

Summary
Non-small-cell lung cancer remains a deadly form of human cancer even in the era of immunotherapy with existing immunotherapy strategies currently only benefiting a minority of patients. Therefore, the derivation of treatment options, which might extend the promise of immunotherapy to more patients, remains of paramount importance. Here, we define using TCGA lung squamous and lung adenocarcinoma RNAseq datasets a significant correlation between epigenetic therapy actionable interferon genes with both predicted tumor immune score generally, and CD8A specifically. IHC validation using primary sample tissue microarrays confirmed a pronounced positive association between CD8+ T cell tumor infiltration and the interferon-associated targets, CCL5 and MDA5. We next extended these findings to the assessment of clinical trial biopsies from patients with advanced non-small-cell lung cancer treated with epigenetic therapy with and without concurrent immunotherapy. These analyses revealed treatment-associated increases in both CD8+ T cell intratumoral infiltration and microenvironment CCL5 staining intensity.
Subject areas: Immunology, Components of the immune system, Cancer
Graphical abstract
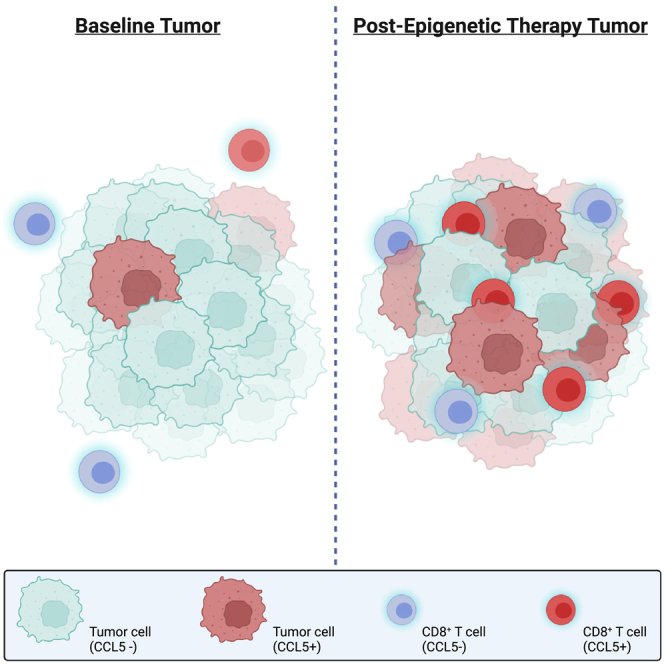
Highlights
-
•
CCL5 protein expression is correlated with CD8+ T cell tumor infiltration in NSCLC
-
•
DNMT+HDAC inhibition increases CCL5 protein and CD8+ T cell tumor infiltration in NSCLC
-
•
Tumors with infiltrative CD8+ T cells have a higher prevalence of CCL5, CD8+/+ cells
Immunology; Components of the immune system; Cancer
Introduction
Type I interferon (IFN)-related signaling has a critical role in responses to a wide variety of pathogens; the most established of which are viral.1,2 The initiation of interferon signaling both potently suppresses viral replication through the cessation of translation and enhances antigen presentation.3,4 Beyond its function within virally infected cells, type I IFN signaling has a demonstrable impact on chemokine and cytokine gradients.5,6 These intra- and extracellular cues can impart significant remodeling of both local and systemic immune environments to impact tumor progression and disease manifestation.7,8 The specific question of whether type I IFN signaling can mediate the accumulation of CD8+ T cells within the tumor microenvironment has been the subject of several inquiries, but the consequences of this inflammatory-related signaling on CD8+ T cell tumor infiltration and direct tumor cell contact have remained far from conclusive.9,10,11,12
Our previous studies have suggested that epigenetic therapies including DNA methyltransferase inhibitor (DNMTi) and DNMTi + histone deacetylase inhibitor (HDACi)-based regimens can augment type I IFN and viral defense signaling accompanied by CD8+ T cell accumulation.13,14 Several recent studies by our group and others have demonstrated that CCL5 (Chemokine (C-C motif) ligand 5) augmentation by DNMTi or DNMTi + HDACi can induce the accumulation of CD8+ T cells within the tumor and associated microenvironment.13,15 CCL5 is a Th1-type chemokine, which binds to three distinct receptors CCR1 (C-C motif chemokine receptor 1), CCR3 (C-C motif chemokine receptor 3), and CCR5 (C-C motif chemokine receptor 5)16 and has a proven function in facilitating the attraction of several immune cell populations including CD8+ T cells, dendritic cells, and macrophages.17,18 While this contribution of CCL5 would suggest a beneficial function in facilitating adaptive immune responses and thus tumor clearance, the role of CCL5 in cancer is complicated and, in some cases, seemingly paradoxical. In two independent studies using colon cancer mouse models, increased expression of CCL5 was associated with immune escape through augmentation of PD-L1 (programmed death ligand 1) and reduced CD8+ T cell tumor infiltration.19,20 Furthermore, CCL5 has been shown to be a potentiator of T regulatory cell and tumor-associated macrophage intratumor infiltration.21,22 In direct contrast to these studies, CCL5 correlative studies in triple-negative breast cancer showed a positive association between CCL5 expression and immune cell tumor association including CD8+ T cells.23 Similarly, Coukos and colleagues demonstrated a significant correlation between CCL5 and an increase in CD8 T cells via the assessment of RNA expression data across several major cancer types including human colorectal cancer.15 Additionally, CCL5 has been shown to be both a facilitator and an indicator of effector T cell recruitment to the tumor microenvironment.24,25 The discrepancies in these findings raise unresolved questions about the roles of CCL5 in specific cancer subtypes and model systems, thus suggesting that more inquiries are needed.
Non-small-cell lung cancer (NSCLC) is the most prevalent type of lung cancer, which is the leading cause of cancer-related death worldwide.26 NSCLC is comprised of two major histologic subtypes lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), which originate from differing anatomical regions of the lung, alveolar, and bronchus, respectively.27 We have previously defined a positive association between viral response signaling and CD8+ T cell intratumoral accumulation in murine models of lung adenocarcinoma in the context DNMTi + HDACi treatment.13 In addition to our findings in murine models, CCL5 has been associated with an active lymphocyte compartment in patients with lung adenocarcinoma.28 Importantly, none of these previous studies defined the consequences of type I IFN signaling on CD8+ T cell infiltration, defined effects in terms of histologic subtype, or specifically studied effects in LUSC.
In the present study, we elucidate the importance of differential viral mimicry responses and interferon signaling on the accumulation and infiltration of CD8+ T cells in the two major NSCLC histologies. We also define the correlation, in both early-stage and advanced-stage lung cancer, between CD8+ T cell tumor dynamics with CCL5 and MDA5 (melanoma differentiation-associated protein 5), a dsRNA helicase type sensor.29 In patients with advanced NSCLC, we derive a significant association, which suggests that epigenetic therapy (DNMTi + HDACi) with or without concurrent administration of anti-PD-1 can augment both the accumulation and infiltration of CD8+ T cells with associative increases in CCL5. Furthermore, through the evaluation of lung tumors with both acquired and primary resistance to immune checkpoint blockade (ICB), we observe the potential of combination epigenetic immunotherapy to enhance CD8+ T cell infiltration in this ICB refractory state.
Results
Analysis of NSCLC TCGA data defines a pronounced association between CD8+ T cell-associated and type I interferon-related genes
Our previous studies have suggested a role for tumor-specific, type I interferon-associated viral response signaling in remodeling the tumor immune environment and augmenting CD8+ T cell tumor attraction.30,31 However, these data were derived in the context of therapeutic responses to DNMTi and HDACi, thereby leaving much to be defined concerning the direct effects of tumor-specific viral response pathways on the associated immune microenvironment. Thus, as an extension to these studies, we first evaluated the basal state association between gene set variation analysis32-derived interferon pathway enrichment scores and the predicted tumor microenvironment immune composition by applying the ESTIMATE algorithm utilizing TCGA (The Cancer Genome Atlas) RNAseq data from both LUAD and LUSC.30,31,33 Querying these data revealed a definite, positive correlation between interferon pathway score with either CD8A normalized counts or inferred immune abundance (immune score) in both LUAD (Figure 1A) and LUSC (Figure 1B). Extending these queries to focus on interferon genes previously associated with epigenetic therapy activity uncovered an overall positive correlation between the viral defense gene panel and immune cell infiltration, which was most distinct for CCL5, STAT1 (signal transducer and activator of transcription 1), and IFIH1 (interferon induced with helicase C domain 1) in both LUAD (Figure 1C) and LUSC (Figure 1D). Given the noted correlation between immune cell infiltration and the viral response gene panel, we expanded our interrogation of TCGA data to include correlation assessment with inferred immune abundance estimates by immune deconvolution-based approaches. Most notably, this analysis revealed a pronounced positive correlation between CCL5 and inferred CD8 abundance across both LUAD and LUSC, while IRF7 had a positive association with inferred T regulatory cell abundance (Figure 2A). Evaluation of CD8A specifically as a marker gene-based assessment confirms the positive correlation between the viral response gene panel components and CD8A expression, which was most evident for CCL5 and STAT1 in both LUSC (Figure 2B) and LUAD (Figure 2C). Interestingly, while we did observe overall conservation of these correlations between the subtypes of NSCLC, histology-specific differences between LUSC and LUAD were apparent in both strengths of correlation and rank order of variables. When evaluating correlations between individual genes in the viral defense gene panel and immune cell infiltration specifically, the most profound difference between the subtypes was the behavior of CD274 (PD-L1), shifting from a strong positive correlation in LUAD to a low-moderate positive correlation in LUSC. Furthermore, we note a larger range of CD8A correlation values in LUAD, which range from near-linear positive for CCL5 to near zero for IRF7 (interferon regulatory factor 7). In contrast, all values have a low to moderate correlation with CD8A in LUSC with a high degree of similarity between marker presentation. These data in total demonstrate a pronounced positive correlation between immune score, CD8A, and type I interferon-associated viral response genes occurring in a conserved manner across the two major subtypes of NSCLC, but with distinct histologic differences in overall strength and range of correlation coefficients noted in our expanded viral-immune response dataset.
Figure 1.

Correlation analyses of TCGA mRNA data for CD8+ T cell-related immune and interferon-related markers
(A) TCGA LUAD (n = 510) Spearman rank correlation analysis and associated scatterplot distribution for comparisons between gene set variation analysis (GSVA) enrichment scores and either RNAseq normalized CD8A counts, or ESTIMATE algorithm derived immune and stromal scores, Spearman Rho value, 95% confidence interval, and p value provided in the figure.
(B) TCGA LUSC Spearman rank correlation analysis (n = 484) and associated scatterplot distribution for comparisons between gene set variation analysis (GSVA) enrichment scores and either RNAseq normalized CD8A counts or ESTIMATE algorithm derived immune and stromal scores, Spearman Rho value, 95% confidence interval, and p value provided in the figure.
(C) TCGA LUAD (n = 510) Spearman rank correlation analysis between targeted interferon-associated genes RNAseq normalized counts, ESTIMATE immune, and ESTIMATE stromal score, Spearman Rho value provided in the figure, correlations with padj >0.05 are depicted as blank in figure. Top panel: Correlogram, Bottom panel: Network plot.
(D) TCGA LUSC (n = 484) Spearman rank correlation analysis between targeted interferon-associated genes RNAseq normalized counts, ESTIMATE immune, and ESTIMATE stromal score, Spearman Rho value provided in the figure, correlations with padj >0.05 are depicted as blank in figure. Top panel: Correlogram, Bottom panel: Network plot.
Figure 2.

Correlation analyses of TCGA mRNA data for CD8+ T cell-related immune and viral defense markers
(A) Heatmap of TCGA LUAD, n = 510 (top panel) and LUSC, n = 484 (bottom panel) RNAseq data derived Spearman correlations between TIMER 2.0 immune deconvolution inferred abundance and normalized counts for targeted interferon gene panel. Color scale based on Z score.
(B) TCGA LUSC forest plot, panel markers vs. CD8A, Pearson correlation (r with 95% confidence interval). ∗ indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli.
(C) TCGA LUAD forest plot, panel markers vs. CD8A, Pearson correlation (r with 95% confidence interval). ∗ indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli.
IHC-based correlation analyses of CD8+, immune and viral defense markers in NSCLC delineate histologic correlates of T cell accumulation and infiltration
Building on these TCGA data, we next used immunohistochemistry (IHC) to define the spatial relationships between tumor compartment-specific viral and type I interferon-associated marker expression on CD8+ T cell tumor accumulation and infiltration. A collection of tissue microarray (TMA) sections of both LUAD and LUSC were pathologist-scored for accuracy of the histologic classification and a modified H-score, hereafter referred to as H-score, was defined to quantitate the localization and infiltration status of CD8α-positive cells, henceforth termed CD8+ T cells. A scale of 0–3 was defined as follows: 0 = absent, 1 = tumor microenvironment (TME) localized, 2 = tumor-TME boundary, and 3 = tumor infiltrative (direct tumor cell-CD8 T cell contact). Interrogation of these IHC data uncovered disparate patterns of CD8+ T cell infiltration distribution between the histologic subtypes of NSCLC. LUAD sections had both higher CD8+ T cell tumor infiltration and lower CD8+ T cell absent presenting sections (Figure S1A), while LUSC tumors had a higher proportion of sections presenting as both absent and non-tumor infiltrative (Figure S1B). Importantly, these data substantiate the concept that our samples capture a diversity of CD8+ T cell localization states within and between LUAD and LUSC histologies.
The CD8 H-score accounts for both abundance and infiltration, a separate H-score quantitation was also deployed across a spectrum of viral, interferon, and proliferation markers as shown in Figure 3A, which is a composite representation of intensity and prevalence, with tumor cells and the TME scored separately. Given the noted histologic differences between LUAD and LUSC by both RNA expression and CD8+ T cell localization, we evaluated IHC datasets in a histotype-specific manner. Correlation-based assessment of these H-score data in LUAD revealed a preponderance of positive correlation between the markers contained in our selected panel (Figures 3A and S2A). These data closely mirror the TCGA data presented in Figure 1. Interestingly, TME-expressed markers, specifically pIRF7 (phosphorylated IRF7), CCL5, and MDA5, demonstrated the most significant positive association with CD8+ T cell H-score (Figures 3B and S2B). When evaluating CD8+ T cell infiltration specifically in association with interferon panel marker H-score, TME-expressed markers MDA5 and CCL5 emerged as the leading candidates in this dataset with a demonstrable positive correlation with intratumor CD8+ T cell infiltration (Figure 3C). In agreement with these H-score data, the examination of high to low CD8+ H-score representative section series recapitulates the overall trend derived by correlation analyses, wherein, both MDA5 and CCL5 intensity demonstrates a positive association with CD8+ T cell tumor infiltration and accumulation (Figure 3D). In total, these data suggest that a positive relationship exists between the presence of CCL5 and MDA5-positive TME-resident cell populations and CD8+ T cell tumor accumulation and infiltration in LUAD.
Figure 3.

IHC-based correlation analyses of CD8+ T cells vs. immune and viral defense markers in LUAD TMAs
(A) LUAD H-score-based Spearman’s rank correlation similarity matrix, one minus Spearman’s rank correlation-based clustering (n = 128).
(B) LUAD H-score-based correlation forest plot for panel markers vs. CD8α H-score, Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ Indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 128).
(C) LUAD H-score-based correlation forest plot for panel markers vs. CD8α infiltration score, Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ Indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 128).
(D) Representative NSCLC LUAD, paraffin archive to show the prioritized 4-marker IHC panel with H&E for morphologic reference. All IHC stains were scored by modified H-score with a primary focus on CD8 T cells and tumor infiltrative pattern. Tumors are arranged top to bottom by high to low CD8α H-scores, respectively. 100x–400x total magnification. Representative scale bar provided in top left image of figure panel.
Similar to the correlation assessments conducted in LUAD, analysis of H-score data in LUSC exhibited a high prevalence of positive correlation between the markers contained in our panel (Figure 4A and S2C) which closely mirrored the TCGA data as presented in Figure 1. Also, in agreement with the LUAD data, TME-expressed factors represent many leading candidates demonstrating a significant positive association with CD8+ H-score; these specific markers included PD-L1, IFI27 (interferon alpha-inducible protein 27), CCL5, and MDA5 (Figures 4B and S2D). When evaluating CD8+ T cell infiltration specifically in association with marker panel H-score, tumor cell-expressed CCL5 emerged as the most significant target, followed by TME-expressed MDA5 and CCL5 (Figure 4C). In agreement with these H-score data, assessment of high to low CD8+ H-score representative section series recapitulates the noted correlation data, wherein, sections with higher MDA5 and CCL5 intensity track with elevated CD8+ T cell tumor infiltration and accumulation. (Figure 4D). These data in total suggest a marked positive association exists between TME accumulation of CCL5 and the acquisition of a CD8 T cell tumor-infiltrative state in LUSC.
Figure 4.

IHC-based correlation analyses of CD8+ T cells vs. immune and viral defense markers in LUSC TMAs
(A) LUSC H-score-based Spearman’s rank correlation similarity matrix, one minus Spearman’s rank correlation-based clustering (n = 92).
(B) LUSC H-score-based correlation forest plot for panel markers vs. CD8α H-score, Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 92).
(C) LUSC H-score-based correlation forest plot for panel markers vs. CD8α infiltration score, Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 92).
(D) Representative NSCLC LUSC, paraffin archive to show the prioritized 4-marker IHC panel with H&E for morphologic reference. All IHC stains were scored by modified H-score and primary focus on CD8 T cells and tumor infiltrative pattern. Tumors arranged top to bottom by high to low CD8α H-scores, respectively. 100x–400x total magnification. See Figure 3D for a representative scale bar applicable to this figure panel.
In summary, a strong positive relationship is evident between our interferon marker panel and CD8+ T cell infiltration in both LUAD and LUSC. When considering T cell infiltration specifically, this association is most apparent for MDA5 and CCL5. For the subsequent analyses to follow, which were deployed in tumor biopsies derived from immunotherapy- or immunotherapy-containing regimen-treated patients, we prioritized a 4-marker IHC panel including CD8, CCL5, MDA5, and PD-L1. These markers provide molecular targets to query, which might facilitate the acquisition of CD8+ T cell attraction and intratumoral deposition in advanced NSCLC tumors, a requisite for these cells to exert antitumor effects. Of note, the selection of PD-L1 as a marker of interest to be queried as part of our prioritized panel in the subsequent sections is based upon the axis targeted (PD-1: PD-L1) by ICB in this setting.
Validation of the selected marker panel for application to clinical trial biopsies
We next sought to confirm our 4-marker panel results in serial tumor samples from patients with metastatic NSCLC. This independent validation sample set of metastatic tumors was derived from patients with advanced NSCLC in whom combination epigenetic and immunotherapy was administered (Azacitidine, Entinostat, and Nivolumab) (NCT01928576). This trial was designed to assess the potential of epigenetic therapies in two different ways to enhance the efficacy of ICB as either sequential therapy prior to anti-PD-1 or concurrently with anti-PD-1 therapy. Consistent with our findings in early-stage lung cancer, we found a markedly positive correlation between the markers contained in our 4-marker panel (Figures 5A and S3A). Furthermore, both CCL5 and MDA5 emerge in this validation dataset as leading candidates with a pronounced positive association with CD8+ T cell H-score (Figures 5B and S3B). Importantly, and in agreement with our TMA data, CCL5 (tumor & TME expressed) and MDA5 TME-specific staining was found to have a statistically significant correlation with CD8+ T cell infiltration (Figure 5C). These data in total serve as both a validation of our discovery dataset and suggest that CCL5 and MDA5 have a positive association with both CD8+ T cell accumulation and infiltration across independent sample sets of LUAD and LUSC.
Figure 5.

IHC-based correlation analyses in NSCLC tumor biopsy validation cohort
(A) H-score-based Spearman’s rank correlation similarity matrix, one minus Spearman’s rank correlation-based clustering (n = 63).
(B) H-score-based correlation forest plot for panel markers vs. CD8α H-score, Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ indicates an adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 63).
(C) H-score-based correlation forest plot for panel markers vs. CD8α infiltration score Spearman’s rank correlation (rho with 95% confidence interval depicted). ∗ Indicates adjusted p value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli (n = 63).
Analysis of paired biopsies reveals epigenetic therapy-induced augmentation of both tumor accumulation and tumor infiltration of CD8+ T cells
Our validated marker panel was utilized for paired IHC analyses of on-treatment versus baseline biopsies from patients in the clinical trial as presented in the previous section. Paired analysis of baseline versus on-treatment biopsies revealed a significant increase in CD8+ H-scores after epigenetic treatment (Figure 6A). This increase in CD8+ H-score encompassed an increase of CD8+ T cell intratumor infiltration as evidenced by a noted gain in post-therapy sections demonstrating direct contact between tumor cells and CD8+ T cells (Figure 6B). This observed augmentation in CD8+ H-score and tumor infiltration occurred in a concordant manner with the enhancement of both tumor and TME CCL5 expression (Figure 6C). In addition to CCL5 and CD8 expression, we also assessed MDA5 and PD-L1 by paired analysis; while both demonstrated modest gains in the on-treatment relative to baseline samples, neither marker achieved statistical significance (Figures S4A and S4B). In agreement with these H-score data, the examination of baseline vs. on-treatment representative sections recapitulates the overall trend derived by pre-post analysis, wherein, increases in both CCL5 staining and CD8+ T cell tumor infiltration and accumulation are evident as a function of treatment. (Figures 6D and S4C).
Figure 6.

Analysis of paired biopsies reveals epigenetic therapy-induced augmentation of both tumor accumulation and tumor infiltration of CD8+ T cells
(A) CD8α H-score comparison baseline vs. on-treatment (n = 8 paired samples). Statistical significance by Wilcoxon Signed Ranked Test, ∗ indicates p value < 0.05, mean and individual sample values are plotted.
(B) CD8+ T cell infiltration status baseline vs. on-treatment (n = 8 paired samples).
(C) CCL5 tumor and tumor microenvironment H-score comparison baseline vs. on-treatment (n = 8 paired samples). Statistical significance by Wilcoxon Signed Ranked Test, ∗ indicates p value < 0.05, mean and individual sample values are plotted.
(D) Representative CD8α and CCL5 single-plex IHC. Top panel: baseline, bottom panel: on-treatment combination epigenetic treatment (Azacitidine + Entinostat). Left panel and right panel are representatives of two independent patients. Representative scale bar provided in the left image of figure panel.
Given the distinct association of CCL5 with CD8+ T cell behavior, we aimed to define whether these factors may co-localize spatially or perhaps mark the same cell through the pursuit of CD8α and CCL5 multiplex IHC. As an initial assessment, we sought to quantitate the association between CCL5 and CD8 using multiplex IHC. These data revealed a strong correlation between the presence of dual-positive CCL5, CD8 +/+ cells, and both total CD8+ cells and non-CCL5-expressing CD8+ T cells within the tumor and associated microenvironment (Figures 7A and S5A). Interestingly, when evaluating the localization status for dual-positive cells, our data indicate that sections scored as having CD8+ T cells as infiltrative or accumulating at the tumor-TME boundary have a demonstrable difference in the percent of dual-positive cells relative to sections scored as having CD8 T cells distal from tumor cells (tumor distal geometric mean: 0.1155, tumor proximal or infiltrative geometric mean: 1.874, p value: 0.0022) (Figure 7B). Considering these data, we next aimed to define the effect that CCL5 might have on CD8+ T cells outside its well-established role as a Th1-type chemokine. Using the Immunological Genome Project database as a resource,34 we evaluated the distribution of CCL5 within deposited data. These analyses demonstrate CCL5 as being highly expressed across NK, NKT, and CD8+ T cell populations (Figure S5B). When specifically evaluating CD8+ T cell populations, the expression of CCL5 appears most pronounced in non-naive, activated CD8+ T cells from murine models of viral infection (lymphocytic choriomeningitis mammarenavirus) (Figure S5C). Expanding these assessments to the interrogation of baseline vs. on-treatment samples in the setting of combination epigenetic therapy revealed a variable, but an overall increase in CCL5 and CD8 marker expression as a function of treatment, which was most pronounced for total CD8 (effect size 1.7-fold) and CCL5, CD8 +/+ populations (effect size 2.4-fold), although neither change was determined to be statistically significant (Figures 7C and S5A). Provided multiplex IHC representative images capture the heterogeneity of the baseline vs. on-treatment response (Figures 7D). These data in total show an overall conserved increase in CD8+ T cells, while CCL5, CD8 +/+ cells show the largest absolute change.
Figure 7.

CD8+ T cells and CCL5 demonstrate spatial co-localization and correlative behavior in NSCLC clinical trial samples
(A) Spearman rank correlation analysis of CD8α and CCL5 multiplex IHC (n = 24), Spearman rho value provided in the figure, statistical significance of correlation defined as p value <0.05 as denoted by ∗.
(B) Dual-positive cell percentage (CD8α, CCL5 +/+) as binned by localization state of CD8+ T cells within section (tumor infiltrative or proximal vs. tumor distal) (n = 24), ∗∗ is a representation of Mann Whitney p value = 0.0022, the dotted line represents the median value.
(C) Quantitative analysis of baseline vs. on-treatment samples, geometric mean and standard deviation of geometric mean provided in figure, each symbol indicative of a single patient sample, n = 8 total paired comparisons.
(D) Top panel: baseline, bottom panel: on-treatment combination epigenetic treatment (Azacitidine + Entinostat). Panels left to right: CD8α +CCL5 dual chromogen, CD8α +CCL5 multiplex IHC pseudo-colored (green = CD8α, red = CCL5, and yellow = CD8α+CCL5 co-localization). Images presented at 200x magnification. Table below figure panel provides Aperio algorithm percent population assessment for the indicated populations.
In summary, our data indicate that CCL5 co-localizes with CD8+ T cells in the tumor microenvironment, and data from experimental model systems indicate that co-expression of these markers occurs markedly in non-naive CD8+ T cells. Additionally, these data show that combination epigenetic therapy-treated tumors show an enhancement of both the accumulation and intratumor infiltration of CD8+ T cells. Furthermore, the data contained in this section suggest that epigenetic therapy might be operative in the enhancement of CD8+ T cell tumor infiltration through the specific perturbation of CCL5 signaling in TME-resident populations.
Concurrent administration of epigenetic therapy with immune checkpoint blockade can potentiate CD8+ T cell tumor attraction in the setting of de novo or acquired resistance to immunotherapy.
The results presented in the preceding section were obtained from patients in our ongoing trial wherein we could chart responses from epigenetic therapy alone. We next expanded our assessment to include ICB-naive and ICB-resistant patients receiving concurrent administration of epigenetic therapy combined with anti-PD-1. Four individual cases in these trial arms, focused on evaluation of this therapeutic approach on addressing the unmet need of ICB resistance in patients with NSCLC, are shown.
Case 1 is from an 83-year-old, male, former smoker, with PD-L1+ LUAD histology without previous exposure to immune checkpoint therapy. This patient achieved a partial response per RECIST v4 criteria to the combination regimen, with associated durable clinical benefit noted for now greater than 3.5 years. The IHC results for CD8+ T cells and CCL5-associated behavior outlined in the previous section from epigenetic therapy alone are more prominent in patients who received simultaneous administration of the triplet therapy. Evaluation of triplet therapy-derived IHC data reveals a marked increase in CD8+ T cells with an associated increase in CCL5 (Figure 8A). Interrogation of multiplex CD8/CCL5 data discerns significant marker spatial co-localization, tumor-specific staining, and dual-population labeling, suggesting that CCL5 might be a facilitator of CD8+ T cell tumor infiltration (Figures 8B and S6A). Importantly, similar results are observed in the next 3 cases all involving patients who had previously developed resistance to immune checkpoint blockade.
Figure 8.

Concurrent epigenetic immunotherapy in advanced NSCLC clinical trial specimen case studies
(A, C, E, G) CD8α and CCL5 single-plex across case studies of concurrent epigenetic immunotherapy as described in the associated text, top panel baseline, bottom panel on-treatment combination epigenetic + Immunotherapy (Azacitidine + Entinostat + Nivolumab). Figure-embedded table (right) provides section H-score depicted as Baseline/On-Tx (on-treatment).
(B, D, F, H) CD8α and CCL5 multiplex IHC across case studies of concurrent epigenetic immunotherapy as described in the associated text. Top panel: baseline, bottom panel: on-treatment combination epigenetic + Immunotherapy (Azacitidine + Entinostat + Nivolumab). Figure-embedded table (right) provides section Aperio software-derived population percentages depicted as Baseline/On-Tx (on-treatment). Multiplex IHC pseudo-colored (green = CD8α, red = CCL5, and yellow = CD8α+CCL5 co-localization). All figure images are presented at 200x magnification. Representative scale bar provided in top left image of figure panel.
Case 2 is from a 55-year-old, male, never smoker, with LUAD histology who had progressed after more than 6 months on first-line combination chemotherapy ICB. This patient achieved a stable disease response with durable clinical benefit and overall survival of over 2 years after the initiation of this combination regimen. Assessment of IHC data demonstrates a sparse CD8+ T cell content in the baseline section, which is markedly increased in the on-treatment biopsy and this infiltration occurred concurrently with an associated increase in CCL5 (Figure 8C). Critically, as shown for results in the previous section, assessment of multiplex CD8/CCL5 data delineates significant marker spatial co-localization in lymphocytes (Figures 8D and S6B).
Case 3 is from a 64-year-old, male, former smoker, harboring LUSC histology previously treated with first-line combination chemotherapy-immunotherapy, followed by single-agent immunotherapy over seven months. On this clinical trial, the patient achieved stable disease response with overall survival of 11 months, even though tumor progression occurred by 5 months post-treatment initiation. Evaluation of IHC data reveals an increase in CD8+ T cells and an associated increase in CCL5, leftmost, and second to the left panels (Figure 8E). Interrogation of multiplex CD8/CCL5 data defines in the on-treatment sample, significant marker spatial co-localization, with tumor-specific CCL5 staining present in both baseline and on-treatment samples (Figures 8F and S6C).
Case 4 is from a 69-year-old, female, former light smoker, with poorly differentiated NSCLC who had progressed within 4 months of initiating single-agent consolidation ICB therapy post concurrent chemotherapy and radiation, prior to trial enrollment. On the combination regimen, the first response assessment was measured as stable disease, but the disease was noted to progress shortly thereafter, and subsequent overall survival was only 6 months from the time of trial treatment initiation. IHC data demonstrate an increase in CD8+ T cells as a function of treatment and an overall gain but not intense CCL5 staining (Figure 8G). Multiplex CD8/CCL5 IHC uncovers sparse marker spatial co-localization (Figure 8H and S6D).
A summary evaluation of these case study data delineates a trend toward enhanced tumor: T cell reactivity inclusive of tumor infiltration (Figures S7A, S8A, and S8B), increased tumor and TME CCL5 (Figures S7A and S8C), decreased PD-L1 (Figures S7A and S8D), and increased MDA5 (Figures S7A and S8E) in the on-treatment biopsies. These case study data in total suggest the potential of combination epigenetic immunotherapy to induce both CCL5 and MDA5 signaling, known T cell attraction facilitators, and critical interferon pathway effectors, respectively. The accumulation of these factors occurs concurrently with the enhancement of CD8+ T cell tumor attraction and infiltration in the setting of both de novo and prior resistance to immunotherapy.
Discussion
Our results presented herein support an expanded view of tumor-associated viral mimicry response signaling as a driver of CD8+ T cell intratumor infiltration across both LUAD and LUSC. Specifically, we demonstrate through interrogation of large-scale TCGA datasets, a definitive positive correlation between interferon and viral response signaling-related gene expression and CD8A, which is suggestive of increased tumor accumulation of CD8+ T lymphocytes. Expanding these initial inquires to the assessment of protein localization and disposition by IHC validates these findings in 2 independent sample cohorts and discerns both histotype-specific behavior of markers, and importantly tumor and expression impart strikingly different consequences on accumulation and infiltration of CD8+ T cells in NSCLC.
The role of CCL5 and MDA5 in our present observations is particularly intriguing and merits special consideration. Several other studies, including our own, have linked the presence of CCL5 to perturbation of CD8+ T cell disposition within the TME, but with seemingly divergent findings that suggest there may be context dependency for the role of this chemokine in cancer.15,19,20,23 Our present study expands on this body of literature and provides clarification for the function in NSCLC, as herein we demonstrate a clear association connecting CCL5 accumulation in the TME with the facilitation of direct contact between CD8+ T cells and tumor cells. While there is a body of research on the role of type I IFN signaling effect on CD8+ T cell biology, much of this has been focused on effector molecules such as interleukins and chemokines35,36,37,38 leaving the impact of this pathways’ requisite components unclear. Thus, our observations that high MDA5 expression within the TME is correlated with CD8+ T cell infiltration are intriguing.
As an extension to our previous pre-clinical studies, our current study confirms the potential of combination DNMTi + HDACi to both increase CCL5 and CD8+ T cell tumor attraction in clinical samples from advanced NSCLC. Importantly, we also propose that this therapeutic combination can enhance the infiltration of CD8+ T cells and facilitate direct tumor cell contact. Interestingly, this potentiation of CD8+ T cell intratumoral deposition is operative in both the immune checkpoint naive and resistant settings. This raises the question of what tumor-resident populations might be able to facilitate T cell infiltration in response to epigenetic therapy in these disparate situations. Broadly, these observations suggest synergy between these compounds and immune checkpoint blockade might be resultant in both the ICB-naive and -resistant states, thus allowing patients more therapeutic options beyond the standard of care chemotherapy + ICB.
There are several additional implications of our present findings from both a basic and translational perspective. First, it is interesting to speculate on the functional status of dual-labeled CCL5-CD8+ T cells within the TME as this population is proposed to be more cytolytic and perhaps might act in a self-recruiting manner, as defined previously in a gastric inflammatory model.39 Second, the uncovering of specific tumor-resident populations, which are positive for CCL5 and MDA5, beyond CD8+ T lymphocytes might define additional targeted therapeutic agents to synergize with standard immunotherapy approaches. Third, the enhancement of CD8+ T cell tumor infiltration in the setting of NSCLC by epigenetic therapy provides evidence that this combinatorial therapeutic paradigm can remodel the immune microenvironment, enabling a more permissive state for T cell infiltration even in the setting of resistance to immunotherapy. Fourth, our results provide additional correlative markers to query in our ongoing trial of concurrent administration of combination epigenetic plus immunotherapy for the treatment of ICB-naive and -resistant advanced NSCLC and may aid in the delineation of factors driving clinical response.
Limitations of the study
The results presented in this study are entirely correlational in nature and thus do not demonstrate causation between the various markers depicted. Additionally, the clinical trial immunohistochemistry analysis is conducted on a small number of patients; therefore, any conclusions made will require validation in subsequent cohorts of combination epigenetic immunotherapy-treated patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CCL5 | R&D Systems | AF-278 RRID:AB_354440 |
| MDA 5 | Invitrogen | 700360 |
| CD8 | Bio Rad | MCA1817 RRID:AB_322868 |
| PD-L1 SP263 | Ventana | 740–4907 |
| RIG-I | Bio Rad | AHP1776 RRID:AB_2175702 |
| IRF7 | Aviva | OABF01189 |
| IFI27 | Thermo fisher | BS-15549R |
| ds RNA J2 | Scicons | 1510 |
| ds RNA K1 | Scicons | 1502 |
| IFIT1 | abcam | ab118062 RRID:AB_10901878 |
| IRF-9 | abcam | ab126940 RRID:AB_11129364 |
| Ki-67 | abcam | ab16667 RRID:AB_302459 |
| pSTAT1 | Cell Signaling | 9167 RRID:AB_561284 |
| Critical commercial assays | ||
| Dako envision Flex Kit | Agilent | K8023 |
| Ventana DAB | Roche | 760–500 |
| Dako High pH retrieval buffer | Agilent | GV804 |
| Dako Low pH retrieval buffer | Agilent | GV805 |
| Ventana CC1 | Roche | 950–224 |
| Software and algorithms | ||
| ESTIMATE | R -Forge | N/A |
| TIMER 2.0 | http://timer.cistrome.org/ | N/A |
| Gene Set Variation Analysis | R Bioconductor | RRID:SCR_021058 |
| Corrr | Github | N/A |
| Hmisc | CRAN | RRID:SCR_02249 |
| Corrplot | Github | RRID:SCR_023081 |
| Pheatmap | R Bioconductor | RRID:SCR_016418 |
| Morpheus | Broad Institute | RRID:SCR_017386 |
| Prism | GraphPad | RRID:SCR_002798 |
| Aperio ImageScope | Leica | RRID:SCR_020993 |
| STATA SE | STATA | RRID:SCR_012763 |
| Other | ||
| Clinical trial | NCT01928576 | N/A |
| TCGA LUAD RNAseq | Broad GDAC Firehose | RRID:SCR_003193 |
| TCGA LUSC RNAseq | Broad GDAC Firehose | RRID:SCR_003193 |
| ImmGen | Immunological Genome Project Consortium | RRID:SCR_021792 |
Resource availability
Lead contact
Furthermore information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael J. Topper (mtopper1@jhmi.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
The protocol was approved by local Institutional Review Boards (IRB), and all participating subjects signed informed consent. For clinical trial samples derived from non-small cell lung cancer patients, age range: 46-83 years of age, Sex: n=23 males, n=21 females.
Method details
Tissue samples
Archival paraffin blocks of non-small cell lung cancer (NSCLC) resections provided preliminary validation in archived, post-retention NSCLC surgical resections from western Michigan hospitals to include lung squamous cell carcinoma (LUSC), n=92, and lung adenocarcinoma (LUAD), n=128. Representative tumor blocks were sampled with 1.0 mm cores, double-punched, arranged in tissue microarray (TMA) format, and constructed with common control cores across all TMA blocks. All tissues were fixed in 10% formalin-fixed and standard processing and paraffin-embedded. The clinical trial tissues (n=63) for primary analysis were biopsies provided as whole sections and freshly cut by the histology department at JHU, sent to VAI, and subsequently, IHC stained promptly. Distant archival samples in TMA format and biopsy slides were IHC stained under the same parameters and antibody dilutions. The clinical trial design included patient-matched metastatic lung tumor biopsies with sample origin coding unique to the JHU team and independent of assigned VAI sample numbers. The VAI pathologist (GH) was blinded at multiple levels to individual and sample pair identity.
Immunostaining
TMA sections and biopsy whole sections were cut at 5-micron thickness onto standard charged slides. Due to the value of biopsy slides, H&E was not performed but cellular adequacy was judged on hematoxylin counterstain, best seen with the MDA5 slide. Minimum criteria of >25 tumor cells are required for analyses and assessment by the primary pathologist (GH). See table below for complete information for each antibody immunostaining procedure, including primary dilution, primary incubation time, secondary type, secondary incubation time, detection reagent, antigen retrieval method, antigen retrieval temperature, and antigen retrieval time.
| Staining platform | Primary | Primary dilution | Primary incubation time | Secondary type | Secondary incubation time | Detection | Antigen Retrieval (AR) | AR temp | AR time |
|---|---|---|---|---|---|---|---|---|---|
| Dako Autostainer Link 48 | CCL5 | 1:40 | 30 min | Goat on Rodent HRP Polymer | 15 min goat probe, 20 min Goat HRP | Dako envision Flex Kit | Dako Low pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | MDA 5 | 1:700 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | CD8 | 1:35 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Ventana Discovery Ultra | PD-L1 SP263 | Ready to use | 60 min | UMAP anti-Rb HRP | 20 min | Ventana DAB | Ventana CC1 | 100C | 92 min |
| Dako Autostainer Link 48 | RIG-I | 1:175 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako Low pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | IRF7 | 1:150 | 30 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako Low pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | IFI27 | 1:100 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako Low pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | ds RNA J2 | 1:150 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | ds RNA K1 | 1:35 | 25 min | Dako Flex/HRP Polymer | 25min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | IFIT1 | 1:50 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako Low pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | IRF-9 | 1:100 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | Ki-67 | 1:100 | 20 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
| Dako Autostainer Link 48 | pSTAT1 | 1:75 | 40 min | Dako Flex/HRP Polymer | 20 min | Dako envision Flex Kit | Dako High pH retrieval buffer | 97C | 20 min |
IHC scoring
Single markers
Single stained slides were scanned at 20x by digital imaging Aperio scanner (Leica, Inc.) and scored by study pathologist (G. Hostetter) using a modified H-Score method comprised of two components of stain intensity (0-3) multiplied by stain prevalence (0-6) with total H-score of range 0-18. All markers except CD8, ki67, and pSTAT1 were scored by tumor and stroma compartment separately. CD8 has a relatively uniform intensity score and received a stromal score only. The intensity score was modified from 1 to 3 to capture the pattern of CD8 infiltration: lack of directionality tumor or stroma=1, aggregating at tumor edge=2, and actively infiltrating tumor=3. Clinical trial samples were reviewed and scored by the pathologist (G. Hostetter) in a double-blind manner and were analyzed at separate institutions. Independent pathology review of separate tumor set of the same IHC single stained markers, and this modified H-Score method showed >90% concordance.
Multiplex marker
CD8/CCL5 slides were scanned at 20x by Aperio scanner. Hematoxylin slide lightly stained a representative case for each stain to be multiplexed (CCL5, CD8) to provide the most robust threshold levels in downstream analyses. Multiplexed slides reviewed post scanning and regions marked to be excluded from analysis were debris, necrosis, and fresh blood/serum. All cellular tissue regions were analyzed and color signal thresholds separate Dapi (nuclear counterstain), DAB chromogenic brown of CD8 marked cells, and magenta chromogenic of CCL5. Pseudo-coloring of the above chromogenic signals was designated blue (Dapi), green(brown), and red(magenta) with overlying red and green producing yellow signal pseudo-color to indicate co-localized expression of CD8 and CCL5. The chromogenic multiplexed images are analyzed using Aperio algorithms based on cell-specific nuclear stains and pseudo color designations of 1) dark blue for counterstained nuclei, 2) green for CD8 (brown chromogen) and 3) red for CCL5 (magenta chromogen). Multiplex data were analyzed using Aperio Colocalization Algorithm. This procedure was used to calculate chromogen staining by pixel location and classifies each pixel represented by single stain or combination of stains and provides precise information on the distribution of CD8 and CCL5 at the pixel level. Aperio algorithms utilize cell-specific nuclear stains and pseudo color designations of 1) dark blue for counterstained nuclei, 2) green for CD8 (brown chromogen), and 3) red for CCL5 (magenta chromogen) The Dapi stained nuclei function is used to determine all cells in the tissue sample and provides metrics on how many cells are analyzed and the percentage of cells stained with a biomarker of interest.
TCGA analysis
For ESTIMATE and Gene Set Variation Analysis (GSVA) correlation analyses: lung adenocarcinoma and lung squamous cell carcinoma normalized RNAseq counts were retrieved from the Broad firehose portal. The ESTIMATE R package was used for the derivation of immune and stromal scores, while GSVA querying was used for the derivations Hallmark interferon alpha, and Hallmark interferon gamma enrichment scores. Correlation analyses (spearman rank) were performed and plotted using the Corrr, Hmisc, and Corrplot R packages. Pearson correlation analyses were conducted on normalized RNAseq counts using GraphPad Prism version 8.4.2. For correlation analyses, significance was defined based on an adjusted p-value less than 0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli. Immune deconvolution analysis was performed using TIMER 2.0 and the resulting inferred abundance values correlated (spearman rank) with normalized TCGA RNAseq counts and then used as input for heatmap visualization using the pheatmap R package.
Quantification and statistical analysis
Statistical analysis RNAseq data
Gene set variation analysis (GSVA) enrichment scores, RNAseq normalized CD8A counts, or ESTIMATE algorithm derived immune and stromal scores, were compared using spearman rank correlation analysis, statistical significance was defined where applicable as either raw- or adjusted p-value <0.05.
Statistical analysis IHC data
Similarity matrix data were derived using a modified H-score as input for the Broad MORPHEUS, using similarity matrix followed by unsupervised hierarchical clustering using one minus Spearman’s rank or one minus Kendall tau. Spearman’s rank correlation analyses were conducted on H-score data using GraphPad Prism version 8.4.2. Kendall Tau correlation analyses were conducted using STATA SE version 16.0. Statistical significance was defined based on an adjusted p-value <0.05 after FDR 0.05 multiple comparisons correction two-stage linear step-up procedure of Benjamini, Kriegerm, and Yekutieli. CD8α and CCL5 H-score comparison baseline vs on-treatment statistical significance was determined by Wilcoxon Signed Ranked Test, the asterisk indicates p-value <0.05. Dual-positive cell percentage (CD8α, CCL5 +/+) as binned by localization state statistical significance was determined by Mann-Whitney.
Additional resources
Clinical registry number: NCT01928576
Acknowledgments
We thank the Van Andel Institute Pathology and Biorepository Core, especially Lisa Turner and Kristin Feenstra, for their assistance in multiplex staining, digital imaging, and analyses. Additionally, we thank Gavin Pereira and Alexandria Curry in the Upper Aerodigestive Biorepository at Johns Hopkins University, School of Medicine for sample procurement. Research funding for this work was provided by Van Andel Institute through the Van Andel Institute–Stand Up To Cancer Epigenetics Dream Team, Stand Up To Cancer is a division of the Entertainment Industry Foundation (S.B.B., P.A.J, and G.H.). The Evelyn Grollman Glick Scholar Award (M.J.T.), The Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (V.V., S.B.B.), US National Institutes of Health grants CA121113 (V.A. and V.V.), CA006973 (V.V), and P30CA006973 (SKCCC Core Grant) (The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health), the V Foundation (V.A., V.V., and S.B.B.), the Stand Up To Cancer Jim Toth Sr. Breakthrough Prize in Lung Cancer (J.B., S.B.B.), the Rising Tide Foundation for Clinical Research (J.B., S.B.B.), Samuel Waxman Cancer Research Foundation Collaboration for a Cure Grant (S.B.B.), and the LUNGevity Foundation (V.A.).
Author contributions
M.J.T. and S.B.B. conceptualized the idea and wrote the original manuscript. M.J.T. and G.H.H. analyzed data and generated visualizations. J.R.B. and K.A.M. provided clinical data. M.J.T., S.B.B., G.H.H., J.R.B., K.A.M., P.A.J., V.A., and V.E.V. edited the manuscript and provided feedback on analysis approaches.
Declaration of interests
V.A. receives research funding to her institution from Astra Zeneca and Personal Genome Diagnostics, has received research funding to Johns Hopkins University from Bristol-Myers Squibb and Delfi Diagnostics in the past 5 years and is an advisory board member for Neogenomics. V.A is an inventor on patent applications (63/276,525, 17/779,936, 16/312,152, 16/341,862, 17/047,006 and 17/598,690) submitted by Johns Hopkins University related to cancer genomic analyses, ctDNA therapeutic response monitoring and immunogenomic features of response to immunotherapy that have been licensed to one or more entities. Under the terms of these license agreements, the University and inventors are entitled to fees and royalty distributions. KAM has received consulting/advisory fees from AstraZeneca, Amgen, Janssen, Mirati Therapeutics, Daiichi Sankyo/Lilly and Puma Biotechnology, as well as Honoraria from AstraZeneca. KAM receives research funding to Johns Hopkins University from Bristol-Myers Squibb and Mirati Therapeutics. S.B.B. is on the Scientific Advisory Board for Mirati Therapeutics Inc. S.B.B. is a consultant for MDxHealth. MSP is licensed to MDxHealth in an agreement with Johns Hopkins University (JHU). S.B.B. and JHU are entitled to royalty shares received from sales. S.B.B. is an inventor of MSP which is licensed to MDxHealth in agreement with Johns Hopkins University (JHU). S.B.B. and JHU are entitled to royalty sales shares. P.A.J. is a paid consultant for Zymo Research Corporation. V.E.V. is a founder of Delfi Diagnostics, serves on the Board of Directors and as an officer for this organization, and owns Delfi Diagnostics stock, which is subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics. V.E.V. divested his equity in Personal Genome Diagnostics (PGDx) to LabCorp in February 2022. V.E.V. is an inventor on patent applications submitted by Johns Hopkins University related to cancer genomic analyses and cell-free DNA for cancer detection that have been licensed to one or more entities, including Delfi Diagnostics, LabCorp, Qiagen, Sysmex, Agios, Genzyme, Esoterix, Ventana and ManaT Bio. Under the terms of these license agreements, the University and inventors are entitled to fees and royalty distributions. V.E.V. is an advisor to Viron Therapeutics and Epitope. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107095.
Contributor Information
Michael J. Topper, Email: mtopper1@jhmi.edu.
Stephen B. Baylin, Email: sbaylin@jhmi.edu.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. Information specific to these datasets are listed in the key resources table. Tissue microarray-based IHC data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snell L.M., McGaha T.L., Brooks D.G. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38:542–557. doi: 10.1016/j.it.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrat F.J., Crow M.K., Ivashkiv L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019;20:1574–1583. doi: 10.1038/s41590-019-0466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M.M.H., MacDonald M.R., Rice C.M. To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol. 2015;25:320–329. doi: 10.1016/j.tcb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sistigu A., Yamazaki T., Vacchelli E., Chaba K., Enot D.P., Adam J., Vitale I., Goubar A., Baracco E.E., Remédios C., et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014;20:1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 6.Denton A.E., Innocentin S., Carr E.J., Bradford B.M., Lafouresse F., Mabbott N.A., Mörbe U., Ludewig B., Groom J.R., Good-Jacobson K.L., Linterman M.A. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 2019;216:621–637. doi: 10.1084/jem.20181216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrales L., Matson V., Flood B., Spranger S., Gajewski T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108. doi: 10.1038/cr.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajewski T.F., Schreiber H., Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuertes M.B., Kacha A.K., Kline J., Woo S.R., Kranz D.M., Murphy K.M., Gajewski T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu C., Klement J.D., Ibrahim M.L., Xiao W., Redd P.S., Nayak-Kapoor A., Zhou G., Liu K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer. 2019;7:157. doi: 10.1186/s40425-019-0635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J., Cao Y., Markelc B., Kaeppler J., Vermeer J.A., Muschel R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Invest. 2019;129:4224–4238. doi: 10.1172/JCI127458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacquelot N., Yamazaki T., Roberti M.P., Duong C.P.M., Andrews M.C., Verlingue L., Ferrere G., Becharef S., Vétizou M., Daillère R., et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29:846–861. doi: 10.1038/s41422-019-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topper M.J., Vaz M., Chiappinelli K.B., DeStefano Shields C.E., Niknafs N., Yen R.W.C., Wenzel A., Hicks J., Ballew M., Stone M., et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284–1300.e21. doi: 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone M.L., Chiappinelli K.B., Li H., Murphy L.M., Travers M.E., Topper M.J., Mathios D., Lim M., Shih I.M., Wang T.L., et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA. 2017;114:E10981–E10990. doi: 10.1073/pnas.1712514114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dangaj D., Bruand M., Grimm A.J., Ronet C., Barras D., Duttagupta P.A., Lanitis E., Duraiswamy J., Tanyi J.L., Benencia F., et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell. 2019;35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marques R.E., Guabiraba R., Russo R.C., Teixeira M.M. Targeting CCL5 in inflammation. Expert Opin. Ther. Targets. 2013;17:1439–1460. doi: 10.1517/14728222.2013.837886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schall T.J., Bacon K., Toy K.J., Goeddel D.V. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 18.Kawai T., Seki M., Hiromatsu K., Eastcott J.W., Watts G.F., Sugai M., Smith D.J., Porcelli S.A., Taubman M.A. Selective diapedesis of Th1 cells induced by endothelial cell RANTES. J. Immunol. 1999;163:3269–3278. [PubMed] [Google Scholar]
- 19.Zhang S., Zhong M., Wang C., Xu Y., Gao W.Q., Zhang Y. CCL5-deficiency enhances intratumoral infiltration of CD8. Cell Death Dis. 2018;9:766. doi: 10.1038/s41419-018-0796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cambien B., Richard-Fiardo P., Karimdjee B.F., Martini V., Ferrua B., Pitard B., Schmid-Antomarchi H., Schmid-Alliana A. CCL5 neutralization restricts cancer growth and potentiates the targeting of PDGFRβ in colorectal carcinoma. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melese E.S., Franks E., Cederberg R.A., Harbourne B.T., Shi R., Wadsworth B.J., Collier J.L., Halvorsen E.C., Johnson F., Luu J., et al. CCL5 production in lung cancer cells leads to an altered immune microenvironment and promotes tumor development. OncoImmunology. 2022;11 doi: 10.1080/2162402X.2021.2010905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larroquette M., Guegan J.P., Besse B., Cousin S., Brunet M., Le Moulec S., Le Loarer F., Rey C., Soria J.C., Barlesi F., et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araujo J.M., Gomez A.C., Aguilar A., Salgado R., Balko J.M., Bravo L., Doimi F., Bretel D., Morante Z., Flores C., et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018;8:4899. doi: 10.1038/s41598-018-23099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong H., Xi Y., Yuan Z., Wang B., Hu S., Fang C., Cai Y., Fu X., Li L. IFN-γ activates the tumor cell-intrinsic STING pathway through the induction of DNA damage and cytosolic dsDNA formation. OncoImmunology. 2022;11 doi: 10.1080/2162402X.2022.2044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J., Ping Y., Zhao Q., Guo R., Shan J., Liu F., Wang J., Zhang Y. BPIFB2 is highly expressed in "cold" lung adenocarcinoma and decreases T cell chemotaxis via activation of the STAT3 pathway. Mol. Cell. Probes. 2022;62 doi: 10.1016/j.mcp.2022.101804. [DOI] [PubMed] [Google Scholar]
- 26.Molina J.R., Yang P., Cassivi S.D., Schild S.E., Adjei A.A. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung W.K.C., Nguyen D.X. Lineage factors and differentiation states in lung cancer progression. Oncogene. 2015;34:5771–5780. doi: 10.1038/onc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moran C.J., Arenberg D.A., Huang C.C., Giordano T.J., Thomas D.G., Misek D.E., Chen G., Iannettoni M.D., Orringer M.B., Hanash S., Beer D.G. RANTES expression is a predictor of survival in stage I lung adenocarcinoma. Clin. Cancer Res. 2002;8:3803–3812. [PubMed] [Google Scholar]
- 29.Brisse M., Ly H. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol. 2019;10:1586. doi: 10.3389/fimmu.2019.01586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heng T.S.P., Painter M.W., Immunological Genome Project Consortium The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 35.Aichele P., Unsoeld H., Koschella M., Schweier O., Kalinke U., Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 36.Kolumam G.A., Thomas S., Thompson L.J., Sprent J., Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curtsinger J.M., Valenzuela J.O., Agarwal P., Lins D., Mescher M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal P., Raghavan A., Nandiwada S.L., Curtsinger J.M., Bohjanen P.R., Mueller D.L., Mescher M.F. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 2009;183:1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohtani N., Ohtani H., Nakayama T., Naganuma H., Sato E., Imai T., Nagura H., Yoshie O. Infiltration of CD8+ T cells containing RANTES/CCL5+ cytoplasmic granules in actively inflammatory lesions of human chronic gastritis. Lab. Invest. 2004;84:368–375. doi: 10.1038/labinvest.3700039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. Information specific to these datasets are listed in the key resources table. Tissue microarray-based IHC data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.