

Abstract
We report a severe form of osteogenesis imperfecta (OI) type VIII from a lower-middle income country. This is the first case report of this type in Tanzania. The term neonate was delivered normally via spontaneous vaginal delivery and presented at the neonatal unit with features of shortened limb girdles and macrocephaly. The long bones had multiple fractures. He was diagnosed clinically to have OI or a type of metaphysial dysplasia. A plain X-ray showed multiple fractures of the long bones. The eyes did not have blue sclerae. Clinically, the generic diagnosis of OI was made.
Genetic testing revealed typical prolyl 3-hydroxylase 1 (P3HI) gene mutations and a variant coordinate NM_001243246.1:c.1095C>G p, indicating a severe, fatal form of autosomal-recessive OI type VIII which presents with white sclerae. This rare variant is described here for the first time in our setting. This case highlights the need for genetic testing.
Keywords: Orthopaedics, Paediatrics, Congenital disorders, Genetics
Background
This is an important case because it is not uncommonly misdiagnosed. Neonates who present with this diagnosis generally do not undergo genetic testing. Therefore, the diagnosis of osteogenesis imperfecta(OI) type VIII rests solely on the clinical features. Since it is prevalent, this is a wake-up call for more surveys to be undertaken for the same genetic variant and to identify potential couples who may transmit the gene to their offspring, especially among those who have relatives diagnosed with Osteogenesis Imperfecta.
Type VIII OI is autosomal-recessive, characterised by white sclerae, severe growth deficiency, extreme skeletal under-mineralisation and bulbous metaphysis.1 Its prevalence is approximately 1 in 10 000–20 000 births.2
We report a case that required genetic testing for confirmation, the results of which helped greatly in counselling the baby's parents. The case in question required a multidisciplinary approach to management.
Case presentation
The term neonate was born to non-consanguineous parents. He was the second born- with an 18 month older female sibling. The mother’s pregnancy was uneventful, and she had attended all her antenatal clinic appointments. She was given routine haematinics and folic acid, and the tetanus vaccine as per standard of care. She progressed well, although she noted poor weight gain in the third trimester.
Antenatal ultrasound was not done. The baby was delivered by spontaneous vaginal delivery and admitted to the neonatal unit with severe respiratory distress and malformed limbs, both upper (figure 1A) and lower (figure 1B). The head circumference was 35 cm at birth. Clinically, the features appeared similar to OI, but a possible metaphysical dysplasia was considered because the sclerae were not blue (figure 2).
Figure 1.

(A) Right upper limb with shortened humerus and curved ulna (arrows). (B) Left lower limb with curved femur and tibia with small left inguinal hernia and congenital hydrocele (arrows).
Figure 2.

Facial features showing the flat nasal bridge and the absence of obvious blue sclera.
On examination, all four limbs were curved, the neonate looked short- with a length of 51 cm, and he had a small, left-sided, reducible, inguinal hernia and congenital hydrocele (figure 1B). There were signs of severe pneumonia with severe chest drawing and nasal flaring.
Investigations
A plain X-ray of the chest and a whole-body scout X-ray were done (figure 3). These revealed multiple fractures of the long bones, horizontal ribs and small rib cage size. Bilateral diffuse non-homogenous opacifications showing bronchopneumonia were also observed.
Figure 3.
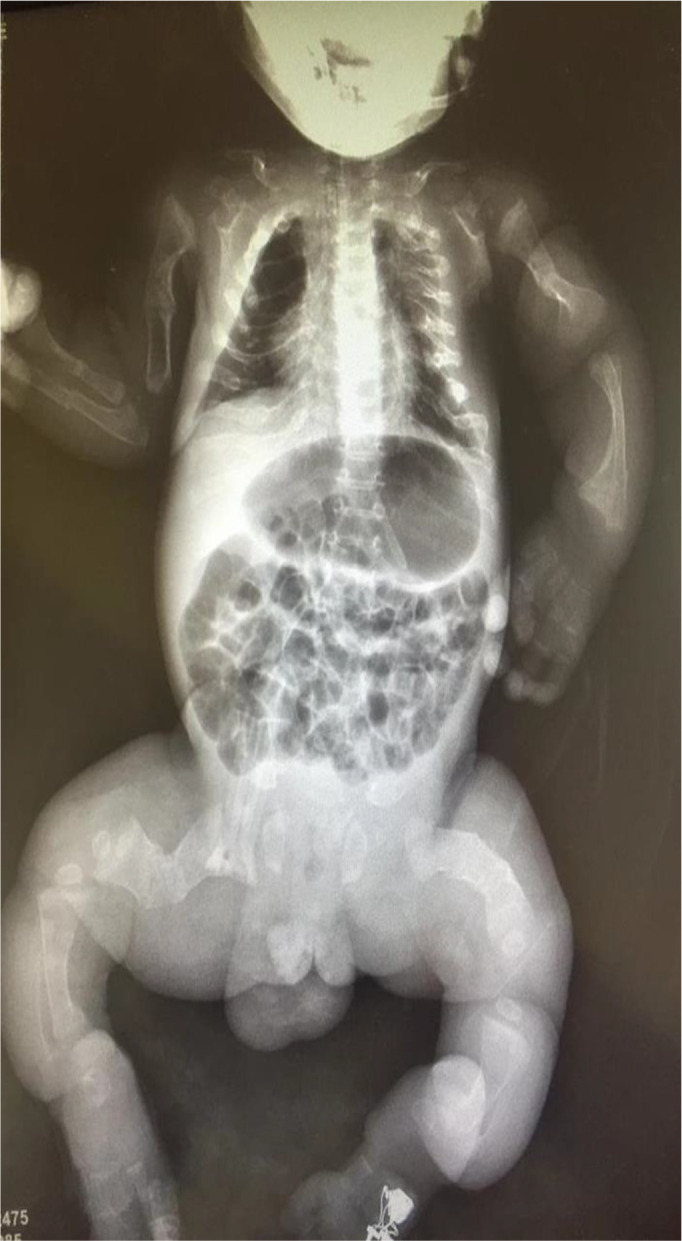
Skeletal survey showing fractures of the ribs, both humeri and femora.
A cranial ultrasound scan (figure 4) showed features of cystic encephalomalacia in the occipital lobe, and surrounding echoic cortex indicating an infarct in resolution, leaving a jagged margin of cystic encephalomalacia (that was probably of intrauterine origin).
Figure 4.

Cranial ultrasound scan with occipital lobe showing features of cystic encephalomalacia (black arrow) and surrounding echoic cortex indicating an infarct in resolution, leaving a jagged margin of cystic encephalomalacia (white arrows).
Genetic testing: Dried blood spots were taken for whole genomic sequencing at CENTOGENE (a company specialising in the diagnosis and treatment of rare diseases based in Rostock, Germany). A homozygous, likely pathogenic variant was identified in the P3H1 gene. The result is consistent with the genetic diagnosis of autosomal-recessive OI type VIII. The variant coordinate was NM_001243246.1: c.1095C>G p with amino acid change: (Tyr365*), recessive homozygous, nonsense, likely pathogenic (class 2).
Echocardiography revealed a structurally normal heart.
Ultrasound of the abdomen showed normal renal and hepatic morphology.
Serum alkaline phosphatase was 652 mmol/dL (normal level up to 125 mmol/dL).
Lactate dehydrogenase was also raised to 585 mmol/dL.
Differential diagnosis
The differential diagnosis was metaphysial dysplasia on account of the symmetrical short bones, macrocephaly and absence of blue sclerae.
Treatment
The baby was treated with appropriate antibiotics for pneumonia and paracetamol for pain. Supportive care was given, including feeding by tube and providing oxygen during the pneumonia episode.
Outcome and follow-up
The baby was discharged after 3 weeks when his condition was stable. He was readmitted at 8 weeks with severe pneumonia. At this time the highest saturations (sPaO2) were in the mid-70s, and he was acidotic with a blood pH of 7.27 and carbon dioxide retention. The baby succumbed to severe pneumonia aged 10 weeks.
The results of genetic analysis were available prior to the baby's death. The mother was counselled, and both parents returned after 6 weeks (the customary period of bereavement) to discuss the test results in more detail.
Discussion
OI, an inherited connective tissue disorder, is recognised by excessive brittleness of the bones. It is the most common genetic disorder of bones with an estimated prevalence of approximately 1 in 10 000–20 000 births.1 2 It has several classification types, and infants with the worst form of the disease die in the perinatal period.
Type VIII OI, a rare autosomal-recessive disorder characterised by white sclerae, severe growth deficiency and bone fragility, was added to the classification in 2007 as a severe and lethal type.1 Type VIII OI accounts for fewer than 10% of patients with OI. DNA analysis is considered a definitive test for confirming the diagnosis.3
A similar case of type VIII OI due to a homozygous mutation of c.1914+1G>C (NM_001243246.1) in P3H1 exhibited retinal detachment, emphasising the need for fundoscopic examination as part of a multidisciplinary approach.4
The variant P3H1, and variant coordinate 1095C>G p.(Tyr365*), create a premature stop codon.5 6 Pathogenic P3H1 variants are causative for autosomal-recessive OI type VIII. In 2007, Cabral et al described a form of autosomal-recessive OI that they designated OI type VIII, characterised by white sclerae, severe growth deficiency, extreme skeletal under-mineralisation and bulbous metaphysis.1
However, this variant coordinate is rare, as compared with the c.1080+1G>T described in West Africa and the descendants of West Africans in the USA.5 In Cabral et al’s study, the screening of contemporary cohorts in West Africa revealed that nearly 1.5% of West Africans are carriers for this mutation and 0.4% of African Americans of Western African descent are carriers.7
The carrier rate in our setting is not known and the variant coordinates are different from what was found in our case. As this is a homozygous-recessive variant, there is a need for genetic counselling and further survey of the families. Although the parents in our case were non-consanguineous, the recessive nature of this condition indicates the need for further screening of the families after genetic counselling in the familial context if possible, similar to the scenario in individauls of Karen descent. Due to resource constraints, however, this is not possible.1 8
Type VIII OI patients present with severe to lethal OI, recessive inheritance, white sclerae, rhizomelia, and poor trabeculation of long bones, with most of the affected individuals dying early from respiratory causes, as in our case. Those who survive into childhood have extremely low bone mineral density, severe growth deficiency and bulbous metaphysis.
A multidisciplinary team approach is essential for management, with the aim of providing the maximum long-term function and autonomy that the disease allows.3 9 Clinical management encompasses physical rehabilitation and surgical procedures, and management of hearing, dental and pulmonary abnormalities.
Fratzl-Zelman et al studied five individuals with type VIII OI.9 All the patients had white sclerae, normal dentition, relative macrocephaly, triangular facies, and skeletal findings that included rhizomelia, radiographic “popcorn” calcifications at epiphyses, and extremely low bone density. Bisphosphonate treatment was initiated, but progressive skeletal deterioration with age was observed.
Our case demonstrates the importance of genetic testing, which revealed typical P3HI gene mutations. P3H1, cartilage-associated protein (CRTAP) and cyclophilin B (CyPB) assemble into a 1:1:1 complex within the endoplasmic reticulum that post-translationally modifies specific proline residues in unfolded collagen alpha chains.10 Recessive OI (types VII and VIII) is caused by deficiency of any of the three collagen prolyl 3-hydroxylation complex components, increasing modification of the collagen helix, supporting delayed collagen folding. In type VIII OI there is the absence of alpha1 (I)Pro986 3-hydroxylation and over-mediation of the collagen helical region, supporting delayed folding.11
A multidisciplinary approach to management was used in our case, which included neonatologists, medical officers, endocrinologists and geneticists. This case demonstrates the challenge in confirming the diagnosis in our setting due to limited resources, and the possibility that most cases of type VIII OI are missed.
Patient’s perspective.
Although my baby died, I am very grateful to the doctors that took care of him. I am glad that it was possible to do a genetic test, despite this being expensive and not available in our country. The test was done for free. The doctors told me that my baby had a rare condition for which there is no cure, and I thank God for the few weeks I spent with my baby before his death.
Learning points.
Pathogenic P3H1variants are causative for autosomal-recessive OI type VIII.
Genetic testing is important for the diagnosis of Type VIII OI.
A multidisciplinary approach in management is important to maximise the long-term survival of the patient.
Acknowledgments
We would like to thank the parents and also the doctors and nurses in the Neonatal Unit and General Pediatrics. We are also grateful to CENTOGENE AG (Rostock, Germany; www.centogene.com) for the free genetic testing and report.
Footnotes
Contributors: The following authors were responsible for drafting of the text, sourcing and editing of clinical images, investigation results, drawing original diagrams and algorithms, and critical revision for important intellectual content. HM: Refined the manuscript, added references and reviewed the final manuscript; also managed the child in the ward, and provided all the clinical care required. KM: reviewed the manuscript, provided input and approved the final manuscript. FM: Reviewed the manuscript and provided technical assistance. KPM: Initiated the first draft, supervised the subsequent drafts, provided technical assistance, responded to reviewers' comments and reviewed the final manuscript. HM, KM, FM and KPM: All the authors gave final approval of the manuscript.
Funding: This case report has not been funded by any institution or organisation and its an efforts collectively done by the authors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Consent obtained from parent(s)/guardian(s)
References
- 1.Cabral WA, Chang W, Barnes AM, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007;39:359–65. 10.1038/ng1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monti E, Mottes M, Fraschini P, et al. Current and emerging treatments for the management of osteogenesis imperfecta. Ther Clin Risk Manag 2010;6:367–81. 10.2147/tcrm.s5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro JR, Sponsellor PD. Osteogenesis imperfecta: questions and answers. Curr Opin Pediatr 2009;21:709–16. 10.1097/MOP.0b013e328332c68f [DOI] [PubMed] [Google Scholar]
- 4.de Souza LT, Nunes RR, de Azevedo Magalhães O, et al. A new case of osteogenesis imperfecta type VIII and retinal detachment. Am J Med Genet A 2021;185:238–41. 10.1002/ajmg.a.61934 [DOI] [PubMed] [Google Scholar]
- 5.Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 2008;10:294–300. 10.1097/GIM.0b013e31816b5cae [DOI] [PubMed] [Google Scholar]
- 6.Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature 2015;526:68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabral WA, Barnes AM, Adeyemo A, et al. A founder mutation in LEPRE1 carried by 1.5% West Africans and 0.4% African Americans causes lethal recessive osteogenesis imperfecta. Genet Med 2012;14:543–51. 10.1038/gim.2011.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kantaputra PN, Angkurawaranon S, Intachai W, et al. A founder intronic variant in P3H1 likely results in aberrant splicing and protein truncation in patients of Karen descent with osteogenesis imperfecta type VIII. Genes (Basel) 2023;14:322. 10.3390/genes14020322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fratzl-Zelman N, Barnes AM, Weis M, et al. Non-lethal type VIII osteogenesis imperfecta has elevated bone matrix mineralization. J Clin Endocrinol Metab 2016;101:3516–25. 10.1210/jc.2016-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vranka JA, Sakai LY, Bächinger HP. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J Biol Chem 2004;279:23615–21. 10.1074/jbc.M312807200 [DOI] [PubMed] [Google Scholar]
- 11.Forlino A, Cabral WA, Barnes AM, et al. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011;7:540–57. 10.1038/nrendo.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]