

Summary
Classification systems reflect our technical abilities in the investigation of tumors and our current theories on tumor development. Herein, by providing a historical perspective on the evolution of classifying renal tumors, we assess the current WHO classification highlighting the novelties and the implications of these changes in daily clinical practice.
Key words: classification, WHO, renal cell carcinoma, molecular, clinical relevance, therapy, management
Historical perspective
The classification of renal cell tumors has changed in the last five decades. The previous classifications have been based on various clinical-pathological findings, including cytological (clear cell and chromophobe renal cell carcinoma) or architectural (papillary renal cell carcinoma) characteristics, tumor location (collecting duct and renal medullary carcinomas), correlations with underlying renal disease (acquired cystic disease-associated renal cell carcinoma), the similarity of tumors to embryological structures (metanephric adenoma), or a specific hereditary background (hereditary leiomyomatosis and renal cell carcinoma syndrome associated renal cell carcinoma) 1-3 (Fig. 1).
Figure 1.
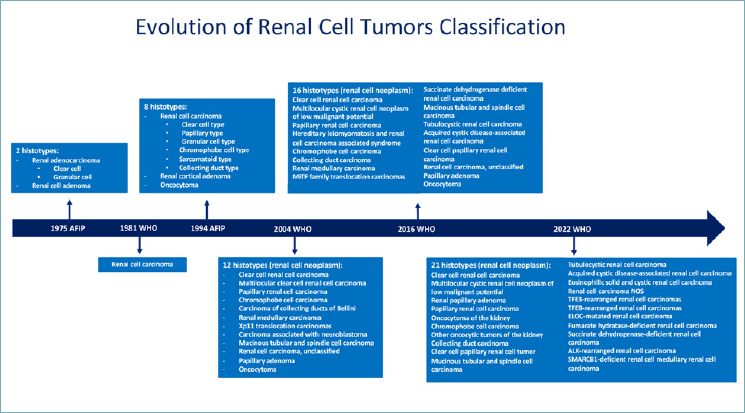
Flow diagram showing the evolution of the classification of renal cell tumor, with progressive implementation of newly recognized entities in the different editions of the AFIP and WHO blue books.
While the two most important categories of renal tumors in adults were only carcinoma and angiomyolipoma acknowledged in the second series of Armed Forces Institute of Pathology fascicle (AFIP) published in 1975 4, the modern era of renal tumor classification started in 1996 with a consensus of an expert group held in Heidelberg 5. It was the first time that genetic alterations were considered with morphological criteria and that the sarcomatoid features were recognized as dedifferentiation of all types of renal cell carcinoma with prognostic implication rather than a histotype per se. Moreover, the introduction in this consensus of unclassified renal cell carcinoma as a diagnostic category has afterward led to the recognition of specific entities initially belonging to it. In fact, the result of this process was evident in the 2004 World Health Organization (WHO) 1 classification in which several important different tumors were introduced such as mucinous tubular and spindle renal cell carcinoma, medullary renal cell carcinoma, epithelioid angiomyolipoma and, for the first time, a tumor defined based on a specific molecular alteration: Xp11 translocation renal cell carcinoma.
The recognition of peculiar tumors in end-stage renal disease is one of the main novelties in the 2016 WHO classification 2. While acquired cystic disease renal cell carcinoma arose only in this clinical setting 6, clear cell papillary renal cell carcinoma has been observed in sporadic scenarios 7 and became the fourth kind of carcinoma occurring in the kidney 8. On the other hand, new molecular-driven histotypes were introduced such as MiTF family translocation carcinoma, succinate dehydrogenase (SDH)-deficient renal cell carcinoma, and hereditary leiomyomatosis and renal cell carcinoma syndrome associated renal cell carcinoma 2. The relevance of molecular data for the improvement of the kidney tumor classification has been confirmed in the last WHO classification in which a category of molecularly defined renal cell carcinoma has been introduced, including TFE3-rearranged renal cell carcinoma, TFEB-rearranged, and TFEB-amplified renal cell carcinoma, FH-deficient renal cell carcinoma, SDH-deficient renal cell carcinoma, ALK-rearranged renal cell carcinoma, ELOC (formerly TCEB1)-mutated renal cell carcinoma and SMARCB1 (INI1)-deficient renal cell carcinoma 3. Finally, recent studies have described several renal tumors with oncocytic features and mTOR gene pathway alterations. Among those, eosinophilic solid and cystic renal cell carcinoma is the only one nowadays recognized as a nosographic entity 9.
Since the qualities of a good classification system are based on clinical relevance, histopathological clues to guide the differential diagnosis, inter- and intraobserver reliability, and ultimately feasibility of the diagnosis in different worldwide laboratories 10, the question is: what is relevant in the WHO 2022 classification of renal tumors?
We are aware that expanding a classification is important from a biological point of view for academic advancement; however, in daily routine practice, the relevance mainly concerns the following aspects: prognosis, clinical management of patients and their families, and therapy.
Changes relevant in prognosis
CLEAR CELL PAPILLARY RENAL CELL TUMOR
Firstly recognized in end-stage kidney disease 6, clear cell papillary renal cell carcinoma was later described in the sporadic setting as well 7. After the identification of this entity in the sporadic scenario, it was initially chosen to designate this neoplasm as a carcinoma for several reasons: 1) in 2006, Tickoo and coauthors named the tumor as carcinoma in end-stage kidney disease; 2) two years later, when it was observed in the sporadic setting, the data regarding of follow-up and knowledge of biology underling were limited; 3) the distinction with low-grade clear cell renal cell carcinoma was subtle, especially when a tubular pattern was observed 11. However, the absence of VHL mutation and 3p loss by gene expression profiling analysis in those tumors supported their distinction from low-grade clear cell renal cell carcinoma 12,13.
Grossly, these neoplasms are usually small well-circumscribed encapsulated masses sometimes with cystic changes. Conversely to clear cell renal cell carcinoma, when the cystic changes are prominent the color is not yellow but grayish and translucent. Morphologically, the tumor is made up of clear cells arranged in a variable mixture of cystic, branched tubular, solid, and papillary components (Fig. 2). Characteristically, the nuclei are oriented towards the lumen of the tubules and papillae. The immunohistochemical phenotype, characterized by diffuse cytokeratin 7 staining, “cup-shaped” expression of carbonic anhydrase 9, GATA3 immunolabelling, and negativity for AMACR and CD10, is distinctive and helpful to properly identify this tumor, even in biopsy samples 6,7,14,15.
Figure 2.
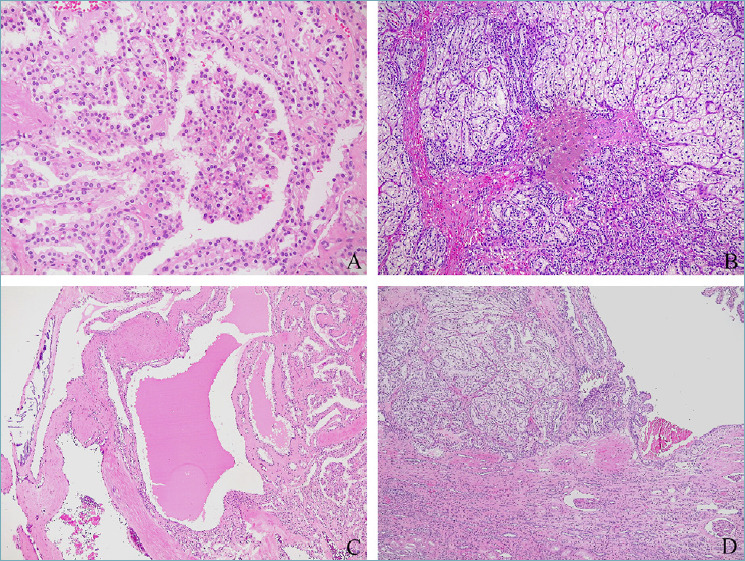
Several possible growth pattern of clear cell papillary renal cell tumor: papillary (A), tubular (B), cystic (C), tubular and cystic (D).
Based on the indolent behavior without, to date, evidence of any recurrent or metastatic case, it was decided to change the name of this neoplasm to tumor rather than carcinoma3.
TFE3 AND TFEB-REARRANGED RENAL CELL CARCINOMA
Previously known as MiT family translocation renal cell carcinomas, TFE3-rearranged and TFEB-rearranged renal cell carcinomas are separate entities in the current classification. Despite both being initially recognized in childhood 16,17, they can occur in adults as well 18. These neoplasms harbor molecular rearrangements involving different genes including the melanocytic inducing transcription factor (MITF), TFE3 gene, and TFEB gene, respectively. Although a spectrum of morphological features has been reported in both entities (Fig. 3), TFE3-rearranged renal cell carcinoma is more commonly a papillary tumor with epithelioid clear cells and psammoma bodies 18, whereas TFEB-rearranged renal cell carcinoma is usually characterized by a distinctive biphasic appearance made up of larger epithelioid cells and smaller cells clustered around eosinophilic spheres formed by basement membrane material 19. By immunohistochemistry, both tumors generally underexpress epithelial markers,TFE3-rearranged renal cell carcinoma stains for cathepsin K in roughly half of the cases, and occasionally for melanogenesis markers (Melan-A and HMB45) 20. On the other hand, these latter markers and cathepsin K are constantly positive in TFEB-rearranged renal cell carcinoma 21-23. In either TFE3 or TFEB-rearranged renal cell carcinoma, the identification of gene rearrangement by FISH assays or RNA-sequencing 24,25 is considered the gold standard to confirm diagnosis. Concerning clinical behavior, TFE3-rearranged renal cell carcinomas are aggressive in up to 50% of cases, while TFEB-rearranged renal cell carcinoma often displays an indolent clinical course. Furthermore, among renal cell carcinoma with TFEB gene alteration, TFEB-amplified renal cell carcinoma is a high-grade renal cell carcinoma characterized by an aggressive clinical course 26-28.
Figure 3.
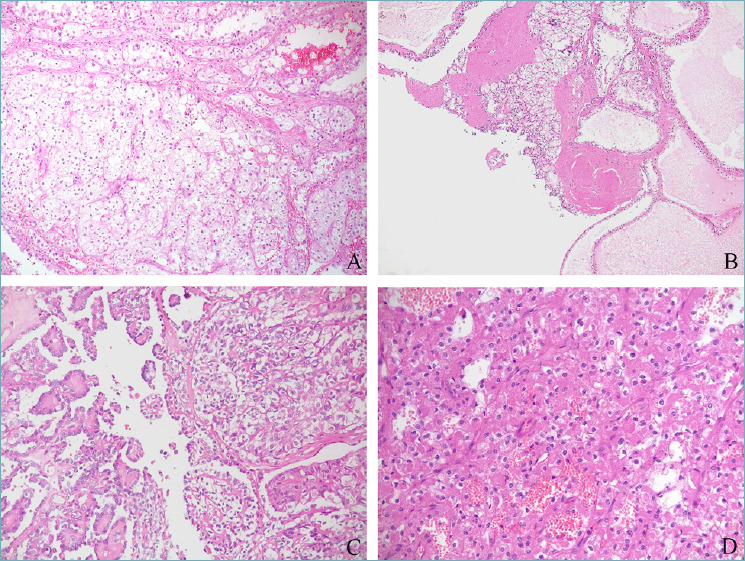
Various challenging morphological appearances of TFE3 and TFEB-rearranged renal cell carcinoma, respectively mimicking clear cell renal cell carcinoma (A), clear cell papillary renal tumor (B), papillary renal cell carcinoma (C), and renal oncocytoma (D).
In conclusion, due to different translocation genes involved, different morphology, and most important different behavior, TFE3-rearranged and TFEB-rearranged renal cell carcinomas are currently considered separate entities.
Changes relevant for clinical management of patients and their family
SDH DEFICIENT RENAL CELL CARCINOMA
Succinate dehydrogenase (SDH)-deficient renal cell carcinoma is a rare neoplasm characterized by a favorable prognosis 29. Germline mutations of any one of the SDH genes are found in the majority of the patients harboring those tumors. Conversely to the paradigm of hereditary tumors, which are usually multiple and bilateral masses where the radiologist is the first to suggest the diagnosis, SDH-deficient renal cell carcinoma is more commonly a solitary tumor and, for this reason, the pathologist could be the first to suspect a hereditary form. Histologically, the tumor is composed of low-grade eosinophilic cells arranged in a nested growth pattern with frequent eosinophilic flocculent cytoplasmic inclusions (Fig. 4a). Loss of immunohistochemical label of SDH, as a surrogate of SDH gene mutation, has been demonstrated to be a useful stain to screen for SDH deficiency 30. Patients with a germline mutation can also develop other tumors such as pituitary adenoma, gastrointestinal stromal tumor (GIST), and paraganglioma/pheochromocytoma 31. Therefore, the role of pathologists to properly recognize these tumors is crucial for patients and their relatives to encourage genetic counseling. Because of the indolent behavior, rather than looking for metastasis the follow-up of this condition mainly deals with early identification of the other neoplastic conditions characterizing the syndrome. To note, SDH-deficient renal cell carcinoma may be misinterpreted with low-grade FH-deficient renal cell carcinoma, representing a challenging differential diagnosis.
Figure 4.
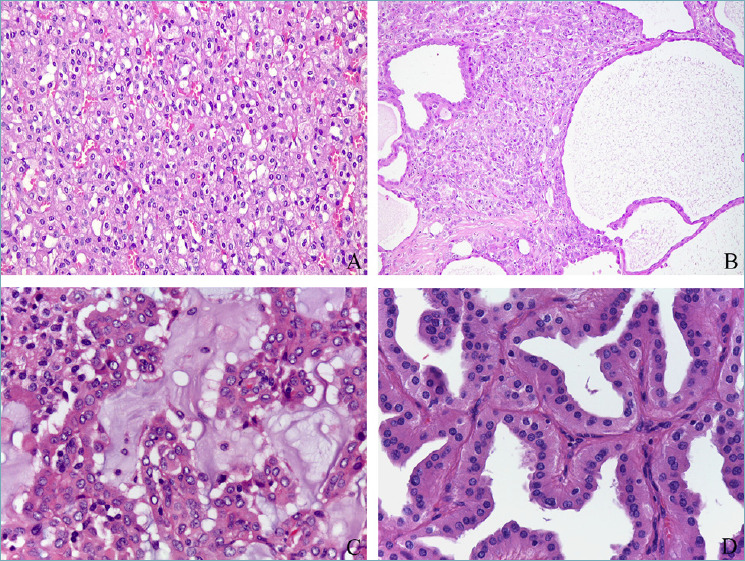
Molecularly defined renal cell tumors: succinate dehydrogenase deficient renal cell carcinoma (A), fumarate hydratase deficient renal cell carcinoma (B), ALK-rearranged renal cell carcinoma (C), papillary neoplasm with inverted polarity (D).
FH DEFICIENT RENAL CELL CARCINOMA
Fumarate hydratase (FH) - deficient renal cell carcinomas are highly aggressive early metastasizing tumors carrying mutations, mainly germline, in the FH gene located at chromosome 1q42. In the beginning, in the 2004 WHO classification 1 they were included among papillary renal cell carcinoma type 2. They were then recognized as a distinctive entity in the 2016 WHO classification where they were designated as hereditary leiomyomatosis and renal cell carcinoma syndrome associated renal cell carcinoma 2, highlighting the hereditary setting. The decision to change the denomination in the current WHO classification is the result of the awareness that roughly 20% of these tumors are sporadic 32. As SDH-deficient renal cell carcinoma, loss of FH protein expression by immunohistochemistry allows the pathologist to identify this entity in the majority of cases.
Microscopically, FH-deficient renal cell carcinoma was originally described as a tumor characterized by a papillary architecture. However, actually, multiple admixed architectural patterns (papillary, solid, tubulocystic, and cystic) have been observed (Fig. 4b). Voluminous eosinophilic cells with high-grade nuclear and nucleolar features are characteristic features. Nevertheless, recently, low-grade FH-deficient renal cell carcinoma cases have been reported 33. Those tumors are composed of cytologically low-grade cells with homogeneous architectural patterns mimicking SDH-deficient renal cell carcinoma, oncocytoma, and, in general, low-grade oncocytic tumors. Conversely to FH-deficient renal cell carcinoma characterized by an aggressive outcome, the low-grade patterns show a more favorable prognosis.
As SDH-deficient renal cell carcinoma, FH-deficient renal cell carcinoma is usually a single solitary mass in the kidney even in the hereditary setting. In these latter cases, cutaneous and uterine leiomyomas are often associated with renal tumors and the role of pathologists to properly recognize them is crucial to encourage genetic counseling of patients and their relatives.
Generally speaking the recommendations for genetic counseling according to the American Urological Association Guidelines are testing all patients ≤ 46 years of age with renal cancer, those with multifocal or bilateral renal masses, or whenever: 1) the personal or family history suggests a familial renal cancer syndrome; 2) there is a first or second-degree relative with a history of renal cell carcinoma (even if a kidney cancer has not been observed); or 3) whenever pathological examination demonstrates histology suggestive for such a syndrome. (https://www.auanet.org//membership/publications-overview/auanews/all-articles/2022/may-2022/aua-guidelines-renal-mass-and-localized-renal-cancer-evaluation-management-and-followup).
Changes relevant for therapy
ALK-REARRANGED RCC
Anaplastic lymphoma kinase (ALK) - rearranged renal cell carcinoma is a tumor harboring chromosomal translocations of the ALK gene located at chromosome 2p23 9,34. It is a rare tumor, as roughly 40 cases have been reported so far without any gender predominance, in both children and adults. Numerous gene fusion partners have been reported: when vinculin (VCL)-ALK gene fusion occurs, they affect young patients with sickle cell trait, and show distinctive morphology. For this reason, this tumor has been proposed as the “eighth sickle cell nephropathy.” Since several morphologies can be observed in this type of tumor, the definitive diagnosis requires the demonstration of ALK gene rearrangement by FISH or immunohistochemistry. However, mucinous background, intracytoplasmic mucin, and myxoid changes have been reported in a subset of cases and can be helpful clues in recognizing these rare tumors (Fig. 4c).
Follow-up of the reported cases is limited to date; however, as 30% of patients demonstrated an aggressive clinical course, they may benefit from targeted ALK inhibitors as a potentially effective treatment 35.
Changes that are biologically relevant but less relevant for prognosis, therapy, and clinical management
PAPILLARY RENAL CELL CARCINOMA
Papillary renal cell carcinoma is the second most common neoplasm arising in the kidney. It is a molecularly heterogeneous entity, ranging from low-grade to high-grade tumors. Traditionally regarded as type 1 and type 2 papillary renal cell carcinoma 36, the former is a quite uniform subgroup on both morphological features and molecular findings (MET gene alterations). On the other hand, the latter is characterized by different genetic alterations so that it has been hypothesized that different tumor entities were included in this initially designed category. For this reason, the diagnostic criteria for type 2 papillary renal cell carcinoma need to be re-evaluated and the subclassification into type 1 and type 2 is no longer recommended 37.
PAPILLARY NEOPLASM WITH INVERTED POLARITY
Papillary neoplasm with inverted polarity is a proposed entity in the new WHO classification currently included as a subtype of papillary renal cell carcinoma with pathogenic mutations of the KRAS gene 38. It is characterized by papillary or tubulopapillary structures covered by a single layer of eosinophilic cells with finely granular cytoplasm and apically located round nuclei with inconspicuous nucleoli 39 (Fig. 4d). Immunohistochemically the expression of GATA3 and, variably, of AMACR suggest that these neoplasms show a differentiation toward distal nephron rather than the proximal tubules 40. The low-grade nuclear feature is reflected in the indolent clinical behavior.
ONCOCYTIC TUMORS
We are aware that the current classification of oncocytic tumors is complex, difficult either for clinicians or pathologists to adopt.
Among these neoplasms, eosinophilic solid and cystic renal cell carcinoma has been introduced in the current WHO classification as a new entity 3. As the name states, it is made up by cells with eosinophilic cytoplasm arranged in a solid and cystic architecture 41 (Fig. 5a). Focal or diffuse immunostaining for cytokeratin 20 and cathepsin K is a characteristic feature 42. Although biallelic losses or mutations in the TSC1/TSC2 genes have been identified in the majority of cases, only a few are associated with tuberous sclerosis 43. This molecular data was interesting one since it was the first renal epithelial tumor identified to harbor mTOR gene pathway alteration 44. Moreover, this finding may open new possible therapeutic approaches (mTOR inhibitors), even though only few metastatic cases have been recorded so far.
Figure 5.
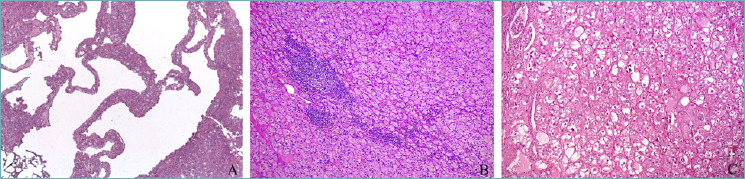
Oncocytic renal tumors: eosinophilic solid and cystic renal cell carcinoma (A), low-grade oncocytic tumor (LOT) (B), eosinophilic vacuolated tumor (EVT) (C).
Other tumors with eosinophilic cells and somatic inactivating mutations of TSC2 gene or activating mutations of mTOR as the primary molecular alteration have been subsequently described. Low-grade oncocytic tumor (LOT) 45,46 and eosinophilic vacuolated tumor (EVT) 47-49 share eosinophilic cells with regular nuclear membranes mainly arranged in an alveolar pattern (Figs. 5b, 5c). While the the former is characterized by few nuclear features, the latter typically display nucleolar prominence. Both neoplasms are usually small and solitary, arising in a sporadic setting with an indolent clinical behavior. These tumors show morphological, molecular, and clinical overlaps and, albeit the distinction is biologically relevant, in the daily routine practice their accurate distinction does not seem to imply any different management. Therefore, in the current WHO classification they are designated as “oncocytic renal neoplasms of low malignant potential NOS”.
Conclusions
In conclusion, the new WHO classification reveals an increasing complexity. The changes and novel implications include: i) distinguishing clear cell papillary renal cell tumor from clear cell renal cell carcinoma due to the indolent behavior of the former; ii) identifying FH-deficient and SDH-deficient renal cell carcinoma for the hereditary implications; iii) recognizing ALK-rearranged renal cell carcinoma for possible targeted therapy.
Finally, the low-grade eosinophilic neoplasia group includes several biologically different tumors but so far sharing the same indolent clinical outcome. On the other hand, the academic position should avoid “lumping” category for better comprehension of oncocytic renal tumors using the descriptive term oncocytic renal neoplasm of low malignant potential EVT type or LOT type.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-to- profit sectors.
AUTHORS’ CONTRIBUTION
Conceptualization: AC and GM. Methodology: AC and GM. Formal analysis and investigation: AC and SM. Writing/original draft preparation: AC and GM. Writing/review and editing: SM and MB. Supervision: GM.
Figures and tables
References
- 1.Eble JN, Sauter G, Epstein JI, et al. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon (France): IARC Press; 2004. [Google Scholar]
- 2.Moch H, Humphrey PA, Ulbright TM, Reuter VE. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon (France): IARC Press; 2016. [DOI] [PubMed] [Google Scholar]
- 3.WHO Classification of Tumours, Urinary and Male Genital Tumours. Lyon (France): IARC Press; 2022. [Google Scholar]
- 4.Bennington JBB. Tumors of the kidney, renal pelvis, and ureter. Atlas of Tumor Pathology. Vol. 12. Washington: Armed Forces Institute of Pathology; 1975. [Google Scholar]
- 5.Kovacs G, Akhtar M, Beckwith BJ, et al. The Heidelberg classification of renal cell tumours. J Pathol 1997;183:131-133. https://doi.org/10.1002/(SICI)1096-9896(199710)183:2<131::AID-PATH931>3.0.CO;2-G [DOI] [PubMed] [Google Scholar]
- 6.Tickoo SK, DePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006;30:141-153. https://doi.org/10.1097/01.pas.0000185382.80844.b1 10.1097/01.pas.0000185382.80844.b1 [DOI] [PubMed] [Google Scholar]
- 7.Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239-1245. https://doi.org/10.1097/PAS.0b013e318164bcbb 10.1097/PAS.0b013e318164bcbb [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Zheng S, Truong LD, et al. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum Pathol 2014;45:59-64. https://doi.org/10.1016/j.humpath.2013.08.004 10.1016/j.humpath.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 9.Trpkov K, Williamson SR, Gill AJ, et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod Pathol 2021;34:1167-1184. https://doi.org/10.1038/s41379-021-00737-6 10.1038/s41379-021-00737-6 [DOI] [PubMed] [Google Scholar]
- 10.Eble JN. Contributions of genetics to the evolution of the diagnostic classification of renal cell neoplasia: a personal perspective. Pathology 2021;53:96-100. https://doi.org/10.1016/j.pathol.2020.10.004 10.1016/j.pathol.2020.10.004 [DOI] [PubMed] [Google Scholar]
- 11.Massari F, Ciccarese C, Hes O, et al. The Tumor Entity Denominated ‘clear cell-papillary renal cell carcinoma’ According to the WHO 2016 new Classification, have the Clinical Characters of a Renal Cell Adenoma as does Harbor a Benign Outcome. Pathol Oncol Res 2018;24:447-456. https://doi.org/10.1007/s12253-017-0271-x 10.1007/s12253-017-0271-x [DOI] [PubMed] [Google Scholar]
- 12.Williamson SR, Eble JN, Cheng L, et al. Re: Brannon A. Rose, Haake Scott M., Hacker Kathryn E., et al. Meta-analysis of clear cell renal cell carcinoma gene expression defines a variant subgroup and identifies gender influences on tumor biology. Eur Urol 2012;62:e81-82. https://doi.org/10.1016/j.eururo.2012.06.056 10.1016/j.eururo.2012.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brannon AR, Haake SM, Hacker KE, et al. Meta-analysis of clear cell renal cell carcinoma gene expression defines a variant subgroup and identifies gender influences on tumor biology. Eur Urol 2012;61:258-268. https://doi.org/10.1016/j.eururo.2011.10.007 10.1016/j.eururo.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantilla JG, Antic T, Tretiakova M. GATA3 as a valuable marker to distinguish clear cell papillary renal cell carcinomas from morphologic mimics. Hum Pathol 2017;66:152-158. https://doi.org/10.1016/j.humpath.2017.06.016 10.1016/j.humpath.2017.06.016 [DOI] [PubMed] [Google Scholar]
- 15.Martignoni G, Brunelli M, Segala D, et al. Validation of 34betaE12 immunoexpression in clear cell papillary renal cell carcinoma as a sensitive biomarker. Pathology 2017;49:10-18. https://doi.org/10.1016/j.pathol.2016.05.014 10.1016/j.pathol.2016.05.014 [DOI] [PubMed] [Google Scholar]
- 16.Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol 2001;159:179-192. https://doi.org/10.1016/S0002-9440(10)61684-7 10.1016/S0002-9440(10)61684-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001;158:2089-2096. https://doi.org/10.1016/S0002-9440(10)64680-9 10.1016/S0002-9440(10)64680-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caliò A, Segala D, Munari E, et al. MiT family translocation renal cell carcinoma: From the early descriptions to the current knowledge. Cancers (Basel) 2019;11:1-12. https://doi.org/10.3390/cancers11081110 10.3390/cancers11081110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caliò A, Brunelli M, Segala D, et al. t(6;11) renal cell carcinoma: a study of seven cases including two with aggressive behavior, and utility of CD68 (PG-M1) in the differential diagnosis with pure epithelioid PEComa/epithelioid angiomyolipoma. Mod Pathol 2018;31:474-487. doi. 10.1038/modpathol.2017.144 10.1038/modpathol.2017.144 [DOI] [PubMed] [Google Scholar]
- 20.Caliò A, Marletta S, Brunelli M, et al. TFE3 and TFEB-rearranged renal cell carcinomas: an immunohistochemical panel to differentiate from common renal cell neoplasms. Virchows Arch 2022. https://doi.org/10.1007/s00428-022-03380-x 10.1007/s00428-022-03380-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martignoni G, Pea M, Gobbo S, et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol 2009;22:1016-1022. https://doi.org/10.1038/modpathol.2009.58 10.1038/modpathol.2009.58 [DOI] [PubMed] [Google Scholar]
- 22.Martignoni G, Gobbo S, Camparo P, et al. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod Pathol 2011;24:1313-1319. https://doi.org/10.1038/modpathol.2011.93 10.1038/modpathol.2011.93 [DOI] [PubMed] [Google Scholar]
- 23.Caliò A, Brunelli M, Gobbo S, et al. Cathepsin K: A Novel Diagnostic and Predictive Biomarker for Renal Tumors. Cancers (Basel) 2021;13. https://doi.org/10.3390/cancers13102441 10.3390/cancers13102441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caliò A, Harada S, Brunelli M, et al. TFEB rearranged renal cell carcinoma. A clinicopathologic and molecular study of 13 cases. Tumors harboring MALAT1-TFEB, ACTB-TFEB, and the novel NEAT1-TFEB translocations constantly express PDL1. Mod Pathol 2021;34:842-850. https://doi.org/10.1038/s41379-020-00713-6 10.1038/s41379-020-00713-6 [DOI] [PubMed] [Google Scholar]
- 25.Harada S, Caliò A, Janowski KM, et al. Diagnostic utility of one-stop fusion gene panel to detect TFE3/TFEB gene rearrangement and amplification in renal cell carcinomas. Mod Pathol 2021;34:2055-2063. https://doi.org/10.1038/s41379-021-00858-y 10.1038/s41379-021-00858-y [DOI] [PubMed] [Google Scholar]
- 26.Caliò A, Brunelli M, Segala D, et al. VEGFA amplification/increased gene copy number and VEGFA mRNA expression in renal cell carcinoma with TFEB gene alterations. Mod Pathol 2019;32;258-268. https://doi.org/10.1038/s41379-018-0128-1 10.1038/s41379-018-0128-1 [DOI] [PubMed] [Google Scholar]
- 27.Argani P. MiT family translocation renal cell carcinoma. Semin Diagn Pathol 2015;32:103-113. https://doi.org/10.1053/j.semdp.2015.02.003 10.1053/j.semdp.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 28.Argani P, Reuter VE, Zhang L, et al. TFEB-amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am J Surg Pathol 2016;40:1484-1495. https://doi.org/10.1097/PAS.0000000000000720 10.1097/PAS.0000000000000720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gill AJ, Patcher NS, Chou A, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol 2011;35;1578-1585. https://doi.org/10.1097/PAS.0b013e318227e7f4 10.1097/PAS.0b013e318227e7f4 [DOI] [PubMed] [Google Scholar]
- 30.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 2011;96:E1472-6. https://doi.org/10.1210/jc.2011-1043 10.1210/jc.2011-1043 [DOI] [PubMed] [Google Scholar]
- 31.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018;72;106-116. https://doi.org/10.1111/his.13277 10.1111/his.13277 [DOI] [PubMed] [Google Scholar]
- 32.Gleeson JP, Nikolovski I, Dinatale R, et al. Comprehensive Molecular Characterization and Response to Therapy in Fumarate Hydratase-Deficient Renal Cell Carcinoma. Clin Cancer Res 2021;27:2910-2919. https://doi.org/10.1158/1078-0432.CCR-20-4367 10.1158/1078-0432.CCR-20-4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith SC, Siroshi D, Ohe C, et al. A distinctive, low-grade oncocytic fumarate hydratase-deficient renal cell carcinoma, morphologically reminiscent of succinate dehydrogenase-deficient renal cell carcinoma. Histopathology 2017;71;42-52. https://doi.org/10.1111/his.13183 10.1111/his.13183 [DOI] [PubMed] [Google Scholar]
- 34.Argani P. Translocation carcinomas of the kidney. Genes Chromosomes Cancer 2022;61:219-227. https://doi.org/10.1002/gcc.23007 10.1002/gcc.23007 [DOI] [PubMed] [Google Scholar]
- 35.Pal SK, Bergerot P, Dizman N, et al. Responses to Alectinib in ALK-rearranged Papillary Renal Cell Carcinoma. Eur Urol 2018;74:124-128. https://doi.org/10.1016/j.eururo.2018.03.032 10.1016/j.eururo.2018.03.032 [DOI] [PubMed] [Google Scholar]
- 36.Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997;10:537-544. [PubMed] [Google Scholar]
- 37.Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med 2016;347:135-145. https://doi.org/10.1056/NEJMoa1505917 10.1056/NEJMoa1505917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SS, Cho YM, Kim GH, et al. Recurrent KRAS mutations identified in papillary renal neoplasm with reverse polarity-a comparative study with papillary renal cell carcinoma. Mod Pathol 202;33:690-699. https://doi.org/10.1038/s41379-019-0420-8 10.1038/s41379-019-0420-8 [DOI] [PubMed] [Google Scholar]
- 39.Chang HY, Hang JF, Wu CY, et al. Clinicopathological and molecular characterisation of papillary renal neoplasm with reverse polarity and its renal papillary adenoma analogue. Histopathology 2021;78:1019-1031. https://doi.org/10.1111/his.14320 10.1111/his.14320 [DOI] [PubMed] [Google Scholar]
- 40.Zhou L, Xu J, Wang S, et al. Papillary renal neoplasm with reverse polarity: a clinicopathologic study of 7 cases. Int J Surg Pathol 2020;28:728-734 https://doi.org/10.1177/1066896920918289 10.1177/1066896920918289 [DOI] [PubMed] [Google Scholar]
- 41.Trpkov K, Hes O, Bonert M, et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol 2016;40:60-71. https://doi.org/10.1097/PAS.0000000000000508 10.1097/PAS.0000000000000508 [DOI] [PubMed] [Google Scholar]
- 42.Munari E, Settanni G, Caliò A, et al. TSC loss is a clonal event in eosinophilic solid and cystic renal cell carcinoma: a multiregional tumor sampling study. Mod Pathol 2022;35:376-385. https://doi.org/10.1038/s41379-021-00816-8. 10.1038/s41379-021-00816-8 [DOI] [PubMed] [Google Scholar]
- 43.Trpkov K, Abou-Ouf H, Hes O, et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol 2017;41:1299-1308. https://doi.org/10.1097/PAS.0000000000000838 10.1097/PAS.0000000000000838 [DOI] [PubMed] [Google Scholar]
- 44.Mehra R, Vats P, Cao X, et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol 2018;74:483-486. https://doi.org/10.1016/j.eururo.2018.06.007 10.1016/j.eururo.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trpkov K, Williamson SR, Gao Y, et al. Low-grade oncocytic tumour of kidney (CD117-negative, cytokeratin 7-positive): a distinct entity? Histopathology 2019;75:174-184. https://doi.org/10.1111/his.13865 10.1111/his.13865 [DOI] [PubMed] [Google Scholar]
- 46.Morini A, Drossart T, Timsit MO, et al. Low-grade oncocytic renal tumor (LOT): mutations in mTOR pathway genes and low expression of FOXI1. Mod Pathol 2022;35:352-360. https://doi.org/10.1038/s41379-021-00906-7 10.1038/s41379-021-00906-7 [DOI] [PubMed] [Google Scholar]
- 47.Tjota M, Chen H, Parilla M, et al. Eosinophilic Renal Cell Tumors With a TSC and MTOR Gene Mutations Are Morphologically and Immunohistochemically Heterogenous: Clinicopathologic and Molecular Study. Am J Surg Pathol 2020;44:943-954. https://doi.org/10.1097/PAS.0000000000001457 10.1097/PAS.0000000000001457 [DOI] [PubMed] [Google Scholar]
- 48.He H, Trpkov K, Martinek P, et al. ‘High-grade oncocytic renal tumor’: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch 2018;473:725-738. https://doi.org/10.1007/s00428-018-2456-4 10.1007/s00428-018-2456-4 [DOI] [PubMed] [Google Scholar]
- 49.Chen YB, Mirsadrei L, Jayakumaran G, et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol 2019;43:121-131. https://doi.org/10.1097/PAS.0000000000001170 10.1097/PAS.0000000000001170 [DOI] [PMC free article] [PubMed] [Google Scholar]