

Abstract
Transfection is a critical step for gene editing and cell-based therapies. Nanoscale technologies have shown great promise to provide higher transfection efficiency and lower cell perturbation than conventional viral, biochemical and electroporation techniques due to their small size and localized effect. Although this has significant implications for using cells post-transfection, it has not been thoroughly studied. Here, we developed the nano-electro-injection (NEI) platform which makes use of localized electric fields to transiently open pores on cell membrane followed by electrophoretic delivery of DNA into cells. NEI provided two-folds higher net transfection efficiency than biochemicals and electroporation in Jurkat cells. Analysis of cell doubling time, intracellular calcium levels and mRNA expression changes after these gene delivery methods revealed that viruses and electroporation adversely affected cell behavior. Cell doubling times increased by more than 40% using virus and electroporation methods indicative of higher levels of cell stress, unlike NEI which only minimally affected cell division. Finally, electroporation, but not NEI, greatly altered the expression of immune-associated genes related to immune cell activation and trafficking. These results highlight that nanoscale delivery tools can have significant advantages from a cell health perspective for cell-based research and therapeutic applications.
Keywords: transfection, nano-electro-injection, nano-electroporation, RNA transcriptomics
Graphical Abstract
Like all of us, cells also appreciate an efficient, reliable and stress-free option for delivering biomolecules into their intracellular spaces. However, existing methods using viruses, biochemicals and bulk electroporation has low efficiency while causing high cell stress and mortality. Nano-scale techniques provide a more efficient and minimally perturbative route for biomolecule delivery into cells.
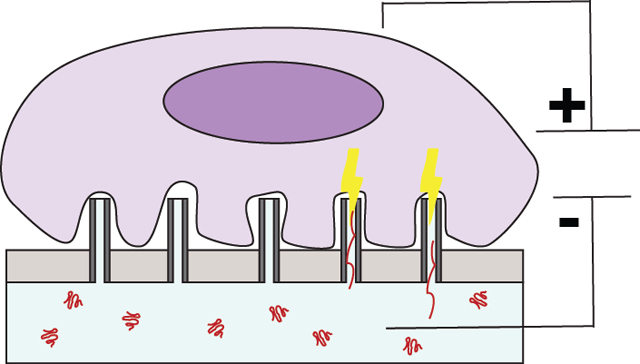
Introduction
Delivery of exogenous biomolecules such as DNA and proteins has become a common yet critical step to manipulate genome and cellular fates for promising biomedical applications like chimeric antigen receptor (CAR) T cell therapy.[1] Viruses are currently the most commonly used transfection method, but challenges with safety, cost, and limited cargo size have led to strong motivations to replace it with physical or biochemical approaches.[2] However, methods such as electroporation and biochemicals like lipofectamine[3] have unique concerns and limitations, with some cell types even being resistant to electroporation-mediated transfection.[4]
These limitations have inspired the development of nanoscale technologies like nanowires,[5] nano-spears,[6] nano-needles[7] and nano-straws platforms[8–10] to improve transfection. Unlike viruses that are evolved to infect specific cell types or biochemicals that have to fuse with the cell membrane for endocytic delivery, nano-structures do not depend on biological factors like membrane receptor density and membrane charges for intracellular delivery. The cargo agnostic nature of nanostructures also enables them to deliver multiple cargo species simultaneously unlike viral and biochemical methods. For example, Shalek et al. first demonstrated the use of nano-wires to deliver different nucleic acids to diverse primary immune cells including T cells, B cells, Natural Killer cells and macrophages.[11] Nano-structures have also been integrated with electrical control to improve transfection efficiency with possibly less cell perturbation than in a electroporation system due to localized nanostructure-cell interactions. For instance, Lee et al. developed the dielectrophoresis nano-electroporation method to transfect immune cells with high cell viability.[12] Recently, our group has also developed the nano-straw platform to transfect a variety of primary cells with high mRNA transfection efficiency between 60–85%.[10]
However, not all successfully transfected, viable cells are ideal for subsequent use in cell-based therapies, as the transfection method itself can induce various degrees of perturbation which can alter gene expression and cell function. For example, DiTommaso et al. found that electroporation induced significant gene expression changes and aberrant cytokine secretions in primary T cells, which subsequently dampened the long-term effector function of T cells in vivo.[13] This is supported by another recent report by Cromer et al. on the deleterious effects of viruses and electroporation on gene expressions in primary stem cells.[14] A transfection-induced downstream effect of great scientific and clinical interest is cell proliferation,[15] as lengthening of doubling time creates much longer preparation times, increasing cost and chance for contamination.
Conceptually, nanoscale delivery systems, with their highly localized physical mechanisms, may induce considerably less perturbation. Transient, small cell membrane ruptures or nanomaterial penetration could avoid significant cell stress and immune response, leading to overall healthier cells. Recent clinical studies show that a single, highly functional T-cell was responsible for 94% of the therapeutic response, suggesting that fewer, healthier cells may be just as or more effective than a large number of compromised cells.[16] Characterizing transfection-induced cellular stress, phenotypical changes, and alteration in mRNA expression is thus critical to understand the biological consequences of transfection, yet these are typically unreported for nanomaterials systems.
In this paper, we first compared the transfection performance of different widely-used transfection methods including virus, biochemicals, electroporation, and the nano-electro-injection (NEI) system. We found that the NEI platform provided about two-folds higher net transfection efficiency than commercially available biochemical and electroporation techniques. In order to assess the degree of cell perturbation, we performed acute/chronic calcium imaging as an indicator of stress levels,[17] measured cell proliferation rate, and performed RNA transcriptomics to measure changes in gene expression.[14] These studies revealed that viral and electroporation methods, but not NEI, resulted in calcium-associated cell stress, and significant lengthening of cell doubling times. Similarly, electroporation greatly altered the expression of immune-associated genes crucial for immune cell activation and trafficking, while NEI induced minimal changes in gene expression. This study provides one of the first head-to-head comparisons of the effects of bulk versus nanoscale electroporation on cell proliferation, calcium stress responses and gene regulation.
Results
Comparing net transfection efficiencies across common transfection techniques
The transfection efficiencies and cell viabilities were first assessed for viral, biochemical, electroporation, and NEI transfection techniques in Jurkat cells, a popular model for human T-lymphocytes.[18] The NEI platform consisted of numerous alumina hollow nano-channels fabricated on a polyethylene membrane.[8,10,19] The dimensions of the hollow nano-channels were optimized to be ~150 nm in diameter and 1.5–2 μm in height (see scanning electron microscopic image in Fig. S1b). During NEI, localized electric fields passing through the hollow nano-channels transiently created pores on cell membrane and electrophoretically drove charged DNA plasmid cargo into cells resting on hollow nanochannels (see schematic of mechanism in Fig. S1c).[10]
While challenging to transfect,[20] a number of transformation protocols and commercial kits are available for Jurkat cells, allowing head-to-head comparisons of efficiency and cell perturbation. Jurkat cells were transfected with eGFP plasmid using commercially-available biochemical agents (Fugene-Promega and Lipofectamine-Thermo Fisher), bulk electroporators (BioRad and Lonza), and lentivirus with Ubiquitin or EF1-α promoter (Stanford Viral Core). In each case, delivery was performed following manufacturers’ instructions without further optimization or according to literature protocols.[21]
The transfection efficiency, cell viability, and net efficiency of each method are tabulated in Fig. 1. Here, the net efficiency is defined as the product of cell viability and transfection efficiency, representing the percentage of the original cell population that are viable and successfully transfected after DNA delivery. As expected, there was a trade-off between cell viability and aggressiveness of the transfection method, particularly at higher multiplicity of infection (MOI), DNA-polymer concentrations, and voltages (Fig. 1a–d, Fig. S1b). For instance, higher voltages reduced cell viability with a noticeable drop at 1000 V for bulk electroporation and 40 V for NEI (Fig. 1c–d, Fig. S1b). On the other hand, higher MOI, DNA-polymer concentrations and voltages enhanced transfection efficiencies (Fig. 1e–h).
Fig. 1.

Cell viability, transfection efficiency, and net efficiencies for different transfection techniques. (a-b) Cell viability was calculated as the percentage of cells that were not stained by propidium iodide, a dye impermeable to live cells, 24 hr after transfection. Generally, higher multiplicity of infection (MOI) i.e. viral particles per cell, concentration of polymer-DNA mixtures and voltages reduced cell viability. (e-h) Transfection efficiency was calculated as the percentage of fluorescent cells 24 hr after transfection. Higher MOI, concentration of polymer-DNA mixtures and voltages boosted transfection efficiencies. (i-l) Net transfection efficiency was calculated as the product of cell viability and transfection efficiency. This parameter illustrates the trade-off between cell viability and transfection efficiency. The results showed that comparable net transfection efficiencies (11–24%) were obtained using different techniques. Error bars shown are ± standard deviations. *: p < 0.05, **: p < 0.001 (statistics were performed relative to NEI, 30 V which provided the highest net transfection efficiency. There were 0.2*106 cells in each transfection condition.).
Here, we found that the NEI platform (30 V, 23.8 ± 2.00%) provided the highest net transfection efficiency and double than that of Fugene (30 uL, 11.2 ± 4.34%) and bulk electroporation (500 V, 16.5 ± 4.36%). Lentivirus with Ubiquitin promoter (MOI = 10) gave the second-best net transfection efficiency around 19.5 ± 0.44% (Fig. 1i–l). These results are consistent with reported transfection efficiencies (ranging from 1–22%) for Jurkat/T cells using manufacturers’ recommended and peer-reviewed published protocols (see list in Table S2/3).[22,23] Note that while some efficiencies of >80% have been reported, these do not account for accompanying high cell mortality, which can be as high as 90–95%.[24] This highlights the importance of comparing net transfection efficiency because only viable, successful transfected cells are useful for subsequent experiments.
Comparing cell proliferation rates across different transfection techniques
Next, we investigated how different transfection techniques affected cell proliferation. Cell doubling time is critical for cellular therapies, as only a limited number of cells are available initially, and cell expansion is essential after transfection. This expansion process can be one of the most time-consuming and expensive steps in cell-based therapy.[25] Therefore, any increase in doubling time is detrimental, and may also indicate significant cell stress, senescence or exhaustion.[15] Table 1 lists the net efficiency for each method that gave the best net transfection efficiencies for each class of transfection technique, together with the number of cell divisions necessary for a scenario starting with 0.2 × 106 cells to reach 109 cells, often used as a target number for cell therapies.
Table 1.
Comparisons of different transfection techniques for cell therapies
| Fugene® (DNA:Reagent = 1:2.5, 30 uL of reagent-DNA mixture) | Viral (lentivirus with ubiquitin promoter, MOI = 10) | Bulk electroporation (Biorad, 1000 μF, 500 V, 30 s) | Nano-electro injection (NEI) system (30 V, 40 Hz, 200 μs, 2 min) | |
|---|---|---|---|---|
| Net efficiency (Enet) | 11.2% | 19.5% | 16.5% | 23.8% |
| Measured cell doubling time (td)+ | 46.8 hr (**) | 53.1 hr (**) | 50.6 hr (**) | 40.0 hr (n.s.) |
| # days to reach Nfinal (D) | 30.1 | 32.4 | 31.6 | 23.9 |
Starting cell # (N0) = 0.2*106 and final cell # (Nfinal) = 109
, where n is the number of cell divisions. Based on this calculation, the ‘n’ value for Fugene, viral, Biorad and NEI are 15.4, 14.6, 15.0 and 14.4 respectively.
tracked for first 4 days after transfection and assuming the value remains unchanged. Our control cells without any transfection treatment doubled every 36.4 hr on average (Table S5).
/n.s. indicates p-value <0.001/non-significant respectively compared to control cells, with starting 0.2*106 cells in each condition.
The average cell doubling time (td) for each transfection technique (averaged for the first four days after treatment) increased dramatically for all methods except NEI (Table 1). Control cells had an average doubling time of 36.4 hr, in line with literature reports. Fugene-treated cell doubling time increased to 46.8 hr, a 29% increase, while that of viral and electroporation increased to 53.1 hr (+46% change) and 50.6 hr (+40% change) respectively. These large increases indicate significant cellular perturbation due to the transfection process. NEI also lengthened the doubling time, though to a much lesser extent i.e. 40.0 hr (<10% increase). The total amount of time required to produce 109 cells could then be calculated by multiplying the doubling time and the number of cell divisions needed, showing correspondingly longer culture time for viral and electroporation. For example, it would take 32.4 days for viral transfection compared to 23.9 days for NEI, a 36% increase.
Comparing calcium stress responses across different transfection techniques
To understand why different transfection techniques affect cell viability and proliferation, we then quantified intracellular calcium (Ca2+) levels in cells, an important second messenger which become elevated during periods of cell stress and apoptosis.[17,26] Ca2+ levels in the cytosol are typically maintained at around 100 nM which is significantly much lower than that in organelles (endoplasmic reticulum, mitochondria) and extracellular environment, which have Ca2+ levels in the μM and mM range respectively. During transfection, Ca2+ levels can increase due to viral entry into cells, endocytosis of cytotoxic biochemical complexes and pore opening due to electrical fields.[26] The bigger the membrane pores and the longer the pores stay open, the greater the increase in intracellular calcium levels. During NEI, electric fields can also open membrane pores, which could also allow an influx of Ca2+ into intracellular spaces. Ca2+ levels in Jurkat cells were measured at 1 hr, 6 hr, and 24 hr post-delivery for each of the transfection conditions tested in Fig. 1. Addition of DNA plasmid to a control cell culture did not cause any significant increase in Ca2+ influx, indicating that Ca2+ influx is attributable to the transfection method (Fig. 2a).
Fig. 2.

Monitoring calcium stresses due to transfection. (a-b) Intracellular Ca2+ fluorescence levels (ΔF/F0) monitored 1 hr after transfection. Higher MOI, concentrations of DNA-polymer mixtures and voltages generally led to greater Ca2+ levels, indicative of higher stress. Low concentration of Fugene-DNA mixture (6 μL) and NEI at 20/30 V) did not significantly cause Ca2+ influx unlike viruses and bulk electroporation. (c) Ca2+ fluorescence images showing high intracellular Ca2+ after transfection with Fugene (30 μL), virus (Ubiquitin promoter, MOI = 10) and bulk electroporation (1000 μF, 1000 V) while NEI (30 V, 2 min) had Ca2+ fluorescence levels similar to control despite achieving similar net transfection efficiencies as the other methods. Enet: Net transfection efficiency. (d) Ca2+ fluorescence levels were monitored at the 1/6/24 hr time points. Although Fugene (6 μL) led to minimal Ca2+ influx 1 hr post-transfection, intracellular Ca2+ fluorescence levels continued to rise until the 6 hr time-point before declining at the 24 hr time-point. On the other hand, NEI led to minimal increase in intracellular Ca2+ fluorescence levels acutely (1 hr) and chronically (> 1 hr). This trend was the opposite with viruses which led to increasing intracellular Ca2+ fluorescence levels even 24 hr post-transfection. n.s.: not significant, *: p < 0.05, **: p < 0.001, with 50 cells analyzed in each condition. Error bars shown are ± standard mean error.
The amount of cell stress measured depended on both transfection technique and conditions. Low concentrations of DNA/Fugene (6 μL) or low NEI voltages (20–30 V) did not significantly elevate intracellular Ca2+ levels. On the other hand, viral and bulk electroporation treatments caused higher cellular stresses regardless of the experimental conditions (Fig. 2a–b). As the concentrations of DNA-Fugene mixtures, MOI, and voltages increased, greater Ca2+ levels were measured, indicative of higher cellular stresses due to transfection conditions. Fig. 2c shows Ca2+ fluorescence images after different treatments with comparable net transfection efficiencies (Enet). Dimethyl-siloxane (DMSO) which created membrane pores was used as a positive control to show that Ca2+ influx is associated with cell stress and apoptosis. Fugene (6 μL) and NEI (30 V) resulted in low Ca2+ influx while virus and bulk electroporation significant heightened intracellular Ca2+ levels.
Ca2+ influx at different time points (6 hr and 24 hr) were also monitored to understand the acute and chronic impact of transfection-induced cell stress. Fig. 2d shows that while Fugene caused low Ca2+ influx at the 1 hr time-point, Ca2+ influx continued to rise until the 6 hr time-point, possibly after endocytosis, before declining at the 24 hr time-point. This could explain why although Fugene led to low Ca2+ influx 1 hr after transfection, it was found to delay cell division significantly (Table 1). On the other hand, intracellular Ca2+ levels in cells treated with NEI quickly returned to levels similar to control at the 6- and 24-hr time points.
These results suggest that NEI generated minimal acute (1 hr) cellular stresses, and that Fugene and viruses led to chronic (>1 hr) cellular stresses. This is the first study, to the best of our knowledge, investigating acute versus chronic calcium-mediated cellular stresses from transfection, and may provide insight into the differential behavior of cells post-transfection at different time-points. It also has implications in the study of calcium-mediated events in cells post-transfection at different time-points.
Analysis of perturbation of mRNA expression
While there is a body of research describing the deleterious impact of biochemical[27] and viral[14] transfection on gene regulation, there is little literature comparing the effects of bulk and nano-scale electroporation delivery methods on gene regulation.[13,14] As bulk electroporation and NEI work through an electrical mechanism, and both techniques primarily caused acute stress responses rather than chronic stresses (unlike viral and biochemical methods), we sought to compare the effects of bulk electroporation and NEI on gene expression through mRNA transcriptomics. We were also motivated by the increasing use of electroporation methods as a non-viral alternative for immune cell transfection (Table S6).[28]
Cells were treated with bulk electroporation or NEI without DNA plasmid to avoid potential complicating effects of the DNA cargo.[14] The same experimental protocol was used by DiTommaso et al. and Cromer et al. who found that the presence of cargo confounded the results of genome-wide study comparing delivery methods.[13,14] Furthermore, as bulk electroporation (16.5%) and NEI (23.8%) also provided different net transfection efficiencies (Table 1) and DNA-induced cytotoxicity is concentration dependent, we performed transfection without DNA plasmid to eliminate this factor from affecting our result interpretations.
After transfection treatments, RNAseq was performed 24 hr later. This time-point was selected as it was previously reported that gene expressions tended to stabilization 24-hr after transfection, while RNA analysis at 6-hr post-transfection found more than 8000 genes had altered expressions in immune cells and the data was noisy.[13] The principal component (PC) plot in Fig. 3a shows that the technical replicates in each treatment group more closely resembled one another, thus enabling reliable comparisons of different treatments on changes in gene expression. Next, the enriched gene ontologies (GOs) for different biological functions were calculated, and it was found that these categories – metabolism/cellular process, stress, regulation/signaling – were the most highly affected (Fig. 3b, Table S7). Interestingly, we also found that while bulk electroporation affected the expression of immune-associated genes, NEI did not.
Fig. 3.

RNAseq data showing minimal gene perturbation by NEI. (a) Principal component (PC) plot showing technical replicates in each treatment group more closely resembled one another. (b) Genes in the enrich gene ontologies (GOs) which were most highly affected by treatments fell in metabolism/cellular response, stress, regulation/signaling and immune response. (c) Volcano plots showing up- and down-regulated genes after each treatment. The p-value cut off was <0.001 and log2 fold change cut-off was <−2 or >2. The number of genes that were up- or down-regulated are shown in the respective plot. (d-e) Bulk electroporation and NEI resulted in fold increase in DDIT4, a gene involved in regulation of apoptosis in response to DNA damage and ALDOC, a gene involved in glycolysis and ATP synthesis. However, bulk electroporation led to greater gene expression changes than NEI, demonstrating greater cellular perturbation by the former. (f) SLC7A11 which down-regulates the transport of amino acids and glucose out of cells was significantly down-regulated by bulk electroporation and NEI but the fold change between both treatments was not statistically significant. (g) -log10 (p-value) of DDIT4, ALDOC and SLC7A11, showing that the changes in their expressions are statistically significant from the controls. **: p < 0.001, n.s.: not significant.
Fig. 3c shows the volcano plots for genes that were up- or down-regulated due to each treatment. To establish stringent criteria, we only considered genes whose expressions have been changed with p-value < 0.001 and with |log2| fold change of at least 2. It was found that although NEI produced a greater change in gene expression i.e. 336 genes compared to 154 genes for bulk electroporation, most of the affected genes had relatively lower fold changes (log2 of 2–3, compared to 5–8 for bulk electroporation). Next, we decided to focus on the top stress- and metabolic-associated gene that has been affected by transfection. Bulk electroporation resulted in significant up-regulation of the stress-related gene, DDIT4 (DNA damage inducible transcript 4,). DDIT4 is implicated in the regulation of apoptosis in response to DNA damage (Fig. 3d). This could explain why we saw lower cell viability with bulk electroporation than NEI (Fig. 1g–h). Furthermore, bulk electroporation significantly increased the expression of the metabolic gene, ALDOC (aldolase, fructose-biphosphate C) which up-regulates glycolysis and ATP synthesis (Fig. 3e). The fold-increase of DDIT4 and ALDOC due to bulk electroporation was 7.16 and 24.21, and they were significantly higher compared to the respective values of 2.42 and 4.29 due to NEI. These results support that bulk electroporation resulted in greater cellular perturbation measured in terms of altered gene expressions than NEI.
It was also found that both bulk electroporation and NEI led to significant down-regulation of SLC7A11 (solute carrier family 7 member 11) which down-regulates the transport of amino acids/glucose out of cells. However, the difference in fold decrease due to both treatments was not statistically significant (Fig. 3f). Combining with the data on ALDOC, we speculate that pore formation due to electrical fields either with bulk electroporation or NEI, can adversely affect the metabolic states of cells, creating a need to generate more energy such as increased ATP synthesis (with increase expression of ALDOC) and reduced transport of glucose out of the cells (with down-regulation of SLC7A11). We also hypothesize that the greater metabolic perturbation due to bulk electroporation might have contributed to correspondingly greater lengthening of cell doubling time. Fig. 3g shows the -log10 (p-value) of DDIT4, ALDOC and SLC7A11 to demonstrate that the change in their expressions are statistically significant from the controls.
Changes in immune-associated gene expressions after bulk electroporation
Through RNAseq, it was found that bulk electroporation, but not NEI, induced changes in the expressions of immune-associated genes. We calculated FDR q-values at different thresholds to determine the number of immune-associated genes with altered expressions after bulk electroporation and NEI treatments with the untreated controls (Fig. 4a). A total of 43 genes were affected by bulk electroporation with FDR q <0.25. Fig. 4b shows the heatmap of the affected genes. This finding is consistent with a recent report that intracellular delivery techniques can cause unintended and non-specific changes to gene expressions and cell functions of immune cells.[13] To gain insights into the effects of bulk electroporation on immune-associated gene expressions, we narrowed the gene sets to include only those genes that were dramatically affected by applying a filtering criterion of p-value <0.001, log2 fold change cut-off was <−2 or >2 and FDR q <0.05. It was found that five genes that are crucial to immune cell function such as activation and trafficking had highly altered expressions (Fig. 4c). CCR8, SELL and CCR4 which regulate immune cell chemotaxis and migration were significantly upregulated. There were also significant fold increases in the expressions of CD4 and CD48 which regulate immune cell activation. Taken together, this suggests that the choice of intracellular delivery method can have deleterious, non-specific impact on cellular functions. This is especially important to consider as immune cell activation and trafficking is crucial for effective cancer immunotherapy against solid tumors.[29] Further work is necessary in the future to examine how long these immuno-defective effects persist and whether there are any functional consequences on cellular behaviors such as chemotactic migration due to altered gene expressions.
Fig. 4.

Bulk electroporation led to unintended and non-specific expression changes in immune-associated genes. (a) Table showing number of immune-associated genes wit altered expressions due to bulk electroporation with different FDR q-values. (b) Heatmap of immune-associated genes affected by bulk electroporation. 43 genes were affected with FDR q-value < 0.25. (b) Five genes (FDR q-value <0.01) related to immune cell trafficking (CCR8, SELL and CCR4) and activation ( CD4 and CD48) were affected by bulk electroporation. The functions of the genes are also included in the figure.
Discussion
Cell-based therapies such as CAR-T immunotherapy is a promising strategy for treating a wide range of diseases. Nevertheless, it remains challenging to achieve high transfection efficiency using viral and bulk electroporation in immune cells without concomitant cell perturbation. Transfection-associated cell stress is a crucial aspect of cell transformation as it can significantly alter gene expressions and lengthen cell doubling time, leading to delays and cost increase during cell manufacturing. Furthermore, sub-optimal cell growth may also indicate signs of senescence or exhaustion which can greatly compromise the efficacy of immunotherapy, yet cell perturbation due to transfection is typically unreported. Nanomaterials delivery systems may avoid many of these delivery-related perturbations, however these have not been extensively studied.
In this paper, we demonstrated that NEI is a high efficiency transfection technique that minimally perturbs cellular states unlike other commonly used viral, biochemical and electroporation techniques. NEI provided the highest net transfection efficiency compared to all methods tested, almost two-fold higher than biochemicals and bulk electroporation. Through analyses of acute/chronic Ca2+ stress signals and RNA transcriptomics, NEI also resulted in the lowest Ca2+-mediated cellular stress and generated minimal perturbation on gene expressions. Viral and bulk electroporation, which are mainstream techniques for T-cell transformation, generated substantial calcium-associated cell stress and significantly lengthened cell doubling times. Bulk electroporation treatment also led to statistically more significant changes in stress- and metabolic-associated genes. Bulk electroporation also resulted in unintended and non-specific changes in expressions of immune-associated genes regulating leukocyte migration and activation.
Although there are increasing number of studies to optimize delivery efficiency and viability, characterizing the downstream functional consequences such as gene expressions and cell proliferation are equally, if not more important. Nanoscale delivery systems appear to minimally perturb cellular function, and could provide healthier cells for future research and clinical therapies. In the future, similar approaches to those used here can be leveraged to study the impact of transfection on cellular states. From these results, the NEI platform may also transfect a variety of other sensitive cell types including primary immune cells and stem cells without negatively impacting cellular physiology, providing a non-viral route to highly functional, transformed cells.
Materials and Methods
Nano-electro injection channel fabrication
The NEI membrane was based on 8-μm-thick (±15%) track-etched polyethylene terephthalate (PET) membranes (GVS Life Sciences) with 80 nm diameter pores and density of 1 × 108 pores/cm2, typically used for water filtration and cell culture. The membrane was first treated with 10% O2/90% Ar plasma, pressure maintained at 100 kPa using rotary vane pump (SPI Supplies) with RF power of 100 W, reverse power between 0–3, for 3 mins to expand the pore to about 200 nm (Plasma PrepIII™ Solid State, SPI Supplies). Next, a 20-nm-thick Al2O3 layer was deposited on the membrane using atomic layer deposition (ALD, Savannah, Cambridge Nanotech) at 100 °C with an initial delay of 30 mins to remove potential contaminants. An exposure mode was used. However, the precursor does not flow rapidly from cylinder through valve through manifold over substrate into pump, but sits in the reactor for expo seconds, and some of the precursor can migrate to all valves. This can cause some deposition in the valves, especially if the expo time is long. To reduce this effect, we pulsed in cycles in the exposure mode to prevent the reactor pressure from getting too high during the expo time. Pulse cycles of the form a/b/c/a/b/c, where a is the precursor exposure time, b is the exposure time, and c is the N2 purge time, each in seconds were implemented. To ensure conformal coverage over high aspect ratio nanopores, a pulse cycle of 0.025/5/45/0.025/5/45. 80 cycles were performed in total. After this step, the interior of the track-etched pores was also coated with Al2O3. The NS were then formed by reactive ion etching (RIE) of the Al2O3 with BCl3 and Cl2 using PlasmaTherm Metal Etcher (Versaline LL-ICP Metal Etcher) in Ar [400 W, 40 standard cm3/min BCl3, 30 standard cm3/min Cl2, 5 mTorr, 1 min] from the top surface (see Table S8 for full details). This is followed treatment with 10% O2/90% Ar plasma, pressure maintained at 100 kPa using rotary vane pump (SPI Supplies) with RF power of 100 W, reverse power between 0–3 (Plasma PrepIII™ Solid State, SPI Supplies) for 10 mins to remove the PET polymer and expose the inorganic hollow NEI tubes with height 1.5–2 μm. The membrane with NEI channels was then cut into circular shapes (diameter of 0.9 cm) and adhered onto a transparent tube (outer diameter of 0.9 cm, inner diameter of 0.6 cm, height of 1.3 cm) with a double-sided tape (3M VHB™).
Cell culture
Jurkat cells were maintained in RPMI 1640 + GlutaMAX™ (Gibco) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and 1% penicillin-streptomycin (PenStrep, Gibco) and cultured in incubator with 5% CO2 at 37°C. Jurkat cells used for this paper was between passage #3–20 to ensure valid comparisons due to possibility of cell senescence after repeated culture.
Plasmid preparation
The Pmax-eGFP plasmid (~5.5 kbp) encoding for the enhanced green fluorescent protein (eGFP)(Addgene) were propagated in competent NEB 10-Beta competent Escherichia coli. The plasmid was extracted and purified using the PureLink HiPure Plasmid Filter Maxiprep Kit (Invitrogen). Plasmids were dissolved with distilled water to obtain a final concentration between 0.5–1 μg/μL.
Transfection protocols
Transfection with lentiviruses: Lentiviruses with either Ubiquitin (~8.9 kbp, titer: 7×109/mL) or EF1-α (~7.5 kbp, titer: 3.4×108/mL) promoter containing eGFP were generous gifts from the Stanford Gene Vector and Viral Core. The Multiplicity of Infection (MOI, 2.5–10) was decided before mixing the viruses with Jurkat cells. The mixture was then subject to spin-trifugation at 5 g for 20 mins before overnight infection with 2 mL Jurkat cell suspension. The MOI was decided based on commonly reported protocol of 5–20 viral particles per Jurkat cell. Transfection with LipofectamineTM: A Lipofectamine 2000 DNA transfection kit (Life Technologies) was used. The DNA–lipid complex was prepared by combining 2 μl of Lipofection 2000 reagent in 100 μl of Opti-MEM medium with 1.25/2/2.75 ug of DNA plasmid in 100 μl of Opti-MEM medium in a 1:1 ratio, followed by 5 mins of incubation at room temperature. 200 μL of the DNA–lipid complex solution was added to 2 mL of Jurkat cell suspension. The DNA plasmid concentration was used based on manufacturer’s recommendation (see page 2 of product protocol). Transfection with Fugene® HD: DNA plasmid was diluted to a concentration of 20 μg/mL with sterile water. The appropriate amount of FuGENE® HD Transfection Reagent was added to achieve a DNA: Reagent ratio of 1:2.5. 6/15/30 μL of FuGENE® HD Transfection Reagent:DNA mixture was added to 2 mL of Jurkat cell suspension for overnight transfection. More details can be found in the product technical manual. Transfection with Biorad Gene Pulser XcellTM: 1 mL of Jurkat cell suspension with 10 μg plasmid DNA was pipetted into the Gene Pulser®/MicroPulser™ Electroporation Cuvette (0.4 cm gap) and subject to electroporation for 30 s. Table S4 lists the actual values during electroporation. Note that there was time decay for field decay as exponential wave was used as suggested by protocols frequently used by others (see Biorad Protocol Finder for a list of compiled literature and their associated electroporation protocols). Transfection with Lonza 4D NucleofectorTM: Transfection was performed by pipetting 1 mL of Jurkat cell suspension with 10 μg plasmid DNA into the 1 mL Nucleofector cuvette, followed by using manufacturer’s protocol for Jurkat cells (Clone EG-1, ATCC® TIB-152™). Transfection with nano-electro injection (NEI) platform: To perform intracellular delivery with NEI, 0.2 × 106 cells in 400 μL were pipetted onto the NS tube and centrifuged. Jurkat cells (Clone E6–1, ATCC® TIB-152™) were centrifuged at 300 g for 6 mins. The tubes containing cells were then placed in a 24-well plate for incubation (5% CO2, 37°C) with additional media around the NEI tubes to prevent drying. During NEI, the NS tubes were placed in the Navan 100 box that provided electrical control with square waves at 400 Hz for electroporation for 2 min. The cargo (10 μg plasmid DNA) were suspended in 104-dilution of 1x Phosphate Buffered Saline (PBS) to maintain optimal ionic salt concentration. Cell viability was determined by calculating the percentage of cells not stained by propidium iodide (Thermo Fisher) at a dilution of 1:1000. Transfection efficiency was determined by calculating the percentage of cells that were fluorescent. Proliferation was determined by counting the cell numbers with a hemocytometer (Hausser Scientific, catalog #3200) from day 1 to day 4.
Calcium dye incubation
Jurkat cells were incubated with Fluo-4 Direct™ calcium assay kit (Life Technologies) according to manufacturer’s protocol with 5 mM stock solution of probenecid. Briefly, 5 mL of calcium assay buffer was mixed and vortexed with 100 μL of probenecid stock solution to create a 2x loading dye solution. The dye solution was then added to the cells with media in a 1:1 ratio and incubated for 1 hr before imaging. For experiments involving DMSO (25%, v/v), the chemical was added before calcium dye incubation for 15 mins. The relative fluorescence change ΔF/F0 (change in fluorescence over background fluorescence) of somatic fluorescence signals was acquired using ImageJ.
RNA isolation
Jurkat cells (0.6 × 106) were subject to (all without cargo) Biorad Gene Pulser Xcell™ electroporation at 1000 μF, 500 V for 30 s or NEI at 30 V for 2 min. The cells were then cultured overnight, lysed the next day and RNA was extracted using the RNeasy Mini kit (QIAgen). There was a total of 3 samples per test group as shown in the PCA plot in Fig. 3a. RNA sequencing was performed at the Stanford Protein and Nucleic Acid Facility. RNA quality was analyzed with an Agilent Bioanalyzer QC, with each sample returning an RNA Integrity Number (RIN) at least 8.5 out of 10. Gene expression was analyzed on Applied Biosystems™ Clariom S Human. The cut-off fold change was set at ±2 (log2) and the cut-off p-value was set at < 0.001. Principal component and volcano plots were generated using the Applied Biosystems™ Transcriptomics Analysis Console software. The list of most significantly up- and down-regulated genes were also obtained through the software. Enriched gene ontologies (GOs) were calculated by identifying the number of genes in each GO divided by the total number of genes in the respective GO (biological function) as identified by the Panther Classification System.
Image acquisition, analysis and statistical evaluations
Fluorescent and bright-field images were acquired using the Axiovert 200M upright microscope (Zeiss) with (20x or 40x air objective from Zeiss) with the Micromanager 1.4 22 software. The NS membrane was prepared for SEM imaging by sputter coating with about 10 nm of Au/Pd. Samples were imaged in the FEI Sirion scanning electron microscope (Stanford Nano Shared Facilities). Statistical significance in Fig. 1 was evaluated using Wilcoxon Mann Whitney Test with null hypothesis = 0 and n = 60,000 (3 tests, 20,000 cells/test). Statistical significance in Fig. 2a/b/d was evaluated using the Student’s t-test after testing for normality using either one-way ANOVA, p < 0.05 (no rejection of normality), or nonparametric Kruskal-Wallis ANOVA, p < 0.05 (normality rejected) with n = 50 cells/condition across triplicates.
Supplementary Material
Acknowledgments
The research was supported by the NIH R21 EB02533201 and NSF STTR grant 1549696 and Bio-X Interdisciplinary Initiatives Program. NEI channels were fabricated in the Stanford Nanofabrication Facility (SNF). Nanoparticle characterizations and SEM imaging of NEI membrane were performed at the Stanford Nano Shared Facilities (SNSF. Research performed at SNF/SNSF was supported by the National Science Foundation under award ECCS-1542152. Lentiviruses with either Ubiquitin or EF1-α promoter containing GFP were generous gifts from the Stanford Gene Vector and Viral Core. AAV viruses containing GCaMP with constitutive expression using an EF1-α promoter (AAV-DJ EF1a-DIO-GCaMP 6f) was deposited by the Deisseroth’s lab in the Stanford Gene Vector and Viral Core. RNAseq was performed at the Stanford Protein and Nucleic Acid Facility. The authors acknowledge the help of S.L.O in media preparation and demonstrating the use of Navan100, R.S./A.J./E.S. in optimizing NEI channels fabrication and T. B.G. in SEM imaging of NEI channels. Andy Tay is supported by the National University of Singapore Overseas Postdoctoral Fellowship.
Footnotes
Competing interests
Andy Tay and N. Melosh have filed a patent for this technology with US Provisional Patent Application Number 62/534511. N. Melosh is a co-founder in Navan Technologies Inc.
References
- [1].Restifo NP, Dudley ME, Rosenberg SA, Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol 2012, 12, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sun S, Rao VB, Rossmann MG, Genome packaging in viruses. Curr. Opin. Struct. Biol 2010, 20, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tay A, Melosh N, PostDoc JJ Postdr. Res 2018, 6. [Google Scholar]
- [4].Stewart MP, Sharei A, Ding X, Sahay G, Langer R, Jensen KF, In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [DOI] [PubMed] [Google Scholar]
- [5].Boukany PE, Morss A, Liao WC, Henslee B, Jung H, Zhang X, Yu B, Wang X, Wu Y, Li L, Gao K, Hu X, Zhao X, Hemminger O, Lu W, Lafyatis GP, Lee LJ, Nat. Nanotechnol 2011, 6, 747. [DOI] [PubMed] [Google Scholar]
- [6].Xu X, Hou S, Wattanatorn N, Wang F, Yang Q, Zhao C, Yu X, Tseng H-R, Jonas SJ, Weiss PS, ACS Nano 2018, acsnano.8b00763. [DOI] [PubMed] [Google Scholar]
- [7].Chiappini C, Martinez JO, De Rosa E, Almeida CS, Tasciotti E, Stevens MM, ACS Nano 2015, 9, 5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xie X, Xu AM, Leal-Ortiz S, Cao Y, Garner CC, Melosh NA, ACS Nano 2013, 7, 4351. [DOI] [PubMed] [Google Scholar]
- [9].Xu AM, Kim SA, Wang DS, Aalipour A, Melosh NA, Lab Chip 2016, 16, 2434. [DOI] [PubMed] [Google Scholar]
- [10].Cao Y, Chen H, Qiu R, Hanna M, Ma E, Hjort M, Zhang A, Lewis RS, Wu JC, Melosh NA, Sci. Adv 2018, 4, eaat8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shalek AK, Gaublomme JT, Wang L, Yosef N, Chevrier N, Andersen MS, Robinson JT, Pochet N, Neuberg D, Gertner RS, Amit I, Brown JR, Hacohen N, Regev A, Wu CJ, Park H, Nano Lett. 2012, 12, 6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chang L, Gallego-Perez D, Zhao X, Bertani P, Yang Z, Chiang C-L, Malkoc V, Shi J, Sen CK, Odonnell L, Yu J, Lu W, Lee LJ, Lab Chip 2015, 15, 3147. [DOI] [PubMed] [Google Scholar]
- [13].DiTommaso T, Cole JM, Cassereau L, Buggé JA, Hanson JLS, Bridgen DT, Stokes BD, Loughhead SM, Beutel BA, Gilbert JB, Nussbaum K, Sorrentino A, Toggweiler J, Schmidt T, Gyuelveszi G, Bernstein H, Sharei A, Proc. Natl. Acad. Sci 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cromer MK, Vaidyanathan S, Ryan DE, Curry B, Lucas AB, Camarena J, Kaushik M, Hay SR, Martin RM, Steinfeld I, Bak RO, Dever DP, Hendel A, Bruhn L, Porteus MH, Mol. Ther 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ben-Porath I, Weinberg RA, When cells get stressed: An integrative view of cellular senescence. J. Clin. Invest 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, Hwang Y, Gohil M, Kulikovskaya I, Nazimuddin F, Gupta M, Chen F, Everett JK, Alexander KA, Lin-Shiao E, Gee MH, Liu X, Young RM, Ambrose D, Wang Y, Xu J, Jordan MS, Marcucci KT, Levine BL, Garcia KC, Zhao Y, Kalos M, Porter DL, Kohli RM, Lacey SF, Berger SL, Bushman FD, June CH, Melenhorst JJ, Nature 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Berridge MJ, Bootman MD, Lipp P, Calcium--a life and death signal. Nature 1998, 395, 645–648. [DOI] [PubMed] [Google Scholar]
- [18].Abraham RT, Weiss A, Nat. Rev. Immunol 2004, 4, 301. [DOI] [PubMed] [Google Scholar]
- [19].Vandersarl JJ, Xu AM, Melosh NA, Nano Lett. 2012, 12, 3881. [DOI] [PubMed] [Google Scholar]
- [20].Basiouni S, Fuhrmann H, Schumann J, Biotechniques 2012, 53. [DOI] [PubMed] [Google Scholar]
- [21].Cao Y, Chen H, Qiu R, Hanna M, Ma E, Hjort M, Zhang A, Lewis RS, Wu JC, Melosh NA, Sci. Adv 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Im DJ, Jeong SN, Biochem. Eng. J 2017, 122, 133. [Google Scholar]
- [23].Przybylski S, Gasch M, Marschner A, Ebert M, Ewe A, Helmig G, Hilger N, Fricke S, Rudzok S, Aigner A, Burkhardt J,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lonza, Nucleofector™ Kits for Human T Cells. [Google Scholar]
- [25].Wang X, Rivière I, Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther - Oncolytics 2016, 3, 16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang SKY, Marshall WF, Self-repairing cells: How single cells heal membrane ruptures and restore lost structures. Science (80-. ). 2017, 356, 1022–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yamano S, Dai J, Moursi AM, Mol. Biotechnol 2010, 46, 287. [DOI] [PubMed] [Google Scholar]
- [28].Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, Tobin V, Nguyen DN, Lee MR, Putnam AL, Ferris AL, Chen JW, Schickel JN, Pellerin L, Carmody D, Alkorta-Aranburu G, Del Gaudio D, Matsumoto H, Morell M, Mao Y, Cho M, Quadros RM, Gurumurthy CB, Smith B, Haugwitz M, Hughes SH, Weissman JS, Schumann K, Esensten JH, May AP, Ashworth A, Kupfer GM, Greeley SAW, Bacchetta R, Meffre E, Roncarolo MG, Romberg N, Herold KC, Ribas A, Leonetti MD, Marson A, Nature 2018. [Google Scholar]
- [29].Slaney CY, Kershaw MH, Darcy PK, Trafficking of T cells into tumors. Cancer Res. 2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.