

Abstract
Background:
Comparisons of late-onset Alzheimer disease (LOAD) and autosomal dominant AD (ADAD) are confounded by age.
Methods:
We compared biomarkers from cerebrospinal fluid (CSF), magnetic resonance and amyloid imaging with the Pittsburgh Compound-B (PiB) across 4 groups of 387 cognitively normal participants of 42 to 65 years in the Dominantly Inherited Alzheimer Network (DIAN) and the Adult Children Study (ACS) of LOAD: DIAN mutation carriers (MC) and non-carriers (NON-MC), and ACS participants with a positive (FH+) and negative (FH-) family history of LOAD.
Results:
At baseline, MC had the lowest age-adjusted level of CSF Aβ42 and the highest levels of total and phosphorylated Tau-181, and PiB uptake. Longitudinally, MC had similar increase in PiB uptake to FH+, but drastically faster decline in hippocampal volume than others, and was the only group showing cognitive decline.
Discussion:
Preclinical ADAD and LOAD share many biomarker signatures, but cross-sectional and longitudinal differences may exist.
Search term: [26] Alzheimer Disease, [36] Cognitive Aging
Keywords: Autosomal dominant Alzheimer disease (ADAD), CSF analytes, imaging biomarkers, late onset Alzheimer disease (LOAD), longitudinal rate of change, Alzheimer disease (AD), Cerebrospinal fluid (CSF), Magnetic resonance imaging (MRI), Positron emission tomography (PET), Pittsburgh Compound-B (PiB)
1. Introduction
Molecular biomarkers of Alzheimer disease (AD) from cerebrospinal fluid (CSF) and neuroimaging have been well established and validated, thanks to major biomarker studies such as the Alzheimer Disease Neuroimaging Initiative (ADNI1) for late-onset Alzheimer disease (LOAD) and the Dominantly Inherited Alzheimer Network (DIAN2) for autosomal dominant AD (ADAD). A recent comparative study between ADAD and LOAD largely focused on the symptomatic individuals of LOAD3. However, converging research evidence suggests that the neuropathological course of AD begins decades prior to symptom onset4–7. Comparative studies of biomarkers at the asymptomatic stage of ADAD and LOAD are needed to understand whether the preclinical progression of AD pathophysiology is similar.
Age is the greatest risk factor of AD, and essentially all established molecular biomarkers of AD are functions of age at the asymptomatic stage for both LOAD and ADAD2,8. Age hence confounds the comparison between LOAD and ADAD and complicates its interpretation. For example, if biomarkers were found different between ADAD and LOAD, the differences can not be solely attributed to the AD subtypes3. Methodologically, it is therefore important to compare ADAD and LOAD when age is matched so that its effect can be controlled and the differences may then be attributed to the two AD subtypes. Most longitudinal biomarker studies of preclinical LOAD focused on the middle to old age windows (e.g., >=45 years), whereas the mean age at symptomatic onset in ADAD is ~46 years9–10. An age-matched study of ADAD and LOAD at the asymptomatic stage, therefore, implies that ADAD mutation carriers (MC) are likely much closer to the expected symptom onset than their middle-aged counterparts in LOAD. Recent studies suggested potentially diverging longitudinal AD biomarker trajectories between ADAD and LOAD among middle-aged individuals. Attenuated longitudinal changes for some AD biomarkers were reported in ADAD when age was close to the expected symptom onset (~46 years)11, whereas accelerated longitudinal changes for almost all major AD biomarkers were reported among cognitively normal individuals of 45 to 65 years at risk of LOAD8. It remains unknown, however, whether the longitudinal biomarker trajectories in age-matched ADAD and LOAD at the asymptomatic stage are similar. Shared pathobiological constructs among cognitively normal individuals would support the rationale that mechanism-based prevention therapies that delay the symptom onset of ADAD also are likely to be efficacious in slowing early progression of LOAD. Findings may also inform the design and analyses of secondary prevention trials both at late preclinical stages of ADAD and at early preclinical stages of LOAD among middle-aged individuals.
The objective of this study is to compare, both cross-sectionally and longitudinally, the CSF and imaging biomarkers as well as cognition in asymptomatic individuals who had a parental history of either ADAD or LOAD and were aged-matched at baseline, and further to assess whether age interacts with AD subtypes in the comparisons. Our hypotheses are that, cross-sectionally, age-matched asymptomatic ADAD MC will have the more advanced molecular biomarker profiles, but longitudinally, they will have similar trajectories of molecular AD biomarkers but faster cognitive decline, in comparison to those with a positive family history (FH) of LOAD.
2. Methods
2.1. Participants
Participants are from the DIAN and the Adult Children Study (ACS) at Washington University (WU) School of Medicine. The DIAN is an international, multicenter network of individuals of 18 years or older from families whose parents have a known causative mutation of AD in the amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) genes2. Both MC and non-carriers (NON-MC) were enrolled. The presence or absence of an ADAD mutation was determined using PCR-based amplification of the appropriate exon followed by Sanger sequencing. Since 2005, the ACS has enrolled a cohort of cognitively normal individuals, 42 to 77 year old, with and without a FH of LOAD12–13. A positive FH (FH+) is defined as at least one biological parent with age at onset for AD dementia below 80 years, and a negative FH (FH-) is defined as both biological parents living to age 70 years or greater without AD dementia. Members in families with an ADAD and/or a known causative mutation for ADAD were excluded in the ACS. In both studies, in addition to clinical/cognitive measures, a comprehensive spectrum of biomarkers for AD were assessed through largely consistent protocols longitudinally, including magnetic resonance imaging (MRI)-based regional brain volumes, cerebrospinal fluid (CSF) analytes13, and molecular imaging of cerebral fibrillar amyloid with positron emission tomography (PET) using the [11C] benzothiazole tracer, Pittsburgh Compound-B (PiB)14.
The main inclusion criterion for the current study is the baseline age of 42 to 65 years, the overlapping window between the ACS and DIAN. Only cognitively normal individuals at baseline, operationalized as a global Clinical Dementia Rating™® (CDR™®)15 of 0, were included. Data were obtained from fully quality-controlled data freeze twelve of the DIAN, and included 33 MC and 58 NON-MC. A total of 176 FH+ and 120 FH- ACS participants were included in the analyses.
The WU Human Research Protection Office approved both the ACS and DIAN, the Institutional Review Boards/Independent Ethics Committees approved the DIAN protocol at all DIAN sites, and all participants gave written informed consent.
2.2. Clinical and cognitive assessments
The clinical assessments of both the DIAN and ACS are similar and have been described previously2,12. In brief, the primary clinical assessment protocol is that of the National Alzheimer Coordinating Center Uniform Data Set (UDS)16, which includes standard definitions and diagnostic criteria for detection of dementia and its etiologic diagnosis15–16. The presence or absence of dementia and, when present, its severity were operationalized with the CDR. Participants completed comprehensive psychometric testing 1 to 2 weeks after they received the clinical assessment in both the DIAN and the ACS, as previously described2,12. The cognitive batteries, however, were designed to be quite different between the two studies. Five cognitive tests were shared: Animal Fluency, Trailmaking A (Trails A), Trailmaking B (Trails B), and Simon and Switch test2,12. A global cognition score was computed by averaging the z-scores from all 5 tests, each of which was obtained by first subtracting the mean of all longitudinal scores over the combined cohort from each individual’s score and then dividing the difference by the standard deviation (SD) that was also computed from all longitudinal scores over the combined cohort. This definition of z-scores is mathematically equivalent to the one using the baseline mean and SD, and hence leads to identical statistical inferences (i.e., p-values) and scientific conclusions. Both clinical and cognitive assessments were conducted longitudinally every three years except for participants later becoming symptomatic during the follow-up in the ACS or participants in the DIAN within three years of their expected age of symptom onset who were assessed annually.
2.3. Image acquisition and processing
Baseline and longitudinal MRI scans in both the ACS and the DIAN were similarly obtained with the use of qualified 3-Tesla scanners. T1-weighted MPRAGE images were processed using the FreeSurfer image analysis suite (Athinoula A. Martinos Center for Biomedical Imaging, Charlestown, Massachusetts, version 5.3). Structural MRI processing steps included motion correction and atlas transformation. Determination of the regions-of-interest (ROIs) and pipelines and FreeSurfer quality control (QC) criteria were described elsewhere17. PET and MRI scans that failed QC or required manual processing were not included in analyses. Volumes were obtained for each region and the ROIs were used for PET processing as previously described18. Amyloid deposition in both studies was quantified using PiB. Regional data from the 30–60 min and 40–70 min post-injection window in the ACS and DIAN cohorts respectively were transformed into standardized uptake value ratio (SUVR) using a cerebellar grey reference. Data was corrected for partial volume effects using a geometric transfer matrix approach2, 12. The mean cortical SUVR (MCSUVR) was calculated from FreeSurfer regions within the prefrontal cortex, precuneus, and temporal cortex (left and right lateral orbitofrontal, medial orbitofrontal, precuneus, rostral middle frontal, superior frontal, superior temporal, and middle temporal cortices)18. All MRI and PET imaging processing was conducted by the same lab following a standard protocol similar to that of the ADNI. More in-depth descriptions of imaging parameters and processing have been published previously2, 12–13,19.
2.4. CSF collection and analysis
Baseline and longitudinal CSF was collected in the ACS and DIAN every two to three years. Both implemented a similar assessment protocol, which has been described previously11–12. Briefly, CSF (20–30 mL) was collected by routine lumber puncture (LP) in polypropylene tubes at 8:00 AM after overnight fasting. ACS samples were briefly centrifuged (2000g for 10 min at 4°C) prior to aliquoting in polypropylene tubes (0.5ml) and storing at −80°C until analysis. DIAN samples were flash frozen on dry ice without prior centrifugation, shipped on dry ice to the DIAN Biomarker Core at WU whereupon they were thawed, aliquoted into polypropylene tubes (0.5ml) and stored at −80°C until analysis. For all CSF measures, sample aliquots were continuously kept on ice, and assays were performed on the same aliquot after a single thaw following initial aliquot freezing. For both ACS and DIAN samples, CSF Aβ42, total Tau (Tau) and phosphorylated Tau-181 (pTau181) were measured with a fully automated system, the Elecsys immunoassays on the cobas e 601analyzer. The Elecsys immunoassays are electrochemiluminescence immunoassays employing a quantitative sandwich principle with a total assay duration of 18 minutes. Pristine aliquots were measured according to the Roche study protocol (RD002967) written specifically to measure these samples. Details were described previously19.
2.5. Genotyping
APOE genotyping was performed from DNA obtained via a blood draw or buccal swab specimens using standard techniques as previously described2,12.
2.6. Statistical analysis
All results reported were from adjusted analyses. Baseline biomarkers and cognition were analyzed as a function of four participant groups, DIAN MC and NON-MC, ACS FH+ and FH-, and age through an Analysis of Covariance (ANCOVA) model. General linear mixed models (GLMM) for longitudinal data20 were implemented to compare biomarkers and cognition on the longitudinal rates of change across the four groups. Specifically, for each marker, a random intercept and a random slope (i.e., the rate of change) were assumed across individuals21, allowing the mean intercept and slope to be functions of the participant groups and other covariates: baseline age, gender, APOE ε4 status, education. Unlike a recent study that compared biomarkers from the DIAN and ADNI and aligned the participants by their ages to symptom onset which, for symptomatic ADNI participants (at baseline), was extrapolated using the longitudinal trajectory of CDR sum of box3, we matched the DIAN and ACS by participants’ baseline age, mainly because extrapolated ages of symptom onset for the middle-aged ACS participants may be subject to substantial bias and errors. Statistical tests to compare participant groups were based on approximate F or t tests with denominator degrees of freedom estimated by the Satterthwaite method22, and implemented in PROC MIXED/ SAS22. Pairwise comparisons across 4 participant groups were only conducted after the omnibus test rejected the null hypothesis that all 4 groups shared the same parameter that was tested by ANOVA or GLMM at a significance level of 5%23.
3. Results
Table 1 presents the baseline demographics as a function of the four participant groups, including APOE ε4 status, the mean (SD) of baseline CSF and imaging biomarker values and cognition, and the mean (SD) for the range of the longitudinal follow-ups as a function of data modalities (clinical/cognition, MRI, and PET PIB amyloid imaging). All 387 individuals underwent baseline clinical and cognitive assessments. Of those, 271 individuals (22 in DIAN MC, 32 in DIAN NON-MC, and 92 in ACS FH-, 125 in ACS FH+) underwent longitudinal clinical and cognitive assessments for an average of 6.9 years (SD=4.1 years) (Table 1).
Table 1.
Demographics and biomarker descriptive statistics at baseline (n=387)
| DIAN MC (n=33) | ACS FH+ (n=176) | DIAN NON MC (n=58) | ACS FH− (n=120) | |
|---|---|---|---|---|
| Variables | ||||
| Age (years): mean (SD) | 47..4 (5..18) | 55..7 (5..75) | 50..3 (5..87) | 57..1 (5..17) |
| Female | 22 (67%) | 124 (71%) | 36 (62%) | 76 (63%) |
| Family History Positive | 33 (100%) | 176 (100%) | 58 (100%) | 0 (0%) |
| APOE ε4 Positive | 12 (36%) | 77 (50%) | 23 (40%) | 26 (23%) |
| Education (years) : mean (SD) | 14..6 (3.03) | 16..3 (2..29) | 15..0 (2..91) | 16..2 (2..46) |
| MMSE: mean (SD) | 28.9 (1.16) | 29.5 (0.83) | 29.1 (1.18) | 29.4 (1.02) |
| CSF Aβ42 ( pg/mL) : mean (SD) | 885.90 (479.33) | 1641.42 (733.44) | 1395.17 (439.06) | 1444.05 (591.64) |
| CSF Tau (pg/mL) : mean (SD) | 246.79 (110.60) | 190.62 (68.77) | 199.26 (54.51) | 181.67 (62.71) |
| CSF pTau181 (pg/mL) : mean (SD) | 24.01 (13.55) | 17.25 (6.37) | 16.54 (5.18) | 16.42 (6.09) |
| PiB MCSUVR: mean (SD) | 1.97 (0.88) | 1.09 (0.30) | 1.10 (0.25) | 1.07 (0.27) |
| Hippocampal Volume (mm3) : mean (SD) | 8710.41 (687.64) | 8188.54 (754.63) | 8589.34 (688.08) | 8214.12 (802.93) |
| Cortical Thickness, (mm) : mean (SD) | 4.60 (0.27) | 4.60 (0.23) | 4.67 (0.25) | 4.60 (0.24) |
| Global Cognition: mean (SD) | −0.06 (0.53) | 0.04 (0.39) | 0.15 (0.44) | −0.03 (0.60) |
| CSF length of follow-up (years) : mean (SD) | 2..10 (1.11), (0.80 – 5.00) n=27 |
5.90 (3.59), (2.93 – 12.45) n=112 |
2.81 (1.17), (1.11 – 5.94) n=31 |
5.20 (2.71), (1.86 – 12.12) n=86 |
| PIB length of follow-up (years) : mean (SD) | 2.59 (1.35), (0.96 – 6.26) n=22 |
4.01 (1.65), (2.61 – 6.72) n=110 |
2.59 (1.35), (0.96 – 6.26) n=31 |
4.47 (1.33), (3.27 – 5.74) n=88 |
| MRI length of follow-up (years) : mean (SD) | 2.50 (1.29), (1.00 – 5.78) n=22 |
6.04 (3.81), (2.58 – 12.53) n=118 |
3.31 (1.64), (1.05 – 6.25) n=29 |
5.17 (2.89), (1.64 – 12.01) n=80 |
| Cognitive length of follow-up (years) : mean (SD) | 2.64 (1.41), (1.08 – 5.78) n=22 |
8.72 (3.95), (2.94 – 15.34) n=125 |
3.26 (1.52), (1.11 – 6.11) n=32 |
6.85 (3.53), (2.20 – 13.02) n=92 |
| Years from parental age of symptom onset: mean (SD) | −6.32 (7.17) (−24.3 – 11.8) |
−16.51 (7.94), (−35.9 – 16.2) |
3.59 (8.49) (−15.4 – 23.9) |
---- |
3.1. Cross-sectional comparisons
The cross-sectional analyses at baseline compared the regression parameters of each marker against baseline age across the four participant groups (Table 2). Older age was significantly associated with higher levels of CSF Tau, pTau181, and PiB MCSUVR, and lower levels of MRI hippocampal volume and cortical thickness, and cognition in ACS FH+ (all p’s<0.05). Both PiB MCSUVR and normalized cortical thickness in DIAN MC were associated with baseline age, and the association of PiB MCSUVR with age in DIAN MC was the strongest among the four groups (Figure 1). No significant association was found between any of the biomarkers/cognition measures and baseline age in DIAN NON-MC, or in ACS FH- with the exception of MRI cortical thickness and cognition. At the mean baseline age of DIAN MC (47.4 years), DIAN MC had the lowest levels of CSF Aβ42 and the highest levels of CSF Tau, pTau181, as well as PET PiB load, after adjusting for the effect of other covariates (APOE ε4, sex, and additionally education for cognition, Table 3), in comparison to the other three groups.
Table 2.
Biomarker association with baseline age (regression parameter (standard error (SE) in each cell) and their cross-sectional comparisons across groups (last column)
| DIAN MC (n=33) |
ACS FH+ (n=176) |
DIAN NON MC (n=58) |
ACS FH− (n=120) |
P-values for Comparisons | |
|---|---|---|---|---|---|
| CSF Aβ42 (pg/mL) |
−26.24 (22.02) *p=0.2343 |
−16.13 (9.14) *p=0.0787 |
−25.11 (14.64) *p=0.0873 |
−13.46 (11.96) *p=0.2613 |
Non-sig |
| CSF Tau ( pg/mL) |
2.88 (2.50) *p=0.2510 |
2.15 (1.05) *p=0.0412 |
1.48 (1.70) *p=0.3834 |
2.07 (1.37) *p=0.1298 |
Non-sig |
| CSF pTau181 ( pg/mL) |
0.46 (0.25) *p=0.0742 |
0.24 (0.11) *p=0.0284 |
0.14 (0.17) *p=0.4095 |
0.25 (0.14) *p=0.0741 |
Non-sig |
| PiB MCSUVR | 0.05 (0.01) *p=<0.0001 |
0.01 (0.01) *p=0.0267 |
0.01 (0.01) *p=0.2533 |
0.01 (0.01) *p=0.1666 |
0.0063; 0.0061; 0.0062 |
| MRI Hippocampal Volume (mm3) |
−16.43 (25.48) *p=0.5195 |
−25.66 (10.56) *p=0.0156 |
−27.59 (17.11) *p=0.1078 |
−29.79 (14.16) *p=0.0362 |
Non-sig |
| MRI Cortical Thickness (mm) |
−0.02 (0.01) *p=0.0120 |
−0.01 (0.003) *p=0.0121 |
0.01(0.01) *p=0.0870 |
−0.01 (0.004) *p=0.0041 |
0.0026; 0.0064; 0.0022 |
| Cognition | −0.01 (0.02) *p=0.5216 |
−0.02 (0.01) *p=0.0048 |
−0.02 (0.01) *p=0.1393 |
−0.02 (0.01) *p=0.0081 |
Non-sig |
DIAN MC vs. ACS FH+;
DIAN MC vs. DIAN NON MC;
DIAN MC vs. ACS FH−;
ACS FH+ vs. DIAN NON MC;
ACS FH+ vs. ACS FH−;
DIAN NON MC vs. ACS FH−
P-value for testing whether each regression parameter equals to 0.
Fig. 1.
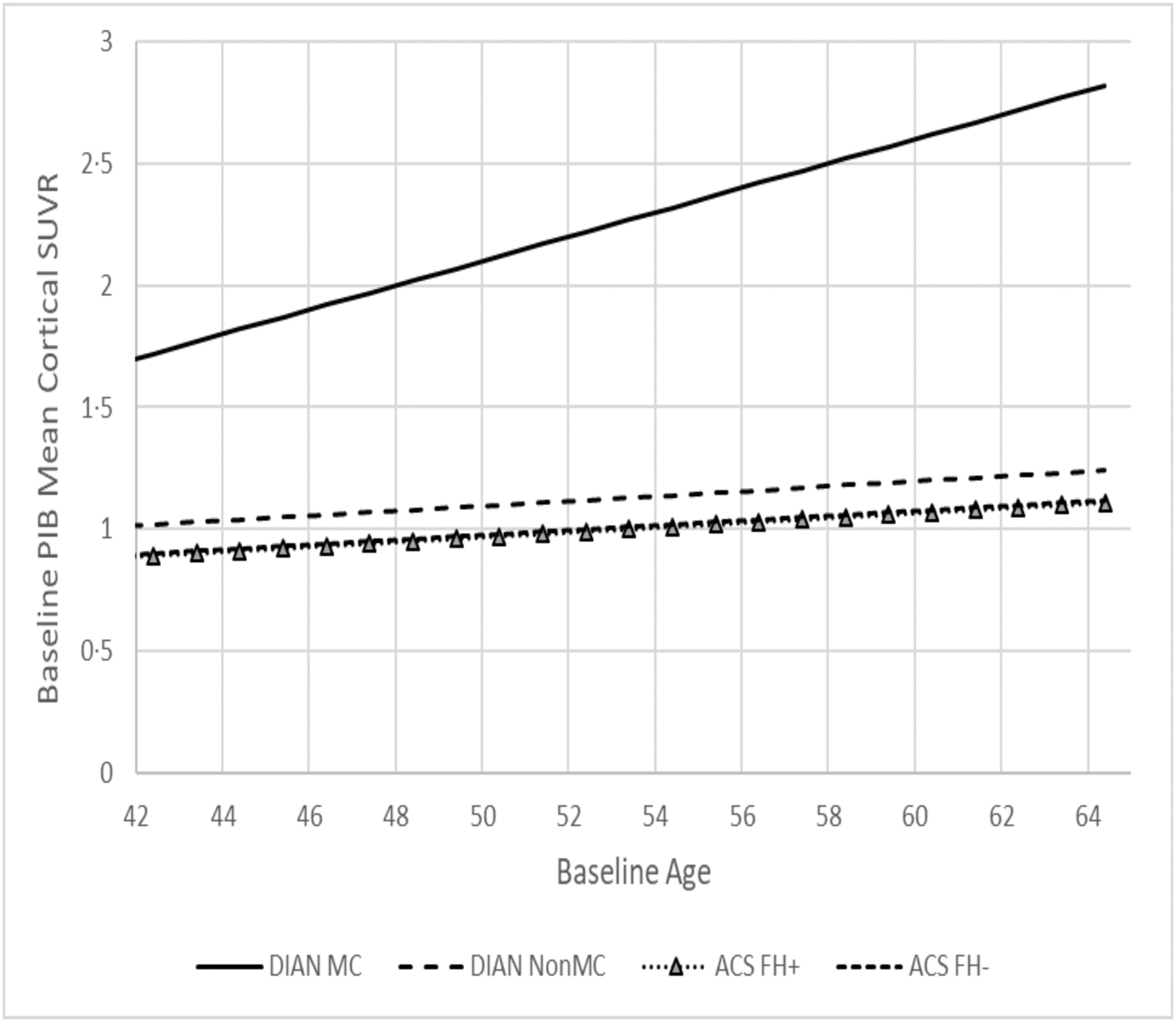
Estimated cross-sectional regression of baseline PiB PET mean cortical SUVR as a function of baseline age.
Table 3.
Adjusted biomarker levels (SE) and cognitive performance (SE) at the mean baseline age of DIAN MC (47.4 years) and their cross-sectional comparisons
| DIAN MC (n=33) |
ACS FH+ (n=176) |
DIAN NON MC (n=58) |
ACS FH− (n=120) |
Comparisons | |
|---|---|---|---|---|---|
| CSF Aβ42 ( pg/mL) |
799.69 (110.58) | 1726.25 (70.61) | 1432.46 (88.43) | 1501.35 (82.67) | 0.0055; <0.00013 <0.00011; 0.0054; <0.00012 |
| CSF Tau ( pg/mL) |
245.96 (12.87) | 168.56 (8.44) | 190.81 (10.37) | 162.10 (9.83) | <0.00013; 0.036; <0.00011; 0.00082 |
| CSF pTau181 ( pg/mL) |
23.45 (1.31) | 14.69 (0.87) | 15.72 (1.05) | 14.42 (1.01) | <0.00013; <0.00011; <0.00012 |
| PIB MCSUVR | 1.97 (0.07) | 0.94 (0.06) | 1.07 (0.06) | 0.95 (0.08) | <0.00013; <0.00011; <0.00012 |
| MRI Hippocampal Volume (mm3) |
8708.58 (130.42) | 8447.12 (88.35) | 8654.18 (101.98) | 8475.84 (102.42) | Non-sig |
| MRI Cortical Thickness (mm) |
4.60 (0.04) | 4.67 (0.03) | 4.68 (0.03) | 4.68 (0.03) | Non-sig |
| Cognition | −0.02 (0.09) | 0.12 (0.05) | 0.23 (0.07) | 0.09 (0.06) | 0.022 |
DIAN MC vs. ACS FH+;
DIAN MC vs. DIAN NON MC;
DIAN MC vs. ACS FH−;
ACS FH+ vs. DIAN NON MC;
ACS FH+ vs. ACS FH−;
DIAN NON MC vs. ACS FH−
3.2. Longitudinal comparisons
All longitudinal comparisons across the 4 groups adjusted for the effects of baseline age, gender, APOE ε4 status, education. Table 4 presents the estimated annual rate of longitudinal change since baseline for PiB MCSUVR (Figure 2), MRI hippocampal volume (Figure 3), cortical thickness, and the cognitive composite (Figure 4). ACS FH+ and FH- both showed longitudinal changes in CSF Aβ42 and PET PiB load. DIAN NON-MC showed no significant longitudinal changes in any biomarkers or cognition, whereas DIAN MC showed significant longitudinal changes in all except CSF biomarkers. The annual rate of increase in PiB PET load for DIAN MC was higher than, but not significantly different from that for ACS FH+. The rate of decline in hippocampal volume for DIAN MC was, however, drastically faster than that for any other groups. Only DIAN MC showed a significant longitudinal decline in cognition. The analyses also revealed that asymptomatic individuals with at least one APOE ε4 allele had a faster rate of increase in CSF Tau and pTau181 than those without APOE ε4 alleles, and that women had a faster longitudinal decrease in CSF Aβ42 than men. Finally, we aligned the ACS and DIAN participants by their self-reported parental age of onset and then compared the rates of change in biomarkers and cognition. These analyses were limited by the smaller sample sizes because not all individuals in ACS FH+ and none in ACS FH- had data on parental age-at-symptomatic onset, but the estimated rates of changes are largely consistent with those presented above (Supplemental Table).
Table 4.
Estimated annual rate of longitudinal changes (SE) for biomarkers and cognition after adjustment for covariates and their comparison across groups
| DIAN MC (n=33) |
ACS FH+ (n=176) |
DIAN NON MC (n=58) |
ACS FH− (n=120) |
Comparisons | |
|---|---|---|---|---|---|
| CSF Aβ42 ( pg/mL) |
21.98 (42.53) *p=0.6056 |
−43.16 (8.89) *p=<.0001 |
−45.30 (27.86) *p=0.1047 |
−27.67 (11.60) *p=0.0186 |
Non-sig |
| CSF Tau ( pg/mL) |
2.62 (3.33) *p=0.4327 |
1.62 (1.11) *p=0.1462 |
−0.21 (2.39) *p=0.9303 |
0.21 (1.35) *p=0.8785 |
Non-sig |
| CSF pTau181 ( pg/mL) |
0.63 (0.37) *p=0.0862 |
0.15 (0.14) *p=0.2751 |
−0.01 (0.27) *p=0.9792 |
0.08 (0.16) *p=0.6370 |
Non-sig |
| PIB MCSUVR | 0.05 (0.02) *p=0.0016 |
0.03 (0.01) *p=<.0001 |
−0.002 (0.01) *p=0.8592 |
0.02 (0.01) *p=0.0007 |
0.00474; 0.0092 |
| MRI Hippocampal Volume (mm3) |
−113.15 (28.54) *p=<.0001 |
5.84 (7.89) *p=0.4612 |
−19.12 (20.44) *p=0.3501 |
6.98 (9.75) *p=0.4756 |
<0.00013; <0.00011; 0.0082 |
| MRI Cortical Thickness (mm) |
−0.02 (0.01) *p=0.0195 |
−0.005 (0.003) *p=0.0795 |
−0.006 (0.01) *p=0.4128 |
−0.01 (0.003) *p=0.0077 |
Non-sig |
| Cognition | −0.08 (0.03) *p=0.0155 |
−0.01 (0.01) *p=0.0863 |
0.01 (0.02) *p=0.6992 |
0.002 (0.01) *p=0.8168 |
0.023; 0.041; 0.032 |
DIAN MC vs. ACS FH+;
DIAN MC vs. DIAN NON MC;
DIAN MC vs. ACS FH−;
ACS FH+ vs. DIAN NON MC;
ACS FH+ vs. ACS FH−;
DIAN NON MC vs. ACS FH−
P-value for testing whether each single annual rate of change equals to 0.
Fig. 2.
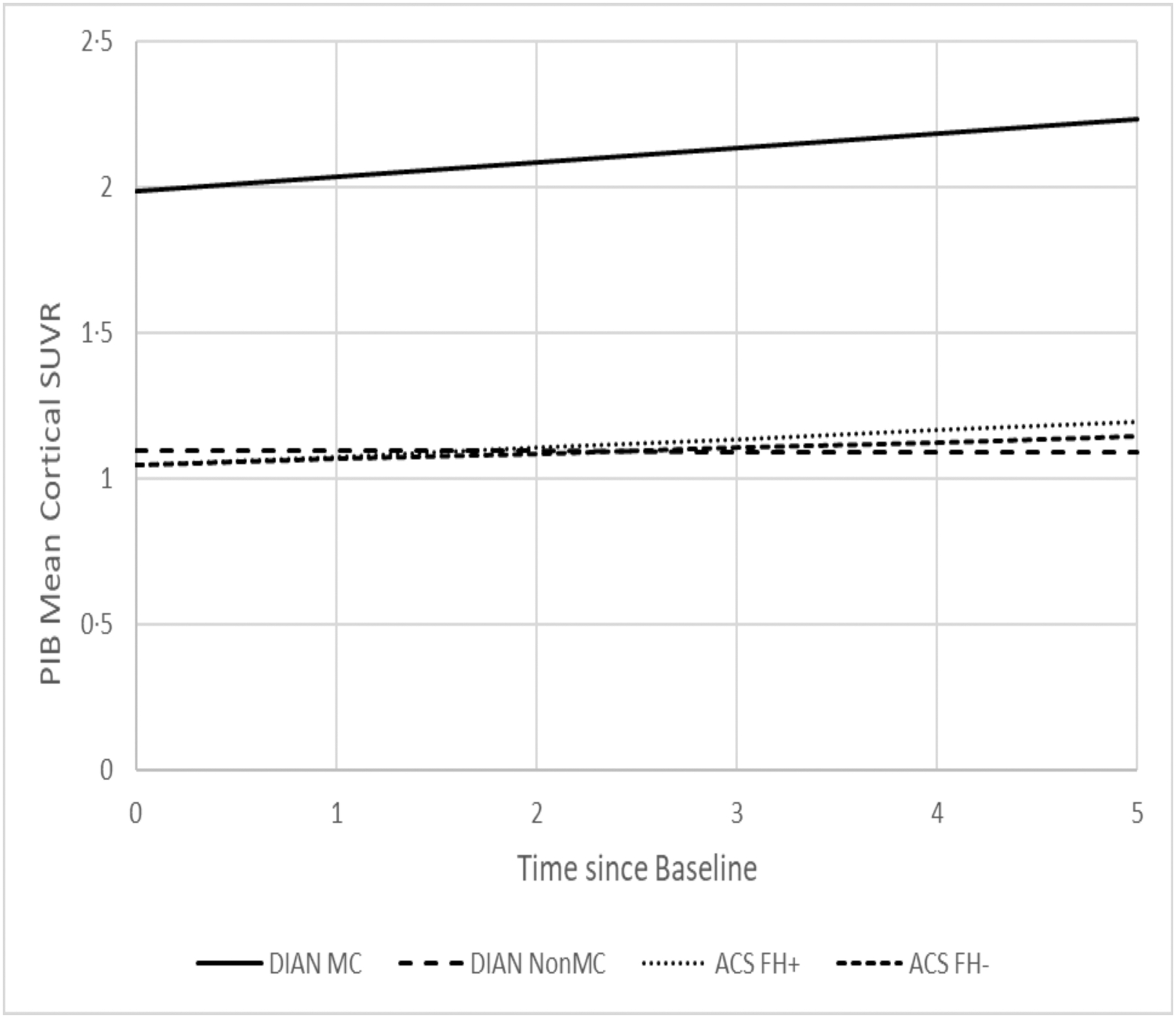
Estimated longitudinal progression of PiB PET cortical mean SUVR as a function of time from baseline.
Fig. 3.
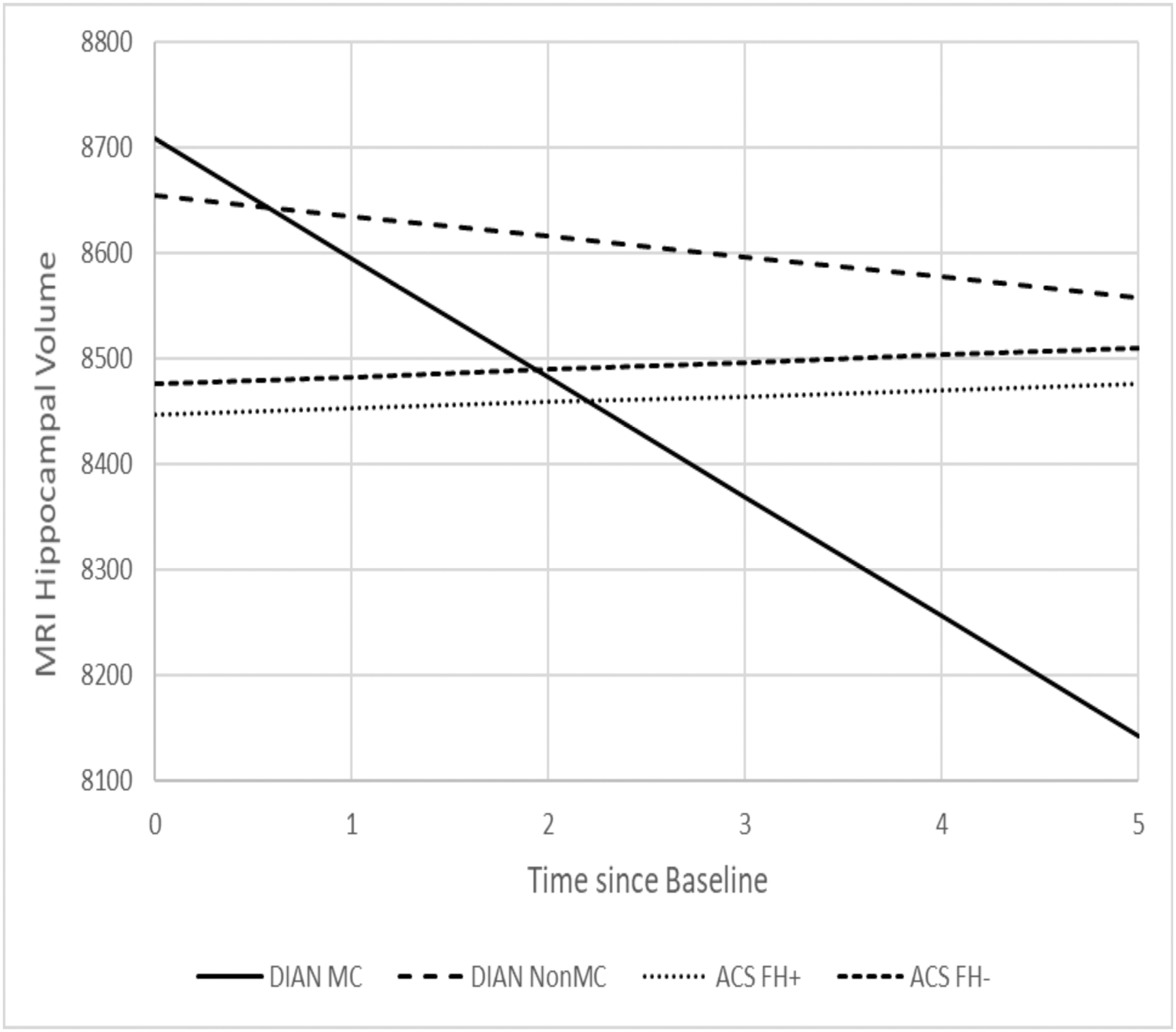
Estimated longitudinal progression of MRI hippocampal volume as a function of time from baseline.
Fig. 4.
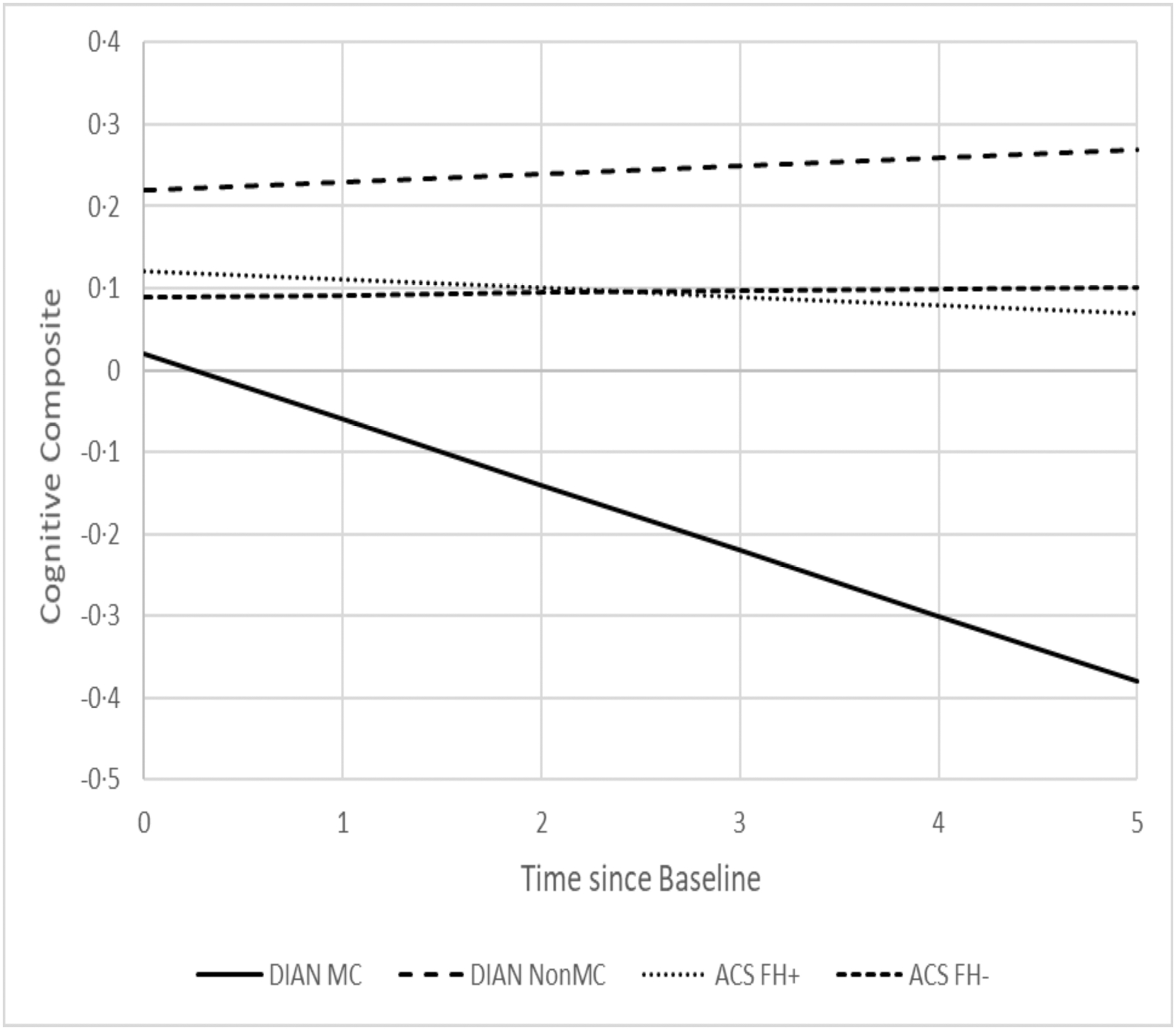
Estimated longitudinal progression of cognition as a function of time from baseline.
4. Discussion
Based on accumulating research suggesting that neurodegenerative processes associated with LOAD may begin in middle age24–27, and almost certainly many years prior to symptom onset, both the ACS and the DIAN have focused on the “preclinical” stage of AD during which no clinical symptoms are present. Biomarkers serve as an effective tool to measure disease progression so that early interventions can be tested and developed. Although ADAD is rare, it is a unique population for studying the preclinical stage of AD and for testing preventive therapies2. The comparison of the underlying pathophysiology of ADAD and LOAD at the asymptomatic stage, through well-established AD biomarkers, may further the understanding of AD and inform future design of prevention strategies. Both similarities and differences between ADAD and LOAD have been reported in the literature from multiple studies of relatively small sample sizes and limited biomarker data or with a primary focus on symptomatic stages3,28–30.
Because of the large age difference between individuals with ADAD and LOAD, the direct comparison of ADAD and LOAD is almost completely confounded by age, which complicates the interpretation of the results. To the best of our knowledge, our study represents the very first longitudinal study seeking to compare asymptomatic individuals within a similar age range who had a FH of either ADAD or LOAD on all major AD biomarkers as well as cognition. We found that, as expected, asymptomatic DIAN MC had the most abnormal AD biomarker profiles at baseline, and their baseline age had the strongest association with PET PiB uptake, but no significant association with any CSF biomarkers, likely due to the small sample size (n=33) and large variations in CSF biomarkers. We also found that at baseline, age was significantly associated with almost all biomarkers and cognition in ACS FH+. Longitudinally, ACS FH+ showed significant changes over time only in CSF Aβ42 and mean cortical PET PiB load, whereas DIAN MC showed a similar rate of increase in PET PiB load as FH+ but much faster decrease in MRI hippocampal volume, suggesting that the brains of DIAN MC, albeit cognitively normal at baseline, went through dramatic changes (i.e., atrophy) during a relatively short longitudinal follow-up of 1 to 5.8 years (Table 1). These findings may be partially explained by the fact that the age interval of 42 to 65 years, albeit quite early in the preclinical progression of LOAD where amyloid started to accumulate, is nonetheless very late and close to the expected age of symptom onset for ADAD where changes in downstream markers such as brain atrophy accelerated. These findings are therefore consistent with the reported temporal orderings of biomarker changes during preclinical stages of AD2, 31–32. Although baseline differences were found in the CSF biomarkers across four groups, no significant differences were found on the rates of longitudinal changes, supporting the idea that AD pathophysiologic mechanisms are similar in ADAD and LOAD. In fact, no significant longitudinal changes were found in CSF biomarkers for all four groups with the exception of ACS FH+ and FH- in CSF Aβ42, consistent with previous findings suggesting that changes in some of the CSF biomarkers such as Tau and pTau181 may start to attenuate close to symptomatic onset in ADAD11. Together, these findings support the utility of CSF biomarkers and PET and MRI imaging markers to adequately track the progression of preclinical AD among individuals who are at risk (ACS FH+) or destined to develop AD (DIAN MC). Hence, our findings support the current approaches of multiple ongoing secondary prevention trials of AD (the DIAN Trials Unit [DIAN-TU], and the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease ([A4] trials) that have been designed using these biomarkers as either efficacy endpoints or parts of the inclusion/exclusion criteria.
The contrasts between DIAN NON-MC and the ACS FH+ may be of particular interest. In DIAN NON-MC, we found no association between age and any of the biomarkers/cognition at baseline (Table 2,3), and no significant longitudinal changes over time in any CSF and imaging biomarkers and cognition. These results indicate that the effect of a parental history of ADAD to NON-MC may actually be more benign than that of a family history of LOAD during middle age, suggesting that the higher prevalence of APOE ε4 alleles in ACS FH+ (Table 1), along with multiple susceptibility genes for LOAD12, may play important roles in the preclinical progression of individuals with LOAD. Further, very few differences were found in comparing ACS FH- and DIAN NON-MC on biomarkers and cognition, indicating that these two groups share largely consistent biomarkers and cognitive profiles which mostly remained latent during middle age.
Strengths of the current study include the relatively large sample size of carefully characterized cognitively normal individuals for whom all major CSF and imaging biomarkers of AD as well as clinical and cognitive data were longitudinally and consistently obtained between the ACS and DIAN. The study also has limitations. First, both the DIAN and ACS are observational studies on convenience samples. Second, although the DIAN and the ACS cohorts were restricted to the shared baseline age window, and the analyses were adjusted for age, a difference of ~7 years in age at baseline may still prove too large in such comparisons. Additionally, the effect of age may be different, depending on the mutation in MC. The relatively small sample size of MC (n=33: n=25 with mutation in PSEN1, n=3 in PSEN2, and n=5 in APP) prevented further analyses to take into account of potentially differential effect of age across mutations, which is another limitation of the study. Third, the longitudinal follow-up was relatively short, preventing us from evaluating the cascade of early events in AD pathogenesis in its entirety. Finally, the ACS and the DIAN had limited overlaps in the cognitive batteries that were heavily weighted toward attention/executive domains which may be more affected in early-onset than late-onset AD. Our results may have been different if tests of episodic memory had been included. The lack of episodic memory tests may also have implications in relation to the higher prevalence of APOE ε4 allele in the FH+ group since presence of APOE ε4 alleles has been associated with greater hippocampal pathology. Despite these, this study represents the first effort to compare biomarker changes between ADAD and LOAD among asymptomatic individuals that controlled for the effect of baseline age. Asymptomatic DIAN MC who were very close to symptom onset may be an ideal group for a future prevention trial because of their advanced biomarker profiles at baseline and profound longitudinal changes in some of these biomarkers. Clinical trial results from this unique group may then inform design and analyses of prevention trials of LOAD.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic Review
Most LOAD studies focused on age 65 or older. ADAD on average were much younger. Prior studies to compare the two disease sub-types were completely confounded by age. Findings have been inconsistent, primarily due to the small sample sizes and confounding of age.
Interpretation
This study constitutes the largest study to date that compares the two types of family history, ADAD and LOAD on all major biomarkers of AD and cognition among cognitively normal individuals within the same age interval at baseline. The findings support the hypothesized temporal ordering of biomarker changes, and demonstrate the utility of CSF and imaging biomarkers in secondary prevention trials of AD.
Future Directions
Despite many similarities, significant differences between preclinical ADAD and ACS FH+ were found. The limited overlap in the cognitive battery between the DIAN and ACS prevents a definite comparison. More and larger studies are needed to validate these findings.
Acknowledgement
This study was supported by National Institute on Aging (NIA) grants R01 AG053550 (Dr Xiong), P01 AG026276 (Dr Morris) and UF1AG032438 (Dr Bateman) as well as, the German Center for Neurodegenerative Diseases (DZNE), the Raul Carrea Institute for Neurological Research (FLENI), AMED grants for DIAN-J (PI Hiroshi Mori, PhD)), and the generous support of Fred Simmons and Olga Mohan. The authors thank the Genetics Core (Alison Goate and Carlos Cruchaga, Core Leaders) of the Knight ADRC (P50 AG05681) and the DIAN for the genetic data. The corresponding author (C Xiong) had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study Funding and Acknowledgements.
This study was supported by National Institute on Aging (NIA) grant R01 AG053550 (Dr. Xiong) and NIA grants P50 AG005681, P01AG026276, and P01 AG0399131 (Dr. Morris), and UF1AG032438 (Dr. Bateman). Image processing was supported in part by the Neuroimaging Informatics and Analysis Center (1P30NS098577) and R01 EB009352 (Benzinger). Data collection and sharing for this project was partly supported by The Dominantly Inherited Alzheimer’s Network (DIAN, UF1AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by Japan Agency for Medical Research and Development (AMED) JP21dk0207049 (Dr. Ikeuchi), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI).This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
Competing Interests
All the authors declare no competing interest.
Drs. Xiong, Schindler, Fagan, Benzinger, Hassenstab, Balls-Berry, Moulder, and Morris all have received research funding from the National Institute on Aging of the National Institutes of Health that was made to their institutions.
Dr. Hassenstab also has received BrightFocus grant that was made to his institution.
Dr. Morris received royalties or licenses for CDR registration, and received support for attending meetings and/or travel (Srinivasan 40th Oration, India; World Congress of Neurology; Cure Alzheimer’s Board meeting; CBR Intl’ Advisory Board).
Dr. Xiong consults for Diadem. There are no conflicts.
Dr. Schindler consults for National Institute on Aging Alzheimer Disease Center Clinical (ADC) Task Force, to me National Centralized Repository for Alzheimer Disease.
Dr. Fagan has received research funding from the National Institute on Aging of the National Institutes of Health, Biogen, Centene, Fujirebio and Roche Diagnostics. She is a member of the scientific advisory boards for Roche Diagnostics, Genentech and AbbVie and also consults for Diadem, DiamiR and Otsuka Pharmaceuticals. Dr. Fagan also consults for Seimens Healthcare Diagnostics. There are no conflicts.
Dr. Benzinger consults for Biogen. There are no conflicts.
Dr. Hassenstab consults for Lundbeck, Eisai, Roch, and Parabon Labs. There are no conflicts.
Dr. Morris consults for Barcelona Betabrain Research Center, BBRC SAB meeting, Barcelona Centre for Brain Research meeting, Bangalore, India. There are no conflicts.
Dr. Schindler received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from University of Wisconsin and St. Luke’s Hospital. There are no conflicts.
Dr. Benzinger received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Biogen. There are no conflicts.
Dr. Hassenstab received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: (seminar speaker) from Alzheimer’s Therapeutic Research Institute (ATRI), University of Southern California. There are no conflicts.
Dr. Morris received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: (Grand Rounds lecture) from Montefiore, NY. There are no conflicts.
Drs. Xiong, Benzinger, Hassenstab, and Balls-Berry all served on Data Safety Monitoring Board or Advisory Board for FDA or NIH-funded studies. There are no conflicts.
Dr. Schindler is a Member of the Board of Directors, Alzheimer’s Association Greater Missouri Chapter. Dr. Balls-Berry is President of the Board of Directors for Health Literacy Media. Dr. Morris is a member of Cure Alzheimer’s Board.
Avid Radiopharmacueticals and Life Molecular Imaging have provided reagents and technology transfer agreements to Dr. Benzinger’s institution for the production of radiopharmaceuticals.
TI is supported by Japan Agency for Medical Research and Development (AMED) JP21dk0207049. There are no conflicts with this work.
GSD is supported by National Institutes of Health /National Institute on Aging (K23AG064029). He serves as a topic editor on dementia for DynaMed Plus (EBSCO Industries, Inc), a consultant for Parabon NanoLabs, is the clinical director for the Anti-NMDA Receptor Encephalitis Foundation (uncompensated), has provided record review and expert medical testimony on legal cases pertaining to management of Wernicke encephalopathy, and holds stocks (>$10,000) in ANI Pharmaceuticals (a generic pharmaceutical company). There are no conflicts with this work.
Johannes Levin reports speaker fees from Bayer Vital, Biogen and Roche, consulting fees from Axon Neuroscience, author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers, non-financial support from Abbvie and compensation for duty as part-time CMO from MODAG, outside the submitted work.
Data sharing
The de-identified data with a comprehensive database dictionary and/or protocols on imaging and CSF collections will be available to all qualified investigators, starting 12/31/2021. Data requests should be submitted online (https://biostat.wustl.edu/adrc/pbs_1/register.html). Once the data request is filed, a project manager will contact the data requestor to obtain project information about the scientific rationale and hypotheses to be tested by using the data and the proposed statistical analysis plan. Upon approval, data will be shared securely after the data requestor signs a data use agreement.
References
- 1.Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74(3):201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TL, et al. Dominantly Inherited Alzheimer Network. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012; 367(9):795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris JC, Weiner M, Xiong C, Beckett L, Coble D, Saito N, Aisen PS, Allegri R, Benzinger TLS, Berman SB, Cairns NJ, Carrillo MC, Chui HC, Chhatwal JP, Cruchaga C, Fagan AM, Farlow M, Fox NC, Ghetti B, Goate AM, Gordon BA, Graff-Radford N, Day GS, Hassenstab J, Ikeuchi T, Jack CR, Jagust WJ, Jucker M, Levin J, Massoumzadeh P, Masters CL, Martins R, McDade E, Mori H, Noble JM, Petersen RC, Ringman JM, Salloway S, Saykin AJ, Schofield PR, Shaw LM, Toga AW, Trojanowski JQ, Vöglein J, Weninger S, Bateman RJ, Buckles VD; Dominantly Inherited Alzheimer Network and the Alzheimer’s Disease Neuroimaging Initiative. Autosomal dominant and sporadic late onset Alzheimer disease share a common in vivo pathophysiology. Brain. 2022. May 17:awac181. doi: 10.1093/brain/awac181. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17(2):101–118. [DOI] [PubMed] [Google Scholar]
- 5.Vos SJB, Xiong CJ, Visser PJ, et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12(10):957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price JL, McKeel DW Jr., Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30(7):1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. [DOI] [PubMed] [Google Scholar]
- 8.Luo J, Agboola F, Grant E, Morris JC, Masters CL, Albert MS, Johnson SC, McDade EM, Fagan AM, Benzinger TLS, Hassenstab J, Bateman RJ, Perrin RJ, Wang G, Li Y, Gordon B, Cruchaga C, Day GS, Levin J, Vöglein J, Ikeuchi T, Suzuki K, Allegri RF, Xiong C; Dominantly Inherited Alzheimer Network (DIAN). Accelerated longitudinal changes and ordering of Alzheimer disease biomarkers across the adult lifespan. Brain. 2022. Aug 4:awac238. doi: 10.1093/brain/awac238. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopera F, Ardilla A, Martinez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA.1997;277(10):793–799. [PubMed] [Google Scholar]
- 10.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83(3):253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fagan AM, Xiong CJ, Jasielec MS, et al. Longitudinal Change in CSF Biomarkers in Autosomal-Dominant Alzheimer’s Disease. Science Translational Medicine. 2014;6(226). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong C, Roe CM, Buckles V, et al. Role of family history for Alzheimer biomarker abnormalities in the adult children study. Arch Neurol. 2011; 68(10):1313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong C, Jasielec MS, Weng H, et al. Longitudinal relationships among biomarkers for Alzheimer disease in the Adult Children Study. Neurology. 2016; 86(16):1499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound‐B. Ann Neurol. 55: 2004; 306–319. [DOI] [PubMed] [Google Scholar]
- 15.Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 1993; 43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 16.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alz Dis Assoc Disord 2006; 20:210–216. [DOI] [PubMed] [Google Scholar]
- 17.Gordon BA, Blazey T, Su Y, Fagan AM, Holtzman DM, Morris JC, Benzinger TL. Longitudinal β-Amyloid Deposition and Hippocampal Volume in Preclinical Alzheimer Disease and Suspected Non-Alzheimer Disease Pathophysiology. JAMA Neurol. 2016. Oct 1;73(10):1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. NeuroImage 2015; 107:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schindler S, Gray J, Gordon B, et al. Cerebrospinal fluid biomarkers measured by Elecsys® assays compared to amyloid imaging. Alzheimers Dement. 2018; 14(11):1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diggle PJ, Heagerty P, Liang K-Y, Zeger SL. Analysis of Longitudinal Data (2nd ed.). New York: Oxford University Press. 2002. [Google Scholar]
- 21.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982; 38(4):963–74. [PubMed] [Google Scholar]
- 22.Littell R, Milliken GA, Stroup W, Wolfinger R. SAS System For Mixed Models. Cary NC: SAS Institute Inc. 1996. [Google Scholar]
- 23.Milliken GA, Johnson DE. Analysis of Messy Data, Volume I: Designed Experiments. Chapman and Hall/CRC, 1984 [Google Scholar]
- 24.Kok E, Haikonen S, Luoto T, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009; 65(6):650–7. [DOI] [PubMed] [Google Scholar]
- 25.van der Vlies AE, Koedam EL, Pijnenburg YA, Twisk JW, Scheltens P, van der Flier WM. Most rapid cognitive decline in APOE epsilon4 negative Alzheimer’s disease with early onset. Psychological Medicine 2009; 39:1907–1911. [DOI] [PubMed] [Google Scholar]
- 26.Panegyres PK, Chen HY. Differences between early and late onset Alzheimer’s disease. J Neurodegenerative Dis 2013; 2:300–306. [PMC free article] [PubMed] [Google Scholar]
- 27.Fleisher AS, Chen K, Quiroz YT, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol 2015; 72(3):316–24 doi: 10.1001/jamaneurol.2014.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerritsen AA, Bakker C, Verhey FR, et al. Prevalence of comorbidity in patients with Young-Onset Alzheimer Disease compared With Late-Onset: A comparative cohort Study. J Am Med Dir Assoc 2016; 1:318–323. [DOI] [PubMed] [Google Scholar]
- 29.Reed BR, Mungas DM, Kramer JH, et al. Profiles of neuropsychological impairment in autopsy-defined Alzheimer’s disease and cerebrovascular disease. Brain 2007; 130:731–739. [DOI] [PubMed] [Google Scholar]
- 30.Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 2015; 35:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7(3):280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers & Dementia. 2018;14(4):535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The de-identified data with a comprehensive database dictionary and/or protocols on imaging and CSF collections will be available to all qualified investigators, starting 12/31/2021. Data requests should be submitted online (https://biostat.wustl.edu/adrc/pbs_1/register.html). Once the data request is filed, a project manager will contact the data requestor to obtain project information about the scientific rationale and hypotheses to be tested by using the data and the proposed statistical analysis plan. Upon approval, data will be shared securely after the data requestor signs a data use agreement.