

Abstract
Aim: Given the encouraging results of the p53-Mdm2 inhibitor RG7388 in clinical trials and the vital function of miR-16-5p in suppressing cell proliferation, the aim of the present study was to investigate the combined impact of RG7388 and miR-16-5p overexpression on the childhood acute lymphoblastic leukemia (chALL).
Methods: miRTarBase and miRDB, along with KEGG and STRING databases, were used to predict miR-16-5p target genes and explore protein-protein interaction networks, respectively. B- and T-lymphoblastic cell lines, in addition to patient primary cells, were treated with RG7388. Ectopic overexpression of miR-16-5p in Nalm6 cell line was induced through cell electroporation and transfection of microRNA mimics was confirmed by qRT-PCR. Cell viability was evaluated using the MTT assay. Western blot analyses were performed to evaluate the effects of RG7388 and miR-16-5p upregulation on the protein levels of p53 and its downstream target genes in chALL cells. Paired sample t-test was employed for statistical analyses.
Results: MTT assay showed RG7388-induced cytotoxicity in wild-type p53 Nalm6 cell line and p53 functional patient primary cells. However, CCRF-CEM and p53 non-functional leukemic cells indicated drug resistance. Western blot analyses validated the bioinformatics results, confirming the downregulation of WIP1, p53 stabilization, as well as overexpression of p21WAF1 and Mdm2 proteins in Nalm6 cells transfected with miR-16-5p. Moreover, enhanced sensitivity to RG7388 was observed in the transfected cells.
Conclusion: This is the first study indicating the mechanistic importance of miR-16-5p overexpression in chALL and its inhibitory role in leukemia treatment when combined with the p53-Mdm2 antagonist, RG7388. These findings might be useful for researchers and clinicians to pave the way for better management of chALL.
Keywords: Pediatric ALL, miR-16-5p, RG7388, PPM1D, p53
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is one of the most common blood cancers in children with a low incidence rate of TP53 mutations at diagnosis, which highlights this type of malignancy as an attractive candidate for treatment with p53-Mdm2 antagonists. Although 85%-90% of patients respond to the treatment, there are subsets that are refractory to the therapy and relapse is prevalent amongst individuals who achieve complete remission[1,2]. For this reason, new strategies and treatments are necessary to overcome drug resistance.
Cancer treatment has been recently improving with the introduction of targeted therapies to achieve greater specificity and less cytotoxicity. Owing to the main function of p53 in the enhancement of cell cycle arrest, response to DNA repair and apoptosis, enormous efforts have been made to advance new cancer treatments based on p53-targeted therapy. p21 is a well-known determinant of cell cycle arrest, which increases following induction of p53 stabilization and its activity[3]. The incidence rate of TP53 mutations at diagnosis is low in various types of blood cancer including ALL (5%-10%). Therefore, activation of the p53 pathway by non-genotoxic inhibitors of mouse double minute 2 homolog (Mdm2) is a promising strategy to improve cancer therapy in hematological malignancies, including ALL[4]. Recently, small molecule inhibitors of p53-Mdm2 interaction have been developed and entered into early-phase clinical trials for the treatment of diverse types of cancer comprising blood cancer[5-8]. Amongst those entered into clinical trials, RG7388 has passed phase II clinical trial[8]. Thus, the prediction of sensitivity to Mdm2 inhibitors and identification of mechanisms of resistance toward Mdm2 inhibitors would be helpful in stratifying patients who might benefit from these therapeutic agents.
p53 levels and its activity are regulated under a complex network of proteins and microRNAs (miRNAs) to maintain normal levels and proper function of p53. The oncogene Protein Phosphatase, Mg2+/Mn2+ Dependent 1D (PPM1D), known as wild-type p53-induced phosphatase 1 (WIP1), negatively regulates p53 through different mechanisms, including direct dephosphorylation of p53 and its subsequent inactivation, and dephosphorylation of other proteins involved in the regulation of p53 such as Mdm2 and p53 activating kinases[9]. Consistent with the inhibitory effect of Wip1 on the p53 pathway, PPM1D gene is amplified and overexpressed in different cancer types including leukemia, suggesting that Wip1 may be a potential therapeutic target for leukemia[10]. Recent studies have demonstrated that selective inhibition of Wip1 leads to increased DNA damage response and sensitivity to anti-cancer agents working through the p53 pathway[10-13].
microRNAs are a class of small RNAs ,with an average of 22 nucleotides in length, which interact with the partially complementary sequences in the 3′ untranslated region (3′ UTR) of the target mRNA and negatively regulate its expression[9,14]. Although not proven in childhood ALL, miR-16-5p appears to be a major negative regulator of Wip1 protein expression in some cancers, which affects p53-Wip1 feedback loop[13] and can regulate cell fate[12]. miR-16-5p interacts with the 3′-UTR binding site of the human PPM1D gene and directly represses the Wip1 protein expression, a negative regulator of p53, thereby indirectly promoting p53 activity and its pathway[13]. Previous research showed that miR-16-5p feedback loop with p53 and Wip1 increases sensitivity to doxorubicin[13] and affects cell fate determination[12]. Moreover, aberrant expression of miR-16-5p has been reported in chronic lymphocytic leukemia (CLL)[15,16].
In the present study, it was hypothesized that cell lines and primary samples harboring functional p53 are more sensitive to RG7388 compared to those with dysfunctional p53, and ectopic overexpression of miR-16-5p in cells with functional p53 affects WIP1 mRNA levels, and consequently respond to the p53-Mdm2 antagonist RG7388 in a p53-dependent manner.
METHODS
miR-16-5p-target interaction databases and visualization of protein-protein interaction network
miRTarBase and miRDB validated microRNA-target interactions databases were employed to clarify the interaction between miR-16-5p and PPM1D expression[17,18]. The Kyoto Encyclopedia of Genes and Genomes database (KEGG) (http://www.genome.jp/kegg/) was used as a pathway database to find the p53 pathway map (map04115)[19]. To visualize protein-protein interaction network between the Wip1 and other proteins with strong confidence (0.700 interaction score), the STRING database (https://string-db.org/) was used[20].
Chemicals and reagents
The small-molecule Mdm2 inhibitor RG7388 (Idasanutlin) was purchased from SelleckChem (Cambridge, UK). RG7388 was dissolved in dimethyl sulfoxide (DMSO) to provide a 10 mM stock solution and stored in small aliquots at -20 °C. miR-CURY LNA™ Universal RT microRNA PCR kit and miR-CURY LNA™ miRNA Mimics (HAS-MIR- 16 -5p & NEGATIVE CONTROL 5 MIRCURY LNA) were purchased from QIAGEN (Hilden, Germany).
Cell lines
The leukemic cell lines Nalm6 and CCRF-CEM were sourced from the Pasteur Institute (Iran) authenticated cell bank. The cells were cultured in RPMI-1640 (Gibco, USA) supplemented with 10% (v/v) FBS (Gibco, USA) and 1% (v/v) penicillin/streptomycin and grown at 37 °C, 5% CO2 in a humidified atmosphere. The TP53 status of Nalm6 cell line is wild-type (NALM6 ATCC CRL-3273™) and the CCRF-CEM cell line (CCRF-CEM ATCC CCL-119™) harbors heterozygous TP53 mutations (c.524G>A; p.R175H, and c.743G>A; p.R248Q).
Patients, sampling and cell isolation
Peripheral blood or bone marrow samples (n = 10, 5 females and 5 males) from childhood ALL patients were collected. ALL in these patients was clinically diagnosed and pathologically confirmed by a clinical team through phenotypic, immunologic and cytogenetic techniques in the pediatric department of Sayed-ol-Shohada Hospital (Isfahan, Iran). Informed written consent was obtained in accordance with the Declaration of Helsinki, and with approval from the Ethics Committee of the University of Isfahan (ethics agreement number IR.UI.REC.1397.145). Informed written consent was obtained from the children’s parents prior to participation in the study.
2-5 mL of heparinized bone marrow sample or peripheral blood were collected from patients and sent to the Cellular and Molecular Biology Laboratory of the University of Isfahan on ice. Mononuclear cells were extracted and isolated using density gradient Lymphoprep (Axis-Shailed Diagnostics Ltd, Oslo, Norway) according to the manufacturer's protocol.
Ex vivo cytotoxicity assay
Cytotoxicity of RG7388 was assessed using MTT cell proliferation kit (Alban, Austria). Growth curves were constructed for Nalm6 and CCRF-CEM cell lines (GraphPad Prism statistical analysis software version 8.) to measure cell doubling time and the optimum seeding density for a subsequent growth inhibition assay. Nalm6 (5 × 105/mL) and CCRF-CEM (4.5 × 104/mL) in 100 μL of medium per well of a 96-well plate were treated with a range of concentrations of RG7388 for 96 and 72 h, respectively (according to their cell doubling time). Then, 10 μL of MTT solution was added into the wells. 100 μL DMSO was added after 3 h to dissolve formazan crystals, and absorbance was measured using a stat fax 2000 microplate reader (Awareness Technology, Inc) at 492 nm wavelength. The LC50 values, the required concentration of each compound expected to kill 50% of the population, were determined using the statistical software mentioned above.
For patients` samples, primary cells (2 × 106/mL) in 100 μL of medium per well of a 96-well plate were exposed to 0.5% DMSO or 0.5 µM RG7388 (2 × LC50 concentration for Nalm6) for 72 h. This concentration was used since sensitive cells respond to it, and the sensitivity is not due to off-target effects. The proportion of the viable cells was measured by comparison between the absorbance of cells exposed to DMSO, as a control, or RG7388, and calculated using the following formula: (%) = [100 × (sample absorbance)/ (control absorbance)].
Functional assessment of the p53 pathway
To determine the functional status of p53 in ALL patients’ samples (those with enough amount of protein lysates), the modulation of p53 and its transcriptional target gene protein products including Mdm2 and p21WAF1 were evaluated following short-term exposure to Mdm2-p53 antagonist RG7388[21,22].
Western blotting
Nalm6 and CCRF-CEM (2.5 × 105/mL) were seeded in 2 mL culture media per well of a 6-well plate and treated with 0.5% DMSO and a range of concentrations of RG7388. Primary cells (0.5 × 106/mL) were also seeded in 2 mL culture media per well of a 6-well plate and exposed to 0.5% DMSO or 0.5 µM RG7388 (2 × LC50 concentration for Nalm6). Cells were harvested and lysed at 6 h. Lysis buffer (12.5 mL Tris HCL, 2 g SDS, 10 mL Glycerol, 67.5 mL Distilled Water) was applied to harvest the whole-cell lysates, followed by sonication. Bradford solutions (100 MG Coomassie Blue 250 G, 50 mL ethanol 96%, 100 mL ortho-phosphoric acid 85% and bringing volume to 1000 mL by adding distilled H2O) were used to estimate the concentration of protein in the cell lysates utilizing NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, USA).
Hand-poured gradient gels were prepared using Bio-Rad mini gel casting apparatus, and two different acrylamide solutions were applied to separate proteins[23]. The separated proteins were transferred by perpendicular electrophoresis to a nitrocellulose HybondTM C membrane (Amersham, Buckinghamshire, UK). Monoclonal mouse anti-human primary antibodies Actin 1:250 (#: C4: sc-47778, Santacruz Biotechnology, INC.), Mdm2 1:300 (#: OP46-100UG, Merck Millipore), p21 1:100 (#: OP64, Calbiochem), p53 1:250 (#: 2B2.68: sc-71817, Santacruz Biotechnology, INC.) and Wip1 1:200 (#: F-10: sc-376257, Santacruz Biotechnology, INC.) were used. Secondary goat anti-mouse HRP-conjugated antibodies (#: P0447/P0448, Dako) were applied at 1:1000. 5% milk/1XTBS-Tween (w/v) was used in order to dilute all antibodies. Enhanced chemiluminescence (GE Life Sciences, UK) and X-ray film (Fujifilm, India) were employed to visualize the proteins. Image J software (National Institute of Health, USA) was used to quantify and analyze the intensity of visualized bands, and the results were normalized to DMSO control.
Cell transfection
Harvested Nalm6 cells in the exponential growth phase were resuspended in RPMI-1640 (Gibco, USA) supplemented with 0.5% (v/v) FBS (Gibco, USA). 200 µL cell suspension containing 2.5 × 106 of cells were transferred into sterile electroporation cuvettes (0.2 cm gap, Bio-Rad, USA) separately (miR-16-5p mimic and negative control). miR-16-5p mimic and negative control (200 nm) (miR-CURY LNA™ miRNA Mimics, Qiagen) were separately added to the cuvettes in the hood just before electroporation, and the cuvettes gently swirled. Then, the cuvette was placed in the holder in the electroporation system (Eppendorf Multiporator®, Germany) at room temperature. Electroporation was performed in accordance with Multiporator® Transfection Protocol with minor changes [Voltage: 250 V, Time constant (τ): 40 μs, No. of pulses (n): 1] in order to specifically optimize protocol based on the cell type and genetic modifications. After the pulse, the cell suspension was allowed to stand in the cuvette for 5 to 10 minutes at room temperature. Finally, the cell suspension was transferred from the cuvette to 2 mL normal medium in a well of a 6-well plate and incubated for 48 h. In order to determine the impact of overexpression of miR-16-5p on sensitivity to RG7388, 48 h transfected cells were exposed to RG7388 (0.5 µM) for 6 h. Transfected cells were harvested to evaluate cell viability and silencing assessment.
RNA extraction and cDNA synthesis
Total RNA including preserved miRNAs was extracted from Nalm6 cells and primary samples using TRizol reagents (Invitrogen, California, CA) as per the manufacturer's recommendations. The quality of the RNA and its concentration was assessed with a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, USA) by the ratio of 260nm:280nm. The complementary DNA (cDNA) for miR-16-5p was synthesized on 200 ng of total RNA using miR-CURY LNATM microRNA PCR kit (QIAGEN, Germany) and the thermal cycler (GeneAmp PCR Systems, AB Applied Biosystems) according to the manufacturer’s guidelines.
Quantitative RT-PCR (qRT-PCR)
Ectopic expression of miR-16-5p was confirmed by real-time PCR assay. qRT-PCR for miR-16-5p converted to cDNA was carried out using the miRCURY LNA SYBR Green PCR kit (Qiagen, Germany), with 50 ng/μL of the cDNA samples per 10 μL final reaction volume, on a Chromo4TM system (BioRad, Foster City, California) as described by the manufacturer. Primers for miR-16-5p quantitative RT-PCR were obtained from Qiagen, and RNU6 small nuclear RNA ((Exiqon, Denmark) was employed as endogenous control for data normalization. ΔΔCt Method was applied to perform data analysis.
Statistical Analysis
All the presented statistical tests were performed, applying GraphPad Prism version 8.4.3 software. The statistical paired t-test was employed to compare the mean of 3 paired biological repeats, and significant differences are defined as P < 0.05.
RESULTS
miR-16-5p is proposed to target WIP1protein affecting p53 pathway
The miRTarBase experimentally validated microRNA-target interactions database confirmed PPM1D as a target for miR-16-5p with strong evidence (Reporter assay, qRT-PCR and Western blot). In terms of miRDB target prediction database, miR-16-5p was at the top 20 miRNAs targeting PPM1D expression.
KEGG p53 pathway has shown Wip1 protein as one of the main negative regulators of p53 activity. In addition, Wip1 activates the main negative regulator of p53, Mdm2, and inactivates the kinases which phosphorylate and enhance p53 activity including CHK2 and ATM [Figure 1A]. Furthermore, STRING database confirmed the functional association between Wip1 and p53 and other proteins involved in p53 activity with a high confidence (0.7 score interaction) [Figure 1B].
Figure 1.
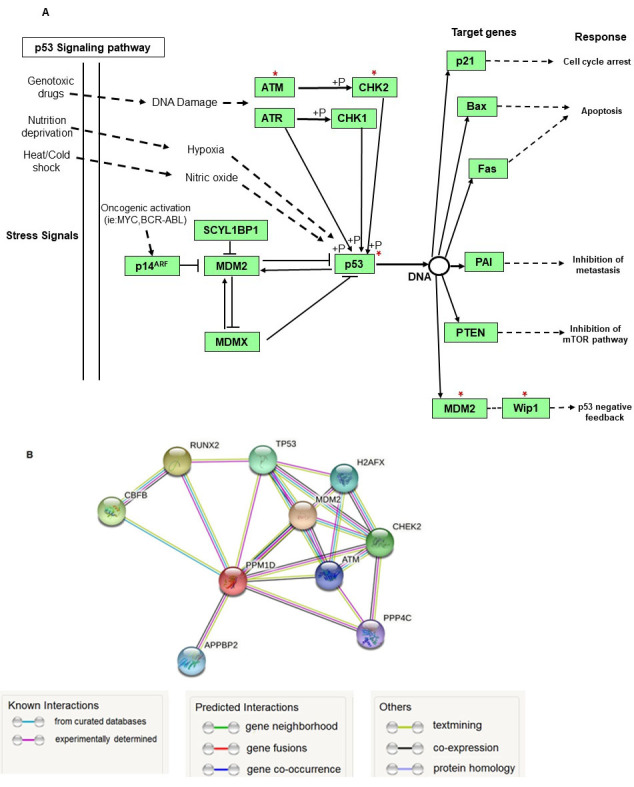
The importance of WIP1 in the p53 signaling pathway and its Protein–protein interaction network. (A) The p53 pathway map (map04115) was provided using the KEGG database. The red stars above each gene represent the genes whose activity is affected by WIP1 via dephosphorylation; (B) WIP1 Protein-protein interaction network was visualized by STRING with high confidence (0.7). The edges represent protein-protein associations which are meant to be specific and meaningful. This does not necessarily mean they are physically binding to each other. MDM2: Mouse Double Minute 2 Homolog; WIP1: Wild-type p53-Induced Phosphatase 1.
The cytotoxic effect of RG7388 on ALL cell lines and primary cultures
The subsequent experiments were carried out to evaluate the sensitivity of two established ALL cell lines and ALL primary cells to RG7388, and the influence of ectopic overexpression of miR-16-5p on this sensitivity. The cytotoxicity of RG7388 and its p53-dependent effect was investigated using the MTT assay on Nalm6 wild-type TP53 and CCRF-CEM mutant TP53 cell lines. The LC50 values (Lethal Concentration 50%) indicated that wild-type TP53 Nalm6 cell line was significantly more sensitive to RG7388 (0.27 ± 0.05 (SEM) μM) compared to CCRF-CEM mutant TP53 cell line (> 2 μM) [Figure 2A]. The cytotoxic effect of RG7388 was also investigated on the cell viability of primary cultures generated from materials donated by ALL patients [Table 1]. 10 ALL samples were incubated with DMSO (0.5%) as control and RG7388 (0.5 μM), and they were examined for viability after 72 h using the MTT assay. RG7388 induced a cytotoxic effect on ALL cells, and 60% of those (6 out of 10) were sensitive and 40% (4 out of 10) were resistant to RG7388 with the delivered dose [Figure 2B]. RG7388 led to a significant reduction in the viability of sensitive ALL primary cells compared to their untreated counterpart (P < 0.05 or P < 0.001), with ALL 278 sample as the most sensitive sample (% viability = 0.66% ± 0.04, P = 0.0005). Overall, the median % survival for primary cultures was 53% [Figure 2C]. Notably, 3 out of 4 RG7388 resistant samples (75%) relapsed [Table 1].
Figure 2.
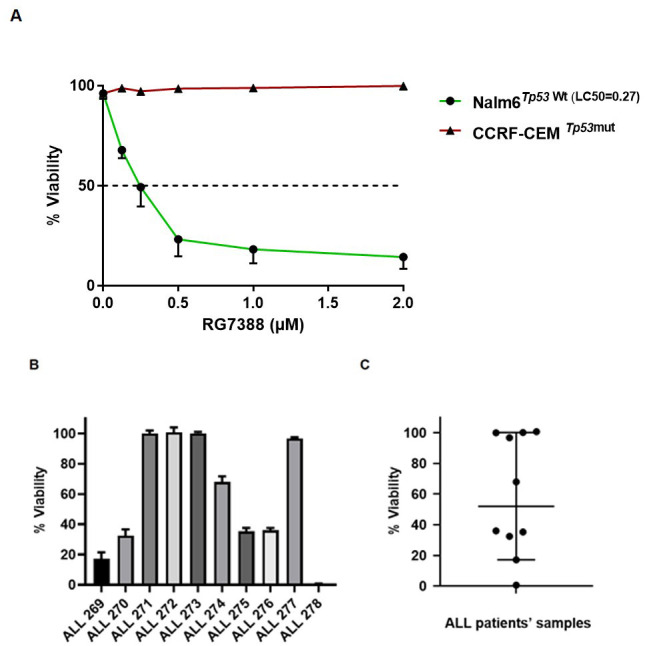
The sensitivity to MDM2 antagonists RG7388 in ALL cell lines and childhood ALL primary cells. (A) Wild-type TP53 Nalm6 cell line is significantly more sensitive to growth inhibition by RG7388 treatment compared to mutant TP53 CCRF-CEM cell line; (B) 10 pediatric ALL samples exposed to RG7388 (0.5 μM) for 72 h. RG7388 markedly decreased cell viability in most samples while assessed by MTT assay; (C) dot-plot of % viability for 10 pediatric ALL samples exposed to RG7388 (0.5 μM) for 72 h. Data shown are the average of three independent experiments and error bars represent SEM. ALL: Acute lymphoblastic leukemia.
Table 1.
Clinicopathological data for 10 samples of pediatric ALL
| Patient's number/Variable* | Sex Age (years) | Peripheral blood/Bone marrow | De novo/ Relapsed | Pre-B cell/ T cell | Cytogenetics** | Response to RG7388 |
| ALL 269 | Boy (2) | Peripheral blood | De novo | Pre-B cell | No | Sensitive |
| ALL 270 | Girl (2) | Bone marrow | De novo | Pre-B cell | No | Sensitive |
| ALL 271 | Boy (3) | Bone marrow | De novo | Pre-B cell | No | Resistant |
| ALL 272 | Girl (5) | Peripheral blood | Relapsed | Pre-B cell | No | Resistant |
| ALL 273 | Boy (9) | Bone marrow | Relapsed | T cell | No | Resistant |
| ALL 274 | Boy (6) | Bone marrow | De novo | Pre-B cell | T (12, 21)/ETV6-RUNX1 | Sensitive |
| ALL 275 | Girl (3) | Peripheral blood | De novo | Pre-B cell | T (1, 19)/TCF3-PBX1 | Sensitive |
| ALL 276 | Girl (5) | Peripheral blood | De novo | Pre-B cell | No | Sensitive |
| ALL 277 | Girl (5) | Peripheral blood | Relapsed | T cell | No | Resistant |
| ALL 278 | Boy (12) | Peripheral blood | De novo | Pre-B cell | No | Sensitive |
*The blast proportion for all patients’ samples was over 70%. **The most common cytogenetics (T (4; 11)/KMT2A-AFF1, T (9; 22)/BCR-ABL1, T (1; 19)/ TCF3-PBX1, T (12; 21)/ ETV6-RUNX1) were only analyzed.
Functional activation of the p53 pathway in ALL cell lines and primary cultures in response to RG7388
Functional assessment of the p53 pathway was evaluated by measuring p53 induction and its stabilization following six hours’ exposure to RG7388, and consequent overexpression of its downstream targets including Mdm2 and p21WAF1 proteins by Western blot. The p53-dependent response to RG7388 showed that RG7388 elevated p53 stabilization and overexpression of p21WAF1 and Mdm2 protein levels 6 h after the commencement of treatment in a concentration-dependent manner, and confirmed functional activation of wild-type TP53 Nalm6 cell line by release from Mdm2. However, as anticipated, it had no impact on the expression of p53-dependent genes in the TP53-mutant CCRF-CEM cell line with the delivered dose range of RG7388 [Figure 3A and Supplementary Figure 1 (362.6KB, pdf) ].
Figure 3.
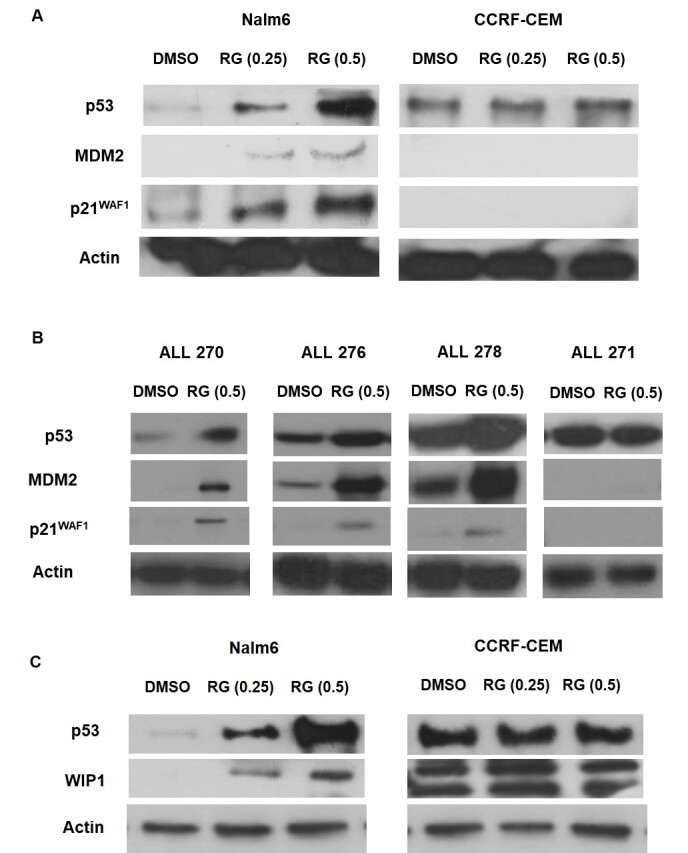
p53 functional stabilization in pediatric ALL cells in response to RG7388. Western blot analysis for (A) established Nalm6 and CCREF-CEM cell lines and (B) representative of patient samples with functional p53 and dysfunctional p53. RG7388 showed stabilization of p53 and upregulation of p53 transcriptional target gene protein levels, MDM2 and p21WAF1, 6 h after the commencement of treatment in wild-type TP53 Nalm6 and p53 functional ALL cells with the indicated doses (μM). However, it had no effect on downstream transcriptional targets of p53 in mutant TP53 CCRF-CEM and dysfunctional p53 patient samples with the delivered dose of RG7388 (μM); (C) western blot analysis indicated stabilization of p53 and induction of WIP1 expression following 6 h treatment with indicated doses of RG7388 (μM) in wild-type TP53 Nalm6 in a concentration manner. Conversely, there was no effect on the p53 stabilization and WIP1 expression in mutant TP53 CCRF-CEM with the delivered dose range of RG7388 (μM). RG, RG7388. ALL: Acute lymphoblastic leukemia; MDM2: mouse double minute 2 Homolog; WIP1: wild-type p53-induced phosphatase 1.
For the primary cultures with sufficient amount of protein lysates for western blot, there was consistency between MTT assay results and p53 function. RG7388 induced functional stabilization of p53 and expression of its downstream target genes, p21WAF1 and MDM2, in ALL samples that showed a significant decrease in their viability rate following treatment with RG7388. Conversely, there was no stabilization of p53 and induction of its downstream targets in primary cultures that were resistant to RG7388 [Figure 3B and Supplementary Figure 2 (362.6KB, pdf) ].
RG7388 induces Wip1 expression in a p53-dependent manner
The basal protein levels of Wip1 and its expression levels following treatment with RG7388 at the 1 × and 2 × LC50 value for Nalm6 were determined in both Nalm6 and CCRF-CEM cell lines [Figure 3C and Supplementary Figure 3 (362.6KB, pdf) ]. RG7388 increased stabilization of p53 protein, with subsequent increased expression of Wip1 protein in Nalm6 cells in a concentration-dependent manner. However, RG7388 failed to stabilize p53 and induce Wip1 in CCRF-CEM cells, indicating p53-dependent expression of Wip1 protein. Notably, both full-length (FL-WIP1) and its previously described shorter isoform (S-WIP1) of Wip1 protein were expressed by CCRF-CEM cell line.
miR-16-5p negatively regulates WIP1 expression and sensitizes Nalm6 cells to RG7388
The p53-Wip1 autoregulatory feedback loop regulates both expression levels of PPM1D gene and p53 activity. Previous studies showed that post-transcriptional regulation influences the expression of Wip1 protein and confirmed that miR-16-5p inhibits WIP1 expression through targeting 3’UTR of WIP1 [Figure 4A]. Thus, it is expected that miR-16-5p affects the response to RG7388 through regulating WIP1 in a p53-dependent manner.
Figure 4.

miR-16-5p suppresses WIP1 expression, affects p53 stabilization and its activity, and enhances sensitivity to RG7388. (A) miR-16-5p sequences and its putative binding sites in the 3′-UTR of PPM1D; (B) Nalm6 cells were transfected with negative control miRNA and miR-16-5p mimic (200nM). Total RNA was extracted from DMSO (0.5%) treated Nalm6, mock control, and those transfected with negative control miRNA or miR-16-5p mimic at 48 h after transfection. miR-16-5p levels were measured by quantitative RT-PCR shown significant ectopic overexpression of miR-16-5p in transfected cells with miR-16-5p compared to DMSO control, mock control and negative control miRNA; (C) ectopic overexpression of miR-16-5p suppresses WIP1 expression, enhances p53 stabilization and upregulates expression of p53 target genes, p21WAF1 and MDM2 in wild-type TP53 Nalm6 treated with RG7388. Nalm6 cells were transfected with miR-16-5p (200nM) or scrambled miRNA. Cells were treated with RG7388 (0.5 μM) at 48 h after transfection and their protein levels analyzed at 6 h after RG7388 treatment. The intensity of p53 blots (P < 0.05 or P < 0.1) (D) and p21WAF1 blots (P < 0.05) (E) was quantified by Image J software and normalized with the DMSO control. Data shown are the average of three independent experiments and error bars represent SEM. Ctrl, Control. *P < 0.05, **P < 0.01
To investigate the hypothesis, firstly, Nalm6 cells were transfected with miR-16-5p mimic or scrambled miRNA oligonucleotides as a negative control. Real-time PCR results confirmed high expression of miR-16-5p in Nalm6 cells resulting from the transfection of miR-16-5p mimic after 48 h. A significant increase was observed in the ectopic expression of miR-16-5p (by ~ 2.5-fold) compared with DMSO control, mock control, and negative control (P < 0.05) [Figure 4B].
We evaluated the protein levels of Wip1, p53, p21WAF1 and Mdm2 in Nalm6 treated with RG7388 in the presence of miR-16-5p mimic and negative control in order to investigate the impact of altered levels of miR-16-5p on the expression of the proteins. The high expression of miR-16-5p in Nalm6 led to a significant increase in p53 protein levels (by ~ 3.0 fold) [Figure 4C and D], p21WAF1 protein levels (by ~ 2.5 fold) [Figure 4C and E] and Mdm2 [Figure 4C] compared to Nalm6 transfected with miRNA oligonucleotides as a negative control and DMSO control (P < 0.05) irrespective of treatment with RG7388. Interestingly, ectopic overexpression of miR-16-5p significantly suppressed induction of WIP1 in Nalm6 treated with RG7388, while scrambled negative control had no effect on the expression levels of WIP1 after RG7388 treatment. Furthermore, overexpression of miR-16-5p caused a significant upregulation of p53 (by ~ 15.5-fold, P < 0.01) [Figure 4C and D], and its target genes, p21WAF1 (by ~ 2-fold, P < 0.05) [Figure 4C and E] and Mdm2, in Nalm6 treated with RG7388 compared to negative control treated with RG7388. These results clearly demonstrated that miR-16-5p negatively regulates Wip1 protein levels, which consequently affects the p53 pathway and response to RG7388.
DISCUSSION
Acute lymphoblastic leukemia (ALL) is the most prevalent type of pediatric blood cancer in which TP53 mutations are infrequent, less than 5%, at diagnosis but rise to about 10% in relapsed ALL[24]. Although complete remission (CR) is achieved for many patients at the end of the induction phase of treatment, relapse and drug resistance are major challenges in treating cancer[25]. Recently, p53-Mdm2 binding antagonists have been advanced to restore wild-type p53 function with subsequent induction of cell cycle arrest and apoptosis. These targeted therapeutic agents have shown in vitro promising results alone and in combined treatments[26-31], and encouraging clinical trial outcomes in diverse types of cancer including blood malignancies[8,32-35]. miR-16-5p was previously reported as a post-transcriptional regulator of Wip1 in some types of cancer[13]. miRTarBase and miRDB microRNA-target interactions databases, KEGG pathway and STRING databases used in this study indicated the critical role of miR-16-5p in p53 activity through targeting PPM1D expression in ALL. Interestingly, the STRING database clearly showed strong interactions between Wip1 and p53 or its regulators comprising ATM, Mdm2 and CHK2 proteins[20].
The present study evaluated, for the first time, the impact of the Mdm2-p53 binding antagonist RG7388 in ALL cell lines, and primary cultures generated from materials donated by ALL patients. Moreover, this is the first study elucidating the positive effect of ectopic miR-16-5p on increasing sensitivity to RG7388 in TP53 wild-type leukemic cells.
Among the individual cell lines studied, wild-type TP53 Nalm6 cell line was significantly more sensitive to RG7388 compared to mutant TP53 CCRF-CEM cell line, which is in line with its mechanism of action[35]. These results partially confirm previous limited previous studies that indicated a significant decrease in the cell viability of wild-type TP53 ALL cell lines following treatment with Mdm2 inhibitor Nutlin-3a[36,37].
Within a panel of primary cultures, RG7388 significantly decreased the viability of most ALL cells (6 out of 7, 86%) isolated from de novo patients. Conversely, primary cultures derived from relapsed patients were resistant to the cytotoxicity effect of RG7388 at the delivered dose. Given the fact that response to RG7388 is dependent on wild-type p53, resistance to RG7388 could be related to TP53 mutations that are infrequent in childhood ALL patients at diagnosis, but they increase at relapse. Since the genomic status of TP53 gene is the major determinant of response to Mdm2 inhibitors, DNA sequencing of TP53, as the gold standard method, is highly recommended for identification of TP53 mutations in primary cells, particularly in the personalized directed use of inhibitors such as RG7388[38]. It is also possible that amplification/overexpression of WIP1 in different types of cancer including hematological tumors[10] results in dysfunctional wild-type TP53 and resistance to p53-dependent treatments comprising Mdm2 inhibitors[11].
In accord with the action mechanism of Mdm2 inhibitors, RG7388 treatment resulted in more stabilization of p53 and increased expression of its downstream targets, p21WAF1 and Mdm2 in wild-type TP53 Nalm6, in a concentration-dependent manner, and functional p53 primary cultures at the delivered dose of RG7388. In contrast, no significant enhancement of p53 downstream target genes was observed in mutant TP53 CCRF-CEM and non-functional p53 ALL samples following treatment with RG7388. These outcomes are consistent with other studies that reported induction of the p53 pathway in wild-type TP53 ALL cell lines after treatment with Nutlin-3a[36,37], CLL patients treated with RG7388[28], and Acute myeloid leukemia patients’ clinical response to RG7388[39].
Given the promising phase 1 results observed in blood cancer patients treated with Mdm2 inhibitors RG73122[40], RG7388[8,39], and AMG-32[5], and the prognostic value of miR-16 expression in childhood ALL[41] and CLL[15], we evaluated the impact of miR-16-5p expression on the induction of p53 pathway and response to Mdm2 inhibitor RG7388.
Wip1 protein levels were measured at the basal level and following treatment with RG7388 in both Nalm6 and CCRF-CEM cell lines. In comparison with CCRF-CEM, in which WIP1 is highly expressed at the basal levels, WIP1 is not detectable at its basal levels in Nalm6. As expected, RG7388 treatment led to more stabilization of p53 and its target, WIP1, in a concentration-dependent manner in wild-type TP53 Nalm6. However, there was no stabilization of p53 and no upregulation of WIP1 in TP53 mutant CCRF-CEM cell line, demonstrating that the effect is p53-dependent. Notably, CCRF-CEM harbors a heterozygous PPM1D mutation (c.1327A>G; p.N443D) reported by the COSMIC (Catalogue of Somatic Mutations in Cancer)[42].
It was also shown that upregulation of miR-16-5p (by ~ 2.5-fold) significantly induced p53 stabilization (by ~ 3-fold) and upregulated its downstream target p21WAF1 (by ~ 2.5-fold). p53 is considered as a representative haploinsufficient tumor suppressor gene in which a small change in its protein levels and/or its activity could immensely affect tumourigenesis in both mice and humans[43]. Interestingly, ectopic overexpression of miR-16-5p dramatically suppressed WIP1 expression after treatment with RG7388, followed by a significant rise in the p53 stabilization (by ~ 15.5-fold) and p21WAF1 upregulation (by ~ 2-fold). These results were in accord with several studies indicating the therapeutic impact of combined treatment between Wip1 inhibitor, GSK2830371, and Mdm2 inhibitors for increasing sensitivity to p53-Mdm2 antagonists in wild-type TP53 cell lines[11,31,44-46]. Mdm2 inhibitors release p53 from its negative regulator, Mdm2, which results in more stabilization of p53. Inhibition of Wip1 leads to increased phosphorylated p53 at serine 15 which activates the p53 pathway playing a critical role in cell cycle arrest mostly via upregulating of p21WAF1, and apoptosis through upregulating proapoptotic genes including PUMA[11,31,46], and downregulating antiapoptotic genes particularly, BCL2 and BIRC5 (survivin)[46,47]. Furthermore, miR-16-5p targets multiple cell cycle genes simultaneously, which leads to the accumulation of cells in G0/G1[47-49] and directly targets the antiapoptotic BCL2 gene which results in enhancing apoptosis[50,51] and modulating multidrug resistance. Therefore, in addition to genomic status of TP53 and its downstream target genes involved in apoptosis and cell cycle arrest, the expression levels of miR-16-5p might be considered as a marker to predict the sensitivity to Mdm2 inhibitors like RG7388 and other drugs working through the p53 pathway[52]. It is of note that local delivery, which is limited to the localized primary tumors, systemic route, viral delivery, and non-viral administration of miRNAs, are choices in delivering miRNAs[53].
In conclusion, the current study indicated the cytotoxic effect of the Mdm2 inhibitor, RG7388, on ALL patients’ primary cells in a p53-dependent manner. Moreover, it was shown that ectopic overexpression of mirR-16-5p increases sensitivity to RG7388 in ALL cells by suppressing WIP1 expression and inducing p53 stabilization. These data indicated, for the first time, the mechanistic importance of miR-16-5p in the pathophysiology of ALL, sensitivity to RG7388, and suggested its combination with RG7388 as a novel strategy for therapeutic targeting of non-mutant p53 pediatric ALL patients.
DECLARATIONS
Acknowledgments
The authors would like to acknowledge Mrs. Saideh Rahmani (University of Isfahan) for purifying mononuclear cells from the collected specimens and all patients and their parents who took part in this study.
Authors’ contributions
Study design: Zanjirband M
Acquisition of data: Zanjirband M, Aberuyi N
Analysis and interpretation of data: Zanjirband M, Rahgozar S, Aberuyi N
Manuscript preparation: Zanjirband M, Rahgozar S, Aberuyi N
Statistical analysis: Zanjirband M, Aberuyi N
Availability of data and materials
All data generated or analyzed during the current study are available from the corresponding author upon reasonable request.
Financial support and sponsorship
This work was supported by the University of Isfahan (96/100000/4000).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This study has been approved by the University of Isfahan review board and ethics committee under ethics agreement number IR.UI.1397.145. The patients’ parents were clearly informed regardnformed written consent was obtained from every patient’s parent before the start of the study by trained medical staff. Participants were assured that they had the right to leave the study at their convenience. Patients' samples were identified through a code number. Clinicopathological data of patients weing the aim of the study, the method and any possible complications related to the bone marrow/blood sampling in written format and verbal explanation. Ire confidentially kept, and the electronic information was stored on a password-protected computer.
Consent for publication
Consents for publication were obtained.
Copyright
© The Author(s) 2023.
Supplementary Materials
References
- 1.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7:e577. doi: 10.1038/bcj.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghodousi ES, Aberuyi N, Rahgozar S. Simultaneous changes in expression levels of BAALC and miR-326: a novel prognostic biomarker for childhood ALL. Jpn J Clin Oncol. 2020;50:671–8. doi: 10.1093/jjco/hyaa025. [DOI] [PubMed] [Google Scholar]
- 3.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 4.Kojima K, Ishizawa J, Andreeff M. Pharmacological activation of wild-type p53 in the therapy of leukemia. Exp Hematol. 2016;44:791–8. doi: 10.1016/j.exphem.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erba HP, Becker PS, Shami PJ, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3:1939–49. doi: 10.1182/bloodadvances.2019030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu H, Gao H, Ji Y et al. Targeting p53–MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J Hematol Oncol. 2022;15:1–23. doi: 10.1186/s13045-022-01314-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Chen F-E. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. European Journal of Medicinal Chemistry. 2022:114334. doi: 10.1016/j.ejmech.2022.114334. [DOI] [PubMed] [Google Scholar]
- 8.Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:1–17. doi: 10.1186/s13045-017-0500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 2018;9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng W, Li J, Dorrah K, et al. The role of PPM1D in cancer and advances in studies of its inhibitors. Biomed Pharmacother. 2020;125:109956. doi: 10.1016/j.biopha.2020.109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esfandiari A, Hawthorne TA, Nakjang S, Lunec J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol Cancer Ther. 2016;15:379–91. doi: 10.1158/1535-7163.mct-15-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Issler MVC, Mombach JCM. MicroRNA-16 feedback loop with p53 and Wip1 can regulate cell fate determination between apoptosis and senescence in DNA damage response. PLoS One. 2017;12:e0185794. doi: 10.1371/journal.pone.0185794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Wan G, Mlotshwa S et al. Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer research. 2010;70:7176–86. doi: 10.1158/0008-5472.can-10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dogan S, Spahiu E, Cilic A. Structural analysis of microRNAs in myeloid cancer reveals consensus motifs. Genes. 2022;13:1152. doi: 10.3390/genes13071152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh CH, Moles R, Nicot C. Clinical significance of microRNAs in chronic and acute human leukemia. Mol Cancer. 2016;15:37. doi: 10.1186/s12943-016-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acunzo M, Croce CM. Downregulation of miR-15a and miR-16-1 at 13q14 in Chronic Lymphocytic Leukemia. Clin Chem. 2016;62:655–6. doi: 10.1373/clinchem.2015.240036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SD, Lin FM, Wu WY, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–9. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–52. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605–12. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pozzo F, Dal Bo M, Peragine N, et al. Detection of TP53 dysfunction in chronic lymphocytic leukemia by an in vitro functional assay based on TP53 activation by the non-genotoxic drug Nutlin-3: a proposal for clinical application. J Hematol Oncol. 2013;6:83. doi: 10.1186/1756-8722-6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin M, Gentles L, Matheson E, et al. Characterization and drug sensitivity of a novel human ovarian clear cell carcinoma cell line genomically and phenotypically similar to the original tumor. Cancer Med. 2018;7:4744–54. doi: 10.1002/cam4.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller AJ, Roman B, Norstrom EM. Protein electrophoretic migration data from custom and commercial gradient gels. Data Brief. 2016;9:1–3. doi: 10.1016/j.dib.2016.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leeuwen FN. Therapeutic targeting of mutated p53 in acute lymphoblastic leukemia. Haematologica. 2020;105:10–1. doi: 10.3324/haematol.2019.234872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang FL, Liao EC, Li CL, Yen CY, Yu SJ. Pathogenesis of pediatric B-cell acute lymphoblastic leukemia: Molecular pathways and disease treatments. Oncol Lett. 2020;20:448–54. doi: 10.3892/ol.2020.11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zanjirband M, Edmondson RJ, Lunec J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget. 2016:7. doi: 10.18632/oncotarget.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanjirband M, Curtin N, Edmondson RJ, Lunec J. Combination treatment with rucaparib (Rubraca) and MDM2 inhibitors, Nutlin-3 and RG7388, has synergistic and dose reduction potential in ovarian cancer. Oncotarget. 2017:8. doi: 10.18632/oncotarget.19266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciardullo C, Aptullahoglu E, Woodhouse L, et al. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica. 2019;104:2429–42. doi: 10.3324/haematol.2018.206631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seipel , Miguel A. T. Marques , Corinne Sidler , Pabst BUMaT. The cellular p53 inhibitor MDM2 and the growth factor receptor FLT3 as biomarkers for treatment responses to the MDM2-inhibitor idasanutlin and the MEK1 inhibitor cobimetinib in acute myeloid leukemia. Cancers. 2018:10. doi: 10.3390/cancers10060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berberich A, Kessler T, Thomé CM, et al. Targeting resistance against the MDM2 inhibitor RG7388 in glioblastoma cells by the MEK inhibitor trametinib. Clin Cancer Res. 2019;25:253–65. doi: 10.1158/1078-0432.ccr-18-1580. [DOI] [PubMed] [Google Scholar]
- 31.Wu CE, Koay TS, Esfandiari A, Ho YH, Lovat P, Lunec J. ATM dependent DUSP6 modulation of p53 involved in synergistic targeting of MAPK and p53 pathways with trametinib and MDM2 inhibitors in cutaneous melanoma. Cancers (Basel) 2018;11:3. doi: 10.3390/cancers11010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58:1038–52. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holzer P, Masuya K, Furet P, et al. Discovery of a Dihydroisoquinolinone derivative (NVP-CGM097): a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58:6348–58. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- 34.Ferretti S, Rebmann R, Berger M, et al. Abstract 1224: Insights into the mechanism of action of NVP-HDM201, a differentiated and versatile Next-Generation small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. Cancer Research. 2016;76:1224–1224. doi: 10.1158/1538-7445.am2016-1224. [DOI] [Google Scholar]
- 35.Zanjirband M, Rahgozar S. Targeting p53-MDM2 interaction using small molecule inhibitors and the challenges needed to be addressed. Curr Drug Targets. 2019;20:1091–111. doi: 10.2174/1389450120666190402120701. [DOI] [PubMed] [Google Scholar]
- 36.Trino S, Iacobucci I, Erriquez D, et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: a new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget. 2016;7:12951–61. doi: 10.18632/oncotarget.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia. 2008;22:730–9. doi: 10.1038/leu.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiaretti S, Brugnoletti F, Tavolaro S, et al. TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica. 2013;98:e59–61. doi: 10.3324/haematol.2012.076786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reis B, Jukofsky L, Chen G, et al. Acute myeloid leukemia patients' clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica. 2016;101:e185–8. doi: 10.3324/haematol.2015.139717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreeff M, Kelly KR, Yee K, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in Leukemia. Clin Cancer Res. 2016;22:868–76. doi: 10.1158/1078-0432.ccr-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaddar T, Chien WW, Bertrand Y, et al. Prognostic value of miR-16 expression in childhood acute lymphoblastic leukemia relationships to normal and malignant lymphocyte proliferation. Leuk Res. 2009;33:1217–23. doi: 10.1016/j.leukres.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–7. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue K, Fry EA. Haploinsufficient tumor suppressor genes. Adv Med Biol. 2017;118:83–122. [PMC free article] [PubMed] [Google Scholar]
- 44.Sriraman A, Radovanovic M, Wienken M, Najafova Z, Li Y, Dobbelstein M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget. 2016;7:31623. doi: 10.18632/oncotarget.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fontana MC, Nanni J, Marconi G, et al. Abstract 2964: Pharmacological inhibition of WIP1 by GSK2830371 sensitizes AML cells to MDM2 inhibitor Nutlin-3a. Cancer Research. 2019;79:2964–2964. doi: 10.1158/1538-7445.am2019-2964. [DOI] [Google Scholar]
- 46.Pechackova S, Burdova K, Benada J, Kleiblova P, Jenikova G, Macurek L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget. 2016;7:14458. doi: 10.18632/oncotarget.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Q, Wang X, Li Z, et al. microRNA-16 represses colorectal cancer cell growth in vitro by regulating the p53/survivin signaling pathway. Oncol Rep. 2013;29:1652–8. doi: 10.3892/or.2013.2262. [DOI] [PubMed] [Google Scholar]
- 48.Liu Q, Fu H, Sun F, et al. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008;36:5391–404. doi: 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai CK, Zhao GY, Tian LY, et al. miR-15a and miR-16-1 downregulate CCND1 and induce apoptosis and cell cycle arrest in osteosarcoma. Oncol Rep. 2012;28:1764–70. doi: 10.3892/or.2012.1995. [DOI] [PubMed] [Google Scholar]
- 50.Pekarsky Y, Balatti V, Croce CM. BCL2 and miR-15/16: from gene discovery to treatment. Cell Death Differ. 2018;25:21–6. doi: 10.1038/cdd.2017.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia L, Zhang D, Du R, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123:372–9. doi: 10.1002/ijc.23501. [DOI] [PubMed] [Google Scholar]
- 53.Forterre A, Komuro H, Aminova S, Harada M. A comprehensive review of cancer microRNA therapeutic delivery strategies. Cancers (Basel) 2020;12:1852. doi: 10.3390/cancers12071852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during the current study are available from the corresponding author upon reasonable request.