3K pathway.
Results
The mutation pattern is determined by the predominance of A > C mutations. Genes with a significant non-silent mutation frequency (FDR) < 0.05) include TP53 (47.5%), EGFR (13.6%) and PTEN (12.1%). Moreover, PI3K signaling (including EGFR, FGRG4, ITGA1, ITGA5, and ITGA2B) is the most mutated pathway, influencing 61.9% (73/118) of the LCLC samples. The cell function test confirmed that the potential carcinogenic mutation of PI3K pathway had a more malignant cell function phenotype. Multivariate analysis further revealed that patients with the PI3K signaling pathway mutations have a poor prognosis (P = 0.007).
Conclusions
These results initially identified frequent mutation of PI3K signaling pathways in LCLC and indicate potential targets for the treatment of this fatal type of LCLC.
Subject terms: Diagnostic markers, Prognostic markers
Introduction
Lung cancer remains the major cause of cancer-associated mortality worldwide, which is mainly categorized into small-cell lung cancer (SCLC, 10–15%) and non-small-cell lung cancer (NSCLC, 80–85%) [1]. NSCLC is composed of these main histological subtypes: adenocarcinoma, squamous cell carcinomas, large cell neuroendocrine carcinoma (LCNEC) and large cell lung carcinoma (LCLC) [2]. The World Health Organization (WHO) classify LCNEC as a neuroendocrine carcinoma, while its management and clinical phenotype have often been considered to be consistent with small cell lung carcinoma (SCLC) [3]. The incidence of LCLC, as a rare neoplasm, is 2.5 per 100,000 people. In 2021, the WHO new classification of lung tumors defined LCLC as an undifferentiated NSCLC, which lacks the characteristics of small cell carcinoma, adenocarcinoma, squamous cell carcinoma, and neuroendocrine carcinoma in terms of cytology, histological structure, and immunophenotype [4]. After auxiliary immunohistochemical (IHC) analysis, the resected, undifferentiated NSCLC cases without pneumocyte marker thyroid transcription factor-1 (TTF-1) or squamous marker p40 expression are classified as LCLC [5].
People with LCLC frequently don’t exhibit early symptoms, which might lead to a late diagnosis and subpar treatment. Although LCLC tends to spread widely, little is known about their pathogenesis. In addition, little is understood about the ideal treatment for LCLC and potential therapeutic molecular targets. Most research on potential genes for LCLC up to this point has concentrated on a small number of genes [6]. Exome sequences from nine LCLC tumors were recently investigated, and the gene TP53 was shown to be highly altered [7]. The range of somatic mutations in LCLC is still not fully understood. Therefore, additional research into the causes and progression of LCLC is crucial to raising the survival rate for lung cancer.
Methods
Study population
From 2009 to 2015, 118 Chinese participants with LCLC who had not received radiation or chemotherapy were included in the study. Of them, 22 were recruited for whole-exome sequencing, 90 for targeted deep sequencing, and 6 were recruited for both (Supplementary Figs. S1 and S2a, and Supplementary Table S1). Tissue was divided in two, with half being kept for histological confirmation and the other half being preserved in liquid nitrogen. Histopathological analysis was used to confirm all LCLC diagnoses and found no tumor cells in the nearby control samples. DNA samples from tissues with tumor content of more than 75% and their matching normal tissues were used for targeted sequencing and exome sequencing. The study was approved by the Ethics Committee of Tongji University School of Medicine and Shanghai Pulmonary Hospital (K15-199).
Whole-exome sequencing
In order to produce genomic DNA libraries, protocols recommended by Illumina were used. The TruSeq Exome Enrichment kit (Illumina) was used for whole-exome enrichment. The Illumina HiSeq 2500 Genome Analyzer was used to sequence the captured DNA libraries and 200 (2× 100) base pairs were generated from the library fragments.
Ultra-deep targeted gene sequencing
A total of 104 tumor-related genes were included in the target enrichment group of ultra-deep targeted sequencing. The selection criteria of these genes were as followed: (I) recurrently altered genes in the 28 LCLC exomes; (II) genes with high priority in the COSMIC database; and (III) drug sensitivity genes (Supplementary Fig. S1 and Supplementary Table S7).
Sequencing data processing and mutation calling
Trimmomatic was used to trim and filter the sequencing reads [8]. The results were matched with the hg19 reference genome using the Burrows–Wheeler Aligner (BWA) and the recalibration of the basic mass fraction, Indel realignment, and duplication removal was performed with the Genome Analysis Toolkit (GATK). Somatic SNV was found in whole-exome and targeted gene sequencing data using the MuTect method [9]. By Bayesian statistical analysis of bases and their properties in tumor and normal BAM files in certain genomic regions, MuTect found potential somatic SNVs. Exome data were processed using default settings. To adjust the extreme depth focus sequencing, the maximum alternative alleles in the normal number were set to 10 and the maximum alternative allele in the normal score was set to 0.05. We performed an additional filtering step requiring that the percentage of alternative alleles in normal samples be less than 0.30 in tumor samples in order to eliminate possible false positives. To find indels with default settings, the Pindel method was employed [10]. Inversions and large indels (>100 bp) were firstly omitted because they were difficult to confirm. Afterwards, we filtered indels using the following criteria: (I) tumor tissue depth ≥10; (II) normal tissue depth ≥8; (III) alternative allele in normal tissue ≥0; and (IV) alternative allele in tumor tissue ≥5 (only used in targeted sequencing). The findings of the somatic SNV and indel analysis were combined, which were then compared with the COSMIC database. SnpEff [11], PolyPhen [12], PROVEAN [13], and SIFT [14] were used to predict mutation functions.
Gene and pathway mutational significance analysis
To determine the relevance of a gene mutation, MutSigCV was used to assess all somatic mutations and save them in a MAF file [15]. By using mutational heterogeneity in the studies, MutSigCV, a tool for identifying cancer driver genes, can get rid of the majority of the genes that appear to be false positives. Significant gene mutations were defined as those with q (FDR) < 0.01. We used the path-scan analysis in the MuSiC suite to find known cellular pathways that had substantial accretions with somatic mutations in LCLC. The possibility of pathway mutations, which together promote tumor formation, was tested by path-scan. We made advantage of canonical pathways, which were defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [16]. We carried out the default settings for the study, but due to the excessive number of TP53 mutations, TP53 mutations were not considered.
Sanger sequencing validation
Sanger sequencing was used to confirm the SNV and indels of 46 single cells found in next-generation sequencing. Sanger sequencing was used to confirm a total of 46 somatic SNVs and indels discovered by next-generation sequencing. After excluding the loci where PCR failed, 38 of 42 SNVs (90.5%) and 4 of 4 indels (100%) were verified correctly.
Cell culture and reagents
The human LCLC cell line NCI-H460 was purchased from the Shanghai Institutes for Biological Sciences (Shanghai, China). The American Type Culture Collection (ATCC) provided the HEK293T cells. All cell lines were confirmed as mycoplasma free by mycoplasma PCR tests. The RPMI-1640 medium for cell growth contained 10% FBS, 100 mg/ml of streptomycin, and 100 units/ml of penicillin. RT-PCR testing revealed no evidence of mycoplasma infection when cells were cultured at 37 °C with 5% CO2. Sigma provided the thiazolyl blue tetrazolium bromide (M5655, MTT).
Influence of RNA interference mediated loss of function on cell viability
We employed the targeted siRNA sequences described in Supplementary Table 17 to silence target genes using siRNA. The negative control was a scrambled sequence. Shanghai Shenggong Company created these double-stranded siRNA duplexes. A 96-well plate with approximately 3000 cells per well was used to test the viability of the cells. Subsequently, the cells were transfected with targeted siRNA by the Lipofectamine RNAi MAX assay. Cell viability was assessed using the MTT test on days 0, 2, 4, and 6 following transfection. The absorbance of the resulting purple product obtained at 490 nm was used to measure cell viability. At least three times each trial was conducted.
Cloning and transduction of mutant or wild-type genes into cell lines
Somatic mutations of the EGFR, FGFR4, ITGA1, or ITG2B genes were added to the pCMV6 vector containing their ORF by the Fast Mutagenesis System Kit (Saiye Biotech). Sanger sequencing was used to verify the constructs, and FuGENE HD (Promega) was used to transfect them into NCI-H460 cells or HEK293T cells. The mRNA expressions of these genes were determined with real-time quantitative PCR. GAPDH was used as the internal control. The 2−ΔΔCT method was used to analyze the expression levels of these genes. As mentioned before, 96 h after transduction, cell viability was evaluated.
Statistical analysis
SPSS 18.0 (SPSS Inc., Armonk, NY, USA) was used for all statistical analysis. The Fisher’s exact test or chi-squared test was used to test the relationship between mutations and clinical features. The univariate survival analysis was conducted using the Kaplan–Meier test. To determine the hazard ratios (HR) and 95% confidence intervals (95% CI) of the relationships between risk factors and survival, multivariate Cox proportional hazards models were utilized. Cell viability was assessed using the Student’s t test. P < 0.05 was used to define the statistical significance of the results.
Results
We sequenced the entire exome of 28 pairs of LCLC tissues and case-matched normal tissue to analyze the somatic mutation spectrum in LCLC (Supplementary Fig. S1 and Supplementary Tables S1 and S2). We found 1397 somatic insertions or deletions (indels) and 11,065 somatic single-nucleotide variants (SNVs) that were expected to change the protein-coding sequence using strict criteria, which were defined by high or medium effects in SnpEff7 (Supplementary Table S3). Overall, 1.96 mutations per Mb were found (Supplementary Table S4).
The pattern of nucleotide mutations may reveal particular mutagenesis processes taking place in tumor cells. Here, we discovered that the most prevalent kind of substitution was a C > T/G > A alteration, which accounted for 30.6 percent (3382/11,065) of all somatic SNVs detected in LCLC (Fig. 1). Moreover, C > T mutations at the TCN sites, particularly at the TCA sites, predominated the trinucleotide signature (Fig. 1). The ideal motif for APOBEC3B is TCA. Additionally, malignant tumors of the bladder, breast, head & neck, and lungs also have the APOBEC mutation pattern. Its presence in LCLC suggests that APOBEC cytidine deaminase mutagenesis is important in the pathogenesis of LCLC.
Fig. 1. Somatic SNV signature in LCLC.
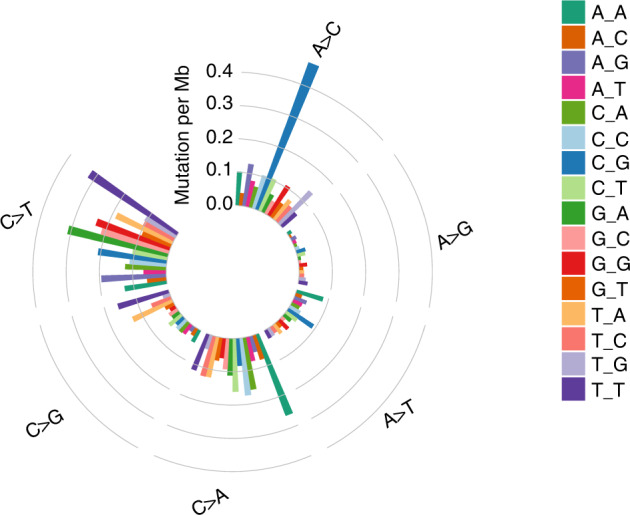
The somatic SNVs found in 28 LCLC exomes were divided into 96 subgroups defined by substitution class and adjacent bases. Each column around the circle represents a subgroup, and the height of each column shows mutation frequency per Mb.
MutSigCV10 determined that 36 genes, including TP53, AHNAK2, MUC5B, PDE4DIP, and MAP3K21, had frequent nonsynonymous somatic mutations that were statistically significant (Fig. 2 and Supplementary Table S5). By using the MuSiC path-scan (P < 0.05; Supplementary Table S6), we also discovered somatic mutations concentrated in PI3K-Akt pathway, ECM-receptor interaction, Circadian entrainment, and R retrograde endocannabinoid signaling linked with cancer cell signaling transduction.
Fig. 2. Significantly mutated genes in LCLC.

The top bar plot shows the somatic mutation frequency for 28 tumors from each case. The bottom middle plot shows the mutation status of the recurrently mutated genes for each tumor. Somatic mutations are colored according to functional class. The bottom right bar plot shows the mutation count for each individual gene. The bottom left plot shows the significance of each gene; The dotted line represents a P value of 0.05.
Genes that recurrently mutated are limited to a small number of cancer-related genes in almost all types of cancer [17]. We created a cancer gene panel that contains 104 candidate genes, which were frequently mutated in exome sequencing (including TP53, ITGA1, PTEN, ITGA2B, STK11, and EGFR) or are often related to a variety of cancer (COSMIC; Supplementary Table S7). Using an average depth of 1938.5× in the ultra-deep targeted sequencing (Supplementary Table S8), these genes were sequenced in 96 pairs of LCLC normal tissue, of which 6 were also examined by whole-exome sequencing (Supplementary Fig. S2 and Supplementary Table S1).
In total, 1232 non-silent SNVs were found (Supplementary Tables S9 and S10), yielding a total mutation rate of 3.29/Mb (Supplementary Tables S10). About 61.9% (73/118) of LCLC tumors were identified to have substantially altered PI3K signaling pathway genes (FDR < 0.05; Fig. 3 and Supplementary Tables S11 and S12).
Fig. 3. Somatic mutations of the PI3K signaling pathway in LCLC.

a Shown are the mutation status of genes of the PI3K pathway in the 73 LCLCs that carry at least one non-silent mutation. b The key genes of the PI3K signaling pathway with mutation frequencies in LCLC are shown. The frequencies for these key genes were estimated from 118 samples analyzed by whole exome and/or targeted sequencing. c The impact of mutations in the PI3K pathway on clinical outcome is shown. Cases with mutations in the PI3K pathway (n = 73) demonstrate worse overall survival than cases without mutations in the PI3K pathway (n = 45).
Mutation rates for the genes TP53, EGFR, PTEN, STK11, and FGFR4 were 47.5%, 13.6%, 12.1%, and 11.1%, respectively (Fig. 3 and Supplementary Fig. S3). Among them, TP53 mutations are a frequent genetic alteration that is thought to be present in more than 50% of human cancers, including LCLC (Fig. 3). PTEN and STK11 tumor suppressor gene inactivating mutations have been investigated in the etiology of lung cancer and have been found to favor the development of carcinomas. Similar to this, we discovered seven mutations of STK11 (encoding p.Lys41X, p.Gly56Arg, p.Glu130X, p.Met136Leu, p.Asp194Glu, and p.Phe354Leu) and six mutations of PTEN (encoding p.Gly4Arg, p.Cys65Ser, p.His122Asp, p.Arg130X) (Supplementary Fig. S3).
Notably, the PI3K signaling pathway has significant somatic mutations (Fig. 3). We discovered that 61.9% of LCLCs (73/118) had mutations that altered the protein sequence in the PI3K signaling pathway after combining the data from targeted sequencing and whole-exome sequencing generated by 118 samples. we found 47 non-silent somatic mutations in 25 PI3K pathway genes (TP53, EGFR, PTEN, STK11, FGFR4, COL4A1, ITGA5, TNN, HGF, SGK2, COL4A2, THBS4, IFNA10, IL3, ITGA2B, RPS6KB2, ITGA1, PHLPP2, TCL1A, GNG10, PIK3CA, GNG8, EIF4B, AKT2, and PDK1) in LCLC (Fig. 4a, b, Supplementary Fig. S3, and Supplementary Table S13). While certain alterations in the route were not mutually exclusive, the majority of them were, particularly those in STK11, FGFR4, and COL4A1.
Fig. 4. Somatic alterations of four genes and their oncogenic effects on normal and LCLC cells.

a Evaluation of cell viability in response to small interfering RNA (siRNA) for the four genes in three types of LCLC cells NCI-H460 at the indicated times after transfection. The data shown are representative of values from three independent experiments (mean ± s.e.m.; *P < 0.05, **P < 0.01 relative to the control siRNA group). b The four genes somatic non-silent alterations (inverted triangles) are depicted over the affected protein domains. RLD Receptor L domain, FLCRR Furin-like cysteine rich region, GF Growth factor receptor domain IV, Pkinase Protein tyrosine kinase, VWA von Willebrand factor type A domain, FG FG-GAP repeat. c Non-malignant HEK293T and LCLC NCI-H460 cells were transiently transfected with vector expressing wild-type (WT) or mutant EGFR, FGFR4, ITGA1 and ITG2B. The cell viability was detected by thiazolyl blue tetrazolium bromide (MTT) assay 96 h after transduction (bottom). Ctrl, pCMV6 control vector. Data represent mean ± s.e.m. (*P < 0.05, **P < 0.01 with respect to cells transfected with the control vector, as indicated). Throughout the figure, unpaired Student’s t tests were used to calculate all P values.
It was determined that there was no significant correlation between PI3K pathway mutations and any of the individual clinicopathological characteristics evaluated in patients with LCLC, including age, sex, histopathological subtypes, TNM stage, lymph node metastasis, vascular invasion, pulmonary membrane invasion, margin status, maximum diameter, and smoking (P > 0.05; Supplementary Table S13).
Univariate analysis with the Cox regression model was used to examine whether clinical factors such as age, sex, histopathological subtypes, TNM stage, lymph node metastasis, vascular invasion, pulmonary membrane invasion, margin status, maximum diameter, smoking, and PI3K pathway mutation status affect LCLC prognosis (Table 1 and Fig. 3c). In patients with LCLC, it was shown that TNM stage, tumor diameter, and PI3K pathway mutation status were significantly linked with shorter OS duration (P < 0.05; Table 1). In addition, we discovered that PI3K pathway mutation was significantly linked with the shorter OS duration in LCLC patients (P = 0.007; Fig. 3c) when Kaplan–Meier survival curves were compared by log-rank test. Additionally, multivariate analysis employing the Cox regression model (Table 2) demonstrated that PI3K pathway mutation was strongly linked with a poor prognosis [hazard ratio (HR) = 6.681; confidence interval (CI): 2.253–9.669; P = 0.001].
Table 1.
Univariate log-rank analysis of overall survival.
| Factor | Category | No. of cases | Median survival time 95% CI (month) | P value |
|---|---|---|---|---|
| Age | ≥60 | 76 | 28.63 (24.29–30.67) | 0.217 |
| <60 | 42 | 34.59 (28.76–41.59) | ||
| Sex | Male | 98 | 32.38 (19.19–43.68) | 0.565 |
| Female | 20 | 28.57 (20.19–45.34) | ||
| Histopathological subtypes | Middle-High | 71 | 29.66 (24.32–31.25) | 0.683 |
| Low | 47 | 25.47 (22.38–32.39) | ||
| TNM stage | I-II | 68 | 36.98 (28.84–44.58) | 0.046* |
| III-IV | 50 | 26.12 (18.59–29.65) | ||
| Lymph node metastasis | Positive | 36 | 29.65 (24.21–35.56) | 0.239 |
| Negative | 82 | 32.33 (28.49–36.73) | ||
| Vascular invasion | Positive | 31 | 27.46 (24.33–34.91) | 0.197 |
| Negative | 85 | 33.56 (29.51–38.83) | ||
| Pulmonary membrane invasion | Positive | 32 | 28.94 (25.45–32.28) | 0.068 |
| Negative | 86 | 36.98 (28.93–38.33) | ||
| Margin status | R0a | 102 | 29.46 (20.19–36.48) | 0.441 |
| R1b | 16 | 27.64 (24.73–32.98) | ||
| Maximum diameter | ≥5 cm | 96 | 27.16 (24.56–31.86) | 0.016* |
| <5 cm | 22 | 39.25 (26.14–43.68) | ||
| Smoking | Positive | 68 | 29.28 (26.43–33.68) | 0.588 |
| Negative | 50 | 32.17 (28.25–34.65) | ||
| PI3K pathway mutation status | Positive | 73 | 28.12 (25.43–30.28) | 0.002* |
| Negative | 45 | 42.68 (29.85–44.62) |
*Statistically significant.
aR0 Resected, negative margin.
bR1 Resected, positive margin.
Table 2.
Multivariate analysis of overall survival.
| Factor | Category | HRa | 95% CI | P value |
|---|---|---|---|---|
| Age | ≥60 | 1.212 | 0.968–1.635 | 0.336 |
| <60 | ||||
| Sex | Male | 0.825 | 0.634–0.963 | 0.628 |
| Female | ||||
| Histopathological subtypes | Middle-High | 0.742 | 0.458–1.263 | 0.558 |
| Low | ||||
| TNM stage | I-II | 0.471 | 0.337–0.625 | 0.024* |
| III-IV | ||||
| Lymph node metastasis | Positive | 1.524 | 0.885–1.899 | 0.186 |
| Negative | ||||
| Vascular invasion | Positive | 1.802 | 0.697–2.335 | 0.154 |
| Negative | ||||
| Pulmonary membrane invasion | Positive | 1.987 | 1.154–3.652 | 0.079 |
| Negative | ||||
| Margin status | R0b | 1.395 | 0.786–2.221 | 0.628 |
| R1c | ||||
| Maximum diameter | ≥5 cm | 3.568 | 2.263–5.415 | 0.011* |
| <5 cm | ||||
| Smoking | Positive | 1.684 | 1.254–3.337 | 0.365 |
| Negative | ||||
| PI3K pathway mutation status | Positive | 6.681 | 2.253–9.669 | 0.001* |
| Negative |
*Statistically significant.
aHR hazard ratio.
bR0 Resected, negative margin.
cR1 Resected, positive margin.
We initially employed RNA interference to cause functional loss to validate the oncogenic tendencies of PI3K family members in the NCI-H460 in order to ascertain the oncogenic impacts of the PI3K pathway mutations. The findings demonstrated that time-dependent silencing of TP53, PTEN, and STK11 hindered the development of NCI-H460 cells (Supplementary Fig. S4). Notably, time-dependent growth inhibition of NCI-H460 cells (Fig. 4a) was caused by the silencing of EGFR, FGFR4, ITGA1, and ITGA2B (Supplementary Fig. S5).
Next, we overexpressed the mutants of EGFR (p.Ser177Leu, p.Arg254Lys, p.Asn261Asp, p.Leu591Arg, p.Glu631Lys, and p.Ser660Thr), FGFR4 (p.Arg154His, p.Thr179Ala, p.Ile197Thr, p.Ala346Thr, and p.Ser384Phe), ITGA1 (p.Thr480Met) and ITGA2B (p.Gly462Arg) in LCLC cell line NCI-H460 and HEK293T cell line (Fig. 4b). Compared with the mock group, a significant increase in proliferation in NCI-H460 or HEK293T cell lines was found (P < 0.05; Fig. 4c) after overexpression of each EGFR, FGFR4, ITGA1 or ITGA2B mutant Moreover, mutants of EGFR (p.Arg254Lys, p.Asn261Asp, and p.Leu591Arg), FGFR4 (p.Arg154His, p.Ile197Thr, and p.Ala346Thr), ITGA1 (p.Thr480Met) or ITGA2B (p.Gly462Arg) induced an extremely significant increase of cell growth in HEK293T or NCI-H460 cell lines when compared to transfection of wild-type EGFR, FGFR4, ITGA1, or ITGA2B (P < 0.001; Fig. 4c). It is clear from these data that mutations in the PI3K genes show oncogenic potential, and the presence of these activating PI3K pathway gene alterations indicates that PI3K pathway is implicated in the initiation and progression of LCLC.
Discussion
Up to now, little is understood about somatic mutations, the ideal treatment for LCLC and potential therapeutic molecular targets. EGFR mutation is reported to be the most common driver mutation in NSCLC [18]. Most NSCLC patients with EGFR mutation will develop resistant mutations in EGFR-sensitive mutation is the EGFR tyrosine kinase inhibitor (EGFR-TKI) therapy [19].
In the publicly available validation NCSLC cohorts, FGFR4 alterations correlated with higher ORR, longer median overall survival and PFS. FGFR4 alterations were confirmed as an independent predictor of superior PFS and OS [20]. Moreover, FGFR4 alterations associated with a higher TMB levels, more CD8 T cells in the tumor stroma, and a higher M1/M2 ratio for tumor-associated macrophages in the tumor center and stroma, which suggesting that FGFR4 alterations may serve as a potential independent predictor of ICI efficacy in NSCLC [21].
Recent studies showed that the genes of the ITGA subfamily play a fundamental role in various cancers [22]. However, little is known about the expression and roles of distinct ITGA proteins and mutations in NSCLCs. A high mutation rate (44%) of the ITGA family was observed in the NSCLCs. Gene Ontology functional enrichment analyses results revealed that the differentially expressed ITGAs could be involved in roles related to extracellular matrix (ECM) organization, collagen-containing ECM cellular components, and ECM structural constituent molecular functions [23]. The expression of ITGAs was significantly correlated with the infiltration of diverse immune cells in NSCLCs. As potential prognostic biomarkers in NSCLCs, ITGAs may fulfill important roles in regulating tumor progression and immune cell infiltration [24].
Overall, the current study revealed a high incidence of A > C/T > G conversions in LCLC, identified LCLC driver genes, and clarified a central PI3K signaling network. It also established the somatic mutation framework of LCLC for the first time in a systematic manner. It is important to remember that patients with LCLC have a poor prognosis when PI3K pathway gene alterations are present. These findings imply that patients with PI3K pathway mutations may benefit from currently existing or future targeted medicines.
Supplementary information
Acknowledgements
We would like to thank Prof. Wen Li for data analysis and critical discussion of the manuscript.
Author contributions
LKH, JBL, CYW and DF conceived and directed the study. JHG, YSM, JWL, JH, GXJ, LKH, HML, CYW and DF contributed to the project design. JHG, YSM, LKH, JH, GXJ, HML, WW, XD, QYF, LKH and DF performed experiments. YSM, JWL, JH, HML, JBL, CYW and DF performed bioinformatics data analysis. LKH, YSM, GXJ, JBL, CYW, and DF contributed samples, data and comments on the manuscript. JHG, YSM, JWL, LKH, JH, HML, CYW, and DF analyzed and interpreted data. YSM, LKH, JH, JWL, GXJ, JBL, CYW, and DF contributed reagents, materials and/or analysis tools. LKH, YSM, and DF wrote the manuscript. JHG, YSM, JWL, GXJ, JH and HML contributed equally to this work. All authors contributed to the final version of the manuscript and approved the final manuscript.
Funding
his study was supported partly by the National Natural Science Foundation of China (82272766, 81702243 and 81472202), Construction of Clinical Medical Center for Tumor Biological Samples in Nantong (HS2016004), Natural Science Foundation of Shanghai (21140903500), Basic Medical Research Program of Navy Military Medical University Affiliated Changhai Hospital (2021JCMS11), Program of Navy Military Medical University (2022MS019), Program of Key Research and Development Program of Hunan Province (2021NK2026), and Key Program of Hunan Provincial Department of Science and Technology (2021JJ30060 and 2020WK2020).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Whole-exome sequencing data and target sequencing data from this study are available for download through the NCBI Sequence Read Archive under accession number PRJNA639383, PRJNA639657, and PRJNA643364. These submission will be released upon publication. Release of BioProject or BioSamples is also triggered by the release of submitted data.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Tongji University School of Medicine and Shanghai Pulmonary Hospital (K15-199). Each participant provided their written informed consent to participate in this study.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jun-Hong Guo, Yu-Shui Ma, Jie-Wei Lin, Geng-Xi Jiang, Juan He, Hai-Min Lu.
Contributor Information
Chun-Yan Wu, Email: wuchunyan581@163.com.
Ji-Bin Liu, Email: tians2008@ntu.edu.cn.
Da Fu, Email: fu800da900@126.com.
Li-Kun Hou, Email: hlk9575@163.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-023-02301-2.
References
- 1.Schwendenwein A, Megyesfalvi Z, Barany N, Valko Z, Bugyik E, Lang C, et al. Molecular profiles of small cell lung cancer subtypes: therapeutic implications. Mol Ther Oncolytics. 2021;20:470–83. doi: 10.1016/j.omto.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu SH, Hsu KW, Lai YL, Lin YF, Chen FH, Peng PH, et al. Systematic identification of clinically relevant miRNAs for potential miRNA-based therapy in lung adenocarcinoma. Mol Ther Nucleic Acids. 2021;25:1–10. doi: 10.1016/j.omtn.2021.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson AG, Tsao MS, Beasley MB, Borczuk AC, Brambilla E, Cooper WA, et al. The 2021 WHO classification of lung tumors: impact of advances since 2015. J Thorac Oncol. 2022;17:362–87. doi: 10.1016/j.jtho.2021.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Lin G, Qi K, Liu B, Liu H, Li J. A nomogram prognostic model for large cell lung cancer: analysis from the Surveillance, Epidemiology and End Results Database. Transl Lung Cancer Res. 2021;10:622–35. doi: 10.21037/tlcr-19-517b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sugár S, Bugyi F, Tóth G, Pápay J, Kovalszky I, Tornóczky T, et al. Proteomic analysis of lung cancer types-a pilot study. Cancers. 2022;14:2629. doi: 10.3390/cancers14112629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos-Paradas J, Gómez-Sánchez D, Rosado A, Ucero AC, Ferrer I, García-Luján R, et al. Comprehensive characterization of human lung large cell carcinoma identifies transcriptomic signatures with potential implications in response to immunotherapy. J Clin Med. 2022;11:1500. doi: 10.3390/jcm11061500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlsson A, Brunnström H, Micke P, Veerla S, Mattsson J, La Fleur L, et al. Gene expression profiling of large cell lung cancer links transcriptional phenotypes to the new histological WHO 2015 classification. J Thorac Oncol. 2017;12:1257–67. doi: 10.1016/j.jtho.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.do Valle ÍF, Giampieri E, Simonetti G, Padella A, Manfrini M, Ferrari A, et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinforma. 2016;17:341. doi: 10.1186/s12859-016-1190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cingolani P. Variant annotation and functional prediction: SnpEff. Methods Mol Biol. 2022;2493:289–314. doi: 10.1007/978-1-0716-2293-3_19. [DOI] [PubMed] [Google Scholar]
- 12.Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomark. 2010;14:533–7. doi: 10.1089/gtmb.2010.0036. [DOI] [PubMed] [Google Scholar]
- 13.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–85. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian X, Gu T, Lee MH, Dong Z. Challenge and countermeasures for EGFR targeted therapy in non-small cell lung cancer. Biochim Biophys Acta Rev Cancer. 2022;1877:188645. doi: 10.1016/j.bbcan.2021.188645. [DOI] [PubMed] [Google Scholar]
- 19.Yu N, Hwang M, Lee Y, Song BR, Kang EH, Sim H, et al. Patient-derived cell-based pharmacogenomic assessment to unveil underlying resistance mechanisms and novel therapeutics for advanced lung cancer. J Exp Clin Cancer Res. 2023;42:37. doi: 10.1186/s13046-023-02606-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Ren Z, Yu B, Tang J. Development of nomogram based on immune-related gene FGFR4 for advanced non-small cell lung cancer patients with sensitivity to immune checkpoint inhibitors. J Transl Med. 2021;19:22. doi: 10.1186/s12967-020-02679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao G, Cui L, Zhou F, Jiang T, Wang W, Mao S, et al. Special issue “The advance of solid tumor research in China”: FGFR4 alterations predict efficacy of immune checkpoint inhibitors in nonsmall cell lung cancer. Int J Cancer. 2023;152:79–89. doi: 10.1002/ijc.34239. [DOI] [PubMed] [Google Scholar]
- 22.Zhou C, Li S, Bin K, Qin G, Pan P, Ren D, et al. ITGA2 overexpression inhibits DNA repair and confers sensitivity to radiotherapies in pancreatic cancer. Cancer Lett. 2022;547:215855. doi: 10.1016/j.canlet.2022.215855. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Wren JD, Ding Y, Chen J, Mittal N, Xu C, et al. EWI2 promotes endolysosome-mediated turnover of growth factor receptors and integrins to suppress lung cancer. Cancer Lett. 2022;536:215641. doi: 10.1016/j.canlet.2022.215641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei S, Xu Q, Hu Y, Tang R, Feng J, Zhou Y, et al. Integrin β3-PKM2 pathway-mediated aerobic glycolysis contributes to mechanical ventilation-induced pulmonary fibrosis. Theranostics. 2022;12:6057–68. doi: 10.7150/thno.72328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Whole-exome sequencing data and target sequencing data from this study are available for download through the NCBI Sequence Read Archive under accession number PRJNA639383, PRJNA639657, and PRJNA643364. These submission will be released upon publication. Release of BioProject or BioSamples is also triggered by the release of submitted data.