

Abstract
Extracellular vesicles (EVs) are continually released from cancer cells into biofluids, carrying actionable molecular fingerprints of the underlying disease with considerable diagnostic and therapeutic potential. The scarcity, heterogeneity and intrinsic complexity of tumor EVs present a major technological challenge in real-time monitoring of complex cancers such as glioblastoma (GBM). Surface-enhanced Raman spectroscopy (SERS) outputs a label-free spectroscopic fingerprint for EV molecular profiling. However, it has not been exploited to detect known biomarkers at the single EV level. We developed a multiplex fluidic device with embedded arrayed nanocavity microchips (MoSERS microchip) that achieves 97% confinement of single EVs in a minute amount of fluid (<10 μL) and enables molecular profiling of single EVs with SERS. The nanocavity arrays combine two featuring characteristics: (1) An embedded MoS2 monolayer that enables label-free isolation and nanoconfinement of single EVs due to physical interaction (Coulomb and van der Waals) between the MoS2 edge sites and the lipid bilayer; and (2) A layered plasmonic cavity that enables sufficient electromagnetic field enhancement inside the cavities to obtain a single EV level signal resolution for stratifying the molecular alterations. We used the GBM paradigm to demonstrate the diagnostic potential of the SERS single EV molecular profiling approach. The MoSERS multiplexing fluidic achieves parallel signal acquisition of glioma molecular variants (EGFRvIII oncogenic mutation and MGMT expression) in GBM cells. The detection limit of 1.23% was found for stratifying these key molecular variants in the wild-type population. When interfaced with a convolutional neural network (CNN), MoSERS improved diagnostic accuracy (87%) with which GBM mutations were detected in 12 patient blood samples, on par with clinical pathology tests. Thus, MoSERS demonstrates the potential for molecular stratification of cancer patients using circulating EVs.
Keywords: single extracellular vesicle analysis, surface-enhanced Raman spectroscopy, plasmonic nanocavity, monolayer MoS2, glioblastoma cancer
Introduction
Extracellular vesicles (EVs) are heterogeneous lipid-bilayer encapsulated vesicular structures shed into biofluids from cancer cells. EVs are an attractive option for liquid biopsy because they contain a wide range of molecular cargo, such as proteins and nucleic acids, that may serve as cancer-specific markers.1−5 Critically, as each EV represents a specific cell, the cancer markers contained in the tumor-derived EV landscape relate to the properties of a given cellular population while reflecting its diversity.6 Consequently, the information contained in a population of individual EVs can capture the clinically important traits reflective of cellular heterogeneity,7,8 and mutational and epigenetic driver events1−4 in cancer. In particular, extracellular vesicles represent a promising platform for liquid biopsy application in glioblastoma (GBM), the most common and aggressive primary astrocytic brain tumor.9−12
Notably, EVs carry signatures paradigmatic for GBM molecular subtypes and traits,13−17 including the mutant oncogenic variant of epidermal growth factor receptor (EGFR), known as EGFRvIII,7,8 as well as O6-methylguanine-DNA methyltransferase (MGMT), a marker of resistance to temozolomide (TMZ) chemotherapy. These changes are important due to their functional impact and prevalence as EGFRvIII is expressed in approximately 20–35% of GBM cases and impacts cellular aggressiveness as well as vesiculation,18 while MGMT expression is normally silenced by promoter methylation in approximately 45% of GBM cases, a factor that renders these tumors sensitive to treatment with TMZ, which may be lost during disease progression. As only 2–3% of blood-circulating EVs are believed to carry GBM signatures due to the blood–brain barrier, EVs carrying these mutations can be easily missed against a large background of EVs released from healthy cells.19 Thus, there is great interest in developing technologies with single EV resolution for early detection and monitoring of GBM-derived EVs for the emergence of resistance during chemotherapy.
Classic EV analysis techniques based on benchtop protocols, e.g.,
polymerase chain reaction (PCR), Western blot or ELISA, average over
very large numbers of EVs (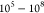 EVs contained in a μL to
mL of sample).20 More recent approaches
utilize microfluidic
mixing and filtration combined with chemical capture,20 for example exploiting device surfaces or microspheres
functionalized with antibodies21 or aptamers22 specific to common EV markers. Such approaches
average over at least 10 EVs bound per bead or surface element.21,23 A third wave of technologies has extended existing fluorescence-based
single-cell and single-molecule analytical approaches to perform individualized
EV analysis. Nano flow cytometry can now profile populations of 1000s
of EVs down to 100 nm in size stained for a limited number of surface
biomarkers.24,25 Alternatively, single EVs can
be surface immobilized and then exposed sequentially to a series of
fluorescent antibody probes targeting specific markers,26−28 achieving higher multiplexing and signal at the expense of throughput.
This latter approach has been extended to the detection of EV-encapsulated
RNA via fusing of surface bound EVs to liposomes containing molecular
beacon probes that hybridize to given RNA/microRNA targets.29 These existing single EV approaches are already
showing promise in research and clinical application.26,27 However, they have two intrinsic disadvantages. First, current methods
inevitably preselect biomarkers of interest from the pool of biomarkers
for which reliable fluorescent probes exist. Second, these methods
entail a trade-off between high marker multiplexing and throughput,
with high multiplexing necessitating many sequential probe washing
cycles (with each cycle taking as long as 1 h).26
EVs contained in a μL to
mL of sample).20 More recent approaches
utilize microfluidic
mixing and filtration combined with chemical capture,20 for example exploiting device surfaces or microspheres
functionalized with antibodies21 or aptamers22 specific to common EV markers. Such approaches
average over at least 10 EVs bound per bead or surface element.21,23 A third wave of technologies has extended existing fluorescence-based
single-cell and single-molecule analytical approaches to perform individualized
EV analysis. Nano flow cytometry can now profile populations of 1000s
of EVs down to 100 nm in size stained for a limited number of surface
biomarkers.24,25 Alternatively, single EVs can
be surface immobilized and then exposed sequentially to a series of
fluorescent antibody probes targeting specific markers,26−28 achieving higher multiplexing and signal at the expense of throughput.
This latter approach has been extended to the detection of EV-encapsulated
RNA via fusing of surface bound EVs to liposomes containing molecular
beacon probes that hybridize to given RNA/microRNA targets.29 These existing single EV approaches are already
showing promise in research and clinical application.26,27 However, they have two intrinsic disadvantages. First, current methods
inevitably preselect biomarkers of interest from the pool of biomarkers
for which reliable fluorescent probes exist. Second, these methods
entail a trade-off between high marker multiplexing and throughput,
with high multiplexing necessitating many sequential probe washing
cycles (with each cycle taking as long as 1 h).26
Surface-enhanced Raman spectroscopy (SERS) is a conceptually distinct alternative that in principle avoids the need for marker preselection and the multiplexing/throughput trade-off. SERS is a label-free approach that converts the entire biochemical signature of an EV, including surface chemistry and molecular cargo, into a single spectroscopic pattern, or “fingerprint”31,32 (see Supporting Table S1). SERS is inherently noninvasive, fast, reliable, and can be adapted for use in low-resource clinical settings, especially with the emergence of hand-held instruments and fiberoptic probes.33,34 SERS applied at an ensemble level, coupled with machine learning methods, can already differentiate between EVs in blood samples collected from healthy and diseased donors of cancers.32,36 The working principle of SERS is based on the adsorption of analyte molecules to a plasmonic surface that allows for a strong enhancement of the Raman signal from the analyte. Nanocavities with independently tunable harmonics achieve strong electromagnetic (EM) field enhancement at distinct wavelengths.37 At the same time, monolayer direct gap semiconductors of transition metal dichalcogenides (like monolayer MoS2) ensure relatively strong light–matter interactions when coupled with the EM field enhancements at their absorption wavelength.38
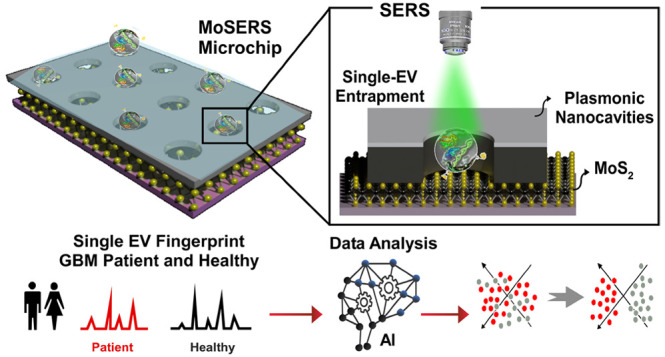
Here we demonstrate a SERS-based nanosurface microfluidic approach for molecular profiling of single EVs that can output information on EV surface properties and encapsulated cargo for early detection and monitoring of the cell transformation events during cancer progression and therapy (Figure 1a). Our MoSERS microchip embedded in a simple microfluidic device for sample delivery is produced via a combined bottom-up and top-down fabrication strategy. The MoSERS microfluidic is a user-friendly platform with attached accessories for flow actuation that automates the assay process when placed for SERS interrogation (Figure 1b). The MoSERS microchip assimilates the SERS signal from EVs trapped in nanocavities constructed from a plasmonic silver/nonplasmonic ZnO bilayer with an embedded MoS2 floor. It can achieve single EV resolved SERS spectrum due to three main factors. First, our optimized photonic design achieves a sufficiently strong in-cavity field enhancement to produce a sufficient signal to obtain SERS spectra from single EVs. Second, relying on attractive interactions between the EV lipid membrane and the MoS2 monolayer, we obtain EV entrapment in nanocavities without requiring biological recognition elements. Third, as the cavities are made sufficiently small to accommodate only a single EV, we ensure that the spectrum obtained from a single cavity in fact corresponds to the spectrum of a single EV (Figure 1c).
Figure 1.

The concept of using single EV SERS approach in liquid biopsy of GBM patients. (a) Possibilities of using MoSERS microchip embedded microfluidic device for liquid biopsy of blood EVs for early detection and monitoring of cancer (i): A microfluidic prototype embedded with MoSERS microchips with the size of 0.1 cm × 0.1 cm for sample delivery and detection (ii). Single EV SERS spectrum analyzed by machine learning algorithm (CNN) to stratify molecular alterations (iii). (b) The nanosurface microfluidic embedded with MoSERS microchip perform for SERS identification of single EVs of Glioblastoma cancer. Single EVs are entrapped in the plasmonic nanocavities, with one EV per cavity, and SERS is performed utilizing high-field enhancement at the nanocavity edges. (c) Single EV entrapment in the MoS2 encapsulated nanocavities of MoSERS microchip. Schematic of MoSERS microchip fabrication and related SEM images of each step.
The MoSERS microchip is benchmarked by identifying and distinguishing signals from single EVs derived from noncancerous glial cells (NHA), glioma cells (U373, U87), and glioma stem cells (GSC83, GSC1005, and GSC1123). These cell lines were either wild-type or manipulated to express relevant molecular alterations, such as oncogenic epidermal growth factor receptor variant III (EGFRvIII), phosphatase and tensin homologue (PTEN), which is naturally lost during GBM progression, and MGMT, a marker and effector of GBM resistance to TMZ. Next, to maximize our technique’s ability to discriminate between SERS spectra with subtle differences and to achieve more accurate and sensitive multidimensional identification of EVs, we combined single EV SERS readouts with deep learning techniques based on a computational neural network (CNN). CNN machine learning was then applied to discriminate between EVs derived from GBM patient blood samples. The CNN approach outputs the probability that each EV carries specific oncogenic markers including EGFR amplification, EGFRvIII mutation and MGMT expression. This information can then be combined to differentiate EVs into marker negative and positive classes and render information that can be built into a diagnostic workflow (Table 1).
Table 1. Single EV Technologies Validated by Clinical Samples.
| Technology | Cancer | Media | Biomarker | Patients Number | Ref. |
|---|---|---|---|---|---|
| Immunoaffinity Labeling | |||||
| Nanoplasmon-enhanced (n-PLEX) | Pancreatic | Plasma | Protein-conjugated primary antibodies | 104 Malignant, 31 benign | 2014,1 201739 |
| Single EV analysis (SEA): multiplex immunostaining and fluorescent imaging | Pancreatic | Plasma | Optimized mutation-conjugated antibodies | 20 Patient, 5 healthy | 2018,26 202227 |
| Single large EV characterization: immunostaining and fluorescent imaging | Glioblastoma | Plasma | Protein-conjugated primary antibodies | 8 Patient, 2 healthy | 201941 |
| TIRF microscopy ∼80 EVs: sequential detection of DNA-PAINT | Breast, ovarian, liver | Plasma | DNA-conjugated primary antibodies | 7 Patient per cancer | 201942 |
| Label-Free | |||||
| Single EV SERS (MoSERS) microchip: label-free SERS and ML assisted data analysis of mutations | Glioblastoma | Plasma | – | 12 Patient, 8 healthy | This work |
Results
Plasmonically Active MoS2 Nanocavities
The MoSERS microchip consists of arrays of circular 250 nm diameter nanocavities embedded in an Ag/ZnO bilayer. The cavities have a pitch of 3 μm to ensure each cavity can be scanned independently. The bilayer is placed on top of hole-pierced CVD-grown monolayer MoS2 resting on a SiO2/Si substrate, so that the nanocavity floor contains an exposed MoS2 monolayer (Figure 2a). The nanocavities are produced using a negative e-beam lithography process (the step-by-step process flow is shown in Figure S1).
Figure 2.

Benchmarking of SERS performance. (a) Schematic of MoSERS microchip fabrication and related SEM images of each step. (b) The electric field distribution in 250 nm diameter nanocavities simulated using a 532 nm TSFS light source. (c) The SEM image and correlated EM field distribution in the nanocavities with different diameters via a 532 nm TSFS light source. (d) The simulated EM-field distribution at the nanocavity edges as a function of nanocavity diameter. Simulated (e) EM-field enhancement and (f) reflectance spectra and the of plasmonic nanocavities with different diameters. (g) Spatial variation of SERS response over nanocavity array obtained from averaging the response from spectral peaks in the R6G spectra. (h) The corresponding SERS intensity of the representative areas (blue = flat, red = nanocavity). (i) The average SERS peak intensity at 1650 cm–1, 1510, and 1362 cm–1 obtained from R6G for MoSERS nanocavities (red) and the flat silver thin film (blue). (j) The sensitivity test of R6G on MoSERS substrate showed a linear range from 0.1 to 200 μM with R2 value of 0.996.
Our nanocavity design confers an enhanced SERS
response arising
from the intense electric field generated by the exposed Ag present
at the nanocavity edge (Figure 2b). The collective coherent excitations of free electrons
in the Ag thin film, known as surface plasmon resonance (SPR), lead
to an enhanced localized electromagnetic (EM) field confinement in
the vicinity of the nanocavities. Patterning the Ag thin film with
nanocavities smaller than the laser wavelength gives rise to a localized
strong electronic excitation at the nanocavity edges (LSPR). The SERS
response can be quantified via the EM-field enhancement factor 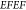 , scaling
as the fourth power of the EM-field
enhancement
, scaling
as the fourth power of the EM-field
enhancement 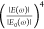 .43 In order
to determine the
.43 In order
to determine the 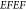 factor
for our nanocavity design, we used
finite-difference time-domain (FDTD) simulation (Lumerical Solutions,
Inc.) incorporating the experimental cavity geometry, material optical
properties and excitation conditions (532 nm diffraction-limited Gaussian
laser beam focus). Figure 2c gives the EM-field distribution in the nanocavities. The
EM-field is enhanced at the cavity edge and maximal for a cavity diameter
of 250 nm (Figure 2d,e), with a corresponding quantitative enhancement of
factor
for our nanocavity design, we used
finite-difference time-domain (FDTD) simulation (Lumerical Solutions,
Inc.) incorporating the experimental cavity geometry, material optical
properties and excitation conditions (532 nm diffraction-limited Gaussian
laser beam focus). Figure 2c gives the EM-field distribution in the nanocavities. The
EM-field is enhanced at the cavity edge and maximal for a cavity diameter
of 250 nm (Figure 2d,e), with a corresponding quantitative enhancement of  800-fold at the cavity edges. The simulated
broad-band reflectance spectra show a sharp peak around the laser
beam wavelength correlated to the diffraction mode (Figure 2f). These EM-field enhancements
correspond to an overall EFEF of 6.4 × 105 (see Figure S2 for more details).
800-fold at the cavity edges. The simulated
broad-band reflectance spectra show a sharp peak around the laser
beam wavelength correlated to the diffraction mode (Figure 2f). These EM-field enhancements
correspond to an overall EFEF of 6.4 × 105 (see Figure S2 for more details).
Next, we benchmarked the experimental SERS response of the MoSERS platform using the well-known SERS marker Rhodamine 6G (R6G). Nanocavity arrays were loaded with 1 μM R6G solution and scanned at 532 nm. Figure 2g gives the spatial variation of the SERS response over the array. Note that SERS hot spots corresponding to cavity positions are arranged along a rectangular lattice (the lattice has a horizontal pitch of 6 μm and vertical pitch of 2 μm). Regions of low response that are observed between cavities in the horizontal direction correspond to cases where the laser spot failed to overlap any cavities. Figure 2h compares SERS spectra from the flat thin film background region (blue) and nanocavities (red). The SERS signal from R6G at the nanocavity sites is over 300 times stronger than the signal from the background region.
To
quantify the SERS enhancement resulting from the nanocavities,
we compared the SERS signal in the presence of the MoSERS nanocavity
with the signal arising from a flat substrate consisting of only an
Ag/ZnO bilayer. The average peak intensity difference was calculated
at the major peak positions (Figure 2i). The SERS intensity (ISERS) is directly proportional to the EFEF according to the formula: ISERS = FSσSCSERSEFEF,44 where FS is an instrumental
factor related to Renishaw micro-Raman, σS is the
Raman cross-section of a particular analyte, and CSERS is the concentration of the test analyte. Quantitatively,
the SERS enhancement factor (EF) is calculated according
to 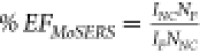 ,45,46 with the quantities
,45,46 with the quantities 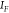 ,
, 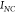 ,
, 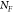 ,
, 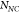 referring respectively to the SERS intensity
of the flat bilayer, the SERS intensity of the MoSERS device, the
number of analytes for the flat thin-film sample and the number of
analytes excited by the localized field enhancement of the MoSERS
structure. The SERS intensity values are computed from the representative
peaks at 1362 cm–1, 1510, and 1650 cm–1, with the exact values obtained from the integrated band areas at
the given peak positions resulting in a linear range from 0.1 to 200
μM with an R2 value of 0.996 and
a limit of detection of 0.1 μM (Figure 2j). The experimental SERS enhancement factor
(EF) was found to be 0.8 × 105, yielding
reasonable order of magnitude agreement with the theoretical estimate
(see Figure S3 for more details).
referring respectively to the SERS intensity
of the flat bilayer, the SERS intensity of the MoSERS device, the
number of analytes for the flat thin-film sample and the number of
analytes excited by the localized field enhancement of the MoSERS
structure. The SERS intensity values are computed from the representative
peaks at 1362 cm–1, 1510, and 1650 cm–1, with the exact values obtained from the integrated band areas at
the given peak positions resulting in a linear range from 0.1 to 200
μM with an R2 value of 0.996 and
a limit of detection of 0.1 μM (Figure 2j). The experimental SERS enhancement factor
(EF) was found to be 0.8 × 105, yielding
reasonable order of magnitude agreement with the theoretical estimate
(see Figure S3 for more details).
Monolayer MoS2 and EVs Interaction
The SERS identification of a single EV is closely related to the ability of trapping the EVs in the plasmonic nanocavities for the test time to enhance the subtle SERS signal from single EVs. The hydrophobic interactive behavior of the MoS2 layer with the EVs as a physical interaction can be one of the reasons for the absorption of EVs in the nanocavities. Theoretically, single crystalline monolayer MoS2 demonstrates a potential interaction with bilayer lipid materials.47,48 Here, we first used electron microscopy to visualize the physical connection between monolayer MoS2 and a phospholipid (Figure 3a–c) after sample preservation. The SEM image showing attachment of EVs to the 2H-phase monolayer MoS2 demonstrated that the majority of the EVs were attracted to the edges of the monolayer. A ratio of 10:1 EVs on average was found at the edges of the monolayer versus at the basal plane of the triangular single crystalline monolayer MoS2. There are substantial atomic defects present at the edges of the monolayer MoS2 that can actively interact with lipid bilayers. In addition, to ensure the edge sites expression in the nanocavities, a pretreatment e-beam positive lithography on monolayer MoS2 was used to introduce defect sites on the basal plane of the monolayer, which in turn promotes the adhesion of the EVs to the MoS2 in the nanocavities (see Figure S4 for more details).
Figure 3.

Interaction of EVs with monolayer MoS2 embedded in MoSERS microchip. (a) The SEM image of the EVs immobilized on a triangular single crystalline monolayer MoS2 demonstrating the attraction of the nanocavity array. (b) High-resolution SEM image of the EVs. (c) High-resolution TEM image of coexisting monolayer MoS2 and EV. (d) Representative simulated trajectory of the restrictive simulation (e) The atom contacts number and the center of mass (COM) distance between the MoS2 nanosheet and the lipid membrane. (f) Representative trajectories of the released simulation. Angle between the lipid bilayer and the MoS2 basal. (g) The interaction energy between the MoS2 nanosheet and the EV’s bilayer lipid. (h) Normalized contact number of each component of the phospholipid bilayer.
We used molecular dynamics (MD) simulation to probe the physical origin of the interactions between monolayer MoS2 and the phospholipid (Figure 3d,e), using a realistically constructed phospholipid bilayer incorporating previously reported components of the EV membrane (cholesterol (CHL), POPC, TSM, POPS, and POPE). When a phospholipid bilayer gets close to a MoS2 layer, the gradient of potential energy resulting from van der Waals and Coulomb energies makes the phospholipid membrane interact with the MoS2 layer. In parallel, understanding the origin of this interaction is essential to further study the inorganic entrapment sites for single EVs biomarkers. The MD simulation analysis confirms a higher attraction force between the phospholipid bilayer of EVs and the edge sites of the monolayer MoS2 compared with the basal plane (Figure 3f). We started the simulation by positioning the MoS2 layer parallel to the constructed phospholipid membrane, and the membrane freely moves. The MoS2 layer is freely flipping, and there is no considerable attraction force between the MoS2 layer and the membrane before 100 ns when the monolayer rotates approximately 90°, and the edge of the layer gets closer to the membrane. Once the phospholipid membrane and the monolayer MoS2 start to interact with each other, the level of energy reaches a minimum level resulted by absorption of the layer inside the membrane. The simulations were carried out (using GROMACS software package) with the design of a phospholipid bilayer to integrate five different lipid components, while the force field parameters of MoS2 were derived from a previous study.49 The PME method was applied to handle the long-range electrostatic interactions, and the van der Waals (vdW) interactions were computed with a cutoff of 1.2 nm (see the Materials and Methods section for more details). To evaluate the free energy changes during the insertion process, we calculated the potential of mean force (PMF) along the direction perpendicular to the phospholipid bilayer surface. The PMF is able to evaluate the binding free energy, which was calculated according to the umbrella sampling simulations. During PMF calculations, the MoS2 nanosheet (selected from the final conformation in the second simulation) was vertically pulled away from the membrane-forming various configurations (see Figure S5 for more details). Then, the perpendicular distance of the center of mass of MoS2 nanosheet with its corresponding initial position was regulated at reference distances (d0) via a harmonic force, F = k × (d – d0), where k is the force constant (2000 kJ mol–1 nm–2). Consistent with our previous findings, we demonstrated an attractive force between the monolayer MoS2 and the phospholipid bilayer (Figure 3g). When a phospholipid bilayer comes close to a MoS2 layer, the gradient of potential energy resulting from van der Waals and Coulomb energies causes the phospholipid membrane to interact with the MoS2 layer. The energies reach a minimum value of −3133.02 kJ mol–1 and −809.04 kJ mol–1 for van der Waals and Coulomb, respectively. A stable membrane comprising the five components listed above served to study the minimum energy level after absorption of a layer inside the membrane and the attraction forces of the phospholipid bilayer interaction and MoS2 (Figure 3h), while the overall contact number of each component of the phospholipid bilayer was also simulated.
Through the combination of theory and experiment, we demonstrated that the interaction forces corroborate the preference of MoS2 edge sites to interact with EVs lipid bilayer which is essential for high-capacity entrapment of EVs.
EV Entrapment in Nanocavities
The EV–nanocavity interaction with EVs that drives the uniform array loading is critical to MoSERS performance. These rectangular arrays of nanocavities with a variety of diameter sizes can be fabricated into arbitrary shapes (see Figure S6 for more details). The EVs are introduced into the array by direct pipetting of a 1–10 μL drop of an EV-containing solution into the MoSERS fluidic device (Figure 4a). The EVs have a size range between 150 and 200 nm in diameter (as measured via NTA Figure S7). Fluorescent labeling is used (Dil) to study EV interaction with the MoS2 monolayer and observe how this interaction affects EV loading in the nanocavity array.
Figure 4.

EVs entrapment in MoSERS nanocavities. (a) Schematic illustrating the EVs introduced to the MoSERS nanocavities and entrapment of the fluorescently labeled EVs in the nanocavities. Fluorescent micrograph of EVs loaded in cavities (b) with embedded monolayer MoS2 and (d) without MoS2. The correlated movement of a single EV through the incubation time in cavities (c) with embedded monolayer MoS2 and (e) without MoS2. (f) Low-magnification SEM image of single EVs entrapped in the nanocavities. (g) High-magnification SEM image of a single cancer EV (U373) entrapped in a nanocavity. (Inset: the TEM image showing the coexistence of MoS2 and a single EV.) (h) The comparison of fluorescent intensity of the EVs over time shows smallest bleaching slope for nanocavities on MoS2, monolayer MoS2, nanocavities, and silica, respectively. (i) Comparison between the mean fluorescence intensity obtained from EVs on SiO2, Nanocavities on SiO2, and nanocavities on MoS2 with different diameters. (j) The averaged SERS spectra from empty cavities (buffer), liposomes, EV populations derived from noncancerous glial cells (NHA) and cultured glioma cells (U373). Each spectrum is obtained from averaging 50 EVs; the SD is indicated in gray. (k) PCA components (i.e., PCA score plot) for single EV recordings obtained with the MoSERS platform. (l) PC1 and PC2 loading Raman bands used to produce the PCA score plot shown in (k). (m) SERS spectra for single EVs released from heterogeneous U373 cancer cells.
Figure 4b–e compares nanocavities that contain monolayer MoS2 (Figure 4b) to nanocavities without MoS2 (Figure 4d). We find that the presence of the MoS2 monolayer strongly enhances cavity loading, with the MoS2 monolayer promoting complete cavity occupancy (Figure 4c,e). While the cavity loading appears to be uniform at the resolution of fluorescence microscopy, we used SEM imaging to gain a higher resolution picture of the EV–nanocavity interaction and confirmed that only one EV binds per well (Figure 4f,g).
One of the important challenges to address is the ability of confining the EVs within the nanocavities during the test time (Figure S6). The normalized fluorescent intensity of the EVs on different substrates has been investigated over time within the same field of view (Figure 4h). This comparison shows that fluorescent EVs fluoresce longer on a MoSERS microchip, compared to single crystal monolayer MoS2, nanocavities without MoS2, and silica, respectively. This shows the fluorescent intensity remains steadier on a MoSERS microchip compared to the other substrates. The mapping of the normalized intensity from fluorophores attached to exosomes in a fixed microscope field of view confirms the longer lifetime of the fluorescent EVs on a MoSERS microchip and the hindrance in the bleaching of fluorophores over test time in the presence of MoS2, which can be due to the nonlinear absorption and large exciton binding energy50 of the MoS2. The normalized fluorescent intensities of the EVs entrapped in the nanocavities with and without MoS2 in different diameter sizes (100–500 nm) were compared with the normalized fluorescent intensity of the EVs on SiO2 (Figure 4i). The normalized fluorescent intensity for each substrate was obtained from over three tests and 10 different random fields of view of the microscope with a 200 pixels × 250 pixels size. The fluorescent intensity collected from nanocavities with MoS2 (i.e., MoSERS nanocavities) was twice the intensity from the nanocavities without MoS2, confirming the higher entrapment efficiency of EVs via MoSERS. The nanocavities with 200–250 nm in diameter, which is a better match with the mean size of the EVs to fit in the cavities, have slightly higher fluorescent intensity compared to the nanocavities with other diameters.
The ability to entrap and isolate single EVs in the MoSERS nanocavities suggests that our approach has the potential to perform SERS on a single vesicle basis. To explore this possibility, we first obtained MoSERS spectra from synthetic liposomes. Liposomes are chemically uniform and provide an estimate of the expected intrinsic variation of the single-vesicle SERS recordings. Next, we analyzed EVs produced by human glial cells (NHA, noncancerous) and then investigated EVs produced by cultured human glioma cells (Figure 4j–m). All recordings were obtained by scanning a vesicle loaded MoSERS device cavity by cavity at 532 nm. For each raw SERS spectrum recorded, the spectrum baseline is subtracted, and the resulting spectrum is normalized and smoothed (Figure S8). The averaged processed spectra obtained from the EV populations (Figure 4j) indicate shared peaks associated with the characteristic Raman bands of bilayer lipids and common proteins in EVs. To characterize the variation of spectra obtained from single EVs, we performed a multivariate principal component analysis (PCA). PCA reveals principal component loadings with clearly separated clusters that quantitatively reflect the major differences in single EV spectra, effectively discriminating among EVs of the human noncancer cells, glioma cells, and liposomes (Figure 4k). The first principal component (PC1) and second principal component (PC2) shown in Figure 4l reveal the peaks enabling differentiation of the distinct vesicle populations. Each point in the plot represents one single EV spectrum with dimension reduction (Figure 4m). As expected, the chemically uniform single liposome recordings show the least scatter between single cavity recordings, the NHA recordings exhibit slightly greater scatter, and the EVs produced by the cultured glioma cells show considerably greater scatter, indicating the increased heterogeneity and molecular complexity of individual EVs.
Single EV Characterization with MoSERS Micro-Chip
The ability to entrap and isolate single EVs in the MoSERS nanocavities suggests that our approach has the potential to perform SERS on a single vesicle basis. To explore this possibility, we first obtained MoSERS spectra from cell lines as a comparison library (Figure 5a).
Figure 5.

Single EV characterization with MoSERS microchip for classifying molecular alterations in GBM cell lines. (a) Schematic showing single EV-resolved SERS analysis of glial, glioma, and glioma stem cell EVs with GBM-derived molecular alterations and their classification using a CNN algorithm (training set 70%, test set 30%). (b) The mean SERS spectra of EVs released from parental and oncogenic EGFRvIII expressing U87 cells. The PCA score plot of the SERS data comparing parental, and EGFRvIII expressed cancer (U87 and U373). (c) The mean SERS spectra and PCA score plot of EVs released from wild type GSC83 glioma stem cells that intrinsically contained EGFRvIII and EGFRvIII knockout (CRISPR/Cas9) GSC83. (d) The mean SERS spectra and PCA score plot of EVs released from wild type GSC1005 glioma stem cells that intrinsically contained EGFRvIII and EGFRvIII knockout (CRISPR/Cas9) GSC1005. (e) The mean SERS spectra and PCA score plot differentiating EVs from GSC1123 glioma stem cells with MGMT molecular alterations (GSC1123 Parental and GSC1123IC9R). (f) Quantitative PCR Study of EVs demonstrating gene expression levels in U87 EGFRvIII and U373 EGFRvIII EVs in a heat map. (g) The standard Western blotting test, revealing the EGFRvIII phenotypic transformation in EVs population. Antibodies against total EGFR (both wild type and mutant - top lanes) or specific to EGFRvIII (bottom lanes) were used to visualize the presence of the respective signals in EVs. (h) Accuracy and loss versus epoch number for training and validation of the cell line classification. (i) The classification accuracy of the cell-lines based on single EV analysis using CNN algorithm and (j) the classification of the molecularly altered cell lines over 9 iterations. (k) The ROC curve of classification of the single EV spectra according to the cell lines with an area under the curve of 0.91.
To determine if the MoSERS spectra are sensitive to the presence of mutations, we analyzed well-characterized EVs released from isogenic GBM cells with different patterns of oncogene expression.7,51,52 In particular, we obtained EVs from glioma and glioma stem cell lines positive or negative for mutant oncogenic variants of EGFR, such as EGFRvIII, or loss of the PTEN tumor suppressor gene. We also examined glioma cells positive and negative for MGMT, a marker of resistance to TMZ. The averaged processed spectra obtained from the EV derived from parental and EGFRvIII expressing U87 cells (Figure 5b) indicate shared peaks associated with their characteristic Raman bands. EVs were obtained from either U87 and U373 parental (control) glioma cell lines or their variants engineered to express this EGFRvIII oncogene. This change led to profound phenotypic transformation and altered molecular composition of GBM EVs, including their content of EGFRvIII18 (Figures S10 and S11). PCA applied to the multiplex cell variant set revealed principal component loadings with clearly separated clusters that recapitulate the fine spectral differences of the individual SERS spectra, effectively discriminating between EVs of the variant cell lines versus controls. While EGFRvIII is a part of the EV cargo, the oncogenic influence of this receptor reprograms molecular composition of cancer cells, impacting processes leading to EV formation7,18 and thereby can give rise to the observed large changes in SERS spectra.
MoSERS was also used to investigate the impact of EGFRvIII on EVs released by patient-derived glioma stem cell lines GSC83 and GSC1005 that naturally express oncogenic EGFRvIII (Figure 5c,d). We also studied the consequences of EGFRvIII knockout (CRISPR/Cas9), and restoration of the naturally lost PTEN tumor suppressor gene in several glioma stem cells via PCA and established cell lines in all of which changes in the MoSERS signal of the respective EVs were detected as a function of cellular genotype. Thus, genes involved in oncogenic transformation may alter the process of formation and cargo loading of tumor EVs in a manner that can be sensitively detected by MoSERS.
Indeed, while EGFRvIII represents an informative molecular paradigm of processes that impact EVs in the course of GBM progression, other molecular changes may also be of interest, especially those that occur in the context of therapy and disease recurrence. In this regard, our earlier study revealed differential EV profiles in glioma stem cells rendered resistant to TMZ during experimental chemotherapy in vivo, including the expression of MGMT. To assess the representation of these changes in EVs by MoSERS, we interrogated this model system in several ways. The effects of MGMT methylation/expression on EV profiles were studied in a series of cell lines derived from GSC1123 glioma stem cells and their variants (Figure 5e). Also, in this case PCA profiles extracted from MoSERS data readily differentiated between EVs generated on the background of therapy susceptible and therapy resistant-cells (GSC1123 and GSC1123IC9R, respectively). However, in this case, the separation occurred with relatively lower efficiency relative to EGFRvIII, as expected for the lower scattering power from the EVs RNA content compared to the surface proteins.
Our results clearly demonstrate that MoSERS can identify EVs released by cells carrying mutant EGFRvIII. We next explored the sensitivity at which MoSERS-based identification of EGFRvIII carrying EVs can be performed. The SERS signal generated by relative SERS spectra of glioma-derived EVs carrying EGFRvIII relative to EVs derived from parental culture carrying wild-type EGFR, often at low levels (see Figure S12 for details), shows an increased intensity at 1430 cm–1 and a decreased intensity at 1345 cm–1. The ratio of the SERS intensity at 1430 cm–1 relative to 1345 cm–1 is therefore a good indicator of the amount of EGFRvIII relative to wild-type EGFR present in a given EV. The increase in this intensity ratio for EGFRvIII relative to wildtype EGFR likely correlates with the more pronounced expression of leucine/histidine over tyrosine in EGFRvIII carrying EVs. The intensity ratio for 1430 cm–1/1345 cm–1 relative wavenumbers, averaged over all EVs measured at each EGFRvIII dilution, is linearly fitted with an R2 value of 0.995, demonstrating a detection limit of 1.23% for EGFRvIII carrying EVs in the pool of wild-type EGFR carrying EVs. This low detection limit of variants in the wild-type population suggests that MoSERS can potentially detect EVs carrying GBM related mutations that are diluted to low concentrations in the bloodstream. In order to assess the extent to which EGFRvIII impacts the cellular machinery involved in EV biogenesis, we evaluated the profiles of vesiculation-related genes in EGFRvIII-positive and -negative cells using polymerase chain reaction (PCR) assays, which revealed considerable differences (Figure 5f) (see Figure S9 for more details). It should be noted that the cell lines used in this study were fully characterized for their molecular profiles including proteomic analysis (see Materials and Methods section for referral). This includes the subsequent Western blotting validation of EVs as a correlative gold standard test, which revealed the EGFRvIII band in EVs population derived from U373vIII and U87vIII with phenotypic transformation and altered molecular composition, whereas this band disappears in EVs derived from GSC83 EGFRvIII-knocked-out glioma stem cells (Figure 5g). The clusters of EVs from each fully characterized cellular model including U87 and U373 systems in PCA graphs demonstrate their differentiation according to the spectral traits that could potentially be assigned to the anomalies connected to the corresponding molecular transition. The potential of the existence of EGFRvIII molecular alteration compared to parental glioma U87 and U373 cells was distinguished from one another with the 95% confidence ellipses. This confidence ellipse defines the region that contains 95% of all samples that can be drawn from the underlying Gaussian distribution. Similarly, for each GSC cells it is possible to distinguish the potential existence of the traits of molecularly altered cells. However, a higher dimension analysis is required to take into account the spectral differences in all cells and classify them simultaneously according to the record of their similarity.
We turned to machine-learning methods to fully exploit the information in the MoSERS spectra for fine-grained discrimination of EVs representative of variant cell lines. The convolutional neural network (CNN) method has been applied to a limited number of spectral studies, but with considerable success,53 while support-vector machine (SVM) was considered as a popular type of machine learning method based on a supervised classification algorithm.53,54 We chose a CNN architecture for a binary algorithm consisting of a first convolution layer, followed by a batch normalization, two residual layers, and a final fully connected layer. The residual layers are composed of 3 blocks, each of two convolutional layers followed by a batch normalization and an activation function and contain a shortcut connection. The residual blocks connected to a shortcut connection boost solving the exploding/vanishing gradient problems recurrent in deep networks. The convergence of training and validation values for loss and accuracy verify that the algorithm has been successfully trained (Figure 5h). Here, we collected 946 SERS fingerprint spectra from cell lines and divided them into two data sets, i.e. the training data set with 70% and the test data set with 30% of SERS spectra from each EV population. The average accuracy of the algorithm in correctly classifying single EV spectra into the corresponding cell types over 8 times of repetition demonstrated an overall accuracy of 89.3% (Figure 5i). Further classifying the single EV spectra data sets into the parental and molecularly altered cell lines demonstrated a relatively higher global accuracy toward EGFRvIII compared with MGMT methylation, which is in correlation with the multivariate studies (Figure 5j). The ROC curve of classification of the single EV spectra according to the cell lines indicates the sensitivity of the algorithm in determining the true positive rate of the prediction over the selectivity of it based on the true negative rate (Figure 5k). The area under the cure demonstrates a 91% global accuracy in classifying the cell lines according to the MoSERS single EV SERS approach.
Single-EV MoSERS Profiles in Biofluids Reflect Tumor Fingerprints in Patients
While experiments with molecularly defined cancer cells document the ability of MoSERS to detect distinct EV spectra, we asked whether this approach can be used for more complex biological samples containing EVs from multiple cellular sources, such as blood. Thus, MoSERS spectra were obtained from EVs isolated from blood samples drawn from 8 healthy individuals and 12 patients (Figures S13 and S14) clinically diagnosed with GBM (Figure 6a). We received clinical annotation of 10 patients to correlate with the SERS study in the following section from a pathology study at the Montreal Neurological Institute and Hospital (MNI) (Table 2). Prior to the start of the SERS characterization, the isolated EVs were tested using PCR via EGFR cDNA amplification to correlate with the clinical data (Figure 6b). To analyze the single EV spectra, the residual neural network (Resnet)-based CNN algorithm (Table S3) was used as a proof-of-concept to classify their possible cellular sources and infer molecular hallmarks of the underlying disease.53 To this end the CNN algorithm was trained with the spectra from healthy and cancerous cell lines, as well as an unseen (blinded) spectra set of 2 healthy and 2 patient samples, followed by testing the MoSERS spectra in the remaining samples. The probability that sampled EVs carry one of the three molecular GBM-associated alterations (EGFR amplification, EGFRvIII, and MGMT methylation) was determined based on a Mahalanobis distance. A total of 70 EVs were measured for each blood sample. The probability score for each individual sample shows a higher similarity between the test EVs from patient samples and cancerous variants compared to the ones derived from healthy individuals (Figure 6c). The convergence of training and validation values for loss and accuracy verifies that the algorithm has been successfully trained. We used the ROC curve to assess the overall true positive rate versus the false negative rate of the MoSERS-based single EV prediction of underlying molecular traits, which resulted in an overall area under the curve (AUC) of 85% (Figure 6d).
Figure 6.

MoSERS profiles of blood-borne EVs from GBM patients harboring distinct molecular alterations. (a) Schematic illustrating the classification of patient-derived EVs using MoSERS single EV fingerprints analyzed by machine learning. Each class represents a different GBM marker. (b) RT-PCR agarose gel of EGFR and EGFRvIII cDNA in control and patient-derived circulating EV samples. (c) The probability distribution of belonging to each of the classes based on CNN output, EGFR amplification (blue), EGFRvIII (green), and MGMT methylation (orange). (d) The ROC curve of assessing the single EV spectra prediction accuracy over clinical annotation demonstrates an overall AUC of 0.85. The probability that EVs are positive for (e) EGFR amplification, (f) EGFRvIII, and (g) MGMT partitioned into classes based on clinical read-out: healthy (gray), negative-variant patients (light color), and individual positive-variant patients (dark color). (h–j) ANOVA analysis of all spectra partitioned based on clinical annotations, demonstrating the ability to distinguish samples from negative and positive variant patients as well as healthy patients. (k) Samples with positive variants of EGFR amplification, EGFRvIII and MGMT methylation were pooled and classified by the probability distribution of each sample. (l) The ROC curve of assessing the overall MoSERS prediction accuracy of individual patients carrying one of the three molecular GBM-associated alterations over clinical annotation demonstrates an overall AUC of 0.91.
Table 2. Clinical Annotations Corresponding to Patient Mutation Statusa.
| Patient | Status | EGFR Amplification | EGFRvIII | MGMT Methylation |
|---|---|---|---|---|
| P-1 | Glioblastoma | Negative | Negative | Positive |
| P-2 | Glioblastoma | Negative | ND | Negative |
| P-3 | Glioblastoma | ND | ND | ND |
| P-4 | Glioblastoma | Positive | Positive | Positive |
| P-5 | Glioblastoma | Negative | ND | Negative |
| P-6 | Glioblastoma | Negative | Negative | Negative |
| P-7 | Glioblastoma | Positive | Positive | Negative |
| P-8 | Glioblastoma | Positive | Negative | Negative |
| P-9 | Glioblastoma | Positive | Positive | Negative |
| P-10 | Glioblastoma | Negative | ND | Negative |
| P-11 | Glioblastoma | ND | ND | ND |
| P-12 | Glioblastoma | ND | ND | ND |
| H-1 | Healthy | – | – | – |
| H-2 | Healthy | – | – | – |
| H-3 | Healthy | – | – | – |
| H-4 | Healthy | – | – | – |
| H-5 | Healthy | – | – | – |
| H-6 | Healthy | – | – | – |
| H-7 | Healthy | – | – | – |
| H-8 | Healthy | – | – | – |
Clinical pathology results of the patient diagnosed with GBM. *ND = not determined. EGFR amplification, EGFRvIII and MGMT methylation status was determined by sequencing of the primary tumor.
To achieve the correlation of these results with clinical annotations, we compared the probability scores of positive-variant patients with negative-variant patients and healthy subjects (Figure 6e–g). We assessed the healthy control, negative-variant patient group and individual positive-variant patients using one-way analysis of variance (ANOVA) with a post hoc Tukey’s test (Figure 6h–j). ANOVA detected an overall significant difference among the majority of the positive-variant individuals (P < 0.001) compared to the negative-variant pool.
The samples were grouped as healthy subjects, GBM patients negative for genetic variant, and GBM patients positive for genetic variant. Positive variant patients demonstrate a relatively higher probability of having the variant gene compared to negative variant patients and samples from healthy donors (Figure 6k). The ROC curve for the individual patients based on the accumulative probabilities of the single EVs carrying one of the three molecular GBM-associated alterations demonstrates an overall area under the curve (AUC) of 91% (Figure 6l).
Performing Tumour Diagnosis via Blood Derived EVs
One area where binary determination of patient subsets through an EC-based liquid biopsy would be at the time of recurrence where predicting efficacy or failure of adjuvant treatment could have actionable consequences. To further explore this avenue, we developed a binary benchmark to predict the overall health status (with respect to GBM) of an individual from our single EV spectra classifications, e.g. to determine whether the spectrum analyzed belongs overall to a “GBM-positive” or “GBM-negative” state (Figure 7a). In this case, the prediction of whether an EV belongs to a certain cell line is not sufficient because patient samples might contain EVs from other cells in the body.
Figure 7.

MoSERS-based separation of EV signals from blood samples of healthy subjects and glioblastoma patients. (a) Schematic showing single EV SERS analysis and binary classification of blood EVs into healthy and GBM patient groups using CNN algorithm trained with a library of cell lines and blood samples. (b) The distribution of the probabilities of the single EV spectra from GBM-negative and GBM-positive samples associated with their belonging to healthy and patient classes. (c) The one-way ANOVA comparison analysis of mean probabilities associated with the single EV spectra. (d) The loss and accuracy of the training and validation sets based on the CNN algorithm. (e) The ROC curve of the CNN prediction of probabilities of single EV spectra to belong to the right class resulted in an overall AUC of 0.89. Binary classification of blood sample EVs into patient and healthy classes based on the average similarity scores. (f) The average similarity of blood samples based on CNN prediction results of single EV spectra, differentiating the data sets into healthy and patient classes with a deterministic cutoff value. (g) Binary classification of blood samples into healthy and patient classes based on the average similarity scores via one-way ANOVA comparison.
We used the CNN binary algorithm to classify the EVs into two classes, which rendered a higher accuracy of 83% compared with the SVM binary algorithm (Figure S15). The training data set (Table S3) was composed of 80% of the single EV spectral acquisitions from healthy donors and from the supernatant of noncancerous cells, designated as GBM-negative class (gray). Similarly, this set contained 80% of the single EV spectral acquisitions of 3 GBM patient donors and the EVs isolated from the supernatant of glioma cells, collectively designated as GBM-positive class (red). The EVs from the rest of the healthy (nHtest = 8) and patient (nP = 12) blood donors were used to determine the accuracy of binary classification into the healthy and patient groups. To avoid overfitting of our pattern-learning algorithm to the data, we validated the training process using a validation set (10% of the training set).
The distribution of the probability of belonging to each binary class generated was based on a total of 70 measurements collected from each blood sample (Figure 7b). We used Platt scaling to calculate the probability of each data set belonging to each class.55 The similarity score for each individual sample based on the Mahalanobis distance shows a higher similarity between the test EVs from patient samples and cancerous cellular variants compared to the ones derived from the healthy individuals. The distribution of calculated probabilities associated with each class (i.e., GBM negative and GBM positive) shows that the single EV spectra from healthy samples have a higher probability of belonging to the GBM negative class, while the single EV spectra from patient samples tend to be classified into the GBM positive class (Figure 7c). The post hoc comparisons via Tukey’s honest significant difference demonstrated a P-value below 0.001 for the probability of the single EV spectra correctly classified into healthy and patient classes.
To verify the proficiency of the algorithm competence in training, the loss and accuracy of the training and validation sets (Figure 7d) were studied, demonstrating the convergence of the training data sets after 11 epochs. At this point, the outputs from the fully connected layer of the training and test data were separated into two classes as the number of epochs increased (Figure S16). The convergence of training and validation values for loss and accuracy verifies that the algorithm is successfully being trained. The ROC curve demonstrates the true positive rate as a function of the false positive rate (Figure 7e), to assess the overall dexterity of the true positive rate versus false negative rate of the MoSERS prediction over clinical annotation resulted in an overall AUC of 89%.
The outputs of the CNN also enable us to determine an average relative similarity for each patient spectrum based on the Mahalanobis distance to differentiate healthy and GBM-positive samples. The average relative similarity plot of the ensemble output of the EVs from each blood sample (Figure 7f) indicates successful discrimination of the healthy samples with lower similarity scores (GBM-negative class) and the patient samples with higher similarity scores (GBM-positive class). The average similarity value for each sample was calculated by taking in over 1200 single EV SERS spectra. A high similarity means a high risk of cancer, while a low similarity means a low risk of cancer. We benchmarked a cutoff average similarity value for binary classification of the data set into healthy and patient categories. The GBM-negative class was used as the threshold to predict a measure for the possibility of diagnosis with cancer. According to this designation, all healthy samples belonged to the GBM-negative class rather than the GBM-positive class. The post hoc comparisons demonstrated a P-value below 0.001 for the average similarity of the healthy and patient samples being correctly classified based on the ensembled single EV spectra (Figure 7g).
Conclusion
EVs encapsulate multiple molecular signals reflective of the phenotype of their originating tumor cells. The MoSERS microchip successfully revealed the molecular diversity of GBM cells by converting them into highly granular, actionable physical fingerprints, combining the complexities of individual EVs and their heterogeneous subpopulations. Current EV-based diagnostic approaches average the signals contained in multiple EVs, thereby obscuring the important subtleties of the EV landscape. Here, we were able to observe changes parallel to the expression of MGMT and EGFR VIII, molecular paradigms for therapeutic resistance and aggressiveness in GBM, at the level of individual cell lines and patients. Indeed, the convolutional neural network (CNN) algorithm demonstrated the potential of MoSERS analysis of circulating EVs to determine the MGMT status in GBM patients, an observation that merits validation in a larger clinical cohort. Thus, MoSERS technology may be applicable as a versatile tool for liquid-biopsy diagnostics to track the evolving landscape of GBM in individual patients while providing clinically actionable information.
The first key feature of the MoSERS system is the patented MoSERS microchip platform confined in microfluidics that overcomes key challenges required to perform SERS with single EV resolution. Our optimized photonic design leads to high in-cavity electric field enhancement and can thereby provide sufficient signal to obtain meaningful single EV spectra. The EVs are loaded in the nanocavities at high efficiency due to attractive interactions with the embedded MoS2 layer. Finally, as only one EV can occupy a given cavity, a single EV can be isolated in a single laser focus. When light is concentrated into the subdiffraction volume of a single nanocavity with patterned monolayer MoS2 at the bottom, the electric field in the small volume becomes very intense, leading to enhancement in the local density of the states in the nearby molecules and the formation of hot spots which in turn enhances the incident field intensity. Therefore, the hot spot improves the efficiency of the optical excitation process when placing a single EV in the nanocavity. In addition, when an EV approaches a MoS2 layer, it forms a strong electrostatic interaction comprised of van der Waals and Coulomb forces. MoS2 forms the Coulomb interactions primarily through contact with the polar head regions of lipid molecules, while the van der Waals interactions mainly arise from contact with the hydrophobic tail regions of lipid chains. As proof-of-principle, we show that MoSERS enhanced by machine learning-based prediction can discriminate EVs from healthy and cancer cells in GBM cell-lines, detect EVs released by GBM variant cell-lines, and differentiate EVs carrying GBM genetic variants in patient blood samples. The label-free single EV SERS technique functions on the basis of strong EM-field enhancement that is highly dependent on the physical distance between the biomolecule of interest and the nanocavities. We studied this effect for the impact of the oncogenic transformation involving surface proteins, since those are often better characterized and more relevant for the application, as well as often present on the outer surface of tumor-derived EVs.
The second feature of the MoSERS system is the integrated microfluidic sample delivery system that allows for real-time monitoring of blood circulating EVs. In cancer, diagnostic procedures based on minimally invasive blood sampling are highly attractive. Currently GBM diagnostics follows clinical, imaging, and operative biopsy steps. These protocols are time-consuming and fraught with risks of surgical complications. Even if molecular diagnosis is carried out, it only offers a single snapshot of otherwise dynamic disease, with little opportunity to explore alternative management. In contrast, a blood test is rapid, safe and can be performed multiple times over a disease time course to assess impact of a given prescribed treatment, including distinction between disease subtypes, therapeutic vulnerabilities, and molecular evolution. The main challenge in using blood as the source of EVs is the considerably lower concentration of the tumor-derived EVs amidst other EV sources (platelets, blood cells).6 We demonstrate a detection limit of 1.23% for EVs from variant cells suspended in a wild-type EV population. This allows for processing blood samples where the GBM biomarkers are diluted to 3–10%. In addition, our analysis of plasma EVs captures global differences in the EV populations in blood of GBM patients and healthy counterparts, which may include alterations in EVs in noncancerous cellular populations (e.g., due to horizontal transfer of EVs from cancer cells or other influences).
Determining the dynamics and nature of a particular tumor with minimal invasiveness is equally important. Currently, healthcare providers rely on traditional characteristics, such as tumor appearance, to diagnose a given tumor subtype and determine optimum treatment, with relatively few molecular markers in use (IDH1 mutations, MGMT). For patients diagnosed with GBM, however, if more complex underlying molecular characteristics of the tumor could be defined, it may be possible to explore more personalized care and evaluate treatment approaches in a more controlled manner. To this end, our MoSERS approach has potential to classify different molecular tumor subtypes on the basis of spectroscopic fingerprints drawn from single EVs as a form of liquid biopsy. In addition to that, the single EV SERS approach allows one to statistically investigate the tumor heterogeneity and monitor complex tumor cell alterations for the postsurgery treatment.
The main limitation of the current MoSERS platform is the time required for data acquisition. A large number of single EV SERS spectra are required to identify the fingerprint spectra for the classification algorithm. Considering the heterogeneity of EVs and preferential enhancement of the Raman modes corresponding to molecular vibrations perpendicular to the SERS surface, at least 50–100 single EV SERS spectra are necessary for a conclusive result. Indeed, over 1 h is presently needed to generate 100 data points for each sample. With testing samples in parallel using multiple devices, it is possible to reduce the time to less than 30 min per sample. To this end, within 90 min and using up to 10–100 μL of purified blood serum, it is possible to generate a comprehensive landscape of EVs at a single EV resolution. In the context of a disease as complex and dynamic as GBM, this minimally invasive technology may have considerable diagnostic potential.
Materials and Methods
Fabrication of MoSERS Device
A combined bottom-up and top-down approach was used to develop the single exosome analyzer nanoelectrodes. First, a chemical vapor deposition (CVD) technique was used to grow single-crystalline monolayer MoS2.56 The monolayer was produced through the CVD method by nucleation and growth from vapor phase solidified on the substrate of choice. Second, e-beam lithography (Raith e-line) was used to pattern the nanohole array in a 300 nm thick layer of negative photoresist (MaN-2403). Third, an 80 nm biocompatible ZnO layer and a 10–100 nm Ag layer were deposited (via BJD 1800) as the back-reflector and plasmonic layer, respectively. Fourth, the plasmonic nanohole array was formed via lift-off. The design of the microchip was optimized to be compatible with integration with most existing types of microfluidic devices.33,58−60 Finally, the MoSERS microchips with the size of 0.1 × 0.1 cm2 were embedded in the fluidic sample delivery device fabricated using a 3D printing technique with a 0.2 μm filter for SERS interrogation of the circulating EVs in the patient blood samples.
Characterization
The SEM was performed using the FEI Quanta 450, the AFM with a Bruker, MultiMode8 instrument, while the TEM with a FEI Tecnai G2 F20 200 kV Cryo-STEM. The optical characterizations were performed via a Lambda750 NIR-UV–visible instrument and a NanoSpec reflection spectro-microscope. EVs samples were prepared for SEM in three steps: (1) The purified EVs in PBS solution (100 μL, 1 mM) were introduced to the recognition substrate for 2 min followed by (2) incubation with glutaraldehyde 3% in sodium cacodylate (0.1 mM) overnight. (3) Dehydration of EVs was performed by immersion in ethyl alcohol (30–100%) to exchange their cytoplasm with alcohol (10 min immersion in each). Then, a critical point dryer (Leica Microsystems EM CPD030) was employed to substitute alcohol content with dry CO2 with minimum damage to the EVs morphology.
Finite Difference Time-Domain
The electric-field distribution over the nanocavity array was modeled using the finite difference time-domain (FDTD) module (v8.21.1781, Lumerical Solutions, Inc.). The electric field distribution was simulated under a plane wave and the laser excitation wavelength of 532 nm using a total field scattered-field (TFSF) light source that resembled the laser. Detail simulation parameters are described in the Supporting Information.
Molecular Dynamics Simulation
The MoS2 nanosheet was modeled as a triangle with each side length of about 7.0 nm. The bilayer membrane was constructed based on a realistic EV lipid with dimensions of 10.7 × 10.7 nm. Five different lipid components including cholesterol, POPC, TSM, POPS, and POPE were considered in the bilayer membrane (while ignoring other components with a ratio lower than 5%), yielding a final number of 276, 96, 102, 72, and 36 molecules, respectively. This membrane bilayer was first equilibrated with a 300 ns MD simulation in order to obtain a stable structure. Then, this stable membrane was utilized to explore its interaction with the MoS2 nanosheet. Two simulation setups were constructed. In the first simulation, the MoS2 was perpendicularly placed on top of the membrane with its corner pointing to the membrane surface. During this simulation, the movement of MoS2 along the directions parallel to the membrane surface was fixed whereas the movement along the direction vertical to the membrane surface was free. In the second simulation, the MoS2 was initially placed on the membrane with its basal plane parallel to and touching the membrane surface. During this simulation, all atoms were free for movement. Both systems were dissolved in a 150 mM NaCl solution, which was each conducted for 300 ns MD simulation.
Processing EV
Isolation of EVs from Cell Cultures
EVs were purified from the growth media of cancer cells (Table 3) using an optimized protocol.25 Briefly, the conditioned medium (CM) was collected from cells grown for 72 h in culture media containing 10% EV-depleted FBS (generated by centrifugation at 150,000g for 18 h at 4 °C). CM was centrifuged one time at 400g and then passed through a 0.2 μm pore-size filter. The resulting CM filtrate was concentrated to 500 μL using an Amicon Ultra-15 Centrifugal Filter Unit (EMD Millipore, Billerica, MA) with a 100,000 NMWL molecular cutoff. The concentrated media was further purified using a qEV single SEC column (Izon Science, UK); 500 μL of the sample was loaded, and 4 fractions of 500 μL of eluent were collected after 2.5 mL of the initial eluent. The concentration and size distribution of EVs in the purified media were obtained using the nanoparticle tracking analysis, NanoSight NS500 instrument (NanoSight Ltd., UK). Three recordings of 30 s at 37 °C were obtained and processed using NTA software (version 3.0).
Table 3. List of the EVs Tested for Validation of MoSERS Single EV Approach.
| Cell Lines | Informative GBM Mutations | |||
|---|---|---|---|---|
| U87 | Parental | EGFRvIII | ||
| U373 | Parental | EGFRvIII | ||
| GSC83 | Parental | EGFRvIII knocked out | ||
| GSC1005 | Parental | EGFRvIII knocked out | ||
| GSC1123 | Parental | 1123IC7R | ||
| 1123IC12S | ||||
| 1123IC9R (MGMT) | ||||
The models used in this study include the glioma U373 parental cell line and its isogenic counterpart expressing oncogenic EGFRvIII receptor (U373vIII cells), as well as another unrelated glioma (U87 parental cells) and its corresponding variants obtained by transfection of either EGFRvIII oncogene, or the PTEN tumor suppressor gene. In addition, we also analyzed glioma stem cells (GSC83) naturally expressing wild-type EGFR and EGFRvIII, which were compared to their isogenic subline, in which the EGFRvIII/EGFR locus was disrupted by CRISPR/Cas9 editing technology. Another similar pair of isogenic glioma stem cell lines included GSC1005 (wild type) cells and their counterparts in which EGFRvIII was knocked out.
Furthermore, we interrogated EVs from another series of isogenic glioma stem cell lines comprising GSC1123 (wild type) cells and their several variants rendered resistant to the chemotherapy agent temozolomide (TMZ), which is used extensively in glioblastoma therapy. These cell lines were allowed to evolve naturally in tumor bearing mice exposed to TMZ. Among those cell lines recovered from such recipients, GSC1123-7 acquired TMZ resistance through hypermutation, while GSC1123-9 cells turned on the expression of the O6-methylguanine-DNA methyltransferase (MGMT) gene, a common event in progression of human glioblastoma. In addition, as a control we used another TMZ-sensitive cell line, designated GSC1123-12, which was recovered from mouse tumors in the absence of TMZ treatment. The derivation, biological properties, drug resistance and various aspects of EV characteristics of these cells were, as mentioned, previously extensively described by us20,52,61−64 and others.65,66
In addition to the aforementioned defining features of our model cell lines, some of their proteomic and genomic profiles were further resolved and published, as were the proteomes and RNA expression profiles of their EVs.20,25,52 We used various techniques such as nanoflow cytometry,20 Western blotting7 and other approaches to document these important traits, including the content of EGFRvIII in EVs. Thus, while biomarker gold standards are still aspirational in the EV research space, we entered the MoSERS project with detailed prior knowledge of the expected molecular characteristics of test EV preparations and sought to understand whether they will be reflected in enhanced Raman spectra at the single EV level. As we argue, this turned out to be the case.
EV Isolation from Cerebrospinal Fluid
The patient cerebrispinal fluid (CSF) samples were filtered through 0.2 μm pore-size filters. The filtered samples were further purified using a qEV single SEC column (Izon Science, UK); 500 μL of each sample was loaded, and 4 fractions of 500 μL of eluent were collected after 2.5 mL of the initial eluent.
EV Isolation from Patient Blood Samples
The patient blood samples were centrifuged one time at 200g for 20 min, and then the supernatants were centrifuged at 1,000g for 15 min. The resulting liquid phase was further centrifuged at 1,500g for 20 min to remove platelets and achieve platelet-poor plasma (PPP), which was passed through a 0.2 μm pore-size filter. The EVs contained in filtered PPP samples were further purified using a qEV single SEC column (Izon Science, UK); 500 μL of the sample was loaded, and 4 fractions of 500 μL of eluent were collected after 2.5 mL of the initial eluent.
Labeling EVs
A fluorescent lipid binding dye, DiI, was used to label the EV producing cells (indirect labeling of EVs). A total of 3 × 106 cells were labeled with 5 mL of DiI (Invitrogen) in 2 mL of DMEM without FBS for 20 min at 37 °C. The cells were washed twice with DMEM without FBS, resuspended in 30 mL of DMEM containing 10% EV-depleted FBS (generated by centrifugation for 18 h at 150,000g), and cultured for 72 h at 37 °C, 5% CO2 to generate CM for EV isolation.
Raman Spectroscopy
Spectra Collection
Surface-enhanced Raman spectroscopy (SERS) and mapping SERS spectra were collected in back scattering geometry, using an InVia Raman microscope (Renishaw plc, Wotton-under-Edge, UK) equipped with a 532 nm HeNe laser (Melles-Griot, Voisins Le Bretonneux, France) delivering 15 mW of laser power. The laser was polarized along the x-axis direction. The platform loaded with biosamples was mounted on a ProScan II motorized stage (Prior, Cambridge, UK) under the microscope. A Leica 100× microscope objective (NA 0.95) focused the laser on the sample into a spot of ∼0.8 μm diameter. An 1800 l/mm grating yielded a spectral resolution of 754 cm–1. A thermoelectrically cooled charge coupled device (CCD) camera was used for detection. The spectrograph was calibrated using the Silicon substrate. Single spectra were collected with an exposure time of 35 s at 0.5% of the laser intensity. Mapping was achieved by collecting spectra with steps of 1 μm, with an exposure time of 20 s for each spectrum. Spectra, consisting of 2924 data points each, were obtained in the 100–3200 cm–1 region using the synchro mode of the instrument software WiRE 5.1 (Renishaw). In the synchro mode, the grating is continuously moved to obtain Raman spectra of extended spectral regions. The dimensions of the map depended on the zone of interest investigated. To characterize the SERS signals, we measured Raman spectra on close to dry samples (0.5–10 μL). The label-free SERS single EV analysis is a comparative method. We used over 70 spectra per sample to run a relatively conclusive comparison that separated the groups of interest. For cell lines, we compared the mutated cells with their wild-type counterparts demonstrating the 95% differentiation via multivariate analysis for each cell type. In addition, we used a machine learning method for compiling the spectral differences over 600 spectra taken from the cell lines and categorized them according to their origins based on the similarity they showed to each training set.
Data Preprocessing and Analysis
All data preprocessing and analysis were performed within the Origin Pro 2019b software, ImageJ, WiRE 5.1, and CanvasX 2019 environment for statistical computing and graphics. In particular, we used principal component analysis for spectroscopy v.1.2 App in OriginPro 2019 for PCA analysis. The preprocessing of SERS spectra consisted of four steps: (i) cosmic rays identification and removal, (ii) baseline correction, (iii) intensity vector-normalization according to Si peak, and (iv) outliers detection and removal. For the baseline correction, a linear baseline was fit automatically to the whole spectral range and was subtracted from each spectrum of the data set. Outliers detection was done by identifying suspicious points on the PCA score maps and inspecting the corresponding spectra. In the preprocessing stage, PCA is thus used as a method to identify suspicious spectra, exploiting its sensitivity to outliers. These suspects were then individually examined before deleting them. The heterogeneity of the data indeed presents a challenge. Our strategy to handle it includes the implementation of a CNN algorithm that follows a standard machine learning practice in which the data library, integrated by tens of single EVs spectrum from each cell line, patient samples, and controls, is divided into different data sets for the training, validation, and testing of the CNN model.
PCA
PCA reduces the number of variables by condensing all the spectral information contained in a large number of spectra into fewer latent variables (the principal components or PCs). Hyperspectral data are thus decomposed by PCA into the latent spectra (loadings) and scores. This approach is closely related to describing each spectrum in a Raman study as a product between components concentrations and pure constituents’ spectra, where the latent spectra are used instead of those of the pure constituents, which are unknown. In the present study, PCA was performed on preprocessed data, and the first three principal components PC 1, PC 2, and PC 3, which could be interpreted in terms of the biochemical components of the EVs and were considered for discussion. The loadings and score maps for the principal components are difficult to interpret. The PCA analysis incorporated in this study is a regression model without intercept-computed based on the covariance matrix, which is considered to implicitly perform the centering, demonstrating the distinguishing spectral traits as principal components.
Machine Learning
Cell Classification
As a way to ascertain that our ML pipeline is working properly, we aimed to use a multiclass ML to categorize different spectra into the 6 predetermined classes (NHA, U87, U373, GSC83, GSC1005, GSC1123) in addition to classifying the single EV spectra from molecularly altered cells.
SVM – A Support Vector Machine (SVM) is a classification ML algorithm that finds hyperplanes that best separate the input data points into different classes. In order to find the optimal hyperparameters (C and the kernel) needed for our classification task, we used a Bayesian search,50 and the parameters were tested using 5-fold cross validation on the training set. C was explored from 10–6 to 100 using a log uniform distribution, and 2 different kernels were tested: a linear and a radial basis function kernel. The hyperparameters kept were C:0.4 and a linear kernel, as those that gave the best overall accuracy over the 5 folds. The hyperparameters used were C: 0.4; kernel: linear.
CNN – To compare against a more advanced modeling architecture that can appreciate nonlinear relationships in our data, we subsequently used a Convolutional Neural Network (CNN). This is a type of neural network that uses convolutional layers to extract the distinguishing features between classes. The final outputs of the network are then used to generate a prediction. The CNN here established is derived from Ho et al.,53 which in turn is based on a code from his algorithm. It consists of a first convolution layer, followed by a batch normalization, two residual layers and a final fully connected layer. The residual layers are composed of 3 blocks, each of two convolutional layers followed by a batch normalization and an activation function, rectified linear activation function (ReLU), containing a shortcut connection, which links the input to the output. The implementation of residual blocks facilitates solving exploding/vanishing gradients problems recurrent of deep networks. The hyperparameters (the number of hidden filters, the number of blocks) were determined through a grid search. The number of blocks was chosen between 2 and 3, the hidden filters were explored between 10 and 40 with an incrementation of 10, and the number of layers was between 1 and 6. The batch size was set to 5. The loss function is cross-entropy, and the optimizer function is the Adam optimizer. Training was done on the training set, with 10% kept as a validation set, to monitor the performance of the algorithm through loss and accuracy. Early stopping after 5 iterations with no improvement of the accuracy on the validation set was also implemented.
The CNN algorithm is trained and validated with the corresponding data set, to verify the proficiency of the algorithm competence in training. Two parameters were studied, loss and accuracy for both the training and validation sets. Loss is an indicator that the CNN has been successfully trained since is a parameter indicating the distance from the predictions of the model and the true values. When the CNN neural network converges it refers to a moment in the training process when the loss parameter is at its minimum. At this point, the outputs from the fully connected layer of the training and test data were separated correctly into the number of classes defined in the study. The results presented in the figures of the manuscript are generated by presenting the test set to the CNN algorithm, this set of data has not been seen by the algorithm before and its appropriate classification, suggests no overfitting of the data and further demonstrates the validity of the approach. Further, an ROC curve was used to assess the overall dexterity of the true positive rate versus false negative rate of the CNN prediction.
Binary
CNN – The CNN algorithm was also implemented for the second stage, the binary classification task. The architecture is identical to the previous one, only with the final output layer being 2 neurons instead of 10. Loss and accuracy curves were obtained to verify the appropriate training of the algorithm, and the convergence was reached after 10 epochs.
Human Samples
To study the MoSERS microChip for clinical samples, we analyzed EVs extracted from the blood of 12 GBM positive patient samples [taken in the surgery room at MNI under supervision of Prof. Petrecca], in contrast to 8 healthy donor samples [from Innovative Research: single donor human blood (IPLASK2E50 ML): HMN494604, HMN494605, HMN494606, HMN494607, HMN495738, HMN495739, HMN495740, HMN495741]. The patient samples were collected from individuals with a confirmed diagnosis of glioblastoma multiforme moments prior they underwent surgery.
We collected the EVs from each individual blood samples on the same day of the surgery in 5 fractions (7–11) using the protocol mentioned above. A large set of single EV spectra from each of the EV samples were collected using MoSERS microchip and microRaman spectroscopy, following the above-mentioned protocols. The heterogeneity of EVs must be addressed in our study to ensure the MoSERS Micro-Chip overcomes this challenge and presents a robust solution to assist in clinical decision-making. The spectra acquired from the platform are processed and analyzed using a CNN algorithm (liability of the algorithm was tested via cell-derived single EV spectrum and explained above in more detail).
Initially, the single EV spectra were studied for their molecular alteration classification according to the clinical annotations. The study aimed to identify EVs carrying molecular alterations the EGFR amplification, EGFRvIII, and MGMT while differentiating them from healthy and samples without those molecular alterations. A tailored CNN algorithm classified each single EV spectrum tested based on having a higher probability of belonging to EGFR amplified, EGFRvIII, or MGMT methylation mutations. The collection of the spectra for each patient and healthy samples was then used to obtain a global classification to correlate with the clinical annotations and determine the global accuracy.
Finally, we used a binary diagnostic test to challenge the accuracy of the system to distinguish healthy and patient blood samples. The CNN outputs probabilities for each single EV spectrum tested classifying them based on having a higher probability of belonging to a GBM-negative or a GBM-positive group. The ensemble of spectra for each patient and healthy samples is then used to obtain a global diagnostic output based on the average relative similarity of the collection of spectra.
Acknowledgments
The authors thank the Faculty of Engineering at McGill University, the Canadian Cancer Society (255878 CCSRI), Natural Science and Engineering Research Council of Canada (NSERC, G247765), New Frontiers in Research Fund (250326), Canada Foundation for Innovation (CFI, G248924), and Brain Canada (257083) for financial support. S.M. was supported by the Canada Research Chair (CRC) award. J.R. was supported by Foundation Grant from the Canadian Institutes of Health Research (CIHR), Cancer Research Society (CRS) and Genome Quebec (GQ) Operating Grant, McGill Interdisciplinary Initiative in Infection and Immunity (MI4) seed grant, Canadian Foundation for Innovation (CFI), NET program sponsored by Fondation Charles Bruneau (FCB), and Jack Cole Chair in Pediatric Hematology/Oncology. R.Z. was supported by the National Key Research and Development Program of China (2021YFA1201201 and 2021YFF1200404), the National Natural Science Foundation of China (U1967217), National Independent Innovation Demonstration Zone Shanghai Zhangjiang Major Projects (ZJZX2020014), the Starry Night Science Fund at Shanghai Institute for Advanced Study of Zhejiang University (SN-ZJU-SIAS-003/006/009), and BirenTech Research (BR-ZJU-SIAS-001). The authors acknowledge Nanotools-Microfab and the Facility for Electron Microscopy Research at McGill University, the Laboratoire de microfabrication (LMF) at Polytechnique, Montreal, and the research facilities of NanoQAM at the Université du Québec à Montréal. M.J. and C.D.R.M. appreciate McGill Engineering Award (MEDA) and Fonds du Recherche du Quebec (FRQnet) scholarships as well as the CMC microsystems student research fund.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c09222.
Overview of recent nanotechnologies for detection of extracellular vesicles, design details of MoSERS system, supporting data of SERS enhancement, MoS2–EV interaction, characterization of cell line EVs, and characterization of human circulating EVs (PDF)
Author Contributions
M.J. initiated the idea, design, and fabrication protocols, performed the physical characterizations, sample preparation, SERS data collection, FDTD modeling, data processing and analysis as well as writing the paper. C.D.R.M. contributed to the SERS data collection, running machine learning algorithms, and writing the paper. O.J. contributed with the machine learning algorithms and data analysis. L.M. and N.T. contributed in EVs sample preparation, purification, and running the standard tests. I.I.H. contributed to the fabrication of the fluidic device and theoretical studies with MD. Z.G. and Z.H. contributed to the MD simulations and data collection. C.S. contributed to providing CAS-9 gene altered glioma stem cell lines. Y.L. contributed to the SERS experiments. K.P. provided patient samples and clinical annotations. M.C.G. contributed to patient samples phenotyping and pathological tests. R.Z. provided the design and analyses on the theoretical studies with MD. S.W.H. provided advice on SERS optimization and functionality of the platform. W.R. provided advice on single EVs confinement and contributed to writing of the paper. J.R. provided the EV samples from GBM cell lines for the entire project, advice on the EV’s biological, and contributed to the writing of the paper. S.M. supervised the project from the idea to development, contributed to the design of the figure sets, and writing of the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Im H.; Shao H.; Park Y. Il; Peterson V. M.; Castro C. M.; Weissleder R.; Lee H. Label-Free Detection and Molecular Profiling of Exosomes with a Nano-Plasmonic Sensor. Nat. Biotechnol. 2014, 32 (5), 490–495. 10.1038/nbt.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-Y.; Du S.-L.; Zhang J.; Liang A.-L.; Liu Y.-J. Exosomes and Breast Cancer: A Comprehensive Review of Novel Therapeutic Strategies from Diagnosis to Treatment. Cancer Gene Ther. 2017, 24 (1), 6–12. 10.1038/cgt.2016.69. [DOI] [PubMed] [Google Scholar]
- Boriachek K.; Islam M. N.; Möller A.; Salomon C.; Nguyen N. T.; Hossain M. S. A.; Yamauchi Y.; Shiddiky M. J. A. Biological Functions and Current Advances in Isolation and Detection Strategies for Exosome Nanovesicles. Small 2018, 14 (6), 1702153. 10.1002/smll.201702153. [DOI] [PubMed] [Google Scholar]
- Mollaei H.; Safaralizadeh R.; Pouladi N. A Brief Review of Exosomes and Their Roles in Cancer. Meta Gene 2017, 11, 70–74. 10.1016/j.mgene.2016.11.010. [DOI] [Google Scholar]
- Zhou Y. G.; Mohamadi R. M.; Poudineh M.; Kermanshah L.; Ahmed S.; Safaei T. S.; Stojcic J.; Nam R. K.; Sargent E. H.; Kelley S. O. Interrogating Circulating Microsomes and Exosomes Using Metal Nanoparticles. Small 2016, 12 (6), 727–732. 10.1002/smll.201502365. [DOI] [PubMed] [Google Scholar]
- Zachariah M. A.; Oliveira-Costa J. P.; Carter B. S.; Stott S. L.; Nahed B. V. Blood-Based Biomarkers for the Diagnosis and Monitoring of Gliomas. Neuro. Oncol. 2018, 20 (9), 1155–1161. 10.1093/neuonc/noy074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Nedawi K.; Meehan B.; Micallef J.; Lhotak V.; May L.; Guha A.; Rak J. Intercellular Transfer of the Oncogenic Receptor EGFRvIII by Microvesicles Derived from Tumour Cells. Nat. Cell Biol. 2008, 10 (5), 619–624. 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- Ramirez M. I.; Amorim M. G.; Gadelha C.; Milic I.; Welsh J. A.; Freitas V. M.; Nawaz M.; Akbar N.; Couch Y.; Makin L.; Cooke F.; Vettore A. L.; Batista P. X.; Freezor R.; Pezuk J. A.; Rosa-Fernandes L.; Carreira A. C. O.; Devitt A.; Jacobs L.; Silva I. T.; Coakley G.; Nunes D. N.; Carter D.; Palmisano G.; Dias-Neto E. Technical Challenges of Working with Extracellular Vesicles. Nanoscale 2018, 10, 881–906. 10.1039/C7NR08360B. [DOI] [PubMed] [Google Scholar]
- Wirsching H. G.; Galanis E.; Weller M.. Glioblastoma. In Handbook of Clinical Neurology; Elsevier: Amsterdam, 2016; Vol. 134, pp 381–397. [DOI] [PubMed] [Google Scholar]
- Nawaz M.; Fatima F.; Zanetti B. R.; de Lima Martins I.; Schiavotelo N. L.; Mendes N. D.; Silvestre R. N.; Neder L. Microvesicles in gliomas and medulloblastomas: an overview. J. Cancer Therapy 2014, 05, 182. 10.4236/jct.2014.52023. [DOI] [Google Scholar]
- Wu P.; Cai J.; Chen Q.; Han B.; Meng X.; Li Y.; Li Z.; Wang R.; Lin L.; Duan C.; Kang C.; Jiang C. Lnc-TALC Promotes O6-Methylguanine-DNA Methyltransferase Expression via Regulating the c-Met Pathway by Competitively Binding with MiR-20b-3p. Nat. Commun. 2019, 10 (1), 2045. 10.1038/s41467-019-10025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis D. N.; Perry A.; Reifenberger G.; von Deimling A.; Figarella-Branger D.; Cavenee W. K.; Ohgaki H.; Wiestler O. D.; Kleihues P.; Ellison D. W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol 2016, 131 (6), 803–820. 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- Reifenberger G.; Wirsching H. G.; Knobbe-Thomsen C. B.; Weller M. Advances in the Molecular Genetics of Gliomas-Implications for Classification and Therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. 10.1038/nrclinonc.2016.204. [DOI] [PubMed] [Google Scholar]
- Pan S.; Zhang Y.; Natalia A.; Lim C. Z. J.; Ho N. R. Y.; Chowbay B.; Loh T. P.; Tam J. K. C.; Shao H. Extracellular Vesicle Drug Occupancy Enables Real-Time Monitoring of Targeted Cancer Therapy. Nat. Nanotechnol. 2021, 16 (6), 734–742. 10.1038/s41565-021-00872-w. [DOI] [PubMed] [Google Scholar]
- Shao H.; Chung J.; Lee K.; Balaj L.; Min C.; Carter B. S.; Hochberg F. H.; Breakefield X. O.; Lee H.; Weissleder R. Chip-Based Analysis of Exosomal MRNA Mediating Drug Resistance in Glioblastoma. Nat. Commun. 2015, 6 (1), 1–9. 10.1038/ncomms7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H.; Chung J.; Balaj L.; Charest A.; Bigner D. D.; Carter B. S.; Hochberg F. H.; Breakefield X. O.; Weissleder R.; Lee H. Protein Typing of Circulating Microvesicles Allows Real-Time Monitoring of Glioblastoma Therapy. Nat. Med. 2012, 18 (12), 1835–1840. 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane R.; Simon T.; Vintu M.; Solkin B.; Koch B.; Stewart N.; Benstead-Hume G.; Pearl F. M. G.; Critchley G.; Stebbing J.; Giamas G. Cell-Derived Extracellular Vesicles Can Be Used as a Biomarker Reservoir for Glioblastoma Tumor Subtyping. Commun. Biol. 2019, 2 (1), 1–12. 10.1038/s42003-019-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D.; Montermini L.; Kim D. K.; Meehan B.; Roth F. P.; Rak J. The Impact of Oncogenic Egfrviii on the Proteome of Extracellular Vesicles Released from Glioblastoma Cells. Mol. Cell. Proteomics 2018, 17 (10), 1948–1964. 10.1074/mcp.RA118.000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang M. J. Y.; Yoon J.; Zhou M.; Wei H. L.; Goh Y. Y.; Li Z.; Feng J.; Wang H.; Su Q.; Ong D. S. T.; Liu X. Deciphering Nanoparticle Trafficking into Glioblastomas Uncovers an Augmented Antitumor Effect of Metronomic Chemotherapy. Adv. Mater. 2022, 34 (3), 2106194. 10.1002/adma.202106194. [DOI] [PubMed] [Google Scholar]
- Sauter E. R. Exosomes in Blood and Cancer. Transl. Cancer Res. 2017, 6, 1316–1320. 10.21037/tcr.2017.08.13. [DOI] [Google Scholar]
- Zhao Z.; Yang Y.; Zeng Y.; He M. A Microfluidic ExoSearch Chip for Multiplexed Exosome Detection towards Blood-Based Ovarian Cancer Diagnosis. Lab Chip 2016, 16 (3), 489–496. 10.1039/C5LC01117E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Yue Y.; Wu S.; Liu W.; Shi J.; Zhang Z. Rapid Capture and Nondestructive Release of Extracellular Vesicles Using Aptamer-Based Magnetic Isolation. ACS Sensors 2019, 4 (5), 1245–1251. 10.1021/acssensors.9b00060. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Atiyas Y.; Shen H.; Siedlik M. J.; Wu J.; Beard K.; Fonar G.; Dolle J. P.; Smith D. H.; Eberwine J. H.; Meaney D. F.; Issadore D. A. Ultrasensitive Single Extracellular Vesicle Detection Using High Throughput Droplet Digital Enzyme-Linked Immunosorbent Assay. Nano Lett. 2022, 22 (11), 4315–4324. 10.1021/acs.nanolett.2c00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vlist E. J.; Nolte-’t Hoen E. N. M.; Stoorvogel W.; Arkesteijn G. J. A.; Wauben M. H. M. Fluorescent Labeling of Nano-Sized Vesicles Released by Cells and Subsequent Quantitative and Qualitative Analysis by High-Resolution Flow Cytometry. Nat. Protoc. 2012, 7 (7), 1311–1326. 10.1038/nprot.2012.065. [DOI] [PubMed] [Google Scholar]
- Choi D.; Montermini L.; Jeong H.; Sharma S.; Meehan B.; Rak J. Mapping Subpopulations of Cancer Cell-Derived Extracellular Vesicles and Particles by Nano-Flow Cytometry. ACS Nano 2019, 13 (9), 10499–10511. 10.1021/acsnano.9b04480. [DOI] [PubMed] [Google Scholar]
- Lee K.; Fraser K.; Ghaddar B.; Yang K.; Kim E.; Balaj L.; Chiocca E. A.; Breakefield X. O.; Lee H.; Weissleder R. Multiplexed Profiling of Single Extracellular Vesicles. ACS Nano 2018, 12 (1), 494–503. 10.1021/acsnano.7b07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S.; Yang K. S.; Zelga P.; Liss A. S.; Carlson J. C. T.; del Castillo C. F.; Weissleder R.. Single-EV Analysis (SEVA) of Mutated Proteins Allows Detection of Stage 1 Pancreatic Cancer. Sci. Adv. 2022, 8 ( (16), ). 10.1126/sciadv.abm3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini I. I.; Liu Z.; Capaldi X.; Abdelfatah T.; Montermini L.; Rak J.; Reisner W.; Mahshid S. Nanofluidics for Simultaneous Size and Charge Profiling of Extracellular Vesicles. Nano Lett. 2021, 21 (12), 4895–4902. 10.1021/acs.nanolett.0c02558. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Wu Z.; Hu J.; Yang D.; Chen X.; Wang Q.; Liu J.; Dou M.; Peng W.; Wu Y.; Wang W.; Xie C.; Wang M.; Song Y.; Zeng H.; Bai C.. High-Throughput Single-EV Liquid Biopsy: Rapid, Simultaneous, and Multiplexed Detection of Nucleic Acids, Proteins, and Their Combinations. Sci. Adv. 2020, 6 ( (47), ). 10.1126/sciadv.abc1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Lehrich B. M.; Zheng S.; Lu M. Emerging Methods in Biomarker Identification for Extracellular Vesicle-Based Liquid Biopsy. J. Extracellular Vesicles 2021, 10 (7), e12090 10.1002/jev2.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Hwang M.; Choi B.; Jeong H.; Jung J. H.; Kim H. K.; Hong S.; Park J. H.; Choi Y. Exosome Classification by Pattern Analysis of Surface-Enhanced Raman Spectroscopy Data for Lung Cancer Diagnosis. Anal. Chem. 2017, 89 (12), 6695–6701. 10.1021/acs.analchem.7b00911. [DOI] [PubMed] [Google Scholar]
- del Real Mata C.; Jeanne O.; Jalali M.; Lu Y.; Mahshid S. Nanostructured-Based Optical Readouts Interfaced with Machine Learning for Identification of Extracellular Vesicles. Adv. Healthcare Mater. 2023, 12 (5), 2202123. 10.1002/adhm.202202123. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Zhou X.; He M.; Shang Y.; Tetlow A. L.; Godwin A. K.; Zeng Y. Ultrasensitive Detection of Circulating Exosomes with a 3D-Nanopatterned Microfluidic Chip. Nat. Biomed. Eng. 2019, 3 (6), 438–451. 10.1038/s41551-019-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H.; Oh S.; Hong S.; Kang M.; Kang D.; Ji Y.-g.; Choi B. H.; Kang K.-W.; Jeong H.; Park Y.; Hong S.; Kim H. K.; Choi Y. Early-Stage Lung Cancer Diagnosis by Deep Learning-Based Spectroscopic Analysis of Circulating Exosomes. ACS Nano 2020, 14 (5), 5435–5444. 10.1021/acsnano.9b09119. [DOI] [PubMed] [Google Scholar]
- Akselrod G. M.; Ming T.; Argyropoulos C.; Hoang T. B.; Lin Y.; Ling X.; Smith D. R.; Kong J.; Mikkelsen M. H. Leveraging Nanocavity Harmonics for Control of Optical Processes in 2d Semiconductors. Nano Lett. 2015, 15 (5), 3578–3584. 10.1021/acs.nanolett.5b01062. [DOI] [PubMed] [Google Scholar]
- Eda G.; Maier S. A. Two-Dimensional Crystals: Managing Light for Optoelectronics. ACS Nano 2013, 7 (7), 5660–5665. 10.1021/nn403159y. [DOI] [PubMed] [Google Scholar]
- Yang K. S.; Im H.; Hong S.; Pergolini I.; Del Castillo A. F.; Wang R.; Clardy S.; Huang C. H.; Pille C.; Ferrone S.; Yang R.; Castro C. M.; Lee H.; Del Castillo C. F.; Weissleder R.. Multiparametric Plasma EV Profiling Facilitates Diagnosis of Pancreatic Malignancy. Sci. Transl. Med. 2017, 9 ( (391), ). 10.1126/scitranslmed.aal3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser K.; Jo A.; Giedt J.; Vinegoni C.; Yang K. S.; Peruzzi P.; Chiocca E. A.; Breakefield X. O.; Lee H.; Weissleder R. Characterization of Single Microvesicles in Plasma from Glioblastoma Patients. Neuro. Oncol. 2019, 21 (5), 606–615. 10.1093/neuonc/noy187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zong S.; Liu Y.; Wang Z.; Zhang Y.; Chen B.; Cui Y. Profiling of Exosomal Biomarkers for Accurate Cancer Identification: Combining DNA-PAINT with Machine- Learning-Based Classification. Small 2019, 15 (43), 1901014. 10.1002/smll.201901014. [DOI] [PubMed] [Google Scholar]
- Shin H.; Jeong H.; Park J.; Hong S.; Choi Y. Correlation between Cancerous Exosomes and Protein Markers Based on Surface-Enhanced Raman Spectroscopy (SERS) and Principal Component Analysis (PCA). ACS Sensors 2018, 3, 2637–2643. 10.1021/acssensors.8b01047. [DOI] [PubMed] [Google Scholar]
- Fraire J. C.; Stremersch S.; Bouckaert D.; Monteyne T.; De Beer T.; Wuytens P.; De Rycke R.; Skirtach A. G.; Raemdonck K.; De Smedt S.; Braeckmans K. Improved Label-Free Identification of Individual Exosome-like Vesicles with Au@Ag Nanoparticles as SERS Substrate. ACS Appl. Mater. Interfaces 2019, 11 (43), 39424–39435. 10.1021/acsami.9b11473. [DOI] [PubMed] [Google Scholar]
- Wells S. M.; Retterer S. D.; Oran J. M.; Sepaniak M. J. Controllable Nanofabrication of Aggregate-like Nanoparticle Substrates and Evaluation for Surface-Enhanced Raman Spectroscopy. ACS Nano 2009, 3 (12), 3845–3853. 10.1021/nn9010939. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Blackie E.; Meyer M.; Etchegoin P. G. Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study. J. Phys. Chem. C 2007, 111 (37), 13794–13803. 10.1021/jp0687908. [DOI] [Google Scholar]
- Ye R.; Song W.; Ou X.; Gu Z.; Zhang D. Membrane Insertion of MoS2 Nanosheets: Fresh vs. Aged. Front. Chem. 2021, 9, 499. 10.3389/fchem.2021.706917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Ou X.; Tian R.; Zhang J.; Jin H.; Dong M.; Li J.; Liu L. Membrane Destruction and Phospholipid Extraction by Using Two-Dimensional MoS2 Nanosheets. Nanoscale 2018, 10 (43), 20162–20170. 10.1039/C8NR04207A. [DOI] [PubMed] [Google Scholar]
- Gu Z.; Chen S. H.; Ding Z.; Song W.; Wei W.; Liu S.; Ma G.; Zhou R. The Molecular Mechanism of Robust Macrophage Immune Responses Induced by PEGylated Molybdenum Disulfide. Nanoscale 2019, 11 (46), 22293–22304. 10.1039/C9NR04358F. [DOI] [PubMed] [Google Scholar]
- Splendiani A.; Sun L.; Zhang Y.; Li T.; Kim J.; Chim C. Y.; Galli G.; Wang F. Emerging Photoluminescence in Monolayer MoS2. Nano Lett. 2010, 10 (4), 1271–1275. 10.1021/nl903868w. [DOI] [PubMed] [Google Scholar]
- Jalali M.; Isaac Hosseini I.; AbdelFatah T.; Montermini L.; Wachsmann Hogiu S.; Rak J.; Mahshid S. Plasmonic Nanobowtiefluidic Device for Sensitive Detection of Glioma Extracellular Vesicles by Raman Spectrometry. Lab Chip 2021, 21 (5), 855–866. 10.1039/D0LC00957A. [DOI] [PubMed] [Google Scholar]
- Garnier D.; Meehan B.; Kislinger T.; Daniel P.; Sinha A.; Abdulkarim B.; Nakano I.; Rak J. Divergent Evolution of Temozolomide Resistance in Glioblastoma Stem Cells Is Reflected in Extracellular Vesicles and Coupled with Radiosensitization. Neuro. Oncol. 2018, 20 (2), 236–248. 10.1093/neuonc/nox142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C. S.; Jean N.; Hogan C. A.; Blackmon L.; Jeffrey S. S.; Holodniy M.; Banaei N.; Saleh A. A. E.; Ermon S.; Dionne J. Rapid Identification of Pathogenic Bacteria Using Raman Spectroscopy and Deep Learning. Nat. Commun. 2019, 10 (1), 1–8. 10.1038/s41467-019-12898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A.; März A.; Schumacher W.; Rösch P.; Popp J. Towards a Fast, High Specific and Reliable Discrimination of Bacteria on Strain Level by Means of SERS in a Microfluidic Device. Lab Chip 2011, 11, 1013–1021. 10.1039/c0lc00536c. [DOI] [PubMed] [Google Scholar]
- Platt J. Probabilistic Outputs for Support Vector Machines and Comparisons to Regularized Likelihood Methods. Adv. Large Margin Classif. 1999, 10 (3), 61–74. [Google Scholar]
- Jalali M.; Gao Z.; Yu Y.; Siavash Moakhar R.; Ding Y.; Zhuang M.; Zhou N.; König T.; Fery A.; Mahshid S.; Luo Z. Synergistic Enhancement of Photoluminesent Intensity in Monolayer Molybdenum Disulfide Embedded with Plasmonic Nanostructures for Catalytic Sensing. 2D Mater. 2021, 8 (3), 035049. 10.1088/2053-1583/ac040e. [DOI] [Google Scholar]
- Jalali M.; Moakhar R. S.; Abdelfattah T.; Filine E.; Mahshid S. S.; Mahshid S. Nanopattern-Assisted Direct Growth of Peony-like 3D MoS2/Au Composite for Nonenzymatic Photoelectrochemical Sensing. ACS Appl. Mater. Interfaces 2020, 12 (6), 7411–7422. 10.1021/acsami.9b17449. [DOI] [PubMed] [Google Scholar]
- Siavash Moakhar R.; del Real Mata C.; Jalali M.; Shafique H.; Sanati A.; de Vries J.; Strauss J.; AbdElFatah T.; Ghasemi F.; McLean M.; Hosseini I. I.; Lu Y.; Yedire S. G.; Mahshid S. S.; Tabatabaiefar M. A.; Liang C.; Mahshid S. A Versatile Biomimic Nanotemplating Fluidic Assay for Multiplex Quantitative Monitoring of Viral Respiratory Infections and Immune Responses in Saliva and Blood. Adv. Sci. 2022, 9 (33), 2204246. 10.1002/advs.202204246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Real Mata C.; Siavash Moakhar R.; Hosseini I. I.; Jalali M.; Mahshid S. A Nanostructured Microfluidic Device for Plasmon-Assisted Electrochemical Detection of Hydrogen Peroxide Released from Cancer Cells. Nanoscale 2021, 13 (34), 14316–14329. 10.1039/D0NR07608B. [DOI] [PubMed] [Google Scholar]
- Adnani L.; Kassouf J.; Meehan B.; Spinelli C.; Tawil N.; Nakano I.; Rak J. Angiocrine Extracellular Vesicles Impose Mesenchymal Reprogramming upon Proneural Glioma Stem Cells. Nat. Commun. 2022, 13 (1), 1–14. 10.1038/s41467-022-33235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel P.; Meehan B.; Sabri S.; Jamali F.; Sarkaria J. N.; Choi D.; Garnier D.; Kitange G.; Glennon K. I.; Paccard A.; Karamchandani J.; Riazalhosseini Y.; Rak J.; Abdulkarim B.. Detection of Temozolomide-Induced Hypermutation and Response to PD-1 Checkpoint Inhibitor in Recurrent Glioblastoma. Neuro-Oncology Adv. 2022, 4 ( (1), ). 10.1093/noajnl/vdac076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus N.; Garnier D.; Meehan B.; McGraw S.; Lee T. H.; Caron M.; Bourque G.; Milsom C.; Jabado N.; Trasler J.; Pawlinski R.; Mackman N.; Rak J. Tissue Factor Expression Provokes Escape from Tumor Dormancy and Leads to Genomic Alterations. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (9), 3544–3549. 10.1073/pnas.1314118111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawil N.; Bassawon R.; Meehan B.; Nehme A.; Montermini L.; Gayden T.; de Jay N.; Spinelli C.; Chennakrishnaiah S.; Choi D.; Adnani L.; Zeinieh M.; Jabado N.; Kleinman C. L.; Witcher M.; Riazalhosseini Y.; Key N. S.; Schiff D.; Grover S. P.; Mackman N.; Couturier C. P.; Petrecca K.; Suvà M. L.; Patel A.; Tirosh I.; Najafabadi H.; Rak J. Glioblastoma Cell Populations with Distinct Oncogenic Programs Release Podoplanin as Procoagulant Extracellular Vesicles. Blood Adv. 2021, 5 (6), 1682–1694. 10.1182/bloodadvances.2020002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P.; Joshi K.; Li J.; Kim S. H.; Li P.; Santana-Santos L.; Luthra S.; Chandran U. R.; Benos P. V.; Smith L.; Wang M.; Hu B.; Cheng S. Y.; Sobol R. W.; Nakano I. Mesenchymal Glioma Stem Cells Are Maintained by Activated Glycolytic Metabolism Involving Aldehyde Dehydrogenase 1A3. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (21), 8644–8649. 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastola S.; Pavlyukov M. S.; Yamashita D.; Ghosh S.; Cho H.; Kagaya N.; Zhang Z.; Minata M.; Lee Y.; Sadahiro H.; Yamaguchi S.; Komarova S.; Yang E.; Markert J.; Nabors L. B.; Bhat K.; Lee J.; Chen Q.; Crossman D. K.; Shin-Ya K.; Nam D. H.; Nakano I. Glioma-Initiating Cells at Tumor Edge Gain Signals from Tumor Core Cells to Promote Their Malignancy. Nat. Commun. 2020, 11 (1), 1–17. 10.1038/s41467-020-18189-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.