

Abstract
Sample preparation for mass spectrometry‐based proteomics has many tedious and time‐consuming steps that can introduce analytical errors. In particular, the steps around the proteolytic digestion of protein samples are prone to inconsistency. One route for reliable sample processing is the development and optimization of a workflow utilizing an automated liquid handling workstation. Diligent assessment of the sample type, protocol design, reagents, and incubation conditions can significantly improve the speed and consistency of preparation. When combining robust liquid chromatography‐mass spectrometry with either discovery or targeted methods, automated sample preparation facilitates increased throughput and reproducible quantitation of biomarker candidates. These improvements in analysis are also essential to process the large patient cohorts necessary to validate a candidate biomarker for potential clinical use. This article reviews the steps in the workflow, optimization strategies, and known applications in clinical, pharmaceutical, and research fields that demonstrate the broad utility for improved automation of sample preparation in the proteomic field.
Keywords: automation, high throughput, optimization, proteomics, quantitative mass spectrometry, robot, sample preparation
1 |. INTRODUCTION
The recent rapid improvements in liquid chromatography mass spectrometry (LC‐MS) instrumentation and informatics have empowered new advancements in MS‐based protein and peptide quantification methodology (Aslam et al., 2017). As an applied bioanalytic tool in basic and clinical research, MS‐based approaches offer the ability to quantify 1000s of proteins in a single MS run (Geyer et al., 2016; Pappireddi et al., 2019). Biofluids, such as blood or urine, provide an easily obtained source of biomolecules that can offer insight into a patient’s health or disease status. The analytical power of MS can be harnessed to identify biomarkers from tissues or biofluids that diagnose, stage, monitor or predict pathology and/or clinical outcomes in a wide variety of diseases.
Developing a biomarker for clinical use requires two distinct phases: discovery and validation. The discovery phase is characterized by comprehensive analysis of patient samples to develop candidate biomarkers. This is often performed using data‐dependent or data‐independent MS approaches (discussed below). In this phase, reliable quantitation of specific proteoforms is essential to be able to discriminate potentially diagnostic signals from noise. When one or more candidate biomarkers have been identified, each needs to be validated using much larger patient cohorts to establish the specificity and sensitivity of the markers for potential clinical use. This utilizes targeted MS approaches like multiple/selective reaction monitoring (M/SRM), parallel reaction monitoring (PRM) or sure quant‐mass spectrometry (SQ‐MS) (discussed below). These approaches provide thorough and accurate quantitation of peptides in complex biological samples.
Regardless of the technique in discovery‐based or targeted‐MS workflows (Li et al., 2020; Vidova & Spacil, 2017; Zhang et al., 2019), the common fundamental step before MS analysis is sample preparation. Protein samples are digested into peptides before MS analysis following a series of labor‐intensive steps.However, reproducibility, time, and cost remain longstanding barriers to large‐scale and high‐throughput MS sample processing (Rogers & Bomgarden, 2016). To address this, the development of fast and accurate MS protein sample preparation workflows is necessary to improve the throughput of MS analysis and help speed the translation from biomarker discovery to validated clinical assay. An emerging innovation to improve sample preparation has been the utilization of robotic liquid handling workstations that can perform the most labor‐intensive aspects of sample preparation accurately and reliably. In the following sections, we outline considerations for adapting and optimizing steps in the MS sample preparation protocol for automation and review examples of how automation has been implemented in discovery, validation, and clinical proteomic workflows.
2 |. ESSENTIAL STEPS AND OPTIMIZATION IN LC‐MS SAMPLE PREPARATION
To prepare samples for proteomic analysis there are several core steps that need to be performed on each sample and, depending on the needs of the experiment, additional specialized processing steps that may be required. The core steps include (1) protein concentration measurement; (2) protein denaturation; (3) reduction of disulfide bonds; (4) alkylation with a cysteine‐blocking agent; (5) enzymatic digestions, for example, trypsin; (6) quenching; (7) desalting by solid phase extraction to remove interfering components (Figure 1). Optimization of each step during sample preparation is essential to obtain a robust, reproducible workflow for quantitative proteomics. For clinical grade validation of biomarkers, the total imprecision should be <20% including inter‐ and intra‐day assessments (Grant & Hoofnagle, 2014). Table 1 outlines general recommendations required to achieve the desired performance and reproducibility. Characteristics like denaturation agents, trypsin‐ (or other enzyme)‐to‐substrate ratio, buffer composition, incubation time, temperature, should each be assessed to understand the implications on MS sensitivity and reliability across the proteome in question. This is frequently performed in a series of sequential evaluations comparing various aspects of the protocol either manually or using an automated liquid handling workstation. Using an automated workstation, the user can achieve much greater control over liquid handling, mixing, incubation times and temperatures for uniform reactions.
FIGURE 1.
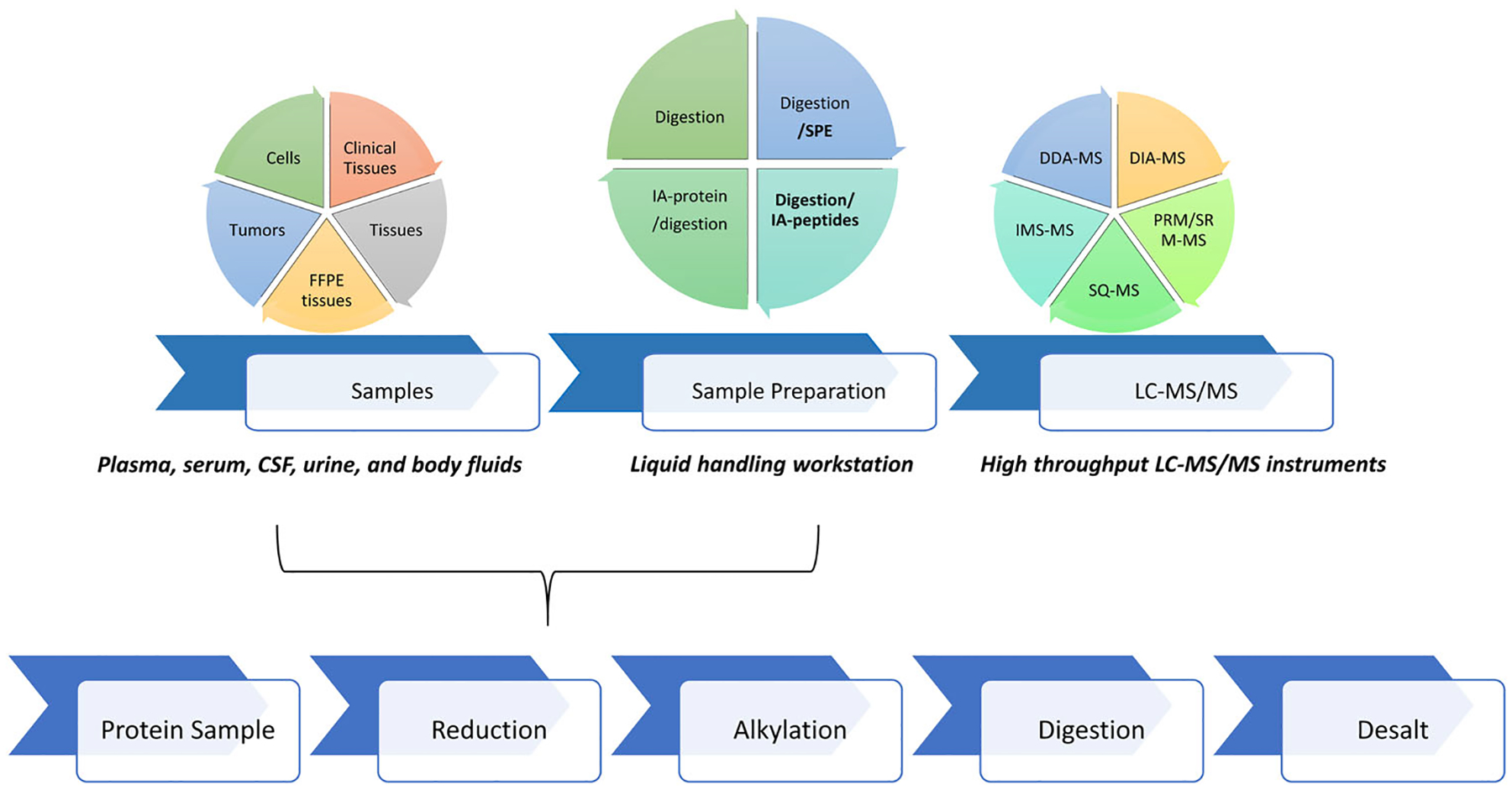
Schematic of the proteomic sample preparation process. Top panel summarizes sample type, different automated processes, and MS acquisition methods. Bottom panel illustrates the basic steps in proteomic sample preparation
TABLE 1.
Optimization of automated sample preparation techniques
| Automation method | Step | Parameters that may be optimized | Recommendations |
|---|---|---|---|
| Aliquoting/plating | From protein samples to a 96-well plate aliquoting | Pipetting techniques (e.g., plasma and serum are viscous); dispensing of samples into the bottom of wells | Slowly aspirate and dispense protein samples |
| Labware (tubes and plates) | Accurately define the plate (X, Y, Z axials, spacing and well offset) | ||
| Digestion | Buffer | Final concentration | Tris-HCl (pH 8.3), NH4HCO3 (pH 8.5) |
| Denaturation/reduction | Chemistry, temperature, incubation time and final concentration of selectants and/or denaturants | Denaturants (OGS, TFE, Rapigest, urea, guanidine HCl; and reducing agents (TCEP, DTT) | |
| Cysteine blocking | Chemistry and concentration | MMTS, iodoacetamide | |
| Trypsin | Substrate to enzyme ratio, incubation time, choice of vendor and grade. Note, other enzymes can be used (e.g., AspN, GluC, etc.) | Trypsin from Promega, Sciex, Thermo Fisher Scientific, Sigma | |
| Quenching | Acid choice and concentration | Formic, acetic, or trifluoroacetic acid | |
| Solid phase extraction | Positive pressure apparatus integrated into and controlled by the automated workstation | Pressure, time, SPE (desalting) chemistry, sorbent to digested peptide ratio), organics | Oasis HLB |
2.1 |. Protein denaturation
Protein denaturation is an initial and important step in preparing the sample for digestion. Denaturation unfolds the secondary structure of the protein to permit preferential access by downstream reagents and proteases. Effective denaturation will increase proteolytic efficiency and ultimately reproducibility of the sample preparation (Proc et al., 2010). The use of detergents, most notably sodium dodecyl sulfate (SDS), is widely reported for efficient digestion of proteins to peptides in the context of complex biological matrices. However, detergents can have serious interference issues for subsequent LC‐MS/MS analysis (Funk et al., 2005; Loo et al., 1996). Other chaotropic agents, such as urea or guanidine‐HCl, can also be used to denature proteins but also interfere with the MS analysis and require specific removal from the sample after proteolysis. As an alternative, MS compatible acid‐labile surfactants (ALS), such Rapigest SF, have been introduced (Meng et al., 2002). Rapigest SF is an SDS analog that is an effective surfactant at physiological pH for proteolysis but is cleaved to noninterfering components when the peptides are acidified for MS analysis (Yu et al., 2003). Several versions of ALSs have been introduced and found to be effective (Chen et al., 2007) although the cost of using these reagents can be prohibitive for large scale cohorts.
A landmark study by Proc et al. (2010) demonstrated that, while indeed SDS yields the most efficient digestion of the plasma proteome, 2‐2‐2 trifluoroethanol (TFE) 50% (v/v), a volatile solvent, can be used in its place and give comparable data (Proc et al., 2010). Many studies describe the effect of TFE on protein structure and it has been shown to melt the hydrophobic core of globular proteins suggesting it is effective in denaturing membrane‐bound proteins. Further, Reiersen et al. demonstrated that TFE concentration ≥30% (v/v) and temperature of 58.6°C induced desirable denaturing conditions (Reiersen & Rees, 2000). Finally, the use of TFE has also been described in the context of many matrices including blood, plasma, urine, tissue and cells (Adachi et al., 2006; Coscia et al., 2020; Yu et al., 2003). Given the breadth of applications, instrument compatibility, and the ease of automation due to its volatile nature, there are significant advantages for using TFE to denature proteins in MS sample preparation however, the effect of any denaturant should be assessed to determine the optimal reagent for a particular workflow.
2.2 |. Reduction and alkylation
Following denaturation, proteins are further reduced and alkylated to break the covalent disulfide bonds that maintain a protein’s tertiary structure and cap the free cysteines to prevent disulfides from reforming. The unlinking of a protein’s cysteine residues improves access for proteolysis and simplifies database searching by eliminating any disulfide linked peptide complexes (Sechi & Chait, 1998). For this process, several reagents have been used. Reduction can be achieved by dithiothreitol (DTT), 2‐mercaptoethanol (2‐ME), tris(2‐carboxyethyl)phosphine (TCEP) or tris(3‐hydroxypropyl)phosphine (THPP). This reaction is usually done at elevated temperatures. Alkylation has been performed using iodoacetamide (IAA), acrylamide (AA), methyl methanethiosulfonate (MMTS), N‐ethylmaleimide (N‐EM), or 4‐vinylpyridine (4‐VP) (Crankshaw & Grant, 2001; Gundry et al., 2009; Hamdan et al., 2001; Hill et al., 2009; Righetti, 2006). The sequential pairing of two of these reagent types can provide reduction and alkylation however, each reaction needs to be evaluated for completeness or the accumulation of side reactions that will alter the anticipated mass of resulting peptides. Comprehensive assessments of reduction and alkylation conditions by Suttapitugsakul et al. (2017) and Muller and Winter (2017) suggest that optimization is necessary to achieve effective reactions in this aspect of sample processing.
2.3 |. Enzymatic digestion
The most common application of bottom‐up proteomics requires that proteins in a sample be digested with a specific protease to facilitate database searching. Trypsin has been the most popular protease in proteomic experiments because of its cleavage specificity (after lysine and arginine residues) and high proteolytic activity. Digestion with trypsin produces many peptides within an analyzable range, 500–5000 Da, and has been found to give preferential sequence coverage. There are several other proteases that can be used such as chymotrypsin, Glu‐C, Asp‐N or proteinase K, each with their own specificity. These have been used on their own or in combination with trypsin to increase sequence coverage of trypsin incompatible regions (Fan et al., 2014). With any proteolysis, effective digestion depends on optimizing the ratio of enzyme to protein, temperature, and time to ensure complete and reliable digestion conditions.
Previous studies have shown that the proteolysis step is a major source for variability in the LC‐MS analysis (van den Broek et al., 2015; Fu et al., 2018; Martin et al., 2013). One of the primary drivers of variability is the inherent complexity of most proteomes. The human proteome arises from >20,000 genes and exhibits additional chemical diversity via the ability of each gene to produce multiple isoforms along with the potential of numerous posttranslational modifications (PTMs). Collectively, the proteoforms are highly regulated in time and space in response to an individual’s physiologic or pathophysiologic states (Figure 2). Beyond the regulation of individual proteins there can also be an enormous dynamic range in protein concentration, where highly abundant proteins can be up to 10 orders of magnitude greater than lowest abundance proteins in a sample (Alpi et al., 2015). In one report, 14 different protein denaturation conditions were investigated for an optimal human plasma digestion efficiency (Proc et al., 2010). Proteolytic cleavage reactions in different matrices (plasma, serum, cerebral spinal fluid [CSF] and other proteomic samples) are complicated as each protein has its own tertiary and quaternary structure as well as unique cleavage site accessibility. For example, in our hands the manual processing of 171 human plasma samples had a coefficient of variation (CV) between 18% and 25% when monitored by spiked exogenous bacterial protein beta‐galactosidase (β‐gal). For comparison, we used an automated workstation to perform all the steps in the preparation of 5 μl of plasma without optimization. Using two proteins (exogenous β‐gal and endogenous human serum albumin) as representative analytes in monitoring the automated digestion, the variability of the peak area ratios (native/stable isotope labeled [SIL] peptides signal) for albumin and β‐gal were 30% CV and 20% CV before optimization, respectively. After the optimization process for each step in the automated workflow, CVs of 3% and 4%–8% were achieved for endogenous human serum albumin and spiked β‐gal protein (Figure 3).
FIGURE 2.
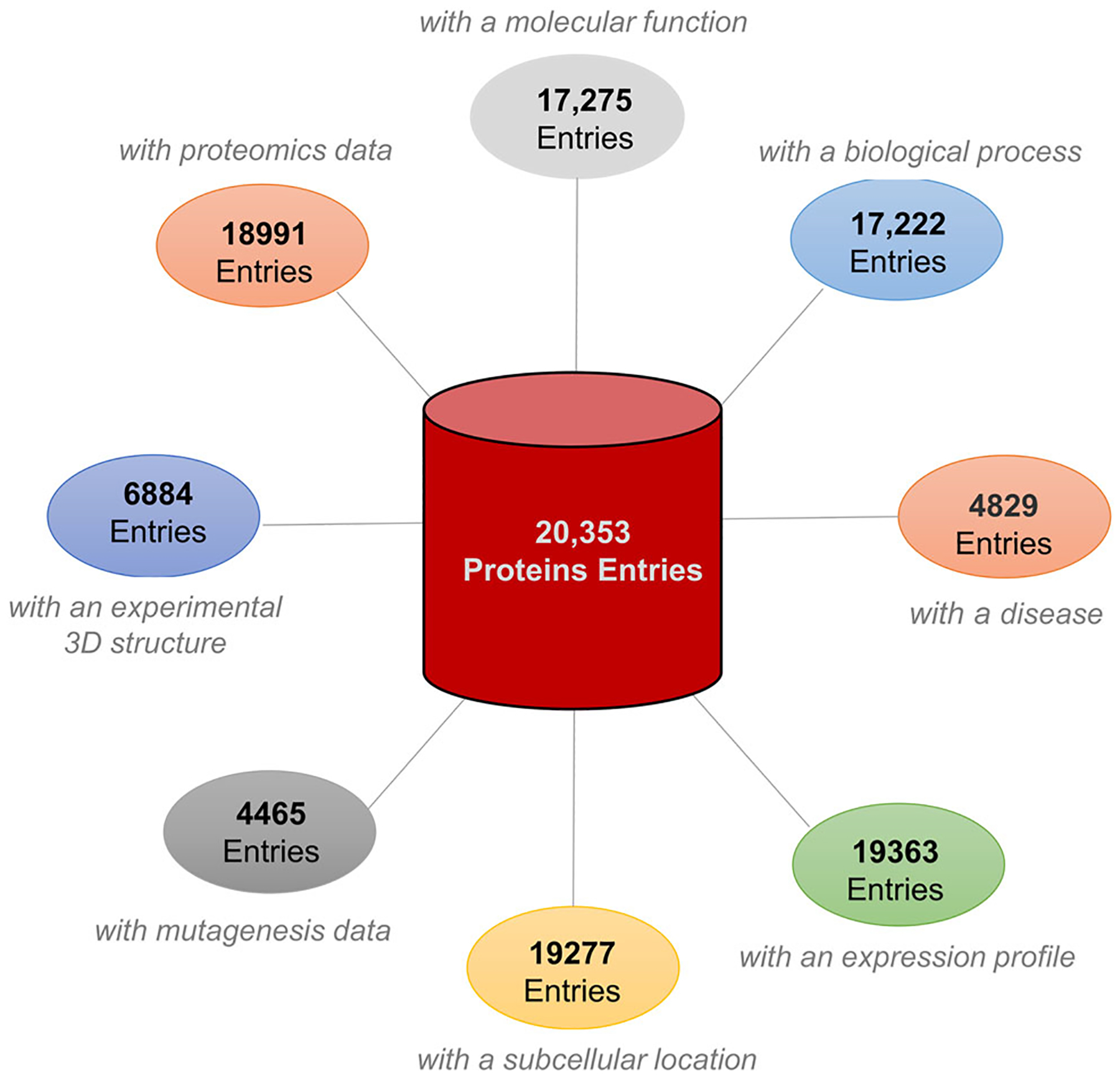
The human proteome illustrated by biological, functional, structural, and sequence complexity. The total number of proteins and their isoforms reported by Swiss‐Prot database; and total proteins associated with eight large categories of biological function (https://www.nextprot.org/about/statistics)
FIGURE 3.
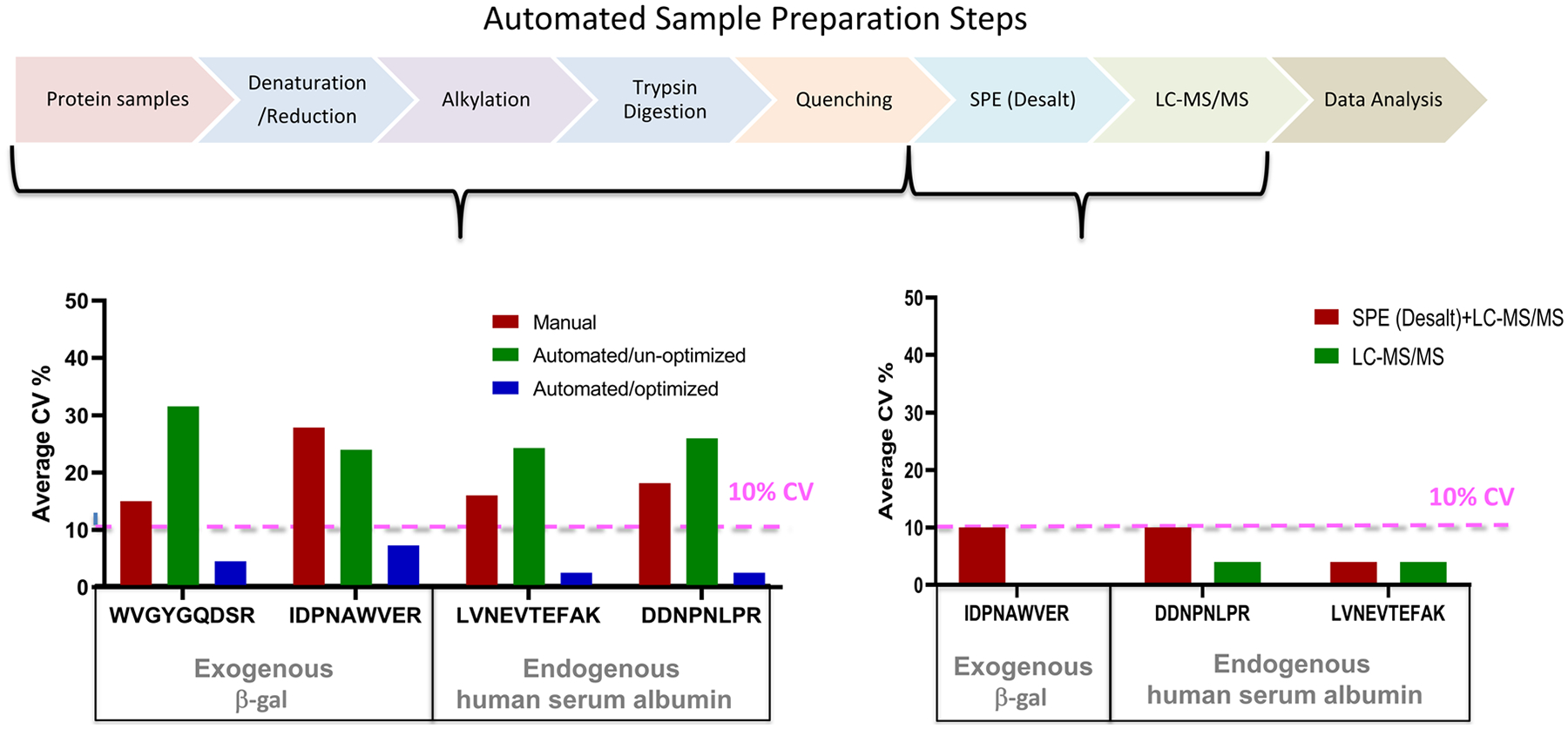
Automated proteomic sample preparation evaluation. Left panel, CV% assessment of three distinct digestion conditions are shown: manual digestion (red), unoptimized automated digestion (green), and optimized automated digestion (blue). The reproducibility was assessed by area ratio (light native/heavy SIL) and the CV% was calculated with 4–8 digestion replicates of β‐gal spiked into pooled healthy human plasma. Plasma samples were denatured, reduced, alkylated, and digested with trypsin. Average area ratio CV% β‐gal and human serum albumin were calculated from peak area light (native/heavy SIL). MRM data was acquired by an 6500QTRAP. Right panel, the CV% assessment of automated solid phase extraction (24‐well) is shown. β‐gal and human serum albumin SIL peptides spiked into predigestion healthy human plasma pool, then desalted by a positive pressure apparatus integrated into a controlled i7 automated workstation. SIL heavy peptide signals after desalting were used to calculate CV%. MRM data was acquired similarly by LC‐MS. CV% was calculated from seven repeated injections. SIL, stable isotope labeled
2.4 |. Quenching
After digestion, the reaction is stopped by quenching in an acidic solution. This step halts the proteolysis and acidifies the peptide solution for desalting. This can be done using several volatile acids: formic, acetic, or trifluoroacetic acids. In case of 18O labeling, quenching is particularly important to prevent trypsin mediated back‐exchange of the heavy label (Petritis et al., 2009).
2.5 |. Desalting
Peptides analyzed by LC‐MS with a high‐resolution mass spectrometer (Orbitrap, TripleTOF or TimsTOF, e.g.) require an extra desalting step postdigestion to remove contaminants and concentrate the peptides. This is usually achieved using a C18 resin to specifically bind peptides while contaminates are removed by wash steps. Solid phase extraction can be achieved with either manual vacuum extraction or a positive pressure apparatus (manually or automated). Administration of a vacuum can be difficult to regulate which would affect the recovery and reproducibility (Psillakis, 2017); use of a positive pressure apparatus is recommended (Johnson et al., 2020). It is possible to integrate a positive‐pressure manifold for peptide desalting on an automation workstation. With an integrated and automated positive pressure apparatus, the pressure is regulated by clean nitrogen gas, and the gas flow and time applied can be pre‐programed based on sample and solvent type. Therefore, more uniform recovery and reproducible solid phase extraction can be achieved.
Of note, there have been recent innovations to integrate the discrete steps of sample preparation into a single self‐contained column or tip. The S‐trap protocol utilizes a porous material to trap denatured protein solutions permitting removal of contaminates, reduction, alkylation and digestion (HaileMariam et al., 2018; Ludwig et al., 2018). This approach has been found to be particularly effective at removing contaminates and denaturing surfactants during sample preparation (Zacchi et al., 2020). A similar approach has been proposed and extended by Chen et al. (2016). Their simple and integrated spin‐tip proteomics technology (SISPROT) utilizes strong cation exchange beads and C18 resin to fully integrate all the steps of sample digestion, including the final desalting, into a single tip that can be rapidly processed achieving high sensitivity with little sample loss (Chen et al., 2016). This approach is also compatibles with some PTM enrichment protocols (Chen et al., 2018). The integration and simplification of sample preparation is an important aspect in the potential to automate these protocols.
A further consideration for the development of an automated workflow is the requirement to select reagents which can be stored as aliquots to minimize the reagent batch effects. Exogenous protein β‐gal can be used for quality control (Fu et al., 2018; Grant & Hoofnagle, 2014) to monitor the reproducibility and accuracy of the automated proteomics sample preparation workflow within a 96‐well plate format. The β‐gal protein and a mixture of its corresponding SIL peptide standards can be added before the reduction and alkylation reactions before enzymatic digestion. Once an optimized automation workflow is established, it is important to evaluate the reproducibility across multiple days. For a standardized automated workflow, multisite validation to ensure the validity of the results is good laboratory practice (Fu et al., 2018; Martin et al., 2013).
3 |. QUANTITATIVE MASS SPECTROMETRY
Once samples have been prepared, peptides can be analyzed using a variety of high‐performance mass spectrometry techniques to perform discovery or validation studies of disease biomarkers. Discovery‐based proteomics is commonly performed using either data‐dependent acquisition mass spectrometry (DDA‐MS) (Zhang et al., 2013) or data‐independent acquisition mass spectrometry (DIA‐MS) (Gillet et al., 2012; Krasny & Huang, 2021) to provide a comprehensive and quantitative evaluation of biological samples. In DDA‐MS, all precursor ions are scanned during the survey scan (MS1), then a subset of ions are individually selected for fragmentation, producing a series of tandem (MS/MS or MS2) mass spectra. The collected mass spectra are searched against a database to be matched and assigned to peptides and then proteins. This approach is highly powerful but suffers from sampling only the n most abundant precursors (predefined numbers of precursor with signals above “noise”) for subsequent MS2 fragmentation. This sampling bias can limit the reliable identification and quantification of low abundance peptides and proteins. In DIA‐MS, all precursor ions within a defined mass to charge (m/z) window observed in a MS1 survey scan are subjected to fragmentation. The analysis is repeated, and the fragment ions are accumulated in a fixed number of isolation windows that span entire mass‐to‐charge (m/z) range. Because of all detected precursors are fragmented, the advantage of DIA‐MS is that the accurate peptide quantification without limitation of selecting predefined peptides as in DDA‐MS. DIA‐MS method requires a mass spectrometer capable of high‐resolution MS/MS spectra.
Moving beyond the discovery phase requires targeted validation, usually involving large scale cohorts of patient samples. Currently, the methods of choice for the evaluation of these samples are multiple reaction monitoring mass spectrometry (MRM‐MS), also known as selected reaction monitoring mass spectrometry (SRM‐MS) (Lange et al., 2008), parallel reaction monitoring mass spectrometry (PRM‐MS) (Gallien et al., 2015), and Sure Quant‐Mass Spectrometry (SQ‐MS) (Liebler & Zimmerman, 2013) which provides precise quantitation of proteotypic peptides (unique surrogates for the corresponding protein) in complex biological samples derived from bodily fluids, biopsies, or cultured cells. SRM‐MS is a targeted approach which utilizes the highly reproducible triple quadrupole (QqQ) mass analyzer to select known peptides of choice for quantitation. The advantages of SRM‐MS are selectivity, sensitivity, and multiplexity in peptide/protein quantitation. PRM‐MS is a SRM‐like targeted approach but performed in a high resolution and high mass accuracy (HR/AM) mode on a mass spectrometer. In PRM‐MS, full MS/MS spectra of the selected peptides are acquired with high resolution and high mass accuracy. Peptide and protein quantification is highly selective and specific. SQ‐MS is a recently developed acquisition method for internal standards targeted approach by using HR/AM Orbitrap mass spectrometer. The mass spectrometer programed to monitor referential internal standards in samples using dynamically changed MS parameters. This method enhances endogenous peptides quantitative data quality and may quantify a large number of peptide targets (Stopfer et al., 2021).
4 |. IMPLEMENTED AUTOMATED PROTEOMIC SAMPLE PREPARATION IN LC‐MS ANALYSIS
The success of any discovery or validation proteomics experiment depends greatly on the speed and quality of the sample preparation. An emerging option to increase throughput and accuracy is the use of liquid handling stations to perform many of the seps in the sample preparation protocol. Any automated workstation suitable for full hands‐off sample preparation for MS requires the following functions: (1) time‐controlled liquid transferring with accuracy, (2) liquid transferring from reagents vials/plates to digestion plate, (3) 96/364 pin and/or 8‐span head, (4) grippers to move the digestion plate, (5) temperature and time‐controlled incubator with shaking ability, and (6) an independent plate shaker to be used as a mixer. These features remove human hands from the most repetitive and labor‐intensive steps and increase accuracy and reproducibility for large numbers of samples. There are a growing number of examples in the literature where automated workstations are utilized for proteomic sample preparation for the quantitative MS analysis applications for high‐throughput clinical, pathological, pharmaceutical, and research domains (Table 2). The following section describes several key studies, and the relative performance of the automated workflow.
TABLE 2.
Examples of automated hands-off workflows for quantitative preclinical and clinical research
| Liquid handling system/workstation property | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Analytes | Throughput | Field application | Sample process | Liquid handling system/workstation | Company | Robotic arm (volume) | Channels | Note | MS (Reference) |
| Protein markers panels (Health Surveillance and Apolipoproteins) | High-throughput preclinical laboratory and research assay | Preclinical and research | Whole plasma/serum/blood Mitra direct tryptic digestion | Biomek NXP Automated Liquid Handler | Beckman Coulter Life Sciences | 0.5–1000 μl (Multichannel); 0.5–5000 μl (Span-8) | Multichannel head (96 or 384) or Span-8 pipettting with gripper | Biomek NXP is no longer available and is replaced by Biomek i-Series, such as i5 and i7 | MRM-MS (Fu et al., 2018; van den Broek, 2017) |
| Apolipoproteins panel | High-throughput preclinical laboratory and research assay | Preclinical and research | Volumetric absorptive microsampling (Mitra) | Biomek NXP Automated Liquid Handler | MRM-MS (van den Broek et al., 2020) | ||||
| Apolipoproteins/plasma | High-throughput clinical laboratory assay | Cardiovascular Diseases | Whole plasma/serum direct tryptic digestion | Bravo 96- channel liquid handling platform | Agilent Technologies | 100 nl-200 μl | 8, 16, 96, and 384 Channel Heads | DDA-MS (van den Broek et al., 2016) | |
| Dried blood spots (DBS) | High-throughput/research | Research | Direct tryptic digestion | Triversa Nanomate robotic platform | Advion Ltd | <10 μl | The Nanomate probe | Chip-based electrospray device with the Nanomate probe | DDA-MS (Martin et al., 2013) |
| Amyloid 40 and 42 | High-throughput clinical laboratory assay | Alzheimer’s disease/Clinical Diagnostics | Solid phase extraction | Microlab Star system integrated with a SPEware IP8 positive-pressure manifold | Hamilton | Custom (Inquire) | Custom (Inquire) | MRM-MS (Weber et al., 2019) | |
| Peptide mapping/monoclonal monitoring | Biopharmaceutical industry | Recombinant mAB quality control | Direct tryptic digestion and SPE | KingFisher | Thermo Scientific | 50–1000 nl for 12− pin head 200–5000 μl/6− pin head | 6− or 12− pin head | DDA-MS (Millan-Martin et al., 2020) | |
| Insulin and C-peptide | High-throughput clinical laboratory assay | Diabetes/Clinical Diagnostics | Immuno-capture of intact protein | Microlab Star system (Hamilton) | Hamilton Company | custom (Inquire) | Custom (Inquire) | MRM-MS (Taylor et al. 2016) | |
| Thyroglobulin/plasma | High-throughput clinical laboratory assay | Treatment follow-ups/clinical pathology | Trypsin digestion and peptide immune- capture | epMotion system | Eppendorf | 1–1000 μl | Up to 8 channels | MRM-MS (Kushnir et al., 2013) | |
4.1 |. Direct digestion‐MS
Whole biofluids such as plasma, serum, CSF, urine, as well as blood, dry blood including Whatman paper spots or volumetric absorptive microsampling (VAMS) are complex matrices with rich proteome information. Several studies have utilized automated workstations to develop hands‐off workflows for direct protein digestion coupled with quantitative MS analysis in preclinical and clinical research studies (Table 2). Direct digestion is the simplest form of sample preparation, it does not require any specific extraction, from cells or tissues, or any specific enrichment/depletion steps. For example, an automated workflow with Biomek NXᴾ (Beckman Coulter Life Science) to directly digest dried blood from a Mitra (a volumetric absorptive microsampling device) has been used with high flow LC with a sensitive triple-quadrupole mass spectrometer acquisition method to achieve reasonable precision (CV < 20%) in quantifying high abundant proteins with MRM analysis (van den Broek et al., 2017). In another study, a Bravo (Agilent) 96‐channel liquid handler was used to process dried blood spots (DBS) which was then combined with high flow LC‐MS analysis to quantify each peptide representing Apolipoproteins A‐I, B, C‐II, C‐III and E with the total observed CV < 6% using MRM (van den Broek et al., 2016). A further study used a Tecan Freedom liquid handler automation workstation to quantify 97 blood proteins also from DBS samples achieved an average intra‐ and inter‐assay CV < 12% (including MS CV) for the various peptides (Chambers et al., 2015).
4.2 |. Solid phase extraction
This workflow has been implemented in the clinical laboratory domain, with Quest Diagnostics reporting a high‐throughput MS assay to determine the ratio of β‐amyloid 1–42 (A42) to β‐amyloid 1–40 (A40) in CSF. This assay is able to diagnose and differentiate patients with Alzheimer’s disease from nondiseased healthy controls. This clinical assay utilizes solid phase extraction‐based automated workflow on a Hamilton automation liquid handler following trypsin digestion of CSF (DeMarco et al., 2020; Weber et al., 2019). Our group have integrated a positive pressure apparatus for desalting on HLB plates using the i7 hybrid liquid handling workstation (Beckman Coulter) (Johnson et al., 2020; McArdle et al., 2021). In our lab, the liquid handling %CV was calculated to be around 6% (when the desalting step was included, the total %CV was ~10%; and without desalting the total %CV was ~4%) (Figure 3 bottom right panel).
4.3 |. Quality control
Monoclonal antibody (mABs) therapy has rapidly emerged as an important therapeutic agent for the treatment of various diseases, including cancer, chronic inflammation and pathological infections (Chung, 2017). Recently, the US Food and Drug Administration (FDA) has authorized the use of monoclonal antibodies for the treatment of COVID‐19 (Drozdzal et al., 2020). For the biopharmaceutical industry, it is critical to monitor mAB quality control, including recombinant sequence verification (An et al., 2020). One inter‐laboratory study on mAB quality control utilized an automated sample preparation method using a KingFisher liquid handling workstation (Thermo Fisher Scientifc) to map global mAB peptides by MS (Millan‐Martin et al., 2020). This team has also performed an inter‐laboratory study using the same automated protocol to demonstrate consistency in mAB product quality control (Millan‐Martin et al., 2020).
4.4 |. Immunoaffinity enrichment and depletion
The blood proteome (and resulting subproteomes of plasma and serum) has a substantial range of concentrations among its various proteins; ~1010 proteins from high mg/ml to sub‐pg/ml concentrations. The highly abundant resident proteins can overwhelm the MS analysis, obscuring the observation of less abundant proteins. To increase the analytic sensitivity of the less abundant proteins, immunoaffinity (IA) approaches have been successfully adapted to automated sample preparation. For example, Lassman et al. used specific antibodies to enrich low abundant plasma cholesteryl ester transfer protein (CEPT) and proprotein convertase subtilisin/kexin type 9 (PSCK9) from 1400 plasma samples in under 3 weeks using an optimized and automated IA procedure (Freedom EVO, Tecan Trading) to achieve an intra‐ and inter‐assay CVs below 15% (Lassman et al., 2014).
Peptides have also been targeted in immune‐capture workflows using stable isotope standards and capture by antipeptide antibodies (SISCAPA or immuno‐MRM [iMRM]). This workflow performs the proteolytic digestion first, then captures surrogate peptides with specific antibodies to detect and quantify via MS. iMRM assays have been used to accurately quantify clinical protein panels (>10 analytes) such as Cystatin C, C‐reactive protein, apolipoprotein E, apolipoprotein C III and other proteins (Campino et al., 1987). Plasma, serum, and DBSs have all been found suitable for iMRM analysis (Anderson et al., 2020). Automated iMRM immuno‐MS workflows have been extensively validated. Multiple automated workstations and platforms have been used successfully in iMRM assays for plasma biomarker quantification, such as the Bravo automated liquid handler (Agilent Technologies) (Razavi et al., 2016, 2019), KingFisher magnetic particle processor (Thermo Fisher Scientific) (Whiteaker et al., 2010), and Hamilton liquid handling workstations (Microlab STAR Hamilton) (van den Broek et al., 2015). Each of these liquid handling workstations have demonstrated that automation can achieve rapid tryptic digestion and immunocapture with inter‐ and intra‐day CVs of <10%.
In addition, one advantage of iMRM antipeptide antibody capture is that the proteolysis before capture exposes previously inaccessible epitopes. Therefore, iMRM can be used to quantify clinically relevant plasma biomarkers with minimal autoantibody interference. For example, studies using thyroglobulin, a circulating tumor marker that has been used to evaluate the effectiveness of thyroid cancer treatment, have portrayed false‐negative results in antibody‐based immunoassays due to the presence of antithyroglobulin autoantibodies, which blocked the epitope of interest (Hoofnagle et al., 2008). In response, an automated iMRM assay sample preparation workflow was developed to measure specific thyroglobulin tryptic peptides (Kushnir et al., 2013). After trypsin digestion of the plasma proteins, selective thyroglobulin specific peptides were enriched using a peptide‐specific antibody via an automation liquid handler workstation (epMotion from Eppendorf). After enrichment, the peptides were submitted for LC‐MS for the quantification of endogenous thyroglobulin peptides along with internal peptide standards, and this method was able to satisfy performance criteria required for clinical diagnosis (CV < 20%) (Kushnir et al., 2013).
4.5 |. Protein extraction from cells or tissues
Any automation workstations that include proteolytic digestion of cells, solid tumors, and/or tissues often require several additional processing steps to be included when compared to sample processing of body fluids. This includes steps required for tissues or tumors to be disrupted, followed by cell lysis, DNA shearing, and protein extraction before protein denaturation and subsequent digestion steps as discussed above. Recently, Müller et al. developed a method to integrate an adaptive‐focused acoustics (AFA) Technology and single‐pot solid‐phase enhanced sample preparation (SP3) on liquid handling workstations for automated processing of tissue lysates in a 96‐well format (Muller et al., 2020). AFA ultrasonication is an advanced acoustic technology which enables the mechanical processing of samples through focused ultrasonication in a 96‐well plate format. SP3 utilizes paramagnetic beads in the presence of an organic solvent (>50% ACN or EtOH) to promote protein binding to the beads and allows extensive washes to eliminate contaminants and harsh detergents. The resulting workflow is an end‐to‐end automation of the sample preparation for cells or tissues (Muller et al., 2020). A recently reported SP2 workflow (an alternative adaptation of carboxylate‐modified paramagnetic particles for peptide isolation utilizing a liquid handler (epMotion workstation, Eppendorf)) was efficient in removing contaminants (e.g., detergents and salts) and additionally cleaning up peptides before mass spectrometry injection (Waas et al., 2019).
4.6 |. COVID‐19
The COVID‐19 pandemic highlights the need and urgency for the integration of the automated sample preparation into the clinical laboratory. The pandemic has accelerated the need for rapid and accurate automated RNA based assays in the clinical laboratory (Tauschmann & Hovorka, 2018). Most recently, a few reports were published in using mass spectrometry‐based for COVID‐19 related screening in clinical laboratories (Cardozo et al., 2020; Jayawardena et al., 2021; Rajczewski et al., 2021; Renuse et al., 2021). One study by Cardozo et al. developed an automated sample preparation method using a robotic liquid handler combined with a high throughput targeted LC‐MS/MS assay to detect SARS‐Cov‐2 nucleoprotein peptides from nasopharyngeal and oropharyngeal swabs (Cardozo et al., 2020). The automated digestion of 96 samples could be completed in 4 h and by multiplexing four samples per 10 min LC‐MS/MS run, 500 samples per day could be analyzed. Another study utilized PRM‐MS to develop a test for COVID‐19 spike glycoprotein and nucleoprotein, the sample preparation was accomplished by an automated workflow with immunoaffinity‐based enrichment and in‐solution digestion (Renuse et al., 2021). The mass spectrometry analysis was done with a high resolution PRM‐MS‐based workflow to develop a test for COVID‐19 spike glycoprotein and nucleoprotein.
5 |. AUTOMATION IN CLINICAL PROTEOMICS‐FUTURE PROSPECTIVE
Beyond liquid handling of the proteolytic digestion, there are many other steps in the sample preparation process that could benefit from automation. For example, automated sample receiving processes (bar coding and verification) that could aliquot, store, and maintain an archive would streamline the management of large clinical cohorts. Automated solutions for transport and loading specimens into the sample preparation workstation would also eliminate many time‐consuming (capping and recapping tubes) and error prone steps. Furthermore, the evolution of a fully automated process would include the standardization of high quality, prepackaged, ready‐to‐use reagents. The continued development of robotics, liquid handling, mass spectrometry analyzers, data handling, and reporting in proteomics labs will ultimately reduce time and cost of preparation while improving reliability and precision of the subsequent analysis.
Rapid advances in MS technology combined with various automated sample preparation workflows suggests these techniques will become more prominent in routine clinical laboratories. A prime example is the relatively recent availability of high‐resolution, accurate MS instruments that can accommodate multiplex analysis. Currently, there are multiple automated sample preparation methods available to supply the various discovery (DDA‐MS, Zhang et al., 2013; DIA‐MS, Gillet et al., 2012) and targeted (PRM‐MS, Gallien et al., 2015; MRM/SRM‐MS, SQ‐MS, Liebler & Zimmerman, 2013) MS acquisition methods. However, automated tools and workflows will need to remain flexible to meet the challenges presented by emerging MS methods. Developing assays that combine automated sample preparation and LC‐MS/MS that utilize high multiplexing capacity, high analytical specificity, and high sensitivity are key for moving proteomic technologies from the basic research realm to the clinical laboratory and into precision medicine.
A fundamental requirement in the evaluation of biological material is its refinement to an analysis ready state. In proteomics, automation of the sample perpetration will increase the uniformity and precision of that process, which is critical for reliable quantification of proteins for biomarker discovery, validation, preclinical, clinical, and pharmaceutical studies. Developing and optimizing sample preparation workflows with precision, reproducibility and robustness is essential in the implementation in routine clinical laboratories. The constant improvement of automation technologies for proteomic sample preparation in a true high‐throughput manner is key in integrating mass spectrometry and proteomics into personalized medicine.
ACRONYMS
- ACN
acetonitrile
- AFA
adaptive focused acoustics
- CSF
cerebrospinal fluid
- CV
coefficient of variation
- DBS
dried blood spot
- DDA‐MS
data‐dependent mass spectrometry
- DIA‐MS
data‐independent mass spectrometry
- EtOH
ethanol
- FDA
US Food and Drug Administration
- IA
immunoaffinity
- iMRM
immuno‐multiple reaction monitoring
- LC
liquid chromatography
- LC‐MS
liquid chromatography mass spectrometry
- mABs
monoclonal antibodies
- MRM
multiple reaction monitoring
- MRM‐MS
multiple reaction monitoring mass spectrometry
- MS
mass spectrometry
- PTMs
posttranslational modifications
- SIL peptides
stable isotope labeled peptides
- SISCAPA
stable isotope standards and capture by antipeptide antibodies
- SISPROT
simple and integrated spin‐tip proteomics technology
- SQ‐MS
sure quant mass spectrometry
- SRM‐MS
selected reaction monitoring mass spectrometry
- TOF
time‐of‐flight
- VAMS
volumetric absorptive microsampling
Biographies
Qin Fu earned her PhD degree in Genetics from the University of Minnesota and received postdoctoral training in Immunology at the University of California, San Francisco. She has research experience in human genetics, autoimmunity, drug resistance, cardiovascular diseases, and biomarker discovery and validation. Her current research interests include automated MS sample preparation, multiplexed targeted MS assays, and biomarker discovery. Dr. Fu is the Director of the High‐Throughput Laboratory in the Advanced Clinical Biosystems Research Institute at Cedars Sinai Medical Center.

Christopher Murray received a PhD in Biological Chemistry from Johns Hopkins in 2012, where he investigated quantitative measures for oxidative posttranslational modifications in heart failure. He did is postdoctoral work in biophysics examining dynamic protein structure in cardiac voltage gated ion channels using unnatural amino acids. His current research interests include MS‐based structural proteomics and automation.

Oleg A. Karpov has received his BSc and MBiol degrees in Medical Biochemistry at the University of Leeds working at the Astbury Centre for Structural Molecular Biology studying novel approaches for the inhibition of cancer angiogenesis. Oleg then went on to receive his PhD in Chemistry from Imperial College London working on the identification of novel kinase substrates using non‐canonical amino acids in tandem with mass spectrometry. Currently, he is based at Jennifer Van Eyk’s group at Cedars‐Sinai Medical Center, specializing in the application of chemical biology approaches paired with mass spectrometry for the elucidation of molecular mechanisms of COVID19 and cardiovascular disease.

Jennifer (Jenny) Van Eyk, PhD is a Professor of Medicine within the Smidt Heart Institute at Cedars‐Sinai Medical Center, in Los Angeles, CA. She is the Director of Advanced Clinical Biosystems Research Institute and of the Precision Biomarker Laboratories and is the inaugural Erika Glazer Endowed Chair in Women’s Heart Health. Dr. Van Eyk is an international leader in the area of proteomics and her lab has focused the developing technical pipelines to precisely quantify proteins and their posttranslational modifications to better define and understand the disease‐induced interplay between cellular pathways. Collectively, these tools are used in many disease contexts to provide better therapeutic interventions and robust biomarkers for the diagnosis, prognosis and risk stratification.

REFERENCES
- Adachi J, Kumar C, Zhang Y, Olsen JV, and Mann M. 2006. ‘The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins’, Genome Biol, 7: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi E, Griss J, da Silva AW, Bely B, Antunes R, Zellner H, Rios D, O’Donovan C, Vizcaino JA, and Martin MJ. 2015. ‘Analysis of the tryptic search space in UniProt databases’, Proteomics, 15: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An B, Zhang M, Pu J, Qu Y, Shen S, Zhou S, Ferrari L, Vazvaei F, and Qu J. 2020. ‘Toward accurate and robust liquid chromatography‐mass spectrometry‐based quantification of antibody biotherapeutics in tissues’, Anal Chem, 92: 15152–61. [DOI] [PubMed] [Google Scholar]
- Anderson L, Razavi M, Pope ME, Yip R, Cameron LC, Bassini‐Cameron A, and Pearson TW. 2020. ‘Precision multiparameter tracking of inflammation on timescales of hours to years using serial dried blood spots’, Bioanalysis, 12: 937–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam B, Basit M, Nisar MA, Khurshid M, and Rasool MH. 2017. ‘Proteomics: Technologies and Their Applications’, J Chromatogr Sci, 55: 182–96. [DOI] [PubMed] [Google Scholar]
- Campino C, Pumarino H, Vidal V, Generini G, and Bruggendieck B. 1987. ‘Effect of sodium valproate and magnesium on the secretion of various pituitary hormones’, Medicina, 47: 328–9. [PubMed] [Google Scholar]
- Cardozo KHM, Lebkuchen A, Okai GG, Schuch RA, Viana LG, Olive AN, Lazari CDS, Fraga AM, Granato CFH, Pintao MCT, and Carvalho VM. 2020. ‘Establishing a mass spectrometry‐based system for rapid detection of SARS‐CoV‐2 in large clinical sample cohorts’, Nat Commun, 11: 6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AG, Percy AJ, Yang J, and Borchers CH. 2015. ‘Multiple reaction monitoring enables precise quantification of 97 proteins in dried blood spots’, Mol Cell Proteomics, 14: 3094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EI, Cociorva D, Norris JL, and Yates JR. 2007. ‘Optimization of mass spectrometry‐compatible surfactants for shotgun proteomics’, J Proteome Res, 6: 2529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Chen L, and Tian R. 2018. ‘An integrated strategy for highly sensitive phosphoproteome analysis from low micrograms of protein samples’, Analyst, 143: 3693–701. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang S, Adhikari S, Deng Z, Wang L, Chen L, Ke M, Yang P, and Tian R. 2016. ‘Simple and integrated spintip‐ based technology applied for deep proteome profiling’, Anal Chem, 88: 4864–71. [DOI] [PubMed] [Google Scholar]
- Chung J 2017. ‘Special issue on therapeutic antibodies and biopharmaceuticals’, Exp Mol Med, 49: e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Doll S, Bech JM, Schweizer L, Mund A, Lengyel E, Lindebjerg J, Madsen GI, Moreira JM, and Mann M. 2020. ‘A streamlined mass spectrometry‐based proteomics workflow for large‐scale FFPE tissue analysis’, J Pathol, 251: 100–112. [DOI] [PubMed] [Google Scholar]
- Crankshaw MW, and Grant GA. 2001. ‘Modification of cysteine’, Curr Protoc Protein Sci, Chapter 15: Unit15 1. [DOI] [PubMed] [Google Scholar]
- DeMarco ML, Nguyen Q, Fok A, Hsiung GR, and van der Gugten JG. 2020. ‘An automated clinical mass spectrometric method for identification and quantification of variant and wild‐type amyloid‐beta 1‐40 and 1‐42 peptides in CSF’, Alzheimers Dement, 12: e12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdzal S, Rosik J, Lechowicz K, Machaj F, Kotfis K, Ghavami S, and Los MJ. 2020. ‘FDA approved drugs with pharmacotherapeutic potential for SARS‐CoV‐2 (COVID‐19) therapy’, Drug Resist Update, 53: 100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Shi Z, Pan Y, Song Z, Zhang W, Zhao X, Tian F, Peng B, Qin W, Cai Y, and Qian X. 2014. ‘Dual matrix‐based immobilized trypsin for complementary proteolytic digestion and fast proteomics analysis with higher protein sequence coverage’, Anal Chem, 86: 1452–8. [DOI] [PubMed] [Google Scholar]
- Fu Q, Kowalski MP, Mastali M, Parker SJ, Sobhani K, van den Broek I, Hunter CL, and Van Eyk JE. 2018. ‘Highly reproducible automated proteomics sample preparation workflow for quantitative mass spectrometry’, J Proteome Res, 17: 420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk J, Li X, and Franz T. 2005. ‘Threshold values for detergents in protein and peptide samples for mass spectrometry’, Rapid Commun Mass Spectrom, 19: 2986–8. [DOI] [PubMed] [Google Scholar]
- Gallien S, Kim SY, and Domon B. 2015. ‘Large‐scale targeted proteomics using internal standard triggered‐parallel reaction monitoring (IS‐PRM)’, Mol Cell Proteomics, 14: 1630‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, and Mann M. 2016. ‘Plasma proteome profiling to assess human health and disease’, Cell Syst, 2: 185–95. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R. 2012. ‘Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: A new concept for consistent and accurate proteome analysis’, Mol Cell Proteomics, 11: 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant RP, and Hoofnagle AN. 2014. ‘From lost in translation to paradise found:Enabling protein biomarker method transfer by mass spectrometry’, Clin Chem, 60: 941–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, and Van Eyk JE. 2009. ‘Preparation of proteins and peptides for mass spectrometry analysis in a bottom‐up proteomics workflow’, Curr Protoc Mol Biol, Chapter 10: Unit10 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HaileMariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, and Yu Y. 2018. ‘S‐trap, an ultrafast sample‐ preparation approach for shotgun proteomics’, J Proteome Res, 17: 2917–24. [DOI] [PubMed] [Google Scholar]
- Hamdan M, Bordini E, Galvani M, and Righetti PG. 2001. ‘Protein alkylation by acrylamide, its N‐substituted derivatives and cross‐linkers and its relevance to proteomics: A matrix assisted laser desorption/ionization‐time of flight‐mass spectrometry study’, Electrophoresis, 22: 1633–44. [DOI] [PubMed] [Google Scholar]
- Hill BG, Reily C, Oh JY, Johnson MS, and Landar A. 2009. ‘Methods for the determination and quantification of the reactive thiol proteome’, Free Radic Biol Med, 47: 675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle AN, Becker JO, Wener MH, and Heinecke JW. 2008. ‘Quantification of thyroglobulin, a low‐abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry’, Clin Chem, 54: 1796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena I, Wilson K, Plebanski M, Grondahl L, and Corrie S. 2021. ‘Morphology and composition of immunodiffusion precipitin complexes evaluated via microscopy and proteomics’, J Proteome Res, 20: 2618–27. [DOI] [PubMed] [Google Scholar]
- Johnson CW, Fu Q, Van Eyk JE, Enane FO, Kowalski M, Wijayawardena B 2020. ‘Fully Automated peptide desalting for liquid chromatography–tandem mass spectrometry analysis using Beckman Coulter Biomek i7 hybrid workstation’, Technical note, Beckman Coulter Life Science, Document # AAG-7941APP09.20. [Google Scholar]
- Krasny L, and Huang PH. 2021. ‘Data‐independent acquisition mass spectrometry (DIA‐MS) for proteomic applications in oncology’, Mol Omics, 17: 29–42. [DOI] [PubMed] [Google Scholar]
- Kushnir MM, Rockwood AL, Roberts WL, Abraham D, Hoofnagle AN, and Meikle AW. 2013. ‘Measurement of thyroglobulin by liquid chromatography‐tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies’, Clin Chem, 59: 982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange V, Picotti P, Domon B, and Aebersold R. 2008. ‘Selected reaction monitoring for quantitative proteomics: A tutorial’, Mol Syst Biol, 4: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassman ME, McAvoy T, Lee AY, Chappell D, Wong O, Zhou H, Reyes‐Soffer G, Ginsberg HN, Millar JS, Rader DJ, Gutstein DE, and Laterza O. 2014. ‘Practical immunoaffinity‐enrichment LC‐MS for measuring protein kinetics of low‐abundance proteins’, Clin Chem, 60: 1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Gonzalez‐Lozano MA, Koopmans F, and Smit AB. 2020. ‘Recent developments in data independent acquisition (DIA) mass spectrometry: Application of quantitative analysis of the brain proteome’, Front Mol Neurosci, 13: 564446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebler DC, and Zimmerman LJ. 2013. ‘Targeted quantitation of proteins by mass spectrometry’, Biochemistry, 52: 3797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo RR, Dales N, and Andrews PC. 1996. ‘The effect of detergents on proteins analyzed by electrospray ionization’, Methods Mol Biol, 61: 141–60. [DOI] [PubMed] [Google Scholar]
- Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, and Aebersold R. 2018. ‘Data‐independent acquisition‐based SWATH‐MS for quantitative proteomics: A tutorial’, Mol Syst Biol, 14: e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NJ, Bunch J, and Cooper HJ. 2013. ‘Dried blood spot proteomics: Surface extraction of endogenous proteins coupled with automated sample preparation and mass spectrometry analysis’, J Am Soc Mass Spectrom, 24: 1242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle A, Binek A, Moradian A, Chazarin Orgel B, Rivas A, Washington KE, Phebus C, Manalo D‐M, Go J, a Venkatraman V, Johnson C, Fu Q, Cheng S, Raedschelders K, Bober J‐F, Pennington SR, Murray CI, and Van Eyk JE. 2021. ‘Standardized workflow for precise mid‐ and high‐throughput proteomics of blood biofluids’, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, and Kelleher NL. 2002. ‘Processing complex mixtures of intact proteins for direct analysis by mass spectrometry’, Anal Chem, 74: 2923–9. [DOI] [PubMed] [Google Scholar]
- Millan‐Martin S, Jakes C, Carillo S, Buchanan T, Guender M, Kristensen DB, Sloth TM, Orgaard M, Cook K, and Bones J. 2020. ‘Inter‐laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes’, Anal Bioanal Chem, 412: 6833–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T, Kalxdorf M, Longuespee R, Kazdal DN, Stenzinger A, and Krijgsveld J. 2020. ‘Automated sample preparation with SP3 for low‐input clinical proteomics’, Mol Syst Biol, 16: e9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T, and Winter D. 2017. ‘Systematic evaluation of protein reduction and alkylation reveals massive unspecific side effects by iodine‐containing reagents’, Mol Cell Proteomics, 16: 1173‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappireddi N, Martin L, and Wuhr M. 2019. ‘A review on quantitative multiplexed proteomics’, Chembiochem, 20: 1210–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petritis BO, Qian WJ, Camp DG, and Smith RD. 2009. ‘A simple procedure for effective quenching of trypsin activity and prevention of 18O‐labeling back‐exchange’, J Proteome Res, 8: 2157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proc JL, Kuzyk MA, Hardie DB, Yang J, Smith DS, Jackson AM, Parker CE, and Borchers CH. 2010. ‘A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin’, J Proteome Res, 9: 5422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psillakis E 2017. ‘Vacuum‐assisted headspace solid‐phase microextraction: A tutorial review’, Anal Chim Acta, 986: 12–24. [DOI] [PubMed] [Google Scholar]
- Rajczewski AT, Mehta S, Nguyen DDA, Gruning B, Johnson JE, McGowan T, Griffin TJ, and Jagtap PD. 2021. ‘A rigorous evaluation of optimal peptide targets for MS‐based clinical diagnostics of Coronavirus Disease 2019 (COVID‐19)’, Clin Proteomics, 18: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi M, Farrokhi V, Yip R, Anderson NL, Pearson TW, and Neubert H. 2019. ‘Measuring the turnover rate of clinically important plasma proteins using an automated SISCAPA workflow’, Clin Chem, 65: 492–94. [DOI] [PubMed] [Google Scholar]
- Razavi M, Leigh Anderson N, Pope ME, Yip R, and Pearson TW. 2016. ‘High precision quantification of human plasma proteins using the automated SISCAPA Immuno‐MS workflow’, N Biotechnol, 33: 494–502. [DOI] [PubMed] [Google Scholar]
- Reiersen H, and Rees AR. 2000. ‘Trifluoroethanol may form a solvent matrix for assisted hydrophobic interactions between peptide side chains’, Protein Eng, 13: 739–43. [DOI] [PubMed] [Google Scholar]
- Renuse S, Vanderboom PM, Maus AD, Kemp JV, Gurtner KM, Madugundu AK, Chavan S, Peterson JA, Madden BJ, Mangalaparthi KK, Mun DG, Singh S, Kipp BR, Dasari S, Singh RJ, Grebe SK, and Pandey A. 2021. ‘A mass spectrometry‐based targeted assay for detection of SARS‐CoV‐2 antigen from clinical specimens’, EBioMedicine, 69: 103465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righetti PG 2006. ‘Real and imaginary artefacts in proteome analysis via two‐dimensional maps’, J Chromatogr B Analyt Technol Biomed Life Sci, 841: 14–22. [DOI] [PubMed] [Google Scholar]
- Rogers JC, and Bomgarden RD. 2016. ‘Sample preparation for mass spectrometry‐based proteomics; from proteomes to peptides’, Adv Exp Med Biol, 919: 43–62. [DOI] [PubMed] [Google Scholar]
- Sechi S, and Chait BT. 1998. ‘Modification of cysteine residues by alkylation. A tool in peptide mapping and protein identification’, Anal Chem, 70: 5150–8. [DOI] [PubMed] [Google Scholar]
- Stopfer LE, Flower CT, Gajadhar AS, Patel B, Gallien S, Lopez‐Ferrer D, and White FM. 2021. ‘High‐density, targeted monitoring of tyrosine phosphorylation reveals activated signaling networks in human tumors’, Cancer Res, 81: 2495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttapitugsakul S, Xiao H, Smeekens J, and Wu R. 2017. ‘Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS‐based proteomics’, Mol Biosyst, 13: 2574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauschmann M, and Hovorka R. 2018. ‘Technology in the management of type 1 diabetes mellitus—Current status and future prospects’, Nat Rev Endocrinol, 14: 464–75. [DOI] [PubMed] [Google Scholar]
- Taylor SW, Clarke NJ, Chen Z, and McPhaul MJ. 2016. ‘A high‐ throughput mass spectrometry assay to simultaneously measure intact insulin and C‐peptide’, Clin Chim Acta, 455: 202–8. [DOI] [PubMed] [Google Scholar]
- van den Broek I, Mastali M, Mouapi K, Bystrom C, Bairey Merz CN, and Van Eyk JE. 2020. ‘Quality Control and Outlier Detection of Targeted Mass Spectrometry Data from Multiplex Protein Panels’, J Proteome Res, 19: 2278–93. [DOI] [PubMed] [Google Scholar]
- van den Broek I, Nouta J, Razavi M, Yip R, Bladergroen MR, Romijn FP, Smit NP, Drews O, Paape R, Suckau D, Deelder AM, van der Burgt YE, Pearson TW, Anderson NL, and Cobbaert CM. 2015. ‘Quantification of serum apolipoproteins A‐I and B‐100 in clinical samples using an automated SISCAPA‐MALDI‐TOF‐MS workflow’, Methods, 81: 74–85. [DOI] [PubMed] [Google Scholar]
- van den Broek I, Romijn FP, Nouta J, van der Laarse A, Drijfhout JW, Smit NP, van der Burgt YE, and Cobbaert CM. 2016. ‘Automated multiplex LC‐MS/MS assay for quantifying serum apolipoproteins A‐I, B, C‐I, C‐II, C‐III, and E with qualitative apolipoprotein E phenotyping’, Clin Chem, 62: 188–97. [DOI] [PubMed] [Google Scholar]
- van den Broek I, Romijn FP, Smit NP, van der Laarse A, Drijfhout JW, van der Burgt YE, and Cobbaert CM. 2015. ‘Quantifying protein measurands by peptide measurements: Where do errors arise?’, J Proteome Res, 14: 928–42. [DOI] [PubMed] [Google Scholar]
- van den Broek I, Qin F, Stuart K, Kowalski PM, Millis K, Andrew Percy, Ronald JH, Vidya V, Jennifer E. 2017. ‘Application of volumetric absorptive microsampling for robust, high‐throughput mass spectrometric quantification of circulating protein biomarkers’, Clinical Mass Spectrometry, 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidova V, and Spacil Z. 2017. ‘A review on mass spectrometry‐based quantitative proteomics: Targeted and data independent acquisition’, Anal Chim Acta, 964: 7–23. [DOI] [PubMed] [Google Scholar]
- Waas M, Pereckas M, Jones Lipinski RA, Ashwood C, and Gundry RL. 2019. ‘SP2: Rapid and automatable contaminant removal from peptide samples for proteomic analyses’, J Proteome Res, 18: 1644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DM, Tran D, Goldman SM, Taylor SW, Ginns EI, Lagier RJ, Rissman RA, Brewer JB, and Clarke NJ. 2019. ‘High‐throughput mass spectrometry assay for quantifying beta‐amyloid 40 and 42 in cerebrospinal fluid’, Clin Chem, 65: 1572–80. [DOI] [PubMed] [Google Scholar]
- Whiteaker JR, Zhao L, Anderson L, and Paulovich AG. 2010. ‘An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry‐based quantification of protein biomarkers’, Mol Cell Proteomics, 9: 184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YQ, Gilar M, Lee PJ, Bouvier ES, and Gebler JC. 2003. ‘Enzyme‐friendly, mass spectrometry‐compatible surfactant for in‐solution enzymatic digestion of proteins’, Anal Chem, 75: 6023–8. [DOI] [PubMed] [Google Scholar]
- Zacchi LF, Recinos DR, Otte E, Aitken C, Hunt T, Sandford V, Lee YY, Schulz BL, and Howard CB. 2020. ‘S‐Trap eliminates cell culture media polymeric surfactants for effective proteomic analysis of mammalian cell bioreactor supernatants’, J Proteome Res, 19: 2149–58. [DOI] [PubMed] [Google Scholar]
- Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, and Paulovich AG. 2019. ‘Clinical potential of mass spectrometry‐based proteogenomics’, Nat Rev Clin Oncol, 16: 256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fonslow BR, Shan B, Baek MC, and Yates JR. 2013. ‘Protein analysis by shotgun/bottom‐up proteomics’, Chem Rev, 113: 2343–94. [DOI] [PMC free article] [PubMed] [Google Scholar]