

Abstract
Phase I oncology clinical trials often comprise a limited number of patients representing different disease subtypes who are divided into cohorts receiving treatment(s) at different dosing levels and schedules. Here, we leverage a previously developed quantitative systems pharmacology model of the anti‐CD20/CD3 T‐cell engaging bispecific antibody, mosunetuzumab, to account for different dosing regimens and patient heterogeneity in the phase I study to inform clinical dose/exposure‐response relationships and to identify biological determinants of clinical response. We developed a novel workflow to generate digital twins for each patient, which together form a virtual population (VPOP) that represented variability in biological, pharmacological, and tumor‐related parameters from the phase I trial. Simulations based on the VPOP predict that an increase in mosunetuzumab exposure increases the proportion of digital twins with at least a 50% reduction in tumor size by day 42. Simulations also predict a left‐shift of the exposure‐response in patients diagnosed with indolent compared to aggressive non‐Hodgkin's lymphoma (NHL) subtype; this increased sensitivity in indolent NHL was attributed to the lower inferred values of tumor proliferation rate and baseline T‐cell infiltration in the corresponding digital twins. Notably, the inferred digital twin parameters from clinical responders and nonresponders show that the potential biological difference that can influence response include tumor parameters (tumor size, proliferation rate, and baseline T‐cell infiltration) and parameters defining the effect of mosunetuzumab on T‐cell activation and B‐cell killing. Finally, the model simulations suggest intratumor expansion of pre‐existing T‐cells, rather than an influx of systemically expanded T‐cells, underlies the antitumor activity of mosunetuzumab.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The complex pharmacology of T‐cell engaging bispecifics like mosunetuzumab presents unique challenges to early clinical development, which can be addressed by leveraging a mechanistic quantitative systems pharmacology (QSP) model. A mosunetuzumab QSP model was previously developed and used to support the identification of an appropriate dosing regimen to mitigate the acute safety risk associated with T‐cell engaging bispecifics. Another key challenge is the characterization of dose/exposure‐response relationship given the limited number of patients, high patient heterogeneity, and variation in dosing regimen in phase I dose escalation clinical trials in oncology.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study applied a QSP digital twin approach to characterize the dose/exposure‐response during dose escalation and identify potential pretreatment biomarkers predictive of response.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
We share a novel approach to leveraging limited phase I clinical data by creating a virtual population of digital twins using a mechanistic QSP model. We show how we have used the virtual population (VPOP) to assess exposure response for different clinical indications. Furthermore, based on exploration in the VPOP, we propose potential baseline tumor characteristics as predictive biomarkers of patient response.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
In the absence of a large sample size from the clinical trial, the dose/exposure‐response predicted by the QSP model identified using the QSP digital twin VPOP can supplement empirical modeling approaches during dose escalation to inform dose optimization or dose justification. The robust analysis of dose level/regimen enabled by the complementary approaches is of particular value given the increased attention on dose‐optimization in oncology. Finally, the use of digital twins based on mechanistic QSP models provides a powerful approach to understanding the biological underpinnings of response and related biomarkers.
INTRODUCTION
Non‐Hodgkin's lymphoma (NHL) is one of the leading causes of cancer death in the United States and Europe. 1 , 2 Whereas lymphomas may originate in either B‐cells or T‐cells, those of B‐cell origin constitute ~80%–85% of all NHL cases. Among the B‐cell NHLs, diffuse large B‐cell lymphoma (DLBCL) and follicular lymphoma (FL) are the most common types. 3 , 4 FL is considered an indolent disease and the histological transformation of FL can lead to more aggressive diseases, such as DLBCL, with poor clinical outcomes. 4
Mosunetuzumab, a full‐length IgG bispecific antibody targeting both CD3 (on the surface of T cells) and CD20 (on the surface of B‐cells), belongs to the class of T‐cell engaging bispecific antibodies that are emerging as novel and important therapeutic modalities in oncology. In this regard, mosunetuzumab is a conditional agonist that redirects patients' endogenous T‐cells to kill malignant B‐cells only upon simultaneous binding to both targets. 5 Simultaneous engagement of both arms of mosunetuzumab results in the formation of an immunologic synapse between a target B‐cell and a cytotoxic T‐cell resulting in T‐cell activation in a target‐ and dose‐dependent manner. 5
The complex pharmacology of T‐cell engaging bispecifics like mosunetuzumab presents many unique challenges to early clinical development, some of which can be addressed by leveraging a mechanistic quantitative systems pharmacology (QSP) modeling to supplement data‐driven pharmacometric approaches:
One key challenge for early clinical drug development of mosunetuzumab was the identification of an appropriate dosing regimen that could improve the therapeutic window, given the known acute safety risks of cytokine release syndrome (CRS). 6 , 7 We previously developed and validated a QSP model for mosunetuzumab in NHL, integrating a diverse set of in vitro, in vivo preclinical, and clinical blinatumomab data to capture the critical interactions related to the drug mechanism of action for both target cell killing (efficacy) and cytokine production (CRS risk). 8 The model was then used to inform the phase Ia/Ib dose escalation design by proposing pharmacologically plausible step‐up dosing regimens to reduce the risk of CRS by balancing efficacy considerations. This model‐based evaluation of the step‐up dosing regimen was used to support the initiation of step‐up dosing regimen cohorts in study GO29781 in patients with relapsing/refractory (r/r) NHL (ClinicalTrials.gov ID: NCT02500407). Clinical data generated from GO29781 (NCT02500407) has since confirmed the CRS mitigation of the QSP model‐informed step‐up dosing regimen. 9 , 10 , 11
In general, another key challenge of early clinical development is the characterization of dose/exposure‐response relationships. During the early‐stage of dose escalation, data are evolving with limited sample size. Further complicating the case for mosunetuzumab, different dosing regimens were used across cohorts (fixed and step‐up dosing administration of mosunetuzumab). Interpretation of potential signals of clinical efficacy with such low number of subjects is further complicated due to patient heterogeneity inherent to phase I trial settings (i.e., variability in disease type, baseline characteristics, and treatment history; e.g., use of prior CD20‐targeting drugs).
The present work applies the concept of “digital twin” 12 using the aforementioned mosunetuzumab QSP model to characterize clinical dose–response relationships and to explore potential underlying biomarkers of mosunetuzumab treatment response. Here, the term digital twin refers to a virtual representation of a mosunetuzumab‐treated clinical patient that integrates patient‐specific clinical data (i.e., pharmacokinetics [PKs], tumor size, and biomarker data) alongside other in vitro/in vivo data within the established mosunetuzumab QSP model. 8 We developed a workflow to generate digital twins, which we refer to in more technical terms as individualized virtual patients (iVPs), whereby the base QSP model was fitted to individual clinical measurements from a single patient in the mosunetuzumab phase I clinical study. Each iVP thus represented one digital twin that reproduced the clinical measurement for the corresponding clinical patient. In order to address biological uncertainty and underspecification of the model, multiple digital twins with different parameterizations of the model were identified for each clinical patient. The collection of all digital twins to all trial patients then forms a virtual population (VPOP) that accounts for the patient heterogeneity from the phase I trial encompassing variability in biological, pharmacological, and tumor‐related parameters. This enabled us to simulate the response of the VPOP to mosunetuzumab at different dosing regimens to more robustly identify the clinical dose–response and propose predictive biomarkers to distinguish responders from nonresponders within the VPOP.
Here, we present the novel QSP digital twin workflow, and its application to (1) characterize the dose/exposure‐response relationship of mosunetuzumab in r/r NHL, (2) assess the impact of tumor characteristics in different subtypes of NHL, and (3) propose underlying factors influencing patient responsiveness to mosunetuzumab. The model predictions for the VPOP were used in conjunction with other modeling approaches, such as pharmacometric exposure‐response modeling, to increase confidence and better inform clinical dose selection decisions.
METHODS
Clinical data
Study GO29781 (NCT02500407) is a first‐in‐human, multicenter, open‐label, phase I/II dose‐escalation and expansion study evaluating the efficacy, safety, tolerability, and PKs of mosunetuzumab in patients with r/r NHL. 13 The study protocol was approved by institutional review boards at each center. The trial was done in accordance with the Declaration of Helsinki, International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable laws and regulations. We obtained written informed consent from all eligible patients.
In this work, iVPs were generated from clinical data from 140 patients in dose escalation cohorts (clinical data cutoff September 2019) who received mosunetuzumab monotherapy (Table S1) in two dosing regimens specified in the Table S2. The dose/exposure‐response were assessed for the two most common subtypes: DLBCL and transformed FL (N = 73), and FL (N = 50). Clinical response in study GO29781 was determined using the Cheson 2007 criteria for patients with NHL where partial response is defined as greater than or equal to 50% decrease in post‐treatment tumor measurement (SPD) of up to six largest dominant masses.
QSP model description
We utilized the mosunetuzumab QSP model developed by Hosseini et al. 8 Briefly, this model describes the dynamics of B‐cells and CD8+ T‐cells and their interactions with mosunetuzumab or blinatumomab in multiple physiological compartments (peripheral blood, tumor, and lymphoid tissues including the spleen, lymph nodes, and bone marrow). Physiological, mechanistic, PK, and pharmacodynamic data were all integrated into the model, including: (1) multiple activation states of CD8+ T‐cells; (2) CD19 + CD20‐ (pro‐B), and CD19 + CD20+ (pre‐ to mature‐B) B‐cells; and (3) the PKs of mosunetuzumab and blinatumomab and their mechanistic effects (activation of CD8+ T‐cells and consequent killing of CD20+ and CD19+ B‐cells, respectively). The model captures T‐cell activation, cytokine release, and target B‐cell killing to enable concurrent prediction of efficacy‐ and safety‐related biomarkers. The QSP model parameters were calibrated based on mosunetuzumab in vitro and cynomolgus monkeys (cyno) 5 data (Figure 1a) and published blinatumomab clinical data (Figure 1b). The work presented here provides novel insights that builds on the cyno virtual cohort generation in Hosseini et al. 8 through the integration of patient‐level clinical data from the mosunetuzumab phase I study GO29781 (Figure 1c). The model was developed in SimBiology and Matlab (MathWorks).
FIGURE 1.
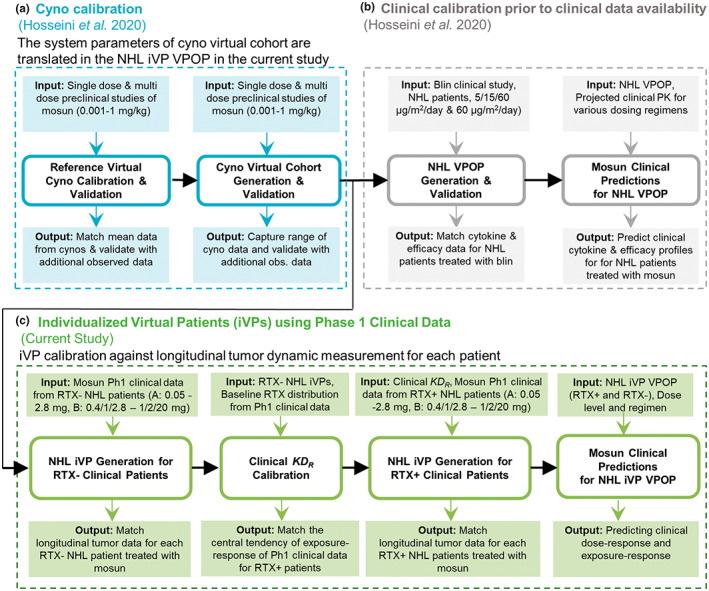
Model development workflow. The (a) cyno calibration and (b) clinical IL‐6 calibration based on published blinatumomab data was presented in Hosseini et al. 8 (c) The current study first translates biological variability from the virtual cynos to healthy individuals and then generates a VPOP consisting of iVPs based on mosunetuzumab phase I clinical data. iVP, individualized virtual patient; Mosun, mosunetuzumab; NHL, non‐Hodgkin's lymphoma; Ph1, phase I; PK, pharmacokinetic; RTX−, baseline rituximab less than lower level of quantification; RTX+, detectable baseline rituximab; VPOP, virtual population.
Transient binding competition of mosunetuzumab and rituximab
The majority of the enrolled patients with NHL in study GO29781 were previously treated with rituximab (RTX), an anti‐CD20 antibody, and were relapsed or refractory (r/r) to the treatment. Thus, at the start of mosunetuzumab treatment, the patients on the trial had varying levels of residual circulating RTX, depending upon their treatment history, dose, and the time elapsed since their last RTX administration. Because mosunetuzumab and RTX bind to overlapping epitopes on CD20, transient competition of binding to CD20 can occur during mosunetuzumab treatment in some patients until RTX washes out. In vitro, the presence of effector‐less RTX (i.e., a RTX variant that binds to CD20 but cannot kill CD20+ B‐cells) increases the half‐maximal effective concentration (EC50) of mosunetuzumab 5 but does not reduce the maximum killing, suggesting that in the presence of RTX, similar target cell killing can be achieved at a higher mosunetuzumab concentration. In the present QSP model, the effect of competitive antagonist action of RTX on mosunetuzumab activity was accounted for using the Schild method. 14 The effective concentration, [M]eff is defined as the adjusted single‐agent mosunetuzumab concentration where the receptor occupancy of mosunetuzumab (ROM) is equivalent to the ROM in the presence of RTX. The methodology to account for competitive binding in the QSP model is described in the Appendix S1.
Parameter estimation for each clinical patient and generation of iVPs
The VPOP generation workflow is shown in Figure 2. The iVPs, which is the technical terminology for digital twins in this paper, were generated for any clinical patient that had baseline and at least one post‐treatment tumor measurement (SPD). The model parameters for the candidate iVPs can be classified into three categories: system parameters, patient‐specific parameters, and tumor parameters. The system parameters were translated from cyno calibration in Hosseini et al. 8 Refer to the Appendix S1 for descriptions of each category.
FIGURE 2.
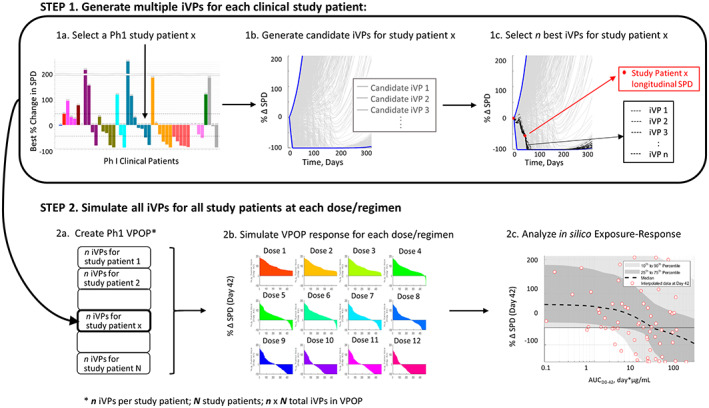
Workflow for VPOP generation, simulation, and exposure‐response analysis from phase I clinical data for the mosunetuzumab QSP model described in Hosseini et al. 8 In step 1, candidate iVPs are generated using the baseline data for each study patient and N final iVPs that best match the study patients' longitudinal response are selected. In step 2, all iVPs for all N study patients are used to create a VPOP that is simulated for each dose regimen, and the simulated data is used to determine the exposure‐response relationship. iVP, individualized virtual patient; Ph1, phase I; QSP, quantitative systems pharmacology; SPD, post‐treatment tumor measurement; VPOP, virtual population.
For each clinical patient, 5000–20,000 candidate iVPs were simulated with the patient's actual dose level and schedule, and the goal was to select 25 iVPs out of all simulated candidate iVPs (Figure 2, step 1). The objective function, defined as the root mean square error between the simulated tumor profiles and observed change from baseline (CFBL) SPD longitudinal data, was computed for each candidate iVP. Out of the candidate iVPs, 25 iVPs with the lowest objective function were chosen. In cases where there were numerous iVPs that produced low objective function (<0.05), 25 iVPs were randomly chosen from this low objective function pool to represent the clinical patient. The iVPs generated for all clinical patients formed the phase I VPOP, allowing the prediction of the VPOP response at a dose level/regimen (Figure 2, step 2).
The VPOP simulation procedure can be found in the Appendix S1.
RESULTS
Generation of iVPs and VPOP
The r/r NHL VPOP consists of iVPs (i.e., digital twins) calibrated to match 140 clinical patients from fixed dosing and step‐up dosing cohorts from the clinical study, GO29781. To address the underspecification of the model, for each clinical patient, 25 iVPs were generated to explore alternate parameterizations of the QSP model that could reasonably match the patient's tumor data. Because the iVPs are generated from multiple sets of system parameters translated from virtual cynos, with patient‐specific parameters and additional variability in tumor superimposed (Appendix S1), they can yield different longitudinal tumor predictions at other dose levels. The iVPs for two clinical patients (one responder and one nonresponder) are used to demonstrate the effect of alternate parameterization (Figure S2). In the two examples, all iVPs for each clinical patient reasonably capture the patient's longitudinal SPD and together yield a range of predictions for the patient's response across exposure levels (Figure S2C,I). The number of SPD observations available per patient was 3 ([2, 7]) ([10th, 90th percentile]). The three tumor parameters in the VPOP appeared to be weakly correlated (Pearson correlation coefficient < 0.4). The iVP generation procedure produced at least 25 iVPs with objective function less than 0.1 for 73% of clinical patients. Fitting to some clinical patients, especially those with an initial response followed by a rebound resulted in higher objective functions because the model does not include representation of potential emergent drug resistance. The iVPs for all 140 clinical patients were included in the VPOP. Our workflow involved generation of a data and fitting dashboard for each patient to allow us to perform visual qualitative checks and monitor the goodness of fit from model calibration. Examples of the dashboard can be found in Figure S2.
Characterization of the exposure‐response and dose–response of mosunetuzumab in the DLBCL and FL patient populations using iVPs
The QSP‐based exposure‐response plot in Figure 3 shows the relationship between area under the curve (AUC) of [M]eff (AUCeff) from 0 to day 42 and the SPD reduction, generated by simulating the VPOP response to a q3w dosing regimen at different dose‐levels. Over the range of dose levels assessed at the time of datacut (target dose of 0.05–27 mg), higher mosunetuzumab exposure leads to a greater simulated decrease in SPD on day 42 for both DLBCL and FL populations. The model simulations capture the variability of response in both populations as highlighted by the 25th–75th and 10th–90th percentile regions. In addition, the median of simulation profiles captured the central tendency of the data. This agreement provides additional confidence that the VPOP was a reasonable representation of the phase I study population. The central tendency of the FL VPOP (Figure 3b) shifted to the left compared to DLBCL (Figure 3a), even after accounting for the competition for CD20 between mosunetuzumab against residual RTX. This trend is consistent with the logistic regression exposure‐response evaluations from Li et al., 13 which found the EC50 of receptor occupancy of CD20 by mosunetuzumab required for tumor response was lower in FL compared to DLBCL for both overall response rate (partial response or better) and for complete response rate. The difference between the DLBCL and FL VPOP exposure‐response relationship results from: (1) differences in the model‐estimated tumor parameters in the iVPs from the two groups (Figure 3c–e), and (2) the higher frequency of patients with measurable baseline RTX concentration in the patients with DLBCL (Figure 3f). In particular, the model estimated higher median tumor B‐cell proliferation rate and higher median tumor B:T‐cell ratio (lower baseline T‐cell infiltration) in the DLBCL VPOP compared to FL VPOP.
FIGURE 3.
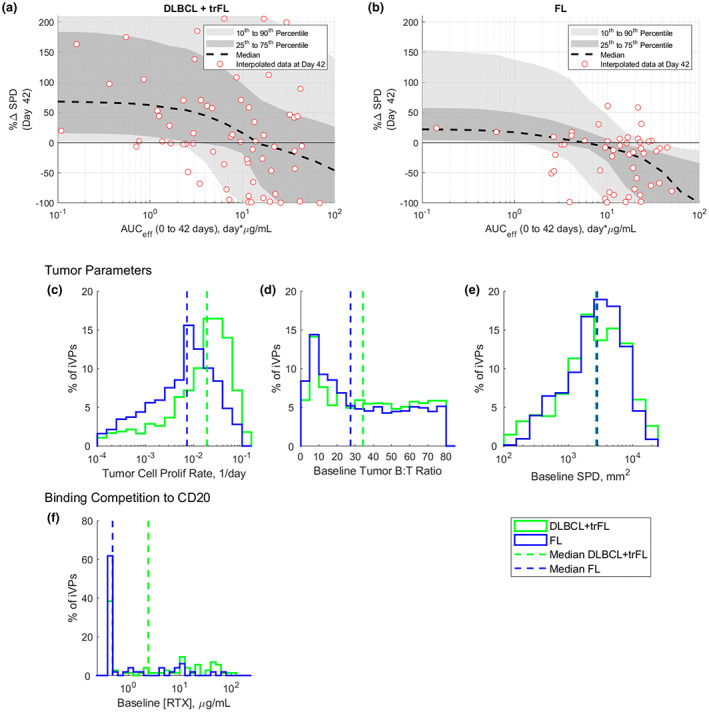
Exposure (effective AUCD0–42) – response (% change in SPD on day 42) for (a) DLBCL and (b) FL populations and the distribution of model estimated tumor parameters, including (c) tumor cell proliferation rate, (d) tumor B:T cell ratio and (e) baseline SPD for DLBCL (green) and FL (blue) populations. Note: (a, b) Red circles are the change from baseline SPD on day 42 for the clinical patients. Black dash line, and dark and light gray shaded regions represent the median, 25th–75th percentile and 10th–90th percentile of the VPOP, respectively. AUCeff, area under the curve (AUC) of [M]eff; DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; iVPs, individualized virtual patients; RTX, rituximab; trFL, transformed follicular lymphoma; SPD, post‐treatment tumor measurement.
Figures 4a and 4b show waterfall plots of the DLBCL and FL VPOP, respectively, at different dose levels. Note that in a clinical study with limited N per cohort, waterfall plots may combine patient data from across several dose levels, whereas, using iVPs, the model can generate a predicted waterfall plot for all patients simulated under any given dosing level/regimen at day 42. As the dose level increases, the waterfall plot shifts to the left. To quantify the extent of the shift, the proportion of iVPs with at least a 50% reduction in SPD was calculated for each dose level and plotted against the cumulative dose (Figure 4c,d). Although the model predicts a steep dose–response relationship at low doses, the increase in the number of iVPs achieving at least 50% reduction in SPD by day 42 gradually diminishes at higher dose levels for both DLBCL and FL populations.
FIGURE 4.
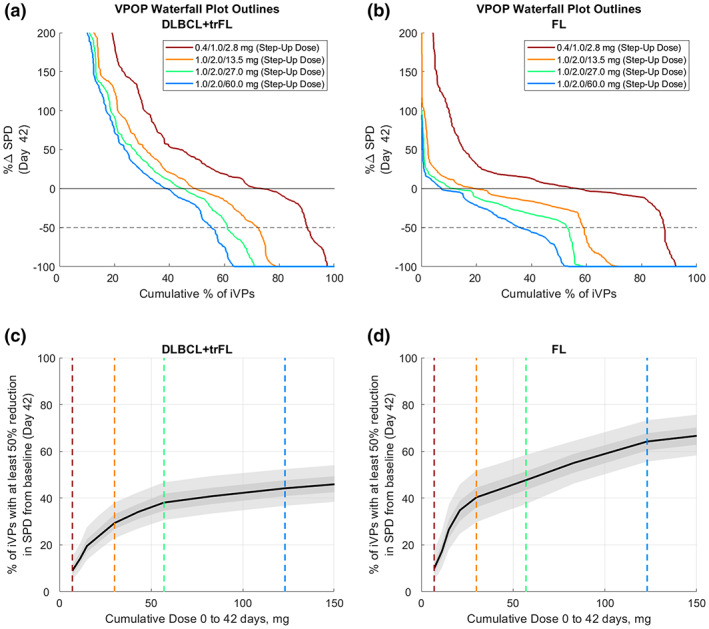
Relationship between dose and response on day 42 for DLBCL+trFL and FL population. Outlines of the waterfall plots of (a) r/r DLBCL+trFL and (b) r/r FL virtual population shows that for higher doses, there is a shift to the left, suggesting that there are more virtual patients achieving 50% reduction in SPD by day 42. (c, d) The plot of percentage of iVPs with at least 50% reduction in SPD further shows the plateau effect with increasing dose levels. Black solid lines and dark and light gray shaded regions represent the VPOP simulation median, 25th–75th percentile and 10th–90th percentile of 500 bootstrap simulations, respectively. DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; iVPs, individualized virtual patients; r/r, relapsing/refractory; SPD, post‐treatment tumor measurement; trFL, transformed follicular lymphoma; VPOP, virtual population.
Although both Figures 3 and 4 show a snapshot of CFBL SPD at day 42, representing the early population response, these results may not capture the maximum tumor reduction as some patients may achieve deeper responses later. We thus evaluated the model‐simulated dose−/exposure‐response at a later timepoint (Figure S3). At day 84, more iVPs achieve at least a 50% reduction in SPD.
Binding competition to CD20 from residual RTX is transient and can be overcome with higher mosunetuzumab exposure
At the beginning of mosunetuzumab treatment, 48% (N = 67/140) of patients showed detectable residual RTX concentration. Until RTX is sufficiently cleared, it lowers the effective concentration of mosunetuzumab due to competitive binding to CD20 (Figure 5a). This transient competition for CD20 between RTX and mosunetuzumab contributes to the shift of the exposure‐response of RTX+ iVPs (Figure 5b) to the right compared to RTX– iVPs (Figure 5d) with AUC of [M] as the exposure metric. It should be noted, however, that the presence of RTX at baseline is not the lone contributor to this shift because the majority of RTX– iVPs are from patients with FL (Figure 3f), and therefore the higher number of FL iVPs in RTX– subset and higher number of DLBCL iVPs in RTX+ subset also contributes to this shift. Because the [M]eff is lower than [M] in the first 42 days of treatment, the AUCeff‐response (Figure 5c) is shifted to the left compared to the AUC‐response curve shown in Figure 5b.
FIGURE 5.
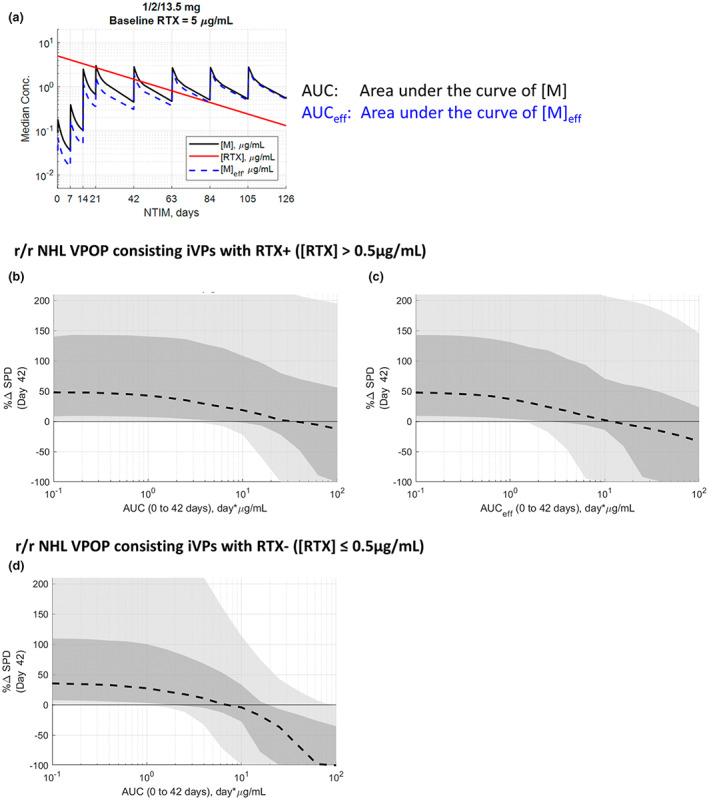
(a) The effective concentration of mosunetuzumab ([M]eff) is dependent on the residual rituximab concentration over time. Exposure‐response curve for r/r NHL VPOP with detectable baseline rituximab concentration ([RTX]) with (b) AUC from mosunetuzumab concentration ([M]) and (c) effective AUC from [M]eff as the exposure metric. (d) Exposure‐response curve for r/r NHL VPOP with undetectable [RTX]. (b–d): Black dash line, and dark and light gray shaded regions represent the VPOP simulation median, 25th–75th percentile and 10th–90th percentile of the VPOP, respectively. AUC, area under the curve; iVPs, individualized virtual patients; NHL, non‐Hodgkin's lymphoma; r/r, relapsing/refractory; RTX, rituximab; SPD, post‐treatment tumor measurement; VPOP, virtual population.
The extent of difference in tumor reduction dynamic is dependent upon the initial RTX concentration of the VPOP (Figure S4), where VPOP simulations were performed for three scenarios: with no, low (5 μg/mL), and high baseline RTX (42.5 μg/mL) at 1/2/13.5 mg of mosunetuzumab. In the VPOP simulated with low and high baseline rituximab (Figure S4E,F), the median % CFBL SPD reaches −50% at later cycles (approximately cycle 5 and >6, respectively) compared to the no RTX scenario (approximately cycle 4; Figure S4D). When a higher dose of 1/2/27 mg is simulated (Figure S4I), the rate of tumor depletion was improved, bringing the low baseline rituximab case to approximately cycle 3. For the high baseline rituximab, 1/2/60 mg is needed for the median % CFBL SPD to reach −50% around cycle 4 (Figure S4N). These simulation results suggest that the effect of the CD20 binding competition can be overcome by the administration of a higher dose of mosunetuzumab.
Dynamics of tumor cells and T‐cells in the tumor microenvironment differ substantially between responders and nonresponders
Having generated patient‐matched iVPs, we sought to examine their in silico characteristics to identify features that might differentiate clinical responders from nonresponders as defined by the Cheson 2007 15 criteria. Figure 6 shows the simulated time course of the tumor B‐cells, T‐cells, and the fraction of T‐ and activated T‐cells in the VPOP stratified by iVPs derived from clinical responders (partial response or complete response) and nonresponders (stable disease or progression of disease) for the combined NHL population. Three different dose levels were simulated: low (0.4 mg fixed dosing regimen), medium (0.4/1.0/2.8 mg step‐up dosing regimen), and high dose (1.0/2.0/13.5 mg step‐up dosing regimen). At low dose level (Figure 6a), the simulated tumor B‐cell counts increase at a faster rate in nonresponders compared to responders, whereas the average tumor T‐cell fractions return to baseline levels in both responder and nonresponder subsets between dosing intervals (Figure 6d). These suggest that neither responders nor nonresponders can overcome the tumor growth at the low dose, and as a result, the proportion of T‐cells in the tumor decrease (Figure 6g). In the medium and high dose simulations (Figure 6b,c), the predicted tumor B‐cells on average decline in responders but increase in the nonresponders. The tumor T‐cell count in both populations increase over time (Figure 6e,f), with the responder population having a greater expansion of T‐cells, likely due to T‐cell activation and proliferation in the presence of target B‐cells at sufficient mosunetuzumab exposure. Thus, the proportion of T‐cells (Figure 6h,i) in the tumor increases in responders due to declining B‐cell count and increasing T‐cell count; the extent of increase is more apparent at the high dose level (Figure 6i). The proportion of activated T‐cells in the tumor (Figure 6k,l) is higher in the responders compared to nonresponders after the simulated target dose administration on day 14. Subsequently, T‐cell activation diminishes in the tumor even though the same target dose administration of 13.5 mg is simulated on days 21 and 42. This is because T‐cell activation by mosunetuzumab is dependent on tumor B‐cell count, for which the median counts in the responders declines at this target dose level.
FIGURE 6.

The time course of (a–c) percent change from baseline for tumor B‐cells, (d–e) percent change from baseline for tumor T‐cells, (f–h) proportion of T cells as of total tumor cells and (i–k) percentage of activated T‐cells in tumor for iVPs derived from clinical responders (median: black dashed line; 90% CI: gray shaded region) and nonresponders (median: red dashed line; 90% CI: red shaded region). CFBL, change from baseline; CI, confidence interval; iVPs, individualized virtual patients.
The simulated proportion of tumor T‐cells was consistent with the tumor biopsy analyses from patients treated with mosunetuzumab, as reported in Hernandez et al., which showed that the extent of increase in the proportion of tumor CD8+ T‐cells post‐treatment appear higher in responders compared to nonresponders. 16 Our simulation results suggest that this trend may be explained by both a reduction in B‐cells in the tumor and the increase of T‐cell number in the tumor. This analysis suggests care be taken in interpreting changes in fractional composition when both populations may be changing.
Model‐inferred baseline tumor characteristics differ substantially between responders and nonresponders
The simulated on‐treatment behavior of the B‐ and T‐cells in the tumor is influenced by the tumor parameters (i.e., tumor B‐cell proliferation rate, B:T‐cell ratio, and baseline SPD), the B‐cell killing parameters of mosunetuzumab (EC50 and maximum effect), T‐cell activation parameters of mosunetuzumab (EC50 and maximum effect), and baseline RTX concentration. Histograms of these model parameters for iVPs derived from clinical responders and nonresponders reveal underlying differences in model‐inferred tumor properties between the two groups. Specifically, the baseline B:T‐cell ratio, tumor B‐cell proliferation rate, and baseline SPD, are generally higher in iVPs derived from nonresponders (Figure 7).
FIGURE 7.
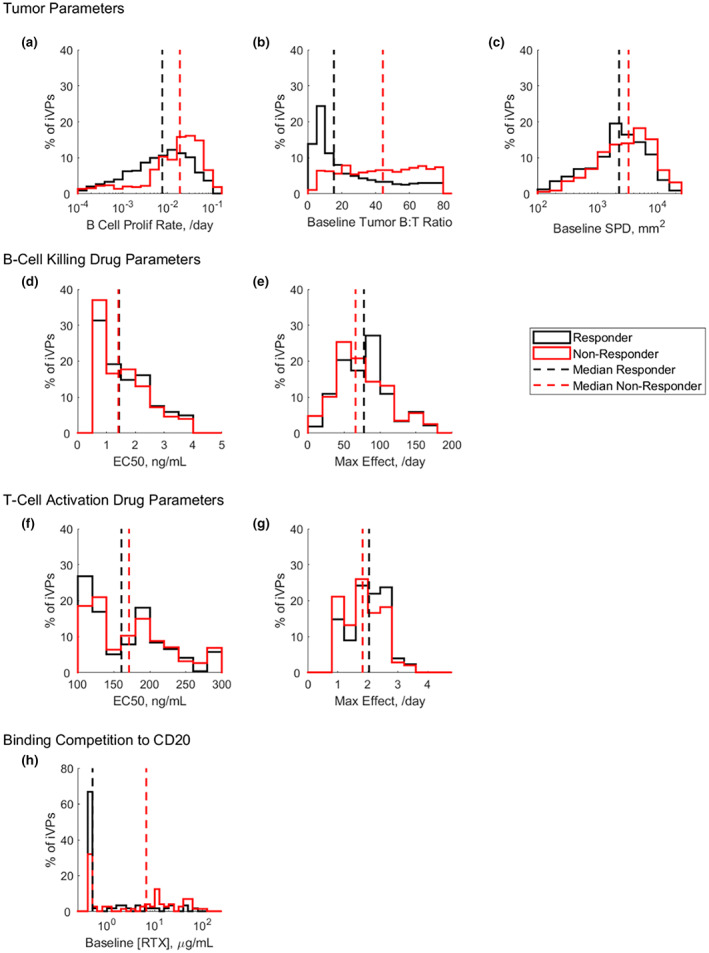
Distribution of model estimated (a–c) tumor parameters, (d, e) B‐cell killing parameters and (f, g) T‐cell activation parameters and (h) patient derived baseline rituximab concentration ([RTX]) for iVPs generated from clinical responders (black) and nonresponders (red). EC50, half‐maximal effective concentration; iVP, individualized virtual patient; RTX, rituximab; SPD, post‐treatment tumor measurement.
The estimated maximum effect of B‐cell killing rate and T‐cell activation rate were higher in responders compared to nonresponders (Figure 7e,g), suggesting that the response in patients could plausibly be attributable to both increased number of activated T‐cells as well as improved killing of B‐cells by the activated T‐cells. In the VPOP, the values and variability of drug‐related parameters from the virtual cyno cohort from Hosseini et al. 8 are assumed to be conserved in patients. This assumption is supported by in vitro data reported by Sun et al. where the target cell killing EC50 for various B lymphoma cell lines by mosunetuzumab was within the range of healthy donor peripheral blood mononuclear cell. 5
As previously discussed, CD20 binding competition to residual RTX may also affect tumor reduction dynamics, in addition to the tumor and drug properties. Figure 7h shows that the responder subset has lower baseline RTX level compared to the nonresponders.
DISCUSSION
In oncology drug development, clinical dose finding of novel therapeutic agents in early phase I studies can be challenging due to the patient heterogeneity, the small sample size per cohort, and variation of dosing regimens. Here, we have developed a digital twin‐based QSP VPOP approach to predict the dose/exposure‐response relationship for mosunetuzumab in patients with r/r NHL. Each clinical patient from the early dose escalation cohorts of study GO29781 is represented by a set of digital twins, and simulation of digital twins over a range of dose regimens synthetically enriches the limited and evolving clinical data. These predictions have supplemented the totality of clinical evidence, including other more empirical and data‐driven approaches, such as logistic regression modeling and provided a complementary, mechanistic interpretation of clinical dose–response relationship. Furthermore, the mechanistic nature of the QSP model allows the identification of potential biological differences in patient subsets that influence response to therapy.
The model‐predicted dose/exposure‐response is generally consistent with the preliminary empirical exposure‐response analysis from Li et al. 13 The two complementary approaches during the dose‐escalation of mosunetuzumab supported and increased confidence in selection of dose for pivotal clinical expansion cohorts. The bottom‐up approach of QSP modeling utilizes the totality of evidence including mechanistic understanding, preclinical, clinical, and competitor data. In the absence of a large sample size from the clinical trial, the QSP digital twin VPOP represents a quantitative way to predict population‐level behaviors by accounting for the biological variability that underlies differential target engagement and disease modulation in the patient population. As the biological underpinnings and response kinetics and are explicitly described, the QSP model also enables extrapolation to novel dose regimens or disease context in the absence of clinical data. Conversely, a limitation of the QSP model is that it does not include mechanisms that can lead to dose‐independent primary resistance or secondary, emergent resistance. On that account, we have focused on predictions for early tumor reduction (day 42), rather than the best response throughout the duration of treatment. Thus, robust analysis of dose level/regimen enabled by complementary approaches is of particular value given the increased attention on dose‐optimization in oncology. 17
The model provided useful insights for designing treatment regimens across NHL histologies with aggressive (e.g., DLBCL) versus indolent (e.g., FL) disease characteristics. Between the DLBCL and FL VPOPs, the model predicts a shift of the exposure‐response relationship (Figures 3a,b and 4c,d), attributable to differences in estimated tumor parameters; specifically the fitting process yielded higher B‐cell proliferation rate and higher baseline tumor B:T‐cell ratios in DLBCL digital twins (Figure 3c–e) despite no a priori assumption of differences between the disease subtypes. The higher estimated tumor proliferation rate in DLBCL digital twins compared to FL (Figure 3c) appear consistent with clinical definition where aggressive NHL subtypes (e.g., DLBCL) progress rapidly compared to indolent subtypes (e.g., FL). The contribution of the higher tumor proliferation rate to lower response in DLBCL compared to FL (Figures 3a,b and 4c,d) is also consistent with a meta‐analysis from NHL studies reported by He et al., 18 where the expression of the cell proliferation marker, Ki67, in NHL negatively correlates with disease‐free survival and overall survival. With respect to the relative abundance of target B‐cells and T‐cells, the B:T‐cell ratio has been demonstrated in vitro 19 and in vivo 20 to alter target cell killing by T‐cell engaging bispecific antibodies, although it is not currently known whether there are clinical differences in baseline T‐cell infiltration between DLBCL and FL.
These same parameters of tumor B‐cell proliferation rate and baseline tumor B:T‐cell ratio were also found to differ between clinical responder and nonresponder subsets of the VPOP (Figure 7). The B:T‐cell ratio of the digital twins is skewed to lower values in responders (i.e., greater T‐cell infiltration at baseline), suggesting a potential diagnostic hypothesis that is supported by preclinical 20 and clinical findings to date. Belmontes et al. 20 observed that both the tumor to T‐cell ratio and tumor doubling time influenced the responsiveness of tumor xenografts to T‐cell engaging bispecifics. From the clinical front, baseline %T‐cell in the tumor derived from tumor biopsy immunohistochemistry assessment from patients receiving mosunetuzumab is trending lower in patients with progressive disease compared to responders, comparable with the model‐inferred values (Figure S5), even though this was not enforced as a constraint in our model calibration workflow. Besides the tumor parameters, the maximum effect of B‐cell killing rate and T‐cell activation rate were found to be higher in responders compared to nonresponders. Given that our analysis showed that multiple independent factors influence the clinical response to mosunetuzumab, a multifactor biomarker diagnostic approach should be considered for further exploration both in silico and in clinical biomarker data.
During treatment with sufficient mosunetuzumab exposure, model‐simulated tumor T‐cell number increases over time with responders having a greater extent of T‐cell growth (Figure 6d–f) and, consequently, improved tumor killing (Figure 6a–c). Increases in on‐treatment T‐cell number can result from either the in situ proliferation of the existing T‐cells in the tumor, or the trafficking of T‐cells from other sites into the tumor. To evaluate the contribution of T‐cell trafficking to tumor cell killing, we simulated the VPOP at a high dose level with and without allowing T‐cell trafficking to/from the tumor (Figure S6). Interestingly, artificial simulations without T‐cell trafficking predicted substantially greater increases in tumor T‐cells (Figure S6H) and faster tumor B‐cell killing (Figure S6L) compared to the physiologically relevant predictions which account for T‐cell trafficking; this reflects a substantial predicted efflux of expanded T‐cells from the tumor (Figure S6B). Systemic T‐cell profiles in cyno, simulated and discussed in Hosseini et al., 8 also highlighted trafficking of tissue‐expanded T‐cells back into the blood. These results suggest that the T‐cell expansion observed in Figure 6d–f was attributable to the proliferation of the pre‐existing T‐cells in the tumor, rather than influx of systemic T‐cells into the tumor. These findings are also consistent with observations by Belmontes et al.20 that the depletion of circulating T‐cells in a syngeneic mouse tumor model treated with a T‐cell engaging bispecific did not affect antitumor efficacy, suggesting that tumor killing response is not dependent upon influx of circulating T‐cells in response to T‐cell engagers.
Even though the mosunetuzumab QSP model structure did not explicitly include CD20 receptors as modeled in previous work, 21 , 22 the effect of binding competition between mosunetuzumab and RTX was evaluated using the Schild method. 14 The transient nature of the binding competition (Figure 5a; Figure S5) delayed the median tumor shrinkage of the VPOP (Figure S5) and partially contributes to the shift of exposure‐response of RTX+ VPOP on day 42 compared to the RTX– VPOP (Figure 5b,d). However, this effect can be overcome by the administration of higher doses of mosunetuzumab, such that the effective mosunetuzumab exposure is close to mosunetuzumab exposure without RTX (Figure S5).
A few VPOP generation methodologies have been previously published. 23 , 24 , 25 Typically, the process begins with an identification of a cohort of virtual subjects whose simulated outputs meet certain prespecified acceptance criteria, based on physiological bounds and observed measurement ranges. The virtual cohort is then refined to a VPOP by further selection or prevalence‐weighting to reproduce statistical features of the outputs/measurements. 24 , 25 In the early stages of a phase I clinical trial, however, statistical features of the clinical data can be unreliable due to the small number of patients per dose escalation cohort. We address this challenge by generating digital twins with alternate inferred model parameterizations that reproduce SPD measurements for each individual clinical patient. One limitation to this approach is the underlying assumption that the clinical patients from the dose escalation study are representative of the overall population of interest. For populations with high variability in treatment response, increasing the number of clinical patients represented in the digital twin VPOP improves the prediction interval (Figure S7).
The current approach to create the digital twin VPOP for mosunetuzumab relies on the translation of system parameters from cyno virtual cohort, 8 population‐level clinical constraints based on the range of pretreatment tumor lymphocyte infiltration observed in pretreatment biopsies (Table S1), and individual data on mosunetuzumab PKs, baseline RTX concentration, baseline circulating T and B cell levels, and longitudinal tumor size measurement from each patient. In the future, other individual patient level biomarkers including pre‐ and/or post‐treatment tumor lymphocyte infiltration, tumor Ki67 expression, or additional longitudinal data may also be used to create the digital twins. In contrast to data‐driven empirical creation of digital twins that often requires rich individualized biomarker data, we suggest that the underlying mechanistic fidelity of the QSP model, biological constraints derived from preclinical and clinical data, and the utilization of multiple digital twins per individual clinical patients together compensate for limited individualized biomarker data. Furthermore, unlike empirical digital twins, the mechanistic nature of the model allows individualized inference and analysis of clinically unmeasured biomarkers in the digital twins, enabling the biomarker analyses we have presented.
In this study and the prior work by Hosseini et al., 8 we have presented the use of the digital twin‐based QSP VPOP approach to evaluate efficacy of mosunetuzumab and to support the identification of dosing regimen to mitigate CRS risk. These demonstrate that a single mechanistic QSP model can be applied to support the molecule's clinical development for both efficacy and safety, as well as the identification of potential biological determinants of clinical response. Other potential applications of the QSP model include the exploration of alternative routes of administration, combination with other treatments, and first‐in‐human dose selection for other novel indications or treatment settings. Whereas the current study focuses on using the digital twins to generate a VPOP for predicting response and biomarker identification, the approach may be further refined to generate digital twins with the capability of forecasting an individual patient's disease trajectory for optimization of personalized therapeutic intervention. 12
AUTHOR CONTRIBUTIONS
M.S., I.H., and S.R. performed the research and analyzed the data. M.S., I.H., S.R., and C.C.L. wrote the manuscript. All authors designed the research.
FUNDING INFORMATION
This project was sponsored by Genentech Inc.
CONFLICT OF INTEREST STATEMENT
M.E.S., C.‐C.L., L.‐Y.H., J.Y.J., S.Y., M.C.W., S.R., and I.H. are all employees of Genentech, Inc. G.H. was an employee of Genentech, Inc. at the time the studies were conducted and is currently an employee at IGM Biosciences. K.G. was an employee of Genentech, Inc. at the time the studies were conducted and is currently an employee at Denali Therapeutics. All authors may be stockholders of F. Hoffmann‐La Roche AG.
Supporting information
Appendix S1
Table S1
Table S2
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
ACKNOWLEDGMENTS
The authors thank E. Penuel, D. Turner, and K. Yoshida for critical reading of the paper and helpful discussions.
Susilo ME, Li C‐C, Gadkar K, et al. Systems‐based digital twins to help characterize clinical dose–response and propose predictive biomarkers in a Phase I study of bispecific antibody, mosunetuzumab, in NHL . Clin Transl Sci. 2023;16:1134‐1148. doi: 10.1111/cts.13501
Saroja Ramanujan and Iraj Hosseini are co‐senior authors.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5‐29. [DOI] [PubMed] [Google Scholar]
- 2. Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B‐cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2015;26(Suppl 5):v116‐v125. [DOI] [PubMed] [Google Scholar]
- 3. Ambinder AJ, Shenoy PJ, Malik N, Maggioncalda A, Nastoupil LJ, Flowers CR. Exploring risk factors for follicular lymphoma. Adv Hematol. 2012;2012:626035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Conconi A, Ponzio C, Lobetti‐Bodoni C, et al. Incidence, risk factors and outcome of histological transformation in follicular lymphoma. Br J Haematol. 2012;157:188‐196. [DOI] [PubMed] [Google Scholar]
- 5. Sun LL, Ellerman D, Mathieu, M , et al. Anti‐CD20/CD3 T cell‐dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015;7:287ra270. [DOI] [PubMed] [Google Scholar]
- 6. Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine‐directed therapy. Blood. 2013;121:5154‐5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turtle CJ, Hay KA, Hanafi LA, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19‐specific chimeric antigen receptor‐modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010‐3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hosseini I, Gadkar K, Stefanich E, et al. Mitigating the risk of cytokine release syndrome in a phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. NPJ Syst Biol Appl. 2020;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartlett NL, Sehn LH, Assouline SE, et al. Managing cytokine release syndrome (CRS) and neurotoxicity with step‐up dosing of mosunetuzumab in relapsed/refractory (R/R) B‐cell non‐Hodgkin lymphoma (NHL). Am Soc Clin Oncol (ASCO). 2019;37:7518. [Google Scholar]
- 10. Budde LE, Sehn LH, Assouline S, et al. Mosunetuzumab, a full‐length bispecific CD20/CD3 antibody, displays clinical activity in relapsed/refractory B‐cell non‐Hodgkin lymphoma (NHL): interim safety and efficacy results from a phase 1 study. Blood. 2018;132:399. [Google Scholar]
- 11. Schuster SJ, Bartlett NL, Assouline S, et al. Mosunetuzumab induces complete remissions in poor prognosis non‐Hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T‐cell (CAR‐T) therapies, and is active in treatment through multiple lines. Blood. 2019;134:6.31273004 [Google Scholar]
- 12. Laubenbacher R, Niarakis A, Helikar T, et al. Building digital twins of the human immune system: toward a roadmap. NPJ Digit Med. 2022;5:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li C‐C, Bender B, Yin S, et al. Exposure‐response analyses indicate a promising benefit/risk profile of mosunetuzumab in relapsed and refractory non‐Hodgkin lymphoma. Blood. 2019;134:1285. [Google Scholar]
- 14. Wyllie DJ, Chen PE. Taking the time to study competitive antagonism. Br J Pharmacol. 2007;150:541‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheson BD, Pfistner B, Juweid ME, et al. International harmonization project on lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579‐586. [DOI] [PubMed] [Google Scholar]
- 16. Hernandez G, Huw LY, Belousov A, et al. Pharmacodynamic effects and immune correlates of response to the CD20/CD3 bispecific antibody mosunetuzumab in relapsed or refractory non‐Hodgkin lymphoma. Blood. 2019;134:1585.31558469 [Google Scholar]
- 17. FDA . Project Optimus – reforming the dose optimization and dose selection paradigm in oncology . 2022 2022. [Google Scholar]
- 18. He X, Chen Z, Fu T, et al. Ki‐67 is a valuable prognostic predictor of lymphoma but its utility varies in lymphoma subtypes: evidence from a systematic meta‐analysis. BMC Cancer. 2014;14:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang X, Chen X, Carpenter TJ, et al. Development of a target cell‐biologics‐effector cell (TBE) complex‐based cell killing model to characterize target cell depletion by T cell redirecting bispecific agents. MAbs. 2018;10:876‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Belmontes B, Sawant DV, Zhong W, et al. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell‐cold solid tumors. Sci Transl Med. 2021;13:eabd1524. [DOI] [PubMed] [Google Scholar]
- 21. Jiang X, Chen X, Jaiprasart P, Carpenter TJ, Zhou R, Wang W. Development of a minimal physiologically‐based pharmacokinetic/pharmacodynamic model to characterize target cell depletion and cytokine release for T cell‐redirecting bispecific agents in humans. Eur J Pharm Sci. 2020;146:105260. [DOI] [PubMed] [Google Scholar]
- 22. Betts A, Haddish‐Berhane N, Shah DK, et al. A translational quantitative systems pharmacology model for CD3 bispecific molecules: application to quantify T cell‐mediated tumor cell killing by P‐cadherin LP DART((R)). AAPS J. 2019;21:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allen RJ, Rieger TR, Musante CJ. Efficient generation and selection of virtual populations in quantitative systems pharmacology models. CPT Pharmacometrics Syst Pharmacol. 2016;5:140‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gadkar K, Kirouac DC, Mager DE, van der Graaf PH, Ramanujan S. A six‐stage workflow for robust application of systems pharmacology. CPT Pharmacometrics Syst Pharmacol. 2016;5:235‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hosseini I, Feigelman J, Gajjala A, et al. gQSPSim: a SimBiology‐based GUI for standardized QSP model development and application. CPT Pharmacometrics Syst Pharmacol. 2020;9:165‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Table S1
Table S2
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7