

Abstract
The recruitment of monocytes and their differentiation into immunosuppressive cells is associated with the low efficacy of preclinical nonconformal radiotherapy (RT) for tumors. However, nonconformal RT (non-CRT) does not mimic clinical practice, and little is known about the role of monocytes after RT modes used in patients, such as conformal RT (CRT). Here, we investigated the acute immune response induced by after CRT. Contrary to non-CRT approaches, we found that CRT induces a rapid and robust recruitment of monocytes to the tumor that minimally differentiate into tumor-associated macrophages or dendritic cells but instead up-regulate major histocompatibility complex II and costimulatory molecules. We found that these large numbers of infiltrating monocytes are responsible for activating effector polyfunctional CD8+ tumor-infiltrating lymphocytes that reduce tumor burden. Mechanistically, we show that monocyte-derived type I interferon is pivotal in promoting monocyte accumulation and immunostimulatory function in a positive feedback loop. We also demonstrate that monocyte accumulation in the tumor microenvironment is hindered when RT inadvertently affects healthy tissues, as occurs in non-CRT. Our results unravel the immunostimulatory function of monocytes during clinically relevant modes of RT and demonstrate that limiting the exposure of healthy tissues to radiation has a positive therapeutic effect on the overall antitumor immune response.
INTRODUCTION
Radiotherapy (RT) commonly induces the infiltration of monocytes to the tumor microenvironment (TME) (1, 2). These monocytes may acquire immunosuppressive phenotypes and, consequently, have been poorly classified as myeloid-derived suppressor cells (MDSCs) (1–4). Monocyte depletion has been shown to improve the therapeutic efficacy of preclinical radiation models (1, 2). Yet depleting monocytes in clinical trials has not improved the therapeutic response in patients undergoing RT (5–8). This discrepancy highlights the need to understand monocyte functions in clinically relevant modes of RT.
RT is the standard of care for >50% of patients with cancer (9). Modern clinical RT uses a variety of technologies to achieve three-dimensional conformal RT (CRT). Volumetric computed tomography (CT) scans are acquired to map the location of the tumor and generate an individual radiation treatment plan (9). This approach has the goal of accurately delivering high radiation doses to tumors while limiting the dose to healthy tissues, thereby minimizing short- and long-term RT toxicity (10, 11). Although CRT has been used in the clinic for over 30 years, the immune responses induced by this treatment remain complex and poorly understood. This is, in part, because most of our current knowledge comes from preclinical non-CRT. In preclinical animal studies, non-CRT uses a shield to cover most of the small animal from radiation exposure (hereafter called shield RT or SRT); however, this approach cannot prevent the un-intentional exposure of healthy tissues to radiation (12). To date, it is unclear whether the exposure of healthy tissues to radiation modifies the acute immune response within tumors.
In addition to the well-documented direct cytotoxic effect on cancer cells, preclinical SRT induces immunostimulatory and immunosuppressive effects in the TME (1, 13–17). Double-stranded DNA released after cell death is sensed via the cyclic guanosine 5′-monophosphate–adenosine 5′-monophosphate synthase (cGAS) stimulator of interferon genes (STING) signaling pathway, which leads to the production of type I interferon (IFN-I) (13, 18). IFN-I recruits and enhances dendritic cell (DC) antigen presentation, resulting in CD4+ and CD8+ T cell priming and the initiation of a systemic antitumor immune response (15, 19, 20). However, SRT also promotes immunosuppression by several mechanisms, including the accumulation of monocyte-derived MDSCs, which is also dependent on STING activation (1, 4). Consequently, monocyte depletion generally improves SRT outcomes (1, 2). Unfortunately, the classification of different monocyte-derived populations as MDSCs has limited the capacity to pinpoint their distinct contribution to the immunostimulatory and immunosuppressive effects of SRT, which has hampered the development of targeted approaches for therapeutic intervention. In addition, it is unclear whether the immune mechanisms reported after SRT also occur in clinically relevant modes of radiation such as CRT.
Here, we investigated the acute immune response induced by CRT. We found that CRT is significantly more efficient than SRT at slowing tumor growth and improving survival, independently of the fractionation regimen. Unexpectedly, CRT efficacy is mediated by the rapid and robust recruitment of monocytes to the TME. These infiltrating monocytes acquire the expression of activation markers and promote polyfunctional effector CD8+ tumor-infiltrating lymphocytes (TILs), which, in turn, reduce tumor burden. Mechanistically, IFN-I produced by monocytes, in a STING-independent manner, is required to promote monocyte accumulation and activation in a positive feedback loop. CRT efficacy is unexpectedly dependent on its capacity to limit radiation exposure to healthy tissues, which cannot be completely achieved by non-CRT. Our study highlights the immune mechanisms underlying the efficacy of clinically relevant modes of RT and pinpoints IFN-I–activated monocytes as potential targets to enhance CRT outcomes.
RESULTS
CRT efficacy depends on CD8+ TILs to rapidly treat tumors
We compared the efficacy of CRT with that of the same dose of SRT. CRT was administered using a small-animal conformal radiation platform, X-Rad SmART image-guided preclinical irradiator, that delivers two focused beams on the basis of CT image guidance (21). SRT was administered using a Polaris SC-500 Series II irradiator that delivers a single beam while most of the healthy tissues are spared by a lead shield (Fig. 1A) (22). Direct dosimetric measurements verified that the same dose of CRT and SRT was administered to tumors (see Supplementary Materials and Methods). CRT or SRT was delivered to subcutaneously engrafted MC38 colon carcinomas using two regimens: a single ablative 20-gray (Gy) dose (20Gy) or hypofractionated 8-Gy doses on three consecutive days (8Gyx3), an approach shown to promote stronger immune responses in preclinical models (Fig. 1B) (17). CRT was more effective at delaying tumor growth and increasing survival versus non-treated (NT) or SRT, independently of the fractionation regimen (Fig. 1C). CRT (20Gy) was also superior to the same dose of SRT when delivered to the Lewis lung carcinoma (LLC) transplanted subcutaneously, a breast adenocarcinoma (E0771) transplanted orthotopically, and the BrafCAxPtenf/fxTyrCreER melanoma (BRaf/Pten) induced by topical tamoxifen application (Fig. 1, D to F). In addition, CRT efficacy was identical when 20Gy was delivered using one or two focused beams (fig. S1A). Given that CRT promoted remission of all MC38 tumors independent of the fractionation regimen, we used this tumor model and a single 20Gy dose to evaluate immunological differences with SRT.
Fig. 1. CRT efficacy depends on CD8+ TILs.
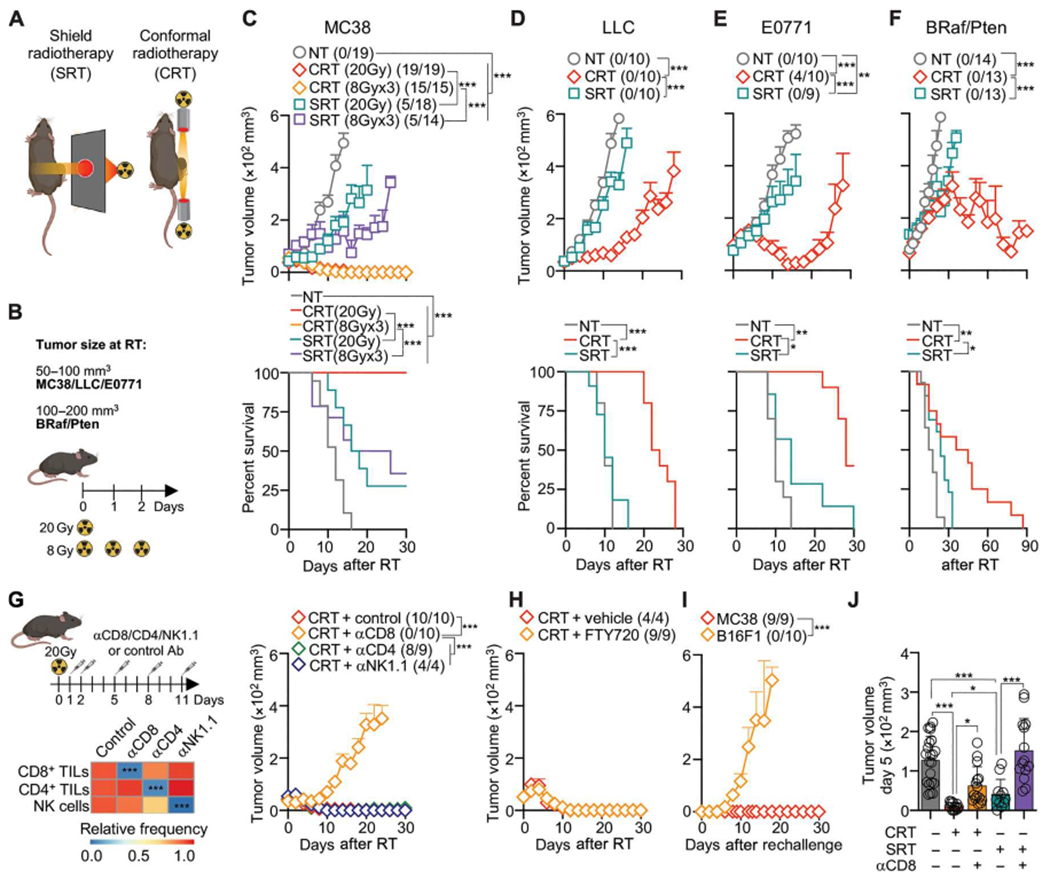
(A and B) Schematic of RT modes and fractionation regimen. (C to F) B6 mice were implanted with MC38 subcutaneously (n = 14 to 19 per group, three to five experiments), LLC subcutaneously (n = 10 per group, two or three experiments), and E0771 orthotopically (n = 9 or 10 per group, two experiments). BRaf/Pten tumors were induced by 4-hydroxytamoxifen (n = 13 or 14 per group, two to four experiments). Top: Mean tumor growth + SEM with ratio of surviving mice in parentheses. Bottom: Survival curves. (C) Tumors were treated with 20 Gy or three 8-Gy (8 Gy ×3) doses of RT. (D to F) Tumors were treated with 20 Gy RT. (G) MC38-bearing mice after CRT and 200 μg of αCD8, αCD4, αNK1.1, or control Ab. Left: Heatmap of relative blood leukocyte frequency relative to control Ab at 3 days after RT (n = 4 to 10 per group, three experiments). Right: Mean tumor growth + SEM (n = 4 to 10 per group, one or two experiments). (H) MC38-bearing mice after CRT and 100 μg of FTY720 inoculated daily from −1 to 10 days (means + SEM; n = 4 to 10 per group, two experiments). (I) MC38-bearing mice that eliminated tumors after 20 Gy CRT were rechallenged 50 days later with MC38 or B16F1 (means + SEM; n = 9 or 10 per group, two experiments). (J) Mean tumor volume + SD at 5 days after RT and αCD8/control Ab inoculation (n = 11 to 21 per group, three to five experiments). Statistics: Two-way ANOVA plus Tukey’s post hoc test for mean tumor growth (C to I); Mantel-Cox test for survival curves (C to F); one-way ANOVA plus Tukey’s post hoc test (J).
CRT efficacy was abrogated by CD8+ TIL depletion, but not a natural killer (NK) or CD4+ TILs (Fig. 1G). However, CD4+ TILs were necessary for long-lasting memory, as shown by reduced protection after rechallenge of surviving animals (fig. S1B). CD8+ TILs were also required after SRT, as previously reported (13), and during an 8Gyx3 delivery regimen (fig. S1, C and D). CRT efficacy was dependent on intratumoral lymphocytes because fingolimod (FTY720), an inhibitor of T cell egress from lymphoid tissues, did not alter the outcome (Fig. 1H). CRT-treated mice developed tumor-specific memory, because mice surviving primary MC38 tumors rejected secondary challenge with MC38, but not B16F1 tumors (Fig. 1I). CRT promoted a significant decrease in tumor size by 5 days after treatment, an effect mediated by CD8+ TILs and more pronounced than SRT (Fig. 1J). In sum, CRT induces more effective acute CD8+ TIL–mediated immune responses than the same dose of SRT, independent of the fractionation regimen.
CRT promotes effector CD8+ TIL function
We performed cytometry by time-of-flight (CyTOF) analysis using X-shift and Uniform Manifold Approximation and Projection (UMAP) (23) to compare lymphocyte differences after CRT and SRT. This revealed an increase in the proportion of CD8+ TILs 5 days after CRT, but not SRT (Fig. 2A). We confirmed these results by flow cytometry analysis: CD8+ TIL proportion was significantly higher 5 days after CRT (Fig. 2B), although the total leukocyte number decreased in parallel to tumor shrinkage (fig. S2A). There were no differences in the proportion of NK and CD4+ TILs. However, the proportion and total number of FOXP3+ regulatory T cells (Tregs) were significantly increased after SRT (Fig. 2B and fig. S2B). Consequently, the ratio of CD8+ TILs to Tregs was greater after CRT (Fig. 2C).
Fig. 2. CRT increases effector CD8+ TIL function.
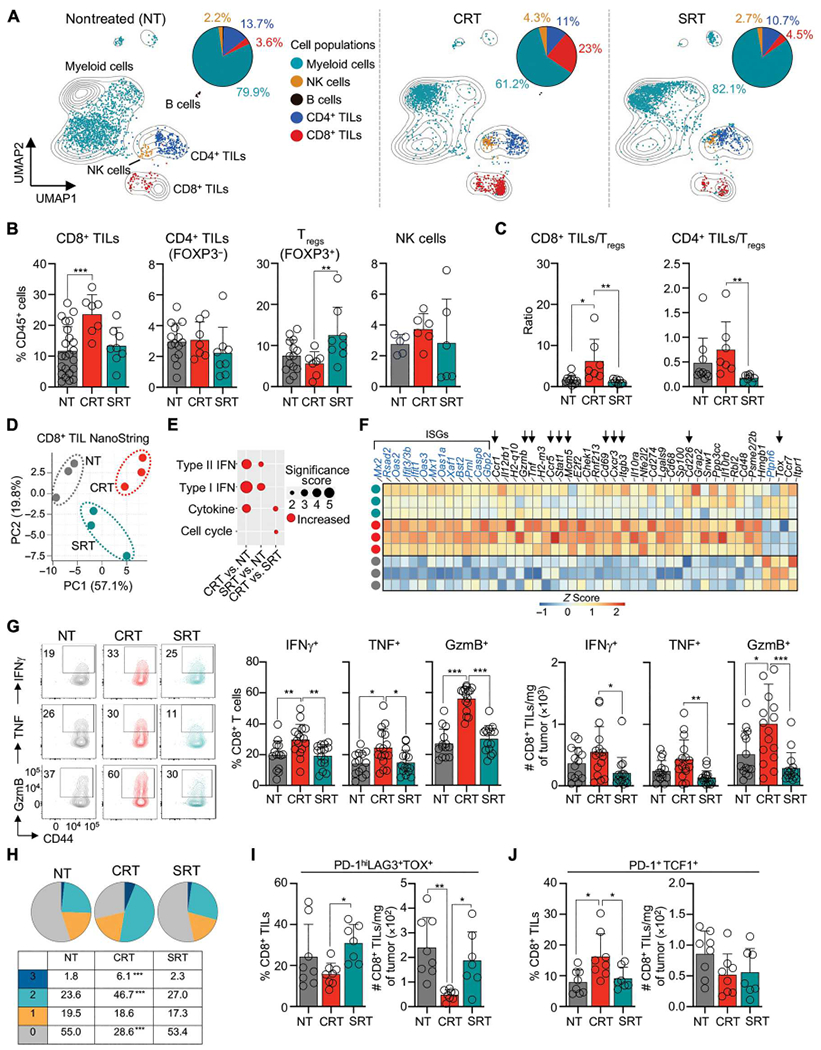
(A) CyTOF analysis of immune cells in MC38 tumors 5 days after RT or NT. UMAP of all cells and treatments (contour plot) were overlaid with cell populations for each treatment. Pie charts show frequency of cells per total leukocytes. (B) TIL frequency 5 days after RT, by flow cytometry (n = 5 to 23 per group, two or three experiments). (C) As in (B), but CD8+ or CD4+ TIL to Treg ratio (n = 7 to 10 per group, two or three experiments). (D to F) NanoString transcriptomic analysis of CD8+ TILs sorted from NT, or 3 days after RT. (D) PCA of DEGs. (E) Pathway enrichment analysis of DEGs. Size indicates significance score. (F) Heatmap of normalized counts of CRT versus NT DEGs (Z-scored). Selected IFN-stimulated genes (blue) and other genes of interest (arrows). (G to J) MC38 tumors from NT or 3 days after RT, by flow cytometry. (G) Representative flow cytometry plots (left), frequency (middle), and total number per milligram of tumor (right) of CD8+ TILs producing IFN-γ, TNF, and GzmB after ex vivo restimulation (n = 13 to 15 per group, three to five experiments). (H) As in (G), but the proportion of CD8+ TILs producing 1, 2, or 3 effector molecules by Boolean gating analysis. (I and J) Frequency and total number per milligram of tumor PD-1hiLAG3+TOX+CD8+ TILs (I) and PD-1+TCF1+CD8+ TILs (J). Statistics: One-way ANOVA plus Tukey’s post hoc test.
We asked whether CRT induced a distinct transcriptional profile in CD8+ TILs, which we reasoned would occur within the first 5 days given differences in tumor volumes (Fig. 1J). NanoString transcriptomic analysis of sorted CD8+ TILs was performed 3 days after RT (fig. S2C). Principal components analysis (PCA) of differentially expressed genes (DEGs) showed that CD8+ TILs from CRT-treated mice clustered separately from NT and SRT-treated mice (Fig. 2D). Pathway analysis suggested that IFN-I and IFN-II, cytokine signaling, and cell cycle were enriched in CRT-treated CD8+ TILs (Fig. 2E). CRT-treated CD8+ TILs had higher expression of transcripts associated with IFN-stimulated genes (ISGs) (e.g., Oas1a, Oas3, Ifit1, Ifit3, and Stat1), effector functions (Gzmb and Tnf), activation (Cd69 and Itga3), cell cycling (Mcm5), trafficking (Cxcr3, Ccr1, and Ccr5), and costimulation (Cd226), suggesting an effector phenotype (24–30). These ISGs were also promoted after SRT but at lower levels. Conversely, NT and SRT-treated CD8+ TILs exhibited higher expression of Tox, a transcript associated with exhausted T cells (Fig. 2F) (31).
We validated T cell phenotype by flow cytometry. CRT promoted higher levels of IFN-γ, tumor necrosis factor (TNF), and granzyme B (GzmB) in CD8+ TILs after ex vivo stimulation (Fig. 2G). CRT increased the proportion of polyfunctional CD8+ TILs producing two or three effector molecules, a feature associated with effective antitumor immune responses (Fig. 2H) (30). Increased effector molecules and polyfunctionality were promoted by CRT independent of the fractionation regimen (fig. S2, D and E). Consistent with their increased effector functions, CRT-treated tumors had lower proportions and numbers of exhausted PD-1hiLAG3+ TOX+CD8+ TILs versus SRT-treated tumors (Fig. 2I). The proportion of stem-like PD-1+TCF1+CD8+ TILs, a precursor population that differentiate into effector cells (32), was also increased after CRT (Fig. 2J). Together, our data show that CRT rapidly promotes effector CD8+ TIL function.
CRT stimulates the rapid infiltration and activation of monocytes
The rapid activation of effector CD8+ TILs prompted the hypothesis that innate immune cells, such as myeloid cells, may determine CRT efficacy, leading us to characterize tumor-infiltrating myeloid cells using CyTOF (Fig. 3A). Using canonical myeloid cell signatures, we identified three DC clusters (DC1, DC2, and pDCs), tumor-associated macrophages (TAMs), eosinophils, neutrophils, and two monocyte clusters (C1 and C2). All monocytes expressed Ly6C, CD14, CD115, and CX3CR1. C1 monocytes harbored higher levels of Ly6C and CD115. C2 monocytes displayed lower levels of Ly6C and CD115 and higher levels of activation markers such as major histocompatibility complex II (MHCII), CD86, and programmed death-ligand 1 (PD-L1); consequently, we refer to C2 as monoACT. Distinct from monoACT, TAMs expressed higher levels of CD64 and F4/80 and lower levels of MHCII. DC1 and DC2 highly expressed CD11c and MHCII, whereas pDCs expressed SiglecH and B220. Last, eosinophils and neutrophils were identified by CD24 and Ly6G expression, respectively.
Fig. 3. CRT induces rapid infiltration and activation of monocytes.
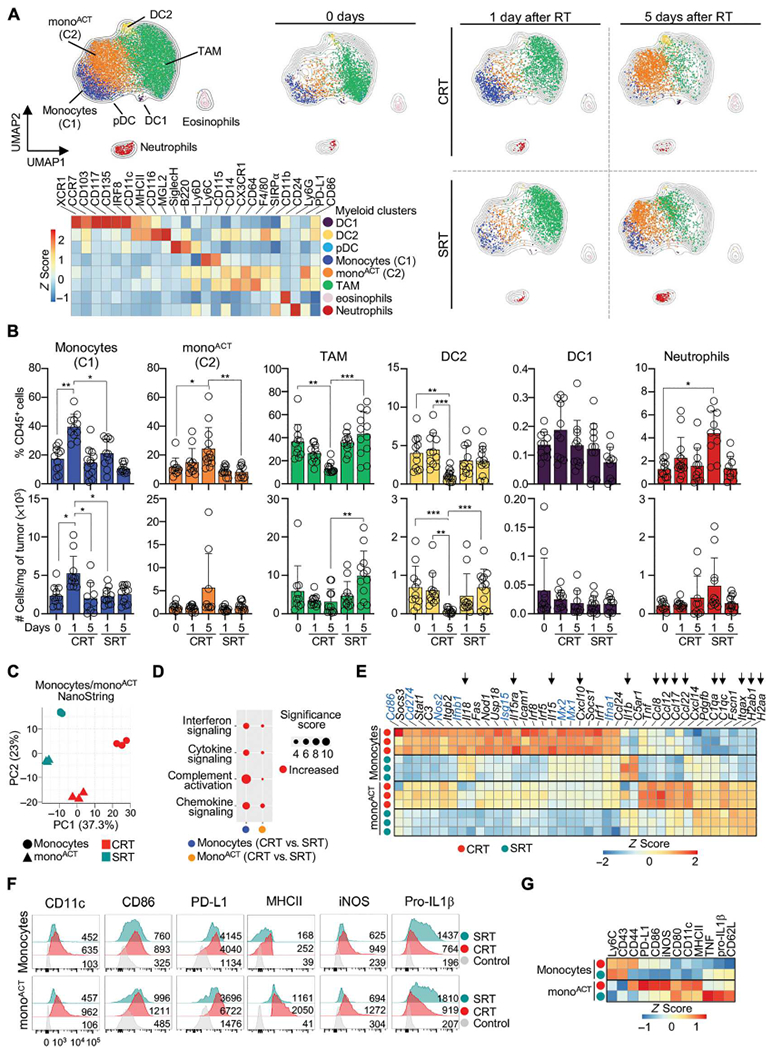
(A) MC38 tumors from NT, 1 and 5 days after RT, by CyTOF. Myeloid cells were gated as live/singlets/CD45+/CD3−/CD19−/CD335−/CD8− (n = 3 per group, two experiments). Top left: UMAP of all cells and treatments. Right: UMAPs of each treatment condition overlaid onto UMAP of all cells. Bottom Left: Heatmap represents Z-scored expression of markers in clusters across all conditions. (B) Frequency (top) and total number per milligram of tumor (bottom) of each population at 1 and 5 days after RT, by flow cytometry (n = 11 to 13 per group, two or three experiments). (C to E) NanoString transcriptomic analysis of monocytes and monoACT 3 days after RT. (C) PCA of normalized cell counts. (D) Pathway enrichment analysis of normalized counts. Size indicates significance score. (E) Heatmap of normalized counts of monocytes after CRT versus all conditions (Z-scored). Selected IFN-associated genes (blue), and other genes of interest (arrows). (F and G) Monocytes and monoACT 3 days after RT, by flow cytometry. (n = 3 to 5 per group, two or three experiments). (F) Representative histograms showing the geometric mean fluorescent intensity. (G) Heatmap of Z-scored marker expression. Statistics: One-way ANOVA plus Tukey’s post hoc test (B).
We compared the dynamic behavior of each myeloid cell population after treatment. CRT promoted significant accumulation of monocytes and monoACT at 1 and 5 days, respectively, whereas NT and SRT maintained high proportions of TAM (Fig. 3A). To confirm these observations in a greater number of animals, we generated a flow cytometry gating strategy based on our unbiased CyTOF analyses (fig. S3, A and B). This approach corroborated myeloid cell dynamics and showed a significant increase in the proportion and number of monocytes at 1 day after CRT (Fig. 3B). CRT also promoted the relative increase in monoACT but decrease in TAMs and DC2. In contrast, SRT did not alter TAM and DC2 proportions or numbers (Fig. 3B). Last, DC1 were unaffected by either treatment, but neutrophil proportions increased after SRT. Similar myeloid cell dynamics were observed when CRT was administered using an 8 Gy ×3 hypofractionation regimen (fig. S3C).
We profiled monocytes and monoACT that increase after CRT using NanoString. Tumor-infiltrating monocytes and monoACT clustered differently after CRT and SRT, suggesting distinct gene expression (Fig. 3C). CRT promoted pathways enriched in interferon and chemokine/cytokine transcripts in both monocytes and monoACT (Fig. 3D). Among these, we observed interferon alpha and beta (Ifna1 and Ifnb1), ISGs (e.g., Isg15, Mx1, Mx2, Cd86, Cd274, and Nos2), and genes that mediate CD8+ T cell recruitment and function (e.g., Il18, Il15, Il15ra, and Cxcl10) (Fig. 3E) (15, 33–36). MonoACT purified from CRT- and SRT-treated tumors shared the expression of MHCII (H2ab1 and H2aa), CD11c (Itgax), and complement genes (C1qa and C1qc); however, CRT promoted transcripts related to monocyte/leukocyte recruitment (e.g., Ccl8, Ccl12, Ccl17, and Ccl22) (37–40). Last, monocytes from SRT-treated tumors expressed higher levels of Il1b.
Expression of some of these genes was confirmed by flow cytometry (Fig. 3, F and G). MonoACT from CRT-treated tumors displayed higher levels of CD11c, MHCII, CD86, and PD-L1, markers known to be induced by IFN-I and -II (34, 35, 41–43). Also, CRT-infiltrating monoACT displayed higher levels of inducible nitric oxide synthase, an IFN-II–regulated small molecule that plays stimulatory and inhibitory roles (36, 44, 45), but lower levels of the immunosuppressive cytokine precursor pro–interleukin-1β (IL-1β) (46). Some molecules that were highly expressed at the protein level in monoACT (e.g., CD86 and PD-L1) were highly expressed at the RNA level in monocytes, favoring the hypothesis that monoACT arises from monocytes. Collectively, our data show that CRT promotes the differential accumulation and activation of monocytes with a strong IFN-associated signature.
Monocytes promote effector CD8+ TIL function after CRT
DC1 has a distinct capacity to prime naïve CD8+ T cells (47); thus, we evaluated their role after CRT. We used Itgax-Cre xIrf8f/f mice in which Irf8 is inactivated in CD11c-expressing cells, resulting in DC1 elimination (48). CRT was ineffective at eliminating tumors engrafted into Itgax-Cre xIrf8f/f mice (fig. S4A). CRT was also ineffective toward tumors engrafted in Irf8Δ32 mice, a recently described model in which deletion of the +32-kb Irf8 enhancer eliminates DC1 without affecting other cells (Fig. 4A and fig. S4B) (49). This lack of response in Irf8Δ32 mice was likewise observed afterSRT, which agrees with the previously reported role of DC1 after non-CRT (fig. S4C) (50). Consistent with DC1’s capacity to prime CD8+ T cells, tumors engrafted in Irf8Δ32 mice had significantly fewer CD8+ TILs (Fig. 4B). However, the few remaining CD8+ TILs were able to up-regulate IFN-γ, TNF and GzmB after CRT (Fig. 4C), suggesting that DC1 may not be the sole driver of effector CD8+ TIL function.
Fig. 4. CRT-recruited monocytes promote effector CD8+ TIL function.
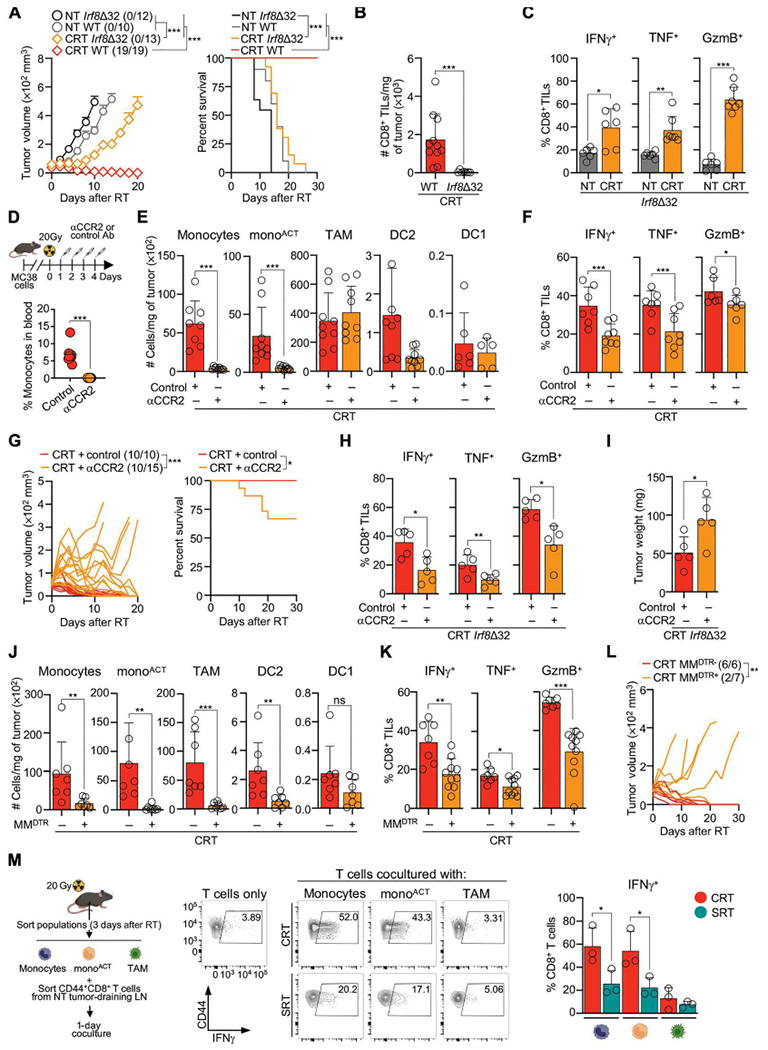
(A to C) MC38-bearing WT or Irf8Δ32 mice after CRT. (A) Mean tumor growth + SEM (left) and survival curve (right) (n = 10 to 19 per group, three or four experiments). (B) CD8+ TIL number per milligram of tumor 3 days after RT by flow cytometry (n = 9 to 11 per group, three experiments). (C) As in (B), but frequency of CD8+ TILs expressing IFN-γ, TNF, and GzmB after ex vivo restimulation (n = 6 or 7 per group, three experiments). (D to G) MC38-bearing WT mice after CRT and αCCR2/control Ab. (D) αCCR2 Ab inoculation schematic (top) and blood monocytes frequency at 3 days (bottom). (E) Number of cells per milligram of tumor 3 days after RT (n = 5 to 9 per group, three or four experiments). (F) Frequency of CD8+ TILs expressing IFN-γ, TNF, and GzmB 3 days after RT, after ex vivo restimulation (n = 7 or 8 per group, two or three experiments). (G) Individual mouse tumor growth (left; ratio of surviving mice in parentheses) and survival curves (right) (n = 10 to 15 per group, three experiments). (H and I) As in (D), but with Irf8Δ32 mice collected at 3 days after CRT (n = 5 per group, two experiments) (H) Frequency of CD8+ TILs expressing IFN-γ, TNF, and GzmB. (I) Tumor weight. (J to L) MC38-bearing MMDTR− and MMDTR+ mice after CRT and 4 ng/g of body weight DT inoculated on day 0 (intravenous) and days 1, 2, 3, and 4 (intraperitoneal). (J) Cells per milligram of tumor 3 days after CRT (n = 7 per group, two experiments). (K) Frequency of CD8+ TILs expressing IFN-γ, TNF, and GzmB 3 days after CRT, after ex vivo restimulation (n = 7 to 10 per group, three experiments). (L) Individual mouse tumor growth after CRT and DT inoculation (n = 6 or 7 per group, two experiments). (M) Ex vivo coculture of tumor-sorted myeloid cell populations (3 days after RT) with effector/memory CD8+ T cells sorted from tumor-draining LNs of NT mice. Experimental schematic (left), representative flow cytometry (middle) and accumulative data (right) of IFN-γ+CD8+ T cells 1 day after coculture (n = 3 per group, three experiments). Statistics: Two-way ANOVA plus Tukey’s post hoc test for mean tumor growth (A, G, and L); Mantel-Cox test for survival curves (A and G); unpaired t test (B to F and H to K); one-way ANOVA plus Tukey’s post hoc test (M).
Next, we evaluated the capacity of other myeloid cells to drive effector CD8+ TIL function after CRT. DC2 can promote CD8+ T cell immunity (51); however, their frequency and numbers are decreased after CRT (Fig. 3B), suggesting that they may not mediate CD8+ TIL effector function. To confirm this, we used Itgax-Cre xIrf4f/del mice that lack DC2 and observed no changes in CRT efficacy compared with control mice (52) (fig. S4D). Monocytes can drive effector CD8+ T cell function during viral infections (33, 53) and significantly infiltrate tumors 1 day after CRT (Fig. 3B). To evaluate monocyte function, we used antibodies against CCR2 (αCCR2 Abs), a chemokine receptor required for monocyte mobilization (54). αCCR2 Ab depleted circulating monocytes, as well as tumor-infiltrating monocytes and monoACT, but not TAMs, DC2, or DC1 in our model (Fig. 4, D and E). αCCR2 Ab decreased the frequency and number of effector CD8+ TILs after CRT (Fig. 4F and fig. S4E), which halted CRT efficacy quickly after-treatment (Fig. 4G). Similarly, αCCR2 Ab decreased the effector function of the remaining CD8+ TILs in Irf8Δ32 mice, resulting in larger tumors 5 days after CRT (Fig. 4, H and I).
To eliminate the possibility of a bystander effect of αCCR2 Ab, we used two additional models. First, we used Ccr2DTR mice in which the diphtheria toxin receptor (DTR) is driven by Ccr2 (54). Monocyte depletion in Ccr2DTR mice limited CRT efficacy (fig. S4, F and G). Second, we used Lyz2Cre xCsf1rLSL-DTR mice, a two-gene approach that targets monocytes and macrophages (MMDTR) (55). DT inoculation into MMDTR+ mice depleted tumor-infiltrating monocytes, monoACT, TAMs, and DC2, but not DC1 (Fig. 4J). Depletion of these cells resulted in the decrease of the effector CD8+ TIL function and, consequently, hampered CRT efficacy (Fig. 4, K and L). Thus, our results using several mouse models point to a key role of monocytes and monocyte-derived cells in promoting the acute effector CD8+ TIL function.
Last, we evaluated monocytes’ capacity to directly modulate CD8+ T cell function through an ex vivo assay. Effector/memory CD44+CD8+ T cells obtained from NT MC38-draining lymph nodes (LNs) were cocultured with tumor-infiltrating monocytes, monoACT, and TAMs purified 3 days after RT (Fig. 4M and fig. S4H). In this culture, T cell activation was solely dependent on tumor-infiltrating myeloid cells because no other cytokines or antigens were added. CD8+ T cells produced significantly higher levels of IFN-γ in response to monocytes and monoACT, but not TAMs, obtained from CRT- versus SRT-treated tumors. Despite monocytes and monoACT having superior capacities to capture tumor antigen after CRT (fig. S4I), these cells did not promote proliferation of effector/memory or naïve CD8+ T cells, suggesting an antigen-independent modulation of T cell function (fig. S4J). Accordingly, monocytes and monoACT obtained from MC38-treated tumors were able to activate T cells purified from either MC38 or B16F1 tumor-draining LNs (fig. S4K). Together, our data support a model in which CRT accumulates and activates monocytes in the TME to quickly promote effector CD8+ TIL function.
CRT promotes monocyte activation rather than differentiation into TAMs or DCs
After their infiltration to the TME, monocytes can acquire immunostimulatory or immunosuppressive functions. To query monocyte fate, we used Ms4a3Cre xRosaLSL-tdTomato mice in which Cre expression under the Ms4a3 gene labels granulocytes, monocytes, and monocyte-derived cells with tdTomato (56). Most tumor-associated mononuclear phagocytes, except for DC1, labeled positively for tdTomato in NT and treated tumors, indicating that they were all derived from monocytes (fig. S5, A and B). We studied monocyte fate through bone marrow (BM) purification and adoptive transfer into congenic tumor-bearing mice (Fig. 5A and fig. S5C). In agreement with the observed myeloid cell dynamics (Fig. 3B), more monocytes infiltrated CRT- versus SRT- treated and NT tumors. Most infiltrating monocytes acquired a monoACT phenotype after CRT, whereas they converted into TAMs after SRT and NT (Fig. 5A). Thus, CRT promotes the rapid accumulation and activation of monocytes.
Fig. 5. CRT, but not SRT, promotes monocyte activation.
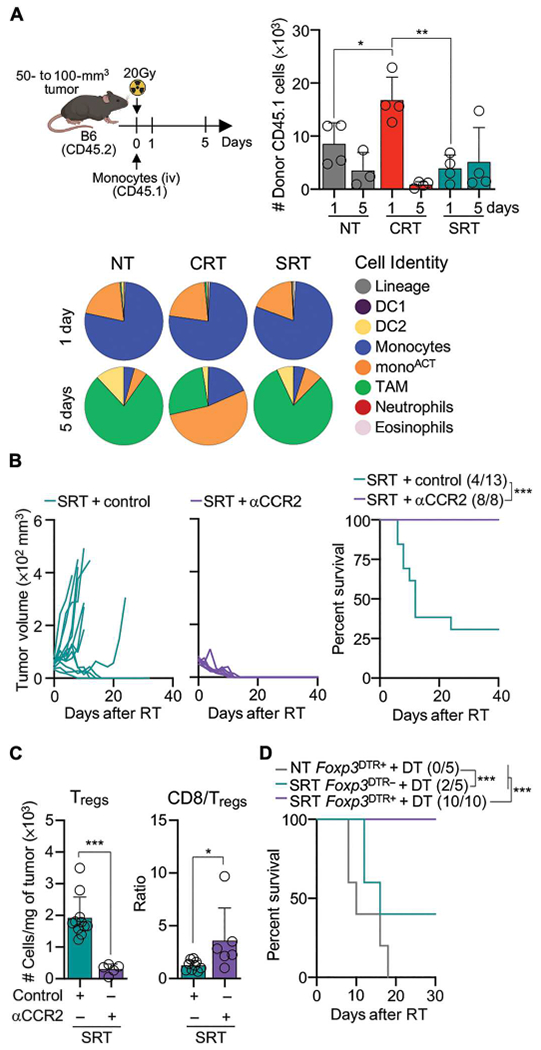
(A) BM CD45.1 monocytes were adoptively transferred into MC38-bearing CD45.2 mice 2 hours after RT. Top left: Experimental design. Top right:Transferred cells detected in tumors 1 and 5 days after RT by flow cytometry. Bottom: CD45.1 cell identity (n = 4 per group, two experiments). (B) MC38-bearing mice after SRT and αCCR2/control Ab (as in Fig. 4D). Left: Individual mouse tumor growth. Right: Survival curves with ratio of surviving mice in parentheses (n = 8 to 13 per group, two or three experiments). (C) Tregs Per milligram of tumor (left) and ratio of CD8+ TILs/Tregs (right) 5 days after SRT. (D) MC38-bearing Foxp3DTR+ and Foxp3DTR− mice NT or after SRT and DT inoculation (1 and 0.5 μg at 3 and 6 days after SRT, respectively) (n = 5 to 10 per group, three experiments). Statistics: Two-way ANOVA plus Tukey’s post hoc test for individual tumor growth curves (B); Mantel-Cox test for survival curves (B and D); unpaired t test (C); one-way ANOVA and Kruskal-Wallis post hoc test (A).
We queried TAM function after SRT by using αCD115 Ab depletion, which did not affect other myeloid cells in our model (fig. S5D). αCD115 Ab had no noticeable impact on animal survival after SRT (fig. S5E), suggesting that the sole elimination of TAM is not enough to promote immune activation. We then depleted all monocyte and monocyte-derived cells after SRT using αCCR2 Ab. Contrary to CRT (Fig. 4G), monocyte and monocyte-derived cell depletion resulted in complete tumor remission after SRT (Fig. 5B), as previously reported (1). Tumor remission correlated with a decrease in Treg numbers (Fig. 5C), suggesting that monocytes and monocyte-derived cells are responsible for Treg accumulation after SRT (Fig. 2B). Accordingly, Treg depletion by DT inoculation into Foxp3DTR+ mice also resulted in tumor remission after SRT (Fig. 5D). The immunosuppressive myeloid cell function after SRT was restricted to monocytes, because neutrophil depletion did not improve treatment outcome (fig. S5, F and G), despite being increased in proportion (Fig. 3B) and suggested to promote SRT resistance (57).
Collectively, our findings demonstrate the superior capacity of CRT to promote monocyte infiltration and activation. Our data also suggest that TAM depletion is not enough to restore immune activation unless it is accompanied by signals that mediate monocyte activation, such as those induced by CRT. Accordingly, TAM depletion promoted even faster tumor reduction after CRT (fig. S5H).
CRT promotes IFN-dependent, STING-independent immune responses
We performed multiplexed quantitative proteomic analysis to evaluate TME factors affecting monocyte activation after RT. CRT-treated tumors clustered separately from NT and SRT by PCA analysis of differentially abundant proteins (Fig. 6A). CRT promoted higher levels of proteins associated with IFN-I signaling, such as ISG20, IFIT3, IFIT2, TRIM26, TRIM14, DHX58, IFNAR2, and NOD1 (Fig. 6B) (58–60). Accordingly, we detected higher expression of Ifna and Ifnb by quantitative real-time PCR (qPCR) and IFN-β protein by enzyme-linked immunosorbent assay (ELISA) 3 days after CRT (Fig. 6, C and D). CRT also promoted higher Cxcl10, a CD8+ T cell recruitment chemokine (15).
Fig. 6. CRT eliminates tumors through an IFN-I–dependent but STING-independent signaling pathway.
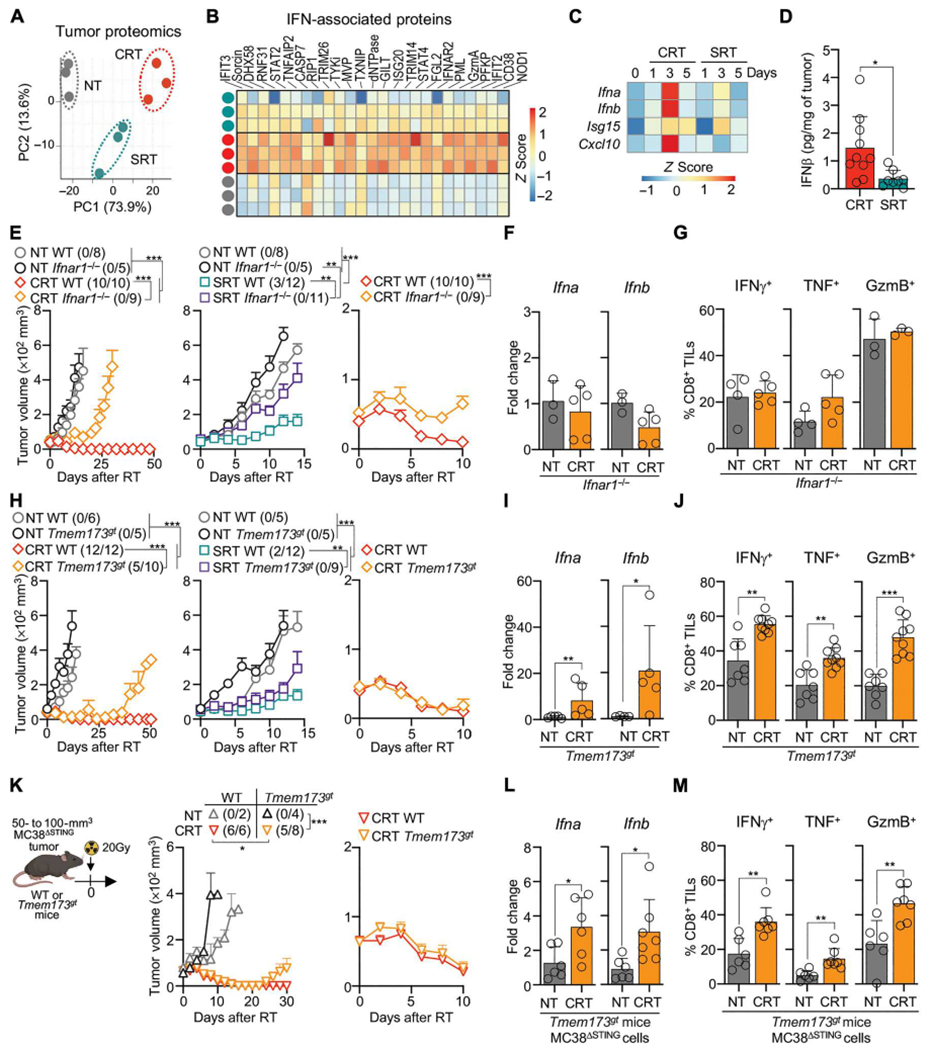
(A to C) MC38 tumors from WT mice, NT or 5 days after RT. (A) PCA of tumor protein differential abundance by mass spectrometry (n = 3 per group, one experiments). (B) Selected IFN-associated proteins, relative abundance (Z-scored) by mass spectrometry. (C) Ifna, Ifnb, Isg15, and Cxcl10 by qPCR (n = 5 to 20 per group, two to four experiments). (D) IFN-β ELISA of tumor lysates 3 days after RT (n = 8 to 10 per group, three experiments). (E to G) MC38-bearing WT and Ifnar1−/− mice after RT. (E) Mean tumor growth + SEM (n = 5 to 10 per group, two or three experiments). Left and middle curves show CRT and SRT, respectively. Right curves show the first 10 days after CRT. (F) Tumor Ifna and Ifnb in NT mice or 3 days after CRT by qPCR (n = 5 per group, two experiments). (G) CD8+ TILs expressing IFN-γ, TNF, and GzmB in NT mice or 3 days after CRT by flow cytometry (n = 3 to 5 per group, two experiments). (H to J) As in (E) to (G) but comparing MC38-bearing WT and Tmem173gt mice. (Kto M) MC38ΔSTING-bearing WT or Tmem173gt mice NT or after CRT. (K) Mean tumor growth + SEM over time (left) or 10 days after CRT (right) (n = 4 to 8, two experiments, except n = 2 for NT WT mice to control for MC38ΔSTING growth). (L) Ifna and Ifnb in tumors 3 days after CRT by qPCR (n = 6 or 7 per group, two experiments). (M) Frequency of CD8+ TILs producing IFN-γ, TNF, and GzmB by flow cytometry. Statistics: Two-way ANOVA plus Tukey’s post hoc test for mean tumor growth (E, H, and K); unpaired t test (D, F, G, I, J, L, and M).
To study the role of IFN-I after CRT, we used IFN-α/β receptor knockout mice (Ifnar1−/−). CRT efficacy was impaired in Ifnar1−/− mice, which was also true for SRT, as previously reported (Fig. 6E) (13). Whereas control wild-type (WT) mice rejected tumors by 5 to 10 days, Ifnar1−/− mice did not (Fig. 6E). This lack of response in Ifnar1−/− mice correlated with their inability to promote Ifna and Ifnb RNA after treatment, suggesting that IFN-I sensing is important to increase its levels in the TME (Fig. 6F). Lack of response in Ifnar1−/− mice also correlated with the inability of CRT to promote effector CD8+ TIL function (Fig. 6G).
Given that STING regulates IFN-I secretion after SRT (13), we tested whether this pathway was necessary after CRT. Unexpectedly, Tmem173gt mice, which harbor a missense mutation in STING, were able to eliminate tumors comparably to WT mice 5 to 10 days after CRT (Fig. 6H), suggesting that STING is not responsible for the acute IFN-I secretion in this model. Accordingly, CRT induced Ifna and Ifnb RNA and promoted effector CD8+ TIL function in Tmem173gt mice (Fig. 6, I and J). Nevertheless, the long-term survival of Tmem173gt mice was decreased, likely due to defects in de novo T cell priming (61). These data suggest that the acute IFN-I secretion after CRT is not regulated by STING activation in nontumor cells.
Some tumors can produce IFN-I through STING activation (62). STING protein was not expressed in our MC38 cell line (fig. S6E). Still, we used two approaches to block STING in tumor cells after RT. First, a STING inhibitor showed no significant decrease in CRT efficacy (fig. S6B). Second, MC38 cells mutated for STING (MC38ΔSTING) were generated using CRISPR-Cas9 (fig. S6, C to E). MC38ΔSTING tumors were quickly eliminated after CRT (Fig. 6K), which correlated with the increase in Ifna and Ifnb RNA, and effector CD8+ TIL function (Fig. 6, L and M). Yet the long-term survival of Tmem173gt mice transplanted with MC38ΔSTING cells was decreased, which resembles our results obtained with MC38WT cells (Fig. 6H).
These findings show that CRT promotes an IFN-I–dependent, STING-independent, acute immune response. We analyzed other pathways known to mediate IFN-I production such as MyD88 and MDA5, but CRT response was independent of these pathways as well (fig. S6, F and G). Further studies are required to pinpoint the immune pathway that promotes the acute IFN-I secretion after CRT.
Monocytes are the main cells producing and responding to IFN-I afterCRT
We next aimed to identify the source of IFN-I after CRT. CD45−, lineage+, and myeloid cell populations were sorted, and their capacity to produce IFN-β was analyzed by ELISA (Fig. 7A). Monocytes were the main source of IFN-β after CRT. Given that low DC numbers impeded their culture, we also used Ifnbmob mice in which enhanced yellow fluorescent protein (EYFP) is coexpressed with Ifnb (Fig. 7B). The frequency of EYFP+ monocytes after CRT was higher than any other cell population, suggesting that they are the main IFN-I source after CRT. Accordingly, monocyte depletion using αCCR2 Ab decreased Ifna and Ifnb in the TME 3 days after CRT (Fig. 7C).
Fig. 7. Monocytes are the main producers and responders to IFN-I after CRT.
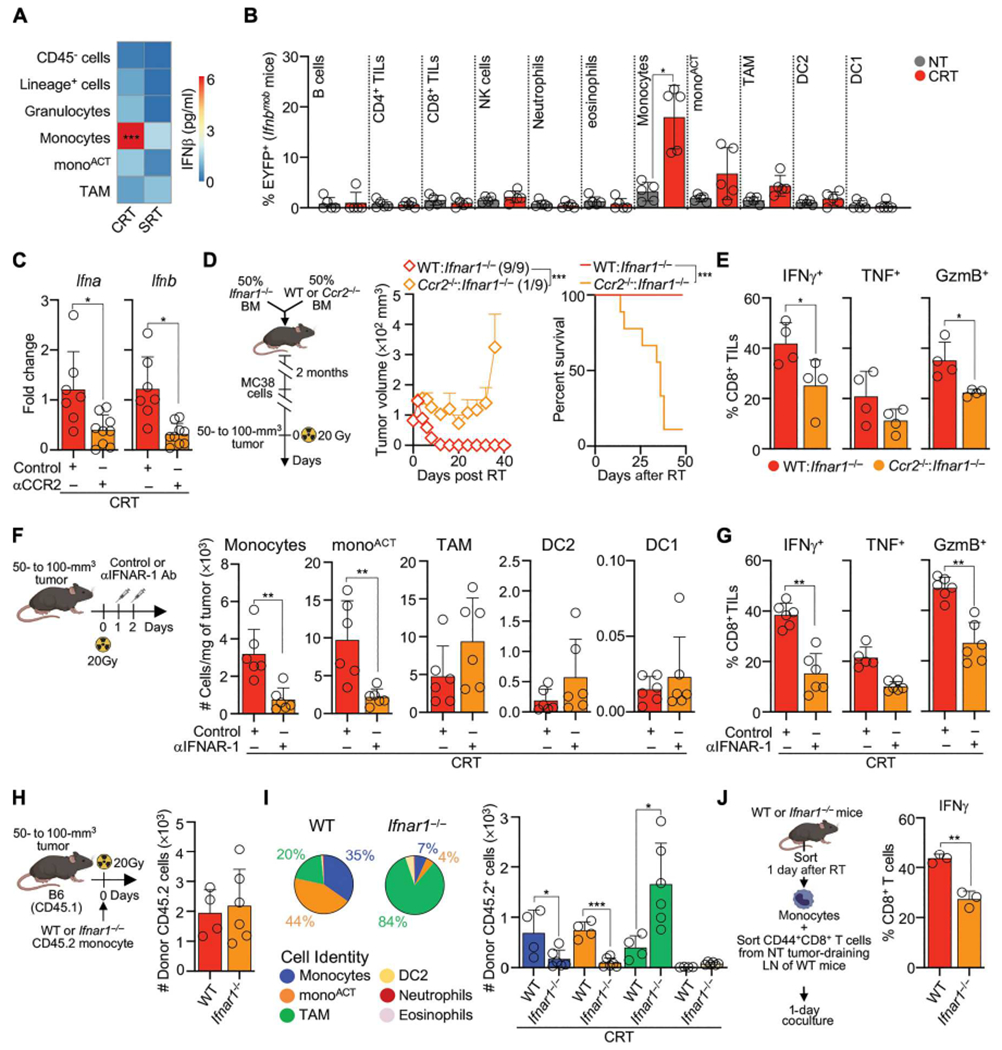
(A) Sorted cells from NT or 3 days after RT from MC38 tumors were cultured for 12 to 15 hours, and supernatant IFN-β was measured by ELISA (n = 3 to 6 per group, three to five experiments) (B) Tumors from MC38-bearing Ifnbmob mice were analyzed 3 days after CRT for EYFP+ cells (n = 5 per group, three experiments) by flow cytometry. Values are normalized to NT controls. (C) Tumor Ifna and Ifnb 3 days after CRT and αCCR2/control Ab by qPCR (n = 7 to 9 per treatment, two or three experiments). (D and E) Mixed BMCs generated by engrafting 50% Ifnar1−/− BM and 50% WT or Ccr2−/− BM into lethally irradiated CD45.1 mice. BMCs were transplanted with MC38 tumors and were CRT-treated. (D) Schematic experimental design (left), mean tumor growth + SEM with ratio of surviving mice in parentheses (middle), and survival curves (right) (n = 9 per group, two experiments). (E) Frequency of CD8+ TILs producing IFN-γ, TNF, and GzmB 3 days after CRT, after ex vivo restimulation (n = 4 per group, one experiments). (F and G) MC38-bearing mice after CRT and αIFNAR-1/control Ab. (F) Cells per milligram of tumor 3 days after CRT by flow cytometry (n = 6 per group, two experiments). (G) Frequency of CD8+ TILs producing IFN-γ, TNF, and Gzmb 3 days after CRT, after ex vivo restimulation (n = 6 per group, two experiments). (H and I) WT or Ifnar1−/− BM monocytes (CD45.2) were adoptively transferred into MC38-bearing CD45.1 mice 1 to 2 hours after CRT. (H) Number of tumor-infiltrating CD45.2 donor cells 3 days after CRT (n = 4 per group, two experiments). (I) Cell type proportion (left) and number (right) of recovered CD45.2 cells (n = 4 per group, two experiments). (J) MC38-infiltrating monocytes sorted from WT or Ifnar1−/− mice 1 day after CRT were cocultured with CD44+CD8+ T cells sorted from MC38 tumor-draining LNs of NT mice. Frequency of IFN-γ+CD8+ T cells 1 day after coculture by flow cytometry (n = 3 per group, two experiments). Statistics: One-way ANOVA plus Tukey’s post hoc test (A, B, and I); unpaired t test (C, E, F, G, H, and J); two-way ANOVA plus Tukey’s post hoc test for tumor growth and Mantel-Cox test for survival (D).
Next, we hypothesized that monocytes were the main cells responding to IFN-I after CRT given their strong IFN-I signature and their impaired accumulation in Ifnar1−/− mice (Fig. 3, D and E, and fig. S6A). To test this, we generated mixed BM chimeras (BMCs) in which monocytes cannot respond to IFN-I by implanting 50% Ccr2−/− and 50% Ifnar1 BM into CD45.1 lethally irradiated mice (Fig. 7D). CRT efficacy was lower in tumors implanted into Ccr2−/−:Ifnar1−/− BMCs, which correlated with a lower effector CD8+ TIL function (Fig. 7E). Given that IFN-I sensing can modulate T cell activation in some cases (63), we also generated Rag1−/−:Ifnar1−/− BMCs (fig. S7); however, survival after CRT was only partially reduced (by 20 to 25%). These data indicate that IFN-I sensing by monocytes is the main signaling pathway that promotes the acute antitumor immune response after CRT.
We queried the consequences of blocking IFN-I signaling in monocytes. We blocked this signaling pathway using αIFNAR-1 Ab 1 day after CRT, which still allowed for monocyte infiltration to the TME. Blocking IFN-I receptor decreased monocyte and monoACT numbers by 3 days (Fig. 7F), suggesting that this pathway is critical for the maintenance and activation of these cells in the TME. Decreased monocytes and monoACT after αIFNAR-1 Ab correlated with a decline in effector CD8+ TIL function (Fig. 7G). To evaluate whether IFN-I signaling led to monocyte activation, we compared the fate of WT and Ifnar1−/− monocytes after their adoptive transfer into WT CRT-treated mice. Similar donor cell numbers were recovered 3 days after CRT (Fig. 7H). Whereas WT monocytes accumulated as monocytes and monoACT, Ifnar1−/− monocytes accumulated as TAMs (Fig. 7I), showing that IFN-I signaling modulates monocyte fate. Last, to evaluate the consequences of IFN-I signaling in the intrinsic capacity of monocytes to activate CD8+ T cells, we performed an ex vivo experiment (Fig. 7J). CD44+CD8+ T cells cultured with tumor-infiltrating WT monocytes produced significantly greater levels of IFN-γ in comparison with T cells cultured with Ifnar1−/− monocytes. Together, our data support a model in which secretion and sensing of IFN-I by monocytes are critical to mediate their maintenance and activation and, consequently, the efficacy of CRT.
Monocyte recruitment after CRT requires limited damage of healthy tissues
We aimed to identify the reason why monocytes are not rapidly recruited to the TME after SRT. We hypothesized that SRT promoted lower levels of monocyte-recruiting chemokines than CRT, but we observed no differences in the up-regulation of Ccl2 or Ccl5 1 day after treatment (fig. S8A).
SRT delivered the same radiation dose to tumors as CRT (see Supplementary Materials and Methods), and no radiation of healthy tissues outside the radiation beam path was observed because the shield absorbs 99.9% of this radiation (fig. S8B). However, the radiation beam path itself was different. Whereas CRT focuses most of the radiation beam on the tumor, the SRT beam delivered lower radiation doses to healthy tissues behind the tumor (Fig. 1A). To test whether differences in the radiation path modulate a distinct immune response between SRT and CRT, we designed a treatment to mimic SRT using the X-Rad SmART image-guided preclinical irradiator, which we refer to as SRT-like. We reasoned that this approach would allow us to compare both RT modes using the same preclinical irradiator while only modifying the radiation beam path. Whereas the CRT radiation beam ran parallel to the animal, the SRT-like radiation beam traveled perpendicular to the animal, similar to the SRT radiation beam (Fig. 8A). We found that SRT-like recapitulated the limited efficacy of SRT (Fig. 8A). SRT-like did not promote the accumulation of monocytes and monoACT (Fig. 8B) but rather promoted TAMs, which coincided with decreased levels of Ifna and Ifnb (Fig. 8C). PCA analysis of NanoString data showed that CD8+ TILs undergo similar transcriptional changes after SRT and SRT-like treatments, which are different from those promoted by CRT (Fig. 8D). Whereas CD8+ TILs from CRT-treated tumors up-regulated IFN-I and II pathways, cytokines, and cell cycle genes, SRT-like and SRT maintained exhausted transcripts such as Tox (Fig. 8, E and F). Last, functional analysis showed that SRT-like did not promote effector CD8+ TIL function or increased the ratio of CD8+ TILs to Tregs, all features promoted by CRT (Fig. 8, G and H). Thus, differences in the radiation path between CRT and SRT-like (or SRT) are responsible for the induction of distinct immune responses.
Fig. 8. CRT efficacy decreases when radiation affects healthy tissues.
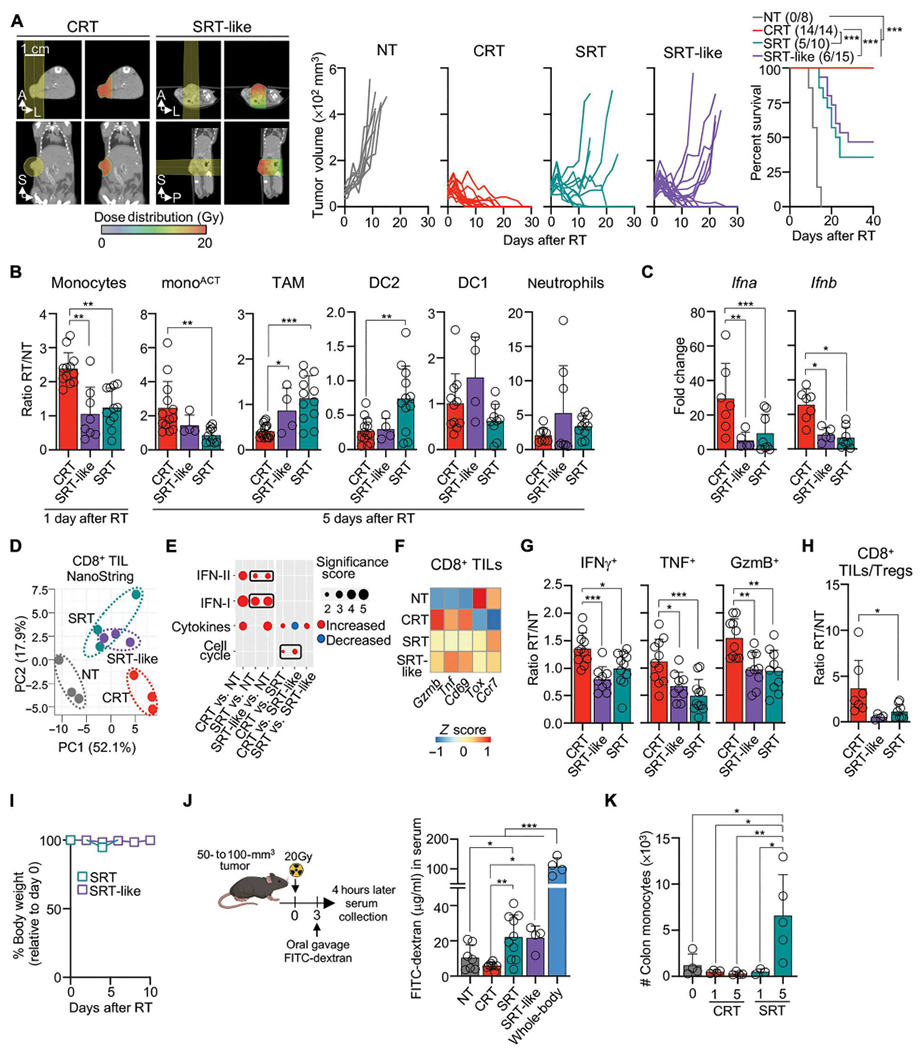
(A) CT scans with beam orientation and dose distribution (left), individual tumor growth for MC38-bearing WT mice NT and after RT (middle), and survival curves with ratio of surviving mice in parentheses (right) (n = 8 to 15 per group, two or three experiments) (B to H) MC38-bearing mice after RT. (B) Cell numbers relative to NT controls for 1 day (monocytes) or 5 days (remaining populations) after RT by flow cytometry (n = 4 to 12 mice, two orthree experiments). (C) Tumor Ifna and Ifnb 3 days after RT relative to NT controls by qPCR (n = 5 to 8 per group, two or three experiments). (D to F) Transcriptomic analysis of CD8+ TILs 3 days after RT by NanoString (n = 3 per group). (D) PCA of the DEGs. (E) Pathway enrichment analysis of DEGs. The size and color indicate the significance score and up-regulated/down-regulated genes, respectively. (F) Normalized counts of selected genes (Z-scored). (G) CD8+ TILs producing IFN-γ, TNF, and GzmB 3 days after RT relative to NT controls by flow cytometry (n = 4 to 16 mice, two to four experiments). (H) Ratio of CD8+ TILs/Tregs. (I) Body weight after RT shown relative to time 0 measurements (n = 5 per group, two experiments). (J) Left: Diagram of FITC-dextran oral gavage of MC38-bearing mice, performed 3 days after RT. Right: Serum FITC-dextran measured 4 hours after gavage by spectrophotometer (n = 4 to 10 mice per group, three experiments). (K) Number of colon monocytes 1 or 5 days after RT by flow cytometry (n = 3 to 5 per group, two experiments). Statistics: Mantel-Cox test for survival curve (A); one-way ANOVA plus Tukey’s post hoc test (B, C, G, H, J, and K); two-way ANOVA plus Tukey’s post hoc test (I).
We hypothesized that the unintended delivery of radiation to tissues localized behind the tumor promoted healthy tissue damage during SRT-like and SRT. Significant radiation doses were observed to normal tissues behind tumors, including gut and kidney (~16.0 to 18.4 Gy), measured by CT scan analysis (Fig. 8A). Nevertheless, SRT-like and SRT did not result in signs of systemic toxicity, i.e., weight loss or blood leukocyte alterations (Fig. 8I and fig. S8C). However, lower doses of radiation received in a small fraction of the intestine behind the tumor resulted in increased gut permeability, a readout of gastrointestinal damage, after SRT-like and SRT but not CRT (Fig. 8J). This gastrointestinal damage was lower than that observed after whole-body radiation, but associated with monocyte infiltration to the colon (Fig. 8K), which was not observed during CRT. Antibiotic treatment did not alter the SRT outcome in our hands (fig. S8D). These observations suggest that SRT-like and SRT promote damage and monocyte infiltration to healthy tissues. This relocalization of monocytes to healthy tissue may explain the distinct acute immune responses after CRT versus SRT.
DISCUSSION
We show that rapid and robust monocyte infiltration into tumors drives the positive therapeutic outcome of RT. These monocytes have an intrinsic capacity to secrete and sense IFN-I, which, in turn, increases monocyte capacity to promote effector CD8+ TIL function and, consequently, control tumor growth. We present evidence that monocyte accumulation and activation in tumors are impaired when the radiation unintentionally affects healthy tissues. Our results demonstrate the positive role of monocytes in the context of clinically relevant RT and suggest that strategies that block monocyte recruitment and function might mitigate the therapeutic potential of focal RT.
Unlike previous reports describing the immunosuppressive nature of tumor-infiltrating monocytes after SRT (1, 2), we show evidence that monocytes have an immunostimulatory function after CRT. This immunostimulatory function depends on the monocyte’s ability to sense IFN-I, which parallels previous observations of monocyte activation during anti-viral immune responses (33, 64). IFN-I–stimulated monocytes acquire the capacity to promote the function of tumor-localized effector/memory CD8+ T cells through a tumor antigen–independent mechanism. It is possible that IFN-I signals monocytes to produce T cell–activating cytokines, as previously observed during viral infections (33, 64). Accordingly, monocytes express higher Il18, Il15r, and Il15 transcripts after CRT (Fig. 3E). Monocytes themselves also acquire signs of increased effector CD8+ TIL functions after CRT. These signs include the expression of IFN-γ–stimulated proteins, i.e., ME1C II, costimulatory markers, and PD-L1. PD-L1 expressed by activated monocytes could inhibit CD8+ TIL function; however, we do not observe a central role of PD-L1–PD-1 (programmed cell death protein-1) inhibitory pathway in our system, perhaps because soluble mediators secreted by monocytes activate CD8+ TILs bypassing the cell-to-cell interactions required for PD-1 inhibition (35). Alternatively, the rapid elimination of tumor cells by effector CD8+ TILs occurs before PD-1–PD-L1 interaction limits the function of these cells. Nevertheless, the observation that PD-L1 is up-regulated in activated monocytes after CRT suggests that this molecule could be a target for tumors that do not respond completely to RT. Past reports show that IFN-I signaling promotes monocytes’ capacity to present antigen, kill tumor cells directly, and activate DCs, suggesting that IFN-I may activate a broad range of immunostimulatory functions in these cells (65–68). This work expands previous observations and provides insight into the role of IFN-I–activated monocytes during acute antitumor immune responses.
Monocytes are both producers and responders of IFN-I after CRT, which modulates their accumulation and activation. This observation implies that the immunostimulatory function of monocytes is promoted by their sheer number. This density-based cellular communication, so-called quorum sensing, is known to modulate myeloid cell functions in other systems, i.e., monocyte survival, IFN-I production by plasmacytoid DCs, and macrophage activation (69–71). In our system, monocytes accumulate in the TME 1 day after CRT, but they seem to redistribute between the TME and irradiated healthy tissues after SRT, explaining the lower IFN-I levels in these tumors. Our data show that lower numbers of IFN-I–secreting monocytes result in their acquisition of immunosuppressive functions. These immunosuppressive monocytes mediate the accumulation of Tregs and the low efficacy of SRT, which agrees with previous reports (1, 2, 50, 72). More studies are needed to discern pathways driving such contrasting monocyte fates; however, our findings illustrate that IFN-I signaling is key for monocyte activation. The accumulation and activation of monocytes are two associated phenomena that depend on IFN-I; thus, it is not possible to discern in vivo whether the immunostimulatory function resides uniquely in monocytes or monoACT. Ex vivo data point to a model in which IFN-stimulated monocytes and monoACT can promote effector CD8+ TIL function. SRT promotes some IFN-I in comparison with NT controls; however, this level of IFN-I is not enough to promote the immunostimulatory functions of monocytes, suggesting that there is a threshold to activate this innate pathway. It is possible that the IFN-I source varies depending on the tumor type and stimulus, and, in some cases, may be produced by tumor cells themselves but, nevertheless, activate monocytes (68).
As shown here, monocytes mediate IFN-I production through a STING-independent pathway after CRT. It is possible that IFN-I production after CRT is mediated by retinoic acid–inducible gene-I/mitochondrial antiviral-signaling protein (RIG-I/MAVS) (73), which requires further investigation. Our findings do not dismiss the importance of the STING signaling pathway after RT. Rather, they suggest that STING is not required for the acute immune response but is needed for the de novo priming of T cells after CRT, which can take 1 to 2 weeks. This observation agrees with the role of STING in DCs, key antigen-presenting cells, during SRT (13, 18, 74). It also agrees with recent observations that tumors engrafted in WT and Tmem173gt mice only diverged in growth after ~15 to 18 days (75). If this model is correct, then STING agonists would aid de novo T cell priming after RT, which seems to be the case for SRT (76, 77). Unfortunately, STING agonists did not improve the RT outcome in early clinical trials even when combined with checkpoint inhibitors (NCT03172936 and NCT03937141), showing that further studies that combine STING agonists with clinically relevant CRT are needed to provide further insights into clinical practice.
The field of RT has progressively enhanced treatment options to limit healthy tissue damage and increase the radiation dose to the target tumor. This is due to the realization that RT induces short- and long-term side effects in patients (10). The long-term effects of RT are gastrointestinal damage, chronic pneumonitis, cardiac toxicity, cognitive impairment, and bone growth impairment (78). Although the short-term toxicities can lead to complications (e.g., mucositis), the acute effect of healthy tissue damage on the therapeutic efficacy of RT is difficult to address. Serendipitously, our study reveals that the acute effect of healthy tissue damage during RT has strong consequences in the activation of TME immune responses (i.e., limited monocyte activation). Reduced tumor-infiltrating monocytes may be caused by the relocalization of these cells into inadvertently radiated healthy tissues, a hypothesis supported by the observation that monocytes increase in the colon after non-CRT. Alternatively, tissue exposure to RT has been associated with vasculopathy, which could limit monocyte infiltration to treated tumors (79, 80). Our findings are critical for understanding RT outcomes in tumors localized within internal organs, such as the prostate, breast, lung, stomach, and brain, where radiation exposure to healthy organs is unavoidable (81, 82). Accordingly, strategies that reduce healthy tissue radiation damage, such as radiosensitizers, may help improve the outcome of internal organ RT. New technologies, such as FLASH RT, which reduces radiation damage to surrounding healthy tissues, could have notable benefits in the clinic (83).
One shortcoming of our study is the lack of human clinical data to support our observations. Analysis of immune responses in patients has only been done weeks after RT; thus, to our knowledge, there is no transcriptomic or public database regarding early immune responses after RT, showing a critical need in the field. Clinical response to anti–PD-1 treatment correlates with an immunostimulatory monocyte signature enriched in IFN-I signaling (68), suggesting that IFN-I–mediated monocyte activation is relevant in human antitumor immune responses.
Together, our data demonstrate that CRT uniquely induces rapid and robust monocyte infiltration, resulting in their downstream activation due to the limited exposure of healthy tissue to radiation. This immune activation mechanism broadens our understanding of clinical RT and provides insights into the development of therapeutic strategies to use in combination with CRT.
MATERIALS AND METHODS
Study design
This study was designed to dissect the acute immune mechanisms induced by a clinically relevant mode of RT, i.e., CRT. The immune mechanism induced by CRT was compared with those induced by a more widely used preclinical mode of RT, i.e., SRT. Tumor-bearing mice were analyzed without excluding outliers. A few mice that randomly presented ulcerations on the day of treatment were excluded from the studies and immediately euthanized per the recommendation of the Institutional Animal Care and Use Committee at Stanford. Treatment planning was done on an animal-by-animal basis to ensure that the dose distributions were consistent across subjects. RT was tailored to treat tumors in different anatomical locations, i.e., MC38 and LLC tumors located on the flank, E0771 tumors located in the mammary fat pad, and BRaf/Pten tumors located mainly in the flank, but the precise location could not be controlled and depended on the spontaneous tumor generation. Individual experiments used mice of the same sex and age. Both male and female mice were used for the experiments unless sex matching was necessary to agree with the sex origin of the tumors. The number of independent experiments and statistical analyses are indicated in the figure legends. The number of mice was estimated using power analysis (G*Power 3) and represents the minimum number to ensure robust statistical data without compromising reproducibility and accuracy. Measurements were not blinded, but experimental animals were randomly assigned to experimental groups, and opposing treatments frequently shared housing conditions. Survival experiments adhered to the recommendation of the Institutional Animal Care and Use Committee at Stanford. Accordingly, mice were euthanized when they lost >10% of body weight, when the tumor mass reached 1.3 cm in diameter, or when the tumor showed any sign of ulceration.
Mice
Mice used in this study are listed in table S1. Mice were maintained under specific pathogen–free conditions and used at 6 to 8 weeks of age, and experiments were overseen by the Institutional Animal Care and Use Committee at Stanford University. BMCs were generated by transplanting BM into lethally irradiated (two doses of 6Gy) CD45.1 mice: (i) WT or Ccr2DTR BM; (ii) 50% Ifnar1−/− BM + 50% WT BM; (iii) 50% Ifnar1−/− BM + 50% Ccr2−/− BM; and (iv) 50% Ifnar1−/− BM + 50% Rag1−/− BM. Sulfamethoxazole and trimethoprim (200 and 40 mg per 5 ml, respectively) were supplemented in the drinking water for 2 weeks. Chimerism was evaluated 2 months later by flow cytometry of blood leukocytes before experimental testing.
Tumor induction
See list of cell lines in table S2. MC38 and E0771 cell lines were cultured in complete RPMI 1640 medium. LLC, RAW264.7, and B16F1 cells were cultured in complete Dulbecco’s modified Eagle’s medium. Complete medium contained 10% heat-inactivated fetal bovine serum (FBS), 1× penicillin/streptomycin, and 1× glutamine. Cells were grown at 37°C and 5%CO2. Cell lines were periodically tested for mycoplasma contamination by PCR (84). Tumor cells were washed and resuspended in phosphate-buffered saline (PBS). A total of 3 × 105 MC38 cells or 5x105 LLC were injected subcutaneously (sc) in the right flank. A total of 5 × 105 E0771 cells were injected in the mammary fat pad (85). For tumor re-challenge, 3x105 tumor cells (MC38 or B16F1) were injected in the left flank 45 to 60 days after RT. BRaf/Pten tumors were induced by topical application of 2 μl of 5 mM 4-hydroxytamoxifen. Tumors were measured every 2 days with calipers, and tumor volume was estimated as follows: [maximum dimension x (minimum dimension)2]/12.
Cell depletion experiments
For lymphocyte depletion, mice were injected with 200 μg of αCD8 Ab (clone 2.43, produced in-house), αCD4 Ab (clone GK1.5, produced in-house), αNK1.1 (clone PK136, BioXCell), or the corresponding isotype control immunoglobulin G2b (IgG2b) (clone LTF-2, BioXCell) or IgG2a Ab (C1.18.4, BioXCell) intraperitoneally (ip) on days 1, 2, 5, 8, and 11 after RT. For TAM depletion, mice were injected with αCD115 (clone AFS98) or IgG2a ip on days 0 (1 mg) and 2 and 5 (0.5 mg). Neutrophils were depleted with a single dose of 200 μg of αLy6G or IgG2a mAb on the day of RT. Monocytes were depleted by inoculating 20 μg of αCCR2 (M.M.; clone MC21) or IgG2b control Ab ip on days 0, 1, 2, 3, and 4 after RT. IFN-I receptor blockade was conducted by inoculating 250 μg of αIFNAR-1 (clone MAR1-5A3; BioXCell) or control Ab on days 1 and 2 after CRT. Ccr2DTR or control WT BMCs were inoculated with 10 ng/g of body weight of DT (Sigma-Aldrich) on days 0,1,3, and 5 after RT. Lyz2Cre+ xCsf1rLSL-DTR+ (MMDTR+) or littermate controls Lyz2Cre+ xCsf1rLSL-DTR− (MMDTR−) mice were inoculated with 4 ng/g of body weight DT on days 0, 1, 2, 3, 4 after RT. Foxp3DTR+ mice or littermate control Foxp3DTR− were inoculated with 50 and 25 ng/g of body weight of DT on days 3 and 6 after RT, respectively.
Radiation therapy
MC38, E0771, and LLC tumors were treated upon reaching a volume of 50- to 100-mm3. BRaf/Pten tumors were treated upon reaching a volume of 100 to 200 mm3. Radiation treatment regimens were either a single 20-Gy or three consecutive doses of 8 Gy (8 Gy ×3). Radiation was delivered using an X-Rad SmART image-guided preclinical irradiator (CRT and SRT-like) (Precision X-ray Inc.) or a Polaris SC-500 Series II orthovoltage irradiator (SRT) (Kimtron). For CRT and SRT-like treatment, x-ray beams were 225 kVp, 13 mA and were filtered with 0.3 mm Cu. Treatment beams were applied for exposures of 4 to 9 min to deliver the prescribed dose, using a measured dose rate for the 1-cm collimator of 2.35 Gy per minute at the isocenter. SRT was done using a 225 kVp, 13.3-mA x-ray beam filtered by 0.5 mm Cu. The prescribed dose was delivered on the basis of quarterly calibration data acquired for the system, involving exposures of 4 min based on the measured dose rate of 5.4 Gy per minute through a 3-mm lead shield. The shield contained 1-cm diameter holes to allow the radiation beam to treat tumors. Calibration of both the X-Rad SmART and the Polaris was performed using ion chamber and film dosimetry (refer to Supplementary Materials and Methods for more details).
Tissue cell suspensions
For a list of key commercial assays and reagents, see table S3. Tumor and LN cell suspensions were obtained by enzymatic digestion using collagenase D (400 U/ml) and deoxyribonuclease I (DNAse I) (5 μg/ml) in Hank’s balanced salt solution (HBSS) buffer with Ca2+ and Mg2+ at 37°C, for 60 or 25 min for tumor and LNs, respectively. Digestions were stopped by adding 5 μM EDTA for 5 min. Blood was collected, diluted 1:1 in PBS/1 mM EDTA, and lysed with ammonium-chloride-potassium lysis buffer. Large intestines were treated with HBSS/2%FBS with 5 mM dithiothreitol (DTT) at 37°C for 12 min. DTT treatment was stopped by washing tissue with 5 mM EDTA. Large intestines were then rinsed in HBSS/2%FBS with 5 mM HEPES and subsequently digested with collagenase IV (50 μg/ml) and DNAse I solution (5 μg/ml) at 37°C for 25 min. Digestion was stopped using 5 μM EDTA. In all cases, single-cell suspensions were filtered using a 70-μm strainer. CD45+ cells were quantified using counting beads. In some experiments, CD45+ cells were enriched using αCD45-biotin Ab (clone 30-F11) and αbiotin beads following the manufacturer’s instructions.
Flow cytometry
Flow cytometry Abs were purchased from BioLegend, Thermo Fisher Scientific, and BD Bioscience (table S4). Cell suspensions were blocked with αCD16/32 Ab (clone 2.4G2; produced in-house) at 4°C for 15 min before staining with primary Abs and LIVE/DEAD Fixable Blue at 4°C for 20 min. For cytokine detection, tumor cell suspensions were stimulated with phorbol 12-myristate 13-acetate (PMA; 100 ng/ml), ionomycin (500 ng/ml), and Brefeldin A (BFA; 10 μg/ml) for 4 hours. Cells were fixed with BD Cytofix/Cytoperm Fixation and Permeabilization solution and stained in perm/wash buffer. For FOXP3 staining, cells were fixed with FOXP3 Transcription Factor Fixation/Permeabilization buffer. Cells were acquired on an LSRFortessa X-20 (BD Biosciences), and data were analyzed using FlowJo software. Boolean gating was conducted using Flowjo software after the identification of single positive (IFN-γ, TNF, and GzmB) cells. Unstained and single-fluorochrome–stained cells were used for compensation. Control samples included fluorescence minus one for cell markers and cytokines.
In vitro coculture
Monocytes, monoACT, and TAMs were sorted from a CD45-enriched MC38 tumor cell suspension using FACSAria Fusion (BD Biosciences) and cultured with effector/memory or naïve CD8+ T cells. CD8+ T cells were sorted from tumor-draining LNs of MC38- or B16F1-bearing mice after enrichment with an in-house Ab cocktail [αB220 (RA36B2), αF4/80 (HB198), αMHCII (TIB120), αCD4 (GK1.5), and αNK1.1 (PK136)] and sheep αRat Dynabeads as previously described (86). CD8+ T cells were stained with CellTrace Violet following the manufacturer’s instructions. A total of 1 × 105 CD8+ T cells were incubated with 2 × 104 myeloid cells in a 96-well U bottom plate for 1 to 3 days before PMA/ionomycin/BFA stimulation.
IFN-I ELISA
A total of 1000 sorted cells per μl were incubated for 15 hours, and culture supernatant was collected for IFN-β ELISA. Tumor lysate was prepared using 50 mM tris (pH 7.4), 250 mM NaCl, 5 mM EDTA, 50 mM sodium fluoride (NAF), 1 mM Na3VO4, 1% NP40, 0.02% NaN3, and cOmplete EDTA-free protease inhibitor cocktail (1 tablet per 50 ml of solution). ELISA on tumor lysate was conducted using 10 μg of total protein measured using Pierce BCA Protein Assay Kit. IFN-β levels were evaluated with the VeriKine-HS mouse IFN-beta Serum ELISA Kit following the manufacturer’s instructions.
CyTOF analysis
Metal-conjugated Abs were obtained from Fluidigm or labeled in-house using the MaxPar X8 labeling kit (Fluidigm) (table S5) (86). CD45-enriched tumor cells were stained with 0.25 μM cisplatin and CD45-barcoding Abs in CyFACS buffer (2 mM EDTA, 1% BSA). Barcoded samples were pooled and stained for surface proteins followed by fixation using eBioscience FOXP3/Transcription Factor Staining Buffer Set and intracellular staining. Stained cells were incubated with 2% paraformaldehyde (Electron) containing 125 nM indium intercalator overnight and acquired using a CyTOF2 (Fluidigm) at the Stanford Shared FACS Facility. CyTOF files were normalized with the ParkerICI/Premessa R package and bead-based normalization. Samples were gated as live, singlets, and CD45+ cells. Myeloid cells were gated as CD3−/CD8−/CD335−/CD19−. UMAP and X-Shift analyses were conducted by concatenating either 10,000 CD45+ cells or myeloid cells. Analyzed files were imported into FlowJo software or R using ggplot2 for visualization.
Quantitative real-time PCR
RNA extractions were conducted using TRIzol. RNA was quantified and diluted to a total of 0.5 to 1 μg, and cDNAwas synthesized using iScript Reverse Transcription Supermix. Itaq Universal SYBR was used for quantitative real-time PCR (qPCR) following the manufacturer’s instructions. Primers were purchased from Integrated DNA Technologies (table S6). Data were analyzed by the 2−ΔΔCT method, and mRNA levels were plotted relative to Rpl13a as a housekeeping control (87).
NanoString gene expression
Tumor cell suspensions were collected at 0 to 3 days after RT, enriched for CD45+ cells, and sorted for CD8+ T cells, monocytes, or monoACT on a FACSAria Fusion (BD Biosciences). Cells were resuspended in 1/3 RNeasy Lysis Buffer RLT (Qiagen) at a concentration of 1000 to 5000 cells per μl and analyzed on the NanoString nCounter Mouse Immune Exhaustion Panel, or the Mouse Myeloid Innate Immunity V2 Standard Platforms, following the manufacturer’s instructions. Samples were processed on the NanoString Digital Analyzer to yield a reporter code count dataset, which was analyzed via ROSALIND (https://rosalind.bio). DEGs were determined by a log2(fold change) of 1.5 and an adj. P value < 0.05 for the indicated comparisons. NanoString gene set analysis was conducted via ROSALIND using directed global significance scores for pathway enrichment analysis.
Gut permeability assay
Gut permeability was determined via fluorescein isothiocyanate (FITC)–dextran 4-kDa oral gavage of 600 mg/kg (diluted in water) in mice that fasted for 8 to 12 hours. Serum FITC-dextran 4 kDa was evaluated 4 hours after gavage by measuring fluorescence at 485/528 nm with a Biotek Neo2 and using a standard curve to determine FITC-dextran concentration.
Quantitative proteomics
Tumors were collected and digested in 500 μl of lysis buffer (1× PBS, 1 mM MgCl2, 0.5% NP-40) containing cOmplete EDTA-free protease inhibitor cocktail (1 tablet per 50 ml of solution) followed by sonication and centrifugation. Samples were processed and analyzed as previously described (88). Briefly, for trypsin digestion and TMT labeling, 25 μg of protein per sample were reduced with 5 mM DTT and alkylated with 12 mM iodoacetamide. The detergent was removed, and peptides were digested with trypsin. Peptides were suspended in 30% acetonitrile and mixed with 5 μl of pre-aliquoted TMT16 reagents (12.5 μg/μl in anhydrous acetonitrile). TMT-labeled peptide samples were analyzed using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). To improve the quantitative accuracy of peptide abundance, the FAIMS-HRMS2 method was used. Spectra were analyzed using MSFragger (version 3.4) implemented in Fragpipe software (version 17.0). Proteins were identified using a mouse proteome database (UniprotKB).
Statistics
Statistical analysis was performed using GraphPad Prism. Statistical analyses for average tumor growth curves were performed using a two-way analysis of variance (ANOVA) plus Tukey’s multiple comparison post hoc test. Statistical analysis for Kaplan-Meyer survival curves was performed using the Mantel-Cox test. Comparisons between more than two groups were performed using one-way ANOVA with Tukey’s or Kruskal-Wallis post hoc tests. Comparisons between two groups were performed using the unpaired t test. P values: nonsignificant (n.s.; are not shown in the figures for clarity), P > 0.05; *P ≤ 0.05; **P < 0.01; ***P < 0.001. Graphics created with Biorender.com.
Supplementary Material
Acknowledgments:
We thank Carette Lab members for assistance during the generation of the CRISPR knockout cell line and Idoyaga Lab members for continuous technical support and discussions.
Funding:
This work was supported by NIH grant to J.I. (R01CA219994). S.T. is a recipient of a Life Sciences Research Foundation fellowship, D.R.C. of a Canadian Institute of Health Research fellowship, R.A.H. and S.T. of a Dean’s fellowship (Stanford), A.G.-B. of a KL2 Mentored Career Development Award (KL2TR003143),and B.J.K. of a K08 award (K08HL159359). J.A.P. is supported by an NIH grant (R01GM132129). CyTOF was performed by a Stanford FACS Facility instrument funded by NIH (S10OD016318-01). Small Animal Radiation was performed by an instrument funded by NIH (S10OD018208-01).
Footnotes
Competing interests: J.I. serves on the scientific advisory board of Immunitas Therapeutics. All the other authors declare that they have no competing interests.
Supplementary Materials
This PDF file includes:
Supplementary Materials and Methods
Other Supplementary Material for this manuscript includes the following:
Data and materials availability:
NanoString data generated in this study are deposited in NCBI GEO (GSE207105 and GSE223400). Mass spectrometry proteomics data have been deposited to MASSIVE and the ProteomeXChange Consortium via the PRIDE repository with the dataset identifier PXD035046. This study did not generate a unique code. The MC38ΔSTING cells are available upon request to J.I. Ms4a3Cre mice are available from F.G. under a material transfer agreement with the Singapore Immunology Network (SIgN), A*STAR Research Entities.
REFERENCES AND NOTES
- 1.Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, Fu Y-X, Weichselbaum RR, Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun 8, 1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G,McBride W, Wu L, CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 73, 2782–2794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegde S, Leader AM, Merad M, MDSC: Markers, development, states, and unaddressed complexity. Immunity 54, 875–884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostrand-Rosenberg S, Horn LA, Ciavattone NG, Radiotherapy both promotes and inhibits myeloid-derived suppressor cell function: Novel strategies for preventing the tumor-protective effects of radiotherapy. Front. Oncol 9, 215 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H-M, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, Ferris R, Kao J, Wang L-H, Pan P-Y, Ko EC, Chen S-H, Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin. Cancer Res 21, 4073–4085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster CC, Fleming GF, Karrison TG, Liao C-Y, Desai AV, Moroney JW, Ratain MJ, Nanda R, Polite BN, Hahn OM, O’Donnell PH, Vokes EE, Kindler HL, Hseu R, Janisch LA, Dai J, Hoffman MD, Weichselbaum RR, Pitroda SP, Chmura SJ, Luke JJ, Phase I study of stereotactic body radiotherapy plus nivolumab and urelumab or cabiralizumab in advanced solid tumors. Clin. Cancer Res 27, 5510–5518 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Kao J, Timmins J, Ozao-Choy J, Packer S, Effects of combined sunitinib and extracranial stereotactic radiotherapy on bone marrow hematopoiesis. Oncol. Lett 12, 2139–2144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pienta KJ, Machiels J-P, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS, Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 31, 760–768 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Citrin DE, Recent developments in radiotherapy. N. Engl. J. Med 377, 1065–1075 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Ruysscher DD, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F, Radiotherapy toxicity. Nat. Rev. Dis. Primers 5, 13 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Scaife JE, Barnett GC, Noble DJ, Jena R, Thomas SJ, West CML, Burnet NG, Exploiting biological and physical determinants of radiotherapy toxicity to individualize treatment. Br. J. Radiol 88, 20150172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bazalova M, Nelson G, Noll JM, Graves EE, Modality comparison for small animal radiotherapy: A simulation study. Med. Phys 41, 011710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu Y-X, Weichselbaum RR, STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G, Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Lim JYH, Gerber SA, Murphy SP, Lord EM, Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol. Immunother 63, 259–271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demaria S, Guha C, Schoenfeld J, Morris Z, Monjazeb A, Sikora A, Crittenden M, Shiao S, Khleif S, Gupta S, Formenti SC, Vikram B, Coleman CN, Ahmed MM, Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 9, e002038 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S, Fractionated but Not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti–CTLA-4 antibody. Clin. Cancer Res 15, 5379–5388 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre M-L, Gajewski TF, STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF, Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med 208, 2005–2016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu Y-X, Auh SL, The efficacy of radiotherapy relies upon induction of type I interferon–Dependent innate and adaptive immunity. Cancer Res. 71, 2488–2496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnstone CD, Lindsay P, Graves EE, Wong E, Perez JR, Poirier Y, Ben-Bouchta Y, Kanesalingam T, Chen H, Rubinstein AE, Sheng K, Bazalova-Carter M, Multi-institutional MicroCT image comparison of image-guided small animal irradiators. Phys. Med. Biol 62, 5760–5776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rafat M, Aguilera TA, Vilalta M, Bronsart LL, Soto LA, von Eyben R, Golla MA, Ahrari Y, Melemenidis S, Afghahi A, Jenkins MJ, Kurian AW, Horst KC, Giaccia AJ, Graves EE, Macrophages promote circulating tumor cell–mediated local recurrence following radiotherapy in immunosuppressed patients. Cancer Res. 78, 4241–4252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samusik N, Good Z, Spitzer MH, Davis KL, Nolan GP, Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL, The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol 10, 48–57 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Nagarsheth N, Wicha MS,Zou W, Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol 17, 559–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platanias LC, Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol 5, 375–386 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Molon B, Gri G, Bettella M, Gómez-Moutón C, Lanzavecchia A, Martínez-A C, Mañes S, Viola A, T cell costimulation by chemokine receptors. Nat. Immunol 6, 465–471 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Li F, Bai X, Li Y, Ni C, Zhao X, Zhang D, ITGA3 is associated with immune cell infiltration and serves as a favorable prognostic biomarker for breast cancer. Front. Oncol 11, 658547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gueguen P, Metoikidou C, Dupic T, Lawand M, Goudot C, Baulande S, Lameiras S, Lantz O, Girard N, Seguin-Givelet A, Lefevre M, Mora T, Walczak AM, Waterfall JJ, Amigorena S, Contribution of resident and circulating precursors to tumor-infiltrating CD8+ T cell populations in lung cancer. Sci. Immunol 6, eabd5778 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Apetoh L, Smyth MJ, Drake CG, Abastado J-P, Apte RN, Ayyoub M, Blay J-Y, Bonneville M, Butterfield LH, Caignard A, Castelli C, Cavallo F, Celis E, Chen L, Colombo MP, Comin-Anduix B, Coukos G, Dhodapkar MV, Dranoff G, Frazer IH, Fridman W-H, Gabrilovich DI, Gilboa E, Gnjatic S, Jäger D, Kalinski P, Kaufman HL, Kiessling R, Kirkwood J, Knuth A, Liblau R, Lotze MT, Lugli E, Marincola F, Melero I, Melief CJ, Mempel TR, Mittendorf EA, Odun K, Overwijk WW, Palucka AK, Parmiani G, Ribas A, Romero P, Schreiber RD, Schuler G, Srivastava PK, Tartour E, Valmori D, van der Burg SH, van der Bruggen P, van den Eynde BJ, Wang E, Zou W, Whiteside TL, Speiser DE, Pardoll DM, Restifo NP, Anderson AC, Consensus nomenclature for CD8+ T cell phenotypes in cancer. Onco. Targets. Ther 4, e998538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siddiqui I, Schaeuble K, Chennupati V, Marraco SAF, Calderon-Copete S, Ferreira DP, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Soudja SM, Ruiz AL, Marie JC, Lauvau G, Inflammatory monocytes activate memory CD8+ T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 37, 549–562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marckmann S, Wiesemann E, Hilse R, Trebst C, Stangel M, Windhagen A, Interferon-β up-regulates the expression of co-stimulatory molecules CD80, CD86 and CD40 on monocytes: Significance for treatment of multiple sclerosis. Clin. Exp. Immunol 138, 499–506 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, Parisi G, Saus CP, Torrejon DY, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Damoiseaux R, Lo RS, Ribas A, Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tötemeyer S, Sheppard M, Lloyd A, Roper D, Dowson C, Underhill D, Murray P, Maskell D, Bryant C, IFN-γ enhances production of nitric oxide from macrophages via a mechanism that depends on nucleotide oligomerization domain-2. J. Immunol 176, 4804–4810 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, Moriyama S, Iida M, Kitamura H, Qiu C-H, Watanabe T, Tanaka M, Intestinal CD169+ macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat. Commun 6, 7802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD, Murine monocyte chemoattractant protein (MCP)-5: A novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med 185, 99–110 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber C, Meiler S, Döring Y, Koch M, Drechsler M, Megens RTA, Rowinska Z, Bidzhekov K, Fecher C, Ribechini E, van Zandvoort MAMJ, Binder CJ, Jelinek I, Hristov M, Boon L, Jung S, Korn T, Lutz MB, Förster I, Zenke M, Hieronymus T, Junt T, Zernecke A, CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Invest 121, 2898–2910 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Argyle D, Kitamura T, Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol 9, 2629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steimle V, Siegrist C-A, Mottet A, Lisowska-Grospierre B, Mach B, Regulation of MHC Class II expression by interferon-γ mediated by the transactivator gene CIITA. Science 265, 106–109 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Tseng S-Y, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H, B7-Dc, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med 193, 839–846 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L, Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med 8, 793–800 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Nathan C, Nitric oxide as a secretory product of mammalian cells. FASEB J. 6, 3051–3064 (1992). [PubMed] [Google Scholar]
- 45.Vannini F, Kashfi K, Nath N, The dual role of iNOS in cancer. Redox Biol. 6, 334–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malik A, Kanneganti T, Function and regulation of IL-1α in inflammatory diseases and cancer. Immunol. Rev 281, 124–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Böttcher JP, e Sousa CR, The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 4, 784–792 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, Van Isterdael G, Toussaint W, Madeira FB, Vergote K, Agace WW, Clausen BE, Hammad H, Dalod M, Saeys Y, Lambrecht BN, Guilliams M, IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity 45, 626–640 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Durai V, Bagadia P, Granja JM, Satpathy AT, Kulkarni DH, Davidson JT, Wu R, Patel SJ, Iwata A, Liu T, Huang X, Briseño CG, Grajales-Reyes GE, Wöhner M, Tagoh H, Kee BL, Newberry RD, Busslinger M, Chang HY, Murphy TL, Murphy KM, Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol 20, 1161–1173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filatenkov A, Baker J, Mueller AMS, Kenkel J, Ahn G-O, Dutt S, Zhang N, Kohrt H, Jensen K, Dejbakhsh-Jones S, Shizuru JA, Negrin RN, Engleman EG, Strober S, Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin. Cancer Res 21, 3727–3739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, Bhutkar A, Wittrup KD, Spranger S, Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 55, 308–323.e9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, Gudjonsson S, Håkansson U, Reizis B, Kotarsky K, Agace WW, IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38, 958–969 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Affandi AJ, Olesek K, Grabowska J, Twilhaar MKN, Rodríguez E, Saris A, Zwart ES, Nossent EJ, Kalay H, de Kok M, Kazemier G, Stöckl J, van den Eertwegh AJM, de Gruijl TD, Garcia-Vallejo JJ, Storm G, van Kooyk Y, den Haan JMM, CD169 defines activated CD14+ monocytes with enhanced CD8+ T cell activation capacity. Front. Immunol 12, 697840 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG, Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6, 470–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, Guermonprez P, Nussenzweig MC, Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J. Exp. Med 210, 2025–2039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, Shin A, Huang W, Dress RJ, Dutertre C-A, Schlitzer A, Chen J, Ng LG, Wang H, Liu Z, Su B, Ginhoux F, Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell 178, 1509–1525.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, Kool R, Bourdeau F, Brimo F, Spicer J, Kassouf W, Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat. Commun 12, 2776 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X-Y, Chen W, Wei B, Shan Y-F, Wang C, IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol. Baltim 187, 2559–2568 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Patil G, Li S, Tripartite motif proteins: An emerging antiviral protein family. Future Virol. 14, 107–122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe T, Asano N, Kitani A, Fuss IJ, Chiba T, Strober W, Activation of type I IFN signaling by NOD1 mediates mucosal host defense against Helicobacter pylori infection. Gut Microbes 2, 61–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woo S-R, Corrales L, Gajewski TF, The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 36, 250–256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S, DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8, 15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Bon AL, Melero I, Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res 17, 2619–2627 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Lee AJ, Chen B, Chew MV, Barra NG, Shenouda MM, Nham T, van Rooijen N, Jordana M, Mossman KL, Schreiber RD, Mack M, Ashkar AA, Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med 214, 1153–1167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tosi D, Valenti R, Cova A, Sovena G, Huber V, Pilla L, Arienti F, Belardelli F, Parmiani G, Rivoltini L, Role of cross-talk between IFN-α-induced monocyte-derived dendritic cells and NK cells in priming CD8+ T cell responses against human tumor antigens. J. Immunol 172, 5363–5370 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Johnson CL, Green DS, Zoon KC, Human monocytes in the presence of interferons Alpha2a and Gamma are potent killers of serous ovarian cancer cell lines in combination with paclitaxel and carboplatin. J. Interferon Cytokine Res 35, 55–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabriele L, Borghi P, Rozera C, Sestili P, Andreotti M, Guarini A, Montefusco E, Foà R, Belardelli F, IFN-α promotes the rapid differentiation of monocytes from patients with chronic myeloid leukemia into activated dendritic cells tuned to undergo full maturation after LPS treatment. Blood 103, 980–987 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Kwart D, He J, Srivatsan S, Lett C, Golubov J, Oswald EM, Poon P, Ye X, Waite J, Zaretsky AG, Haxhinasto S, Au-Yeung E, Gupta NT, Chiu J, Adler C, Cherravuru S, Malahias E, Negron N, Lanza K, Coppola A, Ni M, Song H, Wei Y, Atwal GS, Macdonald L, Oristian NS, Poueymirou W, Jankovic V, Fury M, Lowy I, Murphy AJ, Sleeman MA, Wang B, Skokos D, Cancer cell-derived type I interferons instruct tumor monocyte polarization. Cell Rep. 41, 111769 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Muldoon JJ, Chuang Y, Bagheri N, Leonard JN, Macrophages employ quorum licensing to regulate collective activation. Nat. Commun 11, 878 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Postat J, Bousso P, Quorum sensing by monocyte-derived populations. Front. Immunol 10, 2140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wimmers F, Subedi N, van Buuringen N, Heister D, Vivié J, Beeren-Reinieren I, Woestenenk R, Dolstra H, Piruska A, Jacobs JFM, van Oudenaarden A, Figdor CG, Huck WTS, de Vries IJM, Tel J, Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nat. Commun 9, 3317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mondini M, Loyher P-L, Hamon P, de Thoré MG, Laviron M, Berthelot K, Clémenson C, Salomon BL, Combadière C, Deutsch E, Boissonnas A, CCR2-dependent recruitment of Tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol. Res 7, 376–387 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Ranoa DR, Parekh A, Pitroda S, Huang X, Darga T, Wong A, Huang L, Andrade J, Staley J, Satoh T, Akira S, Weichselbaum R, Khodarev N, Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7, 26496–26515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, Zhang Y, Yu X, Xu M, Mauceri H, Arina A, Weichselbaum RR, Fu Y-X, Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 49, 490–503.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hsieh RC-E, Krishnan S, Wu R-C, Boda AR, Liu A, Winkler M, Hsu W-H, Lin SH, Hung M-C, Chan L-C, Bhanu KR, Srinivasamani A, Azevedo RAD, Chou Y-C, DePinho RA, Gubin M, Vilar E, Chen CH, Slay R, Jayaprakash P, Hegde SM, Hartley G, Lea ST, Prasad R, Morrow B, Couillault CA, Steiner M, Wang C-C, Venkatesulu BP, Taniguchi C, Kim YSB, Chen J, Rudqvist N-P, Curran MA, ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci. Immunol 7, eabl9330 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li L, Yin Q, Kuss P, Maliga Z, Millán JL, Wu H, Mitchison TJ, Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol 10, 1043–1048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yum S, Li M, Chen ZJ, Old dogs, new trick: Classic cancer therapies activate cGAS. Cell Res. 30, 639–648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cytlak UM, Dyer DP, Honeychurch J, Williams KJ, Travis MA, Illidge TM, Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat. Rev. Immunol 22, 124–138 (2022). [DOI] [PubMed] [Google Scholar]
- 79.Halle M, Gabrielsen A, Paulsson-Berne G, Gahm C, Agardh HE, Farnebo F, Tornvall P, Sustained inflammation due to nuclear factor-Kappa B activation in irradiated human arteries. J. Am. Coll. Cardiol 55, 1227–1236 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Weintraub NL, Jones WK, Manka D, Understanding radiation-induced vascular disease. J. Am. Coll. Cardiol 55, 1237–1239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weber DC, Wang H, Bouchardy C, Rosset A, Rapiti E, Schmidlin F, Rouzaud M, Miralbell R, Estimated dose to the rectum and colon in prostate cancer patients treated with exclusive radiation therapy presenting a secondary colorectal malignancy. Clinicaloncology 21, 687–694 (2009). [DOI] [PubMed] [Google Scholar]
- 82.Hoyer M, Roed H, Hansen AT, Ohlhuis L, Petersen J, Nellemann H, Berthelsen AK, Grau C, Engelholm SA, Maase HVD, Phase II study on stereotactic body radiotherapy of colorectal metastases. Acta Oncol. 45, 823–830 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Lin B, Gao F, Yang Y, Wu D, Zhang Y, Feng G, Dai T, Du X, FLASH radiotherapy: History and future. Front. Oncol 11, 644400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cord UC, Hans DG, Comparative PCR analysis for detection of mycoplasma infections in continuous cell lines. In Vitro Cell. Dev. Biol. Anim 38, 79–85 (2002). [DOI] [PubMed] [Google Scholar]
- 85.Ewens A, Mihich E, Ehrke MJ, Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res 25, 3905–3915 (2005). [PubMed] [Google Scholar]
- 86.Leylek R, Alcántara-Hernández M, Lanzar Z, Lüdtke A, Perez OA, Reizis B, Idoyaga J Integrated cross-species analysis identifies a conserved transitional dendritic cell population. Cell Rep. 29, 3736–3750.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Sultonova M, Blackmore B, Du R, Philips O, Paulo JA, Murphy JP, Integrated changes in thermal stability and proteome abundance during altered nutrient states in Escherichia coli and human cells. Proteomics 22, e2100254 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma C-M, Coffey CW, DeWerd LA, Liu C, Nath R, Seltzer SM, Seuntjens JP, AAPM protocol for 40–300 kV x-ray beam dosimetry in radiotherapy and radiobiology. Med. Phys 28, 868–893 (2001). [DOI] [PubMed] [Google Scholar]
- 90.Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, Seeley ES, Winer DA, Reticker-Flynn NE, Engleman EG, An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res. 77, 4158–4170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI, MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NanoString data generated in this study are deposited in NCBI GEO (GSE207105 and GSE223400). Mass spectrometry proteomics data have been deposited to MASSIVE and the ProteomeXChange Consortium via the PRIDE repository with the dataset identifier PXD035046. This study did not generate a unique code. The MC38ΔSTING cells are available upon request to J.I. Ms4a3Cre mice are available from F.G. under a material transfer agreement with the Singapore Immunology Network (SIgN), A*STAR Research Entities.