

Abstract
Adipose tissue signals to brain, liver and muscles to control whole body metabolism through secreted lipid and protein factors as well as neurotransmission, but the mechanisms involved are incompletely understood. Adipocytes sequester triglyceride in fed conditions stimulated by insulin, while in fasting catecholamines trigger triglyceride hydrolysis, releasing glycerol and fatty acids. These antagonistic hormone actions result in part from insulin’s ability to inhibit cAMP levels generated through such G-protein-coupled receptors as catecholamine-activated β-adrenergic receptors. Consistent with these antagonistic signaling modes, acute actions of catecholamines cause insulin resistance. Yet, paradoxically, chronically activating adipocytes by catecholamines cause increased glucose tolerance, as does insulin. Recent results have helped to unravel this conundrum by revealing enhanced complexities of these hormones’ signaling networks, including identification of unexpected common signaling nodes between these canonically antagonistic hormones.
Keywords: Adipose tissue, lipolysis, thermogenesis, lipogenesis, type 2 diabetes
Insulin and cAMP signaling in adipocytes impacts type 2 diabetes
Among the most powerful and broadly relevant cellular signaling pathways in biology are those for insulin[1] and cAMP[2, 3]. Virtually every mammalian cell type responds to insulin or related insulin-like peptides and to catecholamines (see Glossary) or other ligands that activate formation of cAMP. Indeed, even the secretion of insulin itself from pancreatic beta cells, critical for human life, is modulated through the cAMP signaling pathway[4]. Nobel Prizes have further highlighted the importance of these signaling systems, awarded for the discoveries of insulin (1923, to Banting and McCleod), epinephrine as neurotransmitter (1936, Dale and Loewi), and cAMP (1964, Sutherland). Remarkably, additional Nobels have continued to flow in this field, including for achieving the sequence (1958, Sanger) and crystal structure (1964, Hodgkin) of insulin, identification of G proteins that connect receptor activation to adenylate cyclase to catalyze cAMP formation (1994, Rodbell and Gilman) and elucidation of the structure of such receptors (2021, Lefkowitz and Kobilka). In addition to bringing fame to these scientists, this recognition has sparked further intense research activity on insulin and cAMP signaling pathways, as they regulate virtually every organ and cell type, including bacteria in the case of cAMP. Arguably, no other signaling pathways in biology have garnered this level of attention over so many decades.
The adipocyte has similarly gained center stage in the field of metabolic regulation and its dysfunctions in obesity and type 2 diabetes (T2D). The key role of adipose tissue as a master regulator of systemic metabolism is underscored by the fact that both increased adiposity in obesity and decreased adiposity in lipodystrophies similarly cause T2D in mice and humans[5–7]. Conversely, implantations of white adipose tissue reverse diabetes in lipodystrophic rodents[8], while implantations of thermogenic brown adipose tissues reverse diabetes in obese mice[9]. Insulin and cAMP signaling within adipocytes play important roles in these remarkable effects of adipose tissue. Insulin stimulates fat storage through increasing net glucose flux into the pentose shunt and triglyceride in adipocytes, which cAMP signaling inhibits. Catecholamines through cAMP signaling stimulate lipolysis (Box 1), glycogenolysis and fatty acid oxidation, which insulin action inhibits. These antagonisms are thought to largely result from the ability of insulin to attenuate the production of cAMP, although downstream effects are likely also at play. Conversely, cAMP antagonism of insulin action in adipocytes may arise from the ability of the cAMP-dependent protein kinase (PKA) to phosphorylate and inhibit insulin-induced insulin receptor substrate (IRS) signaling and protein kinase B (Akt) activation[10], as well as at the level of downstream targets such as glycogen synthase[11].
Box 1. Mechanisms of adipocyte lipolysis regulation.
During fasting conditions, fatty acid (FA) mobilization and release from TG in white adipocyte lipid droplets (LD) in adipocytes is a fundamental process that supplies fuel to other organs in the body. This process is regulated by norepinephrine (NE) released from adipose sympathetic nerve fibers during fasting or adaptive thermogenesis induced by cold temperature. The LD lipases sequentially degrade TG into FA and glycerol in an orderly fashion: ATGL/PNPLA2 catalyzes hydrolysis of TG to diacylglycerol (DAG) and FA, hormone sensitive lipase (HSL) degrades DAG to monoacylglycerol (MAG) and FA, and MAG lipase (MGL) catalyzes degradation of MAG to glycerol and FA. Multiple LD proteins participate in this process to ensure high efficiency, each exerting a different function. In the fed state in which lipolysis is suppressed by insulin, HSL resides in the cytosol and ATGL/PNPLA2 is bound by the protein inhibitor G0GS2. In this state, perilipin (PLIN1) binds the ATGL activator CGI-58/ABHD5, further preventing activation of ATGL/PNPLA2. Upon NE release in fasting, cAMP stimulates PKA-mediated PLIN1 phosphorylation, releasing CGI-58/ABHD5 to interact with and stimulate ATGL/PNPLA2 activity, initiating TG degradation into diacylglycerol (DAG). Importantly, PKA phosphorylation of PLIN1 also promotes translocation of phosphorylated HSL from the cytosol onto the LD surface, facilitating access of this enzyme to DAGs and their degradation into MAGs and then to glycerol and FA catalyzed by MGL [15].
Beyond these lipolytic enzymes and proteins essential for TG lipolysis, several other LD proteins have been recently identified. Some of these LD proteins have structural functions and exert crucial roles in LD biogenesis from the ER. Others are implicated in cellular dynamics, interactions between LD with other organelles [104–106] and control of mature LD growth. Among these LD proteins, two related proteins that belong to the CIDE (cell death-inducing DFF45-like effector) family, CIDEA and CIDEC, promote LD fusion and enlargement in adipocytes [107, 108]. Accordingly, depletion of CIDE proteins results in numerous small lipid droplets, while their overexpression increases the number of large lipid droplets. Thus, the CIDE proteins are important LD components controlling their size and have emerged as important metabolic regulators.
To date, several human metabolic conditions have been associated with molecular defects in LD components and lipolysis [109, 110]. Some of these pathological conditions are obesity, T2D, non-alcoholic fatty liver disease (NAFLD), lipodystrophy and neutral-lipid storage disease (NLSD). A better understanding of the underlying mechanisms of lipolysis regulation at the level of lipid droplets will likely lead to strategies for treatments of metabolic disease.
Consistent with the antagonistic effects of insulin and cAMP signaling in adipocytes, stimulations of β-adrenergic receptors in fasting are associated with insulin resistance [12]. However, sustained elevations of cAMP signaling specifically within adipocytes, like insulin, is able to improve metabolic health in obese mice[13, 14]. Here we review recent findings showing that, in parallel with the well-known antagonisms between insulin and cAMP, their individual signaling networks also unexpectedly converge at several nodes, including mTORC1, Acly and Chrebp, and they both robustly stimulate palmitate synthesis from glucose (denoted as de novo lipogenesis, DNL) (Box 2). These insights may help to explain how these two normally antagonistic signaling networks in adipocytes can act in concert to alleviate the metabolic derangements in type 2 diabetes (T2D) in obesity.
Box 2. Functions of adipocyte de novo lipogenesis (DNL).
Adipocytes store TGs and other lipid components in lipid droplets containing proteins on their surface that control their functions. FAs in adipocyte TGs derive from two sources: circulating lipoproteins, which account for the vast majority of adipocyte TGs and DNL, which normally accounts for less than 2% of the lipid droplet TG [111], although estimates vary depending on species, diet, and adipose depot. In response to insulin, lipoprotein lipase in adipose tissue hydrolyzes circulating TGs in lipoproteins into free FAs and glycerol that enter adipocytes and are converted to TG [112].
DNL converts carbohydrates, amino acids, and acetate into fatty acids through the sequential activities of enzymes that lead to acetyl CoA synthesis. Carbohydrates or amino acids are metabolized within mitochondria to citrate, which is exported to the cytosol where it is cleaved into oxaloacetate and acetyl-CoA catalyzed by Acly. In an alternate pathway, cytosolic acetyl-CoA is generated by the activation of acetate by acetyl-CoA synthetase 2 (Acss2). Acetyl-CoA is then carboxylated to form malonyl-CoA by the catalytic activity of Acc1. Lastly, Fasn forms a new acyl chain by the sequential elongation of acetyl-CoA with malonyl-CoA, ultimately producing palmitic acid (C16:0) [113]. Palmitic acid itself and its elongation and desaturation products can be stored in TGs or utilized in other ways.
In recent years, it’s been appreciated that DNL serves multifaceted functions in adipocytes [113, 114]. In addition to converting excess nutrients into fatty acids for storage as TGs, adipocyte DNL may be an important source of bioactive lipids, such as palmitoleate and fatty acid esters of hydroxy–fatty acids (FAHFAs), which are reported to exhibit insulin-sensitizing effects [115, 116]. DNL may also be a source of ligands for transcription factors, such as the PPAR family that is important for adipocyte lineage determination and function [117]. Emerging studies also suggest adipocyte DNL supplies an important source of fatty acids for membrane phospholipds[118]. Recent research has revealed the requirement of Fasn for expansion of autophagosome membranes (Rowland et al, unpublished). Therefore, while fatty acid uptake from lipoproteins in the circulation serves mainly for TG storage, it appears that adipocyte DNL additionally supports more specialized functions related to supplying membrane lipids.
Regulators of cAMP signaling in adipocytes
Two major downstream actions of cAMP signaling in adipocytes are the acute stimulation of triglyceride hydrolysis (denoted lipolysis)[15] and the chronic stimulation of thermogenesis with upregulation of the mitochondrial uncoupling protein 1 (UCP1), mitochondrial biogenesis and other thermogenic mechanisms[16, 17] (Figure 1). While adipocytes are highly heterogeneous, they are generally referred to as “white” when they exhibit a single large lipid droplet and high capacity for fat storage versus “brown” or “beige” when containing multiple smaller lipid droplets and thermogenic gene products. The central pathway for catecholamines to stimulate these two outputs is through the activation of cAMP generation and stimulation of PKA activity to phosphorylate multiple targets that mediate lipolysis, e.g., phospho-hormone sensitive lipase (p-HSL) and phospho-perilipin (p-PLIN1), and thermogenic gene regulation, e.g., phospho-protein kinase p38 (p-p38), phospho-mechanistic target of rapamycin complex 1 (p-mTORC1) and phospho-cyclic adenosine monophosphate response element binding protein (p-CREB)[18, 19] (Figure 1). This pathway to generate cAMP and activate PKA includes other downstream elements not shown in Figure 1 for simplicity (also see Figure 4), and cAMP has other effectors in addition to PKA [2, 20]. The physiological impact of white adipocyte lipolysis stimulation is the release of fatty acids to the circulation for uptake by other tissues, while the result of chronic thermogenic stimulation is the oxidation of fatty acids within the brown adipocytes producing heat to maintain body temperature in the cold. In addition, both white and brown adipocytes secrete bioactive lipids and protein factors that can act in autocrine, paracrine and endocrine modes[21–24].
Figure 1. Major regulators of cAMP signaling in adipocytes.
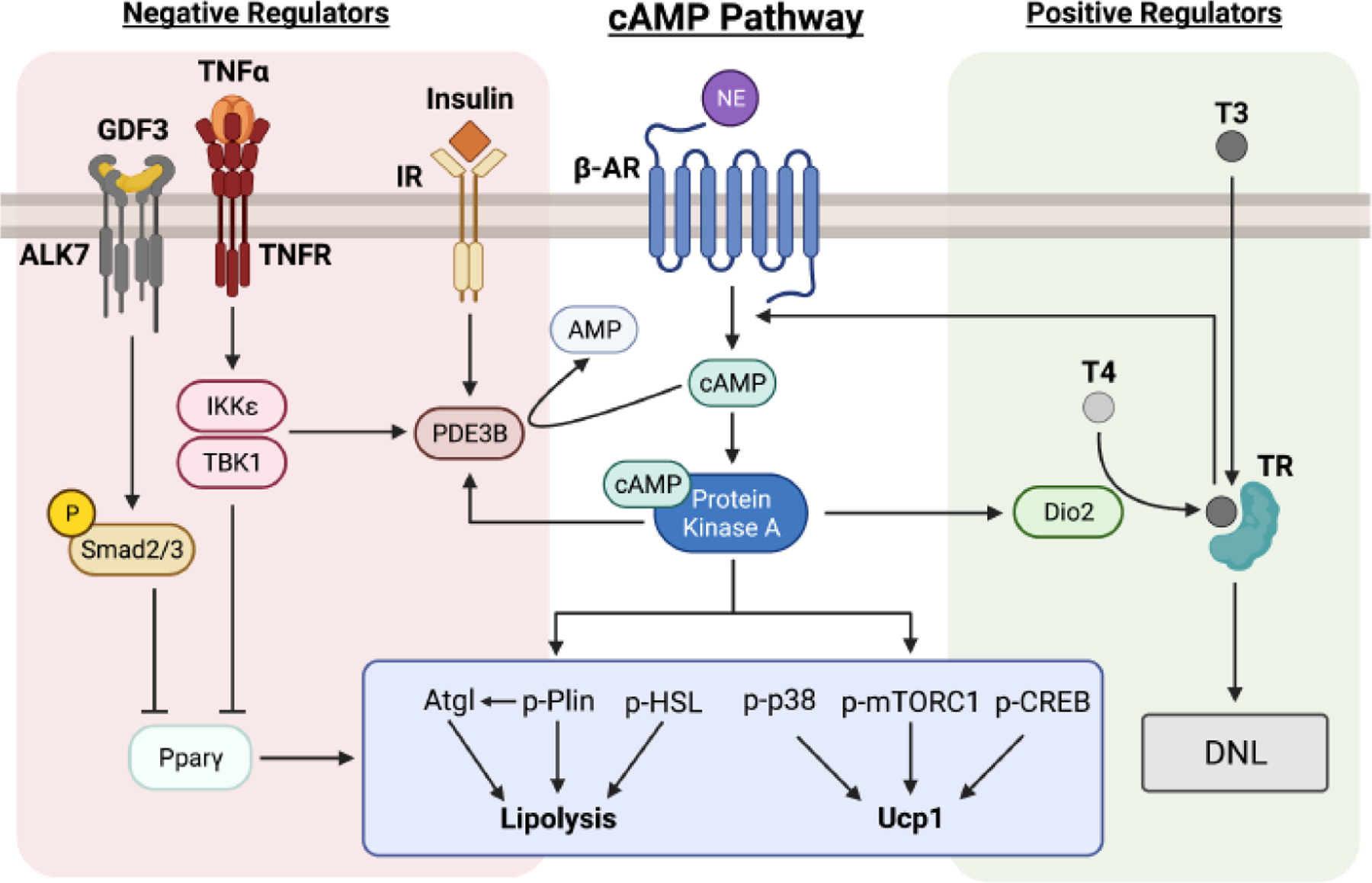
The central cAMP pathway plays a key role in adipocyte lipolysis and thermogenesis. Binding of norepinephrine (NE) to beta-adrenergic receptors (β-AR) induces a rise in cAMP and the sequential stimulation of protein kinase A (PKA), which in relation to lipolysis phosphorylates perilipin (p-Plin) and hormone sensitive lipase (p-HSL). p-Plin releases comparative gene identity-58 (CGI-58, not shown) from lipid droplets to activate adipose triglyceride lipase (ATGL) in cytosol (see Text Box 1). Activated PKA also increases transcription of mitochondrial uncoupling protein 1 (Ucp1), which is stimulated by the PKA-mediated phosphorylation of p38 mitogen-activated protein kinase (p-p38), mechanistic target of rapamycin (mTOR) signaling complex 1 (p-mTORC1), and cAMP response element-binding protein (p-CREB). See refs 2, 3, 18 and 19 for more detailed descriptions of this central pathway, including other elements downstream of cAMP not shown here such as cAMP-dependent exchange factor directly activated by cAMP (Epac1), required for regulation of glucose transport in brown adipocytes (see Figure 4). Major pathways that regulate cAMP levels or its downstream signaling are shown: PKA elevates type 2 iodothyronine deiodinase (Dio2) expression, which catalyzes formation of triiodothyronine (T3) from thyroxine (T4), resulting in thyroid hormone receptor (TR) activation and induction of de novo lipogenesis (DNL). T3 potentiates the cAMP pathway by increasing β-AR expression and coupling to cAMP production through G-coupled proteins and adenylate cyclase (not shown), thereby enhancing cAMP signaling to both thermogenesis and lipolysis. Conversely, PKA activates the cAMP-degrading enzyme phosphodiesterase 3B (PDE3B), which catalyzes the hydrolysis of cAMP to adenosine monophosphate (AMP), as a feedback mechanism. PDE3B is also activated by insulin as well as the tumor necrosis factor (TNF-α) mediated elevation of protein kinases IKKε and TBK1, rendering PDE3B and other phosphodiesterase (PDE) isoforms (not shown) as key nodes of cAMP degradation. The TGF-β superfamily receptor activin receptor-like kinase (ALK7) acts as another negative regulator of cAMP signaling through phosphorylation of Smad2/3, which leads to downregulation of peroxisome proliferator activated receptor gamma (PPARγ and lipolysis, as does TNF-α. This network of cAMP regulators in adipocytes determines the fine tuning of cAMP signaling, controlling major adipocyte functions of lipolysis, thermogenesis and de novo lipogenesis. This figure was created using BioRender (https://biorender.com/).
Figure 4. Convergent signaling nodes in adipocytes regulated by both cAMP and insulin.
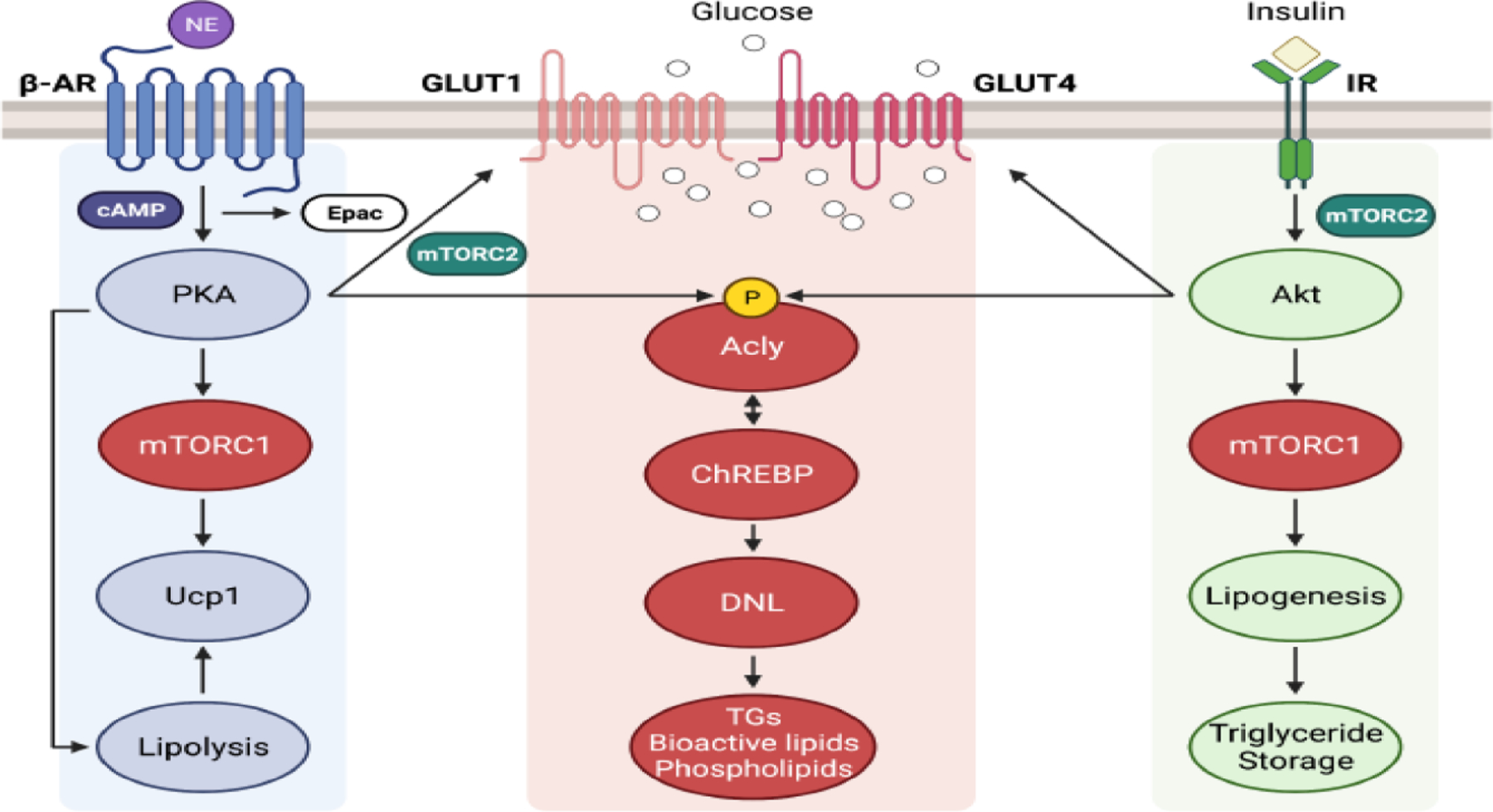
Despite the classical insulin antagonisms of cAMP-stimulated lipolysis and thermogenesis, several adipocyte signaling nodes and pathways are similarly regulated by both insulin and cAMP. These common elements may contribute to the fact that in adipocytes both insulin signaling and chronic elevations of cAMP through activation of adipocyte β–adrenergic receptors or other means both similarly improve systemic glucose tolerance. These common adipocyte signaling nodes for insulin and cAMP are: 1.) mechanistic target of rapamycin complex 1 (mTORC1), which activates the S6 protein kinase and perhaps other common downstream mTORC1 targets by both cAMP and insulin, 2.) mTORC2 in BAT, which is required for stimulation of glucose transport through Glut1, as insulin is able to do through Glut4 regulation, 3.) ATP citrate lyase (Acly) phosphorylation by insulin-mediated Akt activation at the same site as cAMP through activation of protein kinase A, and 4.) Carbohydrate response element binding protein (Chrebp)α and Chrebpβ expression, at least in part through the action of Acly to increase acetyl-CoA and histone acetylation, which regulates many genes related to glucose and lipid metabolism. In addition, the adipocyte de novo lipogenesis pathway is activated by both insulin and β3–adrenergic receptors in vivo, the latter effect requiring triiodothyronine (T3). How these common effects on adipocytes may contribute to increased glucose tolerance are described in the text and are still under investigation. This figure was created using BioRender (https://biorender.com/).
An important feature of this system is the ability of PKA to activate the cAMP degrading enzyme phosphodiesterase 3B (PDE3B) by phosphorylation at Serine 302 (S302), which promotes conversion of cAMP to AMP and promotes feedback inhibition of the overall process [25] (Figure 1). This same site on PDE3B is phosphorylated in response to insulin through the action of the protein kinase Akt, which may be a mechanism for inhibition of lipolysis (Figure 2). Insulin may also antagonize the thermogenesis pathway by this same mechanism since in mice genetic depletion of insulin leads to increased adipose browning of white adipose tissue (WAT)[26]. However, this conjecture needs to be validated by experiments performed in cells in vitro, as the work on mice could also involve effects of insulin on sympathetic flow. The PDE3B node of cAMP degradation and regulation may be further extended to the inflammation pathway in obesity that causes “catecholamine resistance” through phosphorylation of serine 318 (S318) in which adipose tissue undergoes immune cell expansion and secretion of cytokines[27]. Consistent with this concept, blockade of the noncanonical nuclear enhancer of kappa light chain polypeptide gene enhancer in B cells inhibitor (IkB) protein kinases IKKε and TBK1 reversed the attenuation of catecholamine stimulated lipolysis by tumor necrosis factor alpha (TNFα)[27]. Overall, PDE3B phosphorylation and activation, as well as expression changes of additional phosphodiesterase (PDE) isoforms in response to cAMP pathway modulators, appear to be a major general mechanism of cAMP antagonism in biology.
Figure 2. Chronic modulation of insulin signaling strength by gene knockouts selectively in adipocytes drives glucose tolerance in the same direction.
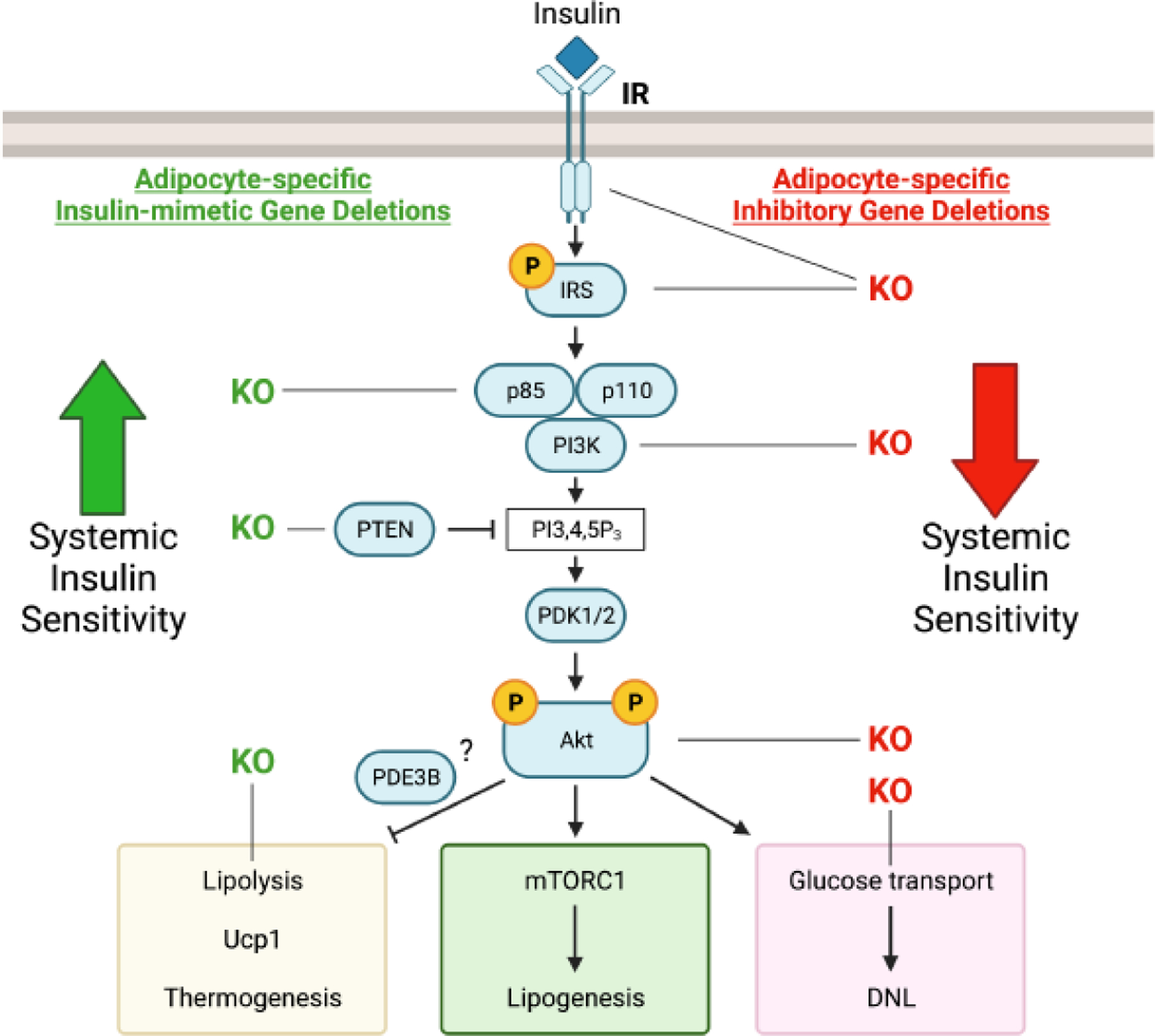
Insulin signaling to metabolic outputs is primarily driven by the phosphpatidlyinositol 3 kinase (PI3K) pathway that leads to activation of the protein kinase Akt. Adipose selective gene knockouts of the insulin receptor (IR) tyrosine kinase, insulin receptor substrate (IRS), catalytic subunit of PI3K p110, Akt, or glucose transporter (Glut4) that reduce insulin actions within adipocytes all cause decreases in whole body glucose tolerance. Conversely, gene knockouts of the negative regulators of insulin signaling, p85 subunit of PI-3kinase or phosphatase and tensin homolog (PTEN), enhance activation of Akt and cause increased systemic glucose tolerance. Likewise, mimicking the inhibition of lipolysis by insulin by knockout of the lipase ATGL also stimulates glucose tolerance in mice. See references 1 and 5 for more details of insulin signaling and its resistance in obesity and type 2 diabetes. This figure was created using BioRender (https://biorender.com/).
While the central pathway of cAMP generation and signaling is well described, many signaling pathways that amplify or dampen cAMP actions have recently been recognized to have profound effects on adipocyte function (Figure 1). A major positive regulator of cAMP signaling is thyroid hormone, mediated by the T3 form of the hormone and thyroid hormone receptors operating in the nucleus to regulate transcription [28–31]. T3 potentiates the cAMP signaling pathway in adipocytes by increasing β3-adrenergic receptor expression and its coupling to adenylate cyclase through protein Gαs [32–35]. Even though T3 enhances thermogenesis in white and brown adipocytes on its own when administered in vivo, its full effects are dependent on cAMP signaling, as deletion of Gαs prevents upregulation of UCP1 by T3 injection[35]. T3 is also required for optimal cAMP stimulation of de novo lipogenesis (DNL) (Figure 1 and Box 2), a process that insulin also activates. Thus, cAMP through PKA activation enhances expression of iodothyronine deiodinase (Dio2) to convert circulating T4 to the active T3[36, 37], which in turn exerts transcriptional stimulation of DNL enzymes Fasn, Acc1 and Acly through the action of nuclear thyroid hormone receptor. Accordingly, DNL upregulation by β3-adrenergic receptor signaling is blocked in hypothyroid mice[35], indicating that T3 is required for this cAMP effect. It should be noted that T3 also acts centrally to increase sympathetic activity in peripheral tissues, which amplifies the effects described above.
Glucocorticoids have also been shown to potentiate catecholamine signaling on adipocyte lipolysis through nuclear glucocorticoid receptor activation[38], and the KO of glucocorticoid receptor in adipocytes in mice blocks the release of glycerol and free fatty acids in response to injection of isoproterenol[39]. However, the effects of glucocorticoids to enhance cAMP signaling seem restricted to the lipolytic pathway in white adipocytes, likely through upregulation of the adipose triglyceride lipase (ATGL, also known as patatin-like phospholipase domain-containing protein 2 lipase), since their effects on thermogenesis are complex and can actually lead to “whitening” of BAT. In addition to the action of glucocorticoid to enhance catecholamine induced lipolysis, a major effect of glucocorticoids at early stages of adipocyte differentiation is well established, and recent data suggest this effect involves the transcription factor heart and neural crest derivatives-expressed protein 2 (HAND2)[40].
Two major negative regulators of cAMP signaling in adipose tissues are mediated by the type 1 transforming growth factor receptor activin receptor-like kinase 7 (ALK7) [41] and inflammation pathways exemplified by TNFα action[42] (Figure 1). ALK7 is highly expressed in adipocytes, and its expression is decreased in obesity. A key ligand for ALK7 is growth differentiation factor 3 (GDF3), which is also produced by adipocytes and acts in an autocrine loop to attenuate the ability of cAMP signaling to stimulate lipolysis[43]. In brown adipocytes, ALK7 expression is under the control of cGMP, which acts through protein kinase G, analogous to the cAMP/PKA system[44], and is highly upregulated during differentiation. However, similar to its effect in white adipocytes, ALK7 activation in brown adipocytes decreases expression of ATGL and lipolysis. Indeed, Alk7 knock-out mice showed enhanced adipose β-adrenergic receptor expression, mitochondrial biogenesis, and lipolysis, leading to elevated energy expenditure and resistance to diet-induced obesity [45]. Mechanistically, ALK7 acts also through phosphorylation of Smad2/3, which, in adipocytes, leads to downregulation of peroxisome proliferator activated receptor gamma (PPARγ) via modulating CCAAT enhancer binding protein alpha (CEBPα[46, 47]. TNFα, which in part mediates the catecholamine resistance caused by adipose tissue inflammation in obesity, also acts to attenuate PPARγ expression[42]. PPARγ in turn drives synthesis of gene products that mediate normal key adipocyte functions, including both lipogenesis and lipolysis. TNFα also acts to diminish β3-adrenergic receptor expression through upregulation of TRIB1, which degrades transcription factors that increase β3-adrenergic receptors[48]. Thus, modulating effects of both ALK7 and TNFα on adipocyte cAMP signaling appear to converge at the downstream target PPARγ (Figure 1).
Antagonism of insulin and cAMP signaling in adipocytes
The insulin signaling pathway is largely driven by the tyrosine kinase activity of the insulin receptor, which has multiple substrates that act as anchors for signaling proteins to activate mitogen activated protein kinase (MAP) kinase pathways and phosphoinositide (PI)-3kinase pathways[1]. It is generally recognized that the relevant metabolic actions of insulin in adipocytes are mostly driven by the PI-3kinase pathway that generates the intermediate PI-3,4,5-P3, the mobilization of 3-PI-dependent kinase (PDK)1 and PDK2 (mTORC2), and their activation of the protein kinase Akt (Figure 2). This insulin signaling pathway antagonizes multiple metabolic effects caused by cAMP, including those on glycogen synthase and glycogen phosphorylase, protein synthesis, and lipolysis. These antagonisms are associated with decreases in cAMP levels by insulin, but the full mechanisms are likely to be more complex and different in each case.
The inhibition of lipolysis by insulin exemplifies the complexities of its mechanisms that antagonize cAMP signaling. Akt catalyzes phosphorylation and activation of PDE at S302, as does PKA itself, which would be expected to lower cAMP levels. Yet, total cAMP levels are not always lowered by insulin under such conditions in adipocytes when lipolysis is inhibited [49], suggesting the possibility that local cAMP concentrations or cAMP-independent pathways may be operating. Furthermore, expression in PDE3B depleted cells of a mutant form of PDE3B in which the Akt-directed site S302 is substituted for alanine did not diminish insulin action on lipolysis [50]. However, knockout of PDE3B in adipocytes does block insulin action, indicating the PDE3B protein is indeed required for insulin’s inhibition on lipolysis. Nonetheless, the idea that PDE3B phosphodiesterase activity is needed for insulin to act is further complicated by apparently divergent results with the use of PDE3B inhibitors. Thus, investigators in one study blocked PDE activity or used cAMP derivatives that were not PDE substrates to stimulate lipolysis in white adipocytes, and still observed insulin-mediated inhibition of lipolysis [51].
Complementing the results above, complete inhibition of detectable Akt activity by small molecule agents that block insulin action on glucose transport do not block its antilipolytic effect [50]. Similarly, deletion of Akt2, the major isoform in adipocytes, also fails to block insulin’s ability to blunt lipolysis. Further complications arise from experiments suggesting that other isoforms such as the PDE4 proteins are critical for lipolysis regulation [52]. The actions of an apparent chaperone-type protein for PDE3B, denoted ABHD15, promotes its stability and seems to be an important regulator of lipolysis and perhaps insulin action [53]. One possible caveat of the experiments described above is the possibility that other as yet unidentified serine or threonine sites on PDE3B are important for insulin to inhibit lipolysis. However, it remains that the PDE3B phosphorylation mutant does not respond to insulin with increased phosphodiesterase activity, showing that its insulin responsiveness was indeed blocked by this mutation [51].
Taking these results together, it appears that the Akt-mediated phosphorylation site of PDE3B is not strictly required for insulin to attenuate adipocyte lipolysis, nor is Akt2 required, and yet PDE3B protein needs to be present. Importantly, these results raise a strong possibility that the antilipolytic action of insulin is not only driven by decreasing cAMP levels, but rather by antagonizing the effects of PKA activity at its downstream substrates. An example of this is the regulation of glycogen synthase, which is activated by insulin through modulation of phosphorylation of the enzyme through protein kinases and phosphatases independent of PKA [11]. Consistent with this concept is the surprising result that the expression of the PDE3B mutant that is unresponsive to Akt and insulin, still allowed insulin to decrease the PKA-mediated phosphorylation of HSL and PLIN1 along with the decrease in lipolysis [51]. Thus, the action of insulin at the level of these lipid droplet protein changes that occur in response to cAMP is likely where insulin signaling meets PKA signaling.
Adipocyte signaling to systemic metabolism
While insulin’s ability to lower blood glucose and increase glucose tolerance results from its combinatorial direct effects on blunting hepatic glucose output and increasing glucose uptake into skeletal and heart muscles, these effects are indirectly promoted by its actions on adipose tissue. For example, lowering adipocyte lipolysis reduces fatty acid availability and oxidation in these other tissues, increasing reliance on glucose utilization. Importantly, fatty acids derived from adipose tissues also stimulate gluconeogenesis in the liver, so lowering fatty acids decrease glucose output into the circulation [54–56]. Thus, chronic activation of the insulin signaling pathway selectively in adipocytes, induced by adipocyte-specific Cre-recombinase directed knockouts of suppressor elements of its signaling, strongly enhances glucose tolerance and systemic insulin sensitivity [57, 58] (Figure 2). This same result of increased glucose tolerance is observed by mimicking insulin’s inhibition of lipolysis in adipocytes by knockout of the ATGL lipase in mice [59]. Conversely, adipose-selective knockouts of key required elements of the insulin signaling pathway, such as the protein kinase Akt, cause systemic glucose intolerance and insulin resistance [60]. These deleterious effects are mimicked by knockout of a downstream target of insulin action in adipocytes—the GLUT4 glucose transporter [61] (Figure 2). Collectively, these results emphasize the important role played by adipocytes in the systemic actions of insulin related to metabolic homeostasis.
Although acute activation of adipocyte functions by catecholamines are generally antagonistic to insulin’s effects, paradoxically, chronic stimulation of the adipocyte cAMP pathway also enhances overall whole-body glucose tolerance. Thus, moderate cold exposure in humans, which activates sympathetic nerve activity in adipose depots, enhances insulin sensitivity without increasing circulating insulin levels [62]. Chronic activation of the β3 adrenergic receptor, which is relatively adipocyte-specific in its expression in mice [63–65] and humans [66–68] also improves glucose tolerance and insulin sensitivity. This intervention causes expansion and activation of brown adipose tissue uncoupled respiration and heat generation, as well as the appearance of brown or “beige” adipocytes within white adipose depots. This can occur in the absence of increases in circulating insulin (62). Beige adipocytes, which also express uncoupling protein UCP1, increased mitochondrial mass and engage in uncoupled respiration in response to cAMP, contribute to the improvement in metabolic health, as evidenced by knockout of transcription regulator PR-domain zinc finger protein 16 (PRMD16), which blocks both appearance of beige adipocytes and glucose tolerance in mice [69]. Furthermore, white adipose tissue browning can be driven by activation of the cAMP pathway in vivo as depletion of PDE3B, which is highly expressed in adipocytes, triggers browning of adipose tissue, increased oxygen consumption and weight loss in mice [70]. Thus, chronic activation of cAMP signaling in adipocytes leads to beneficial systemic metabolic effects that mirror the improvements in glucose tolerance induced by activated insulin signaling in adipocytes.
In spite of this convergence in physiological effects, many of the initial adipocyte events triggered in response to insulin action are quite distinct and even antagonistic from those initiated even by chronic cAMP signaling in adipocytes (Figures 1 and 2). Most strikingly, in addition to the antagonisms at the level of lipolysis noted above, the effects of chronic cAMP signaling on adipocyte browning also appear to be antagonized by insulin. Consistent with this concept, in experiments in mice whereby partial disruption of pancreatic insulin expression was induced by deletion of one allele of the Ins2 gene, increased appearance of beige adipocytes in white adipose tissue occurs [26]. This mutant insulin deficient mouse model also displayed increased energy expenditure, consistent with the expected increase in uncoupled respiration in beige adipocytes in white adipose tissue. Therefore, the mechanisms whereby insulin signaling in adipocytes enhances glucose tolerance does not involve the appearance of beige adipocytes that is associated with the improvement in glucose tolerance induced by chronic adipocyte cAMP signaling.
Convergent insulin and cAMP signals within adipocytes
Recent surprising evidence indicates that there are two levels of convergence of insulin and cAMP signaling within adipocytes that may help to explain the paradox of their similar systemic effects. The first level relates to newly appreciated pathways of convergence within the adipocyte signaling supersystem. As shown in Figure 3, there is a small but significant subset of genes that are modulated similarly by both cAMP and insulin action [71, 72], even though expression of most adipocyte genes are regulated in opposite fashion by these agents. Remarkably, 4 of the 9 genes (Lpin1, Pck1, Ppargc1a, Gpr35) that are increased by either acute insulin or cAMP stimulation are positively associated with systemic insulin sensitivity and glucose tolerance (Figure 3). Lpin1 (Lipin1) overexpression in adipose tissue increases body weight and adiposity upon high fat feeding but preserves glucose levels and insulin sensitivity [73]. Similarly, Pck1 (Pepck) overexpression in adipose tissue increases adiposity without worsening of glucose tolerance and tends to improve insulin sensitivity [74]. While the effects of Lpin1 and Pck1 appear to result from an improved ability of adipose tissue to sequester fatty acids or carbohydrates, the effects of Gpr35 and Ppargc1a are largely due to enhanced adipose tissue thermogenesis. Ppargc1a is a well-established regulator of mitochondrial biogenesis and thermogenesis in brown and white adipose tissue, and adipose specific knockout of Ppargc1a worsens glucose tolerance and insulin sensitivity of obese mice, despite similar body weight gain [75, 76]. Whole body Gpr35 knockout mice develop glucose intolerance on a chow diet, preceding weight gain. These effects are attributed to adipose tissue Gpr35 expression, as Gpr35 activation in adipose tissue enhances thermogenic gene expression and cellular respiration [77]. Thus, the cAMP and insulin signaling pathways may contribute to improved metabolic health through a common set of regulated genes in adipose tissue.
Figure 3. Significant number of genes showing concordant regulation by both insulin and cAMP.
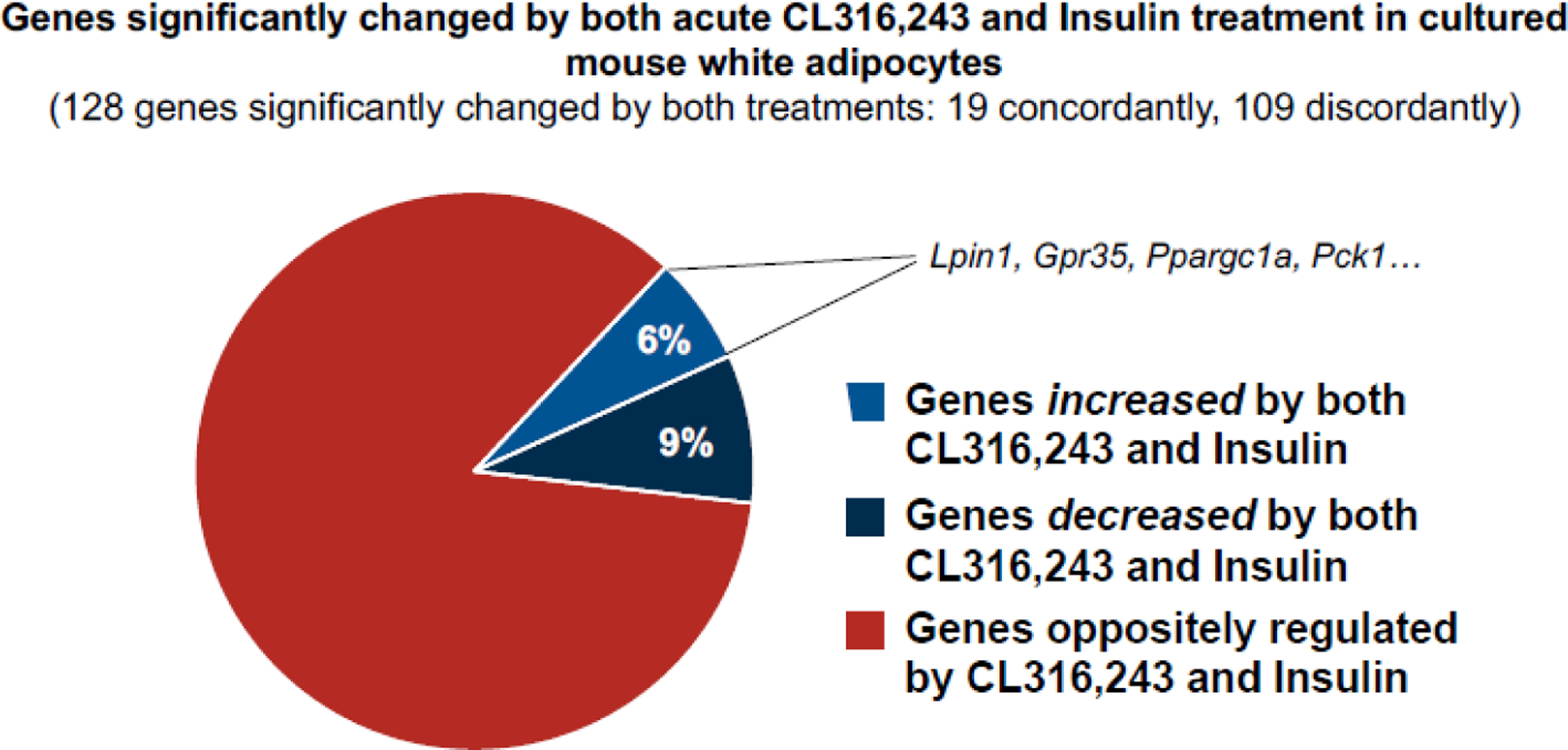
Most genes that are regulated by insulin versus cAMP display opposite expression response profiles. To determine if there are commonly regulated genes, we mined published RNA sequencing datasets from acute insulin stimulation or acute β3-adrenergic stimulation of differentiated white adipocytes cultured in vitro. Preadipocytes from subcutaneous white adipose tissue were induced to differentiate, then treated with 1.72 μM insulin for 8 hours [71] or 10 μM CL316,243, a β3-adrenergic agonist, for 3 hours [72]. Significantly changed gene expressions were defined by the authors. Overall, 128 genes were significantly changed by these treatments, and as expected, most genes (85%) were regulated discordantly by the two treatments. However, 15% (6% upregulated and 9% downregulated) were regulated similarly by insulin and CL316,243. Of note, the expression of 4 of these commonly upregulated genes (Lpin1, Gpr35, Ppargc1a, Pck1) in adipose tissue has been shown to promote glucose homeostasis, insulin sensitivity, and/or metabolic health. This figure was created using BioRender (https://biorender.com/).
Another level of convergence of cAMP and insulin pathways seems to occur at the mTORC1 complex. A first reported indication of this idea was the finding that β-adrenergic stimulation of cAMP signaling causes activation of mTORC1 and its downstream protein kinase (S6 kinase) [78, 79], targets that are classically activated by insulin (Figure 4). These data showed that cAMP acts through PKA to directly phosphorylate mTOR and its regulator regulatory-associated protein of mTOR (RAPTOR), and that blockade of mTORC1 signaling to the S6 protein kinase by rapamycin attenuated the thermogenic response to cold exposure or β3-adrenergic stimulation in vivo. Also, mTOR is involved in mediating cAMP-stimulated glucose uptake in beige and brown adipocytes, which is independent of its effect on UCP1 upregulation[80, 81]. Thus, activation of the mTORC1 signaling node plays a central role in both cAMP and insulin signaling in adipocytes. However, still a mystery is how the outcomes of mTORC1 modulation by these two pathways leads to different adipocyte outcomes, thermogenesis and glucose uptake for cAMP versus lipogenesis for insulin. Presumably, mTORC1 acts in conjunction with other pathways that are selectively modulated by insulin versus cAMP to explain its requirement for the divergent outcomes.
A third recently uncovered signaling node within adipocytes that appears common to both insulin and cAMP signaling is the enzyme ATP citrate lyase (Acly), which initiates the de novo lipogenesis pathway by converting citrate to acetyl-CoA [82]. Curiously, Acly is phosphorylated in response to both insulin and adrenergic agonists at the same site, activating its catalytic activity [83–86]. While this phenomenon was recognized long ago, only more recently has its potential physiological significance been unveiled. Specifically, adipocyte Acly was shown to be required for optimal glycolysis-induced activation of carbohydrate-responsive element-binding protein (Chrebp) [87], a major transcriptional regulator of glucose and fatty acid metabolism-related genes that promotes lipid storage [88, 89]. This Acly-Chrebp axis has been further characterized in BAT to reveal a pathway whereby Akt-mediated phosphorylation of Acly enhances acetyl-CoA generation to activate Chrebp through increased histone acetylation [90]. The increased Chrebp drives glucose transporter and lipogenesis gene transcription to promote synthesis of palmitate as well as esterification of fatty acids into triglyceride [90]. This scenario fits well with the fact that cAMP signaling, like insulin, also enhances DNL, providing a plausible hypothesis of how this could be mediated in part through phosphorylation and activation of Acly. Importantly, this concept illustrated in Figure 4 explains why rapamycin does not block cAMP-stimulated DNL even though it does attenuate induction of thermogenesis, as mTORC1 is downstream of the PKA activation of Acly.
To amplify this Acly-Chrebp-lipogenesis axis, glucose uptake is also similarly upregulated by insulin and cAMP in adipocytes. This is especially robust in thermogenic adipocytes in which expression of the GLUT1 transporter is responsive to cAMP and expression of the GLUT4 transporter is upregulated by insulin [91–93]. Since glucose transport is relatively rate limiting for glycolysis in adipocytes, this convergence of cAMP and insulin signaling similarly enhances levels of glycolytic intermediates that are thought to activate Chrebp transcriptional activity. Additional feed-forward activity in this pathway is represented by the increased transcription of glucose transporter GLUT4 protein promoted by Chrebp [90]. Glucose also serves as precursor for the acetyl-CoA that initiates the DNL pathway, serving as another way by which both cAMP and insulin enhance new palmitate synthesis in adipocytes. Yet another unexpected example of convergence of cAMP and insulin signaling with adipocytes is the recent finding that glycogen synthesis is enhanced by cAMP as it is by insulin[94], in contrast to other tissues in which the signals act in opposite fashion.
Signaling convergence at the systemic level
At a second, systemic level, how might these convergent pathways of adipocyte signaling by chronic cAMP and insulin contribute to metabolic health? One simple possibility is that the stimulatory effects of insulin or cAMP on glucose uptake in thermogenic adipocytes in the cold significantly contribute to increased disposal of circulating glucose and glucose tolerance. While skeletal muscle is the predominant tissue in utilizing glucose in response to insulin, BAT glucose uptake in cold exposed rodents is also highly significant [95, 96]. However, in humans the data available suggest a much smaller role of BAT in mediating glucose disposal, and quantifying this parameter is still under investigation [97].
Another way that chronic cAMP and insulin signaling in adipocytes may both contribute to enhanced systemic glucose tolerance is through their activation of adipocyte DNL. Palmitate synthesis is a source of bioactive lipids that are secreted for action at other tissues. Examples of such bioactive lipids are 12-hydroxyeicosapentaenoic acid (12-HEPE) [98] and 12, 13-dihydroxy-9Z-octadecenoic acid (diHOME) [99] that are secreted by BAT. Cold exposure elevates 12-HEPE in the circulation, while obesity in humans lowers its levels, in excellent correlation with systemic insulin sensitivity. Glucose uptake into BAT and skeletal muscle is stimulated by 12-HEPE, enhancing glucose tolerance [98]. Similarly, diHOME is released into the circulation in mice and humans during exercise, and acts to enhance skeletal [99] and heart [100, 101] fatty acid uptake and oxidation. The signaling mechanisms by which these physiological responses are mediated are, however, currently unknown. Importantly, in these examples, blockade of bioactive lipid synthesis causes deleterious systemic metabolic effects, providing strong support for their physiological relevance. Insulin and cAMP may also stimulate secretion of similar protein factors from adipocytes, as exemplified by adiponectin which enhances insulin sensitivity in liver and muscle [102]. In the case of adiponectin, the cAMP effect to stimulate secretion operates through the cAMP-dependent exchange factor directly activated by cAMP (Epac)1, but PKA-independent, pathway [103]. Collectively, these considerations suggest that a common way that insulin and chronic cAMP signaling may act systemically is through stimulation of similar beneficial secreted factors from adipocytes. However, more work is needed to determine whether insulin also increases the lipid mediators described above in relation to beige adipocytes and exercise.
Finally, a third way that adipocyte regulation by both insulin and chronic cAMP exposure may similarly regulate whole body metabolism is through reducing circulating fatty acids. In this case, the two signaling pathways achieve this end by divergent means. Insulin acts by inhibiting adipocyte lipolysis (Figures 1 and 2), while chronic activation of BAT and beige adipocytes by cAMP can increase fatty acid disposal through increased oxidation and thermogenesis. Fatty acids derived from WAT lipolysis drive fatty liver through esterification to triglyceride and increase hepatic glucose output through their oxidation to acetyl CoA, which activates mitochondrial pyruvate carboxylase to produce oxaloacetate [55, 56]. This metabolite is a starting point for metabolic flux to hepatic gluconeogenesis. High circulating fatty acid levels and perhaps and their oxidation products can also cause insulin resistance in skeletal muscle.
Concluding remarks
The aim of this review was to highlight a paradox in the field of metabolic regulation that has not yet been fully addressed. While the systemic metabolic effects of insulin and catecholamines in vivo result from the integration of many actions of these hormones on multiple tissues, adipocytes play a major role. For example, exposure to cold activates adipose nerve fibers that secrete catecholamines, which improves glucose tolerance, and chronic actions of catecholamine analogs that selectively activate β-adrenergic receptors in adipocytes promote insulin sensitivity. Yet, the thermogenic effects of such chronic catecholamine stimulation, including the appearance of Ucp1-expressing beige adipocytes are not shared by insulin action, even though the same end result of increased glucose tolerance is achieved when insulin action selectively in adipocytes is stimulated. These considerations raise a paramount question: are there hidden similarities between the very different phenotypes of insulin stimulated white adipocytes versus cAMP-activated beige adipocytes that contribute to the common beneficial effects on systemic metabolism?
Over the last few years new data has unexpectedly unveiled similar signaling nodes for insulin and cAMP within adipocytes and two commonly activated metabolic pathways, glucose transport and DNL. As reviewed in this article, both insulin and cAMP also activate the major cell regulator mTORC1 and its downstream target S6 protein kinase, and both signaling pathways phosphorylate and activate Acly, which can modulate global gene transcription through production of acetyl-CoA, the substrate for histone acetylation. Some data already implicate this epigenetic mechanism in elevating expression of Chrebp, a major transcription factor known to regulate adipocytes in a way that improves systemic glucose tolerance. Indeed, insulin and cAMP appear to surprisingly upregulate at least a few common genes that are also reported to modulate whole body metabolism. These and other new insights provide a framework for important future work designed to delve deeper into the above common signaling pathways. How might these signaling nodes cause release of bioactive factors that operate systemically or modulate nerve fibers in adipocytes for transmission to other tissues? Might such common pathways of adipocyte regulation be exploited for developing therapeutic strategies for metabolic disease? The foundation of data reviewed here also suggests that determining whether other signaling elements common to both insulin and cAMP signaling are lurking in adipocytes may well be worth deeper investigation (see Outstanding questions box).
Outstanding Questions.
Do insulin and cAMP signaling pathways that stimulate DNL lead to similar secreted bioactive lipids that regulate systemic metabolism?
To what extent are the adipocyte genes that are concordantly modulated by insulin and cAMP mediated by their similar actions on Chrebpα and Chrebpβ?
Are there other downstream events stimulated by insulin and cAMP that are dependent on their activation of Acly, and are these events solely triggered by acetyl-CoA-mediated epigenetic processes?
Are there divergent signaling outputs from mTORC1 in response to insulin versus cAMP?
To what extent do the common effects of cAMP and insulin in adipocytes contribute to the systemic effects that occur in normal physiological conditions such as HFD feeding?
Highlights.
White adipose tissue lipid storage is promoted by insulin and antagonized by catecholamines that elevate cAMP to stimulate lipolysis and thermogenic adipocytes. Despite these antagonisms, whole body glucose tolerance is similarly enhanced by both insulin and chronic catecholamine stimulation of adipocytes.
We explore the concept that unanticipated convergences of the insulin and cAMP signaling networks in adipocytes contribute to common systemic outcomes. Indeed, the genes Lpin1, Ppargc1a, Gpr35 and Pck1, all known to promote metabolic health, appear to be similarly regulated by both signaling pathways in adipocytes.
Additionally, common signaling nodes for these hormones within adipocytes have recently been revealed, including mTORC1, Acly and Chrebp. Common stimulatory actions of insulin and cAMP in adipocytes also include glucose transport, glycogen synthesis, de novo lipogenesis, and secretion of adiponectin which enhances glucose tolerance.
How such common elements of adipocyte regulation are able to modulate other tissues such as liver and skeletal muscle that control whole body glucose and lipid homeostasis continues to be a key question in the field.
Acknowledgements:
We thank Sheila Collins (Vanderbilt University) for critical reading of the manuscript and helpful comments. We also thank members of our laboratory for helpful discussions and to Kerri Miller for excellent assistance with the manuscript. Funding for our work related to this topic was provided by much appreciated funding from the National Institute of Diabetes and Digestive and Kidney Diseases at the NIH (DK116056, DK030898, DK103047, DK130852). Figures were created with BioRender.com.
Glossary
- Activin A Receptor Type 1C (ALK7)
a type I receptor for the TGFβ family of signaling molecules. In adipocytes, activation of ALK7 reduces β-adrenergic receptor-mediated signaling and lipolysis.
- Adipose triglyceride lipase (ATGL)
an enzyme that catalyzes hydrolysis of triacylglycerols to diacylglycerol and fatty acid.
- ATP citrate lyase (ACLY)
an enzyme that initiates the de novo lipogenesis pathway by converting citrate to acetyl-CoA.
- cAMP response element-binding protein (CREB)
a transcriptional regulator to modulate the transcription of genes with cAMP responsive elements in their promoters.
- Carbohydrate response element binding protein (ChREBP)
glycolytic metabolite activated transcription factors (ChREBPα and ChREBPβ that regulate glycolytic and lipogenic pathways.
- Catecholamines
hormones that are produced in nerve tissues and adrenal glands and function as neurotransmitters.
- Chow Diet
a standard control diet for rodents, generally providing only 7 to 12% of the total energy from fat
- Cyclic adenosine 3’, 5’-monophosphate (cAMP) signaling pathway
a G protein-coupled receptor-triggered signaling cascade in cell communication, which plays a central role in lipolysis and thermogenesis in adipocytes.
- De novo lipogenesis (DNL)
a pathway that converts carbons from nutrients into fatty acids, which are precursors for synthesizing triglycerides or other lipids.
- Deiodinase (Dio2)
an enzyme to convert thyroid hormone from thyroxine (T4) to active form triiodothyronine (T3).
- Fatty Acid Synthase (FASN)
the last enzyme in DNL, catalyzing synthesis of palmitic acid.
- Gs alpha (Gsα)
a subunit of the heterotrimeric G protein Gs that stimulates the cAMP-dependent pathway by activating adenylyl cyclase.
- Histone acetylation
a critical epigenetic modification that changes chromatin architecture and regulates gene expression by modulating chromatin structure.
- Hormone-Sensitive Lipase (HSL)
a rate-limiting enzyme, hydrolyzing diacylglycerol to monoacylglycerol and fatty acids.
- Lipodystrophy
syndromes with abnormal distribution of fat due to the loss of functional adipose tissue depots.
- Lipolysis
a process which breaks down triacylglycerols to glycerol and free fatty acids.
- mTORC1
known as mechanistic target of rapamycin complex 1, is a protein complex that functions as a nutrient/energy/redox sensor and controls protein and lipid synthesis.
- mTORC2
a rapamycin-insensitive protein complex formed by serine/threonine kinase mTOR that regulates cell proliferation and survival, cell migration and cytoskeletal remodeling.
- Phosphodiesterase (PDE)
enzymes that catalyze degradation of cyclic nucleotides cAMP and cGMP to AMP and GMP, respectively, reducing cAMP and cGMP signaling.
- Protein kinase A (PKA)
a family of enzymes whose activity is dependent on cellular levels of cyclic AMP (cAMP).
- Protein Kinase B (Akt)
a group of three insulin-activated serine/threonine-specific protein kinases that play key roles in multiple cellular processes.
- Uncoupling protein 1 (UCP1)
a unique mitochondrial inner-membrane uncoupling protein devoted to heat production (thermogenesis) in beige and brown adipocytes.
- Tumor Necrosis Factor alpha (TNF-α)
an inflammatory cytokine produced by macrophages/monocytes during acute inflammation, leading to necrosis or apoptosis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests: The authors declare no competing interests.
References
- 1.White MF and Kahn CR, Insulin action at a molecular level - 100 years of progress. Mol Metab, 2021. 52: p. 101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou G, et al. , Multifaceted Roles of cAMP Signaling in the Repair Process of Spinal Cord Injury and Related Combination Treatments. Front Mol Neurosci, 2022. 15: p. 808510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calamera G, et al. , Phosphodiesterases and Compartmentation of cAMP and cGMP Signaling in Regulation of Cardiac Contractility in Normal and Failing Hearts. Int J Mol Sci, 2022. 23(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tengholm A and Gylfe E, cAMP signalling in insulin and glucagon secretion. Diabetes Obes Metab, 2017. 19 Suppl 1: p. 42–53. [DOI] [PubMed] [Google Scholar]
- 5.Czech MP, Insulin action and resistance in obesity and type 2 diabetes. Nat Med, 2017. 23(7): p. 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein S, et al. , Why does obesity cause diabetes? Cell Metab, 2022. 34(1): p. 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim K, et al. , Lipodistrophy: a paradigm for understanding the consequences of “overloading” adipose tissue. Physiol Rev, 2021. 101(3): p. 907–993. [DOI] [PubMed] [Google Scholar]
- 8.Gavrilova O, et al. , Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest, 2000. 105(3): p. 271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanford KI, et al. , Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest, 2013. 123(1): p. 215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chai SP and Fong JC, Synergistic induction of insulin resistance by endothelin-1 and cAMP in 3T3-L1 adipocytes. Biochim Biophys Acta, 2015. 1852(10 Pt A): p. 2048–55. [DOI] [PubMed] [Google Scholar]
- 11.Roach PJ, et al. , Glycogen and its metabolism: some new developments and old themes. Biochem J, 2012. 441(3): p. 763–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergman BC, et al. , Effects of fasting on insulin action and glucose kinetics in lean and obese men and women. Am J Physiol Endocrinol Metab, 2007. 293(4): p. E1103–11. [DOI] [PubMed] [Google Scholar]
- 13.McKie GL, et al. , Intermittent cold exposure improves glucose homeostasis despite exacerbating diet-induced obesity in mice housed at thermoneutrality. J Physiol, 2022. 600(4): p. 829–845. [DOI] [PubMed] [Google Scholar]
- 14.Ravussin Y, et al. , Effect of intermittent cold exposure on brown fat activation, obesity, and energy homeostasis in mice. PLoS One, 2014. 9(1): p. e85876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabner GF, et al. , Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat Metab, 2021. 3(11): p. 1445–1465. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda K and Yamada T, UCP1 Dependent and Independent Thermogenesis in Brown and Beige Adipocytes. Front Endocrinol (Lausanne), 2020. 11: p. 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nedergaard J and Cannon B, Brown adipose tissue as a heat-producing thermoeffector. Handb Clin Neurol, 2018. 156: p. 137–152. [DOI] [PubMed] [Google Scholar]
- 18.Ceddia RP and Collins S, A compendium of G-protein-coupled receptors and cyclic nucleotide regulation of adipose tissue metabolism and energy expenditure. Clin Sci (Lond), 2020. 134(5): p. 473–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins S, beta-Adrenergic Receptors and Adipose Tissue Metabolism: Evolution of an Old Story. Annu Rev Physiol, 2022. 84: p. 1–16. [DOI] [PubMed] [Google Scholar]
- 20.Tan YQ, Li J, and Chen HW, Epac, a positive or negative signaling molecule in cardiovascular diseases. Biomed Pharmacother, 2022. 148: p. 112726. [DOI] [PubMed] [Google Scholar]
- 21.Villarroya J, et al. , New insights into the secretory functions of brown adipose tissue. J Endocrinol, 2019. 243(2): p. R19–R27. [DOI] [PubMed] [Google Scholar]
- 22.Guillamat-Prats R, et al. , Endocannabinoid Signalling in Atherosclerosis and Related Metabolic Complications. Thromb Haemost, 2019. 119(4): p. 567–575. [DOI] [PubMed] [Google Scholar]
- 23.Zhao S, Kusminski CM, and Scherer PE, Adiponectin, Leptin and Cardiovascular Disorders. Circ Res, 2021. 128(1): p. 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funcke JB and Scherer PE, Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res, 2019. 60(10): p. 1648–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degerman E, et al. , From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol, 2011. 11(6): p. 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehran AE, et al. , Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab, 2012. 16(6): p. 723–37. [DOI] [PubMed] [Google Scholar]
- 27.Mowers J, et al. , Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife, 2013. 2: p. e01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obregon MJ, Adipose tissues and thyroid hormones. Front Physiol, 2014. 5: p. 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner J, et al. , Thyroid hormones and browning of adipose tissue. Mol Cell Endocrinol, 2017. 458: p. 156–159. [DOI] [PubMed] [Google Scholar]
- 30.Yau WW and Yen PM, Thermogenesis in Adipose Tissue Activated by Thyroid Hormone. Int J Mol Sci, 2020. 21(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sentis SC, Oelkrug R, and Mittag J, Thyroid hormones in the regulation of brown adipose tissue thermogenesis. Endocr Connect, 2021. 10(2): p. R106–R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho SD, Bianco AC, and Silva JE, Effects of hypothyroidism on brown adipose tissue adenylyl cyclase activity. Endocrinology, 1996. 137(12): p. 5519–29. [DOI] [PubMed] [Google Scholar]
- 33.Rubio A, et al. , Effects of thyroid hormone on norepinephrine signaling in brown adipose tissue. I. Beta 1- and beta 2-adrenergic receptors and cyclic adenosine 3’,5’-monophosphate generation. Endocrinology, 1995. 136(8): p. 3267–76. [DOI] [PubMed] [Google Scholar]
- 34.Rubio A, Raasmaja A, and Silva JE, Thyroid hormone and norepinephrine signaling in brown adipose tissue. II: Differential effects of thyroid hormone on beta 3-adrenergic receptors in brown and white adipose tissue. Endocrinology, 1995. 136(8): p. 3277–84. [DOI] [PubMed] [Google Scholar]
- 35.Guilherme A, et al. , Control of Adipocyte Thermogenesis and Lipogenesis through beta3-Adrenergic and Thyroid Hormone Signal Integration. Cell Rep, 2020. 31(5): p. 107598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christoffolete MA, et al. , Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes, 2004. 53(3): p. 577–84. [DOI] [PubMed] [Google Scholar]
- 37.Bianco AC, et al. , Paradigms of Dynamic Control of Thyroid Hormone Signaling. Endocr Rev, 2019. 40(4): p. 1000–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mir N, et al. , Genomic and Non-Genomic Actions of Glucocorticoids on Adipose Tissue Lipid Metabolism. Int J Mol Sci, 2021. 22(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen Y, et al. , Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol Metab, 2017. 6(10): p. 1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giroud M, et al. , HAND2 is a novel obesity-linked adipogenic transcription factor regulated by glucocorticoid signalling. Diabetologia, 2021. 64(8): p. 1850–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibanez CF, Regulation of metabolic homeostasis by the TGF-beta superfamily receptor ALK7. FEBS J, 2021. [DOI] [PubMed] [Google Scholar]
- 42.Guilherme A, et al. , Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol, 2008. 9(5): p. 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersson O, et al. , Growth/differentiation factor 3 signals through ALK7 and regulates accumulation of adipose tissue and diet-induced obesity. Proc Natl Acad Sci U S A, 2008. 105(20): p. 7252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balkow A, et al. , A novel crosstalk between Alk7 and cGMP signaling differentially regulates brown adipocyte function. Mol Metab, 2015. 4(8): p. 576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo T, et al. , Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. Elife, 2014. 3: p. e03245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yogosawa S, et al. , Activin receptor-like kinase 7 suppresses lipolysis to accumulate fat in obesity through downregulation of peroxisome proliferator-activated receptor gamma and C/EBPalpha. Diabetes, 2013. 62(1): p. 115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yogosawa S and Izumi T, Roles of activin receptor-like kinase 7 signaling and its target, peroxisome proliferator-activated receptor gamma, in lean and obese adipocytes. Adipocyte, 2013. 2(4): p. 246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valentine JM, et al. , beta3-Adrenergic receptor downregulation leads to adipocyte catecholamine resistance in obesity. J Clin Invest, 2022. 132(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koren S, et al. , The role of mouse Akt2 in insulin-dependent suppression of adipocyte lipolysis in vivo. Diabetologia, 2015. 58(5): p. 1063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiPilato LM, et al. , The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol Cell Biol, 2015. 35(16): p. 2752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabbay RA and Lardy HA, The antilipolytic effect of insulin does not require adenylate cyclase or phosphodiesterase action. FEBS Lett, 1985. 179(1): p. 7–11. [DOI] [PubMed] [Google Scholar]
- 52.Sancar G, et al. , FGF1 and insulin control lipolysis by convergent pathways. Cell Metab, 2022. 34(1): p. 171–183 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stockli J, et al. , ABHD15 regulates adipose tissue lipolysis and hepatic lipid accumulation. Mol Metab, 2019. 25: p. 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rebrin K, et al. , Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest, 1996. 98(3): p. 741–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perry RJ, et al. , Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell, 2015. 160(4): p. 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Titchenell PM, et al. , Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun, 2015. 6: p. 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mauvais-Jarvis F, et al. , Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest, 2002. 109(1): p. 141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morley TS, Xia JY, and Scherer PE, Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat Commun, 2015. 6: p. 7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schoiswohl G, et al. , Impact of Reduced ATGL-Mediated Adipocyte Lipolysis on Obesity-Associated Insulin Resistance and Inflammation in Male Mice. Endocrinology, 2015. 156(10): p. 3610–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shearin AL, et al. , Lack of AKT in adipocytes causes severe lipodystrophy. Mol Metab, 2016. 5(7): p. 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herman MA, et al. , A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature, 2012. 484(7394): p. 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iwen KA, et al. , Cold-Induced Brown Adipose Tissue Activity Alters Plasma Fatty Acids and Improves Glucose Metabolism in Men. J Clin Endocrinol Metab, 2017. 102(11): p. 4226–4234. [DOI] [PubMed] [Google Scholar]
- 63.Xiao C, et al. , Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity (Silver Spring), 2015. 23(7): p. 1450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michel LYM, Farah C, and Balligand JL, The Beta3 Adrenergic Receptor in Healthy and Pathological Cardiovascular Tissues. Cells, 2020. 9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lowell BB and Flier JS, Brown adipose tissue, beta 3-adrenergic receptors, and obesity. Annu Rev Med, 1997. 48: p. 307–16. [DOI] [PubMed] [Google Scholar]
- 66.Cero C, et al. , beta3-Adrenergic receptors regulate human brown/beige adipocyte lipolysis and thermogenesis. JCI Insight, 2021. 6(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Mara AE, et al. , Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest, 2020. 130(5): p. 2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cypess AM, Reassessing Human Adipose Tissue. N Engl J Med, 2022. 386(8): p. 768–779. [DOI] [PubMed] [Google Scholar]
- 69.Cohen P, et al. , Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell, 2014. 156(1–2): p. 304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chung YW, et al. , White to beige conversion in PDE3B KO adipose tissue through activation of AMPK signaling and mitochondrial function. Sci Rep, 2017. 7: p. 40445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Degirmenci U, et al. , Silencing an insulin-induced lncRNA, LncASIR, impairs the transcriptional response to insulin signalling in adipocytes. Sci Rep, 2019. 9(1): p. 5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abu-Odeh M, et al. , FGF21 promotes thermogenic gene expression as an autocrine factor in adipocytes. Cell Rep, 2021. 35(13): p. 109331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phan J and Reue K, Lipin, a lipodystrophy and obesity gene. Cell Metab, 2005. 1(1): p. 73–83. [DOI] [PubMed] [Google Scholar]
- 74.Franckhauser S, et al. , Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes, 2002. 51(3): p. 624–30. [DOI] [PubMed] [Google Scholar]
- 75.Kleiner S, et al. , Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proc Natl Acad Sci U S A, 2012. 109(24): p. 9635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Puigserver P, et al. , A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell, 1998. 92(6): p. 829–39. [DOI] [PubMed] [Google Scholar]
- 77.Agudelo LZ, et al. , Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab, 2018. 27(2): p. 378–392 e5. [DOI] [PubMed] [Google Scholar]
- 78.Liu D, et al. , Activation of mTORC1 is essential for beta-adrenergic stimulation of adipose browning. J Clin Invest, 2016. 126(5): p. 1704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu D, Ceddia RP, and Collins S, Cardiac natriuretic peptides promote adipose ‘browning’ through mTOR complex-1. Mol Metab, 2018. 9: p. 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olsen JM, et al. , beta3-Adrenergically induced glucose uptake in brown adipose tissue is independent of UCP1 presence or activity: Mediation through the mTOR pathway. Mol Metab, 2017. 6(6): p. 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ikeda K, et al. , UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat Med, 2017. 23(12): p. 1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chypre M, Zaidi N, and Smans K, ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun, 2012. 422(1): p. 1–4. [DOI] [PubMed] [Google Scholar]
- 83.Berwick DC, et al. , The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem, 2002. 277(37): p. 33895–900. [DOI] [PubMed] [Google Scholar]
- 84.Potapova IA, et al. , Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry, 2000. 39(5): p. 1169–79. [DOI] [PubMed] [Google Scholar]
- 85.Pierce MW, et al. , ATP-citrate lyase. Structure of a tryptic peptide containing the phosphorylation site directed by glucagon and the cAMP-dependent protein kinase. J Biol Chem, 1981. 256(17): p. 8867–70. [PubMed] [Google Scholar]
- 86.Pierce MW, et al. , The insulin-directed phosphorylation site on ATP-citrate lyase is identical with the site phosphorylated by the cAMP-dependent protein kinase in vitro. J Biol Chem, 1982. 257(18): p. 10681–6. [PubMed] [Google Scholar]
- 87.Fernandez S, et al. , Adipocyte ACLY Facilitates Dietary Carbohydrate Handling to Maintain Metabolic Homeostasis in Females. Cell Rep, 2019. 27(9): p. 2772–2784 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abdul-Wahed A, Guilmeau S, and Postic C, Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab, 2017. 26(2): p. 324–341. [DOI] [PubMed] [Google Scholar]
- 89.Ortega-Prieto P and Postic C, Carbohydrate Sensing Through the Transcription Factor ChREBP. Front Genet, 2019. 10: p. 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martinez Calejman C, et al. , mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Commun, 2020. 11(1): p. 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Czech MP, Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol Metab, 2020. 34: p. 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Albert V, et al. , mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol Med, 2016. 8(3): p. 232–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Olsen JM, et al. , Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation. J Cell Biol, 2014. 207(3): p. 365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keinan O, et al. , Glycogen metabolism links glucose homeostasis to thermogenesis in adipocytes. Nature, 2021. 599(7884): p. 296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heine M, et al. , Lipolysis Triggers a Systemic Insulin Response Essential for Efficient Energy Replenishment of Activated Brown Adipose Tissue in Mice. Cell Metab, 2018. 28(4): p. 644–655 e4. [DOI] [PubMed] [Google Scholar]
- 96.Jung SM, et al. , In vivo isotope tracing reveals the versatility of glucose as a brown adipose tissue substrate. Cell Rep, 2021. 36(4): p. 109459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zingaretti MC, et al. , The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J, 2009. 23(9): p. 3113–20. [DOI] [PubMed] [Google Scholar]
- 98.Leiria LO, et al. , 12-Lipoxygenase Regulates Cold Adaptation and Glucose Metabolism by Producing the Omega-3 Lipid 12-HEPE from Brown Fat. Cell Metab, 2019. 30(4): p. 768–783 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stanford KI, et al. , 12,13-diHOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab, 2018. 27(5): p. 1111–1120 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagatake T, et al. , 12-Hydroxyeicosapentaenoic acid inhibits foam cell formation and ameliorates high-fat diet-induced pathology of atherosclerosis in mice. Sci Rep, 2021. 11(1): p. 10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pinckard KM, et al. , A Novel Endocrine Role for the BAT-Released Lipokine 12,13-diHOME to Mediate Cardiac Function. Circulation, 2021. 143(2): p. 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hajri T, et al. , Regulation of adiponectin production by insulin: interactions with tumor necrosis factor-alpha and interleukin-6. Am J Physiol Endocrinol Metab, 2011. 300(2): p. E350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Musovic S and Olofsson CS, Adrenergic stimulation of adiponectin secretion in visceral mouse adipocytes is blunted in high-fat diet induced obesity. Sci Rep, 2019. 9(1): p. 10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Olzmann JA and Carvalho P, Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol, 2019. 20(3): p. 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boutant M, et al. , Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J, 2017. 36(11): p. 1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu N, et al. , The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol, 2012. 198(5): p. 895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Puri V, et al. , Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci U S A, 2008. 105(22): p. 7833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishino N, et al. , FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest, 2008. 118(8): p. 2808–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rubio-Cabezas O, et al. , Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med, 2009. 1(5): p. 280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu S, Zhang X, and Liu P, Lipid droplet proteins and metabolic diseases. Biochim Biophys Acta Mol Basis Dis, 2018. 1864(5 Pt B): p. 1968–1983. [DOI] [PubMed] [Google Scholar]
- 111.Smith GI, et al. , Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest, 2020. 130(3): p. 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Czech MP, et al. , Insulin signalling mechanisms for triacylglycerol storage. Diabetologia, 2013. 56(5): p. 949–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wallace M and Metallo CM, Tracing insights into de novo lipogenesis in liver and adipose tissues. Semin Cell Dev Biol, 2020. 108: p. 65–71. [DOI] [PubMed] [Google Scholar]
- 114.Song Z, Xiaoli AM, and Yang F, Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients, 2018. 10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao H, et al. , Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell, 2008. 134(6): p. 933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yore MM, et al. , Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell, 2014. 159(2): p. 318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lodhi IJ, et al. , Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metab, 2012. 16(2): p. 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grunt TW, et al. , Membrane disruption, but not metabolic rewiring, is the key echanism of anticancer-action of FASN-inhibitors: a multi-omics analysis in ovarian cancer. Sci Rep, 2020. 10(1): p. 14877. [DOI] [PMC free article] [PubMed] [Google Scholar]