

Abstract
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is responsible for the regulation of genes involved in inflammation and immune responses. NF-κB may play an important role in cardiovascular diseases (CVDs), atherosclerosis and diabetes. Several therapeutic agents used for the treatment of CVDs and diabetes, such as pimobendan and sodium–glucose cotransporter 2 inhibitors, exert anti-inflammatory effects by inhibiting NF-κB activation; anti-inflammatory therapy may have beneficial effects in CVDs and diabetes. Several pharmacological agents and natural compounds may inhibit NF-κB, and these agents alone or in combination may be used to treat various inflammatory diseases. Immunoglobulin-free light chains could be surrogate biomarkers of NF-κB activation and may be useful for evaluating the efficacy of these agents. This review discusses recent advances in our understanding of how the NF-κB signalling pathway controls inflammation, metabolism and immunity, and how improved knowledge of these pathways may lead to better diagnostics and therapeutics for various human diseases.
Keywords: Atherosclerosis, biomarker, diabetes, immunoglobulin light chains, heart failure, nuclear factor-κB, sodium–glucose cotransporter 2 inhibitor
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) was discovered in 1986 by Sen and Baltimore.1 NF-κB is a dimeric transcription factor, originally found as an inducible B cell-specific factor that binds to the κB motif in the enhancer of the immunoglobulin light chain.2 NF-κB plays a crucial role in the regulation of immune responses and inflammation.2,3 NF-κB consists of five proteins: RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105) and NF-κB2 (p52/p100).2 Heterodimers and homodimers of the five NF-κB proteins are induced and regulated by various signals, including bacterial and viral infections, cytokines, growth factors and different types of stressors.3 In resting cells, NF-κB resides in the cytoplasm, forming a complex with I-κB, which keeps NF-κB in an inactive state, blocking its translocation to the nucleus, binding of DNA and regulation of gene expression.3
There are two signalling pathways for the activation of NF-κB: the classic/canonical and alternative/non-canonical pathways.3 The former is activated by ligands of Toll-like receptors (TLRs), such as lipopolysaccharide (LPS), interleukin (IL)-1 and tumour necrosis factor (TNF)-α, or by engagement with T- and B-cell receptors. This pathway plays an important role in the induction of genes involved in inflammation, cell proliferation and survival, angiogenesis and tumour metastasis. The alternative/non-canonical pathway is activated by lymphotoxin, receptor activator of NF-κB ligand, CD40 ligand and B-cell activating factor belonging to the TNF family, and plays an essential role in the induction of genes associated with the development and maintenance of secondary lymphoid organs.3 Targets for classical NF-κB signalling include genes encoding proinflammatory cytokines, growth factors, chemokines, chemokine ligands, macrophage inflammatory protein (MIP)-1α, MIP-2α, macrophage chemotactic factor (MCP)-1, regulated upon activation, normal T expressed and secreted (RANTES), matrix metalloproteinases (MMPs), cyclo-oxygenase (COX)-2, inducible nitric oxide synthase, vascular endothelial growth factor, vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, E-selectin and inhibitors of NF-κB signalling.3
Recently, cytokines and inflammation have been shown to play an important role in the pathogenesis and progression of cardiovascular diseases (CVDs) such as heart failure, ischaemic heart disease, myocarditis and atrial fibrillation (AF).4 Most of the risk factors for CVDs, such as an unhealthy diet, smoking and infection, induce cellular stress and inflammation.4 NF-κB has been shown to be activated by smoking and viral infections, including adenovirus, hepatitis B virus (HBV), hepatitis C virus (HCV), human herpes virus 6 and HIV, some of which are causes of CVDs.3,5 Thus, it is suggested that NF-κB is activated in CVDs. In addition, there is renewed interest in NF-κB because inflammation is considered to play a significant role in the development of cancer and ageing.3
This review discusses recent advances in our understanding of how NF-κB is related to the pathogenesis of CVDs and their risk factors, as well as how improved knowledge of these factors may lead to better preventive, diagnostic and therapeutic approaches for these diseases.
Nuclear Factor-kB and Inflammation in Cardiovascular Diseases
Heart Failure
Heart failure is a clinical syndrome with signs and symptoms that result from any functional or structural disturbance of ejection or filling of blood by the ventricle.6 Heart failure is considered a disease of the heart resulting from chronic neurohormonal activation. In the 1950s, C-reactive protein (CRP) concentrations were shown to be associated with heart failure, suggesting that inflammation is an important feature of heart failure.4,7 Increased blood endotoxin and cytokine concentrations have been reported in patients with heart failure during exacerbation, suggesting that endotoxins may activate immune reactions during the development of oedema.7 Thereafter, increased circulating inflammatory cytokines were confirmed in heart failure by larger clinical studies.4,8,9 We showed that serum concentrations of TNF-α, IL-1α and IL-1β were increased in patients with myocarditis and that TNF-α levels were frequently elevated in dilated and hypertrophic cardiomyopathies.10 Levels of soluble TNF-α and receptors, IL-1 receptor antagonist (IL-1RA), IL-18, MCP-1, MIP-1α and RANTES have also been shown to be increased in patients with heart failure and cardiomyopathies.4,9
Because NF-κB is activated by endotoxins and cytokines, and this activation leads to the expression of genes encoding cytokines and chemokines, among others, it is suggested that NF-κB may play a crucial role in CVDs and their risk factors, such as atherosclerosis and diabetes.
Ischaemic Heart Disease
Ischaemic heart disease occurs because of thrombotic occlusion of the coronary arteries as a result of atherosclerosis. The occlusion of arteries induces recruitment of immune cells to the vessel wall and myocardium.11 Plaque rupture may cause necrosis of cardiomyocytes, which are then replaced by fibrosis. Inflammation and immune cell infiltration following cardiomyocyte death are critical for cardiac repair after MI. Optimal healing includes an inflammatory and reparative phase that forms a stable scar. In contrast, excessive inflammation and fibrosis lead to cardiac dysfunction.12 In a rat model, the gene expression of TNF-α, IL-1β and IL-6 increased in the infarcted area during the acute stage after coronary occlusion and declined to baseline levels several months after MI.13 In contrast, although the gene expression of cytokines was also increased in the non-infarcted region during the acute stage, it remained elevated throughout the chronic stage. The concentration of IL-1β was correlated with increased collagen deposition in the non-infarcted myocardium during the chronic stages.13 Although timely reperfusion may decrease tissue damage in MI, reperfusion itself also induces inflammation. Therefore, therapeutic manipulation is challenging.14 Because NF-κB is activated by oxidative stress, NF-κB may be activated in MI.3
Viral Myocarditis
Various viral infections activate NF-κB because viruses encode proteins that can activate NF-κB.3 Mice lacking the p50 subunit of NF-κB are resistant to encephalomyocarditis virus (EMCV) infection, and the fibroblasts from these mice show enhanced transcription of interferon (IFN)-β in response to infection with EMCV.2 Thus, the p50 subunit of NF-κB may decrease transcriptional responses, which has important effects on response to pathogens in vivo.2
In our study, EMCV activated NF-κB in cultured non-cardiomyocytes, but did not activate cardiomyocytes in vitro.15 The expression of the proinflammatory cytokines TNF-α and IL-1β and that of the immunoregulatory cytokines IFN-γ and IL-2 was increased in the heart of mice during the acute phase, and persisted for several months after viral infection in a mouse model of EMCV myocarditis.16,17 Activation of NF-κB is considered to play a crucial role in the development of EMCV myocarditis because agents that inhibit NF-κB activation ameliorated myocarditis, as discussed below.15
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes COVID-19. The clinical manifestations of SARS-CoV-2 infection vary from asymptomatic to respiratory failure, multiple organ failure and death. Activation of NF-κB has been reported to be involved in the development of SARS-CoV-2 infection.18 Spike protein, its receptor-binding domain or the integrin-binding tripeptide RGD, translocated NF-κB into the nucleus of endothelial cells and increased the expression of proinflammatory cytokines, adhesion molecules, coagulation factors and angiotensin-converting enzyme 2. Inhibitors of α5β1-integrin prevented these effects. Intravenous administration of spike protein in vivo enhanced the expression of proinflammatory cytokines and adhesion molecules in various tissues.19 These findings showed a direct action of SARS-CoV-2 on endothelial dysfunction and α5β1-integrin as a promising target for treating vascular inflammation in COVID-19.19 The main features of life-threatening cytokine storm syndrome in advanced COVID-19 are spontaneous haemorrhage, thrombocytopenia and systemic inflammation, which are associated with elevated levels of circulating cytokines and chemokines.20 We have reported that, in patients with COVID-19, circulating levels of biomarkers of inflammation, including CRP, IL-6 and immunoglobulin free light chains (FLCs), and biomarkers of myocyte injury, such as cardiac troponins and N-terminal pro-brain B-type natriuretic peptide, are frequently elevated.21 The simultaneous increase in biomarkers of both inflammation and myocardial injury suggests the existence of myocarditis.21,22 Because NF-κB may control the expression of FLCs and IL-6, increased levels of these inflammatory biomarkers may be caused by NF-κB activation. The systemic approach to inhibiting NF-κB activation could be a therapeutic intervention for the cytokine storm. Targeted therapies with proteasome inhibitors, tyrosine kinase inhibitors, nucleotide analogues, monoclonal antibodies against TNF-α, N-acetylcysteine and corticosteroids, which inhibit NF-κB, have demonstrated beneficial effects in patients with severe COVID-19.20
Myocarditis has been reported to occur in some cases after COVID-19 vaccination.23 A recent study on COVID-19 vaccination showed increased circulating levels of full-length spike protein unbound by antibodies, but there was no evidence of excessive antibody or autoantibody production or concomitant viral infection in these patients.24 Because spike protein has been shown to activate NF-κB and enhance the production of proinflammatory cytokines, vaccine-associated myocarditis may be related to the activation of NF-κB.19
Autoimmune Myocarditis
We showed for the first time that disruption of the programmed cell death (PD)-1 receptor led to lethal myocarditis in PD-1-deficient mice.25 PD-1-deficient mice showed prominent cardiac enlargement and dilation of the ventricular cavities, suggesting that the cause of death was heart failure due to myocarditis.25,26 Immune checkpoint inhibitors (ICIs) are monoclonal antibodies against CTLA-4, PD-1 or programmed death-ligand 1 (PD-L1) and activate the immune system against cancer.27 Although ICIs are effective against cancer, ICI therapies can cause immune-related adverse events due to dysregulation of the immune system, leading to various organ injuries, including myocarditis resembling autoimmune diseases.28 Recently, α-myosin was identified as the cognate antigen source for three major histocompatibility complex-I-restricted T-cell receptors derived from mice with fulminant myocarditis.29 α-Myosin-expanded T cells from the peripheral blood of patients with ICI myocarditis shared T-cell receptor clonotypes with the diseased heart and skeletal muscles, indicating that α-myosin may be a clinically important autoantigen in ICI myocarditis.29 These studies clarified the critical role of cytotoxic CD8+ T cells, identified a candidate autoantigen in ICI myocarditis and provided new insights into the pathogenesis of ICI toxicity.29
AF
AF is the most common arrhythmia, and recent evidence shows that immune and inflammatory mechanisms play a critical role in its pathogenesis.30,31 Myocarditis was found histologically in the atria of patients with lone AF, and patients with AF showed higher NF-κB activity and more severe lymphomonocyte infiltration than those with sinus rhythm.30,31 These findings suggest the existence of local immunological and inflammatory responses in the atria of individuals with AF.31
Inflammasomes play a crucial role in the regulation of immune responses. NF-κB signalling plays a critical role in the priming phase of the inflammasome.1 NOD-like-pyrin-domain-containing-protein-3 (NLRP3) is an inflammasome discovered in various cell types.1 Inflammasomes regulate immune responses, and their activation involves priming and triggering. Macrophage infiltration and increased concentrations of active caspase-1 and NLRP3 have been shown in the atria during chronic and postoperative AF, suggesting that activation of the NLRP3 inflammasome may play a role in AF.1 Mechanisms that regulate NF-κB and inflammasome signalling may play critical roles in the development of inflammation in AF. Further investigations regarding NF-κB and inflammasome signalling could identify novel regulatory mechanisms that could help with the development of new diagnostic and therapeutic interventions.30,34
Atherosclerosis
Atherosclerosis is the most common contributing factor to ischaemic heart disease and stroke and is considered a chronic inflammatory disorder.4
Oxidised LDL triggers the immune response and can activate both innate and adaptive immune responses.35 Chronic inflammation results in plaque formation or plaque erosion in advanced atherosclerosis, leading to ischaemic events. Single-cell studies have shown a high heterogeneity of leucocytes in atherosclerotic lesions.36 In addition, recent evidence indicates that atherogenesis is a multiorgan process to which the bone marrow and spleen contribute.37 The presence of clonal haematopoiesis of indeterminate potential has been suggested as a risk factor for CVD.38
NF-κB contributes to many processes involved in atherosclerotic plaque formation. NF-κB activation enhances the expression of genes, including TNF-α, IL-1β, IL-6, MCP-1, and ICAM-1, which initiate and promote atherosclerosis. NF-κB is detected in the nuclei of macrophages in atherosclerotic lesions, suggesting that NF-κB activation is associated with atherogenesis.39 Many genetic studies have shown a critical role for NF-κB in the pathogenesis of atherosclerosis, in which cell-specific actions coordinate its initiation, progression and resolution.39
The benefits of targeting inflammation in atherosclerosis in humans were demonstrated in the Cantos trial, in which treatment with canakinumab, an antibody targeting IL-1β, improved clinical outcomes in patients with a history of MI.40 In addition, clinical trials have shown that the anti-inflammatory agent colchicine reduces the risk of cardiovascular events in patients with recent MI or coronary artery disease.41 Thus, NF-κB inhibition is a promising intervention for the prevention of atherosclerosis and its sequelae.
Diabetes: A Risk Factor for Cardiovascular Diseases
The end products of absorbed advanced glycation and lipid oxidation are linked to obesity and inflammation.42 In addition, high-glycaemic foods increase oxidative stress, which activates inflammatory genes.42 Physical inactivity increases the risk of diabetes, and excessive visceral adipose tissue triggers inflammation.4,42 Circulating CRP, IL-1β, IL-6 and IL-1RA are elevated and are predictive biomarkers for type 2 diabetes.4 The production of TNF-α is increased in adipose tissues by obesity, and TNF-α antagonists improve insulin sensitivity.43 Macrophages may release IL-1β, IL-6, IL-33 and TNF-α, and it is now well known that inflammation plays a crucial role in insulin resistance.44 Mice heterozygous for IκB kinase β (IKKβ) are protected against insulin resistance, and salicylates, which may target IKKβ directly, were reported to inhibit insulin resistance in skeletal muscle.45
Ageing and Nuclear Factor-κB
Ageing is a major risk factor for CVD. Environmental factors, such as overnutrition, smoking and a sedentary lifestyle, may lead to premature disturbances in mitochondrial function, insulin signalling, endothelial homeostasis and redox balance that foster early senescence.46 Molecular investigations have shown that common signalling networks may link the ageing process with deterioration of cardiovascular homeostasis and metabolic disturbances. Suppression of NF-κB in the endothelium has been demonstrated to prolong lifespan and improve obesity-induced insulin resistance in the endothelium of mice.47 Overexpression of the inhibitory NF-κB subunit IκBα in the endothelium of transgenic mice protected against insulin resistance in adipose tissue and skeletal muscle and reduced obesity-induced macrophage infiltration of adipose tissue and circulating markers of oxidative stress.47
Furthermore, age-dependent NF-κB activation is associated with systemic inflammation and impaired endothelium-dependent dilation.48 NF-κB is a potent mediator of age-induced myocardial inflammation and fibrosis.49 These findings suggest that NF-κB could be a preventive and therapeutic target for CVD in the elderly.
Immunoglobulin Free Light Chains as Biomarkers of Nuclear Factor-κB Activation
NF-κB, as its name indicates, was originally found to enhance the transcription of genes of the kappa light chain of immunoglobulin in B cells.1,2 Therefore, increased circulating FLCs have been suggested as biomarkers of NF-κB and B-cell activation in many inflammatory and autoimmune conditions.1,2 FLCs appear in the circulation as excess by-products of antibody synthesis by B cells and plasma cells.50 Polyclonal FLCs have been shown to be predictors of mortality in the general population.51 FLC κ was increased and the FLC κ/λ ratio was higher in patients with rheumatic diseases than in healthy controls.52 FLCs have been correlated with disease activity in inflammatory and autoimmune diseases, suggesting their role as potential therapeutic targets.4
We found that circulating and cardiac FLCs were increased in a murine model of viral myocarditis and heart failure due to EMCV.53 We also showed that circulating FLC λ was increased, whereas the FLC κ/λ ratio was decreased in patients with myocarditis with heart failure compared with healthy controls, and that the FLC κ/λ ratio was an independent prognostic factor for overall survival.54
In patients with lone AF, circulating FLC κ and λ levels were significantly higher than in a healthy volunteer group.31 The mechanism by which FLCs cause AF is not yet fully understood, but the inflammation associated with FLCs may directly induce AF or FLCs may cause a change in membrane fluidity, which, in turn, could alter ion channel function.30,31
Individuals with type 2 diabetes have a lower FLC κ/λ ratio than healthy volunteers, and the sensitivity and specificity of the FLC κ/λ ratio for the diagnosis of diabetes were higher than those of HbA1c.55 These results suggest that FLCs may be a potentially promising biomarker of inflammatory diseases, such as CVD and diabetes, in which NF-κB activation plays a critical role in their pathogenesis.
Modulation of NF-κB Activation in the Management of Cardiovascular Disease and Its Risk Factors
Pimobendan
Phosphodiesterase (PDE) III inhibitors, originally developed as heart failure drugs, improve left ventricular function due to their positive inotropic and vasodilator effects.56,57 Although the short-term effects of PDE III inhibitors were beneficial, multicentre clinical trials of several PDE III inhibitors failed to show an improvement in long-term outcomes.56 However, pimobendan was effective in both acute and chronic heart failure, especially in low doses.57 Compared with other PDE III inhibitors, pimobendan has the unique additional property of increasing the affinity of cardiac contractile proteins for calcium.57 We investigated the effects of pimobendan and other inotropic agents, including PDE III inhibitors, on the activation of NF-κB and found that pimobendan suppressed the expression of luciferase protein in human lung A549 cells transfected with the NF-κB reporter plasmid and stimulated with IL-1β, TNF-α or phorbol 12-myristate 13 acetate.58 Electrophoretic mobility shift assays also showed that pimobendan inhibits NF-κB activation. Thus, pimobendan has a unique property of inhibiting NF-κB, which may be independent of PDE inhibition.58
In addition, we investigated the effects of pimobendan in vivo in a murine model of heart failure due to EMCV myocarditis, finding that pimobendan improved survival and decreased inflammatory lesions of the heart (Figure 1).59 The therapeutic effect of pimobendan appeared to be mediated by inhibition of the production of TNF-α, IL-1β, IL-6 and nitric oxide. Thus, pimobendan may be beneficial for the treatment of viral myocarditis, as well as other diseases in which the enhanced production of cytokines plays a pathogenic role and in which NF-κB activation contributes to the pathogenesis. In a preliminary study, we found that pimobendan decreased inflammation in adjuvant arthritis in rats (Figure 1D), and decreased CRP levels in patients with heart failure with rheumatoid arthritis (unpublished data).
Figure 1: Effects of Pimobendan on NF-κB In Vitro and In Vivo.
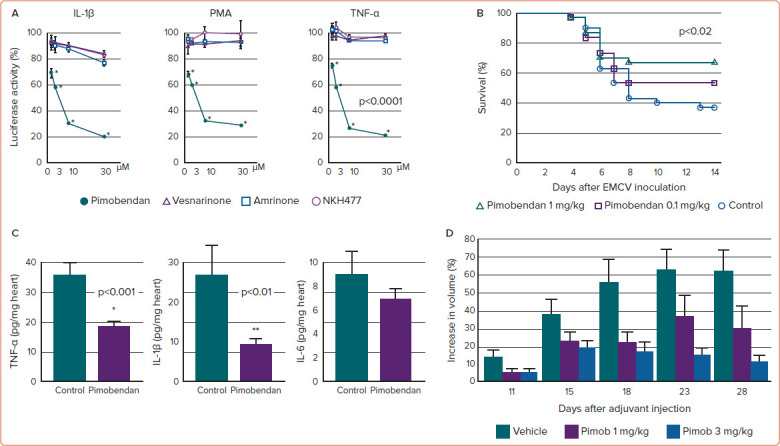
A: Pimobendan significantly decreased the activity of luciferase in A549 cells transfected with the NF-κB reporter plasmid and stimulated with IL-1β, TNF-α or PMA. However, other inotropic agents, namely amrinone, vesnarinone and NKH477, only slightly decreased luciferase activity. B: Effect of pimobendan on the survival of mice after inoculation with EMCV. Treatment with 1 mg/kg/day pimobendan significantly decreased mortality (p<0.02 compared with control). C: Effect of pimobendan on the production of IL-1β, IL-6 and TNF-α in the hearts of mice 7 days after EMCV inoculation. TNF-α and IL-1β concentrations were significantly lower in the hearts of pimobendan-treated mice than in the control group. *p<0.001, **p<0.01 compared with control. D: Effects of pimobendan on adjuvant arthritis in a rat model. Complete Freund's adjuvant was injected into the hind plantars to induce inflammation. Pimobendan was then administered for 28 days. Pimobendan dose-dependently decreased the degree of toe swelling. Data are presented as mean±SEM EMCV = encephalomyocarditis virus; IL = interleukin; NF-κB = nuclear factor-κB; Pimob = pimobendan; PMA = phorbol myristate acetate; TNF-α = tumour necrosis factor-α. Sources: A: Matsumori et al. 2000.58 Reproduced with permission from Elsevier. B,C: Iwasaki et al. 1999.59 Reproduced with permission from Elsevier.
It is important to know that pimobendan is a potent NF-κB inhibitor that is available and is being used for patients with heart failure in Japan without serious adverse effects;57 thus, it can be used for other diseases in which NF-κB plays a critical role in the pathogenesis and pathophysiology. The reason why European and US clinical trials of pimobendan failed to show beneficial long-term outcomes in heart failure may be the use of high doses.56 In a successful trial, pimobendan was used at a lower (approximately one-quarter) dose.57 These findings suggest that the dosage of a NF-κB inhibitor will be critically important for the treatment of diseases in which NF-κB is activated.
SUN C8079
Several inhibitors of NF-κB activation have been described and most have been shown to be effective in vitro, but the therapeutic applications of these inhibitors have not been successful because they cause adverse effects. We investigated the NF-κB inhibitor SUN C8079, which was discovered by random screening (Figure 2).15 SUN C8079 inhibited NF-κB activation induced by LPS in vitro. In addition, treatment of mice with SUN C8079 decreased the production of TNF-α induced by LPS and improved survival. The mechanism of NF-κB inhibition appeared to be downstream of signal transduction in the nucleus because SUN C8079 inhibited NF-κB activation as late as 3 h after stimulation.15 It did not block the direct binding of NF-κB to the consensus oligonucleotide. However, the target of SUN C8079 inhibition remains to be identified.15
Figure 2: Effects of SUN C8079, a NF-κB Inhibitor, on EMCV Infection In Vitro and In Vivo.
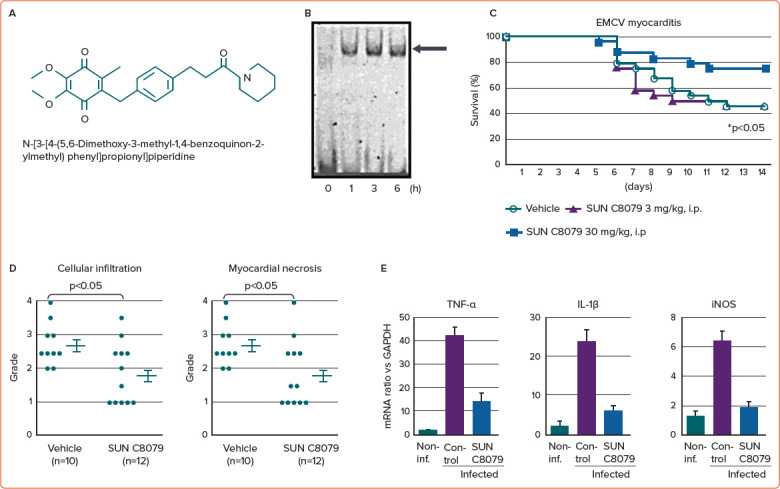
A: Chemical structure of SUN C8079. B: Activation of NF-κB in cardiac fibroblasts of mouse ventricles infected with EMCV in vitro. The arrow indicates NF-κB. C: Effects of SUN C8079 on the survival of mice after EMCV inoculation. Treatment with 30 mg/kg/day SUN C8079 significantly increased the survival of mice. *p<0.05 compared with control. D: Histopathological grade of mouse hearts. Myocardial necrosis and cellular infiltration were significantly less severe in the SUN C8079-treated group than in the vehicle control group (*p<0.05). E: Effect of SUN C8079 on TNF-α, IL-1β and iNOS mRNA levels in the heart after EMCV inoculation. SUN C8079 (30 mg/kg) significantly decreased intracardiac TNF-α, IL-1β and iNOS mRNA levels compared with the control group. Data are the mean ± SEM *p<0.05 compared with control. EMCV = encephalomyocarditis virus; IL = interleukin; iNOS = inducible nitric oxide synthase; NF-κB = nuclear factor-κB; TNF-α = tumour necrosis factor-α. Source: Matsumori et al. 2004.15 Reproduced with permission from Wiley.
As discussed previously, mice lacking the p50 subunit of NF-κB are resistant to EMCV infection, and fibroblasts from mice lacking p50 have enhanced induction of IFN-β transcription following infection with EMCV.2 Therefore, the p50 subunit of NF-κB may downregulate transcriptional responses that have important consequences on the in vivo response to pathogens.2 In our study, EMCV activated NF-κB in cultured non-cardiomyocytes. In a murine model of EMCV myocarditis, SUN C8079 improved the mortality and decreased cellular infiltration, myocardial necrosis and the expression of TNF-α and IL-1β in the heart without a significant effect on viral replication (Figure 2).15
We tested the hypothesis that inhibition of NF-κB activation reduces the expression of proinflammatory molecules and attenuates atherosclerosis in hyperlipidaemic mice. In that study, SUN C8079 significantly suppressed the expression of adhesion molecules and reduced the atherosclerotic area in mice (Figure 3).60 Thus, SUN C8079 is a promising NF-κB inhibitor, but further studies are needed to develop it as a therapeutic agent for human diseases.
Figure 3: Effects of SUN C8079 on the Expression of Proinflammatory Molecules and Atherosclerosis in Hyperlipidaemic ApoE-Deficient Mice.
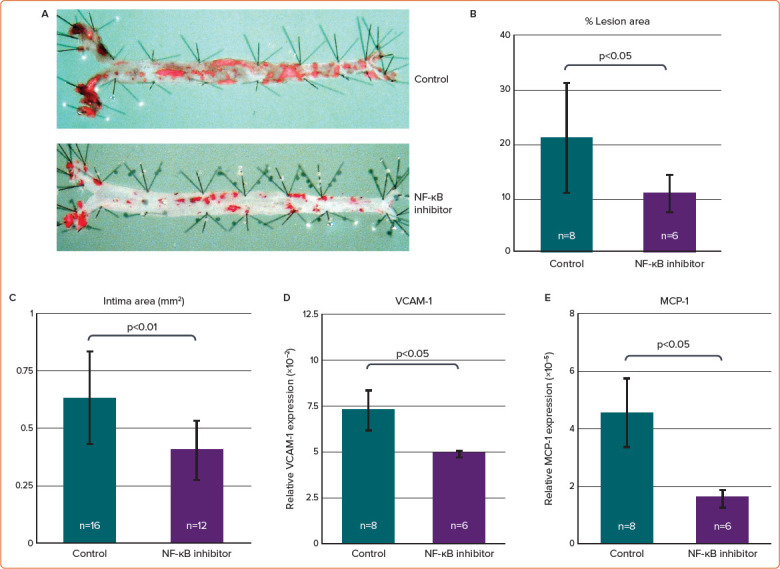
ApoE knockout mice were fed a western-type diet for 5 weeks and the mice were treated with the NF-κB inhibitor SUN C8079 or the vehicle for 10 weeks. A: En face oil red-O staining of the aorta. B: Quantification of the oil red-O staining-positive area. SUN C8079 significantly reduced the positive area (to 10%) compared with control mice (21%). C: SUN C8079 significantly reduced the intima area in transverse sections of aortic roots. D,E: Real-time polymerase chain reaction showed that the expression of VCAM-1 and MCP-1 mRNA in the aorta was significantly decreased by SUN C8079. Data are presented as mean±SD. MCP-1 = macrophage chemotactic factor-1; NF-κB = nuclear factor-κB; VCAM-1 = vascular cell adhesion molecule-1. Source: Furukawa et al. 2002.60
Antidiabetic Agents
Low-grade chronic inflammation has been reported to be associated with diabetes, atherosclerosis, hypertension and obesity.61 Several therapeutic agents for diabetes have been shown to have anti-inflammatory actions, and some anti-inflammatory therapies may, conversely, affect glucose metabolism.4
Sodium–Glucose Cotransporter 2 Inhibitors
Large cardiovascular trials have shown that sodium–glucose cotransporter 2 (SGLT2) inhibitors improve cardiovascular and renal outcomes in patients with diabetes. SGLT2 inhibitors reduce the production of proinflammatory cytokines and inflammation in adipose tissue.62 Canagliflozin, an SGLT2 inhibitor, has been shown to decrease circulating levels of IL-6, TNF receptor 1, fibronectin 1 and MMP7, and to improve inflammation and fibrosis.63 Empagliflozin may have beneficial effects by repleting AMP kinase activation-mediated energy and reducing inflammation.64 SGLT2 inhibitors promote polarisation of M2 macrophages with an anti-inflammatory phenotype, and decrease the production of proinflammatory cytokines in mice.65 Recent studies have shown that the anti-inflammatory effects of empagliflozin are due to the inhibition of NF-κB by downregulation of the IKK/NF-κB, mitogen-activated protein kinase kinase (MKK)/c-Jun N-terminal kinase (JNK) and Janus tyrosine kinase (JAK) 2/signal transducer and activator of transcription (STAT) 1 pathways in macrophages.61
The precise molecular mechanisms underlying the inhibition of NF-κB by SGLT2 inhibitors remain to be clarified, but the anti-inflammatory action of SGLT2 inhibitors may contribute to their beneficial effects in both diabetes and heart failure. Further studies on the effects of SGLT2 inhibitors on inflammatory diseases in which NF-κB is activated are required.
Metformin
In a large-scale treatment trial of newly diagnosed patients with diabetes, metformin was shown to decrease the neutrophil-to-lymphocyte ratio, a marker of systemic inflammation. In a heart failure trial without diabetes, metformin decreased circulating levels of cytokines and chemokines.66 These findings indicate that metformin has anti-inflammatory effects in patients with and without diabetes.66
Many studies have shown that metformin can block the NF-κB signalling pathway in macrophages. Metformin attenuated the LPS-stimulated phosphorylation of p65 and JNK1 and the upregulation of proinflammatory cytokine levels.67 Metformin also inhibited the translocation of NF-κB to the nucleus and reversed the LPS-induced reduction of apolipoprotein E expression in macrophages. Metformin can dose-dependently inhibit the IL-1β-stimulated release of IL-6 and IL-8 by human smooth muscle cells, macrophages and endothelial cells.68 Inhibition of the nuclear translocation of NF-κB occurs through the phosphatidylinositol 3-kinase/Akt pathway.68
Dipeptidyl Peptidase 4 Inhibitors
Dipeptidyl peptidase (DPP)-4 is a transmembrane glycoprotein known as CD26 that is expressed in endothelial cells, lymphocytes and macrophages. DPP-4 regulates the actions of cytokines and chemokines, and is involved in T-cell activation. DPP-4 inhibitors suppress the actions of IL-1, TLR4, NLRP3 inflammasomes and macrophages.69 Sitagliptin and other DPP4 inhibitors reduce the expression or activity of subunit of IκB kinase, JNK1, TNF-α, TLR2, TLR4 and the chemokine receptor CCR2.70 Gemigliptin has been shown to have anti-inflammatory effects by downregulating the IKK/NF-κB, MKK7/JNK and JAK2/STAT1 pathways in macrophages.61
Glucagon-Like Peptide 1 Receptor Agonists
Glucagon-like peptide-1 (GLP-1) receptor agonists inhibit NF-κB activity and reduce inflammatory biomarkers, such as reactive oxygen species, the expression of IL-1β, TNF-α, JNK1, TLR2, TLR4 and SOCS-3 in mononuclear cells and circulating concentrations of IL-6, MCP-1, MMP-9 and serum amyloid A.71
Natural Compounds
Several studies have shown that dietary components such as polyphenols, terpenes, alkaloids and phenolics from different fruits and vegetables have anti-inflammatory effects. NF-κB has been shown to be the target of most of these compounds.72 There is increasing interest in investigating non-toxic natural compounds with fewer side effects for the treatment of cancer. Several studies have indicated that some natural compounds can inhibit NF-κB and may be useful adjuvants for immune-based cancer therapies.72
Pycnogenol
Pycnogenol (PYC), a natural plant extract derived from the bark of French maritime pine (Pinus pinaster ssp atlantica), is a source of flavanols and phenolic and cinnamic acids.73 The anti-inflammatory effects of PYC have been demonstrated in in vitro and ex vivo studies using human plasma following intake of PYC, in which PYC intake decreased NF-κB activation and COX-1, COX-2 and phospholipase A2 activity.73 In another study, PYC inhibited NF-κB activation and decreased the induction of TNF-α, ICAM-1 and VCAM-1 gene expression.4,74
We showed that PYC inhibited the replication of EMCV both in vitro and in vivo in mice, and improved inflammation and preserved myocardial tissue by decreasing necrosis.74
Administration of PYC significantly inhibited the gene expression of proinflammatory cytokines and inhibited the expression of mast cell-related tryptase and stem cell factor. PYC has shown antiviral effects against HCV and synergistic effects with IFN-α or ribavirin in vitro.4 In vivo investigations in HCV-infected chimeric mice revealed that PYC inhibited HCV replication. The addition of PYC to HCV replicon cell lines resulted in a dose-dependent reduction in the production of reactive oxygen species.4 A double-blind placebo-controlled study involving patients with HBV infection showed that circulating HBV levels were reduced and hepatic function was improved after PYC administration.4
Resveratrol
Resveratrol has been shown to improve cardiovascular function in diabetic rats.75 Resveratrol has shown beneficial effects in heart failure by improving left ventricular function, remodelling and interstitial fibrosis.75 Resveratrol inhibited NF-κB activation, decreasing circulating TNF-α and IL-6 levels, MIP-2 and COX-2 activity and the production of reactive oxygen species in a rabbit model of acute pharyngitis.76 Resveratrol modulates the inflammatory response within intestinal cells by downregulating NF-κB activation and preventing mitochondrial dysfunction in vitro.77 This result was confirmed in vivo (rodents and humans), whereby resveratrol inhibited NF-κB activation and TNF-α production, decreased neutrophil infiltration in the intestinal mucosa and repressed intestinal tumour genesis.76 Resveratrol suppressed the TLR4/MyD88/NF-κB signalling pathway in lysophosphatidylcholine-induced damage and inflammation,78 which may be useful for the prevention of atherosclerosis. Thus, resveratrol can prevent inflammation and oxidative stress and may be promising as an anti-inflammatory agent for CVD to improve quality of life and reduce the risk of carcinogenesis.
Curcumin and Others
Nutraceuticals are known to suppress NF-κB activity by modulating several steps, such as IKK activation, IκBα phosphorylation and degradation, p65 nuclear translocation, p65 phosphorylation and acetylation and p65 DNA binding.79 Curcumin, derived from the golden spice turmeric, is known to modulate the production and activity of inflammatory molecules.79 The most common nutraceuticals known to inhibit NF-κB activation include curcumin, capsaicin and caffeic acid phenethyl ester.80 Curcumin and capsaicin are known to prevent the phosphorylation and degradation of IκBα, which is a central point in NF-κB activation.80 In addition to anticancer effects, nutraceuticals are also known to have beneficial effects in other inflammatory diseases. Oral administration of curcumin produces beneficial effects in patients with uveitis and rheumatoid arthritis, as demonstrated in clinical trials.81
Most natural products have merits for use without much concern about adverse effects because nutraceuticals and their sources have been consumed since ancient times, and their safety has been well tested. Nutraceuticals can modulate multiple cell signalling pathways. Therefore, natural products could be used in combination with other pharmacological agents. Clinical trials using these agents with inhibitory effects on NF-κB merit further study in various inflammatory diseases.
Conclusion
Inflammation has been shown to play an important role in the pathogenesis and progression of CVDs such as heart failure, ischaemic heart disease, myocarditis and AF. Most of the risk factors for CVDs, such as an unhealthy diet, smoking and infection, induce cellular stress and inflammation.
Because NF-κB is activated by factors that increase the inflammatory response and this activation leads to the expression of several genes, such as those encoding cytokines and chemokines, NF-κB may play an important role in CVDs, including heart failure and myocarditis, and CVD risk factors, such as atherosclerosis and diabetes (Figure 4). In addition, there is renewed interest in NF-κB because inflammation is considered to play a significant role in the development of cancer and ageing.3
Figure 4: Induction and Regulation of NF-κB.
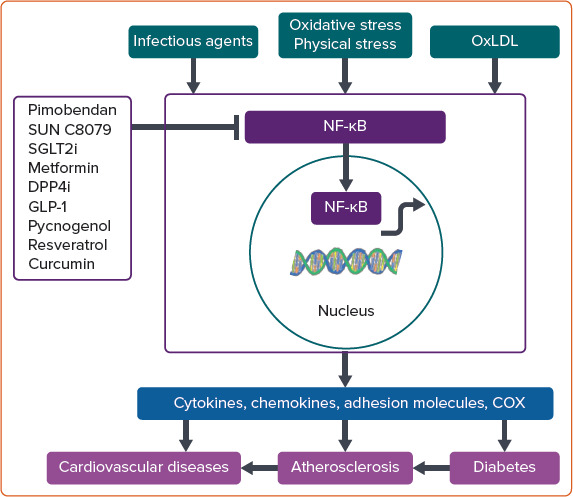
NF-κB is induced by various signals, including bacterial and viral infections, cytokines, growth factors and various stressors, such as oxidative stress and oxidised LDL. In resting cells, NF-κB resides in the cytoplasm, forming a complex with I-κB, which keeps it in an inactive state. When activated, NF-κB translocates to the nucleus, binds DNA and then regulates gene expression. Targets for NF-κB signalling include genes encoding proinflammatory cytokines, growth factors, chemokines, cyclo-oxygenase and inducible nitric oxide synthase. Pimobendan, SUN C8079, SGLT2 inhibitors, metformin, DPP-4 inhibitors, GLP-1 receptor agonists, pycnogenol, resveratrol, curcumin and other agents have inhibitory actions against NF-κB. COX = cyclo-oxygenase; DPP-4i = dipeptidyl peptidase 4 inhibitor; GLP-1 = glucagon-like peptide-1; NF-κB = nuclear factor-κB; OxLDL = oxidised LDL; SGLT2i = sodium–glucose cotransporter 2 inhibitor.
It is clear that NF-κB inflammatory pathways promote CVDs, diabetes, cancer, ageing and other diseases, and may be the target of preventive and therapeutic strategies. Although inhibitors of specific NF-κB pathways are not yet clinically available, progress has been made in the development of more selective anti-NF-κB pharmaceuticals and in understanding the effects of existing therapies on the NF-κB pathway. Several therapeutic agents used for CVDs and diabetes, such as pimobendan, SGLT2 and DPP-4 inhibitors, and GLP-1 receptor agonists may exert anti-inflammatory effects by inhibiting NF-κB activation (Figure 4). Conversely, some anti-inflammatory agents that inhibit NF-κB have beneficial effects on CVDs and diabetes. Despite great expectations following the early development of NF-κB inhibitors for the treatment of inflammatory diseases and cancer, very few of these molecules have reached advanced phases in clinical trials. This is due either to modest efficacy or severe adverse effects.
Although considerable efforts have been made to develop specific NF-κB inhibitors, none of the inhibitors has been approved clinically owing to drug toxicity. Some pharmacological agents and natural compounds that are commercially available without serious adverse effects have an inhibitory effect on NF-κB. These agents, alone or in combination, may be used for the prevention and treatment of various inflammatory diseases in which the NF-κB pathway plays a role. Clinical trials are required to confirm the efficacy of these agents. Because NF-κB activation is tissue or organ specific, the inhibitory action of the NF-κB pathway varies depending on the agent.
Measurement of NF-κB activity is not possible in vivo, and there has been no way to evaluate the efficacy of NF-κB inhibitors. FLCs could be surrogate biomarkers of NF-κB activation and may be useful for evaluating the efficacy of NF-κB inhibitors for CVDs and diabetes.
Improved knowledge of the NF-κB pathway in various diseases may lead to the development of better preventive, diagnostic and therapeutic strategies for various human diseases in which inflammation may play a crucial role in the pathogenesis: not only cardiovascular and metabolic diseases, but also in autoimmune diseases, cancer and neurological diseases.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Am Heart J. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Am Heart J. 1995;80:321–30. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Am Heart J. 2018;18:309–24. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 4.Matsumori A. Targeting inflammation in the diagnosis, management, and prevention of cardiovascular diseases. Am Heart J. 2022;17:80. doi: 10.5334/gh.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anto RJ, Mukhopadhyay A, Shishodia S et al. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Am Heart J. 2002;23:1511–8. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- 6.Heidenreich PA, Bozkurt B, Aguilar D et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Am Heart J. 2022;145:e876–94. doi: 10.1161/CIR.0000000000001062. [DOI] [PubMed] [Google Scholar]
- 7.Niebauer J, Volk HD, Kemp M et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Am Heart J. 1999;353:1838–42. doi: 10.1016/S0140-6736(98)09286-1. [DOI] [PubMed] [Google Scholar]
- 8.Carrillo-Salinas FJ, Ngwenyama N, Anastasiou M et al. Heart inflammation: immune cell roles and roads to the heart. Am Heart J. 2019;189:1482–94. doi: 10.1016/j.ajpath.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumori A. Anti-inflammatory therapy for heart failure. Am Heart J. 2004;4:171–6. doi: 10.1016/j.coph.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Matsumori A, Yamada T, Suzuki H et al. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Am Heart J. 1994;72:561–6. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleet JC, Clinton SK, Salomon RN et al. Atherogenic diets enhance endotoxin-stimulated interleukin-1 and tumor necrosis factor gene expression in rabbit aortae. Am Heart J. 1992;122:294–305. doi: 10.1093/jn/122.2.294. [DOI] [PubMed] [Google Scholar]
- 12.Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. Am Heart J. 2016;91:114–22. doi: 10.1016/j.yjmcc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ono K, Matsumori A, Shioi T et al. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Am Heart J. 1998;98:149–56. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- 14.Libby P, Loscalzo J, Ridker PM et al. Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. Am Heart J. 2018;72:2071–81. doi: 10.1016/j.jacc.2018.08.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumori A, Nunokawa Y, Yamaki A et al. Suppression of cytokines and nitric oxide production, and protection against lethal endotoxemia and viral myocarditis by a new NF-kappaB inhibitor. Am Heart J. 2004;6:137–44. doi: 10.1016/j.ejheart.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Matsumori A. In: Am Heart J. Berhardt LV, editor. Vol. 194. New York NY: Nova Science Publishers, Inc.; 2022. Viral myocarditis from animal models to human diseases. pp. 41–74. [DOI] [Google Scholar]
- 17.Shioi T, Matsumori A, Sasayama S. Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Am Heart J. 1996;94:2930–7. doi: 10.1161/01.cir.94.11.2930. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson-Payant BE, Uhl S, Grimont A et al. The NF-κB transcriptional footprint is essential for SARS-CoV-2 replication. Am Heart J. 2021;95 doi: 10.1128/JVI.01257-21. e0125721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robles JP, Zamora M, Adan-Castro E et al. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin α5β1 and NF-κB signaling. Am Heart J. 2022;298 doi: 10.1016/j.jbc.2022.101695. 101695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Attiq A, Yao LJ, Afzal S, Khan MA. The triumvirate of NF-κB, inflammation and cytokine storm in COVID-19. Am Heart J. 2021;101 doi: 10.1016/j.intimp.2021.108255. 108255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saleh A, Matsumori A, Abdelrazek S et al. Myocardial involvement in coronavirus disease 19. Am Heart J. 2020;45:719–25. doi: 10.1007/s00059-020-05001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komiyama M, Hasegawa K, Matsumori A. Dilated cardiomyopathy risk in patients with coronavirus disease 2019: how to identify and characterise it early? Am Heart J. 2020;15:e49. doi: 10.15420/ecr.2020.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahroum N, Lavine N, Ohayon A et al. COVID-19 vaccination and the rate of immune and autoimmune adverse events following immunization: insights from a narrative literature review. Am Heart J. 2022;13 doi: 10.3389/fimmu.2022.872683. 872683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yonker LM, Swank Z, Bartsch YC et al. Circulating spike protein detected in post-COVID-19 mRNA vaccine myocarditis. Am Heart J. 2023;147:867–76. doi: 10.1161/CIRCULATIONAHA.122.061025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura H, Okazaki T, Tanaka Y et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Am Heart J. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 26.Okazaki T, Tanaka Y, Nishio R et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Am Heart J. 2003;9:1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 27.Rubio-Infante N, Ramírez-Flores YA, Castillo EC et al. A systematic review of the mechanisms involved in immune checkpoint inhibitors cardiotoxicity and challenges to improve clinical safety. Am Heart J. 2022;10 doi: 10.3389/fcell.2022.851032. 851032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haanen JB, Robert C. Immune checkpoint inhibitors. Am Heart J. 2015;42:55–66. doi: 10.1159/000437178. [DOI] [PubMed] [Google Scholar]
- 29.Axelrod ML, Meijers WC, Screever EM et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Am Heart J. 2022;611:818–26. doi: 10.1038/s41586-022-05432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumori A. Management of atrial fibrillation using immunoglobulin free light chains, novel biomarkers of inflammation. Am Heart J. 2022;17:e22. doi: 10.15420/ecr.2022.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumori A, Shimada T, Shimada M et al. Immunoglobulin free light chains as inflammatory biomarkers of atrial fibrillation. Am Heart J. 2020;13 doi: 10.1161/CIRCEP.120.009017. e009017. [DOI] [PubMed] [Google Scholar]
- 32.Capece D, Verzella D, Flati I et al. NF-κB: blending metabolism, immunity, and inflammation. Am Heart J. 2022;43:757–75. doi: 10.1016/j.it.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Kugler S, Onodi Z, Ruppert M et al. Inflammasome activation in end-stage heart failure-associated atrial fibrillation. Am Heart J. 2022;9:2747–52. doi: 10.1002/ehf2.13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gawalko M, Salijic A, Li N Adiposity-associated atrial fibrillation: molecular determinants, mechanisms and clinical significance. Am Heart J. 2022. [DOI] [PMC free article] [PubMed]
- 35.Engelen SE, Robinson AJB, Zurke YX, Monaco C.Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Am Heart J202219522–42. 10.1038/s41569-021-00668-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naghavi M, Libby P, Falk E et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Am Heart J. 2003;108:1664–72. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- 37.Robbins CS, Chudnovskiy A, Rauch PJ et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Am Heart J. 2012;125:364–74. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuster JJ, MacLauchlan S, Zuriaga MA et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Am Heart J. 2017;355:842–7. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brand K, Page S, Rogler G et al. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. Am Heart J. 1996;97:1715–22. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridker PM, Everett BM, Thuren T et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. Am Heart J. 2017;377:1119–31. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 41.Nidorf SM, Fiolet ATL, Mosterd A et al. Colchicine in patients with chronic coronary disease. Am Heart J. 2020;383:1838–47. doi: 10.1056/NEJMoa2021372. [DOI] [PubMed] [Google Scholar]
- 42.Furman D, Campisi J, Verdin E et al. Chronic inflammation in the etiology of disease across the life span. Am Heart J. 2019;25:1822–32. doi: 10.1038/s41591-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Am Heart J. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 44.Donath MY, Meier DT, Böni-Schnetzler M. Inflammation in the pathophysiology and therapy of cardiometabolic disease. Am Heart J. 2019;40:1080–91. doi: 10.1210/er.2019-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan M, Konstantopoulos N, Lee J et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Am Heart J. 2001;293:1673–7. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 46.Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. Am Heart J. 2016;594:2061–73. doi: 10.1113/JP270538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa Y, Saito T, Ogihara T et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Am Heart J. 2012;125:1122–33. doi: 10.1161/CIRCULATIONAHA.111.054346. [DOI] [PubMed] [Google Scholar]
- 48.Tabit CE, Shenouda SM, Holbrook M et al. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Am Heart J. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tas SW, Vervoordeldonk MJ, Tak PP. Gene therapy targeting nuclear factor-kappaB: towards clinical application in inflammatory diseases and cancer. Am Heart J. 2009;9:160–70. doi: 10.2174/156652309788488569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hampson J, Turner A, Stockley R. Polyclonal free light chains: promising new biomarkers in inflammatory disease. Am Heart J. 2014;4:139–49. doi: 10.2147/CBF.S57681. [DOI] [Google Scholar]
- 51.Dispenzieri A, Katzmann JA, Kyle RA et al. Use of nonclonal serum immunoglobulin free light chains to predict overall survival in the general population. Am Heart J. 2012;87:517–23. doi: 10.1016/j.mayocp.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gulli F, Napodano C, Marino M et al. Serum immunoglobulin free light chain levels in systemic autoimmune rheumatic diseases. Am Heart J. 2020;199:163–71. doi: 10.1111/cei.13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsumori A, Shimada M, Jie X et al. Effects of free immunoglobulin light chains on viral myocarditis. Am Heart J. 2010;106:1533–40. doi: 10.1161/CIRCRESAHA.110.218438. [DOI] [PubMed] [Google Scholar]
- 54.Matsumori A, Shimada T, Nakatani E et al. Immunoglobulin free light chains as an inflammatory biomarker of heart failure with myocarditis. Am Heart J. 2020;217 doi: 10.1016/j.clim.2020.108455. 108455. [DOI] [PubMed] [Google Scholar]
- 55.Matsumori A, Shimada T, Shimada M et al. Immunoglobulin free light chains: an inflammatory biomarker of diabetes. Am Heart J. 2020;69:715–8. doi: 10.1007/s00011-020-01357-7. [DOI] [PubMed] [Google Scholar]
- 56.Remme WJ, Krayenbühl HP, Baumann G et al. Long-term efficacy and safety of pimobendan in moderate heart failure: a double-blind parallel 6-month comparison with enalapril. Am Heart J. 1994;15:947–56. doi: 10.1093/oxfordjournals.eurheartj.a060615. [DOI] [PubMed] [Google Scholar]
- 57.Effects of Pimobendan on Chronic Heart Failure Study (EPOCH Study). Effects of pimobendan on adverse cardiac events and physical activities in patients with mild to moderate chronic heart failure: the effects of pimobendan on chronic heart failure study (EPOCH study). Am Heart J. 2002;66:149–57. doi: 10.1253/circj.66.149. [DOI] [PubMed] [Google Scholar]
- 58.Matsumori A, Nunokawa Y, Sasayama S. Pimobendan inhibits the activation of transcription factor NF-kappaB: a mechanism which explains its inhibition of cytokine production and inducible nitric oxide synthase. Am Heart J. 2000;67:2513–9. doi: 10.1016/s0024-3205(00)00834-1. [DOI] [PubMed] [Google Scholar]
- 59.Iwasaki A, Matsumori A, Yamada T et al. Pimobendan inhibits the production of proinflammatory cytokines and gene expression of inducible nitric oxide synthase in a murine model of viral myocarditis. Am Heart J. 1999;33:1400–7. doi: 10.1016/s0735-1097(98)00692-5. [DOI] [PubMed] [Google Scholar]
- 60.Furukawa Y, Kobuke K, Nunokawa Y NF-κB inhibition reduces pro-inflammatory molecule expression and attenuates atherosclerosis in apoE−/− mice. Presented at: 75th Scientific Sessions of the American Heart Association, Chicago, IL, 17–20 November Am Heart J 2002.
- 61.Lee N, Heo YJ, Choi SE et al. Anti-inflammatory effects of empagliflozin and gemigliptin on LPS-stimulated macrophage via the IKK/NF-κB, MKK7/JNK, and JAK2/STAT1 signalling pathways. Am Heart J. 2021;2021 doi: 10.1155/2021/9944880. 9944880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cowie MR, Fisher M. SGLT2 inhibitors: mechanisms of cardiovascular benefit beyond glycaemic control. Am Heart J. 2020;17:761–72. doi: 10.1038/s41569-020-0406-8. [DOI] [PubMed] [Google Scholar]
- 63.Heerspink HJL, Perco P, Mulder S et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Am Heart J. 2019;62:1154–66. doi: 10.1007/s00125-019-4859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koyani CN, Plastira I, Sourij H et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Am Heart J. 2020;158 doi: 10.1016/j.phrs.2020.104870. 104870. [DOI] [PubMed] [Google Scholar]
- 65.Xu L, Nagata N, Nagashimada M et al. SGLT2 Inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. Am Heart J. 2017;20:137–49. doi: 10.1016/j.ebiom.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cameron AR, Morrison VL, Levin D et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Am Heart J. 2016;119:652–65. doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Woo SL, Xu H, Li H et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. Am Heart J. 2014;9 doi: 10.1371/journal.pone.0091111. e91111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Isoda K, Young JL, Zirlik A et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Am Heart J. 2006;26:611–7. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- 69.Dai Y, Dai D, Wang X et al. DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1 beta via GLP-1 receptor in macrophages through protein kinase C pathway. Am Heart J. 2014;28:425–32. doi: 10.1007/s10557-014-6539-4. [DOI] [PubMed] [Google Scholar]
- 70.Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Am Heart J. 2012;33:187–215. doi: 10.1210/er.2011-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang F, Zeng F, Luo X et al. GLP-1 receptor: a new target for sepsis. Am Heart J. 2021;12 doi: 10.3389/fphar.2021.706908. 706908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trung LQ, An DTT. Is resveratrol a cancer immunomodulatory molecule? Am Heart J. 2018;9 doi: 10.3389/fphar.2018.01255. 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maimoona A, Naeem I, Saddiqe Z, Jameel K. A review on biological, nutraceutical and clinical aspects of French maritime pine bark extract. Am Heart J. 2011;133:261–77. doi: 10.1016/j.jep.2010.10.041. [DOI] [PubMed] [Google Scholar]
- 74.Matsumori A, Higuchi H, Shimada M. French maritime pine bark extract inhibits viral replication and prevents development of viral myocarditis. Am Heart J. 2007;13:785–91. doi: 10.1016/j.cardfail.2007.06.721. [DOI] [PubMed] [Google Scholar]
- 75.Riba A, Deres L, Sumegi B et al. Cardioprotective effect of resveratrol in a postinfarction heart failure model. Am Heart J. 2017;2017 doi: 10.1155/2017/6819281. 6819281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou ZX, Mou SF, Chen XQ et al. Anti-inflammatory activity of resveratrol prevents inflammation by inhibiting NF-κB in animal models of acute pharyngitis. Am Heart J. 2018;17:1269–74. doi: 10.3892/mmr.2017.7933. [DOI] [PubMed] [Google Scholar]
- 77.Nunes S, Danesi F, Del Rio D, Silva P. Resveratrol and inflammatory bowel disease: the evidence so far. Am Heart J. 2018;31:85–97. doi: 10.1017/S095442241700021X. [DOI] [PubMed] [Google Scholar]
- 78.Chen J., Cao X., Cui Y., Zeng G., Chen J., Zhang G. Resveratrol alleviates lysophosphatidylcholine-induced damage and inflammation in vascular endothelial cells. Am Heart J. 2018;17:4011–4018. doi: 10.3892/mmr.2017.8300. [DOI] [PubMed] [Google Scholar]
- 79.Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge sword for cancer and other age-related diseases. Am Heart J. 2018;9:2160. doi: 10.3389/fimmu.2018.02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Philip S, Kundu GC. Osteopontin induces nuclear factor kappa B-mediated promatrix metalloproteinase-2 activation through I kappa B alpha/IKK signaling pathways, and curcumin (diferulolylmethane) down-regulates these pathways. Am Heart J. 2003;278:14487–97. doi: 10.1074/jbc.M207309200. [DOI] [PubMed] [Google Scholar]
- 81.Chandran B, Goel A. A randomized, pilot study to assess the efficacy and safety of curcumin in patients with active rheumatoid arthritis. Am Heart J. 2012;26:1719–25. doi: 10.1002/ptr.4639. [DOI] [PubMed] [Google Scholar]