

SUMMARY
Understanding the axis of the human microbiome and physiological homeostasis is an essential task in managing deep-space-travel-associated health risks. The NASA-led Rodent Research 5 mission enabled an ancillary investigation of the gut microbiome, varying exposure to microgravity (flight) relative to ground controls in the context of previously shown bone mineral density (BMD) loss that was observed in these flight groups. We demonstrate elevated abundance of Lactobacillus murinus and Dorea sp. during microgravity exposure relative to ground control through whole-genome sequencing and 16S rRNA analyses. Specific functionally assigned gene clusters of L. murinus and Dorea sp. capable of producing metabolites, lactic acid, leucine/isoleucine, and glutathione are enriched. These metabolites are elevated in the microgravity-exposed host serum as shown by liquid chromatography-tandem mass spectrometry (LC-MS/MS) metabolomic analysis. Along with BMD loss, ELISA reveals increases in osteocalcin and reductions in tartrate-resistant acid phosphatase 5b signifying additional loss of bone homeostasis in flight.
In brief
Bedree et al. demonstrate increases in Lactobacillus murinus and Dorea sp. during microgravity exposure, and pathways capable of producing lactic acid, leucine/isoleucine, and glutathione are enriched. These metabolites are elevated in the serum of the rodent flight cohort within the context of previously demonstrated loss of bone homeostasis.
Graphical Abstract
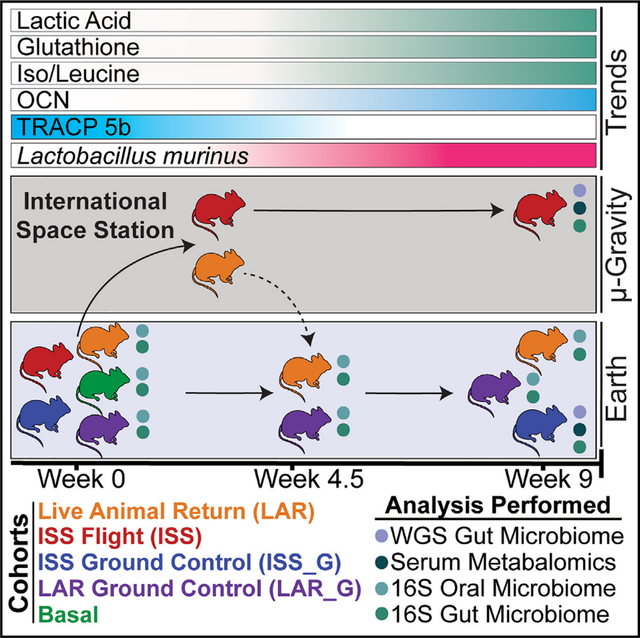
INTRODUCTION
Humankind’s exploration into the cosmos coincides with increasing space travel durations and demands upon the human body, which will require effective strategies for mitigation. The environmental factors of low-Earth-orbit space travel, ranging from galactic cosmic radiation, sleep deprivation, and psychological stress to microgravity, have demonstrable adverse effects on the homeostasis of mammalian physiology, such as reproducible loss of bone mineral density.1 Notably, microgravity is associated with human2–5 and rodent microbiome6,7 alterations; although, the health consequences of microbiome shifts within this environmental context remain vastly underexplored. Furthermore, the difficulty of consistently obtaining microbiome samples at desired time points and reliance of historically limiting culture-dependent assessment, such as during the Skylab program,8,9 hindered rapid exploration. Prior to the Rodent Research (RR) mission series, which was initiated by the International Space Station U.S. National Laboratory (ISSNL), formerly the Center for Advancement of Science in Space, and the National Aeronautics and Space Administration (NASA) Ames Research Center in 2014, animal research in microgravity was limited to less than 3 weeks of spaceflight onboard space shuttles with limited sample size (n = 6–8). This was particularly true for studying bone homeostasis in microgravity.10,11 However, the experimental conditions of these studies were incomparable to human low-Earth-orbit durations, and thus, NASA developed the Rodent Habitat (RH),12,13 which enabled increased-duration rodent research onboard the International Space Station with a larger sample size, animal handling capabilities, and a variety of experimental procedures. Leveraging this technology, the RR-5 mission enabled live animal return (LAR) of rodents from the International Space Station. The primary objective of the RR-5 mission was to investigate if systemic application of BP-NELL-PEG, an engineered form of a bone-formation-promoting natural peptide, NELL-like molecule-1 or NELL-1,14 combats long-duration spaceflight-induced osteoporosis.15
The microbiome-focused ancillary study being presented herein investigated the intersection of the gut and oral microbiome ecology and the bone homeostasis axis. Emerging research has linked changes in the gut microbiome and bone homeostasis16,17 through immune system-modulating effectors,18–20 vitamin and nutrient deficiencies,21 endocrine regulation,22 and energy metabolism through short-chain fatty acids (SCFAs).18,23 Interestingly, in terrestrial rodent models, it has been shown that administration of SCFAs (acetic/propionic/butyric acid), as well as other microbial metabolites, increased serum levels of insulin growth factor-1 (IGF-1)18 and decreased C-terminal telopeptide of type 1 collagen (CTX-1) and nuclear factor κB (NF-κB) factors responsible for osteoclastic activity.18,23 Moreover, SCFAs, primarily produced by gut microbiota,24–27 are known immunomodulators28–30 and regulate systemic bone mass as well as prevent bone mineral density (BMD) loss.23 RR-5 provided a unique opportunity to test the use of a non-ovariectomy BALB/c rodent model as a potential in vivo system for evaluating the effects of microgravity on the host’s microbiome and bone homeostasis, due to the degree and reproducibility of exacerbated bone loss in mammals during microgravity exposure.10,11 An additional rationale for evaluating the specific age range of the RR-5 cohort (30-weeks-old at launch) for microbiome changes is that BMD peaks and stabilizes around 30 weeks of age in BALB/c rodents,31,32 which was confirmed in the RR-5 cohort.15
Understanding the dynamic interactions among the commensal microbiome will be important for developing therapies to maintain healthy physiological homeostasis for future space travel, including that of increased duration.33 Innovations in genomic science and technology have enabled significant leaps in understanding of the effect of the microbiome on both health and disease-associated states. Bacterial dysbiosis of the commensal microbiota is correlated with multiple diseases, ranging from various gastrointestinal (GI) disease states34–38 to various types of cancer.39 In addition, the most recent NASA Twin study2 and other long-term human astronaut studies3 have shown intriguing increases in gut bacterial diversity in flight relative to ground controls. This observation was also reproducibly recapitulated in rodents from NASA’s RR-1 mission.6 While ongoing studies, such as the RR-7 mission and the NASA Astronaut Microbiome project, aim to further clarify the effects of microgravity on the different microbiomes of rodents and humans,33 this study provides new insight into the axis of the murine gut microbiome and bone homeostasis interactions as well as a glimpse into understanding microbiome diversity upon return to Earth in a rodent model.
RESULTS
LAR overall microbiome diversity is similar to LAR_G after return to Earth (week 4.5)
The rodents that did not receive drug therapy, phosphate-buffered saline (PBS)-injected “non-treated” control cohorts, were the focus of this ancillary study within the RR-5 mission. For simplicity, only this control cohort is described in this work (see graphical abstract). The RR-5 PBS control flight cohorts comprised a total of 20 female BALB/c, 30-week-old (at launch) mice that were housed in the RH onboard the International Space Station for 4.5 weeks, after which 10 rodents were returned alive (LAR cohort) to Earth and delivered to the University of California, Los Angeles (UCLA) Animal Care Facility to examine the effects of recovery for an additional 4.5 weeks. The other 10 rodents remained in the International Space Station for a full 9-week duration (full-term flight cohort, or ISS). The 20 cognate ground control rodents, LAR_G (10 rodents) and ISS_G (10 rodents), received a launch and transportation simulation and were then housed in identical RHs at Kennedy Space Center (KSC) in the International Space Station Environmental Simulator (ISSES) chambers. The ISSES mimicked the environmental conditions (temperature, humidity, and CO2 partial pressure) on the International Space Station. The LAR_G cohort was delivered from the ISSES at KSC to the UCLA Animal Care Facility at the same time as the LAR cohort returned via Dragon capsule at week 4.5, and both groups transitioned to standard caging upon arrival at UCLA (see STAR Methods). Furthermore, prior to cohort sorting into RH housing and respective experimental groups, all cohorts remained in standard caging in the Space Station Processing Facility (SSPF) vivarium. The RH hardware system automatically removes urine and fecal waste via constant airflow in microgravity. The ISS, ISS_G, LAR (until week 4.5), and LAR_G (until week 4.5) cohort mice were not coprophagic during flight or in the ISSES (see discussion). The basal cohort vivarium control (20 rodents) was euthanized 24 h post-launch and evaluated to account for environmental variability between the SSPF and the ISSES or the International Space Station and microbiome shifts resulting from lack of coprophagy or other iatrogenic factors. Thus, the basal cohort effectively served as a pre-flight or pre-simulation flight measurement when comparing ISS with ISS_G, LAR with LAR_G, or ISS with LAR cohorts.
Corresponding oral swabs and fresh fecal pellets of the LAR, LAR_G, and basal cohorts were analyzed for microbiome analysis using 16S rRNA amplicon sequencing (Figure 1). To assess the biodiversity of these communities, amplicon sequence variants (ASVs) were taxonomically assigned. Alpha diversity was measured via observed genera, Shannon, and Simpson indices along with beta diversity and Firmicutes to Bacteroidetes (F/B) ratio analysis. In previous human studies, elevated microbial diversity was observed in flight, but returned to pre-flight levels post-Earth return after varying durations in recovery.2,3 Importantly, the previous RR-1 study also recapitulated increases in microbial diversity at 37 days (week 5) in microgravity6 in rodents before euthanasia. Therefore, it was important to evaluate the LAR to Earth (4.5 weeks) time point, which had a similar duration of microgravity exposure relative to the RR-1 mice. However, the LAR flight cohort displayed similarity in community composition as well as in alpha (Figures 1A, 1E, 1I, S1A, and S1D) and beta diversity (Figures 1B, 1F, 1J, S1B, and S1E) relative to LAR_G (measured 24 h after acclimation at UCLA post-Earth return). A community shift was observed from week 4.5 onward relative to the basal cohort, likely influenced by the environmental change from KSC to the UCLA Animal Care Facility and many other iatrogenic factors (see “Limitations of the study”). With respect to the F/B ratio (Figures 1C, 1G, and 1K), a metric for gut dysbiosis in GI disease states,40 we observed the same trend in the gut microbiome (Figure 1). This observation held true until termination (week 9) (Figure 1; Tables S1, S2, and S3). In addition, the LAR to Earth (4.5 weeks) time-point analysis revealed enrichment of the gut microbiome genera Lactobacillus (p = 0.03), Ruminiclostridium 9 (p = 0.004), and Shuttleworthia (p = 0.03), with loss of Escherichia-Shigella (p = 0.04) and Hungatella (p = 0.03) (Figure 1). When assessing oral biodiversity from samples obtained from oral swabs, a modest elevation of observed species in LAR versus LAR_G was found at week 4.5 (Figure S1D; Tables S4, S5, and S6) and overall beta diversity at week 9 (Figure S1H), which could be partially attributed to resuming coprophagy upon return to Earth. Overall, however, no major biodiversity changes were observed throughout the study between these cohorts.
Figure 1. Longitudinal analysis of the fecal microbiome (fresh fecal samples) in the live animal return to Earth (LAR) flight cohort versus the ground control (LAR_G) cohort.
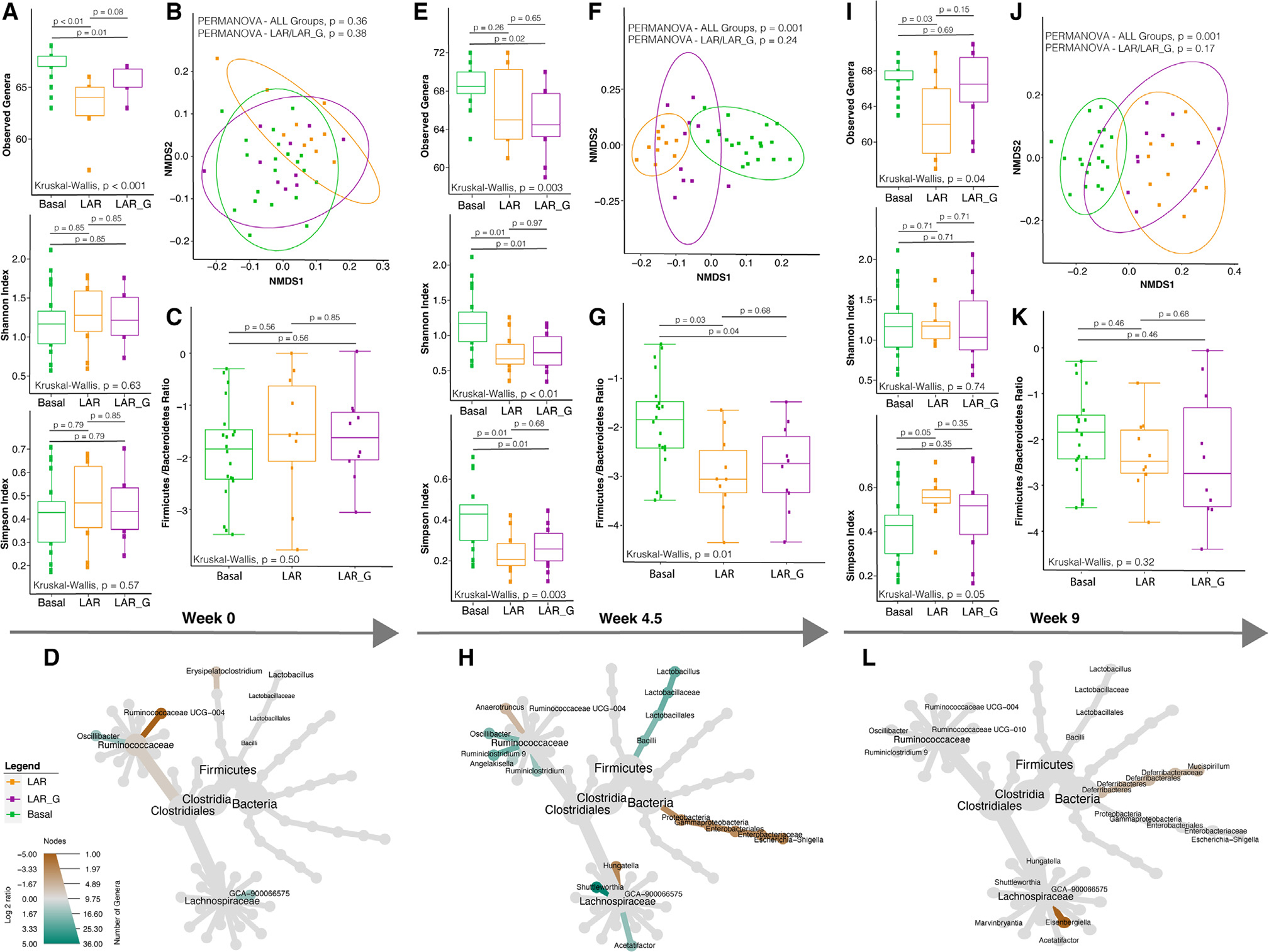
(A, E, I) Longitudinal differences in alpha diversity of observed genera, Shannon, and Simpson diversity indices comparing LAR versus LAR_G cohorts or basal at pre-flight (week 0), live animal return to Earth (week 4.5), and termination (week 9) time points. The non-parametric Kruskal-Wallis statistical test was used to compare groups of two or more. The Wilcoxon rank-sum statistical test was employed for pairwise comparisons. Statistical significance is indicated accordingly.
(B, F, J) Beta diversity analysis comparing LAR versus LAR_G cohorts or basal at pre-flight (week 0), live animal return to Earth (week 4.5), and termination (week 9) time points. Non-parametric multivariate analysis of variance (PERMANOVA) was used to calculate a statistical comparison of variance using distance matrices. Statistical significance is indicated accordingly.
(C, G, K) Comparative analysis of the Firmicutes-to-Bacteroidetes ratio of LAR versus LAR_G or basal cohorts at pre-flight (week 0), live animal return to Earth (week 4.5), and termination (week 9) time points. The non-parametric Kruskal-Wallis statistical test was used to compare groups of two or more. The Wilcoxon rank-sum statistical test was employed for all pairwise comparisons of alpha diversity. Statistical significance is indicated accordingly.
(D, H, L) Comparative analysis of genera enriched or lost in the LAR versus the LAR_G cohorts at pre-flight (week 0), live animal return to Earth (week 4.5), and termination (week 9) time points. Taxa enriched or lost in the LAR cohort at a threshold of p < 0.05 compared with taxa present in the LAR_G cohort are represented in the MetacodeR heat tree by a color intensity log2 median ratio scale. The Wilcoxon rank-sum statistical test was used, and p values are indicated in the text and in Tables S1, S2, and S3.
Microgravity influences microbial diversity and composition of the ISS flight cohort (week 9)
To gain insights into the effects of longer-term exposure to microgravity, the gut microbiome of the ISS cohort was compared with its terrestrial cognate ground control, ISS_G, and basal cohort (experiment 2). Fecal samples were collected during necropsy at termination or week 0 (basal only) after euthanasia, due to pre-determined experimental constraints and RH configuration. Evaluation of the microbiome diversity via 16S rRNA V4 amplicon gene sequencing of the ISS cohort revealed increases in alpha diversity through Shannon and Simpson indices relative to ISS_G, but they were not statistically significant (Figure 2). Furthermore, community structure analysis revealed statistically significant differences in beta diversity (Figure 2B), and the ISS cohort also showed a significant increase in the F/B ratio (Figure 2C) compared with ISS_G, but not basal. These data are consistent with previous reports in both human astronauts2,3 and rodents.6 As such, to further evaluate changes in genera, analysis of differential genera abundance revealed several enriched genera within the ISS cohort, including Clostridium sensu stricto 1 (p = 5.0e−3), Romboutsia (p = 8.0e−4), Ruminiclostridium 9 (p = 0.042), and Shuttleworthia (p = 8.0e−4), and decreases in Hungatella (p = 0.027) (Figure 2D; Table S7). However, Lactobacillus was not found to be statistically significantly enriched.
Figure 2. Alpha diversity, beta diversity, and comparative compositional shift analysis of the fecal microbiome (from necropsy) in the ISS flight cohort versus the ISS_G ground control cohort.
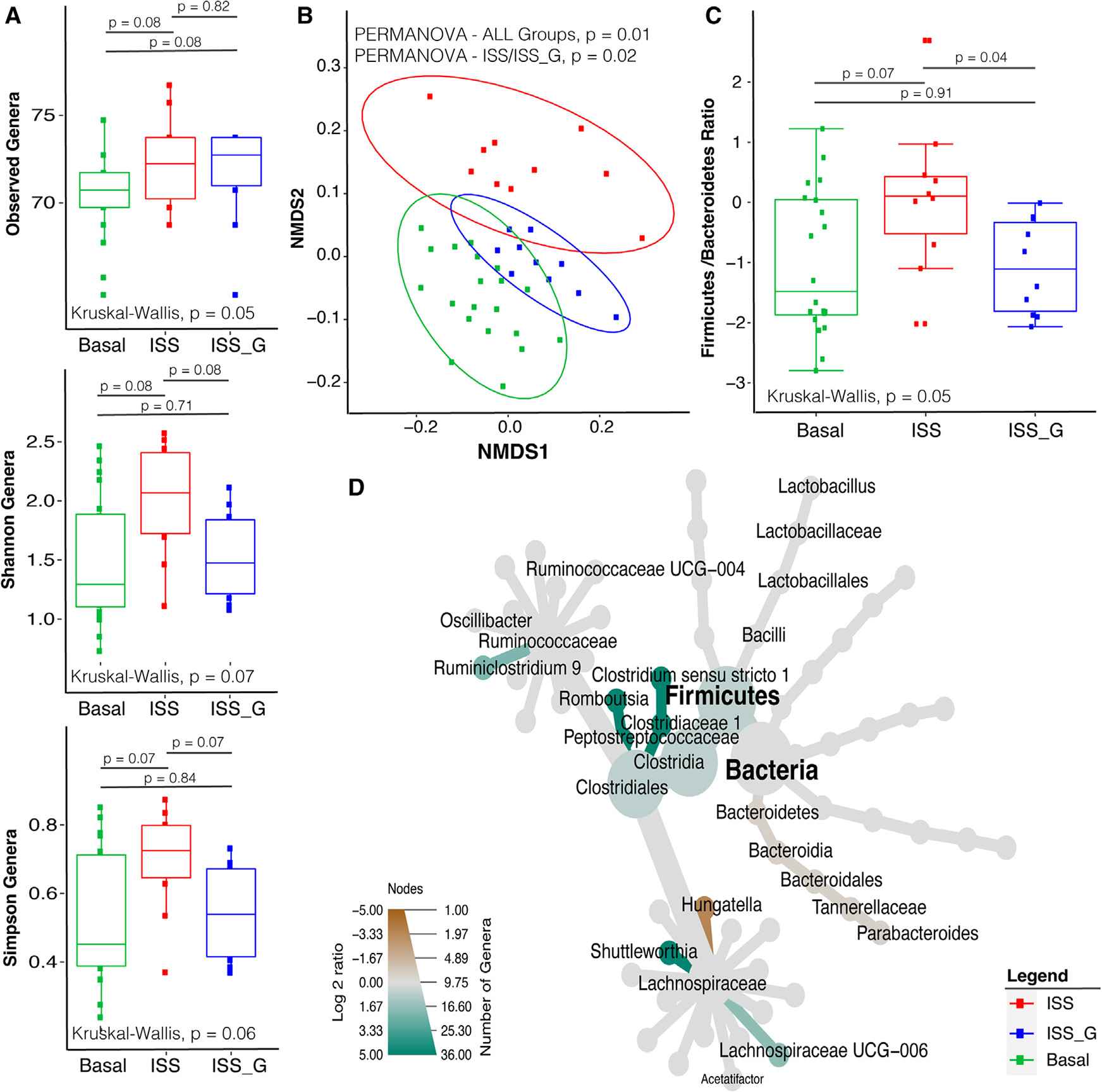
(A) Alpha diversity analysis of observed genera, Shannon, and Simpson diversity indices comparing ISS versus ISS_G or basal cohorts (week 9). The non-parametric Kruskal-Wallis statistical test was used to compare groups of two or more. The Wilcoxon rank-sum statistical test was employed for pairwise comparisons, and statistical significance is indicated accordingly.
(B) Beta diversity analysis comparing the ISS with the ISS_G or basal cohorts (week 9). Non-parametric multivariate analysis of variance (PERMANOVA) was used to calculate a statistical comparison of all cohorts or pairwise using distance matrices. Statistical significance is indicated accordingly.
(C) Comparative analysis of Firmicutes-to-Bacteroidetes ratios between the ISS and the ISS_G or basal cohorts (week 9). The non-parametric Kruskal-Wallis statistical test was used to compare groups of two or more. The Wilcoxon rank-sum statistical test was employed for all pairwise comparisons of alpha diversity. Statistical significance is indicated accordingly.
(D) Comparative analysis of genera enriched or lost in the ISS compared with the ISS_G control cohort (week 9). Taxa enriched or lost in the ISS cohort at a threshold of p < 0.05 compared with taxa present in the ISS_G cohort are represented in the MetacodeR heat tree by a color intensity log2 median ratio scale. The Wilcoxon rank-sum statistical test was used, and p values are indicated in the text.
Significant microbiome compositional structure alterations detected between the ISS and LAR cohorts at termination (week 9)
Understanding how the microbial diversity of the ISS cohort changes with additional microgravity exposure relative to LAR after returning to Earth was highly important. To effectively evaluate the microbiome diversity of the ISS cohort relative to the LAR cohort, additional fecal samples from the LAR and LAR_G cohorts were collected at necropsy after euthanasia at week 9 (Figure S2A; Table S8) to be consistent with ISS, ISS_G, and basal sampling. We observed statistically significant increases in alpha diversity through observed genera, Shannon diversity, and Simpson diversity indices in the ISS versus the LAR cohort (Figure 3A). Community structure analysis revealed statistically significant differences in beta diversity and a significant increase in the F/B ratio (Figures 3B and 3C). Several differential genus abundance comparisons of genera revealed many enriched genera in the ISS cohort relative to LAR (Figure 3D; Table S9) or basal (Figure S2) and intriguingly showed enrichment of Lactobacillus (p = 0.0006).
Figure 3. Alpha diversity, beta diversity, and comparative compositional shift analysis of the fecal microbiome (from necropsy) in the ISS flight cohort versus the LAR flight return cohort.
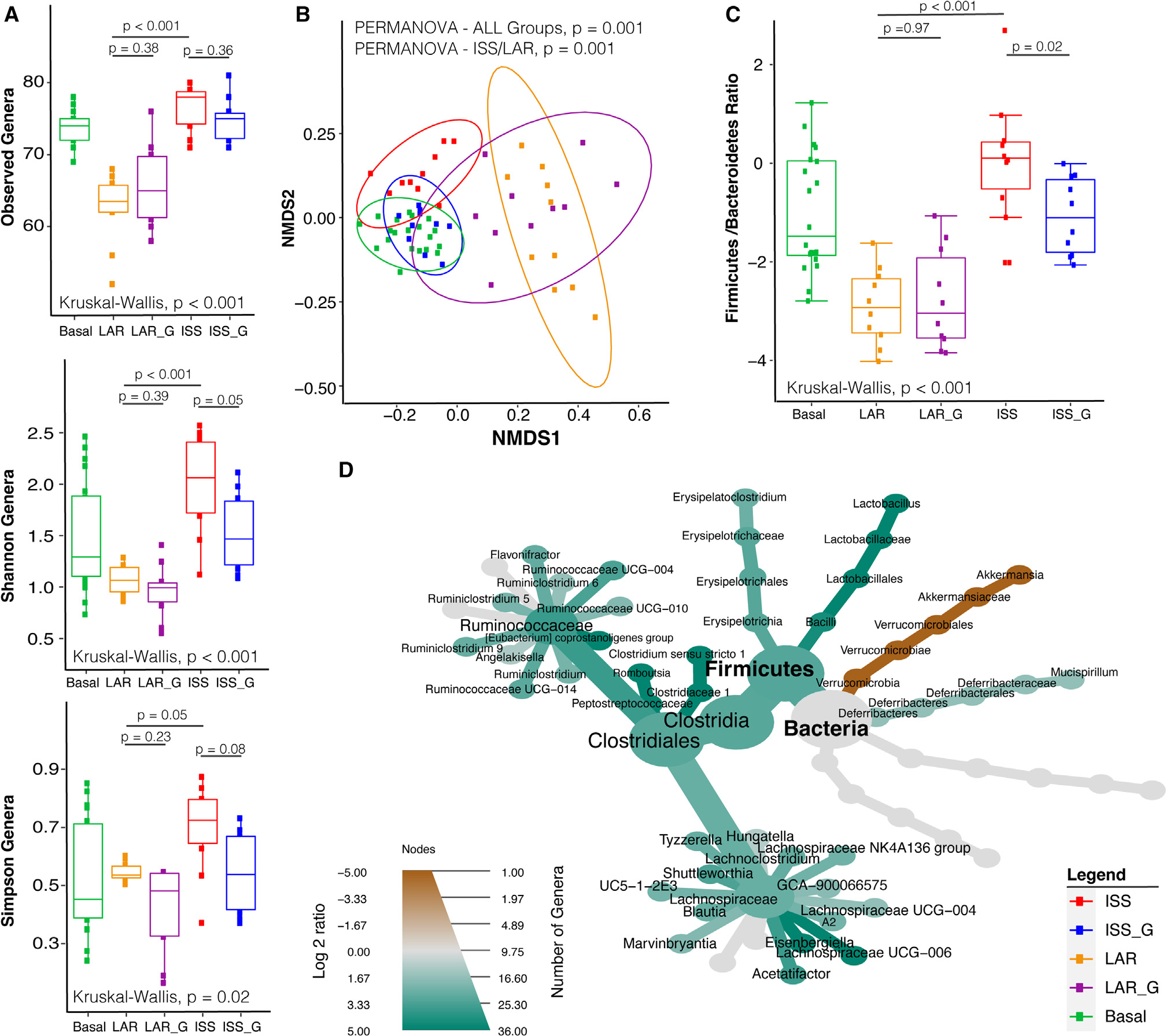
(A) Alpha diversity analysis of observed genera, Shannon, and Simpson diversity indices comparing ISS and LAR or basal cohorts (week 9). The non-parametric Kruskal-Wallis statistical test was used to compare groups of two or more. The Wilcoxon rank-sum statistical test was employed for pairwise comparisons, and statistical significance is indicated accordingly.
(B) Beta diversity analysis between the ISS and the LAR or Basal cohorts (week 9). Non-parametric multivariate analysis of variance (PERMANOVA) was used for statistical comparison using distance matrices between ISS and LAR cohorts. The p values are indicated accordingly.
(C) Comparative analysis of Firmicutes-to-Bacteroidetes ratios between the ISS and the LAR or basal cohorts (week 9). The Wilcoxon rank-sum statistical test was employed to compare ISS versus LAR cohorts, and significance is indicated accordingly.
(D) Comparative analysis of genera enriched or lost in the ISS compared with the LAR control cohort. Taxa enriched or lost in the ISS cohort at a threshold of p < 0.05 compared with taxa present in the LAR cohort are represented in the MetacodeR heat tree by a color intensity log2 median ratio scale. The Wilcoxon rank-sum statistical test was used, and p values are indicated in the text.
Dorea sp. and Lactobacillus murinus are enriched in the ISS gut microbiome, and their specific gene functions are associated with host serum metabolite changes
The above analyses suggested consistent enrichment of genera, including Lactobacillus, in the LAR cohort post-Earth return versus LAR_G (Figure 1H) as well as the ISS cohort versus ISS_G (Figure 2) or LAR (Figure 3). However, to validate the accuracy of the 16S rRNA data analysis and investigate changes in species, deep whole-genome shotgun (WGS) metagenomic sequencing was also performed. Taxonomic differences in the gut microbiome between the ISS (n = 10) and the ISS_G (n = 10) were further investigated as well as elucidation of the functional capacity and differential gene abundance between these cohorts from the same DNA isolates analyzed in Figure 2. Using the Metagenomic Intra-species Diversity Analysis System (MIDAS),41 we carried out species- and gene-level metagenomic analysis of fecal microbiome samples obtained from ISS and ISS_G rodents. MIDAS utilizes the Phy-Eco universal set of single-copy marker genes to determine relative abundance of species-level assignments. Species clusters (from genomes grouped at 95% average nucleotide identity) identified by MIDAS were compiled to generate a set of reference pan-genomes to map reads to obtain gene-level counts. We identified no statistical differences in gene-level alpha diversity, although the ISS was slightly lower than the ISS_G (Figure 4A; Table S10). However, beta diversity analysis showed slight separation (Figure 4B), reflecting compositional changes between the two cohorts. Taxonomic analysis of the MIDAS-assigned pan-genomes resulted in a total of 65 species clusters, with 51 species being shared between the ISS and ISS_G cohorts and 7 species being unique within each cohort. Of the few differentially abundant species between ISS and ISS_G, Dorea sp. and L. murinus species were statistically significantly enriched in the ISS cohort (Figures 4C and 4D). A total of 1,571,490 genes assigned to species clusters were identified by MIDAS, of which 396,080 were assigned a functional annotation. Of those genes assigned functional annotations, 65,547 were identified as differentially abundant when compared by VoomLimma normalized by log2 counts per million (CPM).42 Due to the large number of differentially abundant genes, a strict cutoff was applied (8× coverage, p < 0.001) to highlight the most differentially abundant genes between ISS and ISS_G rodents as well as their associated metabolic functions (Figures 4A and 4D; Table S11). Multiple metabolic pathways were enriched within the ISS cohort compared with the ISS_G cohort (Figure 4E). Lactic acid (L), malic acid (M), glutathione (G), leucine/isoleucine (L/I), and butyric acid metabolism (BM) were all significantly enriched within the ISS cohort (Figure 4F). The functions associated with these pathways from all 396,080 genes were used to identify differentially abundant L-, M-, G-, L/I-, and BM-associated Enzyme Commission (EC) numbers as well as the taxonomic contribution to those EC numbers within each cohort (Figures 4F and S11). Dorea sp. and L. murinus were consistently found to contribute significantly to the enrichment of these pathways in the ISS cohort. Of the five identified differentially abundant EC numbers associated with BM, 60% were found in only one species: L. murinus. Interestingly, deeper analysis of the BM-associated genes also revealed that L. murinus actually provides a unique function within the ISS gut microbiome community; it can convert pyruvate to either meso-(R,S)-2,3-butanediol or para-(R,R)-2,3-butanediol under aerobic and/or anaerobic conditions (Figure 4F; Table S11). The product, 2,3-butanediol, a known end metabolite of pyruvate fermentation in probiotic Lactobacilli, and its role in gut health and gut microbiome research are not well understood, but noted in other studies.43–48
Figure 4. Differentially abundant gene analysis of L. murinus and Dorea sp. within the ISS flight cohort versus the ISS_G ground control cohort.
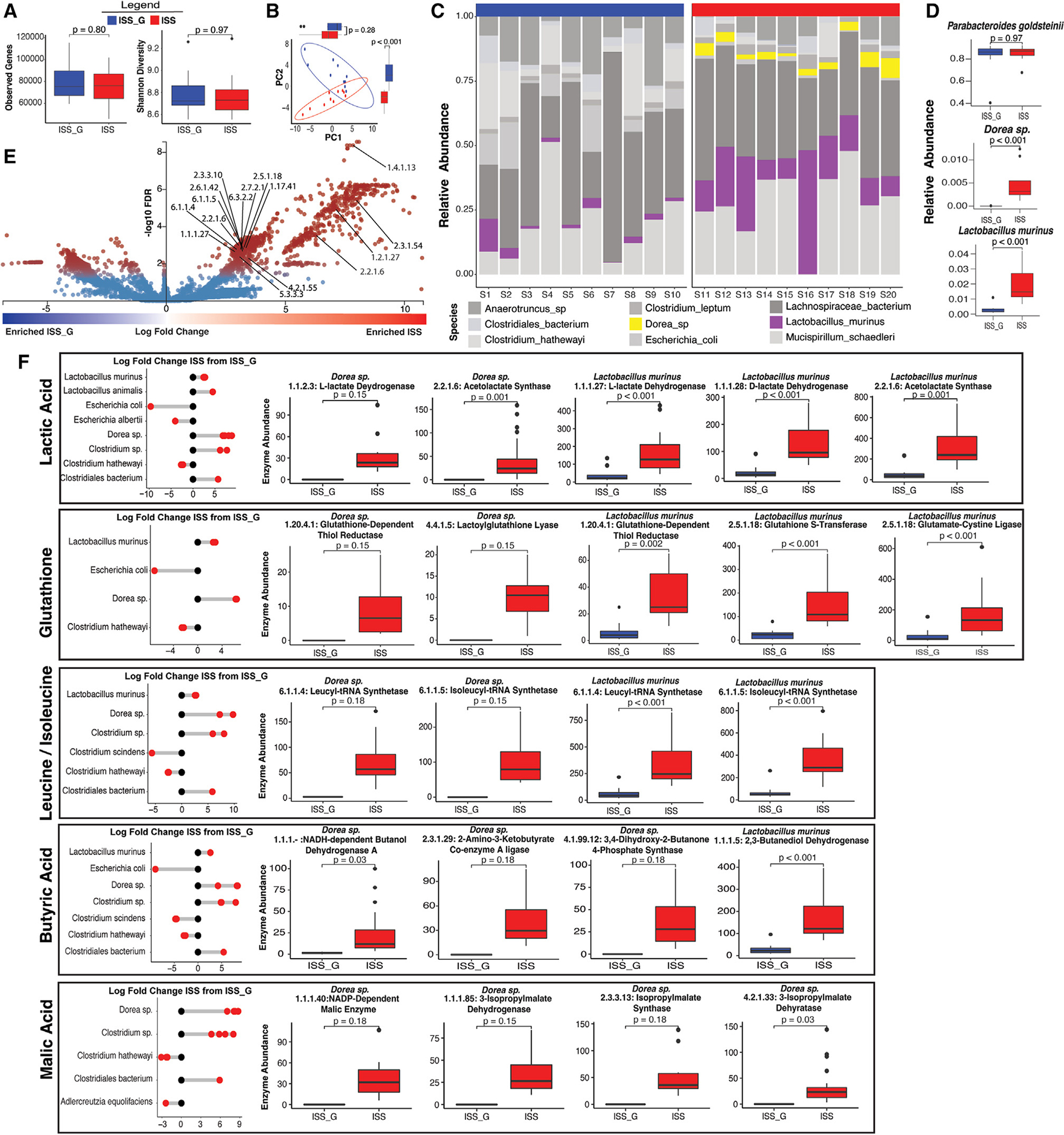
(A) Diversity of genes. A total of 1,571,490 genes were detected with an average of 81,455 (Shannon 8.79) and 76,693 (Shannon 8.78) observed gene hits in the ISS and ISS_G cohorts, respectively. Gene abundance analysis comparing the ISS versus the ISS_G cohort was evaluated through observed and Shannon indices (Wilcox rank-sum test adjusted by false discovery rate (FDR), and p values indicated accordingly).
(B) Principal-coordinate analysis (PCoA) of ordinated CPM-normalized gene counts for ISS_G and ISS. Marginal boxplots show the differences in ordinated distances for PC1 and PC2 by cohort (Wilcoxon test, adjusted by FDR). All boxplots show median and lower/upper quartiles; whiskers show inner fences (see STAR Methods). The p values are indicated accordingly.
(C) Relative abundance of top 9 changing species between ISS and ISS_G. Enriched species L. murinus and Dorea sp. are highlighted.
(D) Relative abundance of select species. Parabacteroides goldsteinii made up nearly 84% of the total relative abundance for both ISS and ISS_G and was excluded from the top 10 most changing bacterial species as no significant difference between ISS and ISS_G for this species was observed. The Wilcox rank-sum statistical test was adjusted by FDR, and p values are indicated accordingly.
(E) Differentially abundant genes between ISS and ISS_G. Log fold changes of 65,547 differentially abundant genes were plotted individually. Significantly changing genes are highlighted in red (padj ≤ 0.05), and non-significantly changing genes are highlighted in blue (padj > 0.05) (see STAR Methods). Select genes with functional annotations of Enzyme Commission numbers (ECs) associated with short-chain fatty acid synthesis (KEGG maps for lactic acid, malic acid, glutathione, leucine/isoleucine, and butyric acid) in mammals were unbiasedly identified and annotated if enriched in the ISS cohort.
(F) Taxonomic contributions to SCFA and differentially abundant metabolite-associated genes between ISS and ISS_G. Log fold changes in EC gene hits within lactic acid, glutathione, leucine/isoleucine, butyric acid, and malic acid pathways (KEGG) are shown for all members that contributed gene hits within the community in relation to ISS_G levels. In addition, individual ECs contributed by enriched species Dorea sp. and L. murinus are highlighted. All boxplots show median and lower/upper quartiles; whiskers show inner fences (see STAR Methods). The Wilcox rank-sum statistical test was adjusted by FDR, and p values are indicated accordingly.
Serum bone remodeling biomarkers and the overall metabolome were altered in spaceflight
The primary RR-5 study demonstrated longitudinal decreases in BMD in the PBS flight group (ISS) relative to PBS ground control (ISS_G).15 Specific decreases in BMD within cortical and trabecular bone of the femur, tibia, and lumbar vertebrae were detected via dual-energy X-ray absorptiometry (DXA) in large overall areas. Decreases in femur trabecular thickness and increases in bone marrow adipogenesis were also found. However, it should be noted that BMD of a small subsection of trabeculae adjacent to the growth plate was also investigated via micro-computed tomography (microCT) scanning and histology to capture changes resulting from PBS or NELL-1 treatment as part of the primary RR-5 objective. MicroCT scanning did not find statistically significant differences between the PBS-treated ISS and ISS_G cohorts, and no histological changes in hydroxyapatite-binding matrix proteins such as osteocalcin (OCN) and tartrate-resistant acid phosphatase (TRAP) were observed in the femur growth plate (see “Limitations of the study”). This significantly smaller region may not reflect the larger areas of BMD decline seen with DXA. This result is not surprising because of discrepancies between DXA and microCT measurements49 as well as variable site-specific responses to microgravity exposure.10,50–53
Terrestrial studies have demonstrated that altered microbiome compositions, including alterations through the use of probiotics,54,55 are implicated in regulating changes observed in BMD and bone matrix16,19,20 through increases in SCFAs, particularly acetic acid, propionic acid, and butyric acid.18,23 In addition, elevated circulation of many osteoblastic formation biomarkers, such as propeptides of type 1 collagen (P1NP) and OCN, and decreased levels of osteoclastic resorption marker CTX-1, as well as histological TRAP staining of osteoclasts, a marker of osteoclastic activity, were found in this association.18,23 Therefore, we hypothesized that these microbiome shifts in the ISS cohort relative to ISS_G could indirectly influence bone homeostasis and investigated these metabolites and biomarkers accordingly. Elevated serum changes in OCN and reductions of TRACP 5b (serum biomarker equivalent of TRAP) were detected when comparing the ISS cohort with ISS_G (Figure 5A), although P1NP abundance was not differentially abundant. When evaluating the abundance of acetic acid, propionic acid, and butyric acid or either of the 2,3-butanediol enantiomers, no differential abundances between the ISS and ISS_G cohorts were detected (Figures 5B and 5C; Tables S12 and S13). Intriguingly, statistically significant increases in abundances of lactic and malic acid were associated with microgravity exposure (Figure 5C; Table S14), as previously observed in human astronauts.2 While cellular metabolism of osteoblasts and mammalian cells certainly produces lactic acid/lactate broadly, gene cluster enrichments encoding lactate dehydrogenase mapped solely to the enriched species L. murinus and Dorea sp. (Figure 4F) and, thus, may have contributed to overall elevated levels detected in serum. Targeted metabolomic analysis (Figures 6A and 6B) demonstrated many other statistically significantly differentially abundant metabolites in the ISS cohort relative to ISS_G (Figure 6; Table S14). For example, the antioxidant Glutathione (Figure 6C), which has been shown to influence redox homeostasis that is essential for osteoblastic function,56 was recently shown to affect the survival of implanted osteoblast precursors in a murine bone regeneration model.57 However, not only was glutathione highly abundant in the ISS cohort sera relative to ISS_G (Figure 6C), but the gene encoding glutamate-cysteine ligase, a crucial enzyme for glutathione synthesis, was functionally enriched in the ISS cohort and associated with the significant increase in L. murinus (Figure 4F), suggesting a possible additive contribution to serum levels of glutathione along with hepatically derived glutathione. In addition, two interesting branched-chain amino acids (BCAAs), leucine and isoleucine, that are actively imported into osteoblasts during chondrogenesis56 were found to be enriched in the ISS cohort serum (Figure 6C). Similarly, gene cluster enrichments for acetolactate synthase and leucyl-tRNA synthetases, responsible for microbial synthesis of BCAAs,58,59 were directly associated with L. murinus and Dorea sp. (Figure 4F). The previous RR-1 predicted relative decreases in microbial-associated putrescine degradation pathways in the gut microbiome; however, the functional outcome was not observed in the ISS cohort relative to ISS_G as shown in the serum metabolite analysis (Figures 6A and 6B).
Figure 5. Bone biomarker and short-chain fatty acid profiling in serum of the ISS flight cohort versus the ISS_G ground control cohort.
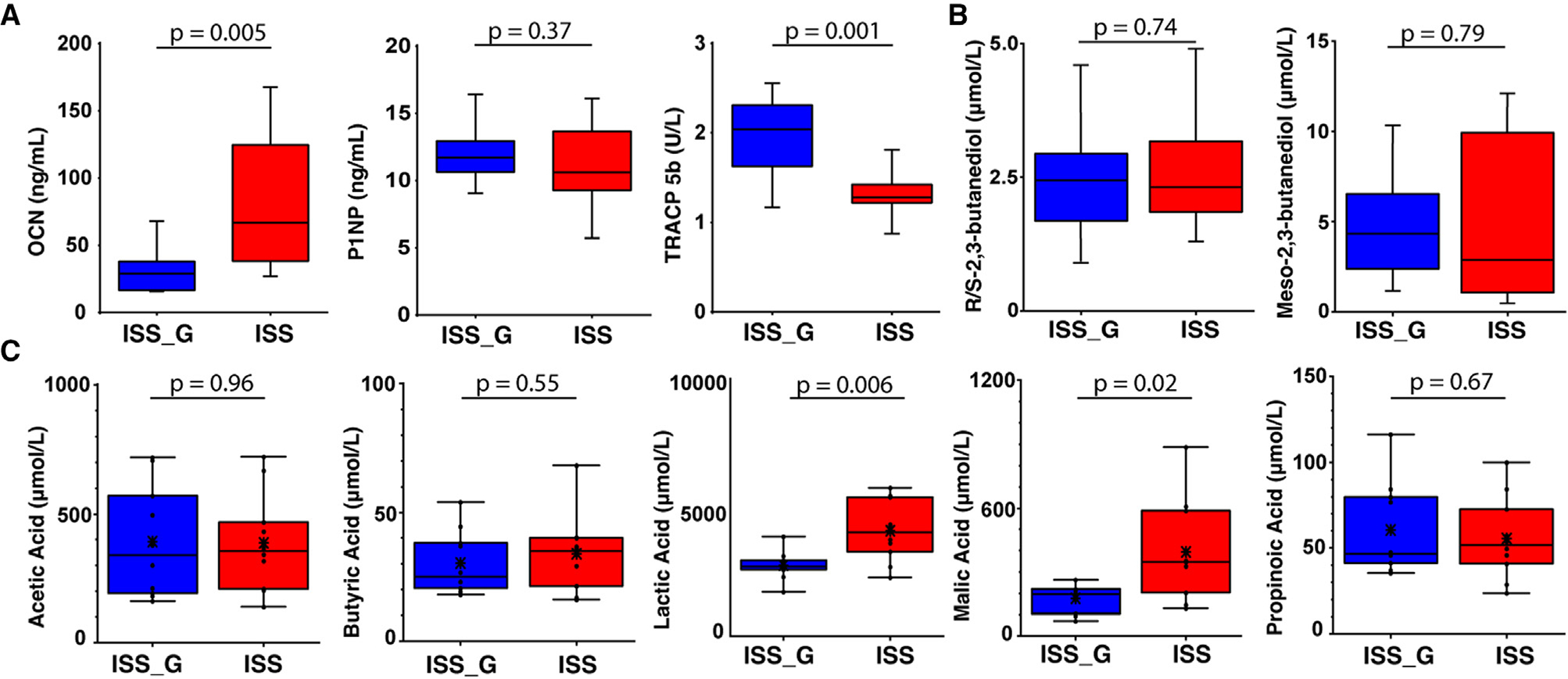
(A) ELISA analysis of bone biomarkers OCN, P1NP, and TRACP in the ISS cohort versus the ISS_G. Unpaired Student’s t test statistical analysis was performed for comparisons of the ISS cohort versus the ISS_G. Statistical significance is indicated accordingly.
(B and C) Absolute quantification of R/S-2,3-butanediol and meso-2,3-butanediol and short-chain fatty acids detected via targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS). Unpaired Student’s t test statistical analysis was performed for comparisons between ISS and ISS_G cohorts. Abundance and individual statistical indications are listed in Tables S12–S14 and statistical significance is indicated accordingly.
Figure 6. Targeted metabolome profiling of serum via liquid-chromatography mass spectrometry in the ISS flight cohort versus the ISS_G ground control cohort.
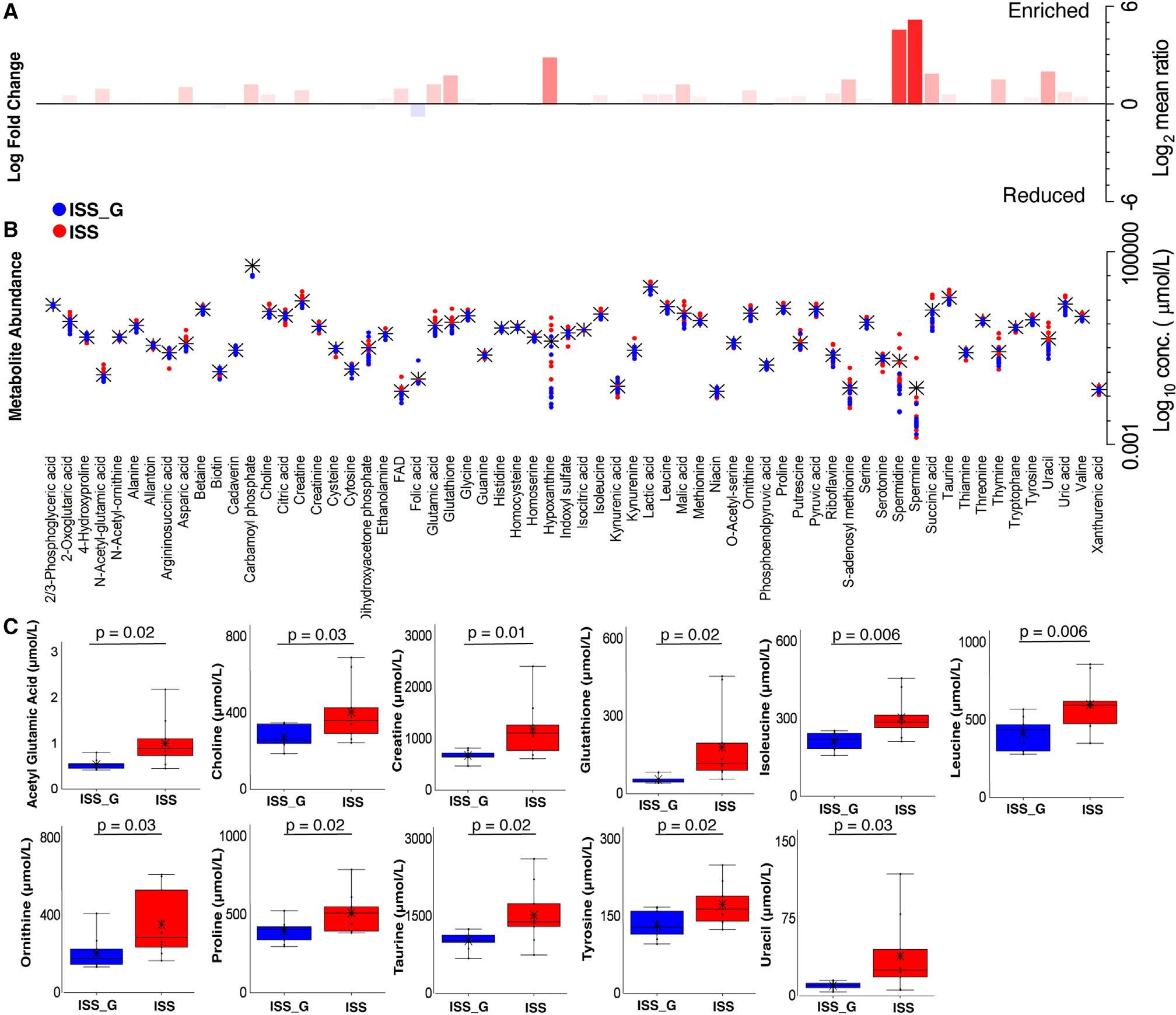
(A and B) Metabolite relative fold change (log2 mean ratios) and abundance distribution in ISS versus ISS_G. Abundance and individual statistical indications are listed in Table S14. Black asterisks indicate the mean abundance, and Student’s t test statistical analysis was employed for pairwise comparisons. Fold changes with log2 mean ratio ≤1 or ≥1 and FDR ≤20 were considered significant.
(C) Enriched metabolites in the ISS versus the ISS_G cohort. Student’s t test statistical analysis was employed for pairwise comparisons. Statistical significance is indicated accordingly.
DISCUSSION
The RR-5 mission enabled evaluation of both the influence of microgravity and the impact of live return (hypergravity) on the rodent gut and oral microbiome. While the gut or oral microbiome diversity of the ISS or LAR cohort could not be sampled on the International Space Station prior to LAR return to Earth due to experimental constraints, the RR-1 study demonstrated a significant increase in alpha and beta diversity at 37 days (~week 5).6 Our data corroborate and further these findings, as elevated diversity was maintained through week 9 of microgravity exposure (Figure 2). Thus, we hypothesized that pre-Earth return, the LAR cohort could display similar diversity profiles. However, when comparing LAR with LAR_G post-Earth return, rapid loss or enrichment of certain existing species was observed (Figures 1E, 1F, 1G, and 1H). In contrast to human studies, which demonstrated increases in richness associated with microgravity and reduction upon returning to Earth within 60 days,3 we observed a reduction in microbial diversity at 24 h post-Earth return when comparing the LAR cohort to LAR_G ground control (see STAR Methods). This phenotype was maintained throughout the remainder of the present study (Figure 1) and could be influenced by resuming coprophagy. However, despite changes in diversity and persistent compositional differences due to caging effects (see “Limitations of the study”), direct conclusions about the health consequences other than the observation of a persistent microbiome shift longitudinally cannot be made. Despite the effects of coprophagy, it is also worth noting that the LAR did not show any significant changes in the diversity within the oral microbiome throughout (Figures S1A–S1I), corroborating the human astronaut tongue3 and saliva60 microbiome analyses.
Ecological gut microbiome dynamics on Earth display a positive correlation between biodiversity and ecosystem stability. Associated decreases in gut microbial diversity are one of the hallmarks of GI disease states, such as inflammatory bowel disease or Crohn’s disease.35,36,61 However, the opposite association is found in oral disease states when evaluating the microbiome,62–65 and oral dysbiosis has empirically been shown to influence GI disease states such as colitis.66 Under these notions, spaceflight effects such as microgravity stress alone should promote dysbiosis within the microbiome, and a decrease in microbial diversity would be expected. Thus, the observed modest increase in gut microbial diversity in the ISS cohort that was exposed to microgravity, shown in this study and other work,2,6 is partially surprising. Despite susceptibility to these spaceflight stressors in rodents and humans alike, it is unclear how these data can be reconciled based solely upon relative abundance or consequential functional outcomes; thus, these paradigms may not hold true in the absence of terrestrial gravitational forces. Nonetheless, long-term exposure to microgravity in low-Earth orbit or beyond the Van Allen radiation belt could impose significant adverse effects, such as systematic and local microenvironmental changes, including known associated dysregulated immune states,67,68 that are likely to have an impact on host microbiomes and induce adaptive and pathophysiological changes in digestive structures and physiology.69,70 Naturally, assessing causal outcomes of dysbiosis, such as inflammation in the colonic epithelial layer, in the ISS cohort versus ISS_G was of high priority, but impossible due to the primary objective of RR-5, which required freezing the carcasses prior to flight return before dissection. Thus, the tissue was compromised from the freeze-thaw event and could not be reliably assessed.
Increasing evidence suggests that an increased F/B ratio is a signature of gut dysbiosis and is correlated with disease states such as inflammatory bowel disease40 and obesity.71 In this study, an increase in relative abundance of Firmicutes, as well as a decrease in Bacteroidetes (F/B ratio >1), was associated with microgravity exposure (Figure 1C), consistent with the pattern observed in the RR-1 study6 and two pivotal human astronaut studies.2,3 Furthermore, when comparing the ISS cohort with LAR to review longitudinal effects of LAR recovery on Earth, we found that the F/B ratio in the ISS cohort was increased relative to the LAR cohort (Figure 3C), as seen previously when astronauts return to Earth from microgravity exposure.2 To evaluate this increase in the F/B ratio along with evidence of elevated genera in the ISS group, deep metagenomic sequencing was performed and revealed that L. murinus, a commensal rodent gut bacterium and of the Firmicutes phylum, was enriched in the ISS rodents in comparison with the ISS_G cohort. Although it is worth noting that, due to the RH Hardware system automatically removing urine and fecal waste via constant airflow in microgravity, the ISS and LAR (until week 4.5) cohort mice were not coprophagic during flight, which may explain some diversity differences, as coprophagy is a known factor influencing the gut microbiome in rodents.72 Conversely, this enabled a rare and true host selection event absent of exogenous microbial colonization as evidenced by a relatively similar microbiome composition, albeit with minor alterations in microbial gene composition in the ISS versus the ISS_G cohort (Figures 4A and 4D). This selection under microgravity could have enabled an opportunistic selection of L. murinus or Dorea sp., which may promote intraspecies growth and indirect benefits to the rodent host. Further mechanistic studies will be required to validate this hypothesis.
We hypothesized that, since the F/B ratio in the ISS versus the ISS_G cohort was increased, a reduction in Bacteroidetes, which are primarily responsible for SCFA production, would present a decrease in SCFAs observed. To validate this hypothesis, we performed metagenomic and targeted metabolomic analyses that unbiasedly displayed statistically significantly enriched predicted gene clusters and metabolites in the ISS cohort relative to ISS_G (Figures 4F, 5C, and 6C; Table S12). Our results showed no differential abundances of acetic/propionic/butyric acid in the ISS cohort serum versus the ISS_G cohort; however, elevated levels of lactic acid and malic acid were enriched in the ISS cohort (Figure 4F), as corroborated by the NASA Twin study.2 Lactate dehydrogenase, which converts pyruvate into lactate, has been shown to be a product of energy metabolism in normal osteoclastic function.56,73,74 Furthermore, the antioxidant glutathione, which is primarily hepatically derived75 and synthesized by some gut microbiota,76 has been shown to influence redox homeostasis, which is essential for osteoblastic function56 and the survival of implanted osteoblast precursors in a murine bone regeneration model.57 Furthermore, the previous RR-1 study indicated a positive correlation between inferred microbial genes encoding glutathione-glutaredoxin redox enzymes and hepatic expression of redox-associated genes, albeit non-significant once accounting for experimental variables that suggest independent regulation between the microbiome and the liver.6 Another important nutritional component of bone maintenance is adequate bioavailability of amino acids. Protein intake was demonstrated to be critical for osteoblast differentiation.77 The BCAAs leucine and isoleucine are actively imported into osteoblasts during chondrogenesis initiated by the osteoblastic transcription factor ATF4, the cognate ligand of RSK2, and mutations in this pathway (Coffin-Lowry syndrome) are associated with skeletal abnormalities.56,78,79 L. murinus and Dorea sp. displayed gene cluster enrichments for acetolactate synthase and leucyl-tRNA synthetases and therefore potentially contribute to a proportion of the increase in leucine/isoleucine detected in ISS sera relative to ISS_G in the current study (Figure 6C).
There is a great need for the expansion of the current arsenal of treatment modalities for prevention and treatment of osteoporosis.17 Previously, probiotic therapy in rodents through the introduction of single Lactobacilli species such as L. helveticus,80 L. reuteri 6475,81 or L. rhamnosus,55 or as part of a cocktail such as VERSUSL#3,55 has been shown to promote increases in BMD. Future mechanistic studies in rodent models are warranted to rigorously evaluate the capacity of L. murinus and Dorea sp. to offset loss of BMD. Most importantly, however, these data encourage more targeted analyses to evaluate the consequences of these observed microbiome shifts and changes in bone homeostasis. This would also enable further insight into the effects of returning to Earth after microgravity exposure, longer durations in low-Earth orbit, and traversing past the Van Allen radiation belt, and beyond.
Limitations of the study
Importantly, there are limitations to the interpreted outcomes worth noting, including behavioral factors like coprophagy72 and iatrogenic effects such as transportation,82–84 vendor source,85–89 environmental psychological stress, caging, bedding, ventilation, and husbandry that can influence the rodent microbiome composition.83,90–92 Accounting for these variables, the RR missions utilize the RH, the next generation of the animal enclosure module (AEM) that was successfully used in rodent spaceflight research in over 26 prior rodent spaceflights. A recent study showed that, with respect to animal health and wellness, including animal growth, organ, body mass, rodent food bar and water consumption, and blood contents, AEM-housed rodents were in normal biological ranges compared with traditional vivarium-housed rodents.93 The RH has since been validated and recapitulated those results as shown in the RR-1 study.12 Therefore, it is important to note that the ISS, ISS_G, LAR (until week 4.5), and LAR_G (until week 4.5) cohorts utilized the RH to standardize conditions across groups. All groups received the same nutrient-upgraded rodent food bar94 throughout the entire study, mitigating diet-induced microbiome effects, as performed in RR-1. Another spaceflight variable worth consideration is the influence of cosmic radiation, which is known to negatively disrupt the GI epithelial integrity and alter the fecal microbiome in simulated spaceflight induced studies.95,96 However, it was eloquently demonstrated in the evaluation of the RR-1 cohort that the gut microbiome diversity increases observed were of a greater magnitude than the space-flight-induced effects,6 and the EXPRESS racks that house the RH on the International Space Station do shield the rodents from cosmic radiation. In addition, the previous STS-135 mission, which had only 13 days of microgravity exposure, did not find significant changes in colonic intestinal injury or inflammatory infiltration.7
Psychological adaptation is another key variable, which can be affected by launch-associated hypergravity and microgravity exposure, but was not within the scope of the RR-5 mission because of previous evaluation. A robust behavior analysis of spaceflight effects and microgravity adaptation of the previous RR-1 mission was conducted using video analysis. Thirty-two-week-old (at launch) female MuRF1 knockout and wild-type (C57BL/NTac-Taconic Biosciences) mice, as well as 16-week-old female wild-type mice (C57BL/6J; The Jackson Laboratory), were assayed for species-typical behavior, including physical activity, grooming, feeding, and circadian rhythm, relative to ground controls for a total of 37 days. The authors showed that both flight groups displayed a full range of species-typical behaviors relative to ground controls, including robust physical activity, effective grooming, and survival of all mice.13 However, “race-tracking” behavior was only detected in the younger flight cohort within 9–11 days of microgravity exposure. Therefore, since the RR-5 mice were similar in age compared with the older mice in this study, we do not believe these behavioral traits had an impact on microbiome compositions. However, it is also worth mentioning that all previous RR missions (RR-1 through RR-4) did not include a nest box (enrichment hut) in the RH. RR-5 was the initial mission to include the nest box, in order to comply with regulatory requirements, and its impacts are still being investigated. As such, future studies would benefit from including an age-matched traditional vivarium box to account for differences resulting from RH nest box utilization by the different cohorts.
We approached evaluation of the taxa present via amplicon sequencing of different variable regions of the 16S rRNA gene in both oral (V1–V3) and gut microbiome samples (V4). It was previously shown that taxa detected in the oral cavity are more distinguishable at the species level than those in the commonly used V3–V4 regions.97 Although this could cause some differences in taxonomic calls, all read sets were assigned using the same classifier and reference database (SILVA-132) and had strict cutoffs for assignment (95%–97%). Therefore, when evaluating differences observed from the 16S rRNA gut microbiome datasets between the ISS and ISS_G cohorts, we utilized high-quality and deeply sequenced WGS datasets, which is where most of this work’s conclusions are supported.
It was recently shown that at week 9, the ISS cohort relative to ISS_G displayed flight-associated longitudinal decreases in BMD within the femur, tibia, and lumbar vertebrae through DXA analysis. Decreases in femur trabecular thickness and increases in bone marrow adipogenesis,15 which is inversely correlated with bone formation,98–100 were also observed. However, the BMD of a small subsection of trabeculae adjacent to the growth plate was also investigated via microCT scanning and histology as part of the experimental design for the primary RR-5 mission. No statistically significant differences between ISS and ISS_G cohorts were found, including when evaluating histological markers, OCN and TRAP, in the analysis of the femur growth plate. These data are surprising, as spaceflight is reproducibly associated with blunted bone formation and increased bone resorption influenced by dysregulated osteoblast and osteoclast function.10 However, this discrepancy could be partially explained by the age of the RR-5 flight cohort (30 weeks old at launch), as previous work has shown that variations in BMD and trabecular microarchitecture are less detectable in older (32-weeks-old) relative to young (9-weeks-old) mice.50 Since mice are considered skeletally mature at ~16 weeks, with BMD peaking and stabilizing at ~30 weeks of age, particularly in the case of BALB/c rodents,31,32 the age of the RR-5 cohort was selected to minimize these variables. The RR-5 mission dataset is the oldest age evaluated for bone homeostasis during spaceflight, compared with 39 days in a recent meta-analysis.101 Bone homeostasis was further evaluated through analysis of the ISS cohort sera versus ISS_G, which revealed decreases in TRACP 5b and increases in OCN, while P1NP remained non-statistically different. Regarding the measurement of OCN, it is important to indicate that the commercial ELISA kit employed cannot distinguish between the γ-carboxylated and uncarboxylated forms of OCN, but rather captures total OCN present. OCN is secreted solely by osteoblasts102 and has high affinity to bone and extracellular matrix in the γ-carboxylated form.103 However, the uncarboxylated form is released into circulation during osteoclastic resorption104 and has been shown to control broad physiological pathways as an endocrine effector.105 Therefore, the P1NP, OCN, and TRACP 5b serum biomarker data may not reflect the overall bone homeostasis in the ISS cohort. Due to experimental design constraints, longitudinal analysis of the bone biomarkers was not possible, and as such, these data at week 9 should not be overinterpreted. The current trends could be due to changes in the ISS_G group; however, that cannot be determined due to the absence of a baseline or an age-matched traditional vivarium control group. Future studies would not only benefit from measuring the two populations (OCN) in relation to BMD measurements to provide further insight into these data, but also include longitudinal assessment throughout. When broadly evaluating the sum of the BMD (DXA and microCT) and biomarker data, we have no empirical evidence to firmly reconcile these phenotypes, of which the underpinnings are unknown. Ultimately, to directly conclude that L. murinus or Dorea sp. harbor any capacity to increase BMD, introduction as individual wild-type species (probiotic therapy), as shown for other Lactobacilli therapies,55,80,81 is required. In future studies, to robustly validate the microbial genetic pathways and the metabolites identified in this study, introducing L. murinus or Dorea sp. mutants overexpressing these metabolites in multiple rodent osteoporotic models would provide the highest quality functional outcome data.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Future inquiries and requests regarding any resources or reagents should be directed to lead contact, Wenyuan Shi (wshi@forsyth.org).
Materials availability
All unique biological samples generated in this study are available from the lead contact with a completed material transfer agreement.
Data and code availability
The un-processed raw 16S rRNA gene and metagenomic WGS sequencing files along with the LC-MS/MS raw data are all deposited in NASA’s GeneLab Open Source Repository (Accession Number: GLDS-417: https://doi.org/10.26030/r6bd-0k20).
All original code scripts used in the pipeline analyses and statistical output files are deposited on Github (https://doi.org/10.5281/zenodo.7620493).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal husbandry
All animals were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the National Aeronautics and Space Administration (NASA) (Protocol number NAS-16-001-Y1) and the University of California, Los Angeles (UCLA) Animal Care Facility (Protocol number 2009-127). Animals were housed in a 12-hr light: dark and temperature-controlled environment. They were given the NASA foodbar (NuRFB)94,107 as food and water ad libitum. The animals used in this study are from the Rodent Research (RR) 5 mission with its primary objective to test an osteoporosis therapy, BP-NELL-PEG.15 In this study, only animals that received control treatment phosphate-buffered saline (PBS) therapy were used and received injections peritoneally, every 2 weeks (0.3 mL). Thus, avoiding influence of the NELL-1 treatment, only these control animals and procedures are described in the methods herein.
25-week-old BALB/c female mice were obtained from Taconic Biosciences (New York, U.S.A.) and acclimated in the Space Station Processing Facility (SSPF) vivarium within Kennedy Space Center (KSC), Florida, U.S.A. Upon receipt, the mice were weighed and then transferred to standard vivarium cages, 5 animals per cage, with standard diet, bedding, and sterilized deionized water via standard sipper tubes ad libitum. The animals were maintained at an appropriate room temperature and placed on a 12-hour light/dark cycle. Cage enrichment (igloos) were also provided. After approximately a week, animals were moved to 10 per cage and acclimation to the NASA Nutrient Upgraded Rodent Foodbars (NuRFB),94 lixit water tubes, and raised wire floors began. 30-week-old experimental animals were assigned to the following cohorts (n=10/cohort) based on average group body mass and DXA measurements: Live-Animal Return cohort (LAR), Full-Term Flight cohort (ISS), Ground control cohort for LAR (LAR_G), and Ground control cohort for ISS (ISS_G). At approximately 48 hours before launch, LAR and ISS groups were loaded into the Rodent Transporter12 and taken to the launch pad. Flight cohorts (LAR and ISS) were flown to space via SpaceX Dragon (CRS-11). Basal animals (n=20) were examined and euthanized 24 hours post rocket launch and carcasses were frozen. Once the Dragon capsule docked to the ISS, the crew members transferred the animals from the Rodent Transporter to the Rodent Habitats12,13 where the animals resided in for the duration of the experiment onboard the International Space Station. Matching ground control cohorts (LAR_G and ISS_G) were housed in identical Rodent Transporter and Habitat units inside the International Space Station Environmental Simulator (ISSES) chambers at NASA KSC, where CO2, temperature, and humidity were matched to that of International Space Station. LAR_G and ISS_G groups were operated on a 3-day offset to allow for ISS environmental telemetry to be received and programmed onto the ISSES chambers. At week 4.5 post-launch, the LAR cohort was returned live to Earth (UCLA) from the International Space Station via Dragon Capsule, and the LAR_G cohort mice were shipped to the UCLA Animal Care Facility from KSC/ISSES. LAR and LAR_G cohorts were both kept at UCLA Animal Care Facility to examine the effects of recovery for 4.5 additional weeks. ISS and ISS_G cohorts remained in the International Space Station and the ISSES at KSC, respectively, for the remaining 4.5 weeks (see Graphical Abstract). All animals were euthanized at termination (week 9 post-launch).
Fecal sample acquisition and harvesting
LAR and LAR_G cohorts were sampled for individual fresh fecal samples from each mouse (in individual, autoclaved cages) at Pre-Flight (week 0), Live Animal Return to Earth (24 hours post arrival: week 4.5), and at Termination (week 9), prior to euthanasia and tissue dissection, for time-point microbiome analysis (Figure 1). The Basal cohort was also sampled (as individual mice, autoclaved cages) for fresh fecal samples at week 0 for comparison prior to euthanasia (Figure 1). ISS and ISS_G cohorts were euthanized at Termination (week 9) in the Space Station and the Kennedy Space Center, respectively. Along with the Basal group (euthanized at week 0), the ISS and ISS_G cohorts were delivered to UCLA Animal Care Facility as frozen carcasses (at −80°C). Thus, fecal pellet samples were acquired at necropsy after carcasses were thawed for tissue harvest and analysis (Figure 2). Additionally, fecal pellets were harvested at necropsy from LAR and LAR_G cohorts to maintain consistent sampling methods enabling true comparison to ISS, ISS_G, and Basal groups (Figure 3). Nominally, two freshly acquired fecal samples or more per mouse were collected and placed in 2.0 mL sterile, nuclease free (RNAase/DNAase), non-pyrogenic, polypropylene Corning Cyrogenic vials. Samples were promptly “flash frozen” with liquid nitrogen for 15 minutes – one hour (within 15 minutes or less of collection) and stored in a −80°C freezer. One fecal pellet (equivalent in mass) was used per mouse for DNA extraction and bacterial DNA was isolated using the QIAGEN Power Fecal DNA isolation kit (QIAGEN, Cat. No. 12955-4) with bead beating. All procedures were approved under IACUC Protocol Number: NAS-16-001-Y1.
Oral sample acquisition and harvesting
LAR and LAR_G cohorts were sampled for individual oral swabs from each mouse (in individual, autoclaved cages) that were acquired at Pre-Flight (week 0), Live Animal Return to Earth (24 hours post arrival week 4.5), and at Termination (week 9), prior to euthanasia and tissue dissection, for time-point microbiome analysis (Figure S1). The Basal cohort was also sampled (as individual mice, autoclaved cages) at week 0 for comparison prior to euthanasia. The molars, cheeks and tongue (lingual and buccal) regions of the LAR and LAR_G rodents (3 swabs per mouse within each cage) were used as a consistent anatomical position for oral microbiome sampling. The rodents were anesthetized via isoflurane and sampled. Specifically, Plasdent Maxaplicator Super fine (2 mm) swabs were used for collection (MPN# 600-R-2). Samples were promptly “flash frozen” with liquid nitrogen for 15 minutes – one hour (within 15 minutes or less of collection) and stored in a −80°C freezer. DNA samples were processed using the Lucigen - Master Pure DNA isolation kit (Ca. No. MC85200). All procedures were approved under IACUC Protocol Number: NAS-16-001-Y1.
METHOD DETAILS
Sequence quality control and noise reduction of fecal microbiome samples
Sequencing of the 16S ribosomal V4 RNA gene was performed as previously described.108 Library preparation for sequencing the V4 region of the 16S rRNA gene consisted of amplification and barcoding using the standard Illumina 515f/806r primer set. After library construction, 2×150bp (paired-end) sequencing was performed on an Illumina HiSeq 2500 platform.109 The reads of amplicons from the V4 region of 16S rRNA were processed using the DADA2 package110 following a standard workflow of quality trimming, de-replicating, DADA2 denoising, read-pair merging and chimera removal steps with the following parameter settings: For quality trimming, truncLen=c(151, 144), maxEE=c(Inf, Inf), minQ=c(0, 0); for error rate learning and DADA2 denoising, selfConsist = TRUE, pool=TRUE; for chimera removal, method = “pooled”. A total of 454 distinct amplicon sequence variants (ASVersus) were identified by DADA2 among all samples. The distinct sequences were sorted according to their total counts in all samples in descending order and assigned a numeric sequence ID, e.g. seq1, seq2, …, seq454, representing from the most abundant sequence to the least abundant sequence in terms of total read count in all samples.
Taxonomy assignment of 16S rRNA amplicons from fecal microbiome samples
Fecal ASV sequences were searched against a reference sequence set containing 16S rRNA gene sequences from all named prokaryotes downloaded from the SILVA high quality ribosomal RNA database(v132)111 using “blastn”. The best hit covering ≥ 95% of the query length was identified for each sequence. If the best hit shares ≥ 98% identity with query sequence, the query sequence is assigned the taxonomy of the hit to the genera level. If the sequence identity between the query and the hit is greater than 97% but less than 98%, the query sequence is assigned taxonomy of the hit to the genus level. If a sequence does not have any hit with ≥ 97% identify, the taxonomy was not assigned. The genus level read count data, generated by the “tax_glom” function of Phyloseq were used in this analysis.
Sequencing noise reduction and sequence quantification of oral samples
The sequencing of the V1-V3 region112 of 16S rRNA gene was completed using a custom protocol113 and on the Illumina Mi-Seq platform. The 100×401 uneven paired-end reads of amplicons from the V1-V3 region of 16S rRNA were processed using the DADA2 package110 for quality trimming, de-replicating, and DADA2 denoising with default settings and following parameters: truncLen=c(100,200), maxN=0, maxEE=c(2,2), truncQ=2. Due to the sequencing gap between the two reads, the read pairs were concatenated by inserting 10 Ns in between two reads with the “justConcatenate=TRUE” option in the DADA2 “mergePairs” function. Chimera were then removed with the “removeBimeraDenovo” function using the “consensus” method. A total of 1,708 ASVersus were identified and were subject to taxonomy assignment.
Taxonomy assignment of 16S rRNA amplicons from oral samples
Oral ASV sequences were searched against a collection of species level full length 16S rRNA reference sequences consisting of the HOMD RefSeq V15.1, HOMD RefSeq Extended V1.11, GreenGene Gold and NCBI 16S rRNA Reference collections, based on a specie-level taxonomy assignment algorithm106 (Detailed algorithm and reference sequences available online https://doi.org/10.5281/zenodo.7620855).
Bacterial diversity and statistical analyses
We profiled the gut microbial abundance and diversity of the PBS flight cohorts ISS and LAR, as well as ground controls, ISS_G and LAR_G, avoiding influence of the NELL-1 treatment. Additionally, only LAR and LAR_G groups were evaluated further for oral microbiome compositions due to experimental constraints. All diversity and statistical analyses were done under the R statistical environment (version 3.6.1).114 ASV count data, along with taxonomy assignment and sample meta information, were imported into R using the PhyloSeq package.115 Count data were sub-sampled into various comparison cohorts (for example, to compare ISS and ISS_G ground control only relevant samples were included). After sub-sampling, ASVs with fewer than 10 reads in at least 2 samples, or fewer than 100 reads across all samples, were excluded, using the “filter_taxa” function.
Alpha diversity analysis was conducted by using the mean ASV counts. To assess the statistical significance of the difference in alpha diversity between samples of two cohorts, the R function “estimate_richness” of the “phyloseq” package was used to calculate alpha diversities using the “Observed”, “Shannon”, and “Simpson” indices. The “kruskal.test” function in R was used as the non-parametric Kruskal-Wallis statistical test to access the significance of differences in samples with more than two groups. The non-parametric Wilcoxon-Rank Sum statistical test (R function “pairwise.wilcox.test”) was then used separately to perform pairwise comparisons with different p value adjustment methods.
Beta diversity was assessed by non-metric multidimensional scaling (NMDS) count data of the comparison cohort were subject to the “ordinate” R function specifying “NMDS” as the ordination method and “bray” specifying Bray-Curtis as the distance calculating function. To assess whether the variation in distances can be explained by the test cohorts, the “adonis” R function (analysis of variance using distance matrices, a form of nonparametric multivariate analysis of variance) were used to partition sums of squares and calculate the R2 and p values (R2 is the portion of the variants that can be explained, and p value indicating the possibility of the result by chance). To identify and illustrate differentially abundant microbes at various taxonomic ranks (i.e., from ASV to domain) the “compare_group” function in the R package metacoder116 was used to determine the differences in median abundances between two cohorts of samples (Figure S2 and Tables S1–S9). The p values were measured using the Wilcoxon Rank-Sum test, followed with adjustment for multiple comparisons using the “fdr” method. Abundance ratios of taxa with non-significant p value (>0.05) were set to zero so that they will not show in the final differential abundance taxonomy tree, which was compiled with the “heat_tree” function in the metacoder. Firmicutes to Bacteroidetes ratios were calculated by agglomerating the read count data to the phylum level using the “tax_glom” function provided by the R PhyloSeq and ggplot2 packages,115 and the log2 count ratios between the two phyla were calculated for each sample. The Wilcoxon Rank-Sum test was then performed to evaluate the difference between the test cohorts.
Metagenome analysis of ISS versus ISS_G cohorts
Whole genome shotgun (WGS) sequencing was performed at the University of Washington’s Northwest Genomics Center (NWGC) on 10 biological replicates for both ISS and ISS_G rodents respectively (n=20). Sequencing libraries were generated using KAPA HTP Library Preparation Kits (07961901001, Roche) and sequenced on an Illumina NovaSeq 6000 System using a S Prime flow cell configured for 300 cycles which resulted in a total of 1,342,813,654 reads with an average of 70,674,403 reads per sample. Raw paired-end reads were then filtered, trimmed for quality, and screened against a mouse (C57BL) reference database using kneadData.117 Filtered reads were then analyzed using the Metagenomic Intra-Species Diversity Analysis System (MIDAS) (database v.1.2, Species Coverage Cutoff 0.01, Merge Sample Depth Cutoff 1.0) (kkerns85/midas_nextflow.git). Species, gene, and single nucleotide polymorphism (SNP) analysis were performed (database v.1.2, Species Coverage Cutoff 0.01, Merge Sample Depth Cutoff 1.0). Species were identified by clustered sub-species (>95% average nucleotide identity) and assigned by a reference genome for that cluster. A total of 67 species clusters were identified. Genes were identified and mapped to pangenomes of these sub-species clusters and then annotated using an in-house annotation pipeline utilizing the Pathosystems Resource Integration Center (PATRIC) Database (v. 3.6.5). Differential species abundance was determined using mean relative abundance between ISS and ISS_G after imposing a cutoff of a row-summed relative abundance ≥ 0.0001 and prevalence ≥ 20% of the total samples (4/20): resulting in a total of 50 species clusters that were then plotted using R (v. 15.6.0; Clustvis118). Differential gene abundance was analyzed using counts per million (CPM) normalized gene counts using the online web server Degust119 which calculated differential abundance by Voom/Limma (Min Gene Read Count 1.0, Min Gen CPM 1.0). Out of 396,080 total genes identified by MIDAS, 64,574 genes were determined to be differentially abundant. Use of a cutoff allowed for more stringent analysis of differentially abundant genes between ISS and ISS_G rodents (FDR p value > 0.001, abs Log Fold Change = 3, > 8X Coverage), which resulted in 651 highly differentially abundant genes. Using the midas_merge function we were able to determine the coverage of genes by taxonomic cohort. In order to better investigate this, we utilized RNASeq2G (unpaired, cohort 0, min read count 10, normalized using trimmed mean of M values (TMM), and log normalized using local polynomial regression, Loess120) within R. TMM normalized gene counts were then used to determine differences in Enzyme Commission numbers (EC’s) for various KEGG annotations. Heatmaps were generated using Clustvis118 in R. TMM normalized gene counts were then converted to using a row z score (x − (mean(x)) / std(x)). Statistical analysis of these differences in EC’s was determined using non-parametric Kruskal-Wallis anova (p values adjusted by FDR). For statistically significant cohorts, the non-parametric Wilcoxon Ranked Sum test was applied (p values adjusted by FDR). EC pathways were mapped using Kyoto encyclopedia of genes and genomes (KEGG) and Metacyc.
LC-MS/MS analysis of rodent sera
Sample preparation
The analysis of the mouse serum metabolome targeted three different classes of molecules that each used different sample preparation and LC-MS/MS protocols: regular polar metabolites (Cohort 1), carbonic acids (Cohort 2), and glycols (Cohort 3). All samples were extracted in a randomized order. First, a stock extract for the three downstream methods was prepared by mixing 5 μL mouse serum with 20 μL methanol. The mixtures were precipitated at −20 °C for 2 hours, 10 min at 20,000 × g and 4 °C and supernatants were transferred into new Eppendorf tubes. For regular polar metabolites (Cohort 1), 2.5 μL extracts were mixed with 50 μL 0.1 % (v/v) formic acid, centrifuged for 10 min at 20,000 × g and 4 °C, transferred into glass vials, and injected (5 μL) directly into the LC-MS/MS system.
Carbonic acids (Cohort 2) were derivatized using the 2-Hydrazinoquinoline-method established by.121 Specifically, 2.5 μL serum extract were mixed with 500 nmol 13C, D4-acetic acid isotope (Cambridge Isotope Laboratories, Ca# CDLM-1581-1) in 50 μL freshly prepared derivatization mix (1 mM 2-hydrazinoquinoline (Alpha Aesar, Ca# H50700MD), 1 mM 2,2′-dipyridil disulfide (Sigma-Aldrich, Ca# 8411090005), and 1 mM triphenylphosphine in acetonitrile (Fischer Scientific, Ca# A998-4,)). The reaction mixture was incubated for 1 h at 60°C, chilled on ice, and quenched with 50 μL H2O. After centrifugation at 20,000 × g and 4°C for 5 min, samples were transferred into glass vials and 10 μL aliquots used for LC-MS/MS-analysis.
Glycols (Cohort 3) were prepared according to.122 2.5 μL blood serum extracts were spiked with 2.5 nmol D4-1,4-butanediol isotope (Cambridge Isotope Laboratories, Inc., Ca# DLM-181-PK) in 7.5 μL water. Then, 10 μL 4 M NaOH were added and mixed with 5 μL benzoyl chloride followed by 5-minute incubation at room temperature. The derivatization reaction was stopped by adding 5 μL 5% (w/v) glycine. Samples were vortexed, incubates for 5 min and the glycol derivatives extracted with 200 mL propane. Samples were centrifuged for 10 min at 4°C and 20,000 × g, supernatants transferred into a fresh Eppendorf tube and evaporated in a SpeedVac to complete dryness. After reconstitution in 50 μL buffer (50% acetonitrile, 50% water, 10 mM ammonium formate pH 2.5), insoluble material was removed by centrifugation and 10 μL aliquots subjected to LC-MS/MS analysis.
Absolute quantitation of polar metabolites, carbonic acids, and glycols in mouse serum by LC-MS/MS
Targeted quantitation was performed on a SCIEX QTRAP 6500 triple quadrupole mass spectrometer (SCIEX, Framingham, MA, USA) equipped with an Ion Drive Turbo V ESI source and a Shimadzu Nexera XR HPLC system (Shimadzu, Kyoto, Japan). Details of the respective high-performance liquid chromatography (HPLC) methods and instrument parameters are summarized in Table S15. For polar metabolites (Cohort 1), a Poroshell 120 EC-C18 (4.6×100 mm, 2.7 μm particle size; Agilent Technologies (Santa Clara, California)) guarded by a Security Guard Ultra C18 precolumn (Phenomenex, Torrance, California) was used for reverse phase separation as described.123 Derivatized carbonic acids were separated using an Acquity UPLC BEH C18 column (2.1×50 mm, 1.7 μm particle size; Waters Corporation (Milford, Massachusetts)) hyphenated to a KrudKatcher Ultra HPLC In-Line Filter (0.2 μm depth × 0.004 inch) as previously described.121 Glycol derivatives were separated using an Atlantis dC18 column (2.1 × 150 mm, 3 μm particle size; Waters Corp.) protected by a KrudKatcher Ultra HPLC In-Line Filter (0.2 μm depth × 0.004 inch) according to.122 The mass spectrometer was optimized for the detection of each molecule to obtain maximum intensity (polarity, collision energy, precursor and product ion selection; see Tables S16, S17, and S18). Samples were analyzed in randomized order and quality control standards were injected at regular intervals during and between analysis batches to monitor instrument performance (analyte separation and detection).
Preparation of calibration standards for quantification
Calibration standards were prepared by serial-dilution of stock solutions containing 64 compounds for polar metabolites, 4 carbonic acids, and 3 glycols. The carbonic acid and glycol standards were derivatized at the same time as the respective sample batches as described above. Calibration curves were collected over a concentration range of 100,000 to 0.0001 nmol using 5 and 10 μL injection volumes (Tables S16–S18). All stock solutions were aliquoted and stored at −20°C until used.
ELISA analysis of rodent serum bone biomarkers
ELISA analysis of rodent serum bone biomarkers was completed using the Mouse propeptide of type 1 procollagen (P1NP) EIA (Immunodiagnostic Systems, East Bolden, United Kingdom) and the Mouse Osteocalcin ELISA (Novus Biologicals, Littleton, CO, USA) by the Multiplex Core at The Forsyth Institute (Cambridge, MA) and have been used in previous studies.124,125 Samples were assayed in duplicate when sufficient volume was available and were diluted 10-fold as per manufacturer’s recommendations for P1NP and 5-fold for OCN analysis. Briefly, samples were thawed on ice, diluted as above, and manufacturer’s protocols followed. The mouse P1NP EIA is a competitive assay where samples, controls, and calibrators are incubated with biotinylated P1NP reagent in wells coated with specific polyclonal anti-P1NP antibody. HRP-labelled avidin is then added, after wash steps, and binds to the biotin complex. TMB colorimetric substrate is added and reaction stopped, absorbance at 450nm with correction at 650nm is read on a microplate reader (SpectraMax 340PC, Molecular Devices, San Jose, CA, USA); the color developed is inversely proportional to the P1NP in the sample. The OCN ELISA is a sandwich ELISA assay, whereby samples and standards are added to a plate pre-coated with anti-mouse OCN. A biotinylated detection antibody and Avidin-HRP conjugate are added in succession. Color substrate is added after all free components are washed away. The reaction is stopped, and optical density is measured at 450nm on a microplate reader. Optical density is proportional to the concentration of mouse OCN. The tartrate-resistant acid phosphatase 5b (TRACP 5b) ELISA is a solid phase immunofixation assay and uses antibody coated microtiter wells to bind TRACP 5b for quantification. After binding and wash, a solution of chromogenic substrate is introduced and incubated for 2 hours. The reaction is chemically halted and absorbance at 405nm is measured on a microplate reader. Color intensity is directly proportional to TRACP 5b amount and activity. All data for P1NP and OCN were calculated based on a 4-parameter logistic curve and adjusted for dilution factors, while data for TRACP 5b were calculated based on linear regression using concentration standards.
QUANTIFICATION AND STATISTICAL ANALYSIS
As microbiome datasets are typically distributed non-normally, thus, all microbial or gene diversity analysis of samples resulting from the 16S rRNA gene or WGS sequencing data were statistically assessed for their differences accordingly: using the R functions to perform the non-parametric Kruskall-Wallis statistical test for comparisons with more than two groups or the Wilcoxon Rank-Sum statistical test for unpaired two-sample statistical comparisons. Beta diversity assessed via NMDS was an analysis of variance using distance matrices, a form of non-parametric multivariate analysis of variance. Statistical analysis of these differences in EC’s, resulting from the metagenomic analysis, was determined using the non-parametric Kruskal-Wallis statistical test (p values adjusted by FDR). For statistically significant cohorts, the non-parametric Wilcoxon Ranked Sum test was applied (p values adjusted by FDR). Statistical output files are available along with all scripts used for pipeline analysis (see key resource table). Raw MS-data were processed using MultiQuant 3.0.1 software (SCIEX) and targeted metabolites were detected using multiple reaction monitoring (MRM) parameters listed in Table S15. Extracted peak areas were used for quantitation based on individual linear calibration curves. For carbonic acids and glycols, the internal isotope-labelled standards were used to normalize analyte recovery and matrix effects. The resulting data was further analyzed in Microsoft Excel for Student t test statistical analysis. Fold changes with log2 mean ratio ≤ or ≥ 1 and FDR ≤ 20 were considered significant. All ELISA data was further analyzed in Graph Pad Prism 6.0 Software (La Jolla, CA), where the parametric un-paired Student’s t test statistical test was utilized for comparison.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Biological samples | ||
|
| ||
| Fecal pellets for gut microbiome sampling | This study | N/A |
| Oral swabs for microbiome sampling | This study | N/A |
| Serum | This study | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetic Acid-D4 Isotope | Cambridge Isotope Laboratories | Ca# CDLM-1581-1 |
| 1,4-Butanediol-2,2,3,3-D4 Isotope | Cambridge Isotope Laboratories | Ca# 1,4-BUTANEDIOL-2,2,3,3-D4 |
| Acetic Acid-D4 Isotope | Cambridge Isotope Laboratories | Ca# CDLM-1581-1 |
| Meso-2,3-Butanediol | Sigma-Aldrich | Ca# 361461-10G |
| (2R, 3R)-2,3-Butanediol | Sigma-Aldrich | Ca# 237639-1G |
| (2S, 3S)-2,3-Butanediol | Sigma-Aldrich | Ca# 300349-1G |
| Butyric acid | Alfa Aesar | Ca# L13189AE |
| 2-Hydrazinoquinoline | Alfa Aesar | Ca# AAH50700MD |
| Folic acid | Alfa Aesar | Ca# AAJ6083314 |
| Lactic acid | Fischer Chemical | Ca# A159-500 |
| Malic acid | Alfa Aesar | Ca# AAJ6322122 |
| Propionic acid | Sigma-Aldrich | Ca# 94425-5MLF |
| Pyruvic acid | SPEX CertiPrep | Ca# S-3242 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse Propeptide of Type 1 Procollagen (P1NP) EIA | Immunodiagnostic Systems, East Bolden, United Kingdom | Ca# AC-33F1 |
| Mouse Osteocalcin (OCN) ELISA | Novus Biologicals, Littleton, CO, USA | Ca# NBP2-68151 |
| Tartrate-resistant acid phosphatase 5b (TRAP 5b) ELISA | Immunodiagnostic Systems, East Bolden, United Kingdom | Ca# SB-TR103 |
|
| ||
| Deposited data | ||
|
| ||
| All raw DNA sequencing and metabolomic datasets | NASA Gene Lab | Accession Number: GLDS-417 https://doi.org/10.26030/r6bd-0k20 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| BALB/c | Shi15 | Taconic Biosciences |
|
| ||
| Software and algorithms | ||
|
| ||
| 16S rRNA gene and metagenomic sequencing pipeline | This study | https://doi.org/10.5281/zenodo.7620493 |
| Forsyth eHOMD NGS pipeline | Al-Hebshi et al.106 | https://doi.org/10.5281/zenodo.7620855 |
Supplementary Material
Highlights.
NASA’s RR-5 mission investigated ancillary effects on the microbiome via live animal return
Abundance of L. murinus and Dorea sp. is elevated under microgravity exposure
Identified pathways capable of producing lactic acid, leucine, and glutathione are enriched
These metabolites are elevated in the serum of flight cohorts, which lose bone homeostasis
ACKNOWLEDGMENTS
The authors would like to thank the NASA Ames Rodent Research Team and the Kennedy Space Center as well as the Space Station Processing Facility Annex Team, especially Ramona Bober, for experimental preparation and facilitation. A special thanks to BioServe Technologies, particularly Shankini Doraisingam, Mark Rupert, and Shannon Floyd, for experimental preparation and facilitation. The authors would also like to thank the ISSNL for their expertise and background support, which enabled high-quality sample acquisition. We would like to thank the UCLA Microbiome Center’s Microbiome Core, particularly the director, Jonathan Jacobs, MD, PhD, and core manager, Venu Lagishetty, PhD, for consultation and performing the gut 16S rRNA V4 gene sequencing data generated in this study. The authors would like to thank the Forsyth Institute Oral Microbiome Sequencing Core for consultation on the oral 16S rRNA V1–V3 gene sequencing methodology and data generated in this study, as well as Yanmei Huang, PhD, for initial consultations on the preliminary gut 16S rRNA V4 gene sequencing analysis. The authors would like to thank Archita Gadkari for processing the ISS and ISS_G DNA samples further and the University of Washington’s Northwest Genomics Center for performing the metagenomic sequencing. We thank the Multiplex Core at The Forsyth Institute, which provided all multiplex ELISA protocol execution and protein analysis services. Last, the authors would also like to thank Lauren Quigley, PhD, and Jacqueline Starr, PhD, for microbial diversity assessment and biostatistical consultation in this study, respectively. Research reported in this study was supported by the Forsyth Pilot Grant Program, by the Forsyth Center for Salivary Diagnostics through the Massachusetts Life Science Center, and partially by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under awards F31DE026057 and R01DE023810.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112299.
REFERENCES
- 1.Afshinnekoo E, Scott RT, MacKay MJ, Pariset E, Cekanaviciute E, Barker R, Gilroy S, Hassane D, Smith SM, Zwart SR, et al. (2020). Fundamental biological features of spaceflight: advancing the field to enable deep-space exploration. Cell 183, 1162–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett-Bakelman FE, Darshi M, Green SJ, Gur RC, Lin L, Macias BR, McKenna MJ, Meydan C, Mishra T, Nasrini J, et al. (2019). The NASA Twins Study: a multidimensional analysis of a year-long human spaceflight. Science 364, eaau8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voorhies AA, Mark Ott C, Mehta S, Pierson DL, Crucian BE, Feiveson A, Oubre CM, Torralba M, Moncera K, Zhang Y, et al. (2019). Study of the impact of long-duration space missions at the International Space Station on the astronaut microbiome. Sci. Rep. 9, 9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urbaniak C, Lorenzi H, Thissen J, Jaing C, Crucian B, Sams C, Pierson D, Venkateswaran K, and Mehta S (2020). The influence of spaceflight on the astronaut salivary microbiome and the search for a microbiome biomarker for viral reactivation. Microbiome 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Luo G, Du R, Sun W, Li J, Lan H, Chen P, Yuan X, Cao D, Li Y, et al. (2020). Effects of spaceflight on the composition and function of the human gut microbiota. Gut Microb. 11, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang P, Green SJ, Chlipala GE, Turek FW, and Vitaterna MH (2019). Reproducible changes in the gut microbiome suggest a shift in microbial and host metabolism during spaceflight. Microbiome 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie LE, Taddeo SS, Weeks BR, Lima F, Bloomfield SA, Azcarate-Peril MA, Zwart SR, Smith SM, and Turner ND (2015). Space environmental factor impacts upon murine colon microbiota and mucosal homeostasis. PLoS One 10, e0125792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor GR, Graves RC, Brockett RM, Ferguson JK, and Mieszkuc BJ (1977). Skylab Environmental and Crew Microbiology Studies. [Google Scholar]
- 9.Johnston RS, and Dietlein LF (1977). Biomedical Results from Skylab (Scientific and Technical Information Office, National Aeronautics and Space). [Google Scholar]
- 10.Coulombe JC, Senwar B, and Ferguson VL (2020). Spaceflight-induced bone tissue changes that affect bone quality and increase fracture risk. Curr. Osteoporos. Rep. 18, 1–12. [DOI] [PubMed] [Google Scholar]
- 11.Vico L, and Hargens A (2018). Skeletal changes during and after spaceflight. Nat. Rev. Rheumatol. 14, 229–245. [DOI] [PubMed] [Google Scholar]
- 12.Choi SY, Saravia-Butler A, Shirazi-Fard Y, Leveson-Gower D, Stodieck LS, Cadena SM, Beegle J, Solis S, Ronca A, and Globus RK (2020). Validation of a new rodent experimental system to investigate consequences of long duration space habitation. Sci. Rep. 10, 2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronca AE, Moyer EL, Talyansky Y, Lowe M, Padmanabhan S, Choi S, Gong C, Cadena SM, Stodieck L, and Globus RK (2019). Behavior of mice aboard the international space station. Sci. Rep. 9, 4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanjaya J, Lord EL, Wang C, Zhang Y, Kim JK, Nguyen A, Baik L, Pan HC, Chen E, Kwak JH, et al. (2018). The effects of systemic therapy of PEGylated NEL-like protein 1 (NELL-1) on fracture healing in mice. Am. J. Pathol. 188, 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi J 2019. Systemic Therapy of Inactivated-Bisphosphonate-Conjugated PEGylated NELL-1 (BP-NELL-PEG) for Spaceflight-Induced Osteoporosis. Doctoral dissertation (University of California, Los Angeles). https://escholarship.org/uc/item/0bk9h299 [Google Scholar]
- 16.Hernandez CJ, Guss JD, Luna M, and Goldring SR (2016). Links between the microbiome and bone. J. Bone Miner. Res. 31, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacifici R (2018). Bone remodeling and the microbiome. Cold Spring Harb. Perspect. Med. 8, a031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan J, Herzog JW, Tsang K, Brennan CA, Bower MA, Garrett WS, Sartor BR, Aliprantis AO, and Charles JF (2016). Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 113, E7554–E7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sjögren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, Bäckhed F, and Ohlsson C (2012). The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 27, 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guss JD, Horsfield MW, Fontenele FF, Sandoval TN, Luna M, Apoorva F, Lima SF, Bicalho RC, Singh A, Ley RE, et al. (2017). Alterations to the gut microbiome impair bone strength and tissue material properties. J. Bone Miner. Res. 32, 1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guss JD, Taylor E, Rouse Z, Roubert S, Higgins CH, Thomas CJ, Baker SP, Vashishth D, Donnelly E, Shea MK, et al. (2019). The microbial metagenome and bone tissue composition in mice with microbiome-induced reductions in bone strength. Bone 127, 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charles JF, Ermann J, and Aliprantis AO (2015). The intestinal microbiome and skeletal fitness: Connecting bugs and bones. Clin. Immunol. 159, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucas S, Omata Y, Hofmann J, Böttcher M, Iljazovic A, Sarter K, Albrecht O, Schulz O, Krishnacoumar B, Krönke G, et al. (2018). Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macfarlane S, and Dillon JF (2007). Microbial biofilms in the human gastrointestinal tract. J. Appl. Microbiol. 102, 1187–1196. [DOI] [PubMed] [Google Scholar]
- 25.Morrison DJ, and Preston T (2016). Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microb. 7, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, Duncan G, Johnstone AM, Lobley GE, Wallace RJ, et al. (2011). High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 93, 1062–1072. [DOI] [PubMed] [Google Scholar]
- 27.Zhao C, Dong H, Zhang Y, and Li Y (2019). Discovery of potential genes contributing to the biosynthesis of short-chain fatty acids and lactate in gut microbiota from systematic investigation in E. coli. NPJ Biofilms Microbiomes 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, and Rudensky AY (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- 30.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlo-oly-y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beamer WG, Donahue LR, Rosen CJ, and Baylink DJ (1996). Genetic variability in adult bone density among inbred strains of mice. Bone 18, 397–403. [DOI] [PubMed] [Google Scholar]
- 32.Buie HR, Moore CP, and Boyd SK (2008). Postpubertal architectural developmental patterns differ between the L3 vertebra and proximal tibia in three inbred strains of mice. J. Bone Miner. Res. 23, 2048–2059. [DOI] [PubMed] [Google Scholar]
- 33.Voorhies AA, and Lorenzi HA (2016). The challenge of maintaining a healthy microbiome during long-duration space missions. Front. Astron. Space Sci. 3, 23. [Google Scholar]
- 34.Saulnier DM, Riehle K, Mistretta TA, Diaz MA, Mandal D, Raza S, Weidler EM, Qin X, Coarfa C, Milosavljevic A, et al. (2011). Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology 141, 1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, and Pace NR (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, Tysk C, and Jansson JK (2009). Twin studies reveal specific imbalances in the mucosaassociated microbiota of patients with ileal Crohn’s disease. Inflamm. Bowel Dis. 15, 653–660. [DOI] [PubMed] [Google Scholar]
- 37.Sartor RB (2008). Microbial influences in inflammatory bowel diseases. Gastroenterology 134, 577–594. [DOI] [PubMed] [Google Scholar]
- 38.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwabe RF, and Jobin C (2013). The microbiome and cancer. Nat. Rev. Cancer 13, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson DA, Frank DN, Pace NR, and Gordon JI (2008). Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 3, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nayfach S, Rodriguez-Mueller B, Garud N, and Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji X-J, Huang H, and Ouyang P-K (2011). Microbial 2, 3-butanediol production: a state-of-the-art review. Biotechnol. Adv. 29, 351–364. [DOI] [PubMed] [Google Scholar]
- 44.Oliphant K, and Allen-Vercoe E (2019). Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome 7, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolte LA, Vich Vila A, Imhann F, Collij V, Gacesa R, Peters V, Wijmenga C, Kurilshikov A, Campmans-Kuijpers MJE, Fu J, et al. (2021). Long-term dietary patterns are associated with pro-inflammatory and anti-inflammatory features of the gut microbiome. Gut 70, 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whiteson KL, Meinardi S, Lim YW, Schmieder R, Maughan H, Quinn R, Blake DR, Conrad D, and Rohwer F (2014). Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2, 3-butanedione fermentation. ISME J. 8, 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen M, Sharma A, Wu W, Gomi R, Sung B, Hospodsky D, Angenent LT, and Worgall S (2016). The fermentation product 2, 3-butanediol alters P. aeruginosa clearance, cytokine response and the lung microbiome. ISME J. 10, 2978–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkataraman A, Rosenbaum MA, Werner JJ, Winans SC, and Angenent LT (2014). Metabolite transfer with the fermentation product 2, 3-butanediol enhances virulence by Pseudomonas aeruginosa. ISME J. 8, 1210–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs AM, Perilli E, Parkinson IH, Wrigley TV, Fazzalari NL, Kantor S, and Wark JD (2010). Novel assessment of subregional bone mineral density using DXA and pQCT and subregional microarchitecture using micro-CT in whole human vertebrae: applications, methods, and correspondence between technologies. J. Clin. Densitom. 13, 161–174. [DOI] [PubMed] [Google Scholar]
- 50.Coulombe JC, Sarazin BA, Mullen Z, Ortega AM, Livingston EW, Bateman TA, Stodieck LS, Lynch ME, and Ferguson VL (2021). Microgravity-induced alterations of mouse bones are compartment- and site-specific and vary with age. Bone 151, 116021. [DOI] [PubMed] [Google Scholar]
- 51.Collet PH, Uebelhart D, Vico L, Moro L, Hartmann D, Roth M, and Alexandre C (1997). Effects of 1-and 6-month spaceflight on bone mass and biochemistry in two humans. Bone 20, 547–551. [DOI] [PubMed] [Google Scholar]
- 52.LeBlanc A, Schneider V, Shackelford L, West S, Oganov V, Bakulin A, and Voronin L (2000). Bone mineral and lean tissue loss after long duration space flight. J. Musculoskelet. Neuronal Interact. 1, 157–160. [PubMed] [Google Scholar]
- 53.McCarthy I, Goodship A, Herzog R, Oganov V, Stussi E, and Vahlensieck M (2000). Investigation of bone changes in microgravity during long and short duration space flight: comparison of techniques. Eur. J. Clin. Invest. 30, 1044–1054. [DOI] [PubMed] [Google Scholar]
- 54.Schwarzer M, Makki K, Storelli G, Machuca-Gayet I, Srutkova D, Hermanova P, Martino ME, Balmand S, Hudcovic T, Heddi A, et al. (2016). Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science 351, 854–857. [DOI] [PubMed] [Google Scholar]
- 55.Li J-Y, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, Darby TM, Weitzmann MN, Mulle JG, Gewirtz AT, et al. (2016). Sex steroid deficiency–associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Invest. 126, 2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee W-C, Guntur AR, Long F, and Rosen CJ (2017). Energy metabolism of the osteoblast: implications for osteoporosis. Endocr. Rev. 38, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stegen S, van Gastel N, Eelen G, Ghesquière B, D’Anna F, Thienpont B, Goveia J, Torrekens S, Van Looveren R, Luyten FP, et al. (2016). HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 23, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Umbarger HE, and He U (1978). Amino Acid Biosynthesis and its Regulation. [DOI] [PubMed] [Google Scholar]
- 59.LaRossa RA, Van Dyk TK, and Smulski DR (1987). Toxic accumulation of alpha-ketobutyrate caused by inhibition of the branched-chain amino acid biosynthetic enzyme acetolactate synthase in Salmonella typhimurium. J. Bacteriol. 169, 1372–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morrison MD, Thissen JB, Karouia F, Mehta S, Urbaniak C, Venkateswaran K, Smith DJ, and Jaing C (2021). Investigation of spaceflight induced changes to astronaut microbiomes. Front. Microbiol. 12, 659179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mosca A, Leclerc M, and Hugot JP (2016). Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front. Microbiol. 7, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao L, Xu T, Huang G, Jiang S, Gu Y, and Chen F (2018). Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell 9, 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng H, Xu L, Wang Z, Li L, Zhang J, Zhang Q, Chen T, Lin J, and Chen F (2015). Subgingival microbiome in patients with healthy and ailing dental implants. Sci. Rep. 5, 10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee W-H, Chen H-M, Yang S-F, Liang C, Peng C-Y, Lin F-M, Tsai L-L, Wu B-C, Hsin C-H, Chuang C-Y, et al. (2017). Bacterial alterations in salivary microbiota and their association in oral cancer. Sci. Rep. 7, 16540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsai C-Y, Tang CY, Tan T-S, Chen K-H, Liao K-H, and Liou M-L (2018). Subgingival microbiota in individuals with severe chronic periodontitis. J. Microbiol. Immunol. Infect. 51, 226–234. [DOI] [PubMed] [Google Scholar]
- 66.Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG Iii, Hayashi A, Imai J, Sugihara K, Miyoshi M, Brazil JC, Kuffa P, et al. (2020). The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell 182, 447–462.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crucian BE, Stowe RP, Pierson DL, and Sams CF (2008). Immune system dysregulation following short-vs long-duration spaceflight. Aviat Space Environ. Med. 79, 835–843. [DOI] [PubMed] [Google Scholar]
- 68.Chang TT, Spurlock SM, Candelario TLT, Grenon SM, and Hughes-Fulford M (2015). Spaceflight impairs antigen-specific tolerance induction in vivo and increases inflammatory cytokines. FASEB J. 29, 4122–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rabot S, Szylit O, Nugon-Baudon L, Meslin J-C, Vaissade P, Popot F, and Viso M (2000). Variations in digestive physiology of rats after short duration flights aboard the US space shuttle. Dig. Dis. Sci. 45, 1687–1695. [DOI] [PubMed] [Google Scholar]
- 70.Arun CP (2004). The importance of being asymmetric: the physiology of digesta propulsion on Earth and in space. Ann. N. Y. Acad. Sci. 1027, 74–84. [DOI] [PubMed] [Google Scholar]
- 71.Ley RE, Turnbaugh PJ, Klein S, and Gordon JI (2006). Human gut microbes associated with obesity. nature 444, 1022–1023. [DOI] [PubMed] [Google Scholar]
- 72.Bo T-B, Zhang X-Y, Kohl KD, Wen J, Tian S-J, and Wang D-H (2020). Coprophagy prevention alters microbiome, metabolism, neurochemistry, and cognitive behavior in a small mammal. ISME J. 14, 2625–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, and Ikeda K (2013). Metabolic regulation of osteoclast differentiation and function. J. Bone Miner. Res. 28, 2392–2399. [DOI] [PubMed] [Google Scholar]
- 74.Ahn H, Lee K, Kim JM, Kwon SH, Lee SH, Lee SY, and Jeong D (2016). Accelerated lactate dehydrogenase activity potentiates osteoclastogenesis via NFATc1 signaling. PLoS One 11, e0153886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu SC (1999). Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 13, 1169–1183. [PubMed] [Google Scholar]
- 76.Loewen PC (1979). Levels of glutathione in Escherichia coli. Can. J. Biochem. 57, 107–111. [DOI] [PubMed] [Google Scholar]
- 77.MacDonell R, Hamrick MW, and Isales CM (2016). Protein/amino-acid modulation of bone cell function. Bonekey Rep. 5, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. (2004). ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: implication for Coffin-Lowry syndrome. Cell 117, 387–398. [DOI] [PubMed] [Google Scholar]
- 79.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, and Karsenty G (2006). ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Narva M, Collin M, Lamberg-Allardt C, Kärkkäinen M, Poussa T, Vapaatalo H, and Korpela R (2004). Effects of long-term intervention with Lactobacillus helveticus-fermented milk on bone mineral density and bone mineral content in growing rats. Ann. Nutr. Metab. 48, 228–234. [DOI] [PubMed] [Google Scholar]
- 81.McCabe LR, Irwin R, Schaefer L, and Britton RA (2013). Probiotic use decreases intestinal inflammation and increases bone density in healthy male but not female mice. J. Cell. Physiol. 228, 1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Montonye DR, Ericsson AC, Busi SB, Lutz C, Wardwell K, and Franklin CL (2018). Acclimation and institutionalization of the mouse microbiota following transportation. Front. Microbiol. 9, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bidot WA, Ericsson AC, and Franklin CL (2018). Effects of water decontamination methods and bedding material on the gut microbiota. PLoS One 13, e0198305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma BW, Bokulich NA, Castillo PA, Kananurak A, Underwood MA, Mills DA, and Bevins CL (2012). Routine habitat change: a source of unrecognized transient alteration of intestinal microbiota in laboratory mice. PLoS One 7, e47416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hart ML, Ericsson AC, Lloyd KCK, Grimsrud KN, Rogala AR, Godfrey VL, Nielsen JN, and Franklin CL (2018). Development of outbred CD1 mouse colonies with distinct standardized gut microbiota profiles for use in complex microbiota targeted studies. Sci. Rep. 8, 10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ericsson AC, Davis JW, Spollen W, Bivens N, Givan S, Hagan CE, McIntosh M, and Franklin CL (2015). Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS One 10, e0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ivanov II, Frutos R.d.L., Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, Karp CL, and Pulendran B (2011). Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J. Immunol. 187, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hufeldt MR, Nielsen DS, Vogensen FK, Midtvedt T, and Hansen AK (2010). Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comp. Med. 60, 336–347. [PMC free article] [PubMed] [Google Scholar]
- 90.Ericsson AC, Gagliardi J, Bouhan D, Spollen WG, Givan SA, and Franklin CL (2018). The influence of caging, bedding, and diet on the composition of the microbiota in different regions of the mouse gut. Sci. Rep. 8, 4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Franklin CL, and Ericsson AC (2017). Microbiota and reproducibility of rodent models. Lab Anim. 46, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bangsgaard Bendtsen KM, Krych L, Sørensen DB, Pang W, Nielsen DS, Josefsen K, Hansen LH, Sørensen SJ, and Hansen AK (2012). Gut microbiota composition is correlated to grid floor induced stress and behavior in the BALB/c mouse. PLoS One 7, e46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moyer EL, Dumars PM, Sun G-S, Martin KJ, Heathcote DG, Boyle RD, and Skidmore MG (2016). Evaluation of rodent spaceflight in the NASA animal enclosure module for an extended operational period (up to 35 days). NPJ microgravity 2, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun G-S, Tou JC, Liittschwager K, Herrera AM, Hill EL, Girten B, Reiss-Bubenheim D, and Vasques M (2010). Evaluation of the nutrient-upgraded rodent food bar for rodent spaceflight experiments. Nutrition 26, 1163–1169. [DOI] [PubMed] [Google Scholar]
- 95.Kumar S, Suman S, Fornace AJ, and Datta K (2018). Space radiation triggers persistent stress response, increases senescent signaling, and decreases cell migration in mouse intestine. Proc. Natl. Acad. Sci. USA 115, E9832–E9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Casero D, Gill K, Sridharan V, Koturbash I, Nelson G, Hauer-Jensen M, Boerma M, Braun J, and Cheema AK (2017). Space-type radiation induces multimodal responses in the mouse gut microbiome and metabolome. Microbiome 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Escapa IF, Chen T, Huang Y, Gajare P, Dewhirst FE, and Lemon KP (2018). New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): a resource for the microbiome of the human aerodigestive tract. mSystems 3, e00187–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dragojevič J, Logar DB, Komadina R, and Marc J (2011). Osteoblastogenesis and adipogenesis are higher in osteoarthritic than in osteoporotic bone tissue. Arch. Med. Res. 42, 392–397. [DOI] [PubMed] [Google Scholar]
- 99.Meunier P, Aaron J, Edouard C, and Vignon G (1971). Osteoporosis and the replacement of cell populations of the marrow by adipose tissue: a quantitative study of 84 iliac bone biopsies. Clin. Orthop. Relat. Res. 80, 147–154. [DOI] [PubMed] [Google Scholar]
- 100.Burkhardt R, Kettner G, Böhm W, Schmidmeier M, Schlag R, Frisch B, Mallmann B, Eisenmenger W, and Gilg T (1987). Changes in trabecular bone, hematopoiesis and bone marrow vessels in aplastic anemia, primary osteoporosis, and old age: a comparative histomorphometric study. Bone 8, 157–164. [DOI] [PubMed] [Google Scholar]
- 101.Fu J, Goldsmith M, Crooks SD, Condon SF, Morris M, and Komarova SV (2021). Bone health in spacefaring rodents and primates: systematic review and meta-analysis. npj Microgravity 7. 19–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hauschka PV, Lian JB, Cole DE, and Gundberg CM (1989). Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol. Rev. 69, 990–1047. [DOI] [PubMed] [Google Scholar]
- 103.Poser JW, Esch FS, Ling NC, and Price PA (1980). Isolation and sequence of the vitamin K-dependent protein from human bone. Under-carboxylation of the first glutamic acid residue. J. Biol. Chem. 255, 8685–8691. [PubMed] [Google Scholar]
- 104.Malashkevich VN, Almo SC, and Dowd TL (2013). X-ray crystal structure of bovine 3 Glu-osteocalcin. Biochemistry 52, 8387–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moser SC, and van der Eerden BCJ (2018). Osteocalcin—a versatile bone-derived hormone. Front. Endocrinol. 9, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Al-Hebshi NN, Nasher AT, Idris AM, and Chen T (2015). Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: application to oral carcinoma samples. J. Oral Microbiol. 7, 28934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun G-S, Tou JC, Yu D, Girten BE, and Cohen J (2014). The past, present, and future of National Aeronautics and Space Administration spaceflight diet in support of microgravity rodent experiments. Nutrition 30, 125–130. [DOI] [PubMed] [Google Scholar]
- 108.Tong M, Jacobs JP, McHardy IH, and Braun J (2014). Sampling of intestinal microbiota and targeted amplification of bacterial 16S rRNA genes for microbial ecologic analysis. Curr. Protoc. Immunol. 107, 7.41.1–7.41.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allen HK, Bayles DO, Looft T, Trachsel J, Bass BE, Alt DP, Bearson SMD, Nicholson T, and Casey TA (2016). Pipeline for amplifying and analyzing amplicons of the V1–V3 region of the 16S rRNA gene. BMC Res. Notes 9, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Escapa I,F, Huang Y, Chen T, Lin M, Kokaras A, Dewhirst FE, and Lemon KP (2020). Construction of habitat-specific training sets to achieve species-level assignment in 16S rRNA gene datasets. Microbiome 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Team RC (2013). R: A Language and Environment for Statistical Computing. [Google Scholar]
- 115.McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Foster ZSL, Sharpton TJ, and Grünwald NJ (2017). Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 13, e1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, and Huttenhower C (2018). bioBakery: a meta’omic analysis environment. Bioinformatics 34, 1235–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heat-map. Nucleic Acids Res. 43, W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Powell D (2015). Degust: interactive RNA-seq analysis. [Google Scholar]
- 120.Zhang Z, Zhang Y, Evans P, Chinwalla A, and Taylor D (2017). RNA-Seq 2G: online analysis of differential gene expression with comprehensive options of statistical methods. Preprint at bioRxiv, 122747. 10.1101/122747. [DOI] [Google Scholar]
- 121.Lu Y, Yao D, and Chen C (2013). 2-Hydrazinoquinoline as a derivatization agent for LC-MS-based metabolomic investigation of diabetic ketoacidosis. Metabolites 3, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Imbert L, Saussereau E, and Lacroix C (2014). Analysis of eight glycols in serum using LC-ESI–MS-MS. J. Anal. Toxicol. 38, 676–680. [DOI] [PubMed] [Google Scholar]
- 123.Schulte F, King OD, Paster BJ, Moscicki A-B, Yao T-J, Van Dyke RB, Shiboski C, Ryder M, Seage G, and Hardt M; Pediatric HIV/AIDS Cohort Study (2020). Salivary metabolite levels in perinatally HIV-infected youth with periodontal disease. Metabolomics 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun W, Chi S, Li Y, Ling S, Tan Y, Xu Y, Jiang F, Li J, Liu C, Zhong G, et al. (2019). The mechanosensitive Piezo1 channel is required for bone formation. Elife 8, e47454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hale LV, Galvin RJS, Risteli J, Ma YL, Harvey AK, Yang X, Cain RL, Zeng Q, Frolik CA, Sato M, et al. (2007). PINP: a serum biomarker of bone formation in the rat. Bone 40, 1103–1109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The un-processed raw 16S rRNA gene and metagenomic WGS sequencing files along with the LC-MS/MS raw data are all deposited in NASA’s GeneLab Open Source Repository (Accession Number: GLDS-417: https://doi.org/10.26030/r6bd-0k20).
All original code scripts used in the pipeline analyses and statistical output files are deposited on Github (https://doi.org/10.5281/zenodo.7620493).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.