

Summary
Autophagy is a dynamic process that degrades subcellular constituents, and its activity is measured by autophagic flux. The tandem proteins RFP-GFP-LC3 and GFP-LC3-RFP-LC3ΔG, which enable the visualization of autophagic vacuoles of different stages by differences in their fluorescent color, are useful tools to monitor autophagic flux, but they require plasmid transfection. In this study, we hence aimed to develop a new method to monitor autophagic flux using small cell-permeable fluorescent probes. We previously developed two green-fluorescent probes, DALGreen and DAPGreen, which detect autolysosomes and multistep autophagic vacuoles, respectively. We here developed a red-fluorescent autophagic probe, named DAPRed, which recognizes various autophagic vacuoles. By the combinatorial use of these green- and red-fluorescent probes, we were able to readily detect autophagic flux. Furthermore, these probes were useful not only for the visualization of canonical autophagy but also for alternative autophagy. DAPRed was also applicable for the detection of autophagy in living organisms.
Subject areas: Optical imaging, Biotechnology, Cell biology, Biological sciences tools
Graphical abstract
Highlights
-
•
Development of a red-fluorescent probe for monitoring autophagy
-
•
Combinational use of the probes allows visualization of autophagy kinetics
-
•
The probes also detect the alternative autophagic compartments
-
•
Zebrafish stained with DAPRed represents autophagic activity in vivo
Optical imaging; Biotechnology; Cell biology; Biological sciences tools
Introduction
Autophagy is a catabolic process in which cellular constituents are degraded by lysosomal lytic enzymes. Autophagy begins from the nucleation of a double-membrane structure (the so-called phagophore), which expands to enclose cytoplasmic cargos, and eventually matures into an autophagosome by the fusion of its leading edge. Then, autophagosome-lysosome fusion occurs to generate an autolysosome, in which cargos are degraded. Autophagy plays essential roles in a wide variety of biological events, including the maintenance of cellular homeostasis, cell differentiation, and stress response.1,2
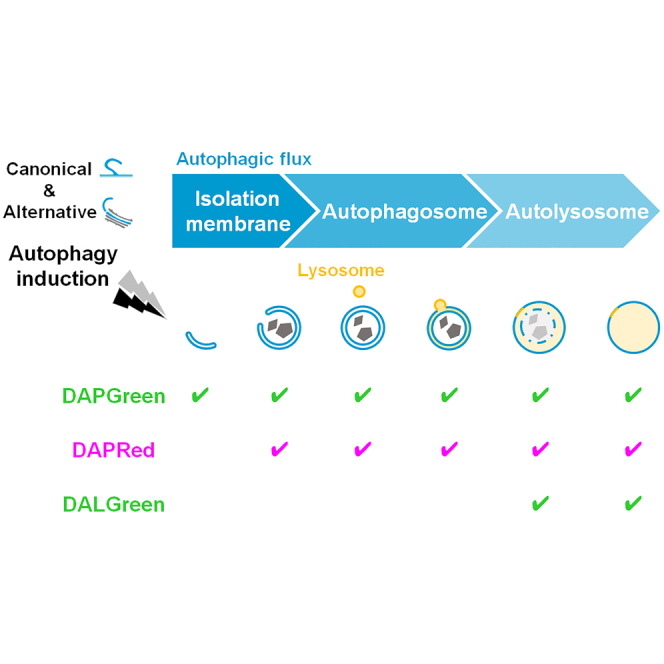
Various techniques have been reported for the detection of autophagy.2,3 The observation of double-membrane autophagosomes and single-membrane autolysosomes by electron microscopy (EM) is a classical but reliable method. Microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) is also used as a marker of autophagy because conjugation of phosphatidylethanolamine (PE) to LC3 occurs with the translocation of LC3 from the cytosol to the phagophore.4 Therefore, puncta formation of LC3 and the detection of LC3-PE are considered to be useful for quantifying autophagy. However, with advancements in autophagy research, these markers are presently considered to be insufficient for the accurate evaluation of autophagy.5 A simple increase in autophagosomes or LC3 modifications does not necessarily indicate increased autophagic activity because these findings may represent either the induction of autophagy or the inhibition of its downstream steps. Therefore, to accurately analyze autophagy activity, it is essential to evaluate the dynamics of autophagy, which is called “autophagic flux”. For the detection of autophagic flux, the tandem proteins red-fluorescent protein (RFP)-GFP-LC3 and GFP-LC3-RFP-LC3ΔG have been developed.6,7 The former protein detects nonacidified and acidified autophagic structures as yellow puncta and red puncta, respectively because GFP is more sensitive to acidic conditions. The latter protein can detect autophagic flux by the GFP/RFP ratio because GFP-LC3 is unstable in autolysosomes, whereas RFP-LC3ΔG remains in the cytosol. By the simultaneous observation of the two fluorescent colors, these two proteins enable the monitoring of autophagic flux. However, the use of this method is limited owing to its requirement for plasmid transfection. Therefore, we aimed to develop small, cell-permeable fluorescent probes that can measure autophagic flux easily.
We recently developed two fluorescent probes, namely, DAPGreen and DALGreen, both of which detect autophagic vacuoles.8 The former probe labels a broad spectrum of autophagic vacuoles, whereas the latter selectively labels autolysosomes. If the detection wavelength of these two probes were different, a combination of these probes would enable the detection of autophagic flux. Therefore, we aimed to develop a red-fluorescent autophagic probe that substitutes for DAPGreen or DALGreen.
Recently, accumulating lines of evidence have indicated the existence of an alternative type of autophagy originating from the Golgi membrane, rather than the endoplasmic reticulum (ER) membrane.9,10,11,12 Unlike canonical autophagy, this Golgi-associated alternative autophagy, also called Golgi membrane-associated degradation (GOMED) (hereafter described as alternative autophagy), occurs via a mechanism dependent on unc-51 like autophagy activating kinase 1 (ULK1) and Beclin-1, but not autophagy-related (ATG) 5, ATG7, ATG9, LC3, and STX17/Syntaxin17. Canonical autophagy and alterative autophagy are activated differently in a stimulus/context-dependent manner, i.e., starvation and rapamycin induce mostly canonical autophagy, whereas genotoxic stress induces both canonical and alternative autophagy.9,13,14 More importantly, these two types of autophagy degrade different cargos, i.e., canonical autophagy degrades SQSTM1/p62 and its binding proteins, whereas alternative autophagy preferentially degrades Golgi-trafficking molecules. Although various methods have been developed to detect canonical autophagy, methods to detect alternative autophagy have not been identified, except for EM and the Lysosomal-associated membrane protein (LAMP) 1 (or LAMP2)-staining assay. LC3 modifications and sequestosome-1 (SQSTM1)/p62 reduction are not markers of alternative autophagy because these molecules are not associated with alternative autophagy. Therefore, the development of a method for monitoring alternative autophagic flux has been anticipated, and we hence tested our small fluorescent probes for this purpose.
In this study, we developed a red-fluorescent autophagic probe named DAPRed. We analyzed the spectrum of autophagic vacuoles that are recognized by this red probe as well as the two green-fluorescent probes. Furthermore, using these green and red probes, we developed a method to evaluate autophagic flux. We also tested the application of this method to evaluate alternative autophagy and to evaluate autophagy in living organisms.
Results
Development of the red-fluorescent autophagy probe DAPRed
We previously developed two compounds, namely, DAPGreen and DALGreen, which can detect autophagic vacuoles.8 DALGreen generates fluorescence in hydrophobic and acidic conditions and hence specifically labels autolysosomes. On the other hand, as DAPGreen is insensitive to low pH, it labels autophagic structures of various stages. If the fluorescence spectrum of these two probes were different from each other, the different fluorescence patterns of DAPGreen and DALGreen could be utilized to observe autophagy dynamics. We thus aimed to develop a red-fluorescent probe from DALGreen.
A red-fluorescent autophagy probe was designed by replacing the naphthalimide scaffold of DALGreen (Figure 1A) by peryleneimide, to shift the π conjugation to longer wavelengths, and to facilitate its ability to intercalate into the membrane of autophagic structures. We synthesized this red-fluorescent probe (Figure 1A) from commercially available perylene-3,4-dicarboxylic anhydride by a five-step reaction (see STAR Methods). The red-fluorescent probe demonstrated much longer fluorescence wavelengths than DALGreen (DALGreen excitation: 405 nm, emission: 525 nm), with an emission peak at 735 nm and an excitation peak at 540 nm, which hence enabled its simultaneous use with DALGreen, DAPGreen, or other green-fluorescent markers. This red-fluorescent probe is membrane permeable and functions only in hydrophobic environments (Figure 1B). Furthermore, its pH sensitivity is minimal, and hence it does not show autolysosome-specific activity (Figure 1C). Therefore, this new probe was expected to detect multiple structures of the autophagic process. We thus named this compound as DOJIN-autophagosome detection red probe (DAPRed). DAPRed showed high photostability in vitro (Figure S1) and in living cells (Figure S2). All three compounds did not affect autophagic activity, which was confirmed by the analysis of autophagic flux using LC3-II and SQSTM1/p62 (Figure S3).
Figure 1.

Development of the red-fluorescent compound DAPRed
(A) Chemical structures of DALGreen and DAPRed.
(B and C) Hydrophobicity and pH dependency of DAPRed fluorescence. DAPRed (5 μM) was excited at 405 nm in various hydrophobic buffers (B) or in various acidic buffers (C), and fluorescence spectra were measured.
(D) HeLa cells were preincubated with DALGreen (1 μM), DAPGreen (0.1 μM), or DAPRed (0.1 μM) for 30 min. Then, cells were starved with or without BafA1 (0.1 μM) for 3 h and analyzed using confocal microscopy. Representative images are shown. Bars = 50 μm. Regions of interest (ROIs) are indicated by the yellow squares and magnified images are shown in the insets. Bars = 10 μm.
In (E–G), the fluorescent area of each probe per cell was calculated from the images. Data are shown as the mean ± S.D. The total number of cells used for analysis is given as the n. ∗p < 0.05, ∗∗∗p < 0.001 (one-way ANOVA followed by the Tukey post hoc test).
(H) Identification of DAPGreen puncta by CLEM analysis. TagRFP-LC3-transfected WT MEFs were preincubated with DAPGreen (0.25 μM) and treated with 0.5 μM rapamycin for 4 h. Cells were then fixed with 2% paraformaldehyde/2% glutaraldehyde, and DAPGreen/TagRFP-LC3 images were acquired. Cells were subsequently fixed using 2% OsO4 and observed by EM. Fluorescent signals are indicated by the dashed circles in the merged image. Red/green puncta were merged with autolysosomes. Bar = 200 nm. Mt, mitochondrion. A whole-cell image is shown in Figure S4.
(I) CLEM analysis shows the labeling of autolysosomes by DAPGreen and DAPRed. A similar experiment to (H) was performed, in which WT MEFs were preincubated with DAPGreen (0.25 μM) and DAPRed (0.1 μM), and then treated with 0.5 μM rapamycin for 2 h. Fluorescent signals are indicated by the dashed circles in the merged image. Bars = 200 nm. A whole-cell image is shown in Figure S5.
We first analyzed the usefulness of DAPRed using starved HeLa cells, in which canonical autophagy is activated. DAPRed showed weak fluorescence in untreated cells, whereas the signal intensity increased upon starvation, similarly to DAPGreen and DALGreen (Figures 1D–1G). Bafilomycin A1 (BafA1) is known to prevent autolysosome maturation, and the treatment of cells with BafA1 almost completely abolished the DALGreen signals, indicating that DALGreen generates fluorescence only in acidic compartments (autolysosomes), as described previously.3,8 In contrast, DAPGreen and DAPRed signals were present even after BafA1 treatment (Figures 1D, 1F, and 1G), suggesting that they recognize autophagic vacuoles of various stages.
To confirm the labeling of autophagic vacuoles by DAPGreen and DAPRed, we analyzed rapamycin-treated mouse embryonic fibroblasts (MEFs) by correlative light and EM (CLEM), using LC3 as a marker of autophagy. As expected, TagRFP-LC3-positive autophagic structures were labeled by DAPGreen (Figures 1H and S4). Furthermore, DAPGreen-positive autophagic structures were also labeled by DAPRed (Figures 1I and S5), confirming that these probes label autophagic vacuoles.
Colocalization of DAPGreen, DALGreen, and DAPRed with various autophagy-associated molecules
We further analyzed the steps of autophagy that are recognized by these three probes. To this end, we investigated the colocalization of these probes with several key autophagy molecules. For the analysis of DAPGreen/DALGreen and DAPRed, we used TagRFP/mCherry-fusion protein and GFP-fusion/Venus-fusion protein, respectively. Because GFP and Venus, but not TagRFP and mCherry, are unstable in autolysosomes owing to their lysosomal degradation and acidic conformational changes,15,16 we analyzed their fluorescence in the presence or absence of chloroquine (CQ) or BafA1, both of which inhibit autolysosome acidification.
WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) localizes on phagophores, which are initial platforms and characteristic membrane structures in autophagy, and dissociates before autophagosome generation.17 However, it was reported that GFP-fused WIPI2 was also present on autophagosomes.18 We found that rapamycin treatment induced WIPI2 puncta formation, and all WIPI2 puncta were labeled by DAPGreen (Figures 2A, 2B, and S6A). Regarding DALGreen, no colocalization was observed with WIPI2 puncta (Figures 2A and 2B). DAPRed labeled 42.1% of WIPI2 puncta, and the ratio was not altered by CQ (Figures 2A and 2B), suggesting that DAPRed labels a proportion of WIPI2 puncta (probably late-phase phagophores and autophagosomes; see discussion section). These data indicated that all and a part of WIPI2-positive structures are labeled by DAPGreen and DAPRed, respectively (see Figure 2G).
Figure 2.

Colocalization of DAPGreen, DALGreen, and DAPRed with various autophagy-associated molecules
(A and B) Colocalization analysis of the three probes with WIPI2. WT MEFs were transiently transfected with mCherry-WIPI2 (for DAPGreen and DALGreen) or GFP-WIPI2 (for DAPRed). Then, cells were preincubated with each probe for 30 min and treated with rapamycin (0.5 μM) in the presence or absence of chloroquine (CQ, 60 μM) for 3 h. Cells were then analyzed by confocal microscopy. Representative images are shown in (A). ROIs are indicated by squares, and their magnified images are shown in the lower panels. Arrows indicate DAPRed puncta colocalized with GFP-WIPI2. Bars = 10 μm and 1 μm in the upper and lower panels, respectively. In (B), the colocalization rate of each probe on WIPI2 puncta were quantified from the microscopic images and calculated using the application “BZ-H4C hybrid cell count” (Keyence).
(C and D) Colocalization analysis of the three probes with LAMP1. Similar experiments to (A, B) were performed using LAMP1-TagRFP and LAMP1-Venus. In (C), the arrow indicates a cup-shaped DAPRed single punctum.
(E and F) Colocalization analysis of the three probes with LC3. Similar experiments to (A and B) were performed using TagRFP-LC3 and GFP-LC3. We also added BafA1 (10 nM) instead of CQ for 3 h.
(G) Schematic summary of the colocalization assays in (A–F). Single-color images of WIPI2, LAMP1, and LC3 are shown in Figures S6A–S6C. In (B, D, and F), error bars indicate S.D. The total cell numbers are given as the n. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA followed by the Tukey post hoc test); ns: no significant difference.
LAMP1, a lysosomal protein, is usually observed as small dots that become large puncta upon the generation of autolysosomes (owing to the fusion of lysosomes with autophagic vacuoles). Upon rapamycin treatment, almost all large Lamp1 puncta were labeled by every probe (Figures 2C, 2D, and S6B), indicating that all of the probes detect autolysosomes. LC3 usually localizes to the cytosol, is recruited to phagophores upon the induction of autophagy, and remains localized to autophagic structures up to late autolysosomes.15 TagRFP-LC3 recognizes autophagic structures from phagophores through to autolysosomes, and DAPGreen almost completely labeled these puncta in rapamycin-treated MEFs (Figures 2E, 2F, and S6C). DALGreen labeled 61.8% of TagRFP-LC3 puncta, and the remaining TagRFP-LC3 puncta are thought to be phagophores and autophagosomes (Figures 2E and 2F). Regarding DAPRed, this probe labeled 65.7% of GFP-LC3 puncta (phagophores, autophagosomes, and early autolysosomes). The remaining GFP-LC3 puncta are thought to be early phagophores. CQ (or BafA1) suppresses the disappearance of GFP from the autolysosomes, and hence the colocalization rate was increased (Figures 2E and 2F). GABARAP showed a similar colocalization to LC3 (Figure S6D). Taken together, these results indicate that DAPGreen recognizes autophagic vacuoles from WIPI2-positive early phagophores to late autolysosomes (Figure 2G). DAPRed recognizes autophagic structures from late WIPI2 puncta to autolysosomes, and DALGreen recognizes only autolysosomes.
DAPGreen and DAPRed label preautolysosomal structures
We further analyzed DAPGreen to confirm its labeling of preautolysosomal structures. For this purpose, we expressed SNAP-tagged Lamp1, mCherry-WIPI2, and Halo-tagged LC3 in MEFs that were starved in the presence of DAPGreen for 2.5 h and then performed four-color imaging. We observed some DAPGreen signals on WIPI2/LC3-positive, Lamp1-negative preautolysosomal structures (Figure 3A, red arrowheads) together with WIPI2-negative, LC3/Lamp1-positive autolysosomes (Figure 3A, white arrows). The colocalization of WIPI2 and DAPGreen was confirmed by line scan analysis (Figure 3A) and by colocalization efficiency analysis (Pearsons, and tM1 and tM2) (Figure S7). These results confirmed the positive staining of preautolysosomal structures by DAPGreen.
Figure 3.

Colocalization of DAPGreen and DAPRed with preautolysosomal structures
(A) LAMP1-SNAP, mCherry-WIPI2, and HaloTag-LC3 were transfected into WT MEFs. The cells were preincubated with DAPGreen (20 nM), SNAP-Cell430 (3 μM), and SaraFluor650T (1 μM) for 30 min, stimulated by nutrient depletion, and spectral imaging was performed using the ZEN application. Bars = 10 μm. ROIs are indicated by the yellow squares, and magnified images are shown in the middle panels. Bars = 1 μm. The fluorescent intensity profiles across the red arrows in the middle panels are shown in the bottom panels. Note that the LAMP1-negative/WIPI2-positive/LC3-positive preautolysosomal structures (preAL; red arrowheads) are colocalized with the DAPGreen signals. LAMP1-positive/LC3-positive/WIPI2-negative autolysosomes (AL; white arrows) are also colocalized with DAPGreen.
(B) Spectral imaging of DAPRed with three makers. Similar experiment to (A) was performed using GFP-WIPI2 and DAPRed instead of mCherry-WIPI2 and DAPGreen. Note that the LAMP1-negative/WIPI2-positive/LC3-positive preautolysosomal structure (preAL; red arrowhead) is colocalized with the DAPRed signal and that the LAMP1-positive/LC3-positive/WIPI2-negative autolysosomes (AL; white arrows) are also colocalized with DAPRed. The colocalization of DAPRed with various autophagic structures is summarized in Figure S8.
(C) Identification of DAPRed puncta as phagophores by CLEM analysis. GFP-LC3-transfected WT MEFs were preincubated with DAPRed (0.1 μM) and treated with 0.5 μM rapamycin for 2 h. Cells were then fixed with 2% paraformaldehyde/2% glutaraldehyde, and DAPRed/GFP-LC3 images were acquired. Cells were subsequently fixed using 2% OsO4 and observed by EM. ROIs are indicated by the blue and yellow squares, and magnified images are shown in the middle and bottom panels, respectively. Bars = 10 μm and 500 nm in the top and bottom panels, respectively. Fluorescent signals are indicated by the dashed circles in the “Merge + DIC” image. CLEM analysis demonstrated that red/green puncta were merged with phagophores.
To confirm the labeling of preautolysosomal structures by DAPRed, we also carried out four-color imaging using GFP-WIPI2 instead of mCherry-WIPI2. As a result, we detected DAPRed signals on WIPI2/LC3-positive, Lamp1-negative preautolysosomal structures (Figure 3B, red arrowheads) together with WIPI2-negative, LC3/Lamp1-positive autolysosomes (Figure 3B, white arrows). Semiquantitative analysis showed that DAPRed labels a proportion of WIPI2 puncta, almost all LC3-positive/Lamp1-negative preautolysosomal structures, and all autolysosomes (LC3-positive/Lamp1-positive puncta) (Figure S8). The labeling of preautolysosomal structures was also validated by CLEM, in which some DAPRed signals were merged with GFP-LC3 signals, and by EM, in which some DAPRed/GFP-LC3-positive signals merged with phagophores (Figure 3C).
We further performed time-lapse imaging of DAPRed, GFP-LC3, and LysoTracker and found that DAPRed signals (Figure 4A; ROI #1, yellow arrowheads, Figure S9) emerged on pre-existing GFP-LC3 signals (lightgreen arrowheads) at 70 s. LysoTracker signals (Figure 4A; ROI #2, white arrowheads, Figure S9) appeared on DAPRed/LC3-positive puncta (yellow arrowheads) at 90 s, showing the sequential labeling of autophagic structures by GFP-LC3, DAPRed, and LysoTracker. These data confirmed the schematic model shown in Figure 2G. Note that there were many DAPRed puncta colocalizing with LC3 signals other than the punctum indicated by the arrowheads (Figure 4A), confirming the usefulness of these probes.
Figure 4.

DAPGreen, DAPRed, and DALGreen label canonical autophagy
(A) GFP-LC3-transfected WT MEFs were preincubated with DAPRed (0.1 μM) and LysoTracker Deep Red (0.5 μM), treated with 1 μM rapamycin for 4 h, and time-lapse three-color images were acquired. Whole-cell images are shown in Figure S9. ROI #1: On the punctum labeled only with GFP-LC3 (lightgreen arrowheads), a DAPRed signal colocalized at 70 s (yellow arrowheads). ROI #2: On the GFP-LC3/DAPRed-positive punctum (yellow arrowheads), a LysoTracker signal (white arrowheads) colocalized at 90 s. Bars = 1 μm.
(B and C) The indicated MEFs were prestained with DAPGreen (0.25 μM) or DALGreen (1 μM) plus DAPRed (0.1 μM), and then starved for 5 h. Then, the cells were analyzed using fluorescence microscopy. Representative images are shown in (B). Bars = 10 μm. In (C), the fluorescent area of each probe per cell was calculated from the images. Data are shown as the mean ± S.D. The total number of images used for analysis is given as the n. Black and red asterisks indicate statistical significance vs. the value of WT and vs. Ulk1/2-deficient cells, respectively (one-way ANOVA followed by the Tukey post hoc test; ∗∗p < 0.01, ∗∗∗p < 0.001, ns, no significant difference).
Labeling of DAPGreen, DAPRed, and DALGreen in various autophagy gene-deficient MEFs
We next used these probes to analyze MEFs lacking specific autophagy genes. Atg5, Atg9, and Ulk1/2 are genes well known to be required for starvation-induced autophagy, and expectedly, MEFs lacking these genes showed substantially low DAPGreen, DALGreen, and DAPRed signals upon their starvation (Figure 4B). Because ATG5 functions at a later step of autophagy than ATG9 and ULK1/2, early phagophores were generated in Atg5-deficient MEFs, but not in Atg9-deficient and Ulk1/2-deficient MEFs, and hence more DAPGreen signals were observed in Atg5-deficient cells than in the other autophagy gene-deficient MEFs (Figures 4B and 4C), confirming the usefulness of these probes to detect autophagy.
Characteristics of DALGreen, DAPGreen, and DAPRed
We next investigated the sensitivity and specificity of these probes. The high specificity of each probe was demonstrated by the high colocalization rate of each probe with LC3 puncta and LAMP1 puncta (Figures 2, 3, and 4). This was also confirmed by multiple CLEM analysis in different conditions (Figures 1H, 1I, and 3C). These data showed the specific staining of autophagic structures by each probe, despite the presence of many nonautophagic organelles, including mitochondria and ER in the cells (Figures 1H and 1I). As expected, these probes did not merge with mitochondria and ER markers (Figure S10). In the case of mitophagy and ER-phagy, which we induced by Parkin gene expression plus a protonophore (CCCP) and rapamycin plus CQ, respectively, we observed the colocalization of our probes and each organelle marker protein (Figure S11). Therefore, these probes are applicable to mitophagy and ER-phagy. Note that in mitophagy and ER-phagy, these probes are assumed to not label the cargos (mitochondria and ER) but label the membranes of the autophagic structures.
To more accurately investigate the specificities of these probes, we added a 4-fold higher dose than normal of these probes into the medium of starvation-treated Ulk1/2-deficient MEFs. As indicated, only weak signals and a small number of puncta were observed by the addition of DALGreen and DAPRed (Figure S12A; Ulk1/2-deficient, middle and bottom panels, respectively) compared with wild-type (WT) MEFs, and they were strictly merged with autophagic structures by CLEM analysis (Figure S12B). Even multivesicular bodies were not labeled by these probes (Figure S12B), indicating that these probes are highly specific for autophagic structures. The existence of a few small autophagic structures in Ulk1/2-deficient MEFs may suggest the possible presence of an Ulk1/2-independent type of autophagy. Regarding DAPGreen, weak signals appeared in a Golgi-like pattern by the addition of a 4-fold higher dose than normal (Figure S12A; top panels). Golgi localization was confirmed by its merging with TagRFP-labeled sialyltransferase (ST6GAL1), which is a marker of the trans-Golgi membrane (Figure S12C). Although we considered the possibility of the staining of ATG9 vesicles, which are known to localize on the Golgi in untreated cells, this is not likely because Golgi-like staining was observed even in Atg9-deficient MEFs (Figure S12A). Therefore, DAPGreen may nonspecifically label the Golgi when excess doses are used.
Regarding sensitivity, a high colocalization rate of DAPGreen/DAPRed and LC3 puncta indicated good sensitivity of these probes. Although the time-lapse images showed that DAPRed puncta emerged later than LC3 puncta (Figure 4A), we considered that this is because of the time-dependent alteration of the nature of autophagic structures. We cannot formally deny the possibility that this difference occurs owing to the low sensitivity of DAPRed, but even if this is the case, the delay time was only minimal (Figure 4A). We further investigated whether these probes also label basal autophagy, and hence we added each probe to untreated WT MEFs and analyzed their colocalization with WIPI2, LC3, and LAMP1. Several fluorescent puncta of each probe were observed (Figure S13), and they were considered to indicate basal autophagy because no fluorescence was observed even in starved Ulk1/2-deficient MEFs upon the addition of the same dose of the probes (Figure S12A). These puncta were much fewer and smaller than those observed in rapamycin-treated MEFs (Figure 2). However, the rate of merging of DAPGreen, DALGreen, and DAPRed with each structure under basal autophagy was almost equivalent to that under rapamycin-induced autophagy (Figures S13 and 2), indicating that these probes label autophagic vacuoles even during basal autophagy. Only the coexistence rate with LAMP1 differs between stimulated and unstimulated cells. This might be owing to the fact that autolysosomes in unstimulated cells are smaller than those in stimulated cells, making it difficult to distinguish from lysosomes. We also performed CLEM analysis to determine whether these probes indeed detect basal autophagy and found that DAPGreen signals merged with mCherry-LC3 puncta and also with autophagic structures on EM (Figure S14A). The labeling of GFP-LC3-positive autophagic vacuoles by DAPRed was also observed (Figure S14B). These data indicated that our probes are sensitive enough to detect basal autophagy.
Analysis of phagophore generation dynamics using DAPGreen and DAPRed
As described, both DAPGreen and DAPRed recognize structures that appear during various steps of the autophagy process, and DAPGreen labels earlier autophagic structures than DAPRed (Figures 2 and 4A; ROI#1). Therefore, treatment with both DAPGreen and DAPRed demonstrated that all DAPRed puncta are also labeled with DAPGreen in rapamycin-treated cells (Figure 5A). In addition, we observed a small number of puncta that were only labeled by DAPGreen (Figures 5A [arrows] and S15), which are thought to be early phagophores. This was confirmed by time-lapse analysis, which showed that DAPRed puncta become bigger to cover the pre-existing DAPGreen puncta within 47 s (Figure 5B, dotted circles). These results suggested that DAPGreen stains earlier autophagic structures than DAPRed, indicating that the former probe has a strong affinity for early phagophores. Therefore, we analyzed the properties of DAPGreen in comparison with DAPRed. When we stained membranes blotted with 14 phosphatidylinositol phosphate species (PIPs) with these probes, we found the specific interaction of phosphatidylinositol 3-phosphate (PI3P) with DAPGreen. In contrast, DAPRed showed nonspecific weak interaction with various PIPs (Figure S16A). Indeed, PI3P plays a key role in the initial step of autophagosome formation. We further found that DAPGreen stains double-membrane structures rather than single-membrane structures (Figure S16B). Therefore, DAPGreen is thought to label early phagophores by recognizing PI3P and double-membrane structures. These results demonstrate that by the combinatorial use of DAPGreen and DAPRed, it should be possible to analyze the kinetics of phagophore generation.
Figure 5.

Analysis of autophagy dynamics using DAPRed with DAPGreen and DALGreen
(A) WT MEFs were prestained with DAPGreen (0.25 μM) and DAPRed (0.1 μM) and then treated with rapamycin (0.5 μM) for 2 h. Then, cells were observed by confocal microscopy. Representative images are shown. Magnified images of the squares are shown in the lower panels. Bars = 10 μm (top), and 1 μm (bottom). Arrows indicate single DAPGreen puncta.
(B) Time-lapse imaging of rapamycin-treated MEFs in the presence of DAPGreen and DAPRed. The same cell as in Figure S15 was analyzed by time-lapse imaging. DAPGreen signals preceded DAPRed signals (dotted circles). Bar = 1 μm.
(C and D) Simultaneous staining of autophagic vacuoles with DAPRed and DALGreen. WT MEFs were prestained with DAPRed (0.1 μM) and DALGreen (1 μM) and treated with rapamycin (0.5 μM) for 8 h. Cells were observed after 0, 2, 4, 5, and 8 h of treatment, by confocal microscopy. In (C), representative images are shown at 5 h of treatment. ROIs are indicated by the squares, and magnified images are shown in the lower panels. Bar = 10 μm (top) and 1 μm (bottom). Arrows and arrowheads indicate DAPRed/DALGreen-merging puncta (autolysosomes) and single DAPRed puncta, respectively. (D) Time-dependent alteration of the number of single DAPRed puncta. The number of single DAPRed puncta per cell was quantified from the images at 0, 2, 4, and 8 h of treatment (n = 6, 4, 8, and 9 cells, respectively). Data are shown as the mean ± S.D. ∗p < 0.05, ∗∗p < 0.01 vs. the value of “0 h” (one-way ANOVA followed by the Tukey post hoc test).
(E) Quantitative time course analysis of fluorescence intensities of total DAPRed area and total DALGreen area. WT MEFs were prestained with DAPRed (0.1 μM) and DALGreen (1 μM) and treated with rapamycin (0.5 μM). Images were obtained using a fluorescence microscope. The ratios of DAPRed (left graph) and DALGreen (right graph) fluorescence area to total cell area were calculated using the application “BZ-H4C hybrid cell count” (Keyence). Data are shown as the mean ± S.D. (n = 3 images). ∗p < 0.05, ∗∗p < 0.01 (Student’s t test for comparison to the control with no treatment).
(F) Time-dependent alteration of the number of single DAPRed puncta. WT MEFs were preincubated with DAPRed (0.1 μM) and DALGreen (1 μM) and starved for 8 h. Cells were observed at the indicated times by confocal microscopy (representative images are shown in Figure S17A), and the number of single DAPRed puncta per cell was quantified from the microscopy images. Data are shown as the mean ± S.D. (n = 6, 4, 8, and 9 cells for 0, 2, 4, and 8 h, respectively).
(G) WT MEFs were transiently transfected with the mCherry-GFP-LC3 plasmid and starved for the indicated hours. Then, cells were observed by confocal microscopy (representative images are shown in Figure S17B), and the number of GFP puncta per cell was quantified from the microscopy images. Data are shown as the mean ± S.D. (n = 10, 9, 11, and 10 cells for 0, 2, 4, and 8 h, respectively). ∗∗p < 0.01 vs. the value of “0 h” (one-way ANOVA followed by the Tukey post hoc test).
Analysis of autophagy dynamics using DAPRed and DALGreen
As the dynamics of phagophore generation can be analyzed by the combinatorial use of DAPGreen and DAPRed, it should be possible to analyze autophagosome/autolysosome kinetics by the combination of DALGreen and DAPRed because they recognize autolysosomes and various autophagic structures, respectively. In fact, in rapamycin-treated MEFs, we observed two types of puncta, i.e., DAPRed/DALGreen-merged puncta (Figure 5C, arrows) and single DAPRed puncta (Figure 5C, arrowheads). The former and latter puncta are thought to correspond to autolysosomes and phagophores/autophagosomes, respectively. Using this method, we analyzed the kinetics of phagophore/autophagosome generation in rapamycin-treated WT MEFs. We found that the number of single DAPRed puncta was increased by 2 h and continued to later time points (Figure 5D). This was confirmed by time course analysis of cellular DAPRed- and DALGreen-fluorescent areas, by which we were able to measure the more detailed kinetics of phagophores/autophagosomes and autolysosomes in the same cells. As shown in Figure 5E, we found that an increase in rapamycin-induced DAPRed signals occurred earlier than an increase in DALGreen signals, and rapamycin-induced DAPRed signal was increased for 8 h, which is consistent with Figure 5D. In the case of starvation treatment, we found that the number of single DAPRed puncta was high at 2 h and decreased at later time points (Figures 5F and S17A), suggesting the transient induction of autophagy at 2 h. Importantly, these kinetics were consistent with those obtained for mCherry-GFP-LC3, which is a tandem protein widely used in autophagic flux assays (Figures 5G and S17B), and were consistent with a previous report.19 Note that the number of puncta labeled with only DAPRed (Figure 5F) was comparable but slightly less than that of GFP puncta labeled with mCherry-GFP-LC3 (Figure 5G), again confirming the high sensitivity of the DAPRed probe and its delayed staining of LC3 puncta (Figure 4A; ROI #1). Although both rapamycin treatment and starvation induced autophagy via mammalian target of rapamycin (mTOR), their autophagy kinetics were different (Figures 5D and 5F), which was confirmed by the phosphorylation of 4E-BP1, a protein downstream of mTOR in the autophagic pathway (Figure S18). We also compared the sensitivity of DAPGreen with that of CytoID, another small autophagy probe, using flow cytometry, by which DAPGreen showed higher sensitivity than CytoID (Figure S19A). Furthermore, the fluorescence of CytoID quickly disappeared, whereas the fluorescence of DAPGreen, DAPRed, and DALGreen remained for more than 2 days, indicating the high stability of these probes (Figure S19B). Taken together, these results show that autophagy dynamics can be measured by the concomitant use of DAPRed and DALGreen.
Staining of DAPRed and DALGreen in etoposide-treated MEFs
We next used these probes to analyze Golgi-mediated alternative autophagy, which is induced by genotoxic stress, but not starvation and rapamycin. Unlike canonical autophagy, alternative autophagy is Ulk1/2 dependent and Atg5/Atg9 independent.9,19 Expectedly, when cells were treated with the DNA-damaging reagent etoposide, substantial suppression of DALGreen and DAPRed signals was observed by addition of the ULK1/2 inhibitor SBI-0206965 (Figure S20), suggesting the usefulness of these probes to detect alternative autophagy. Furthermore, Ulk1/2-deficient MEFs showed substantially low signals of DALGreen and DAPRed, but Atg5-deficient and Atg9-deficient MEFs showed high fluorescence signals similarly to WT MEFs (Figure 6A). Quantitative analysis of DALGreen confirmed no signals in Ulk1/2-deficient MEFs and positive but fewer signals in Atg5-deficient and Atg9-deficient MEFs than in WT MEFs (Figure 6B). In the case of DAPRed, positive signals were not observed in Ulk1/2-deficient MEFs either, and equivalent or higher fluorescence intensity was observed in Atg5-deficient and Atg9-deficient MEFs compared with WT MEFs (Figure 6B). This might be owing to the detection of accumulated phagophores generated by Atg5/Atg9 deficiency, although the precise mechanism remains to be elucidated. In addition to MEFs, we also analyzed HeLa cells. HeLa cell lacking all Atg8 family genes (Atg8 hexa-knockout[KO]) is known to show resistance against canonical autophagy.20 As expected, when cells were starved, Atg8 hexa-KO HeLa cells showed substantially low signals of DAPGreen, DAPRed, and DALGreen (Figure S21A). In contrast, in the case of etoposide treatment, these probes showed almost equivalent fluorescence with HeLa cells (Figure S21B). This is owing to the fact that alternative autophagy is induced independently of ATG8 family proteins and indicates that alternative autophagy is recognized by these three probes. This was further confirmed using STX17/Syntaxin17. STX17 was reported to be involved in autophagosome closure in canonical autophagy and is hence now recognized as a marker of canonical autophagy.21 This was confirmed by the merging of DAPRed signals and GFP-STX17 signals in starved WT MEFs (Figure S22A, left panels). In contrast, DAPRed signals were not merged with GFP-STX17 signals in etoposide-treated Atg5-deficient MEFs (Figure S22A, right panels), suggesting that DAPRed recognizes alternative autophagy in these cells. The detection of alternative autophagy by DAPGreen and DALGreen was also shown by their colocalization with ST6GAL1-TagRFP (Golgi marker) (Figure S22B) because alternative autophagy originates from the Golgi membrane.10,11 DAPRed also colocalized with ST6GAL1-GFP, which was enhanced by the inhibition of GFP disappearance upon CQ treatment (Figure S22B). We also observed the colocalization of DAPRed with Rab9, another marker of alternative autophagy, but not with Atg9, a marker of canonical autophagy, in Atg5-deficient MEFs treated with etoposide (Figure S23). Taken together, these results demonstrate that alternative autophagy can be detected by these probes.
Figure 6.

DAPGreen, DALGreen, and DAPRed also detect alternative autophagy stimulated by genotoxic stress
(A and B) The indicated MEFs were prestained with DALGreen (1 μM) and DAPRed (0.1 μM) and were treated with 10 μM etoposide for 10 h. Then, the cells were analyzed using fluorescence microscopy (A). Bars = 10 μm. From the images, the fluorescent area of DAPRed and DALGreen per cell was calculated (B). Data are shown as the mean ± S.D. The total number of images used for analysis is given as the n. Black and red asterisks indicate statistical significance vs. the value of WT and vs. Ulk1/2-deficient cells, respectively (Mann-Whitney U test; ∗∗p < 0.01, ∗p < 0.05; ns, no significant difference).
(C) Staining of alternative autophagic structures with DAPRed. Atg5-deficient MEFs stably expressing with LAMP1-GFP were preincubated with DAPRed (0.1 μM) and treated with etoposide (10 μM) for 10 h. Then, cells were fixed with 0.75% paraformaldehyde/1.5% glutaraldehyde, and their images were observed by fluorescence microscopy. Cells were subsequently fixed with 1% OsO4 and observed by EM. Bars = 5 μm. ROIs are indicated by the blue and yellow squares, and magnified images are shown in the middle and bottom panels, respectively. Bars = 10 μm in the top panels, and 500 nm in the middle and bottom panels. Fluorescent signals are indicated by the dashed circles in the merged image. Arrows indicate phagophores. “G” indicates the Golgi apparatus. CLEM analysis demonstrated that DAPRed-positive/LAMP1-negative puncta were merged with phagophores. In the middle panel, multiple Golgi-derived vesicles that are not labeled by DAPRed were observed close to the Golgi apparatus.
(D) Quantitative time course analysis of fluorescence intensities of DAPRed and DALGreen. Atg5-deficient MEFs were prestained with DAPRed (0.1 μM) and DALGreen (1 μM), treated with etoposide (10 μM), and images were obtained using a fluorescence microscope. Then, the ratio of fluorescent area to total cell area was calculated using the application “BZ-H4C hybrid cell count” (Keyence). Data are shown as the mean ± S.D. (n = 7). ∗p < 0.05, ∗∗p < 0.01 (Student’s t test for comparison to the no treatment control).
Structures of alternative autophagy are labeled by DAPRed, DAPGreen, and DALGreen
Given that the three probes were applicable for the detection of alternative autophagy, we performed CLEM analysis to determine the structures recognized by these probes. In etoposide-treated Atg5-deficient MEFs, DAPRed-positive/LAMP1-negative puncta indicated phagophores that were located close to the Golgi apparatus (Figures 6C [arrows] and S24). CLEM analysis also showed the labeling of autolysosomes by DAPGreen and DAPRed (Figure S25), confirming the detection of alternative autophagy by these probes. Note that Golgi-derived small vesicles were not labeled by these probes (Figure 6C).
We also analyzed the difference between DAPGreen and DAPRed in detecting alternative autophagy. For this, we labeled Atg5-deficient MEFs with these probes, treated the cells with etoposide, and compared their fluorescence. We found that all the DAPRed-positive puncta were labeled by DAPGreen, whereas there was a small fraction of DAPGreen-positive/DAPRed-negative puncta (Figure S26A). Furthermore, time-lapse analysis showed that the intensity of DAPRed colocalized with the DAPGreen single-positive puncta gradually increased (Figure S26B). Therefore, DAPRed fluorescence appeared later than DAPGreen fluorescence, as in canonical autophagy. We also performed time course analysis of DAPRed and DALGreen fluorescence. These signals robustly increased from 8.5 h after etoposide treatment in Atg5-deficient MEFs (Figure 6D), indicating the successful measurement of the kinetics of autophagosomes and autolysosomes during alternative autophagy using these probes.9 Taken together, these results indicate that these probes are useful for measuring the kinetics of alternative autophagy.
DAPRed is useful for the detection of autophagic structures in zebrafish
Finally, to elucidate whether these probes are useful for the detection of autophagy in vivo, we administered the three probes to zebrafish (Figure 7A). However, DAPGreen and DALGreen signals were substantially disrupted by the autofluorescence from the zebrafish (Figure S27), and only DAPRed was found to be useful. Therefore, we added DAPRed into the water bath of zebrafish for 30 min at 2 days post-fertilization (dpf) and observed zebrafish larvae using confocal microscopy at 3 dpf (Figure 7B). DAPRed signals were observed in some organs, including the heart and eye. These signals were observed even at 7 dpf (120 h after staining), indicating the stability of this probe and its effective application to the long-term observation of zebrafish (Figure S28). To confirm that the signals corresponded to autophagic vacuoles, we injected a synthetic antisense morpholino oligonucleotide (MO) targeting the translation site of Atg5 (Atg5MO) into the fertilized eggs. Depletion of Atg5 was confirmed by immunoblotting (Figure S29A), and decreased DAPRed fluorescence was observed in the heart and eyes of the zebrafish, suggesting the positive labeling of canonical autophagy in these tissues (Figures S29B and S29C). Furthermore, we treated zebrafish with DAPRed together with rapamycin and found strong signals in the cells on the fin surface (Figure 7C).22 Inhibition of autophagy by 3-methyladenine (3-MA) largely suppressed these signals, also confirming the successful detection of autophagic structures by DAPRed in zebrafish (Figure 7C). Recently, the analysis of zebrafish demonstrated that autophagy is activated in the regenerated region of cut fins and that this autophagy is required for proper fin regeneration.23 Accordingly, we cut the fins of zebrafish in a water bath containing DAPRed. As shown in Figure 7D, robust DAPRed signals were observed in the regenerated fins. Furthermore, the suppression of autophagy by BafA1 suppressed the regeneration of fins (Figure 7D), which makes the DAPRed signal appear weak. This result is consistent with previous observations.23 These data indicated that DAPRed is useful for the analysis of autophagy in zebrafish. Taken together, we developed a novel red-fluorescent autophagy probe, DAPRed, and demonstrated that its combinatorial use with DAPGreen or DALGreen is useful for the detection of autophagy dynamics in vitro. DAPRed is also useful for the detection of autophagy in vivo.
Figure 7.

DAPRed is useful for detecting autophagic structures in zebrafish
(A) Schematic flowchart of DAPRed staining in zebrafish. DAPRed (0.1 μM) was added to the water for 30 min at 2 days post fertilization (dpf).
(B) Zebrafish larvae stained with DAPRed were observed by a whole-mount rapid 3D-imaging system (see STAR Methods). Bar = 500 μm. Two ROIs (#1: heart, #2: eye) are indicated by the blue squares. In the lower panels, the magnified images (phase-contrast and DAPRed images) are shown. Magnified images of the yellow squares are shown in the insets. Bar = 10 μm.
(C) Zebrafish larvae at 54 hpf were incubated with 0.1 μM of DAPRed, treated with the indicated chemicals for 24 h, and observed by a whole-mount rapid 3D-imaging system. Autophagic signals were increased by rapamycin (1 μM) treatment and decreased with the concomitant addition of 3-MA (10 mM). DAPRed images (upper panels) and merged images with phase contrast (lower panels) are shown. Bars = 50 μm. ROIs are indicated by the squares and magnified images are shown in the insets. Bars = 5 μm.
(D) Blastemas (2 dpf) were prestained with or without DAPRed. Then, fins were cut and treated with DMSO or 20 nM BafA1. Subsequently, fins were observed by the 3D-imaging system. BafA1 treatment significantly impaired the regeneration of fins and decreased the intensity of DAPRed. The white broken line indicates the amputation plane. Bars = 50 μm.
Discussion
Autophagy is a dynamic process that degrades intracellular cargos. There are two methods to quantify the dynamics of autophagy. One method is to monitor a single parameter both with and without autophagy inhibitors and calculate their difference.3 For example, western blotting analysis of SQSTM1/p62 or LC3-II, with and without BafA1, is a well-accepted method for the detection of autophagic flux. The other method is to monitor two different parameters simultaneously. We here developed such an autophagy dynamics assay using green- and red-fluorescent probes. A similar concept has already been developed as a fluorescent protein-based assay using the tandem proteins RFP-GFP-LC3 and GFP-LC3-RFP-LC3ΔG.6,7 Here, we developed a small-molecule fluorescent probe-based autophagic flux assay, which is a more simple and convenient method with higher resolution for the detection of autophagic flux.
In this study, we developed the red-fluorescent probe DAPRed, which recognizes autophagic structures from various steps, by the modification of DAPGreen and DALGreen, probes that detect autophagic membranes mainly recognizing the thickness of their membrane.8 We first aimed to generate an autolysosome-detecting red-fluorescent probe, and hence we replaced the naphthalimide scaffold of DALGreen to peryleneimide. However, unlike DALGreen, DAPRed showed only weak pH dependency. This might be owing to differences in fluorophore structure. Comparison of DAPRed with DAPGreen showed a difference in the recognition of initial autophagic structures; the latter probe stains earlier phagophore structures than the former probe. Although the precise mechanism remains unclear, we have two hypotheses. First, the difference in PI3P-binding activity might be the reason for the DAPGreen staining of earlier initial autophagic structures (Figure S16A). Second, this might be owing to the small difference between the probes in hydrophobic tail length, which is crucial for the detection of autophagic structures, as reported previously.8
Among the three probes, DAPGreen and DAPRed label preautolysosomal structures, which was clearly shown by their colocalization with WIPI2 puncta (Figures 2 and 3), colocalization with phagophores on CLEM analysis (Figure 3), emergence of LysoTracker signals on DAPRed/LC3-positive puncta on time-lapse imaging (Figure 4A), and the presence of multiple lysosome-negative DAPGreen and DAPRed puncta (Figures 3 and 4). Regarding DAPRed, this probe labeled around 42% of GFP-WIPI2 puncta (phagophores/autophagosomes) and almost all autolysosomes. Then how can the labeling of almost half of the WIPI2 puncta by this probe be explained? One possibility is that two different types of phagophores with different characteristics are generated, and the other possibility is that late-phase phagophores are labeled by DAPRed. Considering that most autophagosomes/autolysosomes are labeled by DAPRed, the latter possibility seems to be more likely. Compared to existing autophagy-monitoring probes, these three probes have three advantages. First, unlike the existing probes, these new probes are suitable for multicolor staining. For example, CytoID only offers the option of green fluorescence, but when designing an experiment using the new probes, the user can choose either green or red fluorescence. Second, DALGreen, DAPGreen, and DAPRed showed high stability in vitro, in cells, and in vivo (Figures S1, S2, S19B, and S28). Finally, flow cytometric detection showed that DAPGreen was more sensitive than CytoID (Figure S19A). Note that when these three probes are actually used, high background fluorescence is detected immediately after staining only for DALGreen (Figures 5E and 6D). Because the background fluorescence of DALGreen decreases within 2 h, additional washing should be performed if autophagy is to be detected in a short time.
These three probes were initially developed to detect canonical autophagy. However, they were found to also be useful for the detection of Golgi-mediated alternative autophagy, also called GOMED. This autophagy originates from trans-Golgi membranes in a manner independent of ATG5 and degrades different substrates to those of canonical autophagy.10,11 The molecules required are also different because alternative autophagy involves ULK1 and PI3K complexes, but not ATG9 complexes, STX17, or the ATG5 or LC3 conjugation system. Canonical autophagy can be readily monitored using LC3 modifications, SQSTM1/p62 degradation, and the fluorescence proteins RFP-GFP-LC3 and GFP-LC3-RFP-LC3ΔG, whereas the monitoring of alternative autophagy is difficult because alternative autophagy-specific protein modifications have not been identified. In this study, we showed that alternative autophagy can also be detected by the DAPGreen, DAPRed, and DALGreen probes. Using Atg5/7-deficient cells, we showed that these probes enable the direct measurement of alternative autophagy dynamics (Figure 6D). Furthermore, we were able to detect total (canonical plus alternative) autophagy in WT cells. By the combinatorial use of these probes and LC3, we detected canonical autophagy as LC3-positive/DALGreen-positive puncta and alternative autophagy as LC3-negative/DALGreen-positive puncta.
Why these probes detect not only canonical autophagy but also alternative autophagy, despite the origin of the autophagic membranes being different, remains unclear. Canonical and alternative autophagic membranes are derived from the ER and Golgi, respectively, which contain different proteins and lipids. Therefore, such lipids and proteins are probably not involved in the detection of the autophagic membranes by these probes. However, a common feature must exist. One candidate factor is PI3P because Beclin-1, a member of the PI3K complex, is required for both types of autophagy. However, as PI3P is present in many types of organelles, such as endosomes, additional factors must be required, which might be a physical factor, such as a double-membrane structure. It is possible that other unknown factors may be involved.
We also tested the usefulness of DAPRed in vivo. Several previous reports have shown the analysis of autophagy in live zebrafish using GFP-LC3 and confirmed the induction of autophagy in rapamycin-treated and regenerated fins.22,23 Consistently, we detected positive DAPRed signals in rapamycin-treated and regenerated fins, indicating that DAPRed can be used to analyze autophagy in live zebrafish (Figure 7). This probe hence enables the simple detection of autophagy during organism development and under stress conditions, which will provide important information toward understanding the role of autophagy in living organisms.
Limitations of the study
It is important to determine the optimal dose for each cell to be used prior to the experiment, as higher doses of DALgreen and other probes can produce nonspecific signals. The effectiveness of these probes in vivo has not been verified in any species other than zebrafish. We believe that they may be effective in mice, such as fetuses, but further validation is needed. There is also a possibility that the probes can be used effectively in adults by devising measurement and administration methods. Furthermore, it is expected that autophagy dynamics can be better analyzed if the fluorescence wavelength of either DAP or DAL can be changed and 3-color imaging can be performed together with DAPred.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LC3 (1:1000) | Nano Tools | Cat# 0231-100/LC-5F10; RRID: AB_2722733 |
| Rabbit polyclonal anti-SQSTM1/p62 (1:1000) | MBL | Cat# PM045; RRID: AB_1279301 |
| Mouse monoclonal anti-TUBA (1:2500) | Sigma-Aldrich | Cat# T9026; RRID: AB_477593 |
| Rabbit monoclonal anti-Phospho4E-BP1(Thr37/46) (1:1000) | Cell Signaling | Cat# 2855; RRID: AB_2097841 |
| Rabbit monoclonal anti-4E-BP1 (1:1000) | Cell Signaling | Cat# 9644; RRID: AB_2097841 |
| Rabbit polyclonal anti-Atg5 (1:1000) | Novus | Cat# NB110-53818; RRID: AB_828587 |
| Chemicals, peptides, and recombinant proteins | ||
| Amylamine | TCI | Cat# A0445 |
| Perylene-3,4-dicarboxylic anhydride | Iwashita et al.8 | N/A |
| 2-(Boc-amino)ethyl bromide | TCI | Cat# B2289 |
| Baffilomycin A1 | Focus Biomolecules | Cat# 10-2060 |
| DMSO | Wako | Cat# 041-29351 |
| Hanks' Balanced Salt Solution | Nacalai | Cat# 09735-75 |
| DALGreen | Dojindo | Cat# D675 |
| DAPGreen | Dojindo | Cat# D676 |
| DAPRed | Dojindo | Cat# D677 |
| Rapamycin | Santa Cruz Biotechnology | Cat# SC-3504 |
| Chloroquine diphosphate | Nacalai | Cat# 08660-62 |
| Etoposide | Sigma-Aldrich | Cat# E1383 |
| SBI-0206965 | Sigma-Aldrich | Cat# SML1540 |
| 3-Methyladenine | Sigma-Aldrich | Cat# M9281 |
| LysoTracker Red DND-99 | Thermo Fisher Scientific | Cat# L7528 |
| LysoTracker Deep Red | Thermo Fisher Scientific | Cat# L12492 |
| MitoTracker Deep Red FM | Thermo Fisher Scientific | Cat# M22426 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| ER-Tracker Red | Thermo Fisher Scientific | Cat# E34250 |
| SNAP-Cell 430 | New England Biolabs | Cat# S9109S |
| HaloTag SaraFluor650T ligand | Goryo Chemical | Cat# A308-01 |
| EYPC | Wako | Cat# 124-05031 |
| PI3P | Echelon | Cat# P-3016 |
| PlasMem Bright Red | Dojindo | Cat# 346-09771 |
| Wortmannin | Merck Calbiochem | Cat# 681675 |
| Dulbecco’s modified Eagle’s medium | Nacalai | Cat# 08458-45 |
| MEM vitamin solution | Gibco | Cat# 11120-052 |
| L-glutamine | Nacalai | Cat# 16948-04 |
| sodium pyruvate | Nacalai | Cat# 06977-34 |
| nonessential amino acids | Gibco | Cat# 11140-050 |
| HEPES | Nacalai | Cat# 17557-94 |
| 2-mercaptoethanol | Nacalai | Cat# 21438-82 |
| penicillin-streptomycin mixed solution | Nacalai | Cat# 26253-84 |
| fetal bovine serum | Biowest | Cat# S1810-500 |
| paraformaldehyde | Nacalai | Cat# 26126-25 |
| glutaraldehyde | TAAB | Cat# 3041 |
| OsO4 | TAAB | Cat# 300 |
| Lead stain solution | Sigma-Aldrich | Cat# 18-0875 |
| Tricaine | Sigma-Aldrich | Cat# E10521 |
| Critical commercial assays | ||
| CytoID | Enzo Life Science | Cat# ENZ-51031-K200 |
| Neon transfection system | Thermo Fisher Scientific | Cat# MPK1096 |
| AMAXA Cell Line Nucleofector Kit V | Lonza | Cat# VCA-1003 |
| Lipid Snoopers, PIPs | Avanti | Cat# 330500 |
| Experimental models: Cell lines | ||
| HeLa cell (Wild type) | Iwashita et al.8 | N/A |
| Atg8 hexa-KO HeLa cell | Nguyen et al.20 | N/A |
| Mouse embryonic fibroblast (WT MEF) | Shimizu et al.24 | N/A |
| Atg5-deficient MEF | Nishida et al.9 | N/A |
| Atg9-deficient MEF | Saitoh et al.25 | N/A |
| Ulk1/2-deficient MEF | McAlpine et al.26 | N/A |
| Experimental models: Organisms/strains | ||
| Zebrafish (Tüpfel long fin) | Asaoka et al.27 | ZFIN: ZDB-GENO-9900623-2 |
| Oligonucleotides | ||
| Wipi2 modification primer F1: 5′-GTACAAGGGATCCATGAACCTGGCGAGCCAGAGC-3′ | This paper | N/A |
| Wipi2 modification primer F2: 5′-TAGATCCATGGTGAGCAAGGGCGAGGAGGATAAC-3′ | This paper | N/A |
| Wipi2 modification primer R1: 5′-CTCACCATGGATCTAATTCCGGCGCCTAGAGAAG-3′ | This paper | N/A |
| Wipi2 modification primer R2: 5′-ATGGATCCCTTGTACAGCTCGTCCATGCCGCCGGTG-3′ | This paper | N/A |
| GABARAP modification primer F: 5′-GCCAGATCTATGAAGTTCGTGTACAAAGAAGAGCATC-3′ | This paper | N/A |
| GABARAP modification primer R: 5′-GCCCTCGAGTCACAGACCGTAGACACTTTCGTCACTG-3′ | This paper | N/A |
| Cytb5104-134 modification primer F1: 5′-taatagatctggctccggaggaggaaagccaatcc-3′ | This paper | N/A |
| Cytb5104-134 modification primer R1: 5′-gagccagatctattaagtttgtgccccagtttgc-3′ | This paper | N/A |
| Cytb5104-134 modification primer F2: 5′-gactgagaattctgcagatatccagcacagtggc-3′ | This paper | N/A |
| Cytb5104-134 modification primer R2: 5′-gcagaattcTCAGTCCTCGGCCATATACAGCCTG-3′ | This paper | N/A |
| STX17 truncation primer F: 5′-CTGCCAGCGGATCCGTTAACTCGCGAACGCGT-3′ | This paper | N/A |
| STX17 truncation primer R: 5′-CGGATCCGCTGGCAGCTCTGCCTGTGGCAGGTG-3′ | This paper | N/A |
| ST6GAL1 cloning primer F: 5′-CTTAAGCTTGAAGATGATTCACACCAACCTGAAGAAAAAG-3′ | This paper | N/A |
| ST6GAL1 cloning primer R: 5′-GTGGATCCTTGCAGTGAATGGTCCGGAAGCCAGGC-3′ | This paper | N/A |
| Tom20 cloning primer F: 5′-CTCGGATCCATGGTGGGCCGGAACAGCG-3′ | This paper | N/A |
| Tom20 cloning primer R: 5′-CATGAATTCTTCCACATCATCCTCACCC-3′ | This paper | N/A |
| Atg5MO: 5′-CCTTGTCATCTGCCATTATCATCGT-3′ | Varga et al.23; GeneTools | N/A |
| custom control oligo (5′-CCTTCTCAGCTCCCATAATCTTCGT-3′ | This paper; GeneTools | N/A |
| Recombinant DNA | ||
| pmCherry-WIPI2 | This paper | N/A |
| pEGFP-WIPI2 | This paper | N/A |
| pEGFP-LC3 | This paper | N/A |
| pmCherry-LC3 | This paper | N/A |
| pmCherry-EGFP-LC3 | This paper | N/A |
| pTagRFP-LC3 | Yamaguchi et al.11 | N/A |
| pGABARAP-Venus | This paper | N/A |
| pGABARAP-TagRFP | This paper | N/A |
| pLamp1-Venus | This paper | N/A |
| pSNAP f vector | New England Bio Labs | Cat# N9183S |
| pLamp1-SNAPtag | This paper | N/A |
| pLamp1-TagRFP | Torii et al.28 | N/A |
| pEGFP-Cytb5104-134 | Tashiro et al.29 | N/A |
| pTagRFP-Cytb5104-134 | This paper | N/A |
| pTom20-EGFP | This paper | N/A |
| pTom20-mRFP | This paper | N/A |
| pEGFP-STX17TM | This paper | N/A |
| pST6GAL1-EGFP | This paper | N/A |
| pST6GAL1-TagRFP | This paper | N/A |
| pEGFP-Rab9 | Nishida et al.9 | N/A |
| pAtg9-EGFP | Takahashi et al.30 | N/A |
| pHaloTag-LC3 | Promega | Cat# FHC05917 |
| Software and algorithms | ||
| ImageJ (Fiji) | Fiji project | https://imagej.net/ |
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/ |
| SnapGene4.1.6 | GSL Biotech | http://www.snapgene.com/ |
| ApE-A plasmid Editor v2.0.47 | EM. Jorgensen laboratory (University of Utah) | https://jorgensen.biology.utah.edu/ |
| ZEN2.1 | Carl Zeiss | https://www.zeiss.com/ |
| BZ-H4C | Keyence | https://www.keyence.com/ |
| THUNDER 3D Cell Culture Imager | Leica | https://www.leica-microsystems.com/ |
| FACSDiva | BD | https://www.bdbiosciences.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shigeomi Shimizu (shimizu.pcb@mri.tmd.ac.jp).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Experimental model and study participant details
Cell lines
The MEFs and HeLa cells listed in the key resources table were kindly gifted as described in the acknowledgments and have been used in many analyses in our laboratory.8,9,10,11,13,14,20,24,25,26,28 To authenticate these defective cell lines, we frequently performed PCR or immunoblotting. We regularly examined mycoplasma contamination by DAPI staining of fixed cells. MEFs and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM high glucose, Nacalai) or Minimum Essential Media (Gibco) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 10 mM HEPES (pH 7.4), 0.05 mM 2-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% (v/v) fetal bovine serum in a humidified 5% or 10% CO2 incubator at 37°C. For the analysis of starvation-induced autophagy, cells were cultured in Hanks’ Balanced Salt Solution (Nacalai) after washing with HBSS(+) three times.
Zebrafish
The TL wild type (WT) strain zebrafish were maintained at 28°C in distilled water, to which a small amount of salts and minerals were added, as described previously.27 Fertilized eggs and embryos were produced by natural matings and staged by standard morphological criteria or by hours or days post-fertilization (hpf or dpf). For staining with probes, fertilized eggs were manually hatched out at 48 hpf and stained with 1 μM DALGreen and 0.1 μM DAPRed added to the water for 30 min at 54 hpf. Following washout of the staining medium, zebrafish larvae were maintained in the water with/without chemicals under constant darkness for 24 h, and anesthetized with 0.02% tricaine (Sigma-Aldrich) in 0.1% agarose for observation using an LSM710 fluorescence microscope. Rapamycin (Santa Cruz) was dissolved in DMSO and diluted to a concentration of 1 μM, and 3-MA (Sigma-Aldrich) was dissolved in PBS (Takara) and diluted to a concentration of 10 mM.
These experiments were carried out in strict accordance with the recommendations of the ethical guidelines of Tokyo Medical and Dental University, and performed in a manner that minimized pain and discomfort. All experimental protocols in this study were approved by the Animal Welfare Committee of Tokyo Medical and Dental University (Permit Number: A2023-001A).
Method details
Development of DAPRed
DAPRed was synthesized by five overall steps. Briefly, the condensation of amylamine with perylene-3, 4-dicarboxylic anhydride produced the N-alkyl peryleneimide compound. The piperazine moiety was introduced via an aromatic substitution reaction after the bromination of the peryleneimide ring, resulting in piperazine-conjugated pelryleneimide. Finally, attachment of the propylamine unit with 2-(Boc-amino)ethyl bromide through an alkylation reaction followed by deprotection of the amine protection group (Boc) resulted in the formation of DAPRed.
Reagents and instruments
Etoposide, SBI-0206965, and 3-MA were purchased from Sigma-Aldrich. Lyso Tracker Deep Red, Lyso Tracker Red, ER-Tracker Red, MitoTracker Green FM, MitoTracker Deep Red FM were purchased from Thermo Fisher Scientific. Rapamycin, BafA1, wortmannin, SNAP-Cell 430, HaloTag SaraFluor650T ligand, CytoID, and PlasMem Bright Red were obtained from Santa Cruz Biotechnology, Focus Biomolecules, Merck Calbiochem, New England Biolabs, Goryo Chemical, Enzo Life Sciences, and Dojindo respectively. Other chemicals were purchased from Nacalai Tesque. Lipid blotted membranes were purchased from Avanti polar lipids. The following antibodies were used for the immunoblot assays: anti-LC3 (nanotools), anti-SQSTM/p62 (MBL), anti-TUBA (Sigma-Aldrich), anti-4E-BP1phos-T37/46, anti-4E-BP1 (Cell Signaling), and anti-Atg5 (Novus). 1H-NMR and 13C-NMR spectra were recorded on a Bruker AVANCE III HD 400 MHz spectroscope. Mass spectra were measured using a Waters SQD2 (Waters) mass spectrometer. Visible UV spectra were obtained on a UV-2450 UV/Vis spectrophotometer (Shimadzu), and fluorescence spectroscopic studies were performed using an FP-6300 fluorescence spectrophotometer (JASCO). Fluorescence images were obtained using LSM710 and LSM800 confocal laser-scanning microscopes (Zeiss). For CLEM observation, a JEM-1400 Plus transmission electron microscope (JEOL) was used with a CCD camera (EM-14830RUBY2; JEOL). Sequential time-lapse imaging was acquired using the fluorescence microscope BZ-X800 and analyzed with BZ-H4C (Keyence). Lipid blotted membranes were detected using Pxi (Syngene). Whole-mount rapid 3D images of zebrafish were obtained using THUNDER 3D Cell Culture Imager (Leica).
Gene transformation and plasmid construction
For transient plasmid expression, MEFs (1 × 106) were transfected with 1 μg plasmid DNA using the Neon transfection system (Thermo) or AMAXA Cell Line Nucleofector Kit V (Lonza) according to the supplier’s protocol (1,300 V, 20 ms, 2 times or program U-20). Atg9-GFP and GFP-Rab9 expression plasmids were kindly gifted.9,29 The expression plasmids mCherry-WIPI2, GFP-WIPI2, GFP-LC3, mCherry-LC3, mCherry-GFP-LC3, LAMP1-Venus, LAMP1-SNAP, TagRFP-Cytb5104-134, GABARAP-Venus, and GABARAP-TagRFP were modified from the plasmids expressing Flag-WIPI2, TagRFP-LC3, LAMP1-TagRFP, SNAP-tag (NEB), GFP-Cytb5104-134, and GABARAP-YFP which were used in a previous study.8,9,11,28,30 The GFP-STX17-expressing plasmid was truncated from the GFP-STX17WT plasmid by 5′-CTGCCAGCGGATCCGTTAACTCGCGAACGCGT-3′ and 5′-CGGATCCGCTGGCAGCTCTGCCTGTGGCAGGTG-3′.21 Sialyltransferase (ST6GAL1) DNA was amplified from human thymus cDNA by 5′-CTTAAGCTTGAAGATGATTCACACCAACCTGAAGAAAAAG-3′ and 5′-GTGGATCCTTGCAGTGAATGGTCCGGAAGCCAGGC-3′, and fused to GFP and TagRFP. Tom20 DNA was amplified from rat cDNA by 5′-CTCGGATCCATGGTGGGCCGGAACAGCG-3′ and 5′-CATGAATTCTTCCACATCATCCTCACCC-3′, and fused to GFP and mRFP. A commercially available plasmid was used for HaloTag-LC3 expression (Promega). The transfection efficiency was more than 75%, as assessed by the GFP fluorescence of the cells.
Staining of autophagic structures
DALGreen, DAPGreen, and DAPRed were used according to the supplier’s protocol as described below. MEFs were preincubated in culture medium with 1 μM DALGreen, 0.25 μM DAPGreen, or 0.1 μM DAPRed for 30 min. The dose was determined according to the differences in fluorescence between untreated cells and cells starved for 2 h. Following washout of the staining medium, canonical autophagy was induced by treatment with 0.5 or 1 μM rapamycin or nutrient depletion, and alternative autophagy was induced by 10 μM etoposide treatment. Fluorescence images were taken under the same conditions (laser power and detector gains) in each experiment.
For flow cytometric analyses, wild-type MEFs (2 × 105) were preincubated with 2 μM DALGreen or 0.1 μM DAPGreen for 30 min. Cells were harvested after induction of canonical autophagy at the indicated time point (Figure S19A) and measured by FACS Canto II (Becton Dickinson Co.). For CytoID analysis, cells were first treated to induce canonical autophagy, then incubated with 1/500 CytoID regent for 30 min according to the manufacture’s protocol. After washing with PBS, cells were measured by FACS Canto II. Data analysis was performed using FACSDiva software.
Spectrum imaging
WT MEFs (1 × 106) were transfected with LAMP1-SNAP, GFP-WIPI2, and HaloTag-LC3-expressing plasmid DNA using the AMAXA Cell Line Nucleofector Kit V according to the supplier’s protocol (program U-20). The cells were preincubated with DAPRed (0.1 μM), DAPGreen (20 nM), SNAP-Cell 430 (3 μM), and SaraFluor650T (1 μM) for 30 min. Then, probes and ligands were washed out with HBSS three times to simultaneously induce autophagy by nutrient depletion. Spectral imaging was carried out using an LSM710 microscope and the ZEN application via individual single-color spectrum data.
CLEM
MEFs cultured on glass-bottom dishes with grids were preincubated in culture medium with 1 μM DALGreen, 0.25 μM DAPGreen, or 0.1 μM DAPRed for 30 min. Following washout of the staining medium, autophagy was induced by the above-mentioned procedure. Samples were fixed with paraformaldehyde (0.75%)/glutaraldehyde (1.5%) for Figures 6C and S24, and paraformaldehyde (2%)/glutaraldehyde (2%) for the others in 0.1 M phosphate buffer (PB) at pH 7.4 at room temperature, and then immediately observed using an LSM710 fluorescence microscope within 30 min. Subsequently, the samples were put into a refrigerator for 30 min to cool them to 4°C. Thereafter, the samples were fixed with 2% glutaraldehyde in 0.1 M PB at 4°C overnight. After fixation, the samples were washed 3 times with 0.1 M PB and were postfixed with 1% OsO4 for Figures 6C and S16 or 2% OsO4 for the other experiments in 0.1 M PB at 4°C for 1 h. After dehydration, ultrathin sections were stained with 2% uranyl acetate and lead stain solution (Sigma-Aldrich), and observed using a JEM-1400Plus electron microscope (JEOL) at 100 kV.
Translational inhibition by morpholino oligos
The morpholino oligo inhibition assay was performed based on the methods of a previous study.27 In detail, Atg5MO (5′-CCTTGTCATCTGCCATTATCATCGT-3′)21 and custom control oligo (5′-CCTTCTCAGCTCCCATAATCTTCGT-3′) were designed and purchased from Gene Tools. For knockdown, fertilized eggs (∼2-cell stage) were injected with the reagent pre-mixed on a coverslip with 3 mM MO, 0.5% phenol red, and 0.5 μg/μL pmaxGFP plasmid (4:1:5). The efficacy of Atg5 translation inhibition was confirmed in 2 dpf embryos by immunoblotting using anti-Atg5 antibody (Figure S29A). Each 20 eggs were lysed in 20 mM HEPES, 100 mM NaCl, 10% glycerol, 0.03% MgCl2, 1 mM EGTA, 10 mM Na4P2O7, and 1% NP-40 without GFP fluorescent screening. For observation, 3 dpf zebrafish larvae were screened for GFP fluorescence and observed by confocal microscopy.
Fin amputation analysis
Fin amputations were performed according to the methods of previous studies.21,31 Fertilized eggs were manually hatched at 48 hpf, and fin amputations were immediately performed. After the surgery, the fish were stained with 0.1 μM DAPRed for 30 min and kept at a constant temperature of 28°C under complete darkness for 48 h. To inhibit the amputated fins, the water containing 20 nM BafA1 was replaced every 24 h. Fish were anesthetized with 0.02% tricaine (Sigma-Aldrich) in 0.1% agarose for observation using an LSM710 fluorescence microscope.
Quantification and statistical analysis
All results are expressed as the mean ± standard deviation (S.D.). Statistical analyses were performed using Prism5 (GraphPad) software. Comparisons of multiple datasets were performed using the Mann-Whitney U test and one-way ANOVA followed by the Tukey post hoc test for multiple comparisons, or the two-tailed unpaired Student t-test with the Smirnov-Grubbs test. A p-value of less than 0.05 was considered to indicate a statistically significant difference between two groups.
Acknowledgments
Atg9-deficient MEFs were kindly provided by Professors T. Saitoh and S. Akira (Osaka University). Ulk1/2-deficient MEFs and Atg8 hexa-KO HeLa cells were kindly provided by Professors S.A. Tooze (The Francis Crick Institute, UK) and M. Lazarou (Monash University), respectively. The GFP-Stx17WT plasmid, the GFP-Cyb5104−134 plasmid, and the Atg9-GFP plasmid were kindly provided by Professor N. Mizushima (The University of Tokyo), Professor Y. Tamura (Yamagata University), and Associate Professor Y. Takahashi (Pennsylvania State University), respectively.
This study was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (S) (23H05480), (A) (20H00467), (C) (20K07353), Grant-in-Aid for Encouragement of Young Scientists (B) (19K16119), and MEXT KAKENHI Grant-in-Aid for Scientific Research on Innovative Areas (17H06414, 22H04639, 23H04773). This study was also supported by AMED-CREST (JP23gm1410012) and the Project for Psychiatric and Neurological Disorders (JP21wm0525028) from the Japan Agency for Medical Research and Development, AMED. This study was also supported by the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University.
Author contributions
Conceptualization, H.T.S., S.S., H.I., and M.I.; Formal analysis. H.T.S.; Investigation, H.T.S., H.I., S.A., A.Y., M.K., and S.K.; Resources, H.T.S., A.Y., M.K., S.K., H.N., Y.U., and H.I.; Writing original draft, S.S. and H.T.S.; Writing review & editing, all authors; Project administration, S.S., H.N., and Y.U.; Supervision, S.S.
Declaration of interests
H. Iwashita, M. Ishiyama, and Y. Ueno are employees of Dojindo Laboratories, which is the manufacturer of DALGreen, DAPGreen, and DAPRed. The authors have no additional competing financial interests associated with this study.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107218.
Contributor Information
Yuichiro Ueno, Email: yueno@dojindo.co.jp.
Shigeomi Shimizu, Email: shimizu.pcb@mri.tmd.ac.jp.
Supplemental information
Data and code availability
-
•
Microscopy images and original western blot images represented in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Kawabata T., Yoshimori T. Beyond starvation: An update on the autophagic machinery and its functions. J. Mol. Cell. Cardiol. 2016;95:2–10. doi: 10.1016/j.yjmcc.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Morishita H., Mizushima N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 2019;35:453–475. doi: 10.1146/annurev-cellbio-100818-125300. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky D.J., Abdel-Aziz A.K., Abdelfatah S., Abdellatif M., Abdoli A., Abel S., Abeliovich H., Abildgaard M.H., Abudu Y.P., Acevedo-Arozena A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) Autophagy. 2021;17:1–382. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuma A., Matsui M., Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 6.Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 7.Kaizuka T., Morishita H., Hama Y., Tsukamoto S., Matsui T., Toyota Y., Kodama A., Ishihara T., Mizushima T., Mizushima N. An autophagic flux probe that releases an internal control. Mol. Cell. 2016;64:835–849. doi: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 8.Iwashita H., Sakurai H.T., Nagahora N., Ishiyama M., Shioji K., Sasamoto K., Okuma K., Shimizu S., Ueno Y. Small fluorescent molecules for monitoring autophagic flux. FEBS Lett. 2018;592:559–567. doi: 10.1002/1873-3468.12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishida Y., Arakawa S., Fujitani K., Yamaguchi H., Mizuta T., Kanaseki T., Komatsu M., Otsu K., Tsujimoto Y., Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi H., Arakawa S., Kanaseki T., Miyatsuka T., Fujitani Y., Watada H., Tsujimoto Y., Shimizu S. Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J. 2016;35:1991–2007. doi: 10.15252/embj.201593191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi H., Honda S., Torii S., Shimizu K., Katoh K., Miyake K., Miyake N., Fujikake N., Sakurai H.T., Arakawa S., Shimizu S. Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration. Nat. Commun. 2020;11:5311. doi: 10.1038/s41467-020-18892-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng H., Wang N., Zhang N., Liao H.H. Alternative autophagy: mechanisms and roles in different diseases. Cell Commun. Signal. 2022;20:43. doi: 10.1186/s12964-022-00851-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torii S., Yamaguchi H., Nakanishi A., Arakawa S., Honda S., Moriwaki K., Nakano H., Shimizu S. Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat. Commun. 2020;11:1754. doi: 10.1038/s41467-020-15577-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda S., Arakawa S., Nishida Y., Yamaguchi H., Ishii E., Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 2014;5:4004. doi: 10.1038/ncomms5004. [DOI] [PubMed] [Google Scholar]
- 15.Katayama H., Yamamoto A., Mizushima N., Yoshimori T., Miyawaki A. GFP-like proteins stably accumulate in lysosomes. Cell Struct. Funct. 2008;33:1–12. doi: 10.1247/csf.07011. [DOI] [PubMed] [Google Scholar]
- 16.Merzlyak E.M., Goedhart J., Shcherbo D., Bulina M.E., Shcheglov A.S., Fradkov A.F., Gaintzeva A., Lukyanov K.A., Lukyanov S., Gadella T.W.J., Chudakov D.M. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- 17.Stavoe A.K., Gopal P.P., Gubas A., Tooze S.A., Holzbaur E.L. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. Elife. 2019;8 doi: 10.7554/eLife.44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proikas-Cezanne T., Robenek H. Freeze-fracture replica immunolabeling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J. Cell Mol. Med. 2011;15:2007–2010. doi: 10.1111/j.1582-4934.2011.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu L., McPhee C.K., Zheng L., Mardones G.A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F., et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen T.N., Padman B.S., Usher J., Oorschot V., Ramm G., Lazarou M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016;215:857–874. doi: 10.1083/jcb.201607039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuboyama K., Koyama-Honda I., Sakamaki Y., Koike M., Morishita H., Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. doi: 10.1126/science.aaf6136. [DOI] [PubMed] [Google Scholar]
- 22.He C., Bartholomew C.R., Zhou W., Klionsky D.J. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy. 2009;5:520–526. doi: 10.4161/auto.5.4.7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varga M., Sass M., Papp D., Takács-Vellai K., Kobolak J., Dinnyés A., Klionsky D.J., Vellai T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ. 2014;21:547–556. doi: 10.1038/cdd.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu S., Kanaseki T., Mizushima N., Mizuta T., Arakawa-Kobayashi S., Thompson C.B., Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 25.Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T., et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McAlpine F., Williamson L.E., Tooze S.A., Chan E.Y.W. Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinase 1 and 2. Autophagy. 2013;9:361–373. doi: 10.4161/auto.23066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asaoka Y., Hata S., Namae M., Furutani-Seiki M., Nishina H. The Hippo pathway controls a switch between retinal progenitor cell proliferation and photoreceptor cell differentiation in zebrafish. PLoS One. 2014;9 doi: 10.1371/journal.pone.0097365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torii S., Yoshida T., Arakawa S., Honda S., Nakanishi A., Shimizu S. Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep. 2016;17:1552–1564. doi: 10.15252/embr.201642565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tashiro S., Kakimoto Y., Shinmyo M., Fujimoto S., Tamura Y. Improved split-GFP systems for visualizing organelle contact sites in yeast and human cells. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.571388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi Y., Tsutsumi Y., Monta O., Ohashi H., Fox T.E., Kester M., Loughran T.P., Wang H.G. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7:61–73. doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poss K.D., Shen J., Nechiporuk A., McMahon G., Thisse B., Thisse C., Keating M.T. Roles for Fgf signaling during zebrafish fin regeneration. Dev. Biol. 2000;222:347–358. doi: 10.1006/dbio.2000.9722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Microscopy images and original western blot images represented in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.